

Abstract
Epigenetic mechanisms including DNA methylation are critical regulators of organismal development and tissue homeostasis. DNA methylation is the transfer of methyl groups to cytosines, which adds an additional layer of complexity to the genome. DNA methylation marks are recognized by the cellular machinery to regulate transcription. Disruption of DNA methylation with aging or exposure to environmental toxins can change susceptibility to disease or trigger processes that lead to disease. In this review, we provide an overview of the DNA methylation machinery. More specifically, we describe DNA methylation in the context of prostate development, prostate cancer, and benign prostatic hyperplasia (BPH) as well as the impact of dietary and environmental factors on DNA methylation in the prostate.
Keywords: Epigenetics, DNMT1, BPH, aging, prostate cancer, development, demethylase
This review has been designed as an introduction to the field of DNA methylation for prostate researchers. We provide a historical perspective by following advances in DNA methylation research from the inception of the field to the recent discoveries involving DNA methylation and prostate cancer. We summarize the functions of key elements of the DNA methylation machinery and their interactions with the histone modification machinery. The role of DNA methylation as a dynamic regulator of development, homeostasis and disease has been described using research carried out in several organ systems including the prostate. Aging is a major risk factor for prostate cancer and benign prostatic hyperplasia (BPH). This review touches on DNA methylation changes occurring during aging, prostate cancer and BPH. We also summarize research on how environmental toxins affect early prostate development with accompanying changes in DNA methylation and the impact of diets enriched with the methyl donor folic acid on prostate homeostasis and susceptibility to prostate cancer.
Epigenetics: historical perspective
Developmental biologists in the early-twentieth century grappled with the question of how cells containing the same genetic code could behave so differently and develop specialized functions. The term epigenetics, meaning ‘above the genome’, was coined by Waddington in 1942 to explain how non-coding changes to genetic material could direct cell specialization during development [1]. In recent times, definitions of epigenetics have moved beyond developmental biology to encompass other fields including cancer biology and toxicology. One widely accepted definition states that epigenetics is “the study of changes in gene function that are mitotically and/or meiotically heritable and that do not entail change in DNA sequence” [2,3]. The modern field of epigenetics has grown to include covalent modifications to histones and DNA as well as non-coding RNAs which act together to finely modulate gene expression.
Introduction to DNA methylation
DNA methylation is the addition of methyl groups to the 5’ position of cytosine bases in DNA, preferentially at CpG dinucleotide sites [4]. 60% of CpG dinucleotides in the mammalian genome are methylated [5]. CpG dinucleotides often reside in clusters, or CpG islands, which are generally unmethylated and associated with gene promoters. DNA methylation is initiated by a class of enzymes called DNA methyltransferases. DNA methyltransferase-1 (DNMT1) preferentially carries out methylation at hemi-methylated DNA sites and preserves methylation patterns during cell replication. DNMT3A and DNMT3B have de novo methylation activity (methyl group addition to unmodified DNA) and are required for mammalian development [6]. Another DNMT3 family member, DNMT3L is catalytically inactive, but cooperates with DNMT3A and DNMT3B during de novo methylation [7,8].
Transfer of methyl groups to cytosines in DNA is a highly conserved process. Methyltransferases bind to DNA, ‘flip’ out cytosine from the DNA helix and attach the methyl group from S-adenosyl methionine to the 5’ position of cytosine. The N-terminal regulatory domain of the DNMTs contains motifs for protein-DNA interactions, protein-protein interactions and nuclear localization. The C-terminal region contains conserved motifs that define the enzyme active site. The N-terminal region of DNMT3L shares similarities with that of DNMT3A and DNMT3B but lacks the catalytic active site in its C-terminal domain.
Adding methylation marks
Broadly, the division of labor between the DNA methyltransferases is clear. DNMT3A and DNMT3B methylate the genome de novo during early development and gametogenesis. DNMT1, with its preference for hemi-methylated DNA, propagates methylation patterns during tissue growth. We will briefly summarize the cellular localization, binding partners and major functions of the DNA methyltransferases below:
DNMT1
DNMT1 is a large protein comprised of ~1620 amino acids. During S-phase, DNMT1 localizes to replication forks and assumes a distinct punctate nuclear staining pattern. During other stages of the cell cycle, DNMT1 nuclear staining is diffuse. The N-terminal region of DNMT1 contains a replication foci targeting sequence (RFTS) that targets it to sites of replication [9]. Autoinhibition of the methyltransferase domain of DNMT1 by interaction with the RFTS domain determines substrate specificity. Conformational changes in the N-terminal region inhibit catalytic activity when DNMT1 is bound to unmethylated CpG substrates. This inhibition is released when DNMT1 is bound to hemi-methylated CpG substrates [10]. Preferential localization to replication foci and substrate specificity for hemi-methylated DNA allows DNMT1 to stably propagate tissuespecific methylation patterns. DNMT1 protein abundance changes during the cell cycle (peaking in S-phase) and is present but not particularly abundant in non-replicating cells [11]. During fetal prostate development, Dnmt1 is initially expressed in the urethral mesenchyme but shifts to the developing epithelial prostate buds at later stages [12].
Knockout, mutation, and inhibition studies have provided key insights into the role of DNMT1 in maintaining global methylation. Targeted mutation of Dnmt1 in mouse embryonic stem (ES) cells reduces 5-methylcytosine (5mC) 3-fold without affecting cell viability. Mouse embryos carrying the same targeted mutation have a similar 3-fold reduction in 5mC levels but are not viable [13]. In contrast to mouse ES cells, DNMT1 knockout in human ES cells triggers apoptosis [14]. Conditional deletion of Dnmt1 in mouse fibroblasts leads to severe hypomethylation, p53-dependent apoptosis and aberrant gene expression [15]. Loss of DNMT1 results in elevated mutation rates, genomic instability [16], microsatellite instability [17], DNA damage [18] and cell cycle arrest [19,20]. DNMT1 activity and expression is upregulated in prostate cancer, where it serves to methylate and suppress key tumor suppressor genes [21].
Mutations in the RFTS domain of DNMT1 have been linked to two neurodegenerative syndromes: autosomal dominant cerebellar ataxia-deafness and narcolepsy (ADCA-DN) and hereditary sensory neuropathy with dementia and hearing loss (HSN1E) [22-25]. These mutations prevent DNMT1 from binding to heterochromatin, leading to aberrant methylation patterns and DNMT1 protein aggregation in the cytosol [25]. There is increasing evidence for the association of single nucleotide polymorphisms of DNA methyltransferases with incidence of different tumor types, including prostate cancer [26-28].
DNMT3 family
The DNMT3 family of methyltransferases is comprised of DNMT3A, DNMT3B and DNMT3L. Except for conserved motifs in the C-terminal catalytic region, the DNMT3 enzymes do not share much sequence similarity with DNMT1 [29]. Their N-terminal domains are considerably shorter than that of DNMT1 and lack the regulatory elements that confer specificity towards hemi-methylated DNA. As a result, DNMT3A and DNMT3B show no preference for hemi-methylated DNA over unmethylated DNA and are called de novo methyltransferases [30,31]. DNMT3L is catalytically inactive but functions as a co-regulator with DNMT3A and DNMT3B. DNMT3A localizes to pericentromeric heterochromatin while DNMT3B is diffusely localized in the nucleus [32]. DNMT3A and DNMT3B expression is abundant in undifferentiated embryonic cells but is barely detectable in differentiated and adult tissues [29]. DNMT3L is highly expressed during gametogenesis [33].
DNMT3A and DNMT3B are essential for de novo methylation, transcriptional repression, and early embryonic development. Inactivation of Dnmt3a and Dnmt3b, simultaneously but not individually, blocks de novo methylation, suggesting that these enzymes have overlapping functions. DNMT3B is essential for early embryonic development as Dnmt3b knockout mice are embryonic lethal. In contrast, Dnmt3a knockout mice live up to 4 weeks of age. Dnmt3a and Dnmt3b double knockout embryos die during mid-gestation. DNMT3A and DNMT3B participate in transcriptional repression in association with histone deacetylases [32]. DNMT3L, although lacking in catalytic activity, stimulates de novo methylation by DNMT3A [7]. In the developing prostate, Dnmt3a and Dnmt3b are predominantly expressed in the urethral mesenchyme during fetal stages but localizes to prostate bud tips postnatally [12].
DNMT3A mutations have been observed in acute myeloid leukemia where they are associated with reduced survival [34,35]. DNMT3B mutations are observed in patients with ICF (immunodeficiency, centromere instability and facial anomalies) syndrome. Loss of DNMT3B function in ICF syndrome is associated with changes in promoter methylation, altered histone marks and aberrant gene expression [36].
Removing methylation marks
DNA methylation is a reversible modification. When DNMT1 is inhibited, cells passively lose methyl marks over several rounds of replication. The ten-eleven translocation (TET) family of methylcytosine dioxygenases catalyze the active removal of methyl marks from DNA independent of replication. During early prostate development, Tet1 and Tet2 are expressed predominantly in the urethral mesenchyme while Tet3 is expressed in the urethral epithelium [12]. TET1, TET2 and TET3 catalyze the conversion of 5mC to 5-hydroxymethylcytosine (5hmC) and subsequently into 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) [37,38]. 5caC is recognized and excised by thymine DNA glycosylase (TDG) [39].
There is increasing evidence that 5hmC participates in transcriptional regulation [40]. 5hmC is highly enriched in brain tissue where it is required for neuronal differentiation [41], learning and memory [42]. 5hmC marks are abundant in embryonic stem cells, where they are enriched at enhancers and gene bodies [43]. A recent study has shown that 5hmC localizes to sites of DNA damage and promotes genome stability [44]. 5hmC levels are dramatically reduced in human cancers, correlating with impaired TET activity [45]. 5hmC is significantly reduced in prostate cancer and has been proposed as a biomarker for prostate cancer detection. In contrast, 5fC and 5caC are increased in prostate cancer [46,47].
Active DNA demethylation can also be catalyzed by 5-methylcytosine deaminases like activation-induced cytidine deaminase (AICDA) and apolipoprotein B mRNA editing enzyme catalytic subunit 1 (APOBEC1) [48]. Deamination of 5mC yields thymine which is immediately replaced with unmethylated cytosine by DNA glycosylases like TDG [49].
Methyl CpG binding domain (MBD) proteins
The MBD proteins are readers of 5mC marks. The mammalian MBD family is comprised of MeCP2, MBD1, MBD2, MBD3 and MBD4 which possess a common Methyl CpG binding domain (MBD). MBD proteins bind to sites of DNA methylation and recruit enzymes for transcriptional repression [50].
DNA methylation and histone modifications
There are two major paradigms of gene silencing by DNA methylation. One, DNA methylation of CpGs in regulatory regions can prevent binding of transcription factor complexes required for active gene expression. Second, methyl CpG binding domain (MBD)-containing proteins recruit chromatin modifiers to sites of DNA methylation to establish or maintain repressed chromatin states. Conversely, DNA methylation activity can be recruited to repressed chromatin to methylate adjacent DNA and reinforce gene silencing.
Chromatin modifiers add post-translational modifications to histones that relax or compact chromatin. Active transcription occurs from relaxed or euchromatin regions while transcription is repressed in compacted heterochromatin regions. Histone acetyltransferases acetylate lysines on histones, which loosens the interaction between DNA and histones, resulting in an accessible, open chromatin configuration. This is reversed by histone deacetylases (HDACs), resulting in gene silencing. Histone methyltransferases add methyl groups to lysines and arginines of Histone H3 and H4. H3K9 and H3K27 di- and tri-methylation and H4K20 tri-methylation repress transcriptional activity by blocking transcriptional factors from accessing DNA. MBD proteins (MECP2, MBD1, MBD2, MBD3, MBD4) bridge the gap between DNA methylation and histone modifications. MECP2 and MBD2 binds to methylated DNA and recruit HDAC activity to silence genes [51,52]. MBD proteins can also recruit histone methyltransferases to sites of DNA methylation. MBD1 recruits the SUV39H1-HP1 heterochromatin complex, which has histone methyltransferase activity, to sites of DNA methylation for transcriptional repression [53]. DNMT1 itself has been shown to interact with chromatin modifiers during DNA replication. DNMT1 forms a repressive complex by direct interaction with the transcriptional repressors HDAC2 and DMAP1 (DNMT1 associated protein). Through this complex, DNMT1 targets the transcriptional repressors to replication foci to maintain heterochromatin regions after DNA replication [54]. Thus, DNA methylation and histone modifications function coordinately in transcriptional repression.
DNA methylation in development
Methylation dynamics in early development
For sexual reproduction, DNA methylation marks need to be erased and reset in early germ cells [55]. Following erasure, sperm- or oocyte-specific methylation patterns are re-established. Imprinted genes, which are expressed from either the maternal or paternal allele, are methylated during gametogenesis by the de novo methyltransferases DNMT3A and DNMT3B [8,33,56].
Following fertilization, the sex-specific methylation patterns of the sperm and oocyte are erased and reset in the zygote. The zygote undergoes a wave of global demethylation until it reaches the blastocyst stage. De novo methylation is carried out by DNMT3A and DNMT3B in the post-implantation embryo. Cells continue to gain DNA methylation as they differentiate. Inheritable methyl marks are maintained by DNMT1 during cell replication.
Genomic imprinting
In mammals, there are over 100 genes that are expressed solely from the maternal or paternal allele. The expression of genes in a parent-of-origin specific manner is regulated by a process called genomic imprinting [57]. Genomic imprinting controls the dosage of key genes involved in growth and metabolism [58].
Methyl marks are erased during gametogenesis and reset to indicate maternal (oocyte) or paternal (sperm) origin. This results in the generation of differentially methylated regions (DMRs) between the maternal and paternal genomes. The role of DNA methylation in genomic imprinting is best illustrated by the well-studied IGF2/H19 gene locus, a paternal DMR. IGF2 expressed from the paternal allele and H19 expressed from the maternal allele are located close to each other. The transcriptional repressor CTCF binds to the unmethylated maternal chromosome while its binding it blocked on the methylated paternal chromosome. CTCF binding blocks IGF2 expression while driving H19 expression from the maternal allele. The absence of CTCF binding allows IGF2 expression from the paternal allele while blocking H19 expression [59].
Loss of imprinting at the IGF2 locus (expression from both alleles instead of one) occurs during aging and tumorigenesis in the prostate [60]. In mouse models, increased expression of the critical paracrine growth factor IGF2 by loss of imprinting drives prostatic neoplastic growth [61]. Reduction of CTCF expression and binding activity as a function of age triggers Igf2 loss of imprinting in the prostate [62,63]. Genomic imprinting is essential for normal development. Prader-Willi syndrome and Angelman syndrome occur from imprinting errors.
X-inactivation
To compensate for gene dosage differences, one X chromosome is inactivated in females in a process called X-inactivation. Re-activation of X-linked genes by pharmacological inhibition of DNA methylation provided the first evidence for X inactivation by DNA methylation [64]. The initiation and maintenance of X inactivation is regulated by the long non-coding RNA XIST. XIST is expressed only from the inactivated X-chromosome where it acts in cis to coat the inactive X-chromosome and repress transcription. The methylation status of the XIST promoter corresponds to its expression levels. The XIST gene is demethylated and its expression upregulated in Dnmt1 male mutant embryos [65].
Silencing of transposable elements
Transposable elements or transposons can change location and copy number within the genome. Approximately 46% of the human genome and 37.5% of the mouse genome are derived from transposable elements [66]. Methylation suppresses the expression of transposons and these elements can be re-expressed when hypomethylated. If unchecked, transposon expression can lead to mutations and genomic rearrangements. The mouse genome contains ~1000 intracisternal A particle (IAP) retroviruses that have Long Terminal Regions (LTRs) comprised of multiple repeating DNA sequences. Methylation of the LTRs causes transcriptional silencing of IAPs in somatic cells. IAP transcripts are upregulated 50-100 fold in Dnmt1 mutant mouse embryos [67]. Demethylation and activation of IAP genes are observed after treatment with DNA methylation inhibitors [68]. Changes in DNA methylation and expression of retrotransposon elements are observed in prostate cancer [69]. Hypomethylation and re-expression of LINE-1 retrotransposons occur in BPH tissues [70].
Transcriptional control of differentiation
As a cell progresses from an undifferentiated to a differentiated state, its DNA methylation landscape changes dramatically. DNMTs play a central role in the transcriptional control of differentiation. DNMTs suppress the expression of differentiation genes to maintain stem cells in an undifferentiated state. Regulation of DNMT1 expression by the pluripotency factors OCT4 and NANOG maintains the undifferentiated state of mesenchymal stem cells [71]. During differentiation, DNMTs methylate pluripotency factors like OCT4 and NANOG to prevent the cell from reverting to an undifferentiated state [72].
Cytidine nucleoside analogs like 5-azacytidine have been used to elucidate the role of DNA methylation in transcriptional control. 5-azacytidine and its 2’-deoxy derivative 5-aza-2’-deoxycytidine can be incorporated into DNA where it inhibits DNA methyltransferase activity and methylation of newly synthesized DNA. Non-myoblast mouse embryo cells change their phenotype drastically to form functional striated muscle cells after 5-azacytidine treatment [73,74]. Swiss3T3 cells treated with 5-azacytidine or 5-aza-2’-deoxycytidine display new mesenchymal phenotypes of contractile muscle, differentiated adipocytes and chondrocytes [75,76]. 5-azacytidine treatment causes hypomethylation and reverts cells to a less differentiated state from which new phenotypes can develop.
5-aza-2’-deoxycytidine (5AzadC) has been used to study the role of DNA methylation in fetal prostate development. The prostate forms during fetal development as solid epithelial buds arising from the urogenital sinus (UGS) epithelium which grow into the surrounding mesenchyme. Epithelial cells of the developing prostate bud downregulate expression of the epithelial adhesion molecule E-cadherin (Cdh1) to achieve invasive growth into the mesenchyme. Treatment of UGS explants with 5AzadC decreased prostate bud elongation and outgrowth without affecting cell viability. 5AzadC treatment decreased Cdh1 promoter methylation and upregulated Cdh1 mRNA expression in prostate epithelial cells. Increased expression of Cdh1 constrained prostate bud elongation and invasive growth into the mesenchyme. Thus, methylation of the Cdh1 promoter downregulates Cdh1 expression to achieve prostate bud outgrowth [77].
Mesenchymal Androgen receptor (Ar) gene expression is required for prostate bud initiation from the UGS epithelium. The onset of mesenchymal Ar expression corresponds to a reduction in DNMT1 expression and a decrease in Ar promoter methylation. Treatment of UGS explants with 5AzadC inhibits DNMT1, reduces Ar promoter methylation and accelerates the onset of Ar expression, thereby increasing the number of prostate buds specified [78].
Tissue growth and organogenesis
The mid-gestational lethality of Dnmt1 knockout embryos prevents the study of organ system development. To overcome this, context-dependent roles of DNMT1 have been studied in several organ systems by employing conditional genetic approaches. This involves the use of tissue-specific Cre drivers to drive Dnmt1 deletion during development. Deletion of Dnmt1 in the developing pancreas results in global hypomethylation, growth arrest and tissue atrophy through p53-dependent apoptosis of pancreatic progenitors [79]. Intestine-specific ablation showed that Dnmt1 is required for proliferation and crypt formation in the perinatal period. Loss of Dnmt1 in the intestinal epithelium led to increased DNA damage and apoptosis [80].
The role of Dnmt1 in the developing lower urinary tract was assessed by conditionally ablating Dnmt1 in the epithelium of the bladder, urethra and prostate. Similar to previous studies on Dnmt1 ablation, widespread DNA damage and p53-mediated apoptosis was observed. This was accompanied by a reduction in prostate bud formation, highlighting the role of Dnmt1 in maintaining cell proliferation and survival of early prostate progenitors [81]. Dnmt1 deletion in the lower urinary tract led to the breakdown of the Wolffian duct-urethra junction which was permissive for the movement of Wolffian duct cells into the urethra and bladder epithelium. These ectopic cells were then reprogrammed to acquire bladder characteristics including Uroplakin expression [82].
DNA methyltransferase deletion studies conducted in several organ systems have been summarized in Table 1.
Table 1.
DNA methyltransferase deletion studies
| Model | Phenotype | Reference |
|---|---|---|
| Global DNMT1 null mice | 3-fold reduction in genomic 5mC levels | [13] |
| Recessive embryonic lethal | ||
| Embryos are developmentally retarded | ||
| Embryos die at mid-gestation | ||
| DNMT3A-/- | DNMT3A-/- mice appear normal at birth but become runted and die at around 4 weeks of age | [6] |
| DNMT3B-/- mice | DNMT3B-/- mice exhibit growth impairment, rostral neural tube defects and die at mid-gestation | |
| DNMT1o knockout mice | Heterozygous embryos from homozygous mutant females die during last third of gestation | [178] |
| Loss of methylation at imprinted loci | ||
| DNMT3L knockout mice | Azoospermia in homozygous males | [33] |
| Heterozygous embryos from homozygous mutant females die during mid-gestation | ||
| Loss of maternal methylation imprints | ||
| Cre-mediated homozygous DNMT1 deletion in fibroblasts | DNMT1 deletion leads to hypomethylation and aberrant expression of 10% of genes | [15] |
| DNMT1 depleted cells undergo p53-mediated apoptosis | ||
| Cre-mediated DNMT1 deletion in neuroblasts | 95% of cells in the brain hypomethylated | [179] |
| (CamK-cre) | Die immediately after birth from respiratory distress; defects in neuronal respiratory control | |
| Cre-mediated DNMT1 deletion in telencephalic precursors | Mutant mice viable but undergo severe neuronal cell death from E14.5-P21 | [180] |
| (Emx1-cre) | Deregulation of neuronal gene expression | |
| Defects in neuronal morphology and excitability | ||
| shRNA knockdown of DNMT1 in primary human keratinocytes | DNMT1 depleted cells exit progenitor compartment, undergo premature differentiation | [92] |
| DNMT1 loss results in upregulation of cyclin-dependent kinase inhibitors, cell cycle arrest and impaired proliferation | ||
| Cre-mediated DNMT1 deletion in hematopoietic stem cells (HSC) | Impaired HSC self-renewal | [181] |
| (Mx-cre) | Increased cell cycling and inappropriate expression of differentiation markers in myeloid progenitor cells | |
| Cre-mediated DNMT1 deletion in pancreatic cells | DNMT1 loss results in de-repression of p53 | [79] |
| (Pdx1-cre) | G2/M cell cycle arrest and apoptosis | |
| Pancreatic agenesis due to apoptosis of progenitors | ||
| Cre-mediated DNMT1 deletion in retinal cells | Defective photoreceptor differentiation | [182] |
| (Chx10-cre) | Altered cell cycle kinetics; increased proportion of G1 phase cells | |
| Increased apoptosis of post-mitotic photoreceptors and other neuronal types | ||
| Cre-mediated DNMT1 deletion in retinal cells | Mice have smaller eyes | [183] |
| (Rx-cre) | Impaired differentiation of retinal pigment epithelium | |
| Defects in photoreceptor outer segment morphogenesis | ||
| Cre-mediated DNMT1 deletion in intestinal cells | Mice die few weeks after birth | [80] |
| (Villin-cre) | Induction of differentiation markers in progenitor cells | |
| Impaired methylation and expression of DNA damage response genes and cell cycle regulators | ||
| Increased DNA damage and apoptosis of progenitor cells | ||
| Cre-mediated DNMT1 deletion in Keratin 14 lineage cells | Uneven epidermal thickness, altered hair follicle size | [184] |
| (K14-cre) | Impaired proliferation at hair follicles, progressive alopecia with age | |
| Cre-mediated DNMT1 deletion in Shh lineage cells | Mice die shortly after birth; respiratory complications arising from severe lung hypoplasia | [81,82] |
| (Shh-cre) | Epithelial depletion of urethral and bladder epithelium | |
| Premature differentiation and loss of bladder progenitors | ||
| Cell cycle arrest and apoptosis of prostate progenitors; reduction in prostate bud number |
DNA methylation in adult homeostasis and disease
Changes in DNA methylation are associated with the formation of specialized tissue types in the mature organism.
Genome-wide methylation patterns
Most methylated CpG nucleotides are scattered throughout the genome and participate in the silencing of retrotransposons and repetitive elements. Clusters of unmethylated CpGs, called CpG islands, are found at the transcription start sites of several housekeeping genes and a few tissue-specific genes. 72% of promoter sequences contain CpG islands [83]. Unmethylated CpG islands are associated with transcriptionally active genes, mostly housekeeping genes and tumor suppressors [84,85]. The majority of CpG islands are unmethylated irrespective of whether the associated gene is transcriptionally active or inactive. However, methylation of CpGs within CpG islands has been observed and this results in the stable silencing of genes [86].
Tissue-specific methylation patterns
Tissue-specific methylation patterns established during development coordinate specialized gene expression programs in multiple tissue types. Demethylation of developmentally methylated regions also contributes to the establishment of tissue-specific methylation patterns. Tissue-specific genes with CpG island promoters are usually unmethylated in all tissue types, even when genes are inactive. In contrast, tissue-specific genes with non-CpG island promoters display tissue-specific methylation patterns. Broadly, DNA methylation at promoter regions is negatively correlated with gene expression [87,88].
Promoters with “weak” CpG islands, which are intermediate between CpG-rich promoters and non-CpG promoters, display a high frequency of methylation. These weak CpG islands are more prone to de novo methylation during development and contribute to the differential methylation observed between somatic and germ cells [89]. Tissue-specific differentially-methylated regions have also been identified in intergenic and intragenic regions of genes with non-CpG island promoters [90].
Expression of DNA methyltransferases in adult tissues
Adult tissues express lower levels of DNA methyltransferases compared to embryonic tissues although DNMT1, DNMT3A and DNMT3B expression is widespread across adult human tissues, with DNMT1 expressed at the highest level [91]. In adult tissues, DNMT1 is highly expressed in rapidly-dividing somatic tissues including epidermis [92] and intestine [93]. DNMT1 regulates the fine balance between self-renewal and differentiation in adult stem cells. Differentiation involves demethylation and activation of tissue-specific genes with concomitant methylation and repression of stem cell genes. DNMT1 expression prevents differentiating cells from reverting back to an undifferentiated state [72]. In self-renewing somatic tissues, DNMT1 methylates and suppresses differentiation of gene promoters and prevents cell cycle exit in tissue progenitors [92]. DNMT1 is highly expressed in developing prostate tissue but its expression is considerably reduced in the adult prostate. DNMT1 expression is upregulated in cycling cells from prostates of castrated mice supplemented with testosterone (Figure 1).
Figure 1.
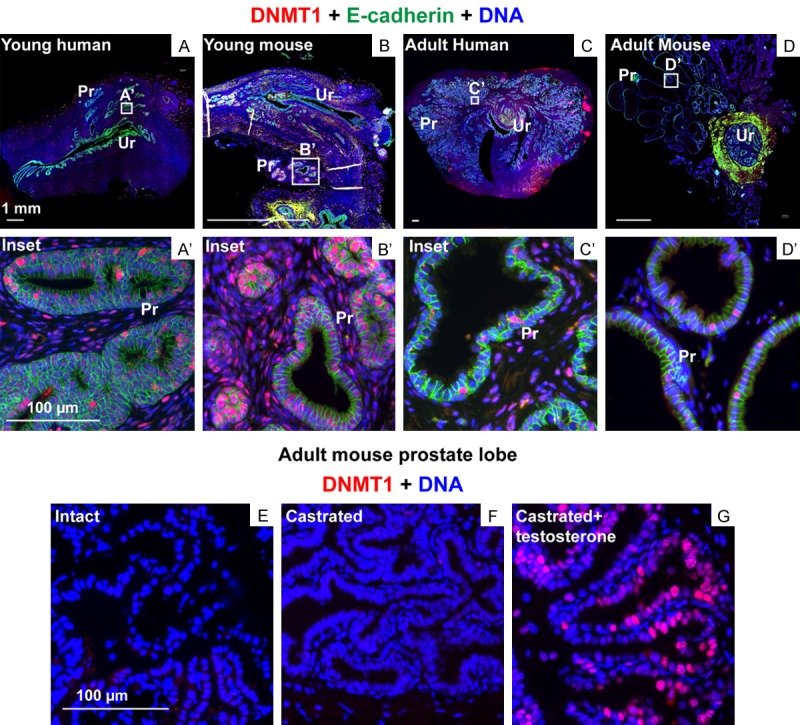
DNMT1 expression in the mouse and human prostate. Tissues sections from (A) young human (21 weeks of gestation), (B) young mouse (Postnatal day 9), (C) adult human (23 years) and (D) adult mouse (7 weeks) were labeled with antibodies to DNMT1 (in red) and E-cadherin (in green). Regions enclosed in white boxes within (A), (B), (C) and (D) are magnified in (A’), (B’), (C’) and (D’). Prostate tissue from (E) intact, (F) castrated and (G) castrated and testosterone supplemented mice were labeled with antibodies to DNMT1 (in red). DAPI staining is shown in blue. Abbreviations Ur: Urethra, Pr: Prostate.
DNA methylation and aging
5’-methyl cytosine (5mC) levels gradually decline over the lifetime of an organism in a process called epigenetic drift. The rate of loss of 5mC is inversely correlated with the lifespan of the organism and is determined by environmental factors like diet [94]. Studies in humans have shown that centenarian DNA has 7% less methylated cytosines than a newborn. Centenarian genomes display loss of methylation at non-CpG island promoters and repetitive DNA sequences like LINE elements and LTR-retrotransposons [95]. Sufficient DNA methylation levels are needed for healthy aging, as demonstrated using DNMT1 haplo-insufficient mice [96].
Alteration of DNA methylation patterns with age occur from cumulative environmental exposures and lifestyle factors [97]. This is best exemplified by the phenotypic changes observed in mono-zygotic twins with age. Monozygotic twins possess identical genomes and epigenomes early in life but display divergence in DNA methylation and histone modifications with age [98]. Aging-related changes in DNMT expression and availability of methyl donors contributes to epigenetic drift [99,100]. Aging fibroblasts show a downregulation of DNMT1 and significant overexpression of DNMT3B. These opposing changes in DNMT expression may explain the global hypomethylation and site-specific hypermethylation observed in aging cells [101,102].
Oxidative stress associated with age has been shown to cause DNA hypomethylation in the prostate [103]. Both global hypomethylation and promoter hypermethylation can result in cellular transformation [97,99]. Aging-related hypermethylation of autophagy genes has been linked to neurodegenerative diseases and chronic inflammation [104]. In the prostate, aging has been linked to loss of imprinting at the IGF2 locus [63]. Despite widespread changes to the DNA methylome with age, it is not fully understood if these changes contribute to aging-related diseases in the prostate. Changes in DNA methylation have been definitively linked to cancer, an age-related disease. However, it is not fully understood whether these DNA methylation changes are causes or consequences of prostate disease.
DNA methylation aberrations in disease
DNA methylation errors occur in several diseases including cancer, syndromes caused by imprinting errors (Prader-Willi, and Angelman syndromes), Fragile X syndrome and ICF syndrome [105]. Among these, the role of DNA methylation in cancer has been extensively studied. DNMT1 and DNMT3A are modestly overexpressed in several tumor tissues while DNMT3B is significantly overexpressed [91,106]. Paradoxically, cancer is associated with global hypomethylation and genomic instability, with significant reduction in methylation at repetitive elements. Several cancer types display aberrant hypermethylation at promoter-associated CpG islands. Hypermethylation of genes involved in apoptosis, cell cycle regulation, cell invasion and DNA repair provide cancer cells with survival advantages over normal cells. De novo methylation of imprint control regions can result in aberrant regulation of imprinted genes. Loss of imprinting, allowing bi-allelic expression of growth promoting genes like Igf2, promotes prostate tumor growth [105].
DNA methylation and prostate disease
In this section we will briefly describe two disease conditions of the prostate: Prostate cancer and Benign Prostatic Hyperplasia (BPH) and the current knowledge of the role of DNA methylation in these diseases.
Prostate cancer and DNA methylation
Prostate cancer is characterized by malignant growth of the prostate and is the third most common cancer type in the United States. Age is a significant risk factor for the development of prostate cancer. Common treatment strategies include anti-androgens (bicalutamide, flutamide, enzalutamide), androgen synthesis inhibitors (ketoconazole, abiraterone acetate), radiation therapy, chemical castration (LHRH agonists) and surgical intervention (www.cancer.gov/types/prostate).
Tissues from metastatic prostate cancer, but not non-metastatic cancer, have drastically reduced 5mC levels compared to normal tissues. This correlation between metastatic capacity and 5mC content suggests that 5mC levels can be used as a biomarker to detect metastatic tumors or that the reduction in 5mC contributes to metastatic progression [107]. Hypermethylation of the regulatory locus of pi-class glutathione S-transferase gene GSTP1 is a recurrent change observed in prostate cancer precursor lesions and prostate adenocarcinoma. GSTP1 encodes an enzyme that catalyzes cellular detoxification reactions [108,109]. CpG island hypermethylation of multiple genes, including APC, RASSF1A, and RUNX3, has been detected in prostate cancer [110-112]. Methylation status of several genes, including APC and RPRM, correlate with biochemical recurrence after radical prostatectomy [113]. Hypermethylation of specific CpG islands in prostate cancer correlates with hypomethylation of Alu and LINE-1 repetitive elements [114]. Genome wide methylation studies have identified differentially-methylated regions (DMRs) in prostate cancer compared to benign tissues. These DMRs are enriched for binding by Enhancer of zeste homolog 2 (EZH2), a key driver of prostate cancer [115,116].
Loss of Androgen receptor (AR) gene expression is a hallmark of castration resistant prostate cancer. CpG methylation of the AR promoter has been observed in prostate cancer cells and is associated with loss of gene expression. Methylation-induced AR inactivation can be reversed by treating cells with DNA methylation inhibitors [117]. The estrogen receptor gene (ESR1) is also frequently methylated and inactivated in prostate cancer [118].
DNMT1 excess has been described in prostate cancer cell lines and tissues [21,119]. DNMT1 expression is significantly upregulated in localized and metastatic prostate cancer tissues compared to normal tissues [120]. Protein and mRNA expression of Dnmt1, Dnmt3a and Dnmt3b are increased in the transgenic adenocarcinoma of mouse prostate (TRAMP) model of prostate cancer [121]. Overexpression of the isoform DNMT3A2 and DNMT3B has also been observed in prostate tumors [122]. DNMT1 inhibition with 5-azacytidine kills prostate tumor cell lines at high doses [123] while promoting metastatic invasion at lower doses [124]. Due to its effectiveness in reversing gene silencing, Azacytidine treatment is being studied in combination with other therapeutic options for the treatment of advanced prostate cancer [125-127]. DNMT inhibition was shown to block tumorigenesis in the TRAMP mouse model of prostate cancer [128]. In a cohort of prostate tissue samples, DNMT1 expression was highest in poorly-differentiated prostate cancer and lowest in well-differentiated prostate cancer. DNMT1 expression was negatively correlated with expression of GSTP1 and APC, which are hyper-methylated in prostate cancer. This suggests a role for DNMT1 in the methylation and repression of key genes during prostate cancer progression [129].
Benign prostatic hyperplasia and DNA methylation
Benign Prostatic Hyperplasia (BPH) is a non-malignant enlargement of the prostate. BPH growth initiates by the age of 30 and continues throughout the individual’s lifetime. The major risk factor for the development of pathological BPH is age. By the age of 50, 50% of men develop pathological BPH [130]. Enlargement of the prostate in BPH results in urethral constriction and manifestation of lower urinary tract symptoms (LUTS). LUTS include storage (increased frequency, urgency, nocturia) and obstructive symptoms (weak stream, incomplete voiding, urinary obstruction, overflow incontinence) [131]. BPH in association with LUTS are a significant financial burden [132,133] and negatively impacts quality of life but does not lead to significant morbidity if treated. BPH is treated with three major classes of drugs, alpha adrenergic receptor blockers, 5-alpha reductase inhibitors and phosphodiesterase-5 inhibitors [134-136].
Prostates with BPH have significantly lower levels of global 5mC compared to normal tissue [107]. Dnmt1 is expressed at low levels in BPH tissue compared to prostate cancer. GSTP1 and APC are hypermethylated in prostate cancer but hypomethylated in BPH [129]. Although BPH is characterized by global hypomethylation, recurrent hypermethylation of the tumor suppressor gene 14-3-3Sigma occurs in BPH tissues [137]. DNMT1 regulates the methylation and repression of SRD5A2 gene expression in BPH samples. DNMT1 expression in SRD5A2-silenced BPH tissues is regulated by the inflammatory mediators IL-6, TNFa and NF-kB [138,139]. Methylation and repression of SRD5A2 results in an androgenic to estrogenic switch. Decreased reliance of androgenic pathways in BPH could be behind the reduced efficacy of 5-alpha reductase inhibitors in some patients [140].
Impact of dietary and environmental factors on DNA methylation
DNA methylation is influenced by environmental factors including diet [141,142], particulate pollution [143-145], carcinogens and benzene exposure [146]. The Developmental Origins of Heath and Disease hypothesis (DOHaD) states that early life environmental influences can impact adult disease. DNA methylation is one mechanism by which signatures of early life exposures can be embedded in the genome. The heritable and stable nature of DNA methylation would allow these early changes to persist into adulthood, where additional insults can trigger disease onset. The DOHaD hypothesis originated from observations made in low birthweight infants. Infants with low birthweight were more prone to cardiovascular disease later in life, suggesting that poor fetal nutrition can increase adult disease susceptibility [147,148].
Maternal diet can influence DNA methylation patterns in offspring. This is best illustrated by the seminal Agouti mouse studies. Transcription from the agouti gene locus is associated with increased obesity and yellow coat color. Maternal diet enriched in methyl donors causes methylation and repression of agouti gene transcription and produces lean mice with brown coat color [149,150]. The Dutch famine studies have demonstrated differential methylation of several metabolism genes, including the IGF2 locus, with prenatal exposure to poor nutrition. Exposed individuals have poor metabolic regulation, increased body mass index and higher risk for developing schizophrenia in later life [151,152].
Commonly occurring environmental toxins like Bisphenol A (BPA), Dioxin (TCDD), and phthalates are capable of interfering with endocrine signaling and are called endocrine disruptors. Developmental exposures to hormones and endocrine-disrupting chemicals have been shown to alter prostate development. Early prostate development is dependent on androgens (testosterone and dihydrotestosterone). Disrupting androgen action or exposure to estrogenic compounds alters early prostate development with long-term consequences to the adult prostate. Exposing pregnant mice to low doses of estrogen or estrogenic compounds (BPA or diethylstilbestrol) increases the number of prostate glands formed during fetal life and increases adult prostate size [153-155]. Developmental exposure to estrogens or BPA increases susceptibility to prostate cancer in an adult rat model of prostate cancer [156]. Prenatal exposure to the endocrine disruptor ethinylestradiol, found in oral contraceptives, pre-disposes male and female gerbils to prostate lesions with aging [157]. Developmental exposure to estradiol and BPA alters DNA methylation patterns of several genes in the prostate including phosphodiesterase type 4 variant 4 (PDE4D4), Pitx3, Wnt10b, Paqr4, Sox2, Chst14, Tpd52 and Creb3l4. These DNA methylome changes persist into adulthood [156,158,159]. Additional research is required to ascertain whether differential regulation of these genes contributes to prostate cancer.
Maternal exposure to the persistent environmental contaminant TCDD alters prostate bud specification by disrupting Aryl hydrocarbon receptor signaling [160,161]. Exposure to TCDD during in utero and lactational stages decreases adult rat prostate size by impairing early prostate proliferation and differentiation [162]. In utero and lactational exposure to TCDD predisposes mice to urinary dysfunction in later life, but the exact mechanism is not known [163]. Early TCDD exposure can alter DNA methylation at imprinted loci in preimplantation embryos [164]. Further research is required to link TCDD induced DNA methylation changes to defects in prostate growth and urinary tract function.
DNA methyltransferase co-factor S-adenosyl methionine (SAM) is the source of methyl groups for DNA methylation. Following removal of the methyl group, SAM is converted to Sadenosyl homocysteine (SAH) and homocysteine. Homocysteine re-methylation to replenish pools of SAM requires several methyl group donors like methyl-folate, methionine and betaine. Availability of methyl donors in the diet can influence DNA methylation. This is elegantly illustrated by studies in the Agouti mouse model. Early life exposure to BPA hypomethylates the IAP retrotransposon element upstream of the agouti gene and shifts coat color to yellow. Maternal diet supplementation with methyl donors reverses BPA-induced hypomethylation and shifts coat color back to brown [165].
Dietary folate is a well-studied methyl donor. Folates are water soluble vitamins that participate in one-carbon transfer, DNA synthesis, cell growth, hematopoiesis and metabolism. While folates occur naturally in the diet, folic acid is the synthetic form that is supplemented in fortified food stuffs or consumed as dietary supplements. Folates and folic acid are converted to 5-methyl tetrahydrofolate which provides methyl groups for the conversion of homocysteine to methionine. Methionine is converted to SAM, the methyl donor for DNA methylation reactions. Adequate consumption of folates is required to maintain SAM pools.
In 1998, the United States mandated folic acid fortification in cereal grains. Folic acid fortification has raised serum folate levels in populations of several age groups, most significantly in children and older populations [166,167]. In addition to participating in methylation, folates are important players in nucleotide biosynthesis and polyamine synthesis. The complex role of folates in the body could yield protective or deleterious effects depending on the context. High gestational folic acid has been shown to have adverse effects on offspring in rodent studies [168,169]. Multiple clinical trials have associated folic acid supplement consumption with increased risk of prostate cancer [170-172]. Moreover, studies in the TRAMP prostate cancer mouse model have shown that dietary folic acid deficiency can suppress tumor growth [173]. However, a few meta-analysis studies using combined data across multiple randomized trials have failed to identify an association between folic acid supplementation and prostate cancer risk [174,175].
Despite contradictory data, the prevalence of folic acid supplement consumption warrants further research into the interaction between folic acid supplementation and prostate disease. Maternal supplementation, food grain fortification and consumption of multi-vitamin supplements have resulted in populations that have been exposed to high levels of folic acid throughout their lifetime. A study modeling this lifetime folic acid exposure in mice, showed that folic acid supplementation from gestation relieves lower urinary tract symptoms in mice with hormone-induced urinary dysfunction [176]. Further, folic acid supplementation slows down prostate shrinkage in mice after castration [177]. It is important to fully understand the consequences of folate supplementation at various stages of development and during disease processes in the prostate. Rodent models and clinical studies examining the impact of folic acid on the prostate are summarized in Table 2.
Table 2.
Studies examining the effects of folic acid on prostate homeostasis and disease
| Study | Findings | Reference |
|---|---|---|
| Animal models of folic acid supplementation | ||
| 4 mg/kg or 24 mg/kg folic acid throughout gestation and lifetime (12-fold higher than recommended) in mice | Folic acid supplemented males mice show improvement of urinary symptoms in a hormone induced model of urinary obstruction | [176] |
| 4 mg/kg or 24 mg/kg folic acid throughout gestation and lifetime (12-fold higher than recommended) in mice | Folic acid supplemented mice show reduced response to castration induced prostate involution. Prostates of castrated folic acid supplemented mice are larger than prostates from castrated control diet mice | [177] |
| 2 mg/kg folic acid (control), 0.3 mg/kg (deficient) and 20 mg/kg (10-fold higher than recommended) fed to TRAMP mice after weaning | Folate deficient diet blocks prostate cancer progression | [173] |
| Folate supplementation had no effect on prostate cancer growth | ||
| Clinical studies on the interaction of folic acid with prostate cancer | ||
| Plasma levels of folates were tested for association with prostate cancer risk (Northern Sweden Health and disease cohort) | Increasing levels of plasma folate positively correlated with prostate cancer risk | [185] |
| Prostate cancer occurrence was assessed after folic acid supplementation (Aspirin/Folate polyp prevention study) | Folic acid supplementation increased the long-term probability of being diagnosed with prostate cancer | [186] |
| Plasma levels of folates were tested for association with prostate cancer risk (ProtecT study) | Folate levels showed a positive correlation with prostate cancer risk | [172] |
| Serum folate levels were tested for association with prostate cancer cell proliferation in biopsies (University of Pittsburgh) | Patients with increased serum folate levels had greater cancer proliferation as measured by Ki67 positivity in biopsy tissue | [187] |
| Oral folic acid supplementation was tested for association with increased cancer risk (Meta-analysis of multiple studies) | Increased prostate cancer risk after folic acid supplementation compared to placebo | [171] |
| Serum folate levels were tested for association with prostate cancer risk (JANUS cohort, Norway) | High serum folate concentration associated with moderate increase in prostate cancer risk | [170] |
| Meta-analysis of published randomized trial data to study the association of folic acid supplementation on cancer risk | Folic acid supplementation does not have a significant association with prostate cancer incidence | [175] |
| Meta-analysis of randomized trial data to study the association of folic acid supplementation with cancer incidence | Folic acid supplementation does not have a significant effect on prostate cancer incidence | [174] |
Conclusions
This review provides an overview of the DNA methylation machinery and the dynamic changes in DNA methylation that occur during development, tissue growth and ultimately aging and disease. DNA methylation is at the confluence of the genome and environment. Early life-changes in DNA methylation patterns are overlaid on our genomes and can affect disease susceptibility later in life. We hope that this review can be used as an introduction to the field by prostate researchers interested in studying DNA methylation.
Acknowledgements
We thank the Health Sciences Tissue Bank, University of Pittsburg (to Dr. Vezina) and the Southwest Transplant Alliance, Dallas TX (to Dr. Strand) for providing the human tissues used in this manuscript. We thank members of the Vezina laboratory for helpful discussions during the preparation of this article. This work is supported by National Institutes of Health R01 DK099328, R01 DK115477, and U54 DK104310. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure of conflict of interest
None.
References
- 1.Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41:10–13. doi: 10.1093/ije/dyr184. [DOI] [PubMed] [Google Scholar]
- 2.Wu C, Morris JR. Genes, genetics, and epigenetics: a correspondence. Science. 2001;293:1103–1105. doi: 10.1126/science.293.5532.1103. [DOI] [PubMed] [Google Scholar]
- 3.Holliday R. Epigenetics: an overview. Dev Genet. 1994;15:453–457. doi: 10.1002/dvg.1020150602. [DOI] [PubMed] [Google Scholar]
- 4.Doskočil J, Šorm F. Distribution of 5-methylcytosine in pyrimidine sequences of deoxyribonucleic acids. Biochim Biophys Acta. 1962;55:953–959. doi: 10.1016/0006-3002(62)90909-5. [DOI] [PubMed] [Google Scholar]
- 5.Bestor TH, Hellewell SB, Ingram VM. Differentiation of two mouse cell lines is associated with hypomethylation of their genomes. Mol Cell Biol. 1984;4:1800–1806. doi: 10.1128/mcb.4.9.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 7.Chédin F, Lieber MR, Hsieh CL. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Natl Acad Sci U S A. 2002;99:16916–16921. doi: 10.1073/pnas.262443999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hata K, Okano M, Lei H, Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129:1983–1993. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- 9.Leonhardt H, Page AW, Weier HU, Bestor TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992;71:865–873. doi: 10.1016/0092-8674(92)90561-p. [DOI] [PubMed] [Google Scholar]
- 10.Zhang ZM, Liu S, Lin K, Luo Y, Perry JJ, Wang Y, Song J. Crystal structure of human DNA methyltransferase 1. J Mol Biol. 2015;427:2520–2531. doi: 10.1016/j.jmb.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szyf M, Kaplan F, Mann V, Giloh H, Kedar E, Razin A. Cell cycle-dependent regulation of eukaryotic DNA methylase level. J Biol Chem. 1985;260:8653–8656. [PubMed] [Google Scholar]
- 12.Keil KP, Altmann HM, Mehta V, Abler LL, Elton EA, Vezina CM. Catalog of mRNA expression patterns for DNA methylating and demethylating genes in developing mouse lower urinary tract. Gene Expr Patterns. 2013;13:413–424. doi: 10.1016/j.gep.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 14.Liao J, Karnik R, Gu H, Ziller MJ, Clement K, Tsankov AM, Akopian V, Gifford CA, Donaghey J, Galonska C, Pop R, Reyon D, Tsai SQ, Mallard W, Joung JK, Rinn JL, Gnirke A, Meissner A. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat Genet. 2015;47:469–478. doi: 10.1038/ng.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson-Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, Csankovszki G, Dausman J, Lee P, Wilson C, Lander E, Jaenisch R. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet. 2001;27:31–39. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- 16.Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- 17.Loughery JE, Dunne PD, O’Neill KM, Meehan RR, McDaid JR, Walsh CP. DNMT1 deficiency triggers mismatch repair defects in human cells through depletion of repair protein levels in a process involving the DNA damage response. Hum Mol Genet. 2011;20:3241–3255. doi: 10.1093/hmg/ddr236. [DOI] [PubMed] [Google Scholar]
- 18.Palii SS, Van Emburgh BO, Sankpal UT, Brown KD, Robertson KD. DNA methylation inhibitor 5-Aza-2’-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol. 2008;28:752–771. doi: 10.1128/MCB.01799-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, Ueda Y, Li E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 20.Unterberger A, Andrews SD, Weaver IC, Szyf M. DNA methyltransferase 1 knockdown activates a replication stress checkpoint. Mol Cell Biol. 2006;26:7575–7586. doi: 10.1128/MCB.01887-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patra SK, Patra A, Zhao H, Dahiya R. DNA methyltransferase and demethylase in human prostate cancer. Mol Carcinog. 2002;33:163–171. doi: 10.1002/mc.10033. [DOI] [PubMed] [Google Scholar]
- 22.Maresca A, Zaffagnini M, Caporali L, Carelli V, Zanna C. DNA methyltransferase 1 mutations and mitochondrial pathology: is mtDNA methylated? Front Genet. 2015;6:90. doi: 10.3389/fgene.2015.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baets J, Duan X, Wu Y, Smith G, Seeley WW, Mademan I, McGrath NM, Beadell NC, Khoury J, Botuyan MV, Mer G, Worrell GA, Hojo K, DeLeon J, Laura M, Liu YT, Senderek J, Weis J, Van den Bergh P, Merrill SL, Reilly MM, Houlden H, Grossman M, Scherer SS, De Jonghe P, Dyck PJ, Klein CJ. Defects of mutant DNMT1 are linked to a spectrum of neurological disorders. Brain. 2015;138:845–861. doi: 10.1093/brain/awv010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winkelmann J, Lin L, Schormair B, Kornum BR, Faraco J, Plazzi G, Melberg A, Cornelio F, Urban AE, Pizza F, Poli F, Grubert F, Wieland T, Graf E, Hallmayer J, Strom TM, Mignot E. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum Mol Genet. 2012;21:2205–2210. doi: 10.1093/hmg/dds035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein CJ, Botuyan MV, Wu Y, Ward CJ, Nicholson GA, Hammans S, Hojo K, Yamanishi H, Karpf AR, Wallace DC, Simon M, Lander C, Boardman LA, Cunningham JM, Smith GE, Litchy WJ, Boes B, Atkinson EJ, Middha S, B Dyck PJ, Parisi JE, Mer G, Smith DI, Dyck PJ. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet. 2011;43:595–600. doi: 10.1038/ng.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Li W, Liu S, Zong S, Wang W, Ren J, Li Q, Hou F, Shi Q. DNMT1, DNMT3 and DNMT3B polymorphisms associated with gastric cancer risk: a systematic review and meta-analysis. EBioMedicine. 2016;13:125–131. doi: 10.1016/j.ebiom.2016.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tao R, Chen Z, Wu P, Liu C, Peng Y, Zhao W, Hu C, Feng J. The possible role of EZH2 and DNMT1 polymorphisms in sporadic triple-negative breast carcinoma in southern Chinese females. Tumour Biol. 2015;36:9849–9855. doi: 10.1007/s13277-015-3754-y. [DOI] [PubMed] [Google Scholar]
- 28.He BS, Pan YQ, Zhu CB. [Polymorphisms of DNA methyltransferases and the risk of prostate cancer] . Zhonghua Nan Ke Xue. 2014;20:1077–1081. [PubMed] [Google Scholar]
- 29.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 30.Gowher H, Jeltsch A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J Mol Biol. 2001;309:1201–1208. doi: 10.1006/jmbi.2001.4710. [DOI] [PubMed] [Google Scholar]
- 31.Lei H, Oh SP, Okano M, Juttermann R, Goss KA, Jaenisch R, Li E. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122:3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- 32.Bachman KE, Rountree MR, Baylin SB. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J Biol Chem. 2001;276:32282–32287. doi: 10.1074/jbc.M104661200. [DOI] [PubMed] [Google Scholar]
- 33.Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 34.Hou HA, Kuo YY, Liu CY, Chou WC, Lee MC, Chen CY, Lin LI, Tseng MH, Huang CF, Chiang YC, Lee FY, Liu MC, Liu CW, Tang JL, Yao M, Huang SY, Ko BS, Hsu SC, Wu SJ, Tsay W, Chen YC, Tien HF. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012;119:559–568. doi: 10.1182/blood-2011-07-369934. [DOI] [PubMed] [Google Scholar]
- 35.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, Schmidt H, Zhang Q, Osborne JR, Lin L, O’Laughlin M, McMichael JF, Delehaunty KD, McGrath SD, Fulton LA, Magrini VJ, Vickery TL, Hundal J, Cook LL, Conyers JJ, Swift GW, Reed JP, Alldredge PA, Wylie T, Walker J, Kalicki J, Watson MA, Heath S, Shannon WD, Varghese N, Nagarajan R, Westervelt P, Tomasson MH, Link DC, Graubert TA, DiPersio JF, Mardis ER, Wilson RK. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin B, Tao Q, Peng J, Soo HM, Wu W, Ying J, Fields CR, Delmas AL, Liu X, Qiu J, Robertson KD. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum Mol Genet. 2008;17:690–709. doi: 10.1093/hmg/ddm341. [DOI] [PubMed] [Google Scholar]
- 37.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan AY, Manley JL. The TET family of proteins: functions and roles in disease. J Mol Cell Biol. 2009;1:82–92. doi: 10.1093/jmcb/mjp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in Mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pfeifer GP, Kadam S, Jin SG. 5-hydroxymethylcytosine and its potential roles in development and cancer. Epigenetics Chromatin. 2013;6:10. doi: 10.1186/1756-8935-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hahn MA, Qiu R, Wu X, Li AX, Zhang H, Wang J, Jui J, Jin SG, Jiang Y, Pfeifer GP, Lu Q. Dynamics of 5-hydroxymethylcytosine and chromatin marks in mammalian neurogenesis. Cell Rep. 2013;3:291–300. doi: 10.1016/j.celrep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin W, Wolf P, Liu N, Link S, Smets M, Mastra FL, Forne I, Pichler G, Horl D, Fellinger K, Spada F, Bonapace IM, Imhof A, Harz H, Leonhardt H. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res. 2015;25:911–929. doi: 10.1038/cr.2015.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen SE. 5-hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54. doi: 10.1186/gb-2011-12-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kafer GR, Li X, Horii T, Suetake I, Tajima S, Hatada I, Carlton PM. 5-hydroxymethylcytosine marks sites of DNA damage and promotes genome stability. Cell Rep. 2016;14:1283–1292. doi: 10.1016/j.celrep.2016.01.035. [DOI] [PubMed] [Google Scholar]
- 45.Pfeifer GP, Xiong W, Hahn MA, Jin SG. The role of 5-hydroxymethylcytosine in human cancer. Cell Tissue Res. 2014;356:631–641. doi: 10.1007/s00441-014-1896-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu J, Xu ZD, Zhu HG, Ling ZQ, Ye D, Guan KL, Xiong Y. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2013;32:663–669. doi: 10.1038/onc.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Storebjerg TM, Strand SH, Hoyer S, Lynnerup AS, Borre M, Orntoft TF, Sorensen KD. Dysregulation and prognostic potential of 5-methylcytosine (5mC), 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) levels in prostate cancer. Clin Epigenetics. 2018;10:105. doi: 10.1186/s13148-018-0540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279:52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 49.Fritz EL, Papavasiliou FN. Cytidine deaminases: aiding DNA demethylation? Genes Dev. 2010;24:2107–2114. doi: 10.1101/gad.1963010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fatemi M, Wade PA. MBD family proteins: reading the epigenetic code. J Cell Sci. 2006;119:3033–3037. doi: 10.1242/jcs.03099. [DOI] [PubMed] [Google Scholar]
- 51.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 52.Ng HH, Zhang Y, Hendrich B, Johnson CA, Turner BM, Erdjument-Bromage H, Tempst P, Reinberg D, Bird A. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet. 1999;23:58–61. doi: 10.1038/12659. [DOI] [PubMed] [Google Scholar]
- 53.Fujita N, Watanabe S, Ichimura T, Tsuruzoe S, Shinkai Y, Tachibana M, Chiba T, Nakao M. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J Biol Chem. 2003;278:24132–24138. doi: 10.1074/jbc.M302283200. [DOI] [PubMed] [Google Scholar]
- 54.Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25:269–277. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- 55.Kagiwada S, Kurimoto K, Hirota T, Yamaji M, Saitou M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J. 2013;32:340–353. doi: 10.1038/emboj.2012.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 57.Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith FM, Garfield AS, Ward A. Regulation of growth and metabolism by imprinted genes. Cytogenet Genome Res. 2006;113:279–291. doi: 10.1159/000090843. [DOI] [PubMed] [Google Scholar]
- 59.Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 60.Jarrard DF, Bussemakers MJ, Bova GS, Isaacs WB. Regional loss of imprinting of the insulin-like growth factor II gene occurs in human prostate tissues. Clin Cancer Res. 1995;1:1471–1478. [PubMed] [Google Scholar]
- 61.Damaschke NA, Yang B, Bhusari S, Avilla M, Zhong W, Blute ML Jr, Huang W, Jarrard DF. Loss of Igf2 gene imprinting in murine prostate promotes widespread neoplastic growth. Cancer Res. 2017;77:5236–5247. doi: 10.1158/0008-5472.CAN-16-3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu VX, Schwarze SR, Kenowski ML, Leblanc S, Svaren J, Jarrard DF. A loss of insulin-like growth factor-2 imprinting is modulated by CCCTC-binding factor down-regulation at senescence in human epithelial cells. J Biol Chem. 2004;279:52218–52226. doi: 10.1074/jbc.M405015200. [DOI] [PubMed] [Google Scholar]
- 63.Fu VX, Dobosy JR, Desotelle JA, Almassi N, Ewald JA, Srinivasan R, Berres M, Svaren J, Weindruch R, Jarrard DF. Aging and cancer-related loss of insulin-like growth factor 2 imprinting in the mouse and human prostate. Cancer Res. 2008;68:6797–6802. doi: 10.1158/0008-5472.CAN-08-1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mohandas T, Sparkes RS, Shapiro LJ. Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science. 1981;211:393–396. doi: 10.1126/science.6164095. [DOI] [PubMed] [Google Scholar]
- 65.Beard C, Li E, Jaenisch R. Loss of methylation activates Xist in somatic but not in embryonic cells. Genes Dev. 1995;9:2325–2334. doi: 10.1101/gad.9.19.2325. [DOI] [PubMed] [Google Scholar]
- 66.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigó R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O’Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Nieder-hausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 67.Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- 68.Davis CM, Constantinides PG, van der Riet F, van Schalkwyk L, Gevers W, Parker MI. Activation and demethylation of the intracisternal a particle genes by 5-azacytidine. Cell Differ Dev. 1989;27:83–93. doi: 10.1016/0922-3371(89)90738-7. [DOI] [PubMed] [Google Scholar]
- 69.Goering W, Ribarska T, Schulz WA. Selective changes of retroelement expression in human prostate cancer. Carcinogenesis. 2011;32:1484–1492. doi: 10.1093/carcin/bgr181. [DOI] [PubMed] [Google Scholar]
- 70.Madigan AA, Sobek KM, Cummings JL, Green WR, Bacich DJ, O’Keefe DS. Activation of innate anti-viral immune response genes in symptomatic benign prostatic hyperplasia. Genes Immun. 2012;13:566–572. doi: 10.1038/gene.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsai CC, Su PF, Huang YF, Yew TL, Hung SC. Oct4 and Nanog directly regulate Dnmt1 to maintain self-renewal and undifferentiated state in mesenchymal stem cells. Mol Cell. 2012;47:169–182. doi: 10.1016/j.molcel.2012.06.020. [DOI] [PubMed] [Google Scholar]
- 72.Schmidt CS, Bultmann S, Meilinger D, Zacher B, Tresch A, Maier KC, Peter C, Martin DE, Leonhardt H, Spada F. Global DNA hypomethylation prevents consolidation of differentiation programs and allows reversion to the embryonic stem cell state. PLoS One. 2012;7:e52629. doi: 10.1371/journal.pone.0052629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Constantinides PG, Jones PA, Gevers W. Functional striated muscle cells from non-myoblast precursors following 5-azacytidine treatment. Nature. 1977;267:364–366. doi: 10.1038/267364a0. [DOI] [PubMed] [Google Scholar]
- 74.Constantinides PG, Taylor SM, Jones PA. Phenotypic conversion of cultured mouse embryo cells by aza pyrimidine nucleosides. Dev Biol. 1978;66:57–71. doi: 10.1016/0012-1606(78)90273-7. [DOI] [PubMed] [Google Scholar]
- 75.Taylor SM, Jones PA. Changes in phenotypic expression in embryonic and adult cells treated with 5-azacytidine. J Cell Physiol. 1982;111:187–194. doi: 10.1002/jcp.1041110210. [DOI] [PubMed] [Google Scholar]
- 76.Taylor SM, Jones PA. Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5-azacytidine. Cell. 1979;17:771–779. doi: 10.1016/0092-8674(79)90317-9. [DOI] [PubMed] [Google Scholar]
- 77.Keil KP, Abler LL, Mehta V, Altmann HM, Laporta J, Plisch EH, Suresh M, Hernandez LL, Vezina CM. DNA methylation of E-cadherin is a priming mechanism for prostate development. Dev Biol. 2014;387:142–153. doi: 10.1016/j.ydbio.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Keil KP, Abler LL, Laporta J, Altmann HM, Yang B, Jarrard DF, Hernandez LL, Vezina CM. Androgen receptor DNA methylation regulates the timing and androgen sensitivity of mouse prostate ductal development. Dev Biol. 2014;396:237–245. doi: 10.1016/j.ydbio.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Georgia S, Kanji M, Bhushan A. DNMT1 represses p53 to maintain progenitor cell survival during pancreatic organogenesis. Genes Dev. 2013;27:372–377. doi: 10.1101/gad.207001.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Elliott EN, Sheaffer KL, Schug J, Stappenbeck TS, Kaestner KH. Dnmt1 is essential to maintain progenitors in the perinatal intestinal epithelium. Development. 2015;142:2163–2172. doi: 10.1242/dev.117341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miranti CK, Koul HK. Meeting report of joint society of basic urologic research (SBUR) and european society of urological research (ESUR) symposium fall 2017. AJCEU. 2017;5(Suppl 1):52. [Google Scholar]
- 82.Joseph DB, Chandrashekar AS, Abler LL, Chu LF, Thomson JA, Mendelsohn C, Vezina CM. In vivo replacement of damaged bladder urothelium by Wolffian duct epithelial cells. Proc Natl Acad Sci U S A. 2018;115:8394–8399. doi: 10.1073/pnas.1802966115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103:1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 85.Ghosh S, Yates AJ, Fruhwald MC, Miecznikowski JC, Plass C, Smiraglia D. Tissue specific DNA methylation of CpG islands in normal human adult somatic tissues distinguishes neural from non-neural tissues. Epigenetics. 2010;5:527–538. doi: 10.4161/epi.5.6.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Han H, Cortez CC, Yang X, Nichols PW, Jones PA, Liang G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum Mol Genet. 2011;20:4299–4310. doi: 10.1093/hmg/ddr356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, Haefliger C, Horton R, Howe K, Jackson DK, Kunde J, Koenig C, Liddle J, Niblett D, Otto T, Pettett R, Seemann S, Thompson C, West T, Rogers J, Olek A, Berlin K, Beck S. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 90.Liang P, Song F, Ghosh S, Morien E, Qin M, Mahmood S, Fujiwara K, Igarashi J, Nagase H, Held WA. Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC Genomics. 2011;12:231. doi: 10.1186/1471-2164-12-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, Jones PA. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999;27:2291–2298. doi: 10.1093/nar/27.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sen GL, Reuter JA, Webster DE, Zhu L, Khavari PA. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature. 2010;463:563–567. doi: 10.1038/nature08683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sheaffer KL, Kim R, Aoki R, Elliott EN, Schug J, Burger L, Schubeler D, Kaestner KH. DNA methylation is required for the control of stem cell differentiation in the small intestine. Genes Dev. 2014;28:652–664. doi: 10.1101/gad.230318.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maegawa S, Lu Y, Tahara T, Lee JT, Madzo J, Liang S, Jelinek J, Colman RJ, Issa JJ. Caloric restriction delays age-related methylation drift. Nat Commun. 2017;8:539. doi: 10.1038/s41467-017-00607-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, Puca AA, Sayols S, Pujana MA, Serra-Musach J, Iglesias-Platas I, Formiga F, Fernandez AF, Fraga MF, Heath SC, Valencia A, Gut IG, Wang J, Esteller M. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A. 2012;109:10522–10527. doi: 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu L, van Groen T, Kadish I, Li Y, Wang D, James SR, Karpf AR, Tollefsbol TO. Insufficient DNA methylation affects healthy aging and promotes age-related health problems. Clin Epigenetics. 2011;2:349–360. doi: 10.1007/s13148-011-0042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jung M, Pfeifer GP. Aging and DNA methylation. BMC Biol. 2015;13:7. doi: 10.1186/s12915-015-0118-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zampieri M, Ciccarone F, Calabrese R, Franceschi C, Bürkle A, Caiafa P. Reconfiguration of DNA methylation in aging. Mech Ageing Dev. 2015;151:60–70. doi: 10.1016/j.mad.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 100.Ciccarone F, Malavolta M, Calabrese R, Guastafierro T, Bacalini MG, Reale A, Franceschi C, Capri M, Hervonen A, Hurme M, Grubeck-Loebenstein B, Koller B, Bernhardt J, Schӧn C, Slagboom PE, Toussaint O, Sikora E, Gonos ES, Breusing N, Grune T, Jansen E, Dollé M, Moreno-Villanueva M, Sindlinger T, Bürkle A, Zampieri M, Caiafa P. Age-dependent expression of DNMT1 and DNMT3B in PBMCs from a large European population enrolled in the MARK-AGE study. Aging Cell. 2016;15:755–765. doi: 10.1111/acel.12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Casillas MA, Lopatina N, Andrews LG, Tollefsbol TO. Transcriptional control of the DNA methyltransferases is altered in aging and neoplastically-transformed human fibroblasts. Mol Cell Biochem. 2003;252:33–43. doi: 10.1023/a:1025548623524. [DOI] [PubMed] [Google Scholar]
- 102.Lopatina N, Haskell JF, Andrews LG, Poole JC, Saldanha S, Tollefsbol T. Differential maintenance and de novo methylating activity by three DNA methyltransferases in aging and immortalized fibroblasts. J Cell Biochem. 2002;84:324–334. doi: 10.1002/jcb.10015. [DOI] [PubMed] [Google Scholar]
- 103.Bhusari SS, Dobosy JR, Fu V, Almassi N, Oberley T, Jarrard DF. Superoxide dismutase 1 knockdown induces oxidative stress and DNA methylation loss in the prostate. Epigenetics. 2010;5:402–409. doi: 10.4161/epi.5.5.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Khalil H, Tazi M, Caution K, Ahmed A, Kanneganti A, Assani K, Kopp B, Marsh C, Dakhlallah D, Amer AO. Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics. 2016;11:381–388. doi: 10.1080/15592294.2016.1144007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Robertson KD. DNA methylation and human disease. Nature Reviews Genetics. 2005;6:597. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 106.Jahangiri R, Mosaffa F, Emami Razavi A, Teimoori-Toolabi L, Jamialahmadi K. Altered DNA methyltransferases promoter methylation and mRNA expression are associated with tamoxifen response in breast tumors. J Cell Physiol. 2018;233:7305–7319. doi: 10.1002/jcp.26562. [DOI] [PubMed] [Google Scholar]
- 107.Bedford MT, van Helden PD. Hypomethylation of DNA in pathological conditions of the human prostate. Cancer Res. 1987;47:5274–5276. [PubMed] [Google Scholar]
- 108.Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, Bova GS, Hsieh WS, Isaacs WB, Nelson WG. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci U S A. 1994;91:11733–11737. doi: 10.1073/pnas.91.24.11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brooks JD, Weinstein M, Lin X, Sun Y, Pin SS, Bova GS, Epstein JI, Isaacs WB, Nelson WG. CG island methylation changes near the GSTP1 gene in prostatic intraepithelial neoplasia. Cancer Epidemiol Biomarkers Prev. 1998;7:531–536. [PubMed] [Google Scholar]
- 110.Kang GH, Lee S, Lee HJ, Hwang KS. Aberrant CpG island hypermethylation of multiple genes in prostate cancer and prostatic intraepithelial neoplasia. J Pathol. 2004;202:233–240. doi: 10.1002/path.1503. [DOI] [PubMed] [Google Scholar]
- 111.Maruyama R, Toyooka S, Toyooka KO, Virmani AK, Zochbauer-Muller S, Farinas AJ, Minna JD, McConnell J, Frenkel EP, Gazdar AF. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin Cancer Res. 2002;8:514–519. [PubMed] [Google Scholar]
- 112.Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, Bova GS, De Marzo AM, Isaacs WB, Nelson WG. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004;64:1975–1986. doi: 10.1158/0008-5472.can-03-3972. [DOI] [PubMed] [Google Scholar]
- 113.Ellinger J, Bastian PJ, Jurgan T, Biermann K, Kahl P, Heukamp LC, Wernert N, Muller SC, von Ruecker A. CpG island hypermethylation at multiple gene sites in diagnosis and prognosis of prostate cancer. Urology. 2008;71:161–167. doi: 10.1016/j.urology.2007.09.056. [DOI] [PubMed] [Google Scholar]
- 114.Cho NY, Kim BH, Choi M, Yoo EJ, Moon KC, Cho YM, Kim D, Kang GH. Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J Pathol. 2007;211:269–277. doi: 10.1002/path.2106. [DOI] [PubMed] [Google Scholar]
- 115.Kirby MK, Ramaker RC, Roberts BS, Lasseigne BN, Gunther DS, Burwell TC, Davis NS, Gulzar ZG, Absher DM, Cooper SJ, Brooks JD, Myers RM. Genome-wide DNA methylation measurements in prostate tissues uncovers novel prostate cancer diagnostic biomarkers and transcription factor binding patterns. BMC Cancer. 2017;17:273. doi: 10.1186/s12885-017-3252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yang YA, Yu J. EZH2, an epigenetic driver of prostate cancer. Protein Cell. 2013;4:331–341. doi: 10.1007/s13238-013-2093-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jarrard DF, Kinoshita H, Shi Y, Sandefur C, Hoff D, Meisner LF, Chang C, Herman JG, Isaacs WB, Nassif N. Methylation of the androgen receptor promoter CpG island is associated with loss of androgen receptor expression in prostate cancer cells. Cancer Res. 1998;58:5310–5314. [PubMed] [Google Scholar]
- 118.Li LC, Chui R, Nakajima K, Oh BR, Au HC, Dahiya R. Frequent methylation of estrogen receptor in prostate cancer: correlation with tumor progression. Cancer Res. 2000;60:702–706. [PubMed] [Google Scholar]
- 119.Gravina GL, Ranieri G, Muzi P, Marampon F, Mancini A, Di Pasquale B, Di Clemente L, Dolo V, D’Alessandro AM, Festuccia C. Increased levels of DNA methyltransferases are associated with the tumorigenic capacity of prostate cancer cells. Oncol Rep. 2013;29:1189–1195. doi: 10.3892/or.2012.2192. [DOI] [PubMed] [Google Scholar]
- 120.Valdez CD, Kunju L, Daignault S, Wojno KJ, Day ML. The E2F1/DNMT1 axis is associated with the development of AR negative castration resistant prostate cancer. Prostate. 2013;73:1776–1785. doi: 10.1002/pros.22715. [DOI] [PubMed] [Google Scholar]
- 121.Morey SR, Smiraglia DJ, James SR, Yu J, Moser MT, Foster BA, Karpf AR. DNA methylation pathway alterations in an autochthonous murine model of prostate cancer. Cancer Res. 2006;66:11659–11667. doi: 10.1158/0008-5472.CAN-06-1937. [DOI] [PubMed] [Google Scholar]
- 122.Kobayashi Y, Absher DM, Gulzar ZG, Young SR, McKenney JK, Peehl DM, Brooks JD, Myers RM, Sherlock G. DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome Res. 2011;21:1017–1027. doi: 10.1101/gr.119487.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Patra A, Deb M, Dahiya R, Patra SK. 5-Aza-2’-deoxycytidine stress response and apoptosis in prostate cancer. Clin Epigenetics. 2011;2:339–348. doi: 10.1007/s13148-010-0019-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee E, Wang J, Yumoto K, Jung Y, Cackowski FC, Decker AM, Li Y, Franceschi RT, Pienta KJ, Taichman RS. DNMT1 regulates epithelial-mesenchymal transition and cancer stem cells, which promotes prostate cancer metastasis. Neoplasia. 2016;18:553–566. doi: 10.1016/j.neo.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gravina GL, Marampon F, Di Staso M, Bonfili P, Vitturini A, Jannini EA, Pestell RG, Tombolini V, Festuccia C. 5-Azacitidine restores and amplifies the bicalutamide response on preclinical models of androgen receptor expressing or deficient prostate tumors. Prostate. 2010;70:1166–1178. doi: 10.1002/pros.21151. [DOI] [PubMed] [Google Scholar]
- 126.Festuccia C, Gravina GL, D’Alessandro AM, Muzi P, Millimaggi D, Dolo V, Ricevuto E, Vicentini C, Bologna M. Azacitidine improves antitumor effects of docetaxel and cisplatin in aggressive prostate cancer models. Endocr Relat Cancer. 2009;16:401–413. doi: 10.1677/ERC-08-0130. [DOI] [PubMed] [Google Scholar]
- 127.Sonpavde G, Aparicio A, Guttierez I, Boehm KA, Hutson TE, Berry WR, Asmar L, von Hoff DD. Phase II study of azacitidine to restore responsiveness of prostate cancer to hormonal therapy. Clin Genitourin Cancer. 2007;5:457–459. doi: 10.3816/CGC.2007.n.036. [DOI] [PubMed] [Google Scholar]
- 128.McCabe MT, Low JA, Daignault S, Imperiale MJ, Wojno KJ, Day ML. Inhibition of DNA methyltransferase activity prevents tumorigenesis in a mouse model of prostate cancer. Cancer Res. 2006;66:385–392. doi: 10.1158/0008-5472.CAN-05-2020. [DOI] [PubMed] [Google Scholar]
- 129.Zhang W, Jiao H, Zhang X, Zhao R, Wang F, He W, Zong H, Fan Q, Wang L. Correlation between the expression of DNMT1, and GSTP1 and APC, and the methylation status of GSTP1 and APC in association with their clinical significance in prostate cancer. Mol Med Rep. 2015;12:141–146. doi: 10.3892/mmr.2015.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Berry SJ, Coffey DS, Walsh PC, Ewing LL. The development of human benign prostatic hyperplasia with age. J Urol. 1984;132:474–479. doi: 10.1016/s0022-5347(17)49698-4. [DOI] [PubMed] [Google Scholar]
- 131.McVary KT, Roehrborn CG, Avins AL, Barry MJ, Bruskewitz RC, Donnell RF, Foster HE Jr, Gonzalez CM, Kaplan SA, Penson DF, Ulchaker JC, Wei JT. Update on AUA guideline on the management of benign prostatic hyperplasia. J Urol. 2011;185:1793–1803. doi: 10.1016/j.juro.2011.01.074. [DOI] [PubMed] [Google Scholar]
- 132.Wei JT, Calhoun E, Jacobsen SJ. Urologic diseases in America project: benign prostatic hyperplasia. J Urol. 2005;173:1256–1261. doi: 10.1097/01.ju.0000155709.37840.fe. [DOI] [PubMed] [Google Scholar]
- 133.Saigal CS, Joyce G. Economic costs of benign prostatic hyperplasia in the private sector. J Urol. 2005;173:1309–1313. doi: 10.1097/01.ju.0000152318.79184.6f. [DOI] [PubMed] [Google Scholar]
- 134.Kaplan SA, Gonzalez RR. Phosphodiesterase type 5 inhibitors for the treatment of male lower urinary tract symptoms. Rev Urol. 2007;9:73–77. [PMC free article] [PubMed] [Google Scholar]
- 135.Roehrborn CG. Current medical therapies for men with lower urinary tract symptoms and benign prostatic hyperplasia: achievements and limitations. Rev Urol. 2008;10:14–25. [PMC free article] [PubMed] [Google Scholar]
- 136.Bechis SK, Otsetov AG, Ge R, Olumi AF. Personalized medicine for management of benign prostatic hyperplasia. J Urol. 2014;192:16–23. doi: 10.1016/j.juro.2014.01.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Henrique R, Jeronimo C, Hoque MO, Carvalho AL, Oliveira J, Teixeira MR, Lopes C, Sidransky D. Frequent 14-3-3 sigma promoter methylation in benign and malignant prostate lesions. DNA Cell Biol. 2005;24:264–269. doi: 10.1089/dna.2005.24.264. [DOI] [PubMed] [Google Scholar]
- 138.Niu Y, Ge R, Hu L, Diaz C, Wang Z, Wu CL, Olumi AF. Reduced levels of 5-alpha reductase 2 in adult prostate tissue and implications for BPH therapy. Prostate. 2011;71:1317–1324. doi: 10.1002/pros.21348. [DOI] [PubMed] [Google Scholar]
- 139.Ge R, Wang Z, Bechis SK, Otsetov AG, Hua S, Wu S, Wu CL, Tabatabaei S, Olumi AF. DNA methyl transferase 1 reduces expression of srd5a2 in the aging adult prostate. Am J Pathol. 2015;185:870–882. doi: 10.1016/j.ajpath.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang Z, Hu L, Salari K, Bechis SK, Ge R, Wu S, Rassoulian C, Pham J, Wu CL, Tabatabaei S, Strand DW, Olumi AF. Androgenic to oestrogenic switch in the human adult prostate gland is regulated by epigenetic silencing of steroid 5alpha-reductase 2. J Pathol. 2017;243:457–467. doi: 10.1002/path.4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hahn O, Grönke S, Stubbs TM, Ficz G, Hendrich O, Krueger F, Andrews S, Zhang Q, Wakelam MJ, Beyer A, Reik W, Partridge L. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 2017;18:56. doi: 10.1186/s13059-017-1187-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yoon AR, Tammen SA, Park S, Han SN, Choi SW. Genome-wide hepatic DNA methylation changes in high-fat diet-induced obese mice. Nutr Res Pract. 2017;11:105–113. doi: 10.4162/nrp.2017.11.2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, Zanobetti A, Sparrow D, Vokonas PS, Schwartz J. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med. 2009;179:572–578. doi: 10.1164/rccm.200807-1097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Madrigano J, Baccarelli A, Mittleman MA, Wright RO, Sparrow D, Vokonas PS, Tarantini L, Schwartz J. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environ Health Perspect. 2011;119:977–982. doi: 10.1289/ehp.1002773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.De Prins S, Koppen G, Jacobs G, Dons E, Van de Mieroop E, Nelen V, Fierens F, Int Panis L, De Boever P, Cox B, Nawrot TS, Schoeters G. Influence of ambient air pollution on global DNA methylation in healthy adults: a seasonal follow-up. Environ Int. 2013;59:418–424. doi: 10.1016/j.envint.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 146.Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun HM, Jiang J, Marinelli B, Pesatori AC, Bertazzi PA, Yang AS. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67:876–880. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- 147.Wadhwa PD, Buss C, Entringer S, Swanson JM. Developmental origins of health and disease: Brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med. 2009;27:358–368. doi: 10.1055/s-0029-1237424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261:412–417. doi: 10.1111/j.1365-2796.2007.01809.x. [DOI] [PubMed] [Google Scholar]
- 149.Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cooney CA, Dave AA, Wolff GL. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr. 2002;132:2393s–2400s. doi: 10.1093/jn/132.8.2393S. [DOI] [PubMed] [Google Scholar]
- 151.Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–4053. doi: 10.1093/hmg/ddp353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.vom Saal FS, Timms BG, Montano MM, Palanza P, Thayer KA, Nagel SC, Dhar MD, Ganjam VK, Parmigiani S, Welshons WV. Prostate enlargement in mice due to fetal exposure to low doses of estradiol or diethylstilbestrol and opposite effects at high doses. Proc Natl Acad Sci U S A. 1997;94:2056–2061. doi: 10.1073/pnas.94.5.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gupta C. Reproductive malformation of the male offspring following maternal exposure to estrogenic chemicals. Proc Soc Exp Biol Med. 2000;224:61–68. doi: 10.1046/j.1525-1373.2000.22402.x. [DOI] [PubMed] [Google Scholar]
- 155.Timms BG, Howdeshell KL, Barton L, Bradley S, Richter CA, vom Saal FS. Estrogenic chemicals in plastic and oral contraceptives disrupt development of the fetal mouse prostate and urethra. Proc Natl Acad Sci U S A. 2005;102:7014–7019. doi: 10.1073/pnas.0502544102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ho SM, Tang WY, Belmonte de Frausto J, Prins GS. Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res. 2006;66:5624–5632. doi: 10.1158/0008-5472.CAN-06-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Perez AP, Biancardi MF, Caires CR, Falleiros-Junior LR, Goes RM, Vilamaior PS, Santos FC, Taboga SR. Prenatal exposure to ethinylestradiol alters the morphologic patterns and increases the predisposition for prostatic lesions in male and female gerbils during ageing. Int J Exp Pathol. 2016;97:5–17. doi: 10.1111/iep.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cheong A, Zhang X, Cheung YY, Tang WY, Chen J, Ye SH, Medvedovic M, Leung YK, Prins GS, Ho SM. DNA methylome changes by estradiol benzoate and bisphenol A links early-life environmental exposures to prostate cancer risk. Epigenetics. 2016;11:674–689. doi: 10.1080/15592294.2016.1208891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tang WY, Morey LM, Cheung YY, Birch L, Prins GS, Ho SM. Neonatal exposure to estradiol/bisphenol A alters promoter methylation and expression of Nsbp1 and Hpcal1 genes and transcriptional programs of Dnmt3a/b and Mbd2/4 in the rat prostate gland throughout life. Endocrinology. 2012;153:42–55. doi: 10.1210/en.2011-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lin TM, Rasmussen NT, Moore RW, Albrecht RM, Peterson RE. Region-specific inhibition of prostatic epithelial bud formation in the urogenital sinus of C57BL/6 mice exposed in utero to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Sci. 2003;76:171–181. doi: 10.1093/toxsci/kfg218. [DOI] [PubMed] [Google Scholar]
- 161.Vezina CM, Allgeier SH, Moore RW, Lin TM, Bemis JC, Hardin HA, Gasiewicz TA, Peterson RE. Dioxin causes ventral prostate agenesis by disrupting dorsoventral patterning in developing mouse prostate. Toxicol Sci. 2008;106:488–496. doi: 10.1093/toxsci/kfn183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Roman BL, Timms BG, Prins GS, Peterson RE. In utero and lactational exposure of the male rat to 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs prostate development. 2. Effects on growth and cytodifferentiation. Toxicol Appl Pharmacol. 1998;150:254–270. doi: 10.1006/taap.1998.8395. [DOI] [PubMed] [Google Scholar]
- 163.Ricke WA, Lee CW, Clapper TR, Schneider AJ, Moore RW, Keil KP, Abler LL, Wynder JL, Lopez Alvarado A, Beaubrun I, Vo J, Bauman TM, Ricke EA, Peterson RE, Vezina CM. In utero and lactational TCDD exposure increases susceptibility to lower urinary tract dysfunction in adulthood. Toxicol Sci. 2016;150:429–440. doi: 10.1093/toxsci/kfw009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wu Q, Ohsako S, Ishimura R, Suzuki JS, Tohyama C. Exposure of mouse preimplantation embryos to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters the methylation status of imprinted genes H19 and Igf2. Biol Reprod. 2004;70:1790–1797. doi: 10.1095/biolreprod.103.025387. [DOI] [PubMed] [Google Scholar]
- 165.Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104:13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pfeiffer CM, Hughes JP, Lacher DA, Bailey RL, Berry RJ, Zhang M, Yetley EA, Rader JI, Sempos CT, Johnson CL. Estimation of trends in serum and RBC folate in the U.S. population from pre- to postfortification using assay-adjusted data from the NHANES 1988-2010. J Nutr. 2012;142:886–893. doi: 10.3945/jn.111.156919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Pfeiffer CM, Johnson CL, Jain RB, Yetley EA, Picciano MF, Rader JI, Fisher KD, Mulinare J, Osterloh JD. Trends in blood folate and vitamin B-12 concentrations in the United States, 1988 2004. Am J Clin Nutr. 2007;86:718–727. doi: 10.1093/ajcn/86.3.718. [DOI] [PubMed] [Google Scholar]
- 168.Barua S, Chadman KK, Kuizon S, Buenaventura D, Stapley NW, Ruocco F, Begum U, Guariglia SR, Brown WT, Junaid MA. Increasing maternal or post-weaning folic acid alters gene expression and moderately changes behavior in the offspring. PLoS One. 2014;9:e101674. doi: 10.1371/journal.pone.0101674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Barua S, Kuizon S, Brown WT, Junaid MA. High gestational folic acid supplementation alters expression of imprinted and candidate autism susceptibility genes in a sex-specific manner in mouse offspring. J Mol Neurosci. 2016;58:277–286. doi: 10.1007/s12031-015-0673-8. [DOI] [PubMed] [Google Scholar]
- 170.de Vogel S, Meyer K, Fredriksen A, Ulvik A, Ueland PM, Nygard O, Vollset SE, Tell GS, Tretli S, Bjorge T. Serum folate and vitamin B12 concentrations in relation to prostate cancer risk--a Norwegian population-based nested case-control study of 3000 cases and 3000 controls within the JANUS cohort. Int J Epidemiol. 2013;42:201–210. doi: 10.1093/ije/dys199. [DOI] [PubMed] [Google Scholar]
- 171.Wien TN, Pike E, Wisløff T, Staff A, Smeland S, Klemp M. Cancer risk with folic acid supplements: a systematic review and meta-analysis. BMJ Open. 2012;2:e000653. doi: 10.1136/bmjopen-2011-000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Collin SM, Metcalfe C, Refsum H, Lewis SJ, Zuccolo L, Smith GD, Chen L, Harris R, Davis M, Marsden G, Johnston C, Lane JA, Ebbing M, Bønaa KH, Nygård O, Ueland PM, Grau MV, Baron JA, Donovan JL, Neal DE, Hamdy FC, Smith AD, Martin RM. Circulating folate, vitamin B12, homocysteine, vitamin B12 transport proteins, and risk of prostate cancer: a case-control study, systematic review, and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2010;19:1632–1642. doi: 10.1158/1055-9965.EPI-10-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bistulfi G, Foster BA, Karasik E, Gillard B, Miecznikowski J, Dhiman VK, Smiraglia DJ. Dietary folate deficiency blocks prostate cancer progression in the TRAMP model. Cancer Prev Res (Phila) 2011;4:1825–1834. doi: 10.1158/1940-6207.CAPR-11-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Vollset SE, Clarke R, Lewington S, Ebbing M, Halsey J, Lonn E, Armitage J, Manson JE, Hankey GJ, Spence JD, Galan P, Bønaa KH, Jamison R, Gaziano JM, Guarino P, Baron JA, Logan RF, Giovannucci EL, den Heijer M, Ueland PM, Bennett D, Collins R, Peto R B-Vitamin Treatment Trialists’ Collaboration. Effects of folic acid on overall and site-specific cancer incidence during the randomised trials: meta-analyses of data on 50,000 individuals. Lancet. 2013;381:1029–36. doi: 10.1016/S0140-6736(12)62001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Qin X, Cui Y, Shen L, Sun N, Zhang Y, Li J, Xu X, Wang B, Xu X, Huo Y, Wang X. Folic acid supplementation and cancer risk: a meta-analysis of randomized controlled trials. Int J Cancer. 2013;133:1033–1041. doi: 10.1002/ijc.28038. [DOI] [PubMed] [Google Scholar]
- 176.Keil KP, Abler LL, Altmann HM, Wang Z, Wang P, Ricke WA, Bjorling DE, Vezina CM. Impact of a folic acid-enriched diet on urinary tract function in mice treated with testosterone and estradiol. Am J Physiol Renal Physiol. 2015;308:F1431–1443. doi: 10.1152/ajprenal.00674.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Joseph DB, Chandrashekar AS, Chu LF, Thomson JA, Vezina CM. A folic acid-enriched diet attenuates prostate involution in response to androgen deprivation. Prostate. 2019;79:183–194. doi: 10.1002/pros.23723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Howell CY, Bestor TH, Ding F, Latham KE, Mertineit C, Trasler JM, Chaillet JR. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell. 2001;104:829–838. doi: 10.1016/s0092-8674(01)00280-x. [DOI] [PubMed] [Google Scholar]
- 179.Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, Biniszkiewicz D, Bates B, Lee PP, Kuhn R, Trumpp A, Poon C, Wilson CB, Jaenisch R. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. 2001;21:788–797. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hutnick LK, Golshani P, Namihira M, Xue Z, Matynia A, Yang XW, Silva AJ, Schweizer FE, Fan G. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum Mol Genet. 2009;18:2875–2888. doi: 10.1093/hmg/ddp222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Trowbridge JJ, Snow JW, Kim J, Orkin SH. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell. 2009;5:442–449. doi: 10.1016/j.stem.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rhee KD, Yu J, Zhao CY, Fan G, Yang XJ. Dnmt1-dependent DNA methylation is essential for photoreceptor terminal differentiation and retinal neuron survival. Cell Death Dis. 2012;3:e427. doi: 10.1038/cddis.2012.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nasonkin IO, Merbs SL, Lazo K, Oliver VF, Brooks M, Patel K, Enke RA, Nellissery J, Jamrich M, Le YZ, Bharti K, Fariss RN, Rachel RA, Zack DJ, Rodriguez-Boulan EJ, Swaroop A. Conditional knockdown of DNA methyltransferase 1 reveals a key role of retinal pigment epithelium integrity in photoreceptor outer segment morphogenesis. Development. 2013;140:1330–1341. doi: 10.1242/dev.086603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Li J. Progressive alopecia reveals decreasing stem cell activation probability during aging of mice with epidermal deletion of DNA methyltransferase 1 (DNMT1) J Invest Dermatol. 2012;132:2681–2690. doi: 10.1038/jid.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hultdin J, Van Guelpen B, Bergh A, Hallmans G, Stattin P. Plasma folate, vitamin B12, and homocysteine and prostate cancer risk: a prospective study. Int J Cancer. 2005;113:819–824. doi: 10.1002/ijc.20646. [DOI] [PubMed] [Google Scholar]
- 186.Figueiredo JC, Grau MV, Haile RW, Sandler RS, Summers RW, Bresalier RS, Burke CA, McKeown-Eyssen GE, Baron JA. Folic acid and risk of prostate cancer: results from a randomized clinical trial. J Natl Cancer Inst. 2009;101:432–435. doi: 10.1093/jnci/djp019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tomaszewski JJ, Cummings JL, Parwani AV, Dhir R, Mason JB, Nelson JB, Bacich DJ, O’Keefe DS. Increased cancer cell proliferation in prostate cancer patients with high levels of serum folate. Prostate. 2011;71:1287–1293. doi: 10.1002/pros.21346. [DOI] [PMC free article] [PubMed] [Google Scholar]