

ABSTRACT
Systemic amyloidosis comprises an uncommon group of disorders caused by the extracellular deposition of misfolded proteins in various organs. Cardiac amyloid deposition, causing an infiltrative/restrictive cardiomyopathy, is a frequent feature of amyloidosis and a major determinant of survival. It may be the presenting feature of the disease or may be identified while investigating a patient presenting with other organ involvement. The need for a high index of suspicion and the critical importance of precise biochemical typing of the amyloid deposits is paramount in light of recent therapeutic advances that can significantly improve prognosis. Most cases of cardiac amyloidosis are of either transthyretin type, which may be acquired in older individuals or inherited in younger patients, or acquired monoclonal immunoglobulin light chain (AL) type. This article aims to review recent developments in diagnosis and management of cardiac amyloidosis.
KEYWORDS: AL amyloidosis, ATTR amyloidosis, cardiac amyloidosis, CMR, infiltrative cardiomyopathy
Introduction
Systemic amyloidosis comprises a family of diseases caused by deposition of misfolded fibrillar proteins in the extracellular space.1 More than 30 different precursor proteins can undergo the substantial molecular transformation to form amyloid fibrils in vivo,2 the resultant diseases being classified and having a clinical phenotype relating to the protein in question.
Two types of amyloid commonly infiltrate the heart: immunoglobulin light chain (AL) amyloidosis and transthyretin (ATTR) amyloidosis. Cardiac involvement is the leading cause of morbidity and mortality in systemic amyloidosis, regardless of the underlying pathogenesis of amyloid production.3 Cardiac amyloidosis is a myocardial disease characterised by extracellular amyloid infiltration throughout the heart. The infiltrative process results in biventricular wall thickening with concentric ventricular remodelling and low cardiac output. The subsequent elevation of pressure in the atria is associated with atrial dilatation. Intramyocardial vessels are frequently infiltrated by amyloid, causing reduced myocardial perfusion.4 The conduction system is also frequently disrupted, with atrial arrhythmias (fibrillation, flutter or atrial tachycardia) and atrioventricular conduction delays being common. Ventricular arrhythmias may also occur although sustained ventricular tachycardia is identified infrequently.
Cardiac AL amyloidosis
Light chain amyloidosis is consequent on a clonal plasma cell proliferative disorder in which misfolded immunoglobulin light chains are deposited as amyloid fibrils in multiple organs, including the heart in about half of cases. Cardiac dysfunction in AL amyloidosis results from extracellular infiltration of the myocardium, but there is often also evidence for a cardiotoxic effect exerted by pre-fibrillar light chain aggregates. The severity of cardiac dysfunction is the major determinant of morbidity and mortality.3
Light chain amyloidosis is the most commonly diagnosed type of cardiac amyloidosis. One or many vital organ systems may be involved, commonly the kidneys, liver, peripheral and autonomic nervous systems and soft tissues. The heart is frequently affected and is the only clinically involved organ in some patients.
Clinical presentation reflects the varying multisystem deposition of amyloid. Examination findings may reflect soft tissue and small vessel amyloid infiltration and include macroglossia, periorbital pupura, submandibular gland enlargement and nail dystrophy. Fatigue and weight loss are common. Hepatic or splenic infiltration may cause palpable organomegaly. Renal dysfunction is common, usually presenting as nephrotic range proteinuria.
Early cardiac amyloidosis is a major diagnostic challenge. The classical features of ‘right-sided’ congestive heart failure may not be evident until cardiac disease is very advanced. Elevated jugular venous pressure, a third heart sound, hepatomegaly and peripheral oedema may be very subtle or absent in patients who have already started diuretics.
Peripheral neuropathy is relatively common, presenting with paraesthesia or dysaesthesia typically in a ‘glove and stocking’ distribution. Autonomic neuropathy is an important diagnostic clue, manifesting as orthostatic hypotension, alternating diarrhoea and constipation and erectile dysfunction.
Monoclonal immunoglobulin or free light chains can be identified in the serum and/or urine of at least 95% of patients using sensitive assays, but are often missed in routine serum electrophoresis. The absence of a detectable clone is problematic for diagnosis and monitoring response to treatment.
Cardiac transthyretin amyloidosis
Transthyretin is a liver-derived plasma protein that in its wildtype (wt) form is the amyloid fibril precursor protein in the non-hereditary, late onset, predominantly male syndrome of cardiac ATTRwt amyloidosis. Genetic variants of transthyretin are associated with the dominantly inherited syndromes of familial amyloid polyneuropathy and familial amyloid cardiomyopathy, which can present from young adulthood onwards. The prognosis of cardiac ATTR amyloidosis is better than cardiac AL amyloidosis, with median survival typically 3–5 years from diagnosis.5
Wildtype transthyretin amyloidosis
Non-hereditary transthyretin-related amyloidosis is commonly referred to as senile systemic amyloidosis (SSA) because of its late age of onset (usually after the seventh decade of life). ATTRwt has a strong male predominance, with between 25 and 50:1 male:female expression.6 The true prevalence of ATTRwt amyloidosis is unknown, but it is likely to be underdiagnosed in many cases. Prevalence is increasing with the increasingly aging population and emerging diagnostic tools, including cardiovascular magnetic resonance imaging and Technetium-99m-3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD) scintigraphy.7,8
Despite being a systemic disease, the heart is generally the only organ involved clinically, with the exception of carpal tunnel syndrome and some other musculoskeletal disorders – including lumbar canal stenosis, which may precede cardiac symptoms by 10–15 years.
An incidental clonal plasma cell dyscrasia is present in up to a quarter of ATTRwt amyloidosis patients reflecting their advanced years and raising the possibility of AL amyloidosis.9 A cardiac biopsy is often needed in such patients to obtain a definitive diagnosis.8
Hereditary transthyretin amyloidosis
The transthyretin (TTR) gene is located on chromosome 18 and contains four exons and five introns. There are more than 120 mutations in the TTR gene, most of which encode pathogenic variants of TTR with increased amyloidogenic potential. However, only a few of these variants are responsible for the majority of cases of hereditary ATTR worldwide, notably Val30Met, Thr60Ala, Ser77Tyr and Val122Ile.10,11
The specific site of an amino acid substitution determines the phenotype of the disease. Inheritance is autosomal dominant with variable penetrance. Correlation between genotype and phenotype is strong for certain mutations, whereas with others the clinical features can vary substantially, even within a given family. The onset occurs from the 3rd decade, most commonly after the age of 40 years. In some forms, peripheral neuropathy or autonomic dysfunction may predominate, with cardiac amyloid being either absent or limited to the conduction system. The prevalent mutation in the UK and the USA, Thr60Ala (substitution of alanine for threonine at position 60), can present with predominant cardiomyopathy characterised by heart failure and conduction system disturbances, autonomic dysfunction or peripheral neuropathy, or any combination of these features.12
The TTR Val30Met variant is the commonest cause of familial amyloid polyneuropathy in the world, with large foci in Portugal, Japan and Sweden. The disease usually presents with peripheral and autonomic neuropathy without cardiac involvement, especially in patients younger than 50 years of age although cardiac amyloidosis does occur in older cases.13
Worldwide, a polymorphism comprising substitution of isoleucine for valine at position 122 (Val122Ile) is particularly notable. Approximately 4% of African Americans and probably a similar proportion of African-Caribbeans are heterozygous for this variant, which appears to be associated with a greatly increased risk of developing late-onset cardiac ATTR amyloidosis that is clinically indistinguishable from the wildtype form.14 There is a high prevalence of hypertensive heart disease in this population, which may lead to a possible diagnosis of amyloidosis being overlooked. The presence of right ventricular thickening, the absence of left ventricular hypertrophy on the electrocardiogram (ECG), and the clinical finding of right heart failure (particularly if there is history of carpal tunnel syndrome) should suggest the diagnosis.
Investigations and diagnosis
The diagnosis of amyloidosis relies on a high index of clinical suspicion. Unfortunately, the disease is frequently asymptomatic until a late stage and, even then, the symptoms can be very nonspecific.
Histology
Diagnosis of systemic amyloidosis usually requires histological confirmation of amyloid deposition through Congo red staining, which produces the pathognomonic green birefringence when viewed under cross-polarised light (Fig 1). Amyloid deposits can usually be identified in biopsies of malfunctioning organs, but relatively non-invasive ‘screening’ biopsies provide an alternative, notably including abdominal fat aspirates that can identify amyloid deposits in 60–80% of patients with systemic AL amyloidosis although fewer with ATTR type.15 Cardiac biopsy provides the most definitive diagnostic evidence in amyloid cardiomyopathy and, if the diagnosis is not confirmed by biopsy of another tissue, endomyocardial biopsy is a safe and relatively simple procedure in skilled hands.
Fig 1.

Congo red staining of myocardial tissue from two patients with amyloid cardiomyopathy. (a) Transthyretin type amyloidosis; (b) immunoglobulin light chain amyloidosis. Top panel = light microscopy; bottom panel = polarised light microscopy.
Serum biomarkers
Cardiac biomarkers may be helpful in raising clinical suspicion of cardiac amyloidosis in a patient with a known plasma cell dyscrasia and should prompt further investigation with ECG and echocardiography. The combination of serum NT-proBNP (N-terminal prohormone of brain natriuretic peptide) and troponin is useful to stratify prognosis and guide treatment strategies in AL amyloidosis.16
Electrocardiography
Because the thickening of the ventricular wall in amyloidosis is due to myocardial infiltration rather than hypertrophy, ECG voltages tend to decrease as the disease progresses. Low voltage on the ECG (defined as all limb leads <5 mm in height) is found in a high proportion of patients and is often associated with extreme left- or right-axis deviation (Fig 2). Although voltage criteria for left ventricular hypertrophy are extremely uncommon in patients with AL amyloidosis, they are present in up to a quarter of patients with ATTR amyloidosis. Other common observations include repolarisation abnormalities, anterior, inferior or lateral pseudoinfarction pattern, left anterior hemiblock, ischemic or nonspecific T wave abnormalities, and rhythm disturbances, particularly atrial fibrillation.
Fig 2.
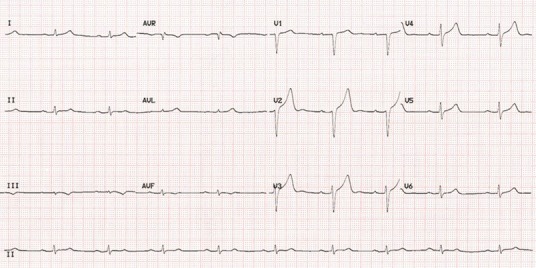
Electrocardiogram of a patient with cardiac transthyretin type amyloidosis, showing sinus bradycardia with first degree atrioventricular block and small QRS voltages (defined as ≤6 mm height), predominantly in the limb leads.
Echocardiography
The cardiac amyloid phenotype is a thick-walled ventricle, small left ventricular chamber volume, valve thickening, atrial enlargement and signs of elevated filling pressures with a restrictive diastolic filling. Interventricular septal thickness of >12 mm, in the absence of aortic valve disease or significant systemic hypertension, is widely used as an echocardiographic criterion to identify cardiac involvement in patients with systemic AL amyloidosis.17
Longitudinal strain measurement by tissue Doppler and echocardiographic speckle tracking have emerged as useful clinical tools for the identification of cardiac amyloidosis and can assist in differentiation from other causes of wall thickening, including hypertension and hypertrophic cardiomyopathy. Two-dimensional (2D) strain mapping shows relative preservation of apical function, which can be an early clue to amyloidosis because it gives rise to a ‘bulls-eye’ pattern when the segmental strain is plotted, which is rare in other cardiomyopathies (Fig 3).
Fig 3.

Echocardiography findings in a patient with cardiac transthyretin type amyloidosis. Parasternal long axis view (a) and four chamber view (b), showing concentric left ventricular hypertrophy; pulse-wave Doppler showing restrictive flow pattern of left ventricular inflow (c); and strain pattern characteristic of an infiltrative process (d).
However, echocardiography alone is often unable to distinguish the numerous pathologies that result in increased wall thickness, prompting multimodality assessment.
Cardiovascular magnetic resonance
Cardiovascular magnetic resonance (CMR) has unique advantages in identifying cardiac involvement in systemic amyloidosis. CMR uses its intrinsic capacity to characterise tissue on the basis of fundamental MR properties (T1 and T2), and these intrinsic properties can be strengthened by administration of gadolinium-based contrast agents (Fig 4).18 The latest MR techniques for evaluating late gadolinium enhancement provide images that are almost pathognomonic in AL and ATTR cardiac amyloidosis with excellent diagnostic accuracy.19 CMR is especially useful in patients with increased left ventricular wall thickness and/or hypertrophy because it can differentiate different cellular and interstitial pathologies, which is not possible on echocardiography.20
Fig 4.

Cardiac magnetic resonance findings in a patient with cardiac immunoglobulin light chain amyloidosis. Four-chamber steady-state free precession cine (a); corresponding native T1 map (b); corresponding phase sensitive inversion recovery reconstruction late gadolinium enhancement image showing subendocardial late gadolinium enhancement (c); and corresponding extracellular volume map.
Fig 5.

Whole-body anterior 99mTc-DPD scintigraphy (a) and single photon emission computerised tomography-CT (b) showing Perugini grade 2 abnormal uptake on a patient with cardiac transthyretin type amyloidosis.
Recently, the role of CMR in systemic amyloidosis has evolved beyond just diagnostic utility. Amyloid infiltration results in expansion of the extracellular space, which can be visualised with late gadolinium enhancement and measured with T1 mapping,21 providing a novel tool to monitor amyloid load in the heart and track response to treatment.7,22
Radionuclide bone scintigraphy
Bone tracer scintigraphy using 99mTc-DPD has recently emerged as a sensitive tool for the identification of cardiac ATTR amyloidosis. Although the basis for localisation of these agents to cardiac amyloid remains unknown, the technique appears to be very sensitive for imaging cardiac ATTR amyloid deposits and is even able to identify presymptomatic disease.
99mTc-DPD scintigraphy is probably positive in all patients with clinically significant cardiac ATTR amyloidosis; however, it is not completely specific for this condition and cardiac localisation does occur in a small proportion of patients with advanced cardiac AL amyloidosis and cardiac ApoA1 amyloidosis.8 Cardiac ATTR amyloidosis can be diagnosed in the absence of histological proof when there is heart failure associated with an echocardiogram or CMR that is consistent with amyloidosis, grade 2 or 3 tracer uptake in the heart on radionuclide bone scintigraphy, and absence of a detectable monoclonal immunoglobulin in the blood and urine. Histological confirmation and typing of amyloid should be pursued in cases that fail to meet these criteria.8
Treatment
Supportive treatment
Patient education and multidisciplinary care are essential. Salt and fluid restriction, daily weights and titrating combinations of different types of diuretic are key to managing heart failure symptoms. Conventional heart failure medications, including beta-blockers, angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are poorly tolerated in cardiac amyloidosis and commonly reduce cardiac output, worsening symptoms.
Orthostatic hypotension can be treated with midodrine and compression stockings, whereas fludrocortisone merely tends to exacerbate fluid retention.
Disease-modifying treatment for AL amyloidosis
The mainstay of treatment in AL amyloidosis is chemotherapy directed towards the underlying clonal plasma cell population. The past decade has seen many new drugs developed for multiple myeloma being adopted for treatment of AL amyloidosis although few randomised controlled trials have been performed in the latter. Frequent monitoring of serum-free light chains enables the haematological response to be ascertained and ineffective treatment to be modified rapidly. While median survival has nearly doubled over the past decade, nearly 25% of patients die within months of diagnosis.
Bortezomib, a proteasome inhibitor used alone or usually in combination with other agents, has emerged as the most widely used first-line treatment in AL amyloidosis and is associated with haematological response rates of up to 90%, with 60% achieving complete response or very good partial response.23 No particular regimen has yet been shown to be superior in very high-risk (stage IIIb) patients.20,24
Other combination regimens may be more appropriate in patients with significant peripheral or autonomic neuropathy, given the potential neurotoxicity of bortezomib, and in patients with underlying lymphoplasmacytic lymphoma.
High-dose melphalan with autologous stem cell transplantation is associated with the most durable remissions, but strict selection of patients is paramount because of the risk of treatment-related mortality among older and sicker patients, including those with advanced cardiac or autonomic nerve involvement.25
Disease-modifying treatment for ATTR amyloidosis
Treatment of patients with hereditary ATTR amyloidosis remains unsatisfactory.
Liver transplantation to remove the source of variant TTR is the treatment of choice in younger patients with early neuropathy associated with the Val30Met variant. It can be beneficial in some patients with other TTR mutations, but cardiac amyloidosis present at the time of liver transplantation continues to progress relentlessly in most cases.
Cardiac disease is the dominant feature ATTRwt and V122I-related ATTR amyloidosis, and rare younger patients can do very well after cardiac transplantation. However, most patients are too old for this to be feasible.
Several promising therapies are in development for ATTR amyloidosis. In vitro studies suggest that the amyloidogenic misfolding of TTR can be inhibited by compounds that bind to TTR in the plasma, by maintaining its normal tetrameric structure. In small phase II and III trials, tafamidis, diflunisal and a combination of doxycycline and tauro-ursodeoxycholic acid have been associated with slower progression of neuropathic disease in ATTR amyloidosis.
Transthyretin is almost exclusively synthesised by the liver, which is the anatomical site most efficiently targeted by novel RNA-inhibiting therapies. Small interfering RNA therapy and anti-sense oligonucleotide therapy are currently in development and both can reduce production of TTR by more than 80%. Phase III trials in familial amyloid polyneurophathy are currently nearing completion.
Immunotherapy
Therapeutic antibodies have potential to target amyloid deposits directly. Mu11-1F4 is a chimeric antibody that binds to many AL fibrils and has lately completed a phase I clinical trial.26 A phase I/II study of another monoclonal antibody, NEOD001 (Onclave Therapeutics Limited, California), has also recently been completed in patients with AL amyloidosis, reporting good safety and tolerability and a suggestion of cardiac improvement. A global phase III randomised trial is currently in progress.
Serum amyloid P (SAP) component binds to all amyloid fibrils in vitro and protects them from degradation. (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl] pyrrolidine-2-carboxylic acid efficiently depletes SAP from the blood but leaves some SAP in the amyloid deposits. SAP in the amyloid deposits can then be specifically targeted by fully human monoclonal anti-SAP antibodies. Results of a phase I trial using the obligate combination of these agents yielded promising results, showing marked and rapid reduction in amyloid deposits in the liver and elsewhere.27 No serious adverse events were observed. A phase II study to investigate patients with cardiac amyloidosis is in progress.
Conclusions
Cardiac amyloidosis is challenging to diagnose and treat. Improved awareness and developments in CMR and DPD scintigraphy are enabling earlier diagnosis. There has been remarkable progress in treatment for AL amyloidosis but the prognosis of the 40% of patients who present with advanced cardiac involvement remains poor. A variety of novel specific therapies are on the near horizon, with potential to both inhibit new amyloid formation and enhance clearance of existing deposits.
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- 1.Lachmann HJ. Hawkins PN. Systemic amyloidosis. Curr Opin Pharmacol. 2006;6:214–20. doi: 10.1016/j.coph.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Sipe JD. Benson MD. Buxbaum JN, et al. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid. 2014;21:221–4. doi: 10.3109/13506129.2014.964858. [DOI] [PubMed] [Google Scholar]
- 3.Fontana M. Banypersad SM. Treibel TA, et al. Differential myocyte responses in patients with cardiac transthyretin amyloidosis and light-chain amyloidosis: a cardiac MR imaging study. Radiology. 2015;277:388–97. doi: 10.1148/radiol.2015141744. [DOI] [PubMed] [Google Scholar]
- 4.Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005;112:2047–60. doi: 10.1161/CIRCULATIONAHA.104.489187. [DOI] [PubMed] [Google Scholar]
- 5.Rapezzi C. Merlini G. Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120:1203–12. doi: 10.1161/CIRCULATIONAHA.108.843334. [DOI] [PubMed] [Google Scholar]
- 6.Ruberg FL. Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126:1286–300. doi: 10.1161/CIRCULATIONAHA.111.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fontana M. Banypersad SM. Treibel TA, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging. 2014;7:157–65. doi: 10.1016/j.jcmg.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 8.Gillmore JD. Maurer MS. Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404–12. doi: 10.1161/CIRCULATIONAHA.116.021612. [DOI] [PubMed] [Google Scholar]
- 9.Pinney JH. Whelan CJ. Petrie A, et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013;2:e000098. doi: 10.1161/JAHA.113.000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connors LH. Lim A. Prokaeva T. Roskens VA. Costello CE. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid. 2003;10:160–84. doi: 10.3109/13506120308998998. [DOI] [PubMed] [Google Scholar]
- 11.Merlini G. Westermark P. The systemic amyloidoses: clearer understanding of the molecular mechanisms offers hope for more effective therapies. J Intern Med. 2004;255:159–78. doi: 10.1046/j.1365-2796.2003.01262.x. [DOI] [PubMed] [Google Scholar]
- 12.Sattianayagam PT. Hahn AF. Whelan CJ, et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J. 2012;33:1120–7. doi: 10.1093/eurheartj/ehr383. [DOI] [PubMed] [Google Scholar]
- 13.Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75:408–27. doi: 10.1093/brain/75.3.408. [DOI] [PubMed] [Google Scholar]
- 14.Connors LH. Prokaeva T. Lim A, et al. Cardiac amyloidosis in African Americans: comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am Heart J. 2009;158:607–14. doi: 10.1016/j.ahj.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Guy CD. Jones CK. Abdominal fat pad aspiration biopsy for tissue confirmation of systemic amyloidosis: specificity, positive predictive value, and diagnostic pitfalls. Diagn Cytopathol. 2001;24:181–5. doi: 10.1002/1097-0339(200103)24:3<181::aid-dc1037>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 16.Comenzo RL. Reece D. Palladini G, et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia. 2012;26:2317–25. doi: 10.1038/leu.2012.100. [DOI] [PubMed] [Google Scholar]
- 17.Gertz MA. Comenzo R. Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol. 2005;79:319–28. doi: 10.1002/ajh.20381. [DOI] [PubMed] [Google Scholar]
- 18.Fontana M. Martinez-Naharro A. Hawkins PN. Staging Cardiac amyloidosis with CMR: understanding the different phenotypes. JACC Cardiovasc Imaging. 2016;9:1278–9. doi: 10.1016/j.jcmg.2016.02.035. [DOI] [PubMed] [Google Scholar]
- 19.Fontana M. Chung R. Hawkins PN. Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20:133–44. doi: 10.1007/s10741-014-9470-7. [DOI] [PubMed] [Google Scholar]
- 20.Wechalekar AD. Schonland SO. Kastritis E, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood. 2013;121:3420–7. doi: 10.1182/blood-2012-12-473066. [DOI] [PubMed] [Google Scholar]
- 21.Fontana M. Pica S. Reant P, et al. Prognostic Value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2015;132:1570–9. doi: 10.1161/CIRCULATIONAHA.115.016567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banypersad SM. Sado DM. Flett AS, et al. Quantification of myocardial extracellular volume fraction in systemic AL amyloidosis: an equilibrium contrast cardiovascular magnetic resonance study. Circ Cardiovasc Imaging. 2013;6:34–9. doi: 10.1161/CIRCIMAGING.112.978627. [DOI] [PubMed] [Google Scholar]
- 23.Venner CP. Lane T. Foard D, et al. Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood. 2012;119:4387–90. doi: 10.1182/blood-2011-10-388462. [DOI] [PubMed] [Google Scholar]
- 24.Venner CP. Gillmore JD. Sachchithanantham S, et al. A matched comparison of cyclophosphamide, bortezomib and dexamethasone (CVD) versus risk-adapted cyclophosphamide, thalidomide and dexamethasone (CTD) in AL amyloidosis. Leukemia. 2014;28:2304–10. doi: 10.1038/leu.2014.218. [DOI] [PubMed] [Google Scholar]
- 25.Gertz MA. Lacy MQ. Dispenzieri A, et al. Refinement in patient selection to reduce treatment-related mortality from autologous stem cell transplantation in amyloidosis. Bone Marrow Transplant. 2013;48:557–61. doi: 10.1038/bmt.2012.170. [DOI] [PubMed] [Google Scholar]
- 26.O’Nuallain B. Hrncic R. Wall JS. Weiss DT. Solomon A. Diagnostic and therapeutic potential of amyloid-reactive IgG antibodies contained in human sera. J Immunol. 2006;176:7071–8. doi: 10.4049/jimmunol.176.11.7071. [DOI] [PubMed] [Google Scholar]
- 27.Richards DB. Cookson LM. Berges AC, et al. Therapeutic clearance of amyloid by antibodies to serum amyloid p component. N Engl J Med. 2015;373:1106–14. doi: 10.1056/NEJMoa1504942. [DOI] [PubMed] [Google Scholar]