

Abstract
Ataxin-3 is a deubiquitinating enzyme and the affected protein in the neurodegenerative disorder Machado–Joseph disease (MJD). The ATXN3 gene is alternatively spliced, resulting in protein isoforms that differ in the number of ubiquitin-interacting motifs. Additionally, nonsynonymous SNPs in ATXN3 cause amino acid changes in ataxin-3, and one of these polymorphisms introduces a premature stop codon in one isoform. Here, we examined the effects of different ataxin-3 isoforms and of the premature stop codon on ataxin-3's physiological function and on main disease mechanisms. At the physiological level, we show that alternative splicing and the premature stop codon alter ataxin-3 stability and that ataxin-3 isoforms differ in their enzymatic deubiquitination activity, subcellular distribution, and interaction with other proteins. At the pathological level, we found that the expansion of the polyglutamine repeat leads to a stabilization of ataxin-3 and that ataxin-3 isoforms differ in their aggregation properties. Interestingly, we observed a functional interaction between normal and polyglutamine-expanded ATXN3 allelic variants. We found that interactions between different ATXN3 allelic variants modify the physiological and pathophysiological properties of ataxin-3. Our findings indicate that alternative splicing and interactions between different ataxin-3 isoforms affect not only major aspects of ataxin-3 function but also MJD pathogenesis. Our results stress the importance of considering isoforms of disease-causing proteins and their interplay with the normal allelic variant as disease modifiers in MJD and autosomal-dominantly inherited diseases in general.
Keywords: alternative splicing, deubiquitylation (deubiquitination), E3 ubiquitin ligase, enzyme degradation, enzyme kinetics, genetic polymorphism, polyglutamine disease, protein aggregation, proteomics, neurodegenerative disease
Introduction
Ataxin-3 (UniProt ID P54252) is a 40–43-kDa protein (1) that is ubiquitously expressed in many different cell types of peripheral and neuronal tissues (2–6). It belongs to the family of cysteine proteases exhibiting a deubiquitinase activity (7, 8). Ataxin-3 consists of a globular N terminus with the so-called Josephin domain, followed by ubiquitin-interacting motifs (UIMs),2 a polyglutamine (polyQ) repeat, and a more flexible C terminus. Moreover, ataxin-3 harbors two nuclear export signals and one nuclear localization signal (9). Ataxin-3 may not be an essential protein, but it is evolutionarily conserved (10, 11), and the exact physiological function is still not completely understood. Ataxin-3 takes part in different cellular pathways and seems to play an important role in the ubiquitin proteasome system (11). However, it is also known to regulate the expression of various genes (12). Moreover, ataxin-3 was found to interact with other ataxin-3 molecules (13). The ataxin-3–encoding gene ATXN3 (HUGO Gene Nomenclature Committee number 7106) consists of 11 exons (14) and is subjected to alternative splicing. Different protein isoforms of ataxin-3 were identified, which mainly differ in their C-terminal sequence (15–17). Alternative splicing creates two different isoforms, which are called ataxin-3a and ataxin-3c (Fig. 1). The third isoform is created by a stop SNP located in exon 10 (the C-terminal part of ataxin-3a), leading to a premature stop codon variant and therefore creating two isoforms: ataxin-3aL (long) and ataxin-3aS (short). The C terminus of ataxin-3aL and -3aS is hydrophobic, whereas the C terminus of ataxin-3c is hydrophilic and contains a third UIM (16). Ataxin-3c seems to be the predominant protein isoform in human and murine brain tissue, but all isoforms were reported to be expressed (19). Interestingly, the expanded CAG repeat in ATXN3 seems to be associated with an increased generation of the ataxin-3a transcript (20).
Figure 1.

The ATXN3/MJD1 gene consists of 11 exons with an alternative splice site in exon 10. Two full-length isoforms were described for ataxin-3 called ataxin-3a (3a, UniProt ID P54252-1) and ataxin-3c (3c, UniProt ID P54252-2), which differ in the third UIM at the C terminus of ataxin-3c encoded by exon 11. ATXN3 is modified by different SNPs; rs1048755 (purple) in exon 8 leads to the missense mutation p.Val212Met N-terminal of UIM1. The SNP rs12895357 (blue) in exon 10 leads to the exchange of p.Gly306Arg directly C-terminal of the polyQ repeat. Last, rs7158733 (ochre) 3′ of the CAG-repeat in exon 10 leads to the nonsense variant p.Tyr349*, which causes a premature stop codon in this isoform and therefore generates two isoforms: ataxin-3a long (3aL, Tyr variant) and ataxin-3a short (3aS, premature stop variant) lacking 16 amino acids at the C terminus. Therefore, three isoforms of ataxin-3 exist, which differ in their C-terminal amino acid sequence and are created by alternative splicing and the stop codon.
A toxic gain-of-function of ataxin-3 (21) leads to a severely progressing neurodegenerative disease called Machado–Joseph disease (MJD (MIM catalog no. 109150), also known as SCA3) caused by a pathological expansion of the CAG repeat in the ATXN3 gene, which is translated into a polyQ repeat within ataxin-3. Normal individuals have 12–44 glutamine repeats, whereas MJD patients have expanded repeats between 61 and 87 glutamines (22, 23). It is known that the majority of Machado–Joseph disease (MJD) patients have a SNP combination that leads to the premature stop codon in ataxin-3a, whereas this variant is uncommon in controls (18). One hallmark of MJD is the formation of macromolecular aggregates containing the pathological expanded ataxin-3 (4, 24). Further, it is known that ataxin-3 is cleaved by calpains and caspases (25–27). Cleavage is required for aggregate formation in vitro and in vivo and may be part of the pathogenesis according to the toxic fragment hypothesis (28). Moreover, the nuclear localization of ataxin-3 turned out to be critical for the manifestation of MJD (29, 30). The exact pathogenic mechanisms of MJD, however, are still not understood in depth, but it is known that the expanded polyQ repeat induces dysfunctions in the cell via different mechanisms. Well-established is an inverse correlation between the age at onset and the number of CAG repeats in ATXN3 (31–33). The expanded CAG repeat size only explains about 50% of the variability of the age at onset (34, 35); the remaining part is thought to be modified by environmental factors (36) as well as further genetic factors (37). In recent years, the normal allele has also been discussed to have some neuroprotective function within the cell (38–42).
In this study, we analyzed the effect of alternative splicing of ATXN3 and the effect of this premature stop codon on physiological and pathophysiological characteristics of ataxin-3 as well as the mutual interaction of ATXN3 allelic variants.
Results
The aim of this study was to show the contribution of different ataxin-3 isoforms to physiological function and to disease modification of MJD. Alternative splicing and multiple isoforms have been recently described for ATXN3 (17, 43); however, only three full-length isoforms exist, which differ in their C termini (16) (Fig. 1). The C terminus of ataxin-3c contains a third UIM, whereas the C terminus of ataxin-3a is more unstructured and highly hydrophobic (16). Exon 10 of the ATXN3 gene contains a SNP that leads to a nonsense mutation creating a premature stop codon and thereby a shorter variant of ataxin-3a. Therefore, we termed these two isoforms ataxin-3aL (long) and ataxin-3aS (short). The SNP leading to a shorter isoform is highly associated with MJD and the expanded allele in MJD (44). Here we studied the physiological and pathophysiological characteristics of the different ataxin-3 isoforms.
To ensure that our analysis of ataxin-3 isoforms is not influenced by endogenously expressed ataxin-3, we generated ATXN3 KO cells. Transcription activator-like effector nucleases (TALENs) targeting exon 2 of ATXN3 were employed in HEK 293T cells followed by an antibiotic selection. Sequencing confirmed frameshift mutations at the expected location (data not shown). We further confirmed that ataxin-3 is no longer expressed in the KO cells by Western blot analysis using two different antibodies flanking the genomic frameshift region (Fig. 2A). All experiments described below were performed in this cell line if not stated differently.
Figure 2.

A, HEK 293T ATXN3 KO cells were generated using TALENs targeting exon 2 of the ATXN3 gene. The successful KO of ataxin-3 was validated on the protein level by Western blotting using two different ataxin-3 antibodies flanking the genomic frameshift region (N′, ARP50507; 1H9, MAB5360). Whereas the original HEK 293T cells (WT) express ataxin-3, it was no longer detected in the ATXN3 KO cells. B and C, Western blot analysis of ataxin-3 isoforms. ATXN3 KO cells were transfected with untagged pTRE ataxin-3 isoforms for 48 h (18Q (black arrowhead)/73Q (red arrowhead)). Immunodetection revealed that normal ataxin-3aS shows a lower protein level than ataxin-3c and ataxin-3aL. Upon a pathological polyQ expansion, ataxin-3aS and ataxin-3aL show weaker signals than ataxin-3c. The polyQ expansion of ataxin-3aL leads to a reduction of its protein level. Scheirer–Ray–Hare test with Conover–Iman and Wilcoxon–Mann–Whitney post hoc tests, adjusted by the Hommel method, were used: *, p < 0.05; **, p < 0.01, n = 5. D and E, the stability of ataxin-3 isoforms was analyzed using the Tet-off system. The expression of ataxin-3 (18Q/73Q) in transfected HEK 293T ATXN3 KO cells was abolished using doxycycline at the indicated time points. F, ataxin-3aS shows a significantly lower half-life than ataxin-3c or ataxin-3aL independent of the polyQ expansion. A polyQ expansion leads to an increase in half-life of all ataxin-3 isoforms. The half-life was calculated for first-order kinetics. One-way ANOVA with Tukey's HSD test was used: ***, p < 0.001, n = 5–9. G, analysis of ATXN3 mRNA stability. Quantitative RT-PCR was performed after expression of pTRE ataxin-3 isoforms (18Q/73Q) in HEK 293 ATXN3 KO cells. No differences in the stability of the mRNA were found 8 h after expression termination by doxycycline (n = 3). The Scheirer–Ray–Hare test was used: isoform, polyQ, and interaction insignificant, n = 3. H, analysis of ataxin-3 solubility. Untagged ataxin-3 isoforms (18Q/73Q) were expressed for 24 h, and expression was afterwards terminated by doxycycline for 32 h. A filter retardation assay for insoluble protein showed that ataxin-3 isoforms do not differ in solubility after 24 h of expression. PC, positive control. I, differences in the stability of non-polyQ–expanded ataxin-3 isoforms arise from different degradation pathways. pTRE-ataxin-3–transfected cells were cultured for 24 h. Expression was terminated 8 h before cells were treated with bafilomycin A1 (50 nm) or lactacystin (10 μm) for 24 h. Western blots were stained for ataxin-3 (1H9), GAPDH, Lys-48–linked ubiquitin (K48), and LC3 (LC3-II). The Lys-48 staining confirmed the inhibition of the proteasome system, whereas the increase in LC3-II confirmed the autophagy inhibition. J, an inhibition of autophagy by a bafilomycin A1 treatment led to a preservation of ataxin-3 for all isoforms. Ataxin-3aS degradation was additionally inhibited after the lactacystin treatment, indicating that this isoform is additionally degraded by the proteasome (one-tailed Wilcoxon signed rank test, Hommel-corrected; *, p < 0.05; n = 6–12). Data are represented as arithmetic mean ± S.E. (error bars).
Physiological function of ataxin-3
In a first step, we investigated differences between isoforms of ataxin-3 at the physiological level and how this is modified by polymorphisms. We therefore expressed non-polyQ–expanded as well as polyQ-expanded isoforms and assessed the protein level using Western blotting (Fig. 2, B and C). For nonexpanded ataxin-3, we found that ataxin-3c and ataxin-3aL show a higher protein level than ataxin-3aS. For expanded ataxin-3, however, the protein level of ataxin-3c was higher than the protein level of both ataxin-3a isoforms. In a next step, we wanted to know whether the stability of the different ataxin-3 isoforms could explain the observed differences in the protein level. To assess the half-life, we employed the Tet-off system (45) and turned off protein expression of ataxin-3 at different time points. Western blotting revealed that ataxin-3c and ataxin-3aL show a similar degradation rate over time, whereas ataxin-3aS is degraded faster independent of the polyglutamine repeat length (Fig. 2, D and E). Ataxin-3aS has a significantly shorter half-life (12 h) than ataxin-3c with 24 h and ataxin-3aL with 26 h, respectively (Fig. 2F), suggesting that the stop codon subjects the protein to fast degradation. Interestingly, we observed an isoform-independent increase in the half-life upon a pathologic expansion of the polyglutamine repeat. Expanded ataxin-3aS, however, is still less stable than expanded ataxin-3c and ataxin-3aL. The observed differences in ataxin-3 half-life could be a result of differences in mRNA stability or an increased insolubility after expression of ataxin-3 isoforms, which leads to a reduction of soluble ataxin-3 levels quantified by Western blotting. We therefore excluded the latter two possibilities; mRNA stability was not significantly different between the isoforms (Fig. 2G), and no insoluble ataxin-3 could be detected after termination of ataxin-3 expression (Fig. 2H).
Ataxin-3aS is therefore probably subjected to a faster degradation. For this reason, we next investigated whether ataxin-3 isoforms are degraded by different pathways. We inhibited either autophagy (bafilomycin A1) or the proteasome system (lactacystin) and observed that indeed the different ataxin-3 isoforms are degraded by different mechanisms (Fig. 2, I and J). We found that ataxin-3c and ataxin-3aL are degraded via autophagy, whereas ataxin-3aS is degraded by both autophagy and the proteasomal pathway. This suggests that the premature stop codon in ataxin-3aS directs the protein to proteasomal degradation, which well explains the observed shorter half-life.
Ataxin-3 is known to be present in different compartments of the cell (3, 46). As the localization of a protein depends on protein-intrinsic factors, such as localization signals, as well as its cellular context, we wanted to know whether ataxin-3 isoforms show differences in their localization as they may carry out different functions in the cell. Therefore, we performed a subcellular fractionation assay separating proteins into a cytoplasmic and nuclear fraction and found that full-length ataxin-3aS was enriched in the nuclear fraction compared with ataxin-3c and -3aL and that this nuclear localization was independent of the polyQ repeat (Fig. 3, A and B). Even more interesting, a fragment of ataxin-3, which was recently described to cause mitochondrial dysfunction (47), was highly enriched in the nucleus as well (Fig. 3C). This is especially important, as we previously observed that ataxin-3's toxicity is linked to its nuclear localization (29, 30).
Figure 3.
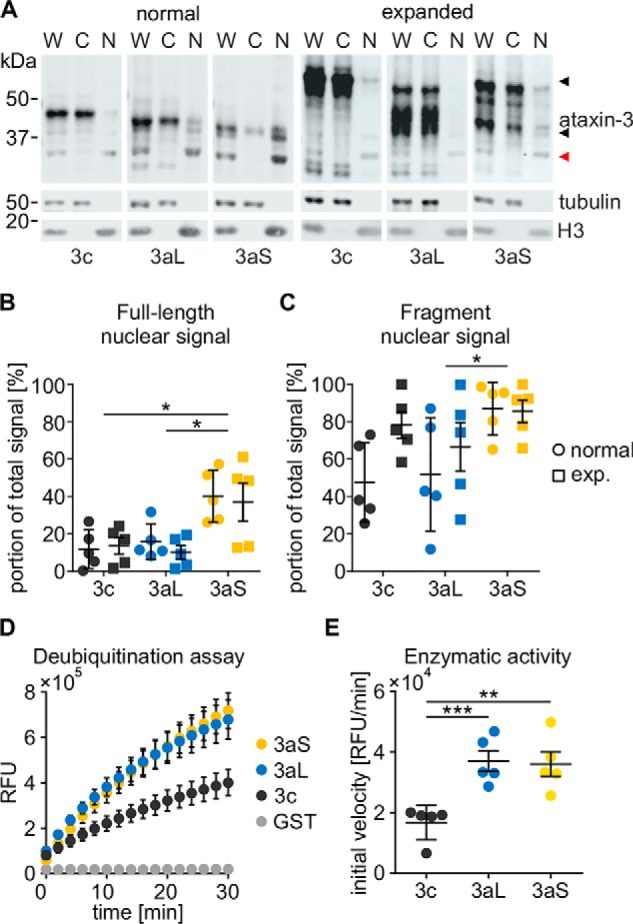
A, subcellular distribution of ataxin-3 isoforms (18Q/73Q). ATXN3 KO cells were transfected with untagged pTRE-ataxin-3 isoforms and the RCA2 promoter construct. After 48 h, cells were harvested and fractionated, generating whole-cell (W), cytoplasmic (C), and nuclear (N) fractions. Ataxin-3 was detected using the antibody 1H9. Tubulin was detected as a cytoplasmic marker, and H3 was detected as a nuclear marker. Full-length ataxin-3 (black arrowhead) as well as ataxin-3 fragments (red arrowhead) could be detected. B and C, quantification of nuclear signals for full-length ataxin-3 isoforms and an N-terminal fragment. Full-length ataxin-3 shows an increased nuclear localization independent of the polyQ expansion (two-way ANOVA; *, p < 0.05). The fragment of ataxin-3aS also shows a stronger nuclear localization than that of ataxin-3aL (two-way ANOVA with Tukey's HSD test; *, p < 0.05, n = 5). D, deubiquitination assay of non-polyQ–expanded ataxin-3 isoforms. GST-ataxin-3 was purified from E. coli and mixed in a 1:5 ratio with ubiquitin-rhodamine-110. Relative fluorescence was measured every 10 s and is displayed for every 2 min. E, calculation of the initial velocity revealed that ataxin-3c shows a significantly reduced activity compared with ataxin-3aL and ataxin-3aS (Kruskal–Wallis test with Conover–Iman post hoc test with Hommel adjustment; **, p < 0.01; ***, p < 0.001; n = 5). Data are represented as arithmetic mean ± S.E. (error bars). RFU, relative fluorescence units.
Ataxin-3 is a deubiquitinating enzyme (7, 8), and it is known that the polyQ expansion close to the C terminus leads to an inefficient deubiquitination (DUB) of cellular proteins (48). Alternative splicing modifies the C terminus as well and even leads to the presence or absence of an additional UIM. We therefore quantified the enzymatic activity of ataxin-3 isoforms using a ubiquitin-rhodamine-110–based deubiquitination assay. To exclude any influence of other proteins, we purified ataxin-3 using a GST tag. Purified ataxin-3 was quantified, and equal molarities were subjected to the DUB assay. We observed that all ataxin-3 isoforms were able to cleave ubiquitin-rhodamine-110 (Fig. 3D). However, the enzymatic activity of ataxin-3c was significantly lower compared with the activity of both ataxin-3aL and -3aS (Fig. 3E).
One major part of a protein's function is characterized by its interactions with other proteins. Multiple interactors are described for ataxin-3 (reviewed Ref. 1), but little is known about the impact of the isoform of ataxin-3 on these interactions. We thus analyzed whether alternative splicing as well as the stop codon have an influence on the protein–protein interaction network of ataxin-3. We assessed the interactions in a high-throughput manner using a stable isotope labeling of amino acids in cell culture (SILAC) approach. After purification of ataxin-3, we performed an MS identification of co-precipitated proteins. From 298 identified interaction partners in our analysis, 32 were already listed in the BioGRID database (49). Common interactors of all isoforms were identified, including the well-described interaction partner VCP (50). Interestingly, also characteristic, isoform-specific interactions were found (Fig. 4A). We validated the MS data by co-immunoprecipitation of the well-known ataxin-3 interaction partners HR23A, HR23B, and VCP. Additionally, UBR2, tubulin, and caspase-7 were used among the newly identified interaction partners (Fig. 5, A–F). Although many common interactors were identified, multiple proteins interact more strongly with one specific isoform. For instance, caspase-7 was identified as a novel interactor strongly interacting with ataxin-3aS. Furthermore, we found HR23B to interact more strongly with ataxin-3c and ataxin-3aL, but more weakly with ataxin-3aS.
Figure 4.

A, volcano plots comparing the interaction of normal ataxin-3 isoforms. HEK 293T cells were transfected with pN-SF-TAP-ataxin-3 isoforms and grown in SILAC medium for 72 h. Ataxin-3 was purified by the Strep-tag. Isoform-specific purifications were combined afterward and concentrated by precipitation before the identification of co-precipitated proteins by MS. Volcano plots show a direct comparison between the p value of interactors of two isoforms (ataxin-3c versus 3aL, ataxin-3c versus 3aS, and ataxin-3aL versus 3aS) and the difference between the isoforms. The plots are divided into four quadrants (small scheme). The bottom two show interactors with insignificant differences (light gray open circle, p > 0.05); the top ones show interactors that interact significantly more strongly with one isoform, depending on the side. Although significant, interactors with p < 0.05 were only considered stronger or weaker upon a difference of at least ±1. Although sharing numerous interactions (light gray open and filled circles), ataxin-3 isoforms show divergent interactions with different proteins. Partners interacting more strongly with ataxin-3c (black filled circles), ataxin-3aL (blue filled circles), and ataxin-3aS (yellow filled circles) could be identified. Ataxin-3c and ataxin-3a show more differences in their interaction than ataxin-3aL and ataxin-3aS. B, Venn diagram comparing proteins that interact more strongly with ataxin-3aL and ataxin-3aS compared with ataxin-3c. 65 interaction partners interact more strongly with ataxin-3aS than ataxin-3c. 38 partners have a stronger interaction with both ataxin-3aS and ataxin-3aL, whereas seven partners have a stronger interaction with ataxin-3aL. A GO annotation of these proteins and analysis of KEGG pathways revealed that these interactors take part in different pathways, whereas proteins associated with Huntington's disease and Parkinson's disease can be found in all three groups. C, interaction network for proteins showing a stronger binding to ataxin-3c. Ataxin-3c shows a common interaction with HR23B and NGLY1 as well as UBR2 and HNRNPL, specifying its role in the ERAD pathway of glycosylated proteins.
Figure 5.

A–F, results from SILAC-MS-MS were validated by performing pulldown assays for selected interaction partners. HEK 293T ATXN3 KO cells were transfected with non-polyQ–expanded pEGFP-C2-ataxin-3 isoforms. In the case of HR23A, HR23B (both V5)- and mUBR2 (FLAG)-tagged constructs with the interaction partner were co-expressed. Cells were harvested 48 h post-transfection, and samples were processed for a GFP-trap interaction assay followed by Western blot analysis. Input as well as immunoprecipitation (IP) was loaded onto the same gel. An interaction could be confirmed for all tested interaction partners. A, VCP; B, HR23A (V5 antibody); C, HR23B (V5 antibody); D, α-tubulin; E, caspase-7; F, mUBR2 (FLAG-antibody). The blot of the top panel of D was split into two different intensities (dashed line) to show input and IP. This panel also shows a redetection of tubulin in the GFP detection. G and H, GFP-trap interaction assay for ataxin-3 isoforms and high-molecular weight ubiquitinated proteins. Western blotting signal was normalized to ataxin-3c. The GFP-trap assay for ubiquitinated proteins shows a stronger interaction of high-molecular weight ubiquitin for ataxin-3aL and ataxin-3aS compared with ataxin-3c, indicating that ataxin-3c is binding lower amounts of ubiquitinated proteins (Wilcoxon signed rank test, Hommel-adjusted; *, p < 0.05, n = 9). I and J, GFP-trap interaction assay for ataxin-3 isoforms and parkin. Western blotting signal was normalized to ataxin-3c. Ataxin-3aL and ataxin-3aS interact significantly more strongly with parkin than ataxin-3c (one-sample t test, Hommel-adjusted; *, p < 0.05, n = 5). Data are represented as arithmetic mean ± S.E. (error bars).
To identify pathways that ataxin-3 isoforms are part of, we performed a gene ontology (GO) KEGG pathway analysis for annotated proteins using the DAVID Bioinformatics Resources version 6.8 (51). GO revealed that proteins that interact more strongly with ataxin-3aL and -3aS were generally associated with the Huntington's disease, Parkinson's disease, spliceosome, and oxidative phosphorylation pathway (Fig. 4B), indicating a role of ataxin-3a within this mechanism. Table 1 summarizes the results with the respective interaction partners. We moreover identified NGLY1 as a new interactor of ataxin-3c, binding more weakly with the ataxin-3a isoforms. Interestingly, four of six proteins interacting more strongly with ataxin-3c are associated with the ERAD pathway (52–55), either by directly being part of it (HR23B and NGLY1) or by interacting with ERAD proteins (UBR2 and HNRNPL), suggesting a role of isoform ataxin-3c within this pathway (Fig. 4C).
Table 1.
KEGG pathways and interactors of ataxin-3aL and -3aS
A KEGG pathway analysis was performed for protein interactors showing a stronger interaction with ataxin-3aL and -3aS compared with ataxin-3c. Annotated proteins as well as their respective KEGG pathway are listed.
| KEGG pathway | Interactors |
||
|---|---|---|---|
| 3aL > 3c | 3aS > 3c | 3aL and 3aS > 3c | |
| Adipocytokine-signaling pathway | IRS4 | ||
| Apoptosis | CASP7 | AIFM1 | |
| Calcium-signaling pathway | SLC25A5, SLC25A6 | ||
| Gap junction | TUBA1C, TUBB, TUBB8, | ||
| Glycolysis/gluconeogenesis | DLD, LDHB | ||
| Huntington's disease | CYC1 | ATP5A1, ATP5B, CLTC | NDUFA4, SLC25A5, SLC25A6 |
| Insulin signaling pathway | IRS4 | ACACA | |
| MAPK signaling pathway | HSPB1 | ||
| Neurotrophin-signaling pathway | IRS4, YWHAE | ||
| Nucleotide excision repair | RPA3 | ||
| Oxidative phosphorylation | CYC1 | ATP5A1, ATP5B | NDUFA4 |
| Parkinson's disease | CYC1 | ATP5A1, ATP5B | NDUFA4, SLC25A5, SLC25A6 |
| Proteasome | PSMA1, PSMA2, PSMB6 | ||
| Spliceosome | HNRNPU, NRPD2, RBM25, SNRPD3 | PCBP1 | |
| Valine | DLD | DBT | |
Due to the differences in the DUB activity and the known difference in the number of UIMs, we also wanted to know whether ataxin-3 isoforms have a differing affinity against ubiquitin-conjugated proteins. We found that ataxin-3c precipitated lower amounts of high-molecular weight ubiquitinated proteins, as assessed by GFP-trap (Fig. 5, G and H).
As we could not identify the well-known interaction partner parkin (56) by the MS-MS approach, possibly due to the low expression level of parkin in HEK 293T cells, we performed a GFP-trap to study its interaction with ataxin-3 isoforms. Western blotting showed that all ataxin-3 isoforms interact with parkin (Fig. 5I). However, ataxin-3c shows a weaker interaction (Fig. 5J), indicating that the C terminus of ataxin-3 is involved in protein binding. Altogether, our interaction analysis demonstrated that ataxin-3 isoforms differ in their interaction networks. A summary table with identified interaction partners can be found in Table S1.
Pathological functions of ataxin-3 isoforms
After observing major differences between ataxin-3 isoforms at the physiological level, we next wanted to assess whether ataxin-3 isoforms differ in pathological mechanisms as well. As protein aggregates are a hallmark of MJD and other polyQ diseases, we analyzed the aggregation kinetics of ataxin-3 isoforms. We found that all ataxin-3 isoforms with a pathologically expanded polyQ repeat form aggregates within 72 h of expression (Fig. 6A). Counting the number of transfected cells with aggregates showed that isoforms ataxin-3aL and ataxin-3aS form significantly more aggregates than isoform ataxin-3c after 48 h (Fig. 6B). We conclude that ataxin-3c needs more time to form visible aggregates, but as soon as seeds are formed, it aggregates quickly. Moreover, we found that ataxin-3aL usually forms multiple aggregates per cell, whereas ataxin-3c and ataxin-3aS tend to form fewer but larger aggregates (Fig. 6C).
Figure 6.

A, microscopic analysis of ataxin-3 aggregation. HEK 293T ATXN3 KO cells were transfected with pEGFP-C2-ataxin-3 isoforms with either normal (18Q) or expanded (151Q) polyQ. Cells were fixed at the respective time points, and the relative number of cells with aggregates was counted. Representative pictures are shown; aggregates are marked with arrowheads; bar, 100 μm. B, quantification of number of cells with aggregates. First differences could be observed 48 h post-transfection. Ataxin-3c with 151Q shows fewer cells with aggregates than both 3a isoforms. The expanded 3a isoform shows significantly more aggregates than the nonexpanded one. After 72 h, all expanded isoforms show a stronger aggregation than the nonexpanded ones (β-regression with estimated marginal means contrasts for each time point, n = 5; *, p < 0.05; **, p < 0.01; ***, p < 0.001). C, the number of aggregates per cell was counted and compared between 151Q ataxin-3 isoforms. Ataxin-3aL shows fewer cells with one aggregate, whereas the number of cells with more than five aggregates is increased (two-way ANOVA with estimated marginal means contrasts; *, p < 0.05; **, p < 0.01; ***, p < 0.001, n = 5). D, analysis of the aggregate size. The aggregate size distribution of ataxin-3c differs from that of 3aL and 3aS 48 h post-transfection. 72 h post-transfection, the size distributions of ataxin-3aS and -3c aggregates differ from 3aL. At both time points, ataxin-3aL produces more small aggregates than ataxin-3c and ataxin-3aS (Kolmogorov–Smirnoff test with Hommel adjustment for multiple comparison; *, p < 0.05; **, p < 0.01; ***, p < 0.001, n = 5). Quartiles are indicated as solid (50%) and dashed lines (25 and 75%). E, comparing the median aggregate sizes of five different experiments, it could be found that ataxin-3aL aggregates are smaller than aggregates from ataxin-3c and ataxin-3aS both at 48 and 72 h. At 48 h, ataxin-3aS aggregates were smaller than aggregates formed by ataxin-3c (two-way ANOVA with Tukey's HSD test; *, p < 0.05; **, p < 0.01; ***, p < 0.001, n = 5). F, solubility analysis of ataxin-3 isoforms. EGFP-C2-ataxin-3 isoforms were expressed for 72 h. Samples were then fractionated into soluble, SDS-soluble, and SDS-insoluble fractions. 18Q and 73Q ataxin-3 were only present in the soluble and SDS-soluble fractions, whereas 151Q ataxin-3 also showed full-length signals in the insoluble fraction. Fragments of all three polyQ expansions were mainly detectable in the SDS-soluble and SDS-insoluble fractions. For technical reasons, it was not possible to specify a loading control in the insoluble fractions. However, we expect that equal protein amounts were loaded onto the gel, as the soluble fraction shows a homogeneous loading. G and H, filter retardation assay of pEGFP-C2-ataxin-3 isoforms 72 h post-transfection. Equal amounts were loaded onto the membrane. 151Q ataxin-3 isoforms show a strong formation of SDS-insoluble aggregates. Quantification revealed lower amounts of aggregates for 151Q ataxin-3aL than for ataxin-3c and ataxin-3aS (one-sample t test (###, p < 0.001) and two-sample t test (*, p < 0.05); p values were Hommel-corrected for multiple comparisons, n = 7–8). Nonexpanded as well as expanded ataxin-3 show lower amounts of aggregates compared with highly expanded ataxin-3 of the same isoform (one-sample t test (###, p < 0.001; p values were Hommel-corrected) and one-way ANOVA with Tukey's HSD test (**, p < 0.01; ***, p < 0.001, n = 6–8)). Data are represented as arithmetic mean ± S.E. (error bars).
Microscopic inspection already revealed that the aggregates formed by the different ataxin-3 isoforms have a different appearance. Therefore, we further characterized the size of aggregates by measuring their area on microscopic images. We found that aggregates formed by ataxin-3 isoforms differ in size. All isoforms formed aggregates of a variety of sizes; however, ataxin-3aL tended to form more and smaller aggregates than the other isoforms, although large aggregates can be found as well (Fig. 6D). This suggests that the stop polymorphism changes the size of aggregates. Both, 48 and 72 h post-transfection, the median aggregate size of ataxin-3aL was smaller compared with ataxin-3c and -3aS (Fig. 6E).
In the next step, we analyzed whether alternative splicing or the stop codon has an influence on the general solubility of ataxin-3. Therefore, we employed solubility fractionation as described by Koch and colleagues (57), separating homogenates into a Triton X-100–soluble, SDS-soluble, and SDS-insoluble fraction. We found that all ataxin-3 isoforms were soluble but also showed SDS-soluble and SDS-insoluble fractions (Fig. 6F). Ataxin-3c was more Triton X-100– and SDS–soluble than ataxin-3aL and -3aS independent of the polyQ expansion. Whereas normal 18Q ataxin-3 and expanded 73Q ataxin-3 isoforms did not differ in their SDS insolubility, highly expanded 151Q ataxin-3 isoforms showed differences in the amount of SDS-insoluble aggregates. Highly expanded ataxin-3aL formed lower amounts of SDS-insoluble aggregates. We conclude that the premature stop codon in ataxin-3aS leads to an increased insolubility of ataxin-3a. Interestingly, proteolytic fragmentation of ataxin-3 also reduced the solubility of ataxin-3 independent of the polyQ expansion and isoform. In general, ataxin-3c showed a higher protein level than ataxin-3aL and -3aS. Despite its lower protein level, ataxin-3aS shows equal amounts of SDS-insoluble aggregates. This shows clearly that this isoform has a stronger tendency to form aggregates.
We further confirmed the differences in insolubility between the isoforms by filter retardation assays (Fig. 6G). Ataxin-3 with 151Q formed SDS-insoluble aggregates, whereas 18Q and 73Q ataxin-3 isoforms were more soluble. Comparison of the 151Q isoforms showed that the formation of SDS-insoluble aggregates is significantly reduced for ataxin-3aL compared with ataxin-3c and ataxin-3aS (Fig. 6H).
Mutual influence of ataxin-3 isoforms
For the characterizations described above, we focused on the individual ataxin-3 isoforms. In patients, however, two alleles (i.e. both a normal and a CAG-repeat expanded allele) are present. We therefore asked the question whether the two allelic variants mutually influence each other, as they are known to be able to interact (13). We were especially interested in knowing whether this mutual interaction depends on the isoforms of ataxin-3. To test whether simultaneous expression has an effect on ataxin-3 stability, we performed co-transfections of ataxin-3 isoforms. In these experiments, the expression of one isoform could be regulated using the Tet-off system, whereas the other isoform was constitutively expressed. For technical reasons, two different backbone vectors were used. However, the results of the pN-SF-TAP vector were confirmed using the pcDNA3.1-FLAG-ataxin-3-V5 vector as well (Fig. S1). We focused on those combinations that are likely to occur in patients based on the literature and our own data (18, 58). Analysis of ataxin-3 stability under the presence of a second ataxin-3 isoform showed that ataxin-3c is stabilized by the presence of normal ataxin-3a. Ataxin-3aS, on the other hand, is stabilized by normal and expanded ataxin-3c (Fig. 7, A and B). This shows that ataxin-3 isoforms can have an influence on each other's physiological characteristics.
Figure 7.

A, HEK 293T ATXN3 KO cells were transfected with nonexpanded 18Q pTRE-ataxin-3 isoforms (black arrowhead) as well as either a constitutively expressing empty vector or ataxin-3 isoform (red arrowhead; nonexpanded 18Q and expanded 65Q/73Q) in combinations that are present in MJD patients. Ataxin-3 expression was abolished using doxycycline 24 h post-transfection for 32 h. Samples were analyzed by Western blotting stained for ataxin-3 (1H9) and GAPDH. For technical reasons, ataxin-3c was co-expressed with a pN-SF-TAP empty vector and isoforms, whereas ataxin-3aL and -3aS were co-expressed with a pcDNA-FLAG-V5 vector. B, quantification of the relative stability under co-expression conditions. Relative ataxin-3 signals (black arrowhead) were quantified for either ataxin-3c, -3aL, or -3aS alone or under a co-expression with another isoform (red arrowhead). Ataxin-3c is slightly stabilized under co-expression of nonexpanded ataxin-3aL compared with an empty vector control. A similar effect could be observed for ataxin-3aS under co-expression with nonexpanded as well as expanded ataxin-3c (one-way ANOVA with Dunnett's test; *, p < 0.05, n = 5–8). The stability of isoform ataxin-3aL is not influenced by the presence of other isoforms. C and D, filter retardation assay of the co-expression of expanded pEGFP-C2-ataxin-3 isoforms. Isoforms were expressed with an empty EGFP vector or with another nonexpanded EGFP-C2-ataxin-3 isoform for 72 h. Samples were analyzed by a filter retardation assay and stained for ataxin-3 (1H9). A co-transfection with nonexpanded ataxin-3 reduces the amount of insoluble ataxin-3 aggregates independent of the expanded isoform as well as of the nonexpanded isoform (one-sample t test with Hommel correction; *, p < 0.05; **, p < 0.01; ***, p < 0.001, n = 6–7). Data are represented as arithmetic mean ± S.E. (error bars).
This raised the question of whether one can observe the same effect also on the important pathophysiological characteristic of aggregation. Indeed, we found that the presence of an unexpanded ataxin-3 allele independent of its isoform reduced the total number of SDS-insoluble aggregates (Fig. 7, C and D). This effect, however, was observed for all combinations of ataxin-3 isoforms, indicating a protective function of the nonexpanded ataxin-3 allele as already observed in a Drosophila model (38).
In summary, we found that ataxin-3 isoforms differ in their protein stability due to differences in their degradation pathway as well as their enzymatic activity. Ataxin-3 isoforms also show different subcellular localizations. All isoforms have their specific interaction network, whereas alternative splicing and the stop codon lead to an increase in or loss of the interaction, depending on the binding partner. Upon a pathological expansion of the polyQ stretch, stability and aggregation propensity of all respective isoforms increased, whereas differences were found in the amount of aggregates, their relative number per cell, and their size. Remarkably, we found a mutual interaction of ataxin-3 isoforms modifying physiological as well as pathophysiological characteristics of the protein. This clearly shows that ataxin-3 isoforms play a differential role in their physiological function in the cell and contribute differently to the pathology of MJD.
Discussion
Alternative splicing plays a crucial role in various neurodegenerative diseases (60), and dysfunction of alternative splicing is known to be one cause for frontotemporal dementia with parkinsonism-17 (61), spinal muscular dystrophy (62), and Alzheimer's disease (63, 64). However, alternative splicing can also act as a modifier of diseases, as previously reported for Parkinson's disease (65, 66), Huntington's disease (67), SCA1 (68), and Alzheimer's disease (69, 70). Alternative splicing of the ATXN3 gene was described only 3 years after its identification (15, 16). However, little is known about ATXN3 transcript diversity (14, 17) and the functional differences between ataxin-3 isoforms on the protein level (4, 19). At least three protein isoforms (16) are generated by alternative splicing (ataxin-3c and -3aL) plus a modification by a nonsense SNP in ATXN3 creating a short variant from ataxin-3aL termed 3aS. Although ataxin-3c seems to be the predominant isoform in murine and human brain (19), the presence of ataxin-3a has a great impact on MJD pathology as well. It was already proposed earlier that the C terminus may lead to functional differences of ataxin-3 isoforms (16). However, these differences, especially the influence of the stop codon, remain elusive. To close this knowledge gap, we analyzed the physiological characteristics of ataxin-3 isoforms as well as their potential significance for MJD pathology.
On a physiological level, we found that ataxin-3 protein isoforms differ in their protein level and stability. Ataxin-3c and -3aL showed comparable protein levels and a half-life that correlates well to the previously reported half-life of ataxin-3c (71). We further expanded and specified the results obtained by Harris and colleagues (19) by differentiating between ataxin-3aL and -3aS as well as the usage of the Tet-off system, which superseded the need of translational inhibition. We observed that the stop codon reduces the stability of the protein as well as the expression level. An expansion of the polyQ repeat within ataxin-3 increases the half-life significantly independent of the isoform. This polyQ-dependent increased half-life was also described previously for expanded ataxin-3 (50, 72), huntingtin (73), ataxin-1 (74), and TATA-box–binding protein (75, 76), showing that this effect is common to polyQ proteins. The observed stability differences between ataxin-3 isoforms are due to their protein degradation rates, as we could not find differences in mRNA stability or aggregation after termination of expression. Although it is known that functional UIMs of ataxin-3 increase the turnover rate of ataxin-3 (77), this obviously does not apply for an exchange of the third UIM by alternative splicing of the whole C terminus.
To analyze why the half-life of ataxin-3aS is reduced, we examined the degradation pathway of ataxin-3 isoforms by inhibition of either autophagy or proteasomal degradation. We found that ataxin-3 isoforms are degraded by autophagy but that the stop codon additionally directs ataxin-3aS to proteasomal degradation. Previous studies presented partially conflicting results for the stability and degradation pathway of ataxin-3 (19, 50, 77–79). These discrepancies may be explained by the isoform applied, the use of tags, and the localization within the protein as well as specific differences in the experiments like translational inhibition and inhibitor concentrations (80–88).
Ataxin-3 was reported to be located in different compartments of the cell (3, 4, 46). Moreover, the nucleus was previously reported to be an essential site for MJD pathology (29, 30). We therefore asked whether ataxin-3 isoforms differ in their subcellular localization. We found a strong localization of ataxin-3aS in the nucleus. Besides the strong nuclear localization of ataxin-3aS, we found a fragment of ataxin-3 to be enriched in the nuclear fractions for all isoforms as well. This is of special interest, as it was previously reported that cleavage of mutant ataxin-3 leads to a nuclear translocation of the polyQ-containing fragment (4, 9). At the same time, these fragments are considered to be more toxic and show an increased propensity to form aggregates compared with the full-length protein (2, 24, 26, 27, 47, 89, 90). Ataxin-3 is known to be a deubiquitinating enzyme (7, 8), and that cellular turnover of ataxin-3, as well as its steady-state level, is regulated by its catalytic activity (13). Therefore, we wanted to investigate whether the stop codon causes changes in ataxin-3's DUB activity and therefore explains its low stability and reduced steady-state level. We found that alternative splicing, but not the stop codon, leads to changes in the DUB activity of ataxin-3 isoforms. The use of a substrate excess in our experiments further specified the previously reported enzymatic activity of ataxin-3 isoforms (19).
The DUB activity of ataxin-3 is modified by ubiquitination of the protein (91). Furthermore, it is known that the catalytic activity influences the stability as well as the steady-state level of ataxin-3 (13). These results fit very well to our observations, as ataxin-3c showed an increased steady-state level.
Ataxin-3 is known to interact with various different proteins, reviewed by Costa and Paulson (1). However, nothing was known about the interaction of particular ataxin-3 isoforms with different proteins. We found that ataxin-3 isoforms share major parts of their interactomes; however, they also have individual protein partners, indicating that the isoforms have their own specific interaction network within the cell and take over functions in different pathways.
Aggregation of the respective polyQ protein is a hallmark of all polyQ diseases (24, 92–97). Interestingly, ataxin-3 isoforms differ in their aggregation behavior. Our results demonstrate that the C terminus is involved in the process of aggregate formation as proposed by Harris et al. (19). It thereby has a great impact not only on aggregation kinetics but also on the properties of aggregates formed.
It was previously reported that ataxin-3 can interact with itself (13). Due to the presence of different ataxin-3 isoforms in one cell, we were interested in understanding how this mutual influence changes physiological and pathophysiological characteristics of ataxin-3. In our study, we only analyzed those combinations that are likely to occur in MJD patients (18, 58). We found that the presence of another ataxin-3 protein isoform is able to change the stability of ataxin-3, depending on the specific isoform combination. However, this change in stability is not a consequence of deubiquitination of one ataxin-3 isoform by another, as it was reported that ataxin-3 is not deubiquitinating other ataxin-3 molecules (13).
Pathophysiologically, the presence of a normal ataxin-3 allele reduced the amount of aggregates formed by a polyQ-expanded ATXN3 allelic variant. These results fit well to a previous report that ataxin-3 is recruited to inclusions and reduces the accumulation of pathogenic protein aggregates (38). Indeed, this mutual interaction of ataxin-3 isoforms may play an important role as a modifier of pathology. The presence of a normal allele was reported to rescue the disease phenotype in Drosophila models of MJD (38–42) but had no effect in mice (98). Nevertheless, our in vitro data clearly show that ataxin-3 isoforms have an influence on each other. In summary, we found that ataxin-3 isoforms differ in physiological as well as pathophysiological characteristics (Fig. 8), creating protein species with specific functions and interactions that, in order to study protein function or MJD, cannot be exchanged by each other. This strongly substantiates the need of a precise annotation in research on neurodegenerative diseases not only of the protein isoform but also of the haplotype that is studied. Further, our results suggest that ataxin-3 isoforms differ in their contribution to MJD pathology. This helps in understanding the pathological differences between genetic subtypes that vary in age at onset and disease severity. Our data provide evidence that the underlying differences are mainly determined by the presence of the stop codon aggravating MJD pathology. Importantly, MJD patients differ in their isoform combination due to different genotypes of the respective polymorphism impacting both the normal and expanded allele of ataxin-3 and, as we demonstrated in this study, ataxin-3's physiological and pathophysiological characteristics. Our results highlight the importance of taking polymorphisms and isoforms of disease-causing proteins as well as mutual interactions with the respective normal protein allele as disease modifiers in MJD and other autosomal-dominantly inherited disorders into consideration.
Figure 8.

Ataxin-3 isoforms show differences on physiological as well as pathophysiological levels. We found that ataxin-3 isoforms have a different stability and degradation pathway as well as a differing enzymatic activity. Moreover, they show differences in their interaction networks. On the pathophysiological level, isoforms show differences in their aggregation kinetics, number of aggregates per cell, and aggregate size. The line type shows effect strength or amount, and the tachometer is an indicator of the kinetics (10 o'clock/green, slow; 12 o'clock/yellow, moderate; 2 o'clock/red, fast).
Experimental procedures
Expression constructs
Ataxin-3–expressing constructs were used with the following backbone vectors. Aggregate analysis was performed using the vector pEGFP-C2 (Takara Bio Europe, Saint-Germain-en-Laye, France), pGEX-6P-1 (GE Healthcare, Munich, Germany) was used to purify GST-ataxin-3, the vector pN-SF-TAP (99) was used for the SILAC experiments, and pcDNA3.1 (Thermo Fisher Scientific, Schwerte, Germany) was used for co-expression experiments. Expression of untagged ataxin-3 using the Tet-off system was performed from the pTRE vector together with pTET (Takara Bio Europe, Saint-Germain-en-Laye, France). Vectors for ataxin-3 interaction partners pcDNA3-V5-hHR23A and pcDNA3-V5-hHR23B were a gift from Steven Grossman (Addgene plasmid 13054) (100), and pcDNA3.1(−)-FLAG-UBR2 was a gift from Alexander Varshavsky (Addgene plasmid 50573). All ataxin-3 isoforms contained those SNP combinations that are highly common according to the literature (18, 58). For this reason, the normal allele was cloned with a haplotype G669G987 (Val212-Gly306), whereas the expanded allele contained the haplotype A669C987 (Met212-Arg306). Isoform ataxin-3a contained either TAA1118 (Stop349) or TAC1118 (Tyr349).
Cloning strategies
Inducible constructs
To generate untagged ataxin-3 constructs for the Tet-off system, ataxin-3 cDNA was amplified from pEGFP-C2-hMJD1c18CAG-987C using the primers 5′-AAGGGATCCGAGCTCCAGCTAGCTGGCGGCCGCCATGGAGTCCATCTTCCAC-3′ and 5′-CTTGGATCCGGGCCCTTATTTTTTTCCTTCTGT-3′ and was cloned into the pTRE-hMJD77 vector by BamHI restriction digestion. Afterward the 18 CAG 3a isoforms as well as the 73 CAG isoforms were cloned by exchanging the 3′ sequence using a SpeI and ApaI fragment from the pEGFP-C2-hMJD vectors. Due to the haplotype, the 73 CAG vectors pTRE-hMJD1c73CAG-AC, pTRE-hMJD1aL73CAGACC, and pTRE-hMJD1aS73CAG-ACA were generated successfully. In a last step, the SNP rs1048755 was exchanged from A to G for the 18 CAG isoforms by a restriction digestion of pGEX-hMJD1c18CAG-GG and the pTRE vectors with NsiI and BglII. Sequencing with the primers 5′-CGCCTGGAGACGCCATCC-3′, 5′-GCTAAGTATGCAAGGTAGTTCC-3′, and 5′-CCACACCTCCCCCTGAAC-3′ confirmed the generated pTRE-hMJD1c18CAG-GG, pTRE-hMJD1aL18CAG-GGC, and pTRE-hMJD1aS18CAG-GGA vectors.
GST-tagged constructs
GST-tagged constructs were generated for the expression and isolation of ataxin-3 isoforms from Escherichia coli to measure the DUB activity of ataxin-3. Ataxin-3 cDNA isoforms were amplified from pEGFP-C2-hMJD1 isoform vectors using the primers 5′-ATCCCGGCGGCCGCCTCGAGGGAAAGTATGAAT-3′ (3c), 5′-TTTTACTCGAGTTAAAGAGGGAATGAAGA-3′ (3aL), and 5′-TTTTACTCGAGTTATGTCAGATAAAGTGT-3′ (3aS) as well as preCAG-fwd (5′-GCTAAGTATGCAAGGTAGTTCC-3′). The isoform-specific 3′ fragments were exchanged in the pGEX-hMJD1 vector by digestion with PpuMI and XhoI to generate the new pGEX-hMJD1 vectors. The sequence was verified by Sanger sequencing using the primers pGEXseq-rev (5′-CCGGGAGCTGCATGTGTCAGAGG-3′) and preCAG-fwd.
In the next step, the CAG repeat was exchanged by cloning the 73 CAG fragment from pGEX-hMJD73 into the new isoform-specific pGEX-hMJD1 vectors using BglI and PpuMI restriction sites, resulting in the new vectors pGEX-hMJD1c73CAG-AC, pGEX-hMJD1aL73CAG-ACC, and pGEX-hMJD1aS73CAG-ACA. Cloning was sequence-verified using the primers pGEXseq-rev, ATXN3-fwd1 (5′-CTCCTGCAGATGATTAGGGT-3′), and preCAG-fwd.
For the exchange of SNP rs1048755 A to G in the 18 CAG vectors, a gBlock (Integrated DNA Technologies, Leuven, Belgium) was ordered, containing the desired SNP modification, which was cloned into the vectors using the EcoRI and PpuMI restriction sites, resulting in the new vectors pGEX-hMJD1c18CAG-GG, pGEX-hMJD1aL18CAG-GGC, and pGEX-hMJD1aS18CAG-GGA. Sequence verification was done with the primers 5′-GCTTCTCGTCTCTTCCGAAG-3′ and preCAG-fwd.
To exchange the SNP rs1048755 A to G in the pEGFP-C2-hMJD1 vectors, the respective ataxin-3 cDNA fragment was cloned from pTRE-hMJD1c18CAG-GG using the NotI and PpuMI restriction sites. Exchange of the SNP was verified by sequencing using the primer ATXN3-fwd1, ATXN3-fwd2 (5′-CAGGTTATAAGCAATGCCTTG-3′), ATXN3-fwd3 (5′-TGGAGTCCACTTCCACGA-3′), and 5′-TTGAGGATAATTCCACAGGGC-3′.
N-SF-TAP constructs
Ataxin-3 constructs for the SILAC-MS identification of ataxin-3 interactors were generated by first exchanging the SNP rs1048755 from A to G. Therefore, ataxin-3 cDNA from pTRE-hMJD1c18CAG-GG was introduced into pN-SF-TAP-hMJD18 by a restriction digestion with Bsu36I and BsmBI. Afterward, the 3′ fragments of ataxin-3 cDNA were exchanged for the 3a isoforms by the isoform-specific variants from the pTRE-hMJD1a vectors using the ApaI and PpuMI restriction sites. Sequencing with preCAG-fwd and ATXN3-fwd1, -fwd2, and -fwd3 confirmed the new vectors pN-SF-TAP-hMJD1c-18CAG-GG, pN-SF-TAP-hMJD1aL18CAG-GGC, and pN-SF-TAP-hMJD1aS18CAG-GGA.
FLAG-V5–tagged constructs
FLAG-ataxin-3-V5 constructs were generated to distinguish co-transfected ataxin-3 from untagged isoforms. Therefore, ataxin-3 was introduced into pcDNA-FLAG-hMJD1-M01-T60-V5-His after PCR amplification from pTRE-hMJD with 18CAG using the forward primer 5′-ACTGGGATCCCTGGAGTCCATCTT-3′ and the isoform-specific reverse primers 5′-AGCATCTAGATTTTTTTCCTTCTGTTTTCA-3′ (3c), 5′-AGCATCTAGAAAGAGGGAATGAAGAATAA-3′ (3aL), and 5′-AGCATCTAGATGTCAGATAAAGTGTGAA-3′ (3aS) and the BamHI and XbaI restriction sites. Sequence verification was done using the primers preCAG-fwd, ATXN3-fwd2, and 5′-TGGACCCGTCAAGAGAGAAT-3′. His-tag was removed afterwards by introduction of a TAA-stop before the tag. V5 was amplified using the primers 5′-AGACTTCTAGAGGTAAGCCTATCCCT-3′ and 5′-GTCACCGGTTTACGTAGAATCGAGAC-3′. PCR product was cloned into the vector using the XbaI and AgeI restriction sites. Sequencing with 5′-GCAGCGGGACCTATCAGG-3′ confirmed the introduced stop. The CAG repeat was exchanged using the restriction sites EcoRI and PpuMI from pEGFP-C2-hMJD1c73CAG-AC and -hMJD1c153CAG-AC. The CAG tract was sequence-verified using the primer preCAG-fwd. All enzymes used for cloning were purchased from New England Biolabs (Frankfurt am Main, Germany).
Generation of ATXN3 knockout cells
Ataxin-3 knockout (KO) cells were generated to avoid interference of endogenous ATXN3 alleles. KO of ataxin-3 in HEK 293T (ATCC, CRL-11268) cells was achieved using TALENS as described previously by Kim et al. (101). HEK 293T cells were transfected with two TALEN plasmids (TALE expression vector), the hygromycin reporter plasmid Hygro RS (102) (TALEN Library Resource, H170773), and a guide oligonucleotide 5′-TAGTGAAGATTATCGCACGTTTTTACAGGTACTGATTTTAAACTCAC-3′ (Metabion International, Planegg, Germany). The specific TALEN target site sequence was 5′-TCAACATTGCCTGAATAACT-TATTGCAAGGAG-AATATTTTAGCCCTGTGGAA-3′ (left-spacer-right). Hygromycin selection (800 μg/ml) was performed 72 h after transfection for 3 days. Cells were then recovered from selection stress for 48 h. Cells were detached and diluted to seed single cells in each well of a 96-well plate. Cells were cultured for 6 days before KO was validated by Sanger sequencing using primers 5′-CTAACACAGGATGAAACCCCG-3′ and 5′-ACGTGCGATAATCTTCACTAGT-3′, next-generation sequencing, and Western blotting.
Cell culture
HEK 293T (ATTC number CRL-11268, RIP ID CVCL_1926) and HEK 293T ATXN3 KO cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS and 1% penicillin/streptomycin (all from Gibco, Fisher Scientific, Schwerte, Germany) at 37 °C and 5% CO2. Mycoplasma tests were performed every 6 months, and cells were negative for an infection in all tests. Transient transfections were performed using Attractene (Qiagen, Hilden, Germany) following the manufacturer's instruction.
Protein extraction and Western blot analysis
To obtain protein lysates, the culture medium was removed, and cells were detached with DPBS and pelleted at 300 × g for 5 min. Cells were lysed in radioimmune precipitation assay buffer (25 mm Tris, 150 mm NaCl, 0.1% SDS, 0.5% sodium doxycholate, 1% Triton X-100, cOmplete protease inhibitor without EDTA (Roche Diagnostics, Mannheim, Germany)) for 20 min, vortexing every 5 min. Homogenates were centrifuged for 10 min at 16,000 × g and 4 °C, and the supernatant was supplemented with glycerol (10% final concentration). For protein homogenates, cell pellets were lysed in DPBS supplemented with 1% Triton X-100 by sonication. Protein concentration was measured using a Bradford protein assay (Bio-Rad Laboratories, Munich, Germany).
Western blotting was performed according to standard procedures. In brief, 30 μg of protein were mixed with 4× LDS sample buffer (1 m Tris base, pH 8.5, 2 mm EDTA, 8% lithium dodecyl sulfate, 40% glycerol, 0.025% phenol red) and 100 mm DTT. Samples were heat-denatured for 10 min at 70 °C. Electrophoresis was performed using BisTris gels (8 or 10%) with either MES buffer (50 mm MES, 50 mm Tris base, 0.1% SDS, 1 mm EDTA) or MOPS buffer (50 mm MOPS, 50 mm Tris base, 0.1% SDS, 1 mm EDTA). Proteins were transferred on Amersham Biosciences Protran Premium 0.2-μm nitrocellulose or Amersham Biosciences Hybond P 0.2-μm polyvinylidene difluoride membranes (GE Healthcare) using a Bicine/BisTris transfer buffer (25 mm Bicine, 25 mm BisTris, pH 7.2, 1 mm EDTA, 15% methanol). Membranes were blocked with 5% skim milk powder (Sigma-Aldrich, Taufkirchen, Germany) for 1 h in TBS (10 mm Tris, 150 mm NaCl) at room temperature. Antibodies were diluted in TBST (TBS supplemented with 0.1% Tween 20), and primary antibody incubation was performed at 4 °C overnight. Secondary antibody was incubated for 1 h at room temperature. Signals were detected using the ODYSSEY FC system (LI-COR Biosciences, Bad Homburg, Germany) and quantified using Image Studio version 4.2 (LI-COR Biosciences). Tris-glycine gels were used with different buffers: 5× TG sample loading buffer (125 mm Tris-HCl, pH 6.8, 8% DTT, 50% glycerol, 2% SDS, 0.05% Coomassie Brilliant Blue G-250), TG running buffer (192 mm glycine, 25 mm Tris base, 0.1% SDS), TG transfer buffer (192 mm glycine, 25 mm Tris base, 15% methanol). Table S2 shows the antibodies used.
Protein stability analysis
For degradation analysis, the Tet-off system was employed. HEK 293T ATXN3 KO cells were transfected with pTRE-ataxin-3 responder constructs and a pTET-RCA2 (103) promoter construct in a 1:1 ratio. Transfected proteins were expressed for at least 24 h before expression was shut off by the addition of doxycycline (4.5 μm). Expression was then turned off at different time points before cells were harvested and prepared for Western blotting. The relative ataxin-3 levels were fit by an exponential function. Protein half-life was then calculated from the exponent of this exponential function following first-order kinetics. For the analysis of the degradation pathway cells were cultured for 24 h after transfection. Doxycycline was added 8 h before application of the inhibitors. Proteasomal degradation was inhibited using 10 μm lactacystin (Enzo Lifescience, Lausen, Switzerland). Autophagy was inhibited by 50 nm bafilomycin A1 (Invivogen, Toulouse, France).
mRNA stability analysis
RNA was extracted using the RNeasy MiniKit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. Cells were lysed by QIAshredder columns, and DNA contaminations were removed using the RNase-free DNase kit (both from Qiagen). 1 μg of RNA was then transcribed to cDNA using the QuantiTect reverse transcription kit (Qiagen) according to the manufacturer's protocol. ATXN3 (forward, 5′-TGGATGAGGAGGAGAGGATG-3′; reverse, 5′-CAAGGCATTGCTTATAACCTGA-3′) and GAPDH (forward, 5′-GCTCTCTGCTCCTCCTGTTC-3′; reverse, 5′-ACGACCAAATCCGTTGACTC-3′) expression was analyzed together with a no-template control, a reverse-transcription control, and a standard curve. Quantitative RT-PCR was performed using the QuantiTect SYBR Green PCR kit (Qiagen) in a LightCycler 480 (Roche Diagnostics). Relative expression levels of ATXN3 were obtained by normalization to GAPDH expression using the advanced relative quantification method of the LightCycler 480 software version 1.5 (Roche Diagnostics). GAPDH was identified to be the most reliable housekeeper among TBP, P4HB, SDHA, TFRC, YWHAZ, PGK1, and ACTB using NormFinder (104).
Analysis of subcellular localization
Subcellular localization of ataxin-3 was analyzed by a separation of homogenates into whole-cell, nuclear, and cytoplasmic fractions using the REAP fractionation protocol (105) with minor modifications (27).
Deubiquitination assay
Ataxin-3 was isolated from a day culture of pGEX-ataxin-3–transformed E. coli BL21 (A600 of 0.6), which was induced with isopropyl 1-thio-β-d-galactopyranoside (100 nm) and grown for 3 h. Bacteria were harvested by centrifugation at 1,500 × g. Cells were washed with PBS and centrifuged again. Lysis was performed in GST-lysis buffer (50 mm Na3PO4, 100 mm NaCl, 10% glycerol, 1 mm DTT, cOmplete protease inhibitor without EDTA (Roche Diagnostics)). 8 mg/ml lysozyme from chicken egg white (Carl Roth, Karlsruhe, Germany) and 1% Triton X-100 were added. Cells were lysed for 30 min in an ice bath and treated with ultrasonication three times for 30 s. Lysate was cleared twice by centrifugation (30 min at 25,500 × g, 4 °C). GST-ataxin-3 was purified using glutathione-Sepharose 4B (GE Healthcare) according to the manufacturer's protocol. Eluates (10 mm GSH in 50 mm Tris, pH 8) were collected over a total of three elution steps. Pooled eluate was concentrated to 500 μl using 10-kDa Amicon Ultra-0.5 centrifugal filter columns (Merck, Darmstadt, Germany) and supplemented with glycerol (10%).
For the deubiquitination assay itself, 2 μm ubiquitin-rhodamine-110 (LifeSensors, Malvern, PA) in DUB assay buffer (50 mm HEPES, 500 μm EDTA, 1 mm DTT, 100 μg/ml BSA) was mixed 1:1 with 400 nm GST-ataxin-3. Emission was immediately measured after mixing at 535 nm (485-nm excitation) in an EnVision reader (PerkinElmer, Rodgau, Germany) every 10 s for 30 min. All measurements were performed in technical duplicates, which were averaged for the analysis. Relative fluorescence units were plotted over time and fitted by a linear function in the first 2 min to determine the initial velocity.
SILAC-MS identification of interaction partners
The SILAC-based quantification by MS was carried out as described previously (99). Briefly, pN-SF-TAP-ataxin-3 isoforms were expressed in HEK 293T cells cultured either in heavy ([13C6-15N2]lysine; [13C6-15N4]arginine), medium (4,4,5,5-d4-lysine; [13C6]arginine), light (lysine; arginine) SILAC medium and purified individually via the tandem Strep-tag II. The eluates were mixed in a 1:1:1 ratio prior to the MS analysis. To allow a robust quantification, labels were switched, allowing each isoform to be expressed in all three SILAC conditions. Samples were precipitated by chloroform methanol and redissolved in 50 mm ammonium bicarbonate buffer containing 0.1% RapiGest (Waters, Eschborn, Germany). Proteolysis was performed by adding 0.1 mg/ml trypsin (sequencing grade; Sigma Aldrich) overnight at 37 °C. The RapiGest surfactant was hydrolyzed and removed, and samples were further purified via C18-StageTips (Thermo Fisher Scientific, Karlsruhe, Germany) following standard protocols. Samples were subsequently analyzed by LC-MS/MS using a nanoflow HPLC system (Dionex Ultimate 3000 RSLC, Thermo Fisher Scientific) coupled to a Q-Exactive Plus tandem mass spectrometer (Thermo Fisher Scientific). The raw data were directly analyzed by the MaxQuant software (version 1.5.2.8) (106), allowing protein identification and quantification with the following Andromeda search engine parameters: database, human subset of the SwissProt database (2015/05, 20198 entries); carbamidomethyl (cysteine) as fixed modification as well as N-terminal protein acetylation as variable modification with the heavy/medium/light (L/M/H) isotope pairs lysine/arginine, 4,4,5,5-d4-lysine/[13C6]arginine and [13C6-15N2]lysine/[13C6-15N4]arginine for quantification. As the enzyme, trypsin was chosen. The initial mass accuracy (MS) for mass recalibration was set to 20 ppm. For MS/MS spectra, the mass accuracy was set to 0.1 Da. Razor peptides and peptides with N-terminal acetylation or methionine oxidation were included in protein quantification. Downstream analysis was carried out using Perseus version 1.5.4.0 (107). Potential contaminations were filtered, and the interaction ratios between 3aL/3c, 3aS/3c, and 3aS/3aL for each interaction partner were transformed by 1/χ followed by log2 χ. Single experiments were grouped, and one-sample t tests were performed to test whether the ratios differed from 0. The false discovery rate threshold was set to 0.05.
GFP-trap assay
C-terminally EGFP-tagged ataxin-3 isoforms were expressed in HEK 293T ATXN3 KO cells for 72 h. Isoforms were immunoprecipitated using GFP-trap A beads (Chromotek, Planegg-Martinsried, Germany) according to the manufacturer's protocol with minor modifications: samples were cleared by centrifugation at 16,000 × g for 10 min at 4 °C. All washing steps were performed using the lysis buffer (10 mm Tris, pH 7.5, 150 mm NaCl, 0.5 mm EDTA, 0.5% Nonidet P-40). Elution was performed in 1× LDS containing 100 mm DTT by denaturation (70 °C, 10 min).
Microscopic analysis of aggregation
pEGFP-C2-ataxin-3–expressing cells were prefixed for 10 min by adding 0.4% paraformaldehyde to the culture medium. Cells were washed in DPBS and fixed for 15 min in 4% paraformaldehyde in DPBS. Cells were washed for 5 min with DPBS. Cells grown in culture dishes were afterward stored in DPBS at 4 °C until analysis. Cells grown on a coverslip were mounted onto a glass slide using VECTASHIELD antifade mounting medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA). Edges were sealed using nail polish. Microscopy was performed with an Axioplan 2 imaging system with ApoTome (Carl Zeiss Microscopy, Jena, Germany) using a ×40/0.75 Zeiss Plan-Neofluar objective (air, room temperature). Images were taken using an AxioCam MRm (Carl Zeiss Microscopy) and AxioVision version 4.8 (Carl Zeiss Microscopy). Microscopy and aggregate counting was performed blinded to prevent an experimenter bias. At least 200 GFP-positive cells were counted per biological replicate.
Aggregate size analysis
About 20 images of fixed cells having more than 100 aggregates in total were taken with a Nikon CFI Ph1 ADL Plan objective (×10/0.25, air, room temperature; Nikon (Düsseldorf, Germany)) with an identical exposure time of 100 ms using an Eclipse TS100 (Nikon) and an AxioCam MRm (Carl Zeiss Microscopy, Jena, Germany) with AxioVision version 4.8 (Carl Zeiss Microscopy). To prevent an experimenter bias, microscopy was performed blinded. The background of images was calculated, and thresholding was performed before a watershed analysis was executed from a distance map of the image using R (version 3.4.2) (108) and the EBIimage package (109). Area, perimeter, and position of aggregates were computed. Objects having a perimeter smaller than 10 pixels were excluded. Numbering of the objects recognized in the image was performed using the magick package (59). Manual quality control was performed comparing the identified objects and the original fluorescence image. Misidentifications were excluded whenever necessary.
Analysis of protein solubility
12.5 μg of protein homogenate were diluted in DPBS containing 2% SDS and 50 mm DTT. Samples were denatured (95 °C, 5 min) and cooled down to room temperature. A 0.45-μm cellulose-acetate membrane (GE Healthcare) was equilibrated by filtering 0.1% SDS in DPBS using a Minifold II slot blot system (GE Healthcare). Samples were filtered onto the membrane, and the membrane was washed twice with DPBS afterward. Membrane was transferred to TBS and washed for 5 min before membrane was detected following the standard immunodetection protocol.
Fractionation based on protein solubility into Triton X-100–soluble, SDS-soluble, and SDS-insoluble protein fractions was performed according to Koch et al. (57).
Statistical analysis
Data were collected randomized and blinded whenever necessary. Statistical tests and comparisons were defined a priori according to the following strategy. Data were tested for normal distribution using the Shapiro–Wilks test for n ≥ 5 and were additionally plotted in a quantile–quantile plot to inspect distribution. In case of a normal distribution, a parametric test was chosen, and in the case of unknown or nonnormal distribution, a nonparametric test was conducted. Data with no independent variable were analyzed by either one-sample t test or one-sample Wilcoxon signed rank test. If not otherwise indicated, compensation for the multiple-comparison problem was performed by the family-wise error rate procedure of Hommel whenever necessary. Independent groups with one independent variable of two levels were compared by either a two-sample t test or a Wilcoxon–Mann–Whitney test. For hypothesis tests with one independent variable within independent groups with more than two levels, either ANOVA (proven homogeneity of variance by a Levene test and normal distribution) followed by Tukey's honest significant difference (HSD) test or a Kruskal–Wallis test (nonnormal distribution) followed by a Conover–Iman multiple-comparison test was performed. If multiple comparisons were performed with a control, Dunnett's multiple-comparison test was used. Data with two independent variables were analyzed in the case of proven homogeneity of variance, normal distribution, and equal sample size by a two-way ANOVA followed by Tukey's HSD test (main effects) or estimated marginal means contrasts (simple main effects). In the case of a nonnormal distribution or heteroscedasticity, a Scheirer–Ray–Here test was performed followed by a Conover–Iman test. Data representing portions were analyzed by β regression in the case of a nonnormal distribution and heteroscedasticity. The significance level α was defined to 5% in all tests. Data analysis was performed in R version 3.4.2 (108). Graphical representation was performed in R and GraphPad Prism version 6.01 (GraphPad Software, Inc., La Jolla, CA). Final adjustments were made using Corel Draw version 17.6.0.1021. If not stated differently, error bars represent S.E.
Author contributions
D. W., O. R., and T. S. conceptualization; D. W. and C. J. G. data curation; D. W. software; D. W. and C. J. G. formal analysis; D. W., J. Schneider, B. P. P., S. M. D., and F. v. Z. investigation; D. W. visualization; D. W. and S. M. D. methodology; D. W. writing-original draft; C. R. validation; C. J. G. resources; C. J. G., J. Schmidt, O. R., and T. S. writing-review and editing; J. Schmidt and T. S. funding acquisition; O. R. and T. S. supervision; O. R. and T. S. project administration.
Supplementary Material
Acknowledgments
We thank Jonasz J. Weber for helpful discussions and ideas. We thank the TALEN Library Resource for providing the TALENs and hygromycin reporter used for creating the ATXN3 KO. We thank the staff of the Core Facility for Medical Bioanalytics at the Institute for Ophthalmic Research (University of Tübingen) for technical assistance in MS.
This work was supported by the National Ataxia Foundation through the Spinocerebellar Ataxia Research Award (to T. S.) and a fellowship from the Landesgraduiertenförderung Baden-Württemberg (to D. W.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1 and S2 and Fig. S1.
- UIM
- ubiquitin-interacting motif
- DUB
- deubiquitination
- ERAD
- endoplasmic reticulum–associated protein degradation
- GO
- gene ontology
- HEK
- human embryonic kidney
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- KO
- knockout
- MJD
- Machado–Joseph disease
- polyQ
- polyglutamine
- SILAC
- stable isotope labeling of amino acids in cell culture
- TALEN
- transcription activator-like effector nuclease
- SCA
- spinocerebellar ataxia
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- DPBS
- Dulbecco's PBS
- ANOVA
- analysis of variance
- HSD
- honest significant difference
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
References
- 1. Costa, Mdo C., and Paulson H. L. (2012) Toward understanding Machado–Joseph Disease. Prog. Neurobiol. 97, 239–257 10.1016/j.pneurobio.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paulson H. L., Das S. S., Crino P. B., Perez M. K., Patel S. C., Gotsdiner D., Fischbeck K. H., and Pittman R. N. (1997) Machado–Joseph disease gene product is a cytoplasmic protein widely expressed in brain. Ann. Neurol. 41, 453–462 10.1002/ana.410410408 [DOI] [PubMed] [Google Scholar]
- 3. Trottier Y., Cancel G., An-Gourfinkel I., Lutz Y., Weber C., Brice A., Hirsch E., and Mandel J. L. (1998) Heterogeneous intracellular localization and expression of ataxin-3. Neurobiol. Dis. 5, 335–347 10.1006/nbdi.1998.0208 [DOI] [PubMed] [Google Scholar]
- 4. Schmidt T., Landwehrmeyer G. B., Schmitt I., Trottier Y., Auburger G., Laccone F., Klockgether T., Völpel M., Epplen J. T., Schöls L., and Riess O. (1998) An isoform of ataxin-3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients. Brain Pathol. 8, 669–679 10.1111/j.1750-3639.1998.tb00193.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ichikawa Y., Goto J., Hattori M., Toyoda A., Ishii K., Jeong S. Y., Hashida H., Masuda N., Ogata K., Kasai F., Hirai M., Maciel P., Rouleau G. A., Sakaki Y., and Kanazawa I. (2001) The genomic structure and expression of MJD, the Machado–Joseph disease gene. J. Hum. Genet. 46, 413–422 10.1007/s100380170060 [DOI] [PubMed] [Google Scholar]
- 6. Costa M. C., Gomes-da-Silva J., Miranda C. J., Sequeiros J., Santos M. M., and Maciel P. (2004) Genomic structure, promoter activity, and developmental expression of the mouse homologue of the Machado–Joseph disease (MJD) gene. Genomics 84, 361–373 10.1016/j.ygeno.2004.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burnett B., Li F., and Pittman R. N. (2003) The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 12, 3195–3205 10.1093/hmg/ddg344 [DOI] [PubMed] [Google Scholar]
- 8. Tzvetkov N., and Breuer P. (2007) Josephin domain-containing proteins from a variety of species are active de-ubiquitination enzymes. Biol. Chem. 388, 973–978 10.1515/BC.2007.107 [DOI] [PubMed] [Google Scholar]
- 9. Antony P. M. A., Mäntele S., Mollenkopf P., Boy J., Kehlenbach R. H., Riess O., and Schmidt T. (2009) Identification and functional dissection of localization signals within ataxin-3. Neurobiol. Dis. 36, 280–292 10.1016/j.nbd.2009.07.020 [DOI] [PubMed] [Google Scholar]
- 10. Rodrigues A.-J., Coppola G., Santos C., Costa, Mdo C., Ailion M., Sequeiros J., Geschwind D. H., and Maciel P. (2007) Functional genomics and biochemical characterization of the C. elegans orthologue of the Machado–Joseph disease protein ataxin-3. FASEB J. 21, 1126–1136 10.1096/fj.06-7002com [DOI] [PubMed] [Google Scholar]
- 11. Schmitt I., Linden M., Khazneh H., Evert B. O., Breuer P., Klockgether T., and Wuellner U. (2007) Inactivation of the mouse Atxn3 (ataxin-3) gene increases protein ubiquitination. Biochem. Biophys. Res. Commun. 362, 734–739 10.1016/j.bbrc.2007.08.062 [DOI] [PubMed] [Google Scholar]
- 12. Evert B. O., Araujo J., Vieira-Saecker A. M., de Vos R. A., Harendza S., Klockgether T., and Wüllner U. (2006) Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J. Neurosci. 26, 11474–11486 10.1523/JNEUROSCI.2053-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Todi S. V., Laco M. N., Winborn B. J., Travis S. M., Wen H. M., and Paulson H. L. (2007) Cellular turnover of the polyglutamine disease protein ataxin-3 is regulated by its catalytic activity. J. Biol. Chem. 282, 29348–29358 10.1074/jbc.M704126200 [DOI] [PubMed] [Google Scholar]
- 14. Bettencourt C., Santos C., Montiel R., Costa, Mdo C., Cruz-Morales P., Santos L. R., Simões N., Kay T., Vasconcelos J., Maciel P., and Lima M. (2010) Increased transcript diversity: novel splicing variants of Machado–Joseph disease gene (ATXN3). Neurogenetics 11, 193–202 10.1007/s10048-009-0216-y [DOI] [PubMed] [Google Scholar]
- 15. Kawaguchi Y., Okamoto T., Taniwaki M., Aizawa M., Inoue M., Katayama S., Kawakami H., Nakamura S., Nishimura M., and Akiguchi I. (1994) CAG expansions in a novel gene for Machado–Joseph disease at chromosome 14q32.1. Nat. Genet. 8, 221–228 10.1038/ng1194-221 [DOI] [PubMed] [Google Scholar]
- 16. Goto J., Watanabe M., Ichikawa Y., Yee S. B., Ihara N., Endo K., Igarashi S., Takiyama Y., Gaspar C., Maciel P., Tsuji S., Rouleau G. A., and Kanazawa I. (1997) Machado–Joseph disease gene products carrying different carboxyl termini. Neurosci. Res. 28, 373–377 10.1016/S0168-0102(97)00056-4 [DOI] [PubMed] [Google Scholar]
- 17. Bettencourt C., Raposo M., Ros R., Montiel R., Bruges-Armas J., and Lima M. (2013) Transcript diversity of Machado–Joseph disease gene (ATXN3) is not directly determined by SNPs in exonic or flanking intronic regions. J. Mol. Neurosci. 49, 539–543 10.1007/s12031-012-9832-3 [DOI] [PubMed] [Google Scholar]
- 18. Gaspar C., Lopes-Cendes I., Hayes S., Goto J., Arvidsson K., Dias A., Silveira I., Maciel P., Coutinho P., Lima M., Zhou Y. X., Soong B. W., Watanabe M., Giunti P., Stevanin G., et al. (2001) Ancestral origins of the Machado–Joseph disease mutation: a worldwide haplotype study. Am. J. Hum. Genet. 68, 523–528 10.1086/318184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harris G. M., Dodelzon K., Gong L., Gonzalez-Alegre P., and Paulson H. L. (2010) Splice isoforms of the polyglutamine disease protein ataxin-3 exhibit similar enzymatic yet different aggregation properties. PLoS One 5, e13695 10.1371/journal.pone.0013695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ramani B., Harris G. M., Huang R., Seki T., Murphy G. G., Costa, Mdo C., Fischer S., Saunders T. L., Xia G., McEachin R. C., and Paulson H. L. (2015) A knockin mouse model of spinocerebellar ataxia type 3 exhibits prominent aggregate pathology and aberrant splicing of the disease gene transcript. Hum. Mol. Genet. 24, 1211–1224 10.1093/hmg/ddu532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mauri P. L., Riva M., Ambu D., De Palma A., Secundo F., Benazzi L., Valtorta M., Tortora P., and Fusi P. (2006) Ataxin-3 is subject to autolytic cleavage. FEBS J. 273, 4277–4286 10.1111/j.1742-4658.2006.05419.x [DOI] [PubMed] [Google Scholar]
- 22. Lima M., Costa M. C., Montiel R., Ferro A., Santos C., Silva C., Bettencourt C., Sousa A., Sequeiros J., Coutinho P., and Maciel P. (2005) Population genetics of wild-type CAG repeats in the Machado–Joseph disease gene in Portugal. Hum. Hered. 60, 156–163 10.1159/000090035 [DOI] [PubMed] [Google Scholar]
- 23. Maciel P., Costa M. C., Ferro A., Rousseau M., Santos C. S., Gaspar C., Barros J., Rouleau G. A., Coutinho P., and Sequeiros J. (2001) Improvement in the molecular diagnosis of Machado–Joseph disease. Arch. Neurol. 58, 1821–1827 10.1001/archneur.58.11.1821 [DOI] [PubMed] [Google Scholar]
- 24. Paulson H. L., Perez M. K., Trottier Y., Trojanowski J. Q., Subramony S. H., Das S. S., Vig P., Mandel J. L., Fischbeck K. H., and Pittman R. N. (1997) Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19, 333–344 10.1016/S0896-6273(00)80943-5 [DOI] [PubMed] [Google Scholar]
- 25. Wellington C. L., Ellerby L. M., Hackam A. S., Margolis R. L., Trifiro M. A., Singaraja R., McCutcheon K., Salvesen G. S., Propp S. S., Bromm M., Rowland K. J., Zhang T., Rasper D., Roy S., Thornberry N., et al. (1998) Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J. Biol. Chem. 273, 9158–9167 10.1074/jbc.273.15.9158 [DOI] [PubMed] [Google Scholar]
- 26. Hübener J., Weber J. J., Richter C., Honold L., Weiss A., Murad F., Breuer P., Wüllner U., Bellstedt P., Paquet-Durand F., Takano J., Saido T. C., Riess O., and Nguyen H. P. (2013) Calpain-mediated ataxin-3 cleavage in the molecular pathogenesis of spinocerebellar ataxia type 3 (SCA3). Hum. Mol. Genet. 22, 508–518 10.1093/hmg/dds449 [DOI] [PubMed] [Google Scholar]
- 27. Weber J. J., Golla M., Guaitoli G., Wanichawan P., Hayer S. N., Hauser S., Krahl A. C., Nagel M., Samer S., Aronica E., Carlson C. R., Schöls L., Riess O., Gloeckner C. J., Nguyen H. P., and Hübener-Schmid J. (2017) A combinatorial approach to identify calpain cleavage sites in the Machado–Joseph disease protein ataxin-3. Brain 140, 1280–1299 10.1093/brain/awx039 [DOI] [PubMed] [Google Scholar]
- 28. Haacke A., Broadley S. A., Boteva R., Tzvetkov N., Hartl F. U., and Breuer P. (2006) Proteolytic cleavage of polyglutamine-expanded ataxin-3 is critical for aggregation and sequestration of non-expanded ataxin-3. Hum. Mol. Genet. 15, 555–568 10.1093/hmg/ddi472 [DOI] [PubMed] [Google Scholar]
- 29. Bichelmeier U., Schmidt T., Hübener J., Boy J., Rüttiger L., Häbig K., Poths S., Bonin M., Knipper M., Schmidt W. J., Wilbertz J., Wolburg H., Laccone F., and Riess O. (2007) Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: in vivo evidence. J. Neurosci. 27, 7418–7428 10.1523/JNEUROSCI.4540-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sowa A. S., Martin E., Martins I. M., Schmidt J., Depping R., Weber J. J., Rother F., Hartmann E., Bader M., Riess O., Tricoire H., and Schmidt T. (2018) Karyopherin α-3 is a key protein in the pathogenesis of spinocerebellar ataxia type 3 controlling the nuclear localization of ataxin-3. Proc. Natl. Acad. Sci. U.S.A. 115, E2624–E2633 10.1073/pnas.1716071115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dürr A., Stevanin G., Cancel G., Duyckaerts C., Abbas N., Didierjean O., Chneiweiss H., Benomar A., Lyon-Caen O., Julien J., Serdaru M., Penet C., Agid Y., and Brice A. (1996) Spinocerebellar ataxia 3 and Machado–Joseph disease: clinical, molecular, and neuropathological features. Ann. Neurol. 39, 490–499 10.1002/ana.410390411 [DOI] [PubMed] [Google Scholar]
- 32. Maciel P., Gaspar C., DeStefano A. L., Silveira I., Coutinho P., Radvany J., Dawson D. M., Sudarsky L., Guimarães J., and Loureiro J. E. (1995) Correlation between CAG repeat length and clinical features in Machado–Joseph disease. Am. J. Hum. Genet. 57, 54–61 [PMC free article] [PubMed] [Google Scholar]
- 33. Schöls L., Vieira-Saecker A. M. M., Schöls S., Przuntek H., Epplen J. T., and Riess O. (1995) Trinucleotide expansion within the MJD1 gene presents clinically as spinocerebellar ataxia and occurs most frequently in german SCA patients. Hum. Mol. Genet. 4, 1001–1005 10.1093/hmg/4.6.1001 [DOI] [PubMed] [Google Scholar]
- 34. van de Warrenburg B. P. C., Hendriks H., Dürr A., van Zuijlen M. C. A., Stevanin G., Camuzat A., Sinke R. J., Brice A., and Kremer B. P. H. (2005) Age at onset variance analysis in spinocerebellar ataxias: a study in a Dutch-French cohort. Ann. Neurol. 57, 505–512 10.1002/ana.20424 [DOI] [PubMed] [Google Scholar]
- 35. Globas C., du Montcel S. T., Baliko L., Boesch S., Depondt C., DiDonato S., Durr A., Filla A., Klockgether T., Mariotti C., Melegh B., Rakowicz M., Ribai P., Rola R., Schmitz-Hubsch T., Szymanski S., Timmann D., Van de Warrenburg B. P., Bauer P., and Schols L. (2008) Early symptoms in spinocerebellar ataxia type 1, 2, 3, and 6. Mov. Disord. 23, 2232–2238 10.1002/mds.22288 [DOI] [PubMed] [Google Scholar]
- 36. Junck L., and Fink J. K. (1996) Machado–Joseph disease and SCA3: the genotype meets the phenotypes. Neurology 46, 4–8 10.1212/WNL.46.1.4 [DOI] [PubMed] [Google Scholar]
- 37. DeStefano A. L., Cupples L. A., Maciel P., Gaspar C., Radvany J., Dawson D. M., Sudarsky L., Corwin L., Coutinho P., and MacLeod P. (1996) A familial factor independent of CAG repeat length influences age at onset of Machado–Joseph disease. Am. J. Hum. Genet. 59, 119–127 [PMC free article] [PubMed] [Google Scholar]
- 38. Warrick J. M., Morabito L. M., Bilen J., Gordesky-Gold B., Faust L. Z., Paulson H. L., and Bonini N. M. (2005) Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol. Cell 18, 37–48 10.1016/j.molcel.2005.02.030 [DOI] [PubMed] [Google Scholar]
- 39. Tsou W.-L., Burr A. A., Ouyang M., Blount J. R., Scaglione K. M., and Todi S. V. (2013) Ubiquitination regulates the neuroprotective function of the deubiquitinase ataxin-3 in vivo. J. Biol. Chem. 288, 34460–34469 10.1074/jbc.M113.513903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tsou W. L., Ouyang M., Hosking R. R., Sutton J. R., Blount J. R., Burr A. A., and Todi S. V. (2015) The deubiquitinase ataxin-3 requires Rad23 and DnaJ-1 for its neuroprotective role in Drosophila melanogaster. Neurobiol. Dis. 82, 12–21 10.1016/j.nbd.2015.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burr A. A., Tsou W. L., Ristic G., and Todi S. V. (2014) Using membrane-targeted green fluorescent protein to monitor neurotoxic protein-dependent degeneration of Drosophila eyes. J. Neurosci. Res. 92, 1100–1109 10.1002/jnr.23395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sutton J. R., Blount J. R., Libohova K., Tsou W. L., Joshi G. S., Paulson H. L., Costa M. D. C., Scaglione K. M., and Todi S. V. (2017) Interaction of the polyglutamine protein ataxin-3 with Rad23 regulates toxicity in Drosophila models of spinocerebellar ataxia type 3. Hum. Mol. Genet. 26, 1419–1431 10.1093/hmg/ddx039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bettencourt C., Santos C., Montiel R., Kay T., Vasconcelos J., Maciel P., and Lima M. (2010) The (CAG)n tract of Machado–Joseph Disease gene (ATXN3): a comparison between DNA and mRNA in patients and controls. Eur. J. Hum. Genet. 18, 621–623 10.1038/ejhg.2009.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maciel P., Gaspar C., Guimarães L., Goto J., Lopes-Cendes I., Hayes S., Arvidsson K., Dias A., Sequeiros J., Sousa A., and Rouleau G. A. (1999) Study of three intragenic polymorphisms in the Machado–Joseph disease gene (MJD1) in relation to genetic instability of the (CAG)(n) tract. Eur. J. Hum. Genet. 7, 147–156 10.1038/sj.ejhg.5200264 [DOI] [PubMed] [Google Scholar]
- 45. Gossen M., and Bujard H. (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U.S.A. 89, 5547–5551 10.1073/pnas.89.12.5547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pozzi C., Valtorta M., Tedeschi G., Galbusera E., Pastori V., Bigi A., Nonnis S., Grassi E., and Fusi P. (2008) Study of subcellular localization and proteolysis of ataxin-3. Neurobiol. Dis. 30, 190–200 10.1016/j.nbd.2008.01.011 [DOI] [PubMed] [Google Scholar]
- 47. Harmuth T., Prell-Schicker C., Weber J. J., Gellerich F., Funke C., Driessen S., Magg J. C. D., Krebiehl G., Wolburg H., Hayer S. N., Hauser S., Krüger R., Schöls L., Riess O., and Hübener-Schmid J. (2018) Mitochondrial morphology, function and homeostasis are impaired by expression of an N-terminal calpain cleavage fragment of ataxin-3. Front. Mol. Neurosci. 11, 368 10.3389/fnmol.2018.00368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Winborn B. J., Travis S. M., Todi S. V., Scaglione K. M., Xu P., Williams A. J., Cohen R. E., Peng J., and Paulson H. L. (2008) The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J. Biol. Chem. 283, 26436–26443 10.1074/jbc.M803692200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chatr-Aryamontri A., Oughtred R., Boucher L., Rust J., Chang C., Kolas N. K., O'Donnell L., Oster S., Theesfeld C., Sellam A., Stark C., Breitkreutz B. J., Dolinski K., and Tyers M. (2017) The BioGRID interaction database: 2017 update. Nucleic Acids Res. 45, D369–D379 10.1093/nar/gkw1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matsumoto M., Yada M., Hatakeyama S., Ishimoto H., Tanimura T., Tsuji S., Kakizuka A., Kitagawa M., and Nakayama K. I. (2004) Molecular clearance of ataxin-3 is regulated by a mammalian E4. EMBO J. 23, 659–669 10.1038/sj.emboj.7600081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang D. W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 52. Havugimana P. C., Hart G. T., Nepusz T., Yang H., Turinsky A. L., Li Z., Wang P. I., Boutz D. R., Fong V., Phanse S., Babu M., Craig S. A., Hu P., Wan C., Vlasblom J., et al. (2012) A census of human soluble protein complexes. Cell 150, 1068–1081 10.1016/j.cell.2012.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wood A., Krogan N. J., Dover J., Schneider J., Heidt J., Boateng M. A., Dean K., Golshani A., Zhang Y., Greenblatt J. F., Johnston M., and Shilatifard A. (2003) Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol. Cell 11, 267–274 10.1016/S1097-2765(02)00802-X [DOI] [PubMed] [Google Scholar]
- 54. Park H., Suzuki T., and Lennarz W. J. (2001) Identification of proteins that interact with mammalian peptide:N-glycanase and implicate this hydrolase in the proteasome-dependent pathway for protein degradation. Proc. Natl. Acad. Sci. U.S.A. 98, 11163–11168 10.1073/pnas.201393498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Medicherla B., Kostova Z., Schaefer A., and Wolf D. H. (2004) A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 5, 692–697 10.1038/sj.embor.7400164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Durcan T. M., Kontogiannea M., Thorarinsdottir T., Fallon L., Williams A. J., Djarmati A., Fantaneanu T., Paulson H. L., and Fon E. A. (2011) The Machado–Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Hum. Mol. Genet. 20, 141–154 10.1093/hmg/ddq452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Koch P., Breuer P., Peitz M., Jungverdorben J., Kesavan J., Poppe D., Doerr J., Ladewig J., Mertens J., Tüting T., Hoffmann P., Klockgether T., Evert B. O., Wüllner U., and Brüstle O. (2011) Excitation-induced ataxin-3 aggregation in neurons from patients with Machado–Joseph disease. Nature 480, 543–546 10.1038/nature10671 [DOI] [PubMed] [Google Scholar]
- 58. Martins S., Calafell F., Wong V. C. N., Sequeiros J., and Amorim A. (2006) A multistep mutation mechanism drives the evolution of the CAG repeat at MJD/SCA3 locus. Eur. J. Hum. Genet. 14, 932–940 10.1038/sj.ejhg.5201643 [DOI] [PubMed] [Google Scholar]
- 59. Ooms J. (2017) magick: Advanced Graphics and Image-Processing in R, R package version 1.4 [Google Scholar]
- 60. Mills J. D., and Janitz M. (2012) Alternative splicing of mRNA in the molecular pathology of neurodegenerative diseases. Neurobiol. Aging 33, 1012.e11–24 10.1016/j.neurobiolaging.2011.10.030 [DOI] [PubMed] [Google Scholar]
- 61. D'Souza I., Poorkaj P., Hong M., Nochlin D., Lee V. M.-Y., Bird T. D., and Schellenberg G. D. (1999) Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci. U.S.A. 96, 5598–5603 10.1073/pnas.96.10.5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cartegni L., and Krainer A. R. (2002) Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN. Nat. Genet. 30, 377–384 10.1038/ng854 [DOI] [PubMed] [Google Scholar]
- 63. De Jonghe C., Cruts M., Rogaeva E. A., Tysoe C., Singleton A., Vanderstichele H., Meschino W., Dermaut B., Vanderhoeven I., Backhovens H., Vanmechelen E., Morris C. M., Hardy J., Rubinsztein D. C., St George-Hyslop P. H., and Van Broeckhoven C. (1999) Aberrant splicing in the presenilin-1 intron 4 mutation causes presenile Alzheimer's disease by increased Aβ42 secretion. Hum. Mol. Genet. 8, 1529–1540 10.1093/hmg/8.8.1529 [DOI] [PubMed] [Google Scholar]
- 64. Singleton A. B., Hall R., Ballard C. G., Perry R. H., Xuereb J. H., Rubinsztein D. C., Tysoe C., Matthews P., Cordell B., Kumar-Singh S., De Jonghe C., Cruts M., van Broeckhoven C., and Morris C. M. (2000) Pathology of early-onset Alzheimer's disease cases bearing the Thr113–114ins presenilin-1 mutation. Brain 123, 2467–2474 10.1093/brain/123.12.2467 [DOI] [PubMed] [Google Scholar]
- 65. West A. B., Maraganore D., Crook J., Lesnick T., Lockhart P. J., Wilkes K. M., Kapatos G., Hardy J. A., and Farrer M. J. (2002) Functional association of the parkin gene promoter with idiopathic Parkinson's disease. Hum. Mol. Genet. 11, 2787–2792 10.1093/hmg/11.22.2787 [DOI] [PubMed] [Google Scholar]
- 66. Beyer K. (2006) α-Synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol. 112, 237–251 10.1007/s00401-006-0104-6 [DOI] [PubMed] [Google Scholar]
- 67. Zuccato C., Liber D., Ramos C., Tarditi A., Rigamonti D., Tartari M., Valenza M., and Cattaneo E. (2005) Progressive loss of BDNF in a mouse model of Huntington's disease and rescue by BDNF delivery. Pharmacol. Res. 52, 133–139 10.1016/j.phrs.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 68. Lim J., Crespo-Barreto J., Jafar-Nejad P., Bowman A. B., Richman R., Hill D. E., Orr H. T., and Zoghbi H. Y. (2008) Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 452, 713–718 10.1038/nature06731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Goedert M., Spillantini M. G., Jakes R., Rutherford D., and Crowther R. A. (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3, 519–526 10.1016/0896-6273(89)90210-9 [DOI] [PubMed] [Google Scholar]
- 70. Sato N., Hori O., Yamaguchi A., Lambert J. C., Chartier-Harlin M. C., Robinson P. A., Delacourte A., Schmidt A. M., Furuyama T., Imaizumi K., Tohyama M., and Takagi T. (1999) A novel presenilin-2 splice variant in human Alzheimer's disease brain tissue. J. Neurochem. 72, 2498–2505 [DOI] [PubMed] [Google Scholar]
- 71. Mueller T., Breuer P., Schmitt I., Walter J., Evert B. O., and Wüllner U. (2009) CK2-dependent phosphorylation determines cellular localization and stability of ataxin-3. Hum. Mol. Genet. 18, 3334–3343 10.1093/hmg/ddp274 [DOI] [PubMed] [Google Scholar]
- 72. Zhou Y. F., Liao S. S., Luo Y. Y., Tang J. G., Wang J. L., Lei L. F., Chi J. W., Du J., Jiang H., Xia K., Tang B. S., and Shen L. (2013) SUMO-1 modification on K166 of polyQ-expanded ataxin-3 strengthens its stability and increases its cytotoxicity. PLoS One 8, e54214 10.1371/journal.pone.0054214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Roscic A., Baldo B., Crochemore C., Marcellin D., and Paganetti P. (2011) Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J. Neurochem. 119, 398–407 10.1111/j.1471-4159.2011.07435.x [DOI] [PubMed] [Google Scholar]
- 74. Riley B. E., Xu Y., Zoghbi H. Y., and Orr H. T. (2004) The effects of the polyglutamine repeat protein ataxin-1 on the UbL-UBA protein A1UP. J. Biol. Chem. 279, 42290–42301 10.1074/jbc.M406284200 [DOI] [PubMed] [Google Scholar]
- 75. Cullen D. A., Leigh P. N., and Gallo J. M. (2004) Degradation properties of polyglutamine-expanded human androgen receptor in transfected cells. Neurosci. Lett. 357, 175–178 10.1016/j.neulet.2003.12.055 [DOI] [PubMed] [Google Scholar]
- 76. Bailey C. K., Andriola I. F. M., Kampinga H. H., and Merry D. E. (2002) Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 11, 515–523 10.1093/hmg/11.5.515 [DOI] [PubMed] [Google Scholar]
- 77. Blount J. R., Tsou W.-L., Ristic G., Burr A. A., Ouyang M., Galante H., Scaglione K. M., and Todi S. V. (2014) Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23. Nat. Commun. 5, 4638 10.1038/ncomms5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang H., Jia N., Fei E., Wang Z., Liu C., Zhang T., Fan J., Wu M., Chen L., Nukina N., Zhou J., and Wang G. (2007) p45, an ATPase subunit of the 19S proteasome, targets the polyglutamine disease protein ataxin-3 to the proteasome. J. Neurochem. 101, 1651–1661 10.1111/j.1471-4159.2007.04460.x [DOI] [PubMed] [Google Scholar]
- 79. Kristensen L. V., Oppermann F. S., Rauen M. J., Hartmann-Petersen R., and Thirstrup K. (2017) Polyglutamine expansion of ataxin-3 alters its degree of ubiquitination and phosphorylation at specific sites. Neurochem. Int. 105, 42–50 10.1016/j.neuint.2016.12.019 [DOI] [PubMed] [Google Scholar]
- 80. Watanabe-Asano T., Kuma A., and Mizushima N. (2014) Cycloheximide inhibits starvation-induced autophagy through mTORC1 activation. Biochem. Biophys. Res. Commun. 445, 334–339 10.1016/j.bbrc.2014.01.180 [DOI] [PubMed] [Google Scholar]
- 81. Kovács J. (1974) Effect of cycloheximide on induced autophagy in epithelial cells of the seminal vesicle of mice. Acta Morphol. Acad. Sci. Hung. 22, 69–75 [PubMed] [Google Scholar]
- 82. Papadopoulos T., and Pfeifer U. (1986) Regression of rat liver autophagic vacuoles by locally applied cycloheximide. Lab. Invest. 54, 100–107 [PubMed] [Google Scholar]
- 83. Lawrence B. P., and Brown W. J. (1993) Inhibition of protein synthesis separates autophagic sequestration from the delivery of lysosomal enzymes. J. Cell Sci. 105, 473–480 [DOI] [PubMed] [Google Scholar]
- 84. Réz G., Pálfia Z., and Fellinger E. (1991) Occurrence and inhibition by cycloheximide of apoptosis in vinblastine-treated murine pancreas: a role for autophagy? Acta Biol. Hung. 42, 133–140 [PubMed] [Google Scholar]
- 85. Zhao J., Zhai B., Gygi S. P., and Goldberg A. L. (2015) mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. U.S.A. 112, 15790–15797 10.1073/pnas.1521919112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shenkman M., Tolchinsky S., Kondratyev M., and Lederkremer G. Z. (2007) Transient arrest in proteasomal degradation during inhibition of translation in the unfolded protein response. Biochem. J. 404, 509–516 10.1042/BJ20061854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Alvarez-Castelao B., Ruiz-Rivas C., and Castaño J. G. (2012) A critical appraisal of quantitative studies of protein degradation in the framework of cellular proteostasis. Biochem. Res. Int. 2012, 823597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Majorek K. A., Kuhn M. L., Chruszcz M., Anderson W. F., and Minor W. (2014) Double trouble: buffer selection and His-tag presence may be responsible for nonreproducibility of biomedical experiments. Protein Sci. 23, 1359–1368 10.1002/pro.2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Warrick J. M., Paulson H. L., Gray-Board G. L., Bui Q. T., Fischbeck K. H., Pittman R. N., and Bonini N. M. (1998) Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 93, 939–949 10.1016/S0092-8674(00)81200-3 [DOI] [PubMed] [Google Scholar]
- 90. Ikeda H., Yamaguchi M., Sugai S., Aze Y., Narumiya S., and Kakizuka A. (1996) Expanded polyglutamine in the Machado–Joseph disease protein induces cell death in vitro and in vivo. Nat. Genet. 13, 196–202 10.1038/ng0696-196 [DOI] [PubMed] [Google Scholar]
- 91. Todi S. V., Winborn B. J., Scaglione K. M., Blount J. R., Travis S. M., and Paulson H. L. (2009) Ubiquitination directly enhances activity of the deubiquitinating enzyme ataxin-3. EMBO J. 28, 372–382 10.1038/emboj.2008.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. DiFiglia M., Sapp E., Chase K. O., Davies S. W., Bates G. P., Vonsattel J. P., and Aronin N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 10.1126/science.277.5334.1990 [DOI] [PubMed] [Google Scholar]
- 93. Skinner P. J., Koshy B. T., Cummings C. J., Klement I. A., Helin K., Servadio A., Zoghbi H. Y., and Orr H. T. (1997) Ataxin-1 with an expanded glutamine tract alters nuclear matrix- associated structures. Nature 389, 971–974 10.1038/40153 [DOI] [PubMed] [Google Scholar]
- 94. Becher M. W., Kotzuk J. A., Sharp A. H., Davies S. W., Bates G. P., Price D. L., and Ross C. A. (1998) Intranuclear neuronal inclusions in Huntington's disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol. Dis. 4, 387–397 10.1006/nbdi.1998.0168 [DOI] [PubMed] [Google Scholar]
- 95. Li M., Miwa S., Kobayashi Y., Merry D. E., Yamamoto M., Tanaka F., Doyu M., Hashizume Y., Fischbeck K. H., and Sobue G. (1998) Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann. Neurol. 44, 249–254 10.1002/ana.410440216 [DOI] [PubMed] [Google Scholar]
- 96. Huynh D. P., Del Bigio M. R., Ho D. H., and Pulst S. M. (1999) Expression of ataxin-2 in brains from normal individuals and patients with Alzheimer's disease and spinocerebellar ataxia 2. Ann. Neurol. 45, 232–241 10.1002/1531-8249(199902)45:2%3C232::AID-ANA14%3E3.0.CO%3B2-7 [DOI] [PubMed] [Google Scholar]
- 97. Holmberg M., Duyckaerts C., Dürr A., Cancel G., Gourfinkel-An I., Damier P., Faucheux B., Trottier Y., Hirsch E. C., Agid Y., and Brice A. (1998) Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions. Hum. Mol. Genet. 7, 913–918 10.1093/hmg/7.5.913 [DOI] [PubMed] [Google Scholar]
- 98. Hübener J., and Riess O. (2010) Polyglutamine-induced neurodegeneration in SCA3 is not mitigated by non-expanded ataxin-3: conclusions from double-transgenic mouse models. Neurobiol. Dis. 38, 116–124 10.1016/j.nbd.2010.01.005 [DOI] [PubMed] [Google Scholar]
- 99. Gloeckner C. J., Boldt K., Schumacher A., Roepman R., and Ueffing M. (2007) A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics 7, 4228–4234 10.1002/pmic.200700038 [DOI] [PubMed] [Google Scholar]
- 100. Brignone C., Bradley K. E., Kisselev A. F., and Grossman S. R. (2004) A post-ubiquitination role for MDM2 and hHR23A in the p53 degradation pathway. Oncogene 23, 4121–4129 10.1038/sj.onc.1207540 [DOI] [PubMed] [Google Scholar]
- 101. Kim Y., Kweon J., Kim A., Chon J. K., Yoo J. Y., Kim H. J., Kim S., Lee C., Jeong E., Chung E., Kim D., Lee M. S., Go E. M., Song H. J., Kim H., et al. (2013) A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 31, 251–258 10.1038/nbt.2517 [DOI] [PubMed] [Google Scholar]
- 102. Kim H., Um E., Cho S. R., Jung C., Kim H., and Kim J. S. (2011) Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat. Methods 8, 941–943 10.1038/nmeth.1733 [DOI] [PubMed] [Google Scholar]
- 103. Boy J., Schmidt T., Wolburg H., Mack A., Nuber S., Böttcher M., Schmitt I., Holzmann C., Zimmermann F., Servadio A., and Riess O. (2009) Reversibility of symptoms in a conditional mouse model of spinocerebellar ataxia type 3. Hum. Mol. Genet. 18, 4282–4295 10.1093/hmg/ddp381 [DOI] [PubMed] [Google Scholar]
- 104. Andersen C. L., Jensen J. L., and Ørntoft T. F. (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250 10.1158/0008-5472.CAN-04-0496 [DOI] [PubMed] [Google Scholar]
- 105. Suzuki K., Bose P., Leong-Quong R. Y., Fujita D. J., and Riabowol K. (2010) REAP: a two minute cell fractionation method. BMC Res. Notes 3, 294 10.1186/1756-0500-3-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Cox J., Matic I., Hilger M., Nagaraj N., Selbach M., Olsen J. V., and Mann M. (2009) A practical guide to the MaxQuant computational platform for silac-based quantitative proteomics. Nat. Protoc. 4, 698–705 10.1038/nprot.2009.36 [DOI] [PubMed] [Google Scholar]
- 107. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 10.1038/nmeth.3901 [DOI] [PubMed] [Google Scholar]
- 108. R Core Team (2017) R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 109. Pau G., Fuchs F., Sklyar O., Boutros M., and Huber W. (2010) EBImage: an R package for image processing with applications to cellular phenotypes. Bioinformatics 26, 979–981 10.1093/bioinformatics/btq046 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.