

Abstract
Elongation factor for RNA polymerase II 2 (ELL2) and ELL-associated factor 2 (EAF2) are two functionally related androgen responsive gene-encoded proteins with prostate tumor suppressor characteristics. EAF2 and ELL2 have both been shown to be down-regulated in advanced prostate cancer, and mice with either Eaf2 or Ell2 deficiency developed murine prostatic intraepithelial neoplasia (mPIN), increased cellular proliferation and increased vascularity. Functional studies have revealed that EAF2 and ELL2 can bind to each other and have similar roles in regulating cell proliferation, angiogenesis and prostate homeostasis. Here, cell line experiments showed that knockdown of EAF2 or ELL2 induced an increase in proliferation and migration in C4-2 and 22Rv1 prostate cancer cells. Concurrent knockdown of EAF2 and ELL2 increased proliferation and migration similarly to the loss of EAF2 or ELL2 alone. Mice with homozygous deletion of Ell2 or heterozygous deletion of Eaf2 developed mPIN lesions characterized by increased epithelial proliferation, intraductal microvessel density, and infiltrating intraductal CD3-positive T-cells compared to wild-type controls. Mice with combined heterozygous deletion of Eaf2 and Ell2 developed mPIN lesions that were similar to those observed in mice with deficiency in Eaf2 or Ell2 alone. These results suggest that EAF2 and ELL2 have similar functions and are likely to require each other in their regulation of prostate epithelial cell proliferation and migration in prostate cancer cells as well as their tumor suppressive properties in the murine prostate.
Keywords: ELL2, EAF2, prostate cancer, C4-2, 22Rv1, mouse
Introduction
Eleven-nineteen lysine-rich leukemia 2 (ELL2) and ELL-associated factor 2 (EAF2) are two interacting proteins that play an important role in transcription elongation and the regulation of gene expression in both mammalian cells as well as in lower eukaryotes. The ELL family proteins suppress transient pausing of RNA polymerase II [1-4]. The EAF family proteins, EAF1 and EAF2, can bind directly to the ELL family proteins (ELL, ELL2 and ELL3) to stimulate elongation activity [5]. In S. pombe, the SpELL-SpEAF complex stimulated the transcription elongation rate, while SpEAF or SpELL alone had a negligible effect on the rate of elongation [6]. The SpELL-SpEAF complex could also stimulate pyrophosphoryolysis, suggesting that SpELL-SpEAF could stimulate transcription by stabilizing the active site of RNA polymerase II in its active configuration [6]. In C. elegans, worm orthologs of the EAF and ELL family were shown to share expression patterns and to have overlapping function in the regulation of fertility, survival and cuticle formation [7]. Loss of either C. elegans ell-1 or eaf-1 resulted in abnormal development, reduced survival and reduced fertility [7]. In mammalian cells, ELL and EAF formed a stable complex that could stimulate the rate of transcription elongation above the transcription elongation rate stimulated by either ELL or EAF proteins alone [5].
EAF2 and ELL2 also appear to play similar and potentially inter-dependent roles in maintaining adult prostate homeostasis. In advanced prostate cancer, downregulation of EAF2 [8-10] and/or ELL2 [11,12] have been shown in human tissue specimens, while EAF2 and ELL2 are up-regulated in the epithelium of benign prostatic hyperplasia nodules [13]. Knockdown of EAF2 in prostate cancer cell lines has been shown to enhance proliferation, migration and invasion [14]; and ELL2 knockdown in LNCaP or C4-2 cells also stimulated proliferation, migration and invasion [11,12]. In murine models, homozygous Eaf2 [10,15] and Ell2 [16] deficiency similarly induced increased proliferation, high-grade murine prostatic intraepithelial neoplasia (mPIN) and increased vascularity in the prostate. Recently, we have shown that in C4-2 prostate cancer cells, EAF2 could bind to ELL2, stabilizing ELL2 protein and inhibiting its polyubiquitination [12]. EAF2 protein levels were reduced in 22Rv1 cells treated with ELL2 knockdown, while EAF2 protein levels in C4-2 and LNCaP were either decreased or unchanged [12]. These results suggest that loss of EAF2 could induce a down-regulation of ELL2 in prostate cells. Cumulatively, these studies suggest that EAF2 and ELL2 may require each other to act as prostate tumor suppressors.
In the current study, the potential functional consequences of combined loss of EAF2 and ELL2 in the prostate were explored in prostate cancer cell lines and in a murine model. Prostate cancer cell lines C4-2 and 22Rv1 were utilized in knockdown studies to determine the combined effects of EAF2 and ELL2 loss on proliferation and migration. Mice with heterozygous deficiency in Eaf2 and mice with combined heterozygous deficiency in Eaf2 and Ell2 knockout mice were generated and analyzed for histological defects.
Materials and methods
Cell culture experiments
Human C4-2 prostate cancer cells were a gift from Dr. Leland W.K. Chung. The 22Rv1 cell line was purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA). All cell lines were maintained in RPMI-1640 medium, supplemented with 10% heat-inactivated fetal bovine serum (FBS). The C4-2 and 22Rv1 prostate cancer cell lines were authenticated in 2016 by the University of Pittsburgh Cell Culture and Cytogenetics Facility using DNA fingerprinting by examining microsatellite loci in a multiplex PCR (AmpFlSTR Identifiler PCR Amplification Kit, Applied Biosystems, Foster City, CA). All experiments using cell lines were performed a minimum of three times.
The effects of EAF2 and ELL2 knockdown were determined in C4-2 and 22Rv1 cells. Small interfering RNAs (siRNAs) targeting EAF2 (siEAF2-1 forward, 5’-AAACAGUUACUGGUGGAGUUGAACCUU-3’, siEAF2-1 reverse, 5’-AAGGUUCAACUCCACCAGUAACUGUUU-3’, siEAF2-2 forward, 5’-CUGUUCACCUUCACCAACCUCAAGGUA-3’, siEAF2-2 reverse, 5’-UACCUUGAGGUUGGUGAAGGUGAACAG-3’); siELL2 (siELL2-1 forward, 5’-CUCACAUCCUCCUCAGAUUGUAAAT, siELL2-1 reverse, 5’-AUUUACAAUCUGAGGAGGAUGUGAGAU-3’, siELL2-2 forward, 5’-AAGCAUUUCACCUUGCACAUUACUGUU-3’, siELL2-2 reverse, CAGUAAUGUGCAAGGUGAAAUGCUU-3’) were purchased from Integrated DNA Technologies (Co-ralville, IA, USA), and negative control (siCtrl) siRNAs were purchased from Dharmacon (ONTARGETplus Non-targeting Pool, # D-001810-10-50). Cells were transfected with siRNA for 72 hours using DharmaFECT 1 Transfection Reagent (Dharmacon) following the manufacturer’s instructions. Control siRNA was added to siEAF2 or siELL2 to make sure that total siRNA amount was the same in different treatment conditions. For the control group, 50 nM control siRNA was added. For the EAF2 knockdown group. 25 nM control siRNA and 25 nM siEAF2 were added. For the ELL2 knockdown group. 25 nM control siRNA and 25 nM siELL2 were added. For double knockdown group, 25 nM siELL2 and 25 nM siEAF2 were added. All knockdown experiments were repeated with 2 different siRNAs to control for off target effects.
BrdU incorporation assay
For the BrdU incorporation assay, cells were seeded on coverslips (Fisher, Pittsburgh, PA, US) and transfected with siELL2 then treated with R1881 at 1 nM for 48 hours. Cells were subsequently cultured in the presence of 10 µM BrdU for two hours and then fixed with Carnoy’s fixative (3:1 volume methanol to glacial acetic acid) for 20 min at -20°C. After treatment with 2 M HCl and wash with 0.1 M boric acid, cells were incubated with 3% hydrogen peroxide (H2O2) for 10 min at room temperature, followed by blocking with 10% goat serum for 1 hour. Cells were then incubated with anti-BrdU antibody (B2531, Sigma) overnight at 4°C and CY3 labeled goat anti-mouse secondary antibody (A10521, Life technologies) for 1 hour at 37°C. The nuclei were stained with SYTOX Green (S7020, Life technologies). Images were acquired using a fluorescence microscope (Nikon TE2000-U). The cells were counted using Photoshop CS5 counting tool (Adobe, San Jose, California, USA) and the percentage of BrdU-positive cells was calculated using the following formula: (BrdU-positive cell number/total cell number) × 100%.
Western blot
Cell samples were lysed in RIPA buffer [50 mM Tris-Cl (pH 7.4), 1 mM EDTA, 1% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, 150 mM NaCl] with 1% protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) and protein concentration was determined by BCA Protein Assay (Thermo Scientific, Rockford, IL). Tissue lysate was boiled with SDS sample buffer, separated on a NEXT GELTM 10% gel (Amresco) under reducing conditions and transferred onto a nitrocellulose membrane. Antibodies used were an internally generated mouse monoclonal anti-EAF2 (1:1000, [10]), rabbit polyclonal anti-ELL2 (1:1000, A302-505A, Bethyl Laboratories, Inc., Montgomery, TX, USA), and rabbit polyclonal anti-GAPDH (1:6000, FL-335, sc25778, Santa Cruz Biotechnology, Dallas, TX, USA). Blotted proteins were probed with primary antibodies followed by HRP (Horseradish peroxidase) labeled secondary antibody (Santa Cruz Biotechnology). Signals were visualized using chemiluminescence (ECLTM Western Blotting Detection Reagents®, GE Healthcare) and imaged using the VersaDoc imaging system (4000 MP, Bio-Rad, Hercules, CA, USA) following the manufacture’s instruction.
Wound healing assays
The wound healing assay was performed in a six-well plate. Following RNA interference, C4-2 and 22Rv1 cells were treated with 2 nM R1881 for 48 hours until they reached a density of about 70%. Straight scratches were made using a 200 μl pipette tip. Debris was removed by washing the cells with phosphate buffered saline (PBS), and 2 ml of fresh medium supplemented with 2% FBS was added. Cells were incubated at 5% CO2 and 37°C for 24 hours. Images were taken using bright field microscopy (TS100, Nikon, Japan) at 0, 12 and 24 hours. Each experiment was performed in triplicate and repeated a minimum of 3 times. Quantification of gap closure was also normalized to the initial gap size and results were plotted as a percentage of the initial gap size at time 0.
Generation of Ell2 and Eaf2 deletion mice
Here, mice with combined heterozygous deletion of Eaf2 and Ell2 were generated by crossing the previously described Eaf2 [10] and Ell2-cko (Ell2loxp/loxpPbsnCre4) [16] knockout mice on a C57BL6/J background. Experimental cohorts included wild-type (Eaf2+/+Ell2+/+PbsnCre4), Eaf2+/- (Eaf2+/-Ell2+/+PbsnCre4), Ell2-cko (Eaf2+/+Ell2-/-PbsnCre4), and Eaf2+/-Ell2+/- (Eaf2+/-Ell2loxp/+PbsnCre4) C57BL/6J male littermates, and all mice were maintained identically. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pittsburgh and were conducted in strict accordance with the standards for humane animal care and use as set by the Animal Welfare Act and the National Institutes of Health guidelines for the use of laboratory animals. Genotyping was determined by PCR analysis of mouse tail and confirmed on muscle DNA when animals were euthanized as previously described for conventional deletion of Eaf2 [15] and conditional deletion of Ell2 [16]. Prostate tissue necropsy was performed and organs were cleaned of excess fat and membrane with phosphate-buffered saline; mass of each prostate lobe was determined after blotting with filtration paper to remove excess water. Samples were fixed in 10% formalin for at least 24 hours, then embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin. All tissues were examined by a board-certified animal pathologist in a blinded fashion (LHR, V.M.D).
Immunohistochemistry
Immunohistochemical stains were performed on five-micron sections of paraffin-embedded murine tissue specimens. The slides were deparaffinized and rehydrated using a standard histology protocol. Antigen retrieval was performed using a citrate retrieval solution and a Decloaking chamber at 120°C. The slides were stained using an Autostainer Plus (Dako, Carpenteria, CA) platform with TBST rinse buffer (Dako). The Ki67 (D3B5) antibody (Cell Signaling Technology, Danvers, MA) and the CD3 (Rabbit Polyclonal) (Dako) were applied at a 1:100 dilutions. The detection used for the Ki67 and CD3 consisted of Envision + Anti-Rabbit HRP polymer (Dako). The CD19 (D4V4B) antibody (Cell Signaling Technology, Danvers, MA) was applied at a 1:800 dilution. The Rabbit Signal Stain Boost Detection Kit (Cell Signaling) was used for the detection. The CD31 (Goat Polyclonal) antibody (Santa Cruz Biotechnology, Dallas, TX) was applied at a 1:250 dilution. The Goat Polymer/Probe system (Biocare Medical, Concord, CA) was used for the detection. The substrate used for all of the above, was 3,3, diaminobenzidine (Dako). Lastly, the slides were counterstained with Hematoxylin (Cell Signaling). Immunostained sections were imaged with a Leica DM LB microscope (Leica Microsystems Inc, Bannockburn, IL) equipped with an Imaging Source NII 770 camera (The Imaging Source Europe GmbH, Bremen, Germany) and NIS-Elements Documentation v 4.6 software (Nikon Instruments, Inc., Mellville, NY). All tissues were examined by a board-certified veterinary pathologist (L.H.R.) using light microscopy.
For murine tissues, Ki-67-positive cell density was determined by analysis of sections from at least 3 independent mice from each genotype from at least 3 different fields per mouse imaged at 20 × magnification with no overlap. Composite images were constructed with Photoshop CS (Adobe Systems, San Jose, CA). Inflammatory cells were counted to determine the average number of CD3-positive and CD19-positive cells per 40 × field for each section. Assessment of intraductal microvessel density was determined based on CD31-positive blood vessel count as previously [14].
Statistical analysis
Comparison between groups were calculated using the Student’s t-test, the two-tailed Fisher’s exact test method of summing small p values, the 1-way and 2-way ANOVA and Bonferonni’s Multiple Comparison Test as appropriate. A value of P < 0.05 was considered significant. GraphPad Prism version 4 was used for graphics (GraphPad Software, San Diego, CA, USA). Values are expressed as means ± S.E.M.
Results
Combined knockdown of EAF2 and ELL2 effect on cellular proliferation
We have previously shown in BrdU incorporation assays that individual knockdown of EAF2 [14] or ELL2 [11] in C4-2 cells increased cell proliferation. Here, cell proliferation in C4-2 cells with combined knockdown of EAF2 and ELL2 was similar to that of either EAF2 or ELL2 alone (Figure 1A, 1B). Similarly, individual knockdown of EAF2 or ELL2 induced a statistically significant increase in proliferation of 22Rv1 cells as measured by BrdU uptake (Figure 1C, 1D). Combined knockdown of EAF2 and ELL2 in 22Rv1 cells did not differ from the increase induced by individual knockdown of EAF2 or ELL2, in agreement with the findings in C4-2 cells. Western blotting analysis verified the knockdown of EAF2 and/or ELL2 in both cell lines (Figure 1E, 1F).
Figure 1.

Cell proliferation in EAF2- and ELL2-deficient prostate cancer cells. (A) BrdU incorporation in C4-2 cells transfected with nontargeted control (siCtrl) siRNA, targeted to EAF2 (siEAF2), ELL2 (siELL2), or concurrent EAF2 and ELL2 (siDouble) knockdown. The upper panel shows BrdU-positive nuclei (red), and the lower panel shows nuclear staining with SYTOX Green (green). Original magnification, × 40. (B) Quantification of BrdU incorporation shown as mean percentage ± standard deviation of BrdU-positive cells relative to the total number of cells (at least 400 total cells were counted for each condition). (C) BrdU incorporation in 22Rv1 cells transfected with nontargeted control (siCtrl) siRNA, targeted to EAF2 (siEAF2), ELL2 (siELL2), or concurrent EAF2 and ELL2 (siDouble) knockdown as above. (D) Quantification of BrdU incorporation. Results are representative of four individual experiments. (E) Western blot analysis of EAF2 and ELL2 protein from C4-2 cell lysates and (F) 22Rv1 cell lysates following siRNA knockdown as in (A). GAPDH served as internal loading control. *P < 0.05, **P < 0.01, ***P < 0.001.
Combined knockdown of EAF2 and ELL2 effect on migration
Migration of C4-2 cells in response to EAF2 and/or ELL2 knockdown was determined in C4-2 cells and in 22Rv1 cells in a scratch assay. Both C4-2 and 22Rv1 cell migration was significantly enhanced after knockdown of EAF2 or ELL2 (Figure 2). Combined knockdown of EAF2 and ELL2 did not significantly increase gap closure compared to individual knockdown of either EAF2 or ELL2 in C4-2 cells (Figure 2A, 2B). Similarly, combined knockdown of EAF2 and ELL2 did not significantly increase gap closure compared to individual knockdown of either EAF2 or ELL2 in 22Rv1 cells (Figure 2C, 2D). These results suggest that EAF2 and ELL2 suppress tumor growth and motility in cultured prostate cancer cells and that their combined loss does not additively promote prostate cancer cell migration.
Figure 2.
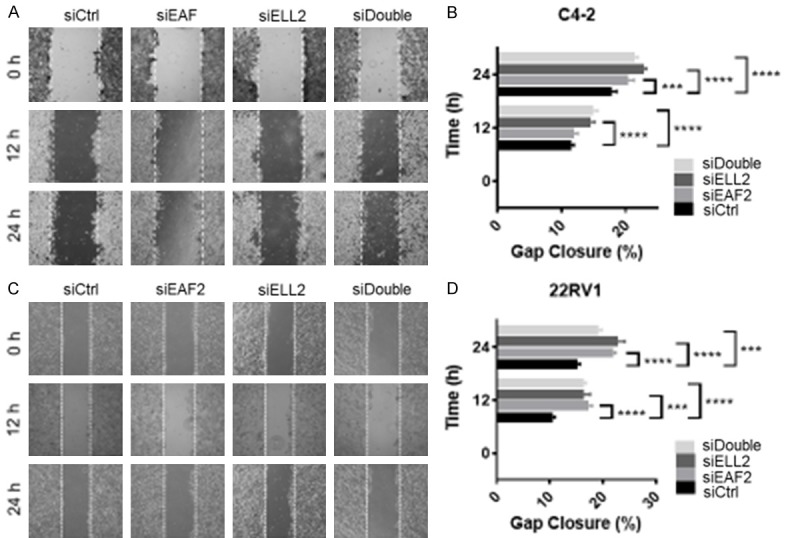
Cell migration in EAF2- and ELL2-deficient prostate cancer cells. (A) Wound healing assay in C4-2 cells transfected with nontargeted control (siCtrl) siRNA, targeted to EAF2 (siEAF2), ELL2 (siELL2), or concurrent EAF2 and ELL2 (siDouble) knockdown. Cells were imaged at 0, 12, and 24 hours after treatment and gap widths were measured using ImageJ software (National Institutes of Health). Original magnification, × 10. (B) Quantification of gap closure in (A), shown as the mean percentage ± standard error of the mean of initial gap and are representative of three individual experiments. (C) Wound healing assay in 22Rv1 cells and (D) quantification of the images in (C). The experimentation and data analysis were performed the same as for C4-2 cells. Results are representative of three individual experiments. ***P < 0.001, ****P < 0.0001.
Concurrent Eaf2 and Ell2-deficiency in a murine model
We have previously reported that Eaf2-/- mice have an increased incidence in murine prostatic intraepithelial neoplasia (mPIN) [10,15]. We also recently reported that mice with prostate-specific conditional deletion of Ell2 displayed prostatic defects including increased epithelial hypertrophy, increased vascularity, stromal defects and murine prostatic intraepithelial neoplasia (mPIN) compared to wild-type controls [16]. Here we generated a cohort of mice with concurrent heterozygous deletion of Eaf2 and Ell2 to determine if there were additive effects on the development and/or severity of these prostatic defects (Table 1). Mice with heterozygous deletion of Eaf2 alone and a small group of three mice with homozygous conditional deletion of Ell2 were also generated and characterized. At 24 mos of age, mice were euthanized and prostates were examined for defects. No wild-type animals displayed mPIN lesions. As previously observed in mice with homozygous deletion of Eaf2 [10], C57BL/6J Eaf2+/- mice developed PIN lesions at 24 mos of age (Figure 3). Previously, we have reported that homozygous deletion of Eaf2 could induced mPIN lesions in 83.3% of C57BL/6J mice at 20-24 mos of age [10]. In the current study, heterozygous deletion of Eaf2 induced mPIN lesions in 67% of animals (4/6) suggesting that heterozygous deficiency in Eaf2 is sufficient to induce mPIN lesions. In our previous study of Ell2 knockout mice on a C57BL/6J background, 42% of animals displayed mPIN lesions at 17-20 mos of age [16]. Here, we generated an additional smaller group of three animals with conditional homozygous deletion of Ell2 and the mice were examined for prostatic defects at 24 mos of age. Of these animals, 3/3 (100%) displayed mPIN lesions at 24 mos of age. The increased mPIN incidence in this group of animals compared to the previous study could be due to the increased age of the animals used in this current study. All of the mice generated with combined heterozygous deletion of Eaf2 and Ell2 displayed mPIN lesions (6/6, 100%). Although the difference was not statistically significant (P = 0.45), possibly due to the small number of animals, there was an increased incidence in the number of Eaf2+/-Ell2+/- animals with mPIN lesions compared to Eaf2+/- mice (4/6, 67%). These results suggested that combined heterozygous deletion of Eaf2 and Ell2 may induce mPIN lesions slightly more efficiently than Eaf2 or Ell2 deficiency alone but was not sufficient to induce prostate tumors in the murine model.
Table 1.
Distribution of mice studied for prostatic defects
| Genotype | Number of animals analyzed | Animals with mPIN (%) | Fisher’s exact p-value |
|---|---|---|---|
| WT | 6 | 0 (0%) | |
| Eaf2+/- | 6 | 4 (67%) | 0.061 |
| Ell2-cko | 3 | 3 (100%) | 0.012 |
| Eaf2+/-Ell2+/- | 6 | 6 (100%) | 0.0022 |
Figure 3.
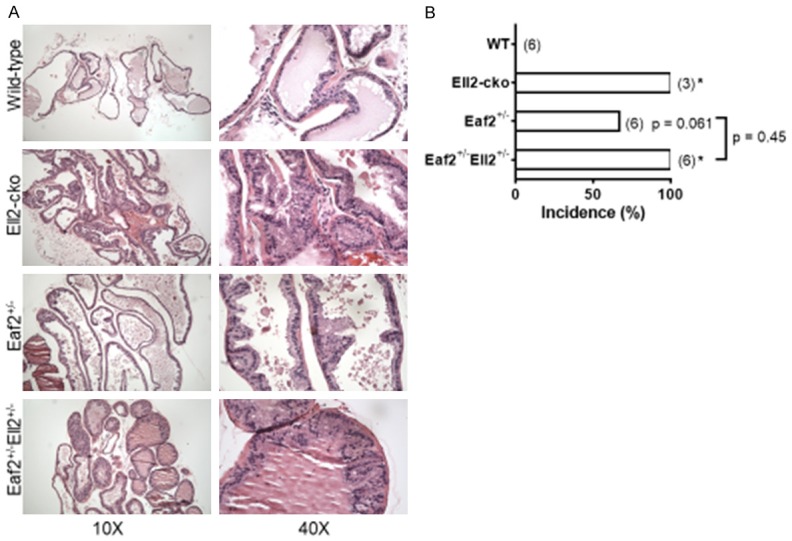
Combined loss of Eaf2 and Ell2 induced murine prostatic intraepithelial neoplasia in the mouse model. A. Histology of mouse ventral prostate from wild-type (Eaf2+/+Ell2+/+PbsnCre4), Eaf2+/- (Eaf2+/-Ell2+/+PbsnCre4), Ell2-cko (Eaf2+/+Ell2-/-PbsnCre4), and Eaf2+/-Ell2+/- (Eaf2+/-Ell2loxp/+PbsnCre4) C57BL/6J male mice aged 24 months stained with hematoxylin and eosin (H&E). Original magnification, × 10, inset, × 40. B. Quantification of mPIN incidence in mice. Data are expressed as percentage from 3 to 6 mice per group. *P < 0.05.
The proliferative marker Ki-67 was used to detect dividing cells in the murine prostates. As we recently reported [16], the number of Ki-67-positive epithelial cells was significantly increased in the prostates of Ell2-cko mice compared to wild-type controls (Figure 4A). The number of Ki-67-positive epithelial cells was also significantly increased in the prostates of Eaf2+/- mice (Figure 4B, 4C) which is in agreement with our previous findings for mice with homozygous deletion of Eaf2 [10]. Mice with combined heterozygosity in Eaf2 and Ell2 displayed an increase in Ki-67-positive cells that was similar to that of mice with Eaf2 or Ell2 deficiency alone (Figure 4B, 4C). These results suggest that both alleles of Eaf2 were required for Ell2 to modulate proliferation and vice versa in the murine prostate. These results were in agreement with the cell proliferation assays in prostate cancer cell lines C4-2 and 22Rv1 treated with knockdown of EAF2 and/or ELL2.
Figure 4.
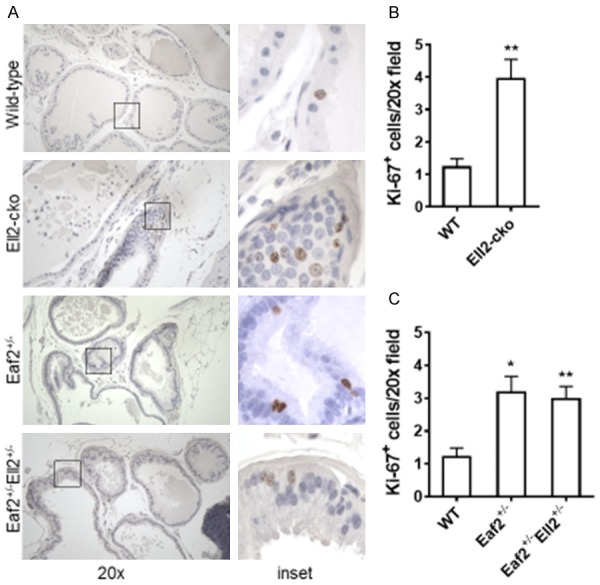
Effects of combined Eaf2 and Ell2 loss on epithelial proliferation in the C57BL/6J mouse prostate at age 24 mos. A. Ki-67 immunostaining in transverse sections of prostate ventral lobes from indicated genotypes at 24 months of age. Original magnification, × 20. B. Quantification of Ki-67+ epithelial cells in mouse prostate from wild-type (Eaf2+/+Ell2+/+PbsnCre4), Eaf2+/- (Eaf2+/-Ell2+/+PbsnCre4), Ell2-cko (Eaf2+/+Ell2-/-PbsnCre4), and Eaf2+/-Ell2+/- (Eaf2+/-Ell2loxp/+PbsnCre4) at 24 months of age. C. Quantification of Ki-67+ epithelial cells in mouse prostate from wild-type and Ell2-cko mice at 24 months of age Data are expressed as mean ± standard error of the mean from 3 to 6 mice per group. *P < 0.05, **P < 0.01.
Concurrent Eaf2 and Ell2-deficiency induces increased vascular density in aged C57BL/6J mice prostates
In previous studies, mice with homozygous deletion of Eaf2 displayed an increase in microvessel density in the prostate [14]. Conditional homozygous deletion of Ell2 in the murine prostate also induced an increase in microvessel density [16]. To investigate whether combined deficiency in Eaf2 and Ell2 further increased microvessel density in the murine prostate, we examined the number of CD31-positive blood vessels by immunostaining in the dorsal-lateral prostate lobes (Figure 5). As expected, the number of CD31-positive intraductal vessels (microvessel density) was significantly increased in the Ell2-cko mice compared to wild-type control animals (Figure 5A). There was also a statistically significant increase in microvessel density in Eaf2+/- mice (Figure 5B, 5C). Interestingly, Eaf2+/-Ell2+/- mice had a seemingly additive, but not statistically significant (P = 0.084) increase in microvessel density compared to Eaf2+/- mice (Figure 5B, 5C). These results suggested that Eaf2 and Ell2 could modulate microvessel density and angiogenesis in the murine prostate when one of them is in heterozygosity.
Figure 5.
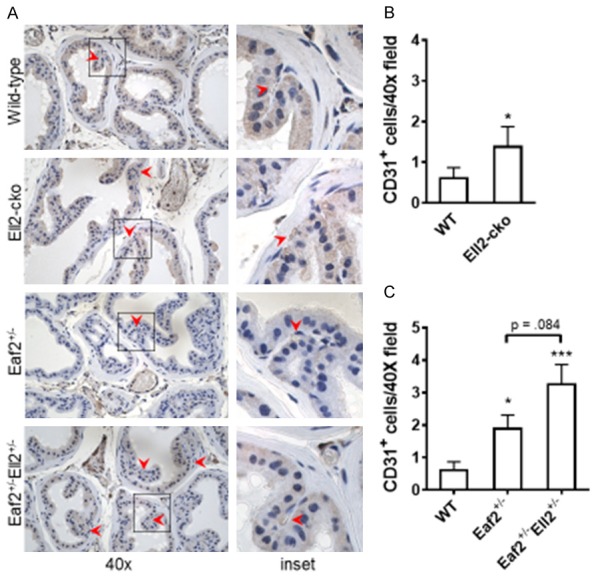
Effects of combined Eaf2 and Ell2 loss on CD31-positive microvessel density in the C57BL/6J mouse prostate at age 24 mos. A. CD31+ immunostaining in transverse sections of prostate dorsal-lateral lobes from indicated genotypes at 24 months of age. Red arrowheads indicate microvessel. Original magnification, × 40. B. Quantification of CD31+ intraductal microvessels in mouse prostate from wild-type and Ell2-cko mice at 24 months of age. C. Quantification of CD31+ vessels in mouse prostate from wild-type and Ell2-cko mice at 24 months of ageData are expressed as mean ± standard error of the mean from 3 to 6 mice per group. *P < 0.05, ***P < 0.001.
Impact of concurrent Eaf2 and Ell2-deficiency on inflammation
In a rat model of prostatic inflammation induced by E. coli, several androgen-responsive genes including EAF2 and ELL2 were found to be up-regulated in the prostate epithelium [17]. Knockdown of EAF2 in C4-2 prostate cancer cells was shown to induce the differential regulation of several genes involved in interferon signaling [12], suggesting that EAF2 and/or ELL2 may play a role in inflammatory response. Previously, CD19Cre-driven deletion of ELL2 in a murine model did not impact the total number of bone marrow or splenic B cells but induced an increase in naïve bone marrow cells and a decrease in splenic transitional 3 (T3) and plasma cells, suggesting that ELL2 may be required for B-cell maturation [18]. We therefore analyzed the number of CD19-positive B-cells in the murine prostates and the number of infiltrating intraductal CD3-positive T-cells within the murine prostate ducts of mice with Eaf2 and/or Ell2 deficiency. There was no statistically significant difference in the number of CD19-positive B cells among the groups (data not shown). However, the number of infiltrating CD3-positive T cells was significantly increased in all groups compared to wild-type controls. Ell2-cko mice displayed an approximate 2-fold increase in the number of infiltrating CD3-positive cells compared to wild-type controls (Figure 6A). Eaf2+/- mice also displayed a statistically significant, similar increase in the number of CD3-positive cells (Figure 6B). While there was also an increase in the number of CD3-positive cells in Eaf2+/-Ell+/-, there was no additive increase in mice with combined deficiency in Eaf2 and Ell2. Others have shown that Eaf2 deficiency did not affect B-cell and T-cell development and maturation, but was an inducer of apoptosis in GC B cells suggesting that Eaf2 could play a role in regulating immune balance [19].
Figure 6.
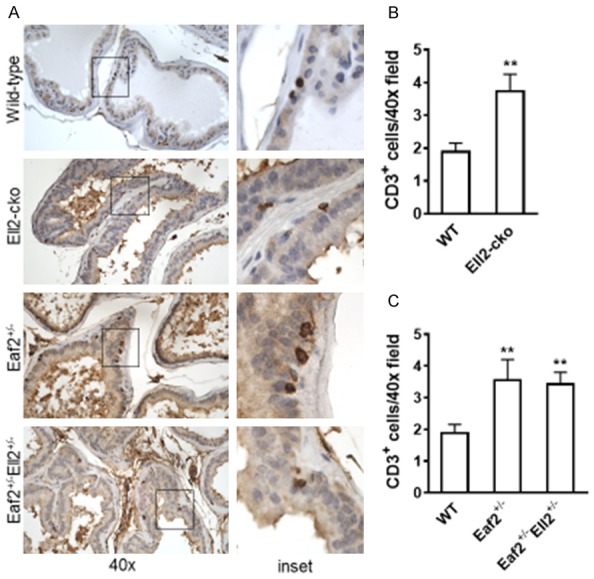
Effects of combined Eaf2 and Ell2 loss on CD3-positive T cells in the C57BL/6J mouse prostate at age 24 mos. A. CD3+ T cells immunostaining in transverse sections of prostate ventral lobes from indicated genotypes at 24 months of age. B. Quantification of infiltrating intraductal CD3+ T cells in mouse prostate from wild-type and Ell2-cko mice at 24 months of age. C. Quantification of CD3+ T cells in mouse prostate from wild-type and Ell2-cko mice at 24 months of age. Data are expressed as mean ± standard error of the mean from 3 to 6 mice per group. **P < 0.01.
Discussion
The EAF and ELL proteins function together to enhance transcription elongation activity in eukaryotic cells and have been shown to individually play important roles in prostate tumor suppression. EAF2 and ELL2 expression have both been demonstrated to be androgen responsive in the prostate [8,16] and both EAF2 and ELL2 loss have individually been associated with increased prostate epithelial proliferation, invasion and migration in prostate cancer cell lines [11,14]. In C57BL/6J mice, deletion of Eaf2 or Ell2 could induce an increased incidence in mPIN lesions, prostate epithelial cell proliferation and prostate intraductal microvessel density [14,16]. In the current study, we examined the potential overlapping and/or inter-dependent function of ELL2 and EAF2 by investigating the impact of their combined knockdown in prostate cancer cell lines and combined heterozygous deletion in a murine model.
Knockdown of EAF2 or ELL2 in C4-2 and 22Rv1 prostate cancer cells increased cellular proliferation as determined by BrdU incorporation assays. Wound healing assays showed an increased migration in both cell lines in response to EAF2 or ELL2 knockdown. Combined knockdown of EAF2 and ELL2 increased proliferation and migration similarly to that observed following knockdown of EAF2 or ELL2 alone. These results suggest that EAF2 and ELL2 require each other for their regulation of proliferation and migration in prostate cancer cells. EAF2 and ELL2 may also be in the same signaling cascades which regulate the proliferation and migration of prostate cancer cells. Thus, knockdown of either protein may be sufficient to induce the phenotype and further reduction of EAF2 or ELL2 does not induce further change.
Since EAF2 knockdown was previously shown to up-regulate interferon signaling genes in C4-2 prostate cancer cells [12], we investigated whether Eaf2 and/or Ell2 deficiency influenced the infiltration of inflammatory B-cells or T-cells in the murine prostate. Ell2-cko mice displayed a significant increase in the number of infiltrating T-cells in the prostatic ducts. Eaf2+/- mice also displayed a similar increase, as did Eaf2+/-Ell+/- mice. The number of B-cells in all groups was similar.
In the mouse model, heterozygous deficiency in Eaf2 induced mPIN lesions characterized by increased proliferation and microvessel density. These mPIN lesions were similar to those previously reported in mice with homozygous deletion of Eaf2 on a C57BL/6J background [10]. Similar to the impact of siRNA knockdown of EAF2 and/or ELL2 in prostate cancer cells, there was no additive increase in Ki-67 positive epithelial cells and a trend of increased mPIN lesions incidence and intraductal microvessel density in mice with combined heterozygous deficiency in Eaf2 and Ell2 compared to Eaf2+/- mice. Although there was an increased incidence in mPIN lesions and intraductal microvessel density in Eaf2+/-Ell+/- mice compared to Eaf2+/- mice, the difference was not statistically significant. The lack of a statistically significant difference could be due in part to the small sample size, or it may be that EAF2 and ELL2 regulate angiogenesis through a different pathway than they regulate proliferation and migration.
The absence of an additive effect in the combined loss of EAF2 and ELL2 in prostate cancer cell lines and in the mouse model suggests that EAF2 and ELL2 are inter-dependent in prostate tumor suppression and require their binding to each other. Loss of ELL2 may also reduce EAF2 levels, which may contribute to the phenotype observed in the prostates of Ell2 knockout mice. Mice with heterozygous deficiency in Eaf2 and Ell2 had a non-significant increase in the incidence of mPIN lesions compared to Eaf2+/- mice. This could be due in part to Ell2 deficiency further decreasing Eaf2 levels. Our data suggests that loss of either EAF2 or ELL2 inhibits the remaining protein’s ability to function in prostate tumor suppression. A limitation of our study was the inability of generating mice with homozygous deletion of Eaf2 and Ell2. We do not know the reason why there were no pups born with homozygous deletion of Eaf2 and Ell2.
Cumulatively, these results suggest that the prostate tumor suppressive properties of EAF2 and ELL2 are inter-dependent and involve their interaction and that the loss of either gene can result in increased proliferation, migration and the development of preneoplastic lesions in the murine model.
Acknowledgements
We are grateful to Dawn Everard, Robin Frederick, Elaine Isherwood, Megan Lambert, Katie Leschak, Kathleen J. O’Malley, Yao Wang, and Aiyuan Zhang for technical support. We thank Camerynn Keahi and Saaz Malhotra for assistance in writing the figure legends. This work was funded in part by National Institutes of Health Grants R01 CA186780 (Z.W.), P50 CA180995 (Z.W.), R50 CA211242 (L.E.P.), and T32 DK007774 (Z.W.) and scholarships from the Tippins Foundation (LEP) and the Mellam Foundation (KZM). This project used the UPCI Animal Facility and Tissue and Research Pathology Services (TARPS) and was supported in part by award P30CA047904.
Disclosure of conflict of interest
None.
References
- 1.Shilatifard A, Haque D, Conaway RC, Conaway JW. Structure and function of RNA polymerase II elongation factor ELL. Identification of two overlapping ELL functional domains that govern its interaction with polymerase and the ternary elongation complex. J Biol Chem. 1997;272:22355–22363. doi: 10.1074/jbc.272.35.22355. [DOI] [PubMed] [Google Scholar]
- 2.Miller T, Williams K, Johnstone RW, Shilatifard A. Identification, cloning, expression, and biochemical characterization of the testis-specific RNA polymerase II elongation factor ELL3. J Biol Chem. 2000;275:32052–32056. doi: 10.1074/jbc.M005175200. [DOI] [PubMed] [Google Scholar]
- 3.Shilatifard A, Duan DR, Haque D, Florence C, Schubach WH, Conaway JW, Conaway RC. ELL2, a new member of an ELL family of RNA polymerase II elongation factors. Proc Natl Acad Sci U S A. 1997;94:3639–3643. doi: 10.1073/pnas.94.8.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shilatifard A, Lane WS, Jackson KW, Conaway RC, Conaway JW. An RNA polymerase II elongation factor encoded by the human ELL gene. Science. 1996;271:1873–1876. doi: 10.1126/science.271.5257.1873. [DOI] [PubMed] [Google Scholar]
- 5.Kong SE, Banks CA, Shilatifard A, Conaway JW, Conaway RC. ELL-associated factors 1 and 2 are positive regulators of RNA polymerase II elongation factor ELL. Proc Natl Acad Sci U S A. 2005;102:10094–10098. doi: 10.1073/pnas.0503017102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banks CA, Kong SE, Spahr H, Florens L, Martin-Brown S, Washburn MP, Conaway JW, Mushegian A, Conaway RC. Identification and characterization of a schizosaccharomyces pombe RNA polymerase II elongation factor with similarity to the metazoan transcription factor ELL. J Biol Chem. 2007;282:5761–5769. doi: 10.1074/jbc.M610393200. [DOI] [PubMed] [Google Scholar]
- 7.Cai L, Phong BL, Fisher AL, Wang Z. Regulation of fertility, survival, and cuticle collagen function by the Caenorhabditis elegans eaf-1 and ell-1 genes. J Biol Chem. 2011;286:35915–35921. doi: 10.1074/jbc.M111.270454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao W, Zhang Q, Jiang F, Pins M, Kozlowski JM, Wang Z. Suppression of prostate tumor growth by U19, a novel testosterone-regulated apoptosis inducer. Cancer Res. 2003;63:4698–4704. [PubMed] [Google Scholar]
- 9.Ai J, Pascal LE, O’Malley KJ, Dar JA, Isharwal S, Qiao Z, Ren B, Rigatti LH, Dhir R, Xiao W, Nelson JB, Wang Z. Concomitant loss of EAF2/U19 and Pten synergistically promotes prostate carcinogenesis in the mouse model. Oncogene. 2013;33:2286–94. doi: 10.1038/onc.2013.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pascal LE, Ai J, Masoodi KZ, Wang Y, Wang D, Eisermann K, Rigatti LH, O’Malley KJ, Ma HM, Wang X, Dar JA, Parwani AV, Simons BW, Ittman MM, Li L, Davies BJ, Wang Z. Development of a reactive stroma associated with prostatic intraepithelial neoplasia in EAF2 deficient mice. PLoS One. 2013;8:e79542. doi: 10.1371/journal.pone.0079542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiu X, Pascal LE, Song Q, Zang Y, Ai J, O’Malley KJ, Nelson JB, Wang Z. Physical and functional interactions between ELL2 and RB in the suppression of prostate cancer cell proliferation, migration, and invasion. Neoplasia. 2017;19:207–215. doi: 10.1016/j.neo.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pascal LE, Wang Y, Zhong M, Wang D, Chakka AB, Yang Z, Li F, Song Q, Rigatti LH, Chaparala S, Chandran U, Parwani AV, Wang Z. EAF2 and p53 co-regulate STAT3 activation in prostate cancer. Neoplasia. 2018;20:351–363. doi: 10.1016/j.neo.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Malley KJ, Dhir R, Nelson JB, Bost J, Lin Y, Wang Z. The expression of androgen-responsive genes is up-regulated in the epithelia of benign prostatic hyperplasia. Prostate. 2009;69:1716–1723. doi: 10.1002/pros.21034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ai J, Pascal LE, Wei L, Zang Y, Zhou Y, Yu X, Gong Y, Nakajima S, Nelson JB, Levine AS, Lan L, Wang Z. EAF2 regulates DNA repair through Ku70/Ku80 in the prostate. Oncogene. 2017;36:2054–2065. doi: 10.1038/onc.2016.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao W, Zhang Q, Habermacher G, Yang X, Zhang AY, Cai X, Hahn J, Liu J, Pins M, Doglio L, Dhir R, Gingrich J, Wang Z. U19/Eaf2 knockout causes lung adenocarcinoma, B-cell lymphoma, hepatocellular carcinoma and prostatic intraepithelial neoplasia. Oncogene. 2008;27:1536–1544. doi: 10.1038/sj.onc.1210786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pascal LE, Masoodi KZ, Liu J, Qiu X, Song Q, Wang Y, Zang Y, Yang T, Rigatti LH, Chandran U, Colli LM, Vencio RZN, Lu Y, Zhang J, Wang Z. Conditional deletion of ELL2 induces murine prostate intraepithelial neoplasia. J Endocrinol. 2017;235:123–136. doi: 10.1530/JOE-17-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Funahashi Y, O’Malley KJ, Kawamorita N, Tyagi P, DeFranco DB, Takahashi R, Gotoh M, Wang Z, Yoshimura N. Upregulation of androgen-responsive genes and transforming growth factor-beta1 cascade genes in a rat model of non-bacterial prostatic inflammation. Prostate. 2014;74:337–345. doi: 10.1002/pros.22668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park KS, Bayles I, Szlachta-McGinn A, Paul J, Boiko J, Santos P, Liu J, Wang Z, Borghesi L, Milcarek C. Transcription elongation factor ELL2 drives Ig secretory-specific mRNA production and the unfolded protein response. J Immunol. 2014;193:4663–4674. doi: 10.4049/jimmunol.1401608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Takahashi Y, Fujii S, Zhou Y, Hong R, Suzuki A, Tsubata T, Hase K, Wang JY. EAF2 mediates germinal centre B-cell apoptosis to suppress excessive immune responses and prevent autoimmunity. Nat Commun. 2016;7:10836. doi: 10.1038/ncomms10836. [DOI] [PMC free article] [PubMed] [Google Scholar]