

Summary
MyD88, an adaptor molecule downstream of innate pathways, plays a significant tumor-promoting role in sporadic intestinal carcinogenesis of the Apcmin/+ model, which carries a mutation in the Apc gene. Here, we show that deletion of MyD88 in intestinal mesenchymal cells (IMCs) significantly reduces tumorigenesis in this model. This phenotype is associated with decreased epithelial cell proliferation, altered inflammatory and tumorigenic immune cell infiltration, and modified gene expression similar to complete MyD88 knockout mice. Genetic deletion of TLR4, but not interleukin-1 receptor (IL-1R), in IMCs led to altered molecular profiles and reduction of intestinal tumors similar to the MyD88 deficiency. Ex vivo analysis in IMCs indicated that these effects could be mediated through downstream signals involving growth factors and inflammatory and extracellular matrix (ECM)-regulating genes, also found in human cancer-associated fibroblasts (CAFs). Our results provide direct evidence that during tumorigenesis, IMCs and CAFs are activated by innate TLR4/MyD88-mediated signals and promote carcinogenesis in the intestine.
Keywords: cancer-associated fibroblasts, innate immunity, tumor microenvironment
Graphical Abstract
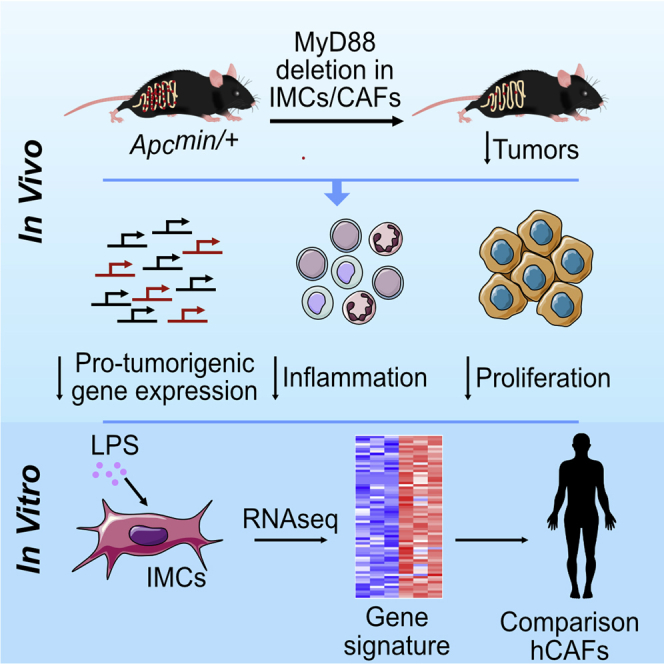
Highlights
-
•
Deletion of MyD88 or TLR4 in IMCs and/or CAFs leads to reduced intestinal tumorigenesis
-
•
The phenotype of the IMC-specific MyD88 mice is similar to the complete knockouts
-
•
MyD88−/− IMCs show a reduced pro-tumorigenic and/or inflammatory gene expression profile
-
•
Human CAFs show upregulation of a similar MyD88-specific gene expression signature
Koliaraki et al. show that MyD88 in mesenchymal cells is responsible for its tumor-promoting role in the Apcmin/+ model. They further show that this is a TLR4-mediated mechanism that leads to the production of pro-tumorigenic molecules, also identified in human CAFs.
Introduction
Intestinal carcinogenesis is the result of multiple mutations, but it is now well established that the microenvironment plays also an important role in cancer growth, progression, and metastasis (Hanahan and Coussens, 2012). One of the most important deregulated pathways in intestinal cancer is the β-catenin pathway. In mice, mutation in the Apc gene, a central regulator of β-catenin signaling, leads to spontaneous intestinal carcinogenesis similar to human familial adenomatous polyposis, characterized by multiple polyps located mainly in the small intestine (Apcmin/+ model) (Dove et al., 1997). The difference in tumor location between mice and humans is due to the different number of stem cell divisions in the colon and small intestine between the two species (Tomasetti and Vogelstein, 2015). Antibiotic treatment or rederivation of Apcmin/+ mice in germ-free conditions results in reduced tumor load, indicating an important tumor-promoting role of the microbiota in intestinal tumorigenesis (Dove et al., 1997, Li et al., 2012, Song et al., 2014). This is mechanistically associated with increased c-Jun and signal transducer and activator of transcription 3 (STAT3) phosphorylation in cancer cells and increased infiltration of inflammatory cells (Li et al., 2012).
MyD88 is a central regulator of innate immunity, as it acts directly downstream of Toll-like receptors (TLRs) and cytokine receptors, while it is also implicated in carcinogenesis (Salcedo et al., 2013). Genetic deletion of MyD88 in Apcmin/+ mice results in reduced number and size of tumors and correlates with suppressed proliferation, enhanced apoptosis, and a deregulated gene expression profile in tumors (Rakoff-Nahoum and Medzhitov, 2007). Bone marrow chimeras have shown that polyp growth in Apcmin/+ mice depends on MyD88 signaling in non-hematopoietic cells, and MyD88 in intestinal epithelial cells (IECs) was shown to stabilize Myc expression through ERK phosphorylation (Lee et al., 2010).
The role of intestinal mesenchymal cells (IMCs) in inflammation and cancer has recently gained momentum. The ability of IMCs to respond to inflammatory stimuli and regulate immune responses and inflammation has been demonstrated in vitro; however, its pathophysiological significance has only recently started to be characterized (Koliaraki et al., 2017, Powell et al., 2011). We have recently dissected intracellular MAPK and nuclear factor κB (NF-κB) signals that modulate the function of IMCs in the development of colitis and colitis-associated cancer (Henriques et al., 2018, Koliaraki et al., 2012, Koliaraki et al., 2015, Roulis et al., 2014). In the present study, we explored the in vivo role of mesenchymal-specific innate sensing in spontaneous intestinal tumorigenesis of the Apcmin/+ mouse model. We reveal that TLR4/MyD88 signaling in IMCs and/or CAFs is a dominant physiological mechanism in the promotion of cancer.
Results
MyD88 Signaling in Mesenchymal Cells Promotes Intestinal Tumorigenesis in the Apcmin/+ Model
Since IMCs have been shown to respond to immune stimuli in vitro, we hypothesized that IMCs could play a significant role in intestinal carcinogenesis via sensing of innate stimuli. To examine the pathophysiological role of IMC-specific MyD88 in disease pathogenesis of the Apcmin/+ model, we crossed MyD88 conditional knockout mice with ColVI-cre mice (Apcmin/+-Myd88IMCko), which target mesenchymal cells in the intestine and intestinal tumors (Armaka et al., 2008, Koliaraki et al., 2015). Deletion efficiency in ColVI+ cells was verified by qRT-PCR analysis (Figure S1A). 4-month-old Apcmin/+-Myd88IMCko mice displayed a significant reduction in both the number and size of tumors in comparison to their littermate Apcmin/+-Myd88F/F controls (Figures 1A–1D). Stratification of tumor frequency by intestinal region showed that the differences were mainly found in the small intestine and particularly in the ileum and jejunum, where the majority of polyps are also located (Figure 1B). It should be noted that Cre itself did not affect tumor development (Figures S2A and S2B). In addition, size-matched tumors from the Apcmin/+-Myd88IMCko mice showed decreased proliferation, while there was no difference in apoptosis, assessed by bromodeoxyuridine (BrdU) incorporation and Tunel staining, respectively (Figures 1E–1G). Histopathological analysis at 6 weeks of age did not show a statistically significant decrease in the number of dysplastic foci in the small intestine of Apcmin/+-Myd88IMCko mice, suggesting a more important role for IMC-specific MyD88 in the progression rather than initiation of carcinogenesis, which is consistent with the role of MyD88 in this model (Rakoff-Nahoum and Medzhitov, 2007) (Figure 1H).
Figure 1.

Deletion of MyD88 in Intestinal Mesenchymal Cells Reduces Tumorigenesis in the Apcmin/+ Model of Sporadic Intestinal Cancer
(A and B) Total number of tumors per mouse (A) and number of tumors per intestinal part (B) in 4-month-old Apcmin/+Myd88IMCko mice (n = 15) and their littermate controls (n = 18). The insert shows the number of colonic tumors.
(C and D) Size of small intestinal tumors presented as mean tumor size (C) and distribution of tumors per size (D) in the two genotypes.
(E) Representative BrdU and Tunel staining in small intestinal tumors of Apcmin/+Myd88IMCko mice and their littermate controls. DAPI was used to stain the nuclei in the Tunel stainings.
(F and G) Quantification of the number of BrdU+ cells per tumor (F) and Tunel+ cells (G) per field in equal-sized tumors (n = 6 mice per genotype).
(H) Number of dysplastic lesions per mouse in 6-week-old Apcmin/+Myd88IMCko and their littermate controls (n = 6–7 mice per genotype).
(I) Number of tumors per mouse in Myd88IMCko mice (n = 8) and their littermate controls (n = 7) at the end of the AOM/DSS protocol (one representative experiment of four performed).
Data represent mean ± SEM. ∗∗p < 0.01, ∗∗∗p < 0.001, ns = not significant. Scale bars represent 50 μm. See also Figures S1 and S2.
Contrary to spontaneous tumorigenesis, mesenchymal-specific deletion of MyD88 did not have a significant effect in the azoxymethane (AOM)/dextran sulfate sodium (DSS) model of inflammation-induced colorectal carcinogenesis (Figure 1I). These results suggest an important tumor-promoting role of the MyD88 signaling pathway in IMCs in spontaneous intestinal tumorigenesis and are in agreement with the phenotype of complete MyD88 knockout mice in this model, which show a significant reduction in tumor load, accompanied by reduced proliferation (Rakoff-Nahoum and Medzhitov, 2007). Differences in the severity of the disease, apoptosis, and the effect on colonic polyps between the complete and conditional knockout mice indicate that MyD88 in other stromal cell types, including other mesenchymal cell populations, could also contribute to the phenotype of the complete MyD88 knockout mice.
Deletion of MyD88 in IMCs Results in Deregulated Gene Expression, Reduced STAT3 Phosphorylation, and Altered Inflammatory Cell Infiltration
To further analyze the similarities between Apcmin/+-Myd88IMCko mice and the complete knockout mice, we next analyzed the gene expression profile of Apcmin/+-Myd88IMCko tumors by measuring the expression level of genes that were differentially regulated in the complete MyD88 knockout mice (Rakoff-Nahoum and Medzhitov, 2007). We found significant deregulation in many genes, especially those encoding pro-inflammatory mediators and matrix metalloproteinases (MMPs), while Igf1 and Igfbp5 were not upregulated in our tumor samples (Figure 2A). Interestingly, among the deregulated genes were Il6 and Il11, two pro-inflammatory cytokines with important functions in enhancing epithelial cell proliferation and tumorigenesis through STAT3 activation (Bollrath et al., 2009, Schmidt et al., 2018). Accordingly, we found that Apcmin/+-Myd88IMCko mice showed decreased pSTAT3 staining both in tumors and normal villi in comparison to Apcmin/+-Myd88F/F controls (Figures 2B and 2C). Since there was a significant differential expression of pro-inflammatory genes in the small intestine of Apcmin/+-Myd88IMCko mice, we also examined inflammatory cell infiltration in tumors by fluorescence-activated cell sorting (FACS) analysis. We did not find statistically significant differences in the numbers of infiltrating CD45+ hematopoietic cells between the two genotypes (Figure 2D); however, we found a reduction in CD45+CD11b+F4/80+ macrophages, CD45+CD11b+Gr1+ neutrophils, and CD45+CD4+ T cells, while CD45+CD8+ T cells were increased in the Apcmin/+-Myd88IMCko tumors (Figures 2E and 2F; Figure S3A). There was no statistically significant difference in the normal small intestine between the two genotypes (Figure S3B). Therefore, Apcmin/+-Myd88IMCko mice display altered balances in immune infiltration toward a less pro-inflammatory microenvironment and interestingly enhanced cytotoxic T cell infiltration, both being directly associated with the decreased number and size of tumors. These results indicate that a MyD88-dependent pathway in IMCs and/or CAFs regulates the infiltration of immune populations creating a pro-tumorigenic inflammatory milieu in the Apcmin/+ model, which potentially acts as an additional mechanism to accelerate tumorigenesis.
Figure 2.

Tumors from Mice with Deletion of MyD88 in IMCs Show Differential Gene Expression, Reduced STAT3 Activation, and Altered Inflammatory Cell Infiltration
(A) Gene expression analysis in the small intestine of Myd88F/F and Myd88IMCko mice (n = 3) and in tumors from Apcmin/+Myd88F/F and Apcmin/+Myd88IMCko mice (n = 6). Hprt was used for normalization.
(B) Representative immunohistochemical staining for pSTAT3 in the normal small intestine and tumors of Apcmin/+Myd88F/F and Apcmin/+Myd88IMCko mice (n = 5 mice per genotype).
(C) Quantification of pSTAT3 staining in tumors from Apcmin/+Myd88F/F and Apcmin/+Myd88IMCko mice (n = 12–14 tumors from 5 mice per genotype).
(D–F) Infiltration of CD45+ cells (D), CD11b+F4/80+ macrophages and CD11b+Gr1+ neutrophils (E), and CD4+ T cells, CD8+ T cells, CD19+ B cells, and CD11c+ dendritic cells (F) in tumors from 4- to 5-month-old Apcmin/+Myd88F/F and Apcmin/+Myd88IMCko mice (n = 4–7), quantified by FACS analysis (from two independent experiments).
(G) Representative confocal images from tumors of Apcmin/+-ColVIcre-Myd88F/+-mTmG and Apcmin/+-ColVIcre-Myd88F/F-mTmG mice (n = 3). Scale bars represent 50 μm.
(H) FACS analysis of tumors from Apcmin/+-ColVIcre-mTmG mice, stained with markers for epithelial (EpCAM), immune (CD45), and endothelial (CD31) cells (n = 3 mice). Unstained controls are presented as gray histograms.
Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ns = not significant. See also Figure S3.
Importantly, deletion of MyD88 did not influence the distribution of ColVIcre+ cells in tumors of the Apcmin/+-Myd88IMCko mice, as assessed by combining these mice with the RosamT/mG (mT/mG) reporter mice, suggesting that MyD88 signaling does not interfere with normal development of these cells (Figure 2G). The specificity of the ColVI-cre mouse for IMCs and/or CAFs in tumors was also verified by FACS analysis, using the lineage-negative markers epithelial cell adhesion molecule (EpCAM), cluster of differentiation 45 (CD45), and CD31 for epithelial, immune, and endothelial cells, respectively (Figures 2H and S3C).
IMC-Specific Genetic Deletion of TLR4, but Not IL-1R, Reduces Tumorigenesis in the Apcmin/+ Model
To further identify the innate molecular mechanisms regulated by MyD88 in IMCs, we next isolated mesenchymal cells from the small intestine and incubated them with various MyD88 inducers, including TLR ligands and interleukins IL-1β (interleukin-1β), IL-18, and IL-33. We used measurement of IL-6 as the readout of this initial analysis. Our results showed that IL-6 was increased in response to IL-1β and ligands for TLR1/2, TLR4, and TLR6 and that lipopolysaccharide (LPS) and IL-1β were the stimuli that produced the most abundant effect (Figure 3A). Moreover, analysis of the dataset GEO: GSE39395, which contains expression profiles of isolated cancer cells, leukocytes, and cancer-associated fibroblasts from tumors of patients with colorectal cancer, revealed enrichment in genes encoding TLR4 and TLR5, as well as receptors for the cytokines IL-1 and IL-33 (IL1R1, IL1RL1, and IL1RAP) in CAFs versus cancer cells or leukocytes (Figure 3B) (Calon et al., 2012).
Figure 3.

Deletion of TLR4 in IMCs Reduces Tumorigenesis in the Apcmin/+ Model of Sporadic Intestinal Cancer
(A) IL-6 quantification by ELISA in the supernatants of IMCs stimulated for 24 h with TLRs and interleukins, as described in STAR Methods. One representative of three independent experiments performed in triplicates is presented.
(B) Heatmap of the differential expression of the indicated genes in CAFs versus cancer cells and leukocytes from the dataset GEO: GSE39395. Log2 fold-change of genes is shown. Red denotes high expression values, and blue denotes low expression values.
(C and D) Total number (C) and size (D) of tumors per mouse in 4-month-old Apcmin/+Il1r1IMCko mice (n = 14) and their littermate controls (n = 24).
(E and F) Total number of tumors per mouse (E) and number of tumors per intestinal part (F) in 4-month-old Apcmin/+Tlr4IMCko mice (n = 13) and their littermate controls (n = 20). The insert shows the number of tumors in the colon.
(G and H) Mean tumor size (G) and distribution of tumors (H) per size in the two genotypes.
(I) Representative BrdU and pSTAT3 staining in small intestinal tumors of Apcmin/+Tlr4IMCko mice and their littermate controls. Scale bar = 50 μm.
(J) Quantification of the number of BrdU+ cells per tumor (n = 6 mice per genotype).
(K) Mean signal intensity (MSI) of pSTAT3 (n = 12–14 tumors from 4 mice per genotype).
(L) Gene expression analysis in tumors from Apcmin/+Tlr4F/F and Apcmin/+Tlr4IMCko mice (n = 6). Hprt was used for normalization.
(M) Number of tumors per mouse in Tlr4IMCko mice (n = 8) and their littermate controls (n = 8) at the end of the AOM/DSS protocol (one representative experiment of three performed).
Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ns = not significant. See also Figure S1.
Based on this analysis, we focused more on LPS and IL-1β stimulation. LPS is known to activate innate immune responses in IMCs in vitro, leading to production of pro-inflammatory molecules, such as IL-6, COX2, and CXCL1, through activation of several signaling pathways, including NF-κB, phosphatidylinositol 3-kinase (PI3K), and mitogen-activated protein kinases (MAPKs) (Walton et al., 2009). IL-1β mediates the activation of a pro-inflammatory signature in CAFs through the NF-κB pathway and has been implicated in carcinogenesis (Erez et al., 2010). To verify if upstream signaling from TLR4 or IL-1R was responsible for the IMC-specific MyD88-induced effect on intestinal carcinogenesis, we next crossed Il1r1F/F (Abdulaal et al., 2016) and Tlr4F/F (Sodhi et al., 2012) mice with ColVI-cre mice (Figures S1B and S1C). We found that 4-month-old Apcmin/+-Il1r1IMCko mice showed no difference in tumor multiplicity and size (Figures 3C and 3D), indicating that IL-1R signaling in IMCs does not play an important role in spontaneous intestinal carcinogenesis. On the contrary, Apcmin/+-Tlr4IMCko showed a significant reduction in both tumor multiplicity and size (Figures 3E–3H). Tumors from these mice also displayed decreased proliferation measured by BrdU staining (Figures 3I and 3J) and decreased phosphorylation of STAT3 (Figures 3I and 3K), as well as reduced expression of inflammatory mediators (Figure 3L). These results are in agreement both with the phenotype observed in Apcmin/+-Myd88IMCko and our in vitro data, suggesting that upstream signaling from TLR4 drives MyD88 activation in IMCs and concomitant modulation of intestinal tumorigenesis.
We also subjected Tlr4IMCko and their controls to the AOM/DSS model of colitis-associated cancer and found no difference in tumorigenesis, in accordance with Myd88IMCko mice, indicating that TLR4 signaling in IMCs and/or CAFs is necessary only for the development of sporadic intestinal carcinogenesis (Figure 3M).
An IMC-Specific TLR4/MyD88-Regulated Gene Signature Is Found Also in Human CAFs
To identify the IMC-specific MyD88-dependent gene expression changes, we next performed 3′ RNA sequencing of ColVIcre+ wild-type (WT) and MyD88 knockout IMCs before and after treatment with LPS for 6 h. Interestingly, even unstimulated MyD88 knockout cells showed a significantly altered gene expression profile in comparison to the unstimulated control cells (Figure S4A). Gene Ontology (GO) analysis of the mostly downregulated genes in non-induced MyD88 knockout cells revealed differences in pathways related to inflammatory and/or immune response and cell proliferation, indicating an intrinsic defect of these cells in acquiring an innate identity under homeostatic conditions (Figures 4A and 4D). Comparisons with the LPS-stimulated control and MyD88 knockout samples further showed a significant number of MyD88-regulated genes, which either remained unchanged or were altered upon LPS stimulation (Figure 4B; Figures S4B and S4C). GO analysis of these MyD88-regulated genes showed enrichment in inflammatory and/or immune response and regulation of cell proliferation (Figure 4C). Related genes included mainly chemokines (Cxcl1, Cxcl2, Cxcl5, Ccl2, Ccl7, Ccl8, Ccl11), cytokines (Il6, Il34), growth factors (Fgf7, Fgf10, Tgfa, Ctgf, Igf1, Igfbp4), and MMPs (Mmp3, Mmp8, Mmp9, Mmp10, Mmp13) (Figure 4D). This MyD88-dependent gene signature is in agreement with the deregulated gene expression of the IMC-specific MyD88 knockout tumors and the accompanied reduced proliferation and altered inflammatory infiltration.
Figure 4.

TLR4/MyD88 Signaling Induces a Pro-Tumorigenic Gene Signature in IMCs
(A) Gene Ontology (GO) terms enriched in differentially expressed genes between untreated control and MyD88 knockout IMCs.
(B) Venn diagram showing the number of overlapping differentially regulated genes between untreated and LPS-treated control and MyD88 knockout IMCs.
(C) GO terms enriched in MyD88-regulated genes indicated as bold in (B).
(D) Heatmap of differentially expressed genes between control and LPS-induced WT and MyD88 knockout IMCs that belong to the GO terms shown in (C). Log2 transformed normalized read counts of genes are shown. Read counts are scaled per column. Red denotes high expression values, and blue denotes low expression values.
(E and F) Venn diagram showing the number of overlapping genes (E) and heatmap of their expression between the IMC-specific MyD88-regulated gene signature and genes overexpressed in CAFs versus cancer cells from the datasets GEO: GSE39395 and GSE35602 (F). Log2 fold-change of genes is shown.
See also Figure S4.
To further validate if this expression profile was also present in human CAFs, we next compared this MyD88-regulated gene profile with two datasets that contained comparisons of CAFs versus cancer cells (GEO: GSE39395 and GSE35602) (Calon et al., 2012, Nishida et al., 2012). We found that out of 120 genes, 68 and 65 were preferentially expressed in CAFs in each dataset, respectively, while 53 were in common between the two lists (Figures 4E and 4F). These included cytokines (IL6), chemokines (CCL2, CCL8, CCL11), growth factors (FGF7, CTGF, IGF1), and MMPs (MMP3, MMP10, MMP13) (Figure 4F), suggesting that a similar TLR4/MyD88-regulated gene signature exists also in human CAFs.
Discussion
MyD88 is known to have a tumor-promoting role in spontaneous intestinal cancer (Rakoff-Nahoum and Medzhitov, 2007). Here we show, through its IMC-specific deletion, that it is MyD88 in IMCs that is responsible for this function, in agreement with previous bone marrow transfer studies, indicating a predominant role of MyD88 in the non-hematopoietic compartment (Lee et al., 2010). It should be noted that the phenotypic differences between the IMC-specific and the published complete MyD88 knockout mice also suggest a possible contribution of MyD88 in other non-hematopoietic cell types, including other mesenchymal cell populations.
In agreement with these data, IMC-specific TLR4, but not ILR1, ablation also ameliorates intestinal tumorigenesis, indicating that microbial sensing by these cells may be a pathogenic mechanism in the development of the disease. Although many ligands for TLR4 have been described and may be (co)responsible for the TLR4 effects described here, a potential source for TLR4 stimulation could be LPS from bacteria that are detected close to neoplastic lesions and within tumors in the intestine (Grivennikov et al., 2012). Recently, a specific species, Fusobacterium nucleatum, which is a Gram-negative bacterium, has been found in tumors from human patients and in tumors from Apcmin/+ mice fed with this strain but not in normal mucosa (Kostic et al., 2013, Yang et al., 2017). Importantly, infection of Apcmin/+ with this bacterial strain led to increased tumorigenesis, associated with increased proliferation, infiltration of CD11b+F4/80+ and CD11b+Gr1+ immune cells, and expression of inflammatory genes, such as Il6, Ptgs2, and Mmp3 (Kostic et al., 2013). These data indicate that microbes can be found in intestinal tumors and are causally linked to cancer progression. Our study therefore offers a direct mechanistic link between intratumoral bacterial sensing and innate activation of IMCs and/or CAFs that together contribute strongly to tumor progression.
Our ex vivo data indicated that the mechanism downstream of innate sensing in IMCs includes the production of effector molecules, which in turn affect both cancer cell proliferation and the immune microenvironment, mainly during the progression rather than the initiation stage of spontaneous tumorigenesis. Among them, IL-6/IL-11 and downstream-regulated STAT3 signaling is an important pro-tumorigenic mechanism (Bollrath et al., 2009, Schmidt et al., 2018). Interestingly, we have recently published that inhibitor of nuclear factor κB subunit β (IKKβ) deletion in IMCs leads to reduced inflammation and carcinogenesis in the AOM/DSS model of colitis-associated cancer, which is mediated by the production of IL-6 and downstream phosphor-STAT3 (pSTAT3) phosphorylation, while it does not affect carcinogenesis in the sporadic Apc1638N/+ or AOM model (Koliaraki et al., 2015). The Myd88IMCko mice and Tlr4IMCko mice show an opposite phenotype, affecting the number and size of tumors in spontaneous, but not in colitis-associated, cancer. These results suggest that production of IL-6 and other inflammatory cytokines and/or chemokines is mediated in IMCs through the NF-κB pathway, which can be activated, but not exclusively, by Myd88-dependent signals in the early stages of colitis associated cancer (CAC). In spontaneous intestinal cancer, this cytokine and/or chemokine production is induced largely by the TLR4/MyD88 pathway, and it is notably not dependent on NF-κB, indicating heterogeneous signaling in the context of IMC and/or CAF innate activation, which is probably regulated by the distinct microenvironment of the tumors. It should be noted that in the Apcmin/+ model, the colon tumor number, although very low, was also not affected, implying that potential site-specific differences, such as the mesenchymal population targeted by the ColVI-cre mouse, could also play a role in the absence of a phenotype.
Besides IL-6, a variety of other effector molecules, such as growth factors, chemokines, and MMPs, are also regulated by the TLR4/MyD88 pathway in IMCs. A pro-inflammatory gene expression profile has been found also in other types of cancer (Erez et al., 2010), and the role of specific cytokines and chemokines in cancer promotion is now well established (Terzic et al., 2010). Interestingly, we have found that many of these are also found enriched in CAFs from human colorectal cancer, suggesting a potential role for innate responses also in human tumors and their possible utility as prognostic markers. For example, MMP3 and CXCL1 have already been suggested as potent stromal protein markers of dysplasia-to-carcinoma transition in sporadic colorectal cancer (Sipos et al., 2014).
In conclusion, our results reveal the cellular specificity of MyD88’s function in spontaneous intestinal carcinogenesis and provide direct evidence for a pathophysiological significant innate immune function of IMCs and CAFs in cancer. This suggests that these cells could directly respond to the microbiota and that this response is crucial for tumor growth, providing a conceptual advance on the regulation, functions, and potential therapeutic targeting of CAFs, including recent efforts to enhance immune targeting of cancer.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Phospho-STAT3 (Tyr705) | Cell Signaling | Cat#9131; RRID: AB_331586 |
| Purified Rat anti-mouse CD16/32 | Biolegend | Cat#101302; RRID: AB_312801 |
| FITC-conjugated Rat anti-mouse CD11b (clone M1/70) | eBioscience | Cat#11-0112-85; RRID: AB_464936 |
| APC/Cy7-conjugated Rat anti-mouse CD45 (clone 30-F11) | Biolegend | Cat#103116; RRID: AB_312981 |
| A700-conjugated Rat anti-mouse CD45 (clone 30-F11) | Biolegend | Cat#103128; RRID: AB_493715 |
| PE/Cy7-conjugated Armenian Hamster anti-mouse CD11c (clone N418) | Biolegend | Cat#117318; RRID: AB_493568 |
| A700-conjugated Rat anti-mouse CD19 (clone eBio1D3 (1D3)) | eBioscience | Cat#56-0193-80; RRID: AB_837082 |
| A700-conjugated Rat anti-mouse CD4 (RM4-5) | eBioscience | Cat#56-0042-82; RRID: AB_494000 |
| APC-conjugated Rat anti-mouse CD8a (clone 53-6.7) | Biolegend | Cat#100712; RRID: AB_312751 |
| A647-conjugated Rat anti-mouse Gr-1 (clone RB6-8C5) | Biolegend | Cat#108418; RRID: AB_389331 |
| PE-conjugated Rat anti-mouse F4/80 (clone BM8) | eBioscience | Cat#12-4801-82; RRID: AB_465923 |
| Streptavidin-conjugated A750 | Life Technologies | Cat#S21384 |
| Biotinylated Goat Anti-Rabbit IgG | Vector Laboratories | Cat#BA-1000; RRID: AB_2313606 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase IV | Sigma-Aldrich | C5138 |
| Collagenase XI | Sigma-Aldrich | C7657 |
| Deoxyribonuclease I from bovine pancreas (DNase I) | Sigma-Aldrich | DN25 |
| Dispase II, powder | Roche | 25766800/04942078001 |
| DMEM | Biochrom | F0455 |
| Fetal Bovine Serum (FBS) | Biosera | FB-1001/500 |
| Antibiotic-antimycotic solution | GIBCO | 15240-062 |
| L-Glutamine | GIBCO | 25030-024 |
| Nonessential amino acids (MEM NEAA) | GIBCO | 11140-035 |
| Penicillin and streptomycin | GIBCO | 15140-122 |
| Amphotericin B | Sigma-Aldrich | A2942 |
| Propidium Iodide Solution | Sigma-Aldrich | 255535-16-4 |
| BrdU, powder | Roche | 10 280 879 001 |
| Vectastain ABC | Vector Laboratories | PK-6100 |
| ImmPACT DAB Peroxidase (HRP) Substrate | Vector Laboratories | SK-4105 |
| MMLV Reverse Transcriptase | Promega | M1701 |
| Platinum SYBR-Green qPCR SuperMix | Invitrogen | 11733046 |
| Oligo-dT primers | New England BioLabs | S1316S |
| Azoxymethane (AOM) | Sigma-Aldrich | A5486 |
| Dextran Sodium Sulfate (DSS) | MP Biomedicals | M9147 |
| LPS from E. coli | Sigma-Aldrich | L2630 |
| Recombinant Murine IL-1β | Peprotech | 211-11B |
| Recombinant Mouse IL-18 | MBL | B002-5 |
| Recombinant Mouse IL-33 | R&D Systems | 3626-ML |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | 74104 |
| RNeasy Micro Kit | QIAGEN | 74004 |
| Mouse IL-6 Duo-Set ELISA | R&D Systems | DY4046 |
| BrdU Proliferation kit | BD Biosciences | 550803 |
| DeadEnd Fluorometric Tunel assay | Promega | G3250 |
| Mouse TLR1-9 Agonist kit | Invivogen | tlrl-kit1mw |
| Agilent RNA 6000 Nano kit | Agilent Technologies | 5067-1511 |
| QuantSeq 3′ mRNA-Seq Library Prep Kit for Ion Torrent | Lexogen | 012 |
| DNA High Sensitivity Kit | Agilent Technologies | 5067-4626 |
| Ion PI™ IC 200 Kit | ThermoFisher Scientific | 4488377 |
| Ion PI™ Sequencing 200 V3 Kit | ThermoFisher Scientific | 4488315 |
| Ion Proton PI™ V2 chips | ThermoFisher Scientific | 4482321 |
| Deposited Data | ||
| Normalized data | Calon et al., 2012 | GEO: GSE39395 |
| Normalized data | Nishida et al., 2012 | GEO: GSE35602 |
| Raw and analyzed data | This study | GEO: GSE119341 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J-ApcMin/J | Jackson Laboratory | https://www.jax.org/strain/002020 |
| Mouse: B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J | Jackson Laboratory | https://www.jax.org/strain/007676 |
| Mouse: Tg(CollagenVI-Cre) | Kollias Laboratory | Armaka et al., 2008 |
| Mouse: Myd88f/f | Pasparakis Laboratory | Vlantis et al., 2016 |
| Mouse: Tlr4f/f | Hackam Laboratory | Sodhi et al., 2012 |
| Mouse: Il1r1f/f | Muller Laboratory | Abdulaal et al., 2016 |
| Oligonucleotides | ||
| See Table S1 for primer sequences | This study; Salcedo et al., 2010, Ding et al., 2016 | N/A |
| Software and Algorithms | ||
| ImageJ | National Center for Microscopy and Imaging Research | RRID: SCR_003070 |
| GraphPad Prism | GraphPad | RRID: SCR_002798 |
| Leica Application Suite X | Leica Microsystems | RRID: SCR_013673 |
| Opticon Monitor | Bio-Rad | RRID: SCR_014241 |
| FACSDiva | BD Biosciences | RRID: SCR_001456 |
| FlowJo | Tree Star Inc | RRID: SCR_008520 |
| Geo2R function | NCBI | N/A |
| InteractiveVenn | Heberle et al., 2015; http://www.interactivenn.net | N/A |
| Functional Annotation tool from DAVID for Gene Ontologies | Huang et al., 2009a, Huang et al., 2009b; https://david.ncifcrf.gov/ | N/A |
Contact or Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, George Kollias (kollias@fleming.gr).
Experimental Model and Subject Details
Mice
Myd88F/F, Tlr4F/F, Il1r1F/F and ColVI-cre mice have been previously described (Abdulaal et al., 2016, Armaka et al., 2008, Sodhi et al., 2012, Vlantis et al., 2016). RosamTmG and Apcmin/+ mice were purchased from the Jackson Laboratory (Dove et al., 1997, Muzumdar et al., 2007). Both male and female mice were used at the ages of 2-4 months. All mice were maintained on a C57/Bl6 background and experiments were performed using littermate and co-housed (according to gender) control and experimental mice. Experiments were performed in the animal facilities of Biomedical Sciences Research Center (BSRC) “Alexander Fleming” under specific pathogen–free conditions. Mice were euthanized by CO2 asphyxiation.
Model Systems and Permissions
All animal studies were approved by the Institutional Committee of Protocol Evaluation in conjunction with the Veterinary Service Management of the Hellenic Republic Prefecture of Attika according to all current European and national legislation and performed in accordance with the guidance of the Institutional Animal Care and Use Committee of BSRC “Alexander Fleming.”
Isolation and Induction of Primary IMCs
Isolation and culture of primary intestinal mesenchymal cells was performed as previously described (Koliaraki and Kollias, 2016). In more detail, the small intestine was removed, flushed with ice-cold HBSS (GIBCO), containing antibiotic-antimycotic solution (GIBCO), opened longitudinally and cut into 1 cm pieces. Fat, adherent connective tissue and Peyer’s patches were removed. Intestinal pieces were incubated with pre-warmed 5mM EDTA (Acros Organics) and 1 mM DTT (Sigma) in HBSS for 20 minutes at 37°C with horizontal shaking to remove the epithelial layer, washed extensively and then digested using 300u/ml Collagenase XI (Sigma) and 0.1mg/ml Dispase II (Roche) for approximately 40 minutes at 37°C. The resulting supernatant was passed through a 70 μm strainer and single cells were pelleted by centrifugation at 300g for 5 minutes and plated in DMEM (Biochrom), supplemented with 10% FBS, (Biochrom), 2 mM L-glutamine (GIBCO) 1% nonessential amino acids (GIBCO), 100 U/ml penicillin and 100 μg/ml streptomycin (GIBCO), and 1 μg/ml amphotericin B (Sigma-Aldrich) until confluent. Cells were used at passages 3-4. For induction assays, cells were plated, serum starved overnight, and stimulated with 2 μg/ml LPS from E. coli (Sigma), 10ng/ml IL1β (Peprotech), IL33 (MBL), IL18 (R&D systems) and the mouse TLR1-9 Agonist kit (Invivogen). The quantities used for the TLR agonists were 100ng/ml Pam3CSK4 (TLR1/2), 2x107/ml HKLM cells (TLR2), 10 μg/ml poly(I:C) (TLR3), 100ng/ml FLA-ST (TLR5), 100ng/ml FSL1 (TLR6/2), 1 μg/ml ssRNA/Lyovec (TLR7) and 1μΜ ODN1826 (TLR9).
Method Details
Induction of Colitis-associated Cancer
Induction of colitis-associated cancer was performed as previously described (Koliaraki et al., 2015, Wirtz et al., 2017). In more detail, littermate and co-housed 6-8 weeks old male and female mice were injected with 10mg/Kg AOM (Sigma), followed by three cycles of 2.5% DSS (MP Biomedicals, colitis grade, MW: 36–50 kDa) administration in the drinking water. Each cycle lasted 5 days and was followed by 16 days of regular drinking water. Body weights and animal well-being were monitored throughout the study. The mice were euthanized 10 days after the end of the last DSS cycle (Day 60), colon was removed and macroscopically visible tumors were counted.
Histology and Immunohistochemistry
Mice were sacrificed, and the intestine was resected, opened longitudinally, and tumors were counted. Tissues were either fixed overnight in 10% formalin and embedded in paraffin or fixed in 4% Paraformaldehyde (PFA) and embedded in OCT (VWR Chemicals). Four μm sections were mounted on slides. Formalin-fixed paraffin-embedded (FFPE) sections were stained with hematoxylin and eosin (Merck) and images were acquired with a Nikon microscope, equipped with a QImaging digital camera. The ImageJ software was used for measuring tumor size. Sections from OCT-embedded tissues were left to dry, washed with PBS and covered with mounting medium containing DAPI (Sigma). Images were acquired with the TCS SP8X White Light Laser confocal system (Leica).
For immunohistochemistry, FFPE sections were de-paraffinized with Xylene and hydrated in 100% and 70% ethanol. Antigen retrieval was performed using heat-mediated Citrate buffer (pH 6.0) for 20 minutes and slides were allowed to cool down. Slides were then incubated at room temperature in 3% hydrogen peroxidase in PBS for 15 minutes to block endogenous peroxidase activity. Tissue sections were washed and blocked with 1% BSA in PBS for 1 hour and incubated with the primary antibody against phospho-STAT3 (Cell Signaling) overnight at 4°C. A secondary biotinylated antibody against rabbit IgG (Vector) was added for 1 hour at room temperature, followed by the Vectastain ABC kit for 30 minutes at room temperature and peroxidase activity was visualized using ImmPACT DAB Peroxidase (HRP) Substrate (Vector Laboratories). The sections were counterstained with hematoxylin (Merck), dehydrated by incubation in 70% and 100% ethanol, permeated with xylene and preserved in mounting medium (Sigma). For analysis of proliferation and apoptosis, FFPE sections were stained using the BrdU proliferation kit (BD Biosciences) and the DeadEnd Fluorometric Tunel assay (Promega), respectively, according to manufacturer’s instructions. Hematoxylin and DAPI (Sigma) were used for counterstain, respectively. For assessment of proliferation, 100mg/ml BrdU (Roche) was administered via i.p. injection 90min before sacrifice. The number of BrdU and Tunel positive cells was quantified in size-matched tumors.
FACS analysis and Cell Sorting
For FACS analysis a part of the small intestine or tumors were removed, washed with HBSS (GIBCO), containing antibiotic-antimycotic solution (GIBCO) and cut into pieces. Intestinal pieces or minced tumors were digested using 400 U/ml Collagenase IV (Sigma-Aldrich), 1 mg/ml Dispase II (Roche) and 100 U/ml Dnase I (Sigma-Aldrich) in DMEM for 40-60 minutes at 37°C. The cell suspension was centrifuged, resuspended in FACS buffer (PBS with 2% FBS) and cells were counted. The anti-Fc Receptor (anti-CD16/32) antibody (Biolegend) was used to prevent non-specific binding. For stainings, 1-2 million cells were incubated with the following antibodies: FITC-conjugated anti-CD11b (eBioscience), APC/Cy7-conjugated or A700-conjugated anti-CD45 (Biolegend), PE/Cy7-conjugated anti-CD11c (Biolegend), A700-conjugated anti-CD19 (eBioscience), A700-conjugated anti-CD4 (eBioscience), APC-conjugated anti-CD8 (Biolegend), A647-conjugated anti-Gr1 (Biolegend), PE-conjugated anti-F4/80 (eBioscience) and streptavidin-conjugated A750 (Invitrogen) (see Key Resources Table). Propidium iodide (PI) was used for live/dead exclusion (Sigma). Analysis was performed using a FACS Canto II Flow cytometer (BD Biosciences) and FACSDiva (BD Biosciences) or FlowJo software (FlowJo, LLC). Cultured cells were used for sorting based on their GFP and Tomato fluorescent protein expression, using a FACSAria III Cell Sorter (BD Biosciences).
ELISA
Supernatants were collected at 24h and IL-6 quantification was performed using the mouse IL-6 Duo-Set ELISA kit, according to the manufacturer’s instructions (R&D Systems).
RNA Isolation and qRT-PCR
RNA was isolated from tumors, intestinal tissue or intestinal mesenchymal cells using the RNeasy mini or micro kit (QIAGEN), according to the manufacturer’s instructions. Isolated RNA was subsequently used either for 3′RNA-seq sequencing and analysis, or for construction of cDNA, using the MMLV Reverse Transcriptase (Promega), according to the manufacturer’s instruction. The cDNA was subsequently used for qRT-PCR using the Platinum SYBR-Green qPCR SuperMix (Invitrogen) and the CFX96 Touch Real-Time PCR Detection System (Biorad). Quantification was performed with the DDCt method. Primer sequences (5′-3′) are provided (see Table S1).
3′ RNA-Seq sequencing
The quantity and quality of RNA samples were analyzed using Agilent RNA 6000 Nano kit with the bioanalyzer from Agilent. RNA samples with RNA Integrity Number (RIN) > 7 were used for library construction using the 3′ mRNA-Seq Library Prep Kit Protocol for Ion Torrent (QuantSeq-LEXOGEN™) according to manufacturer’s instructions. DNA High Sensitivity Kit in the bioanalyzer was used to assess the quantity and quality of libraries, according to the manufacturer’s instructions (Agilent). Libraries were then pooled and templated using the Ion PI™ IC 200 Kit (ThermoFisher Scientific) on an Ion Proton Chef Instrument or Ion One Touch System. Sequencing was performed using the Ion PI™ Sequencing 200 V3 Kit and Ion Proton PI™ V2 chips (ThermoFisher Scientific) on an Ion ProtonTM System, according to the manufacturer’s instructions.
Quantification and Statistical Analysis
Analysis of RNaseq data
Mapping of sequencing reads to reference genome was performed as recommended by the manufacturer and gene differential expression analysis was performed using Bioconductor package DESeq through metaseqR pipeline, as previously described (Moulos and Hatzis, 2015). Downstream bioinformatics analysis was performed using InteractiveVenn for Venn diagrams (http://www.interactivenn.net) (Heberle et al., 2015) and the Functional Annotation tool from DAVID for Gene Ontologies (https://david.ncifcrf.gov/) (Huang et al., 2009a, Huang et al., 2009b), while volcano plots and heatmaps were generated in R using an in-house developed script and packages ggplot and gplots (R Core Team, 2017, Warnes et al., 2016, Wickham, 2016).
Datasets and Statistical analysis
The public datasets GEO: GSE39395 and GSE35602 was analyzed using the Geo2R function from NCBI. For Figure 3B, values with the lowest adjusted p value were selected for each gene. For Figure 4F, genes with adjusted p value ≤ 0.05 and log2 fold change ≥ 1 were used for downstream analysis.
The Student’s t test was used to calculate statistical significance for samples that showed normal distribution and the Mann Whitney test for those that did not pass the normality test. Equal variance was evaluated by the F-test. One-way ANOVA, corrected using Tukey test, was used for multiple comparisons. Information on the value of n, what n represents, the definition of center and dispersion and precision measures can be found in the Figure legends. Mouse sample size was estimated by power analysis. Data were analyzed with GraphPad Prism 6. p values ≤ 0.05 were considered significant.
Data and Software Availability
RNA-seq datasets are accessible through GEO: GSE119341 of NCBI’s Gene Expression Omnibus.
Acknowledgments
We thank Michalis Meletiou, Anna Katevaini, and Spiros Lalos for technical assistance in mouse genotyping and histology. We thank Sofia Grammenoudi for assistance in flow cytometry. We thank the Genomics Facility of BSRC “Alexander Fleming,” specifically Vaggelis Harokopos for performing RNA sequencing and Martin Reczko for initial bioinformatics analyses. We also thank the InfrafrontierGR infrastructure (co-funded by the European Regional Development Fund and Greek NSRF 2007-2013 to G.K.) for providing mouse hosting and phenotyping facilities. This work was supported by a grant from the Stavros Niarchos Foundation to the BSRC “Alexander Fleming” as part of the Foundation’s initiative to support the Greek research center ecosystem and the FP7 Advanced ERC Grant MCs-inTEST (grant 340217) to G.K.
Author Contributions
Conceptualization, V.K. and G.K.; Investigation, V.K., N.C., A.H., and A.P.; Formal Analysis and Visualization, V.K., N.C., A.H., and C.T.; Data Curation, C.T.; Writing – Original Draft, V.K; Writing – Review & Editing, M.P. and G.K; Resources, M.P., D.J.H., A.W., and W.M.; Funding Acquisition, G.K.; Supervision, V.K. and G.K.
Declaration of Interests
The authors declare no competing interests.
Published: January 15, 2019
Contributor Information
Vasiliki Koliaraki, Email: koliaraki@fleming.gr.
George Kollias, Email: kollias@fleming.gr.
Supplemental Information
References
- Abdulaal W.H., Walker C.R., Costello R., Redondo-Castro E., Mufazalov I.A., Papaemmanouil A., Rothwell N.J., Allan S.M., Waisman A., Pinteaux E., Müller W. Characterization of a conditional interleukin-1 receptor 1 mouse mutant using the Cre/LoxP system. Eur. J. Immunol. 2016;46:912–918. doi: 10.1002/eji.201546075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armaka M., Apostolaki M., Jacques P., Kontoyiannis D.L., Elewaut D., Kollias G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J. Exp. Med. 2008;205:331–337. doi: 10.1084/jem.20070906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollrath J., Phesse T.J., von Burstin V.A., Putoczki T., Bennecke M., Bateman T., Nebelsiek T., Lundgren-May T., Canli O., Schwitalla S. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Calon A., Espinet E., Palomo-Ponce S., Tauriello D.V., Iglesias M., Céspedes M.V., Sevillano M., Nadal C., Jung P., Zhang X.H. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571–584. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M., Bruick R.K., Yu Y. Secreted IGFBP5 mediates mTORC1-dependent feedback inhibition of IGF-1 signalling. Nat. Cell Biol. 2016;18:319–327. doi: 10.1038/ncb3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove W.F., Clipson L., Gould K.A., Luongo C., Marshall D.J., Moser A.R., Newton M.A., Jacoby R.F. Intestinal neoplasia in the ApcMin mouse: independence from the microbial and natural killer (beige locus) status. Cancer Res. 1997;57:812–814. [PubMed] [Google Scholar]
- Erez N., Truitt M., Olson P., Arron S.T., Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010;17:135–147. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- Grivennikov S.I., Wang K., Mucida D., Stewart C.A., Schnabl B., Jauch D., Taniguchi K., Yu G.Y., Osterreicher C.H., Hung K.E. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Coussens L.M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- Heberle H., Meirelles G.V., da Silva F.R., Telles G.P., Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics. 2015;16:169. doi: 10.1186/s12859-015-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques A., Koliaraki V., Kollias G. Mesenchymal MAPKAPK2/HSP27 drives intestinal carcinogenesis. Proc. Natl. Acad. Sci. USA. 2018;115:E5546–E5555. doi: 10.1073/pnas.1805683115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Koliaraki V., Kollias G. Isolation of intestinal mesenchymal cells from adult mice. Bio-protocol. 2016;6:e1940. [Google Scholar]
- Koliaraki V., Roulis M., Kollias G. Tpl2 regulates intestinal myofibroblast HGF release to suppress colitis-associated tumorigenesis. J. Clin. Invest. 2012;122:4231–4242. doi: 10.1172/JCI63917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliaraki V., Pasparakis M., Kollias G. IKKβ in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J. Exp. Med. 2015;212:2235–2251. doi: 10.1084/jem.20150542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliaraki V., Pallangyo C.K., Greten F.R., Kollias G. Mesenchymal Cells in Colon Cancer. Gastroenterology. 2017;152:964–979. doi: 10.1053/j.gastro.2016.11.049. [DOI] [PubMed] [Google Scholar]
- Kostic A.D., Chun E., Robertson L., Glickman J.N., Gallini C.A., Michaud M., Clancy T.E., Chung D.C., Lochhead P., Hold G.L. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–215. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Hu L.L., Gonzalez-Navajas J., Seo G.S., Shen C., Brick J., Herdman S., Varki N., Corr M., Lee J., Raz E. ERK activation drives intestinal tumorigenesis in Apc(min/+) mice. Nat. Med. 2010;16:665–670. doi: 10.1038/nm.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Kundu P., Seow S.W., de Matos C.T., Aronsson L., Chin K.C., Kärre K., Pettersson S., Greicius G. Gut microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis. 2012;33:1231–1238. doi: 10.1093/carcin/bgs137. [DOI] [PubMed] [Google Scholar]
- Moulos P., Hatzis P. Systematic integration of RNA-Seq statistical algorithms for accurate detection of differential gene expression patterns. Nucleic Acids Res. 2015;43:e25. doi: 10.1093/nar/gku1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar M.D., Tasic B., Miyamichi K., Li L., Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nishida N., Nagahara M., Sato T., Mimori K., Sudo T., Tanaka F., Shibata K., Ishii H., Sugihara K., Doki Y., Mori M. Microarray analysis of colorectal cancer stromal tissue reveals upregulation of two oncogenic miRNA clusters. Clin. Cancer Res. 2012;18:3054–3070. doi: 10.1158/1078-0432.CCR-11-1078. [DOI] [PubMed] [Google Scholar]
- Powell D.W., Pinchuk I.V., Saada J.I., Chen X., Mifflin R.C. Mesenchymal cells of the intestinal lamina propria. Annu. Rev. Physiol. 2011;73:213–237. doi: 10.1146/annurev.physiol.70.113006.100646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R Foundation for Statistical Computing; 2017. R: A Language and Environment for Statistical Computing.https://www.R-project.org/ [Google Scholar]
- Rakoff-Nahoum S., Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- Roulis M., Nikolaou C., Kotsaki E., Kaffe E., Karagianni N., Koliaraki V., Salpea K., Ragoussis J., Aidinis V., Martini E. Intestinal myofibroblast-specific Tpl2-Cox-2-PGE2 pathway links innate sensing to epithelial homeostasis. Proc. Natl. Acad. Sci. USA. 2014;111:E4658–E4667. doi: 10.1073/pnas.1415762111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo R., Worschech A., Cardone M., Jones Y., Gyulai Z., Dai R.M., Wang E., Ma W., Haines D., O’hUigin C. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J. Exp. Med. 2010;207:1625–1636. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo R., Cataisson C., Hasan U., Yuspa S.H., Trinchieri G. MyD88 and its divergent toll in carcinogenesis. Trends Immunol. 2013;34:379–389. doi: 10.1016/j.it.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S., Schumacher N., Schwarz J., Tangermann S., Kenner L., Schlederer M., Sibilia M., Linder M., Altendorf-Hofmann A., Knösel T. ADAM17 is required for EGF-R-induced intestinal tumors via IL-6 trans-signaling. J. Exp. Med. 2018;215:1205–1225. doi: 10.1084/jem.20171696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipos F., Germann T.M., Wichmann B., Galamb O., Spisák S., Krenács T., Tulassay Z., Molnár B., Műzes G. MMP3 and CXCL1 are potent stromal protein markers of dysplasia-carcinoma transition in sporadic colorectal cancer. Eur. J. Cancer Prev. 2014;23:336–343. doi: 10.1097/CEJ.0000000000000058. [DOI] [PubMed] [Google Scholar]
- Sodhi C.P., Neal M.D., Siggers R., Sho S., Ma C., Branca M.F., Prindle T., Jr., Russo A.M., Afrazi A., Good M. Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology. 2012;143:708–718.e5. doi: 10.1053/j.gastro.2012.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X., Gao H., Lin Y., Yao Y., Zhu S., Wang J., Liu Y., Yao X., Meng G., Shen N. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity. 2014;40:140–152. doi: 10.1016/j.immuni.2013.11.018. [DOI] [PubMed] [Google Scholar]
- Terzic J., Grivennikov S., Karin E., Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138:2101–2114.e5. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- Tomasetti C., Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78–81. doi: 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlantis K., Polykratis A., Welz P.S., van Loo G., Pasparakis M., Wullaert A. TLR-independent anti-inflammatory function of intestinal epithelial TRAF6 signalling prevents DSS-induced colitis in mice. Gut. 2016;65:935–943. doi: 10.1136/gutjnl-2014-308323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton K.L., Holt L., Sartor R.B. Lipopolysaccharide activates innate immune responses in murine intestinal myofibroblasts through multiple signaling pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:G601–G611. doi: 10.1152/ajpgi.00022.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes, G.R., Bolker, B., Bonebakker, L., Gentleman, R., Liaw, W.H.A., Lumley, T., Maechler, M., Magnusson, A., Moeller, S., Schwartz, M., et al. (2016). gplots: Various R programming tools for plotting data. Package version 3.0.1. https://cran.r-project.org/web/packages/gplots/index.html.
- Wickham H. Springer-Verlag; 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- Wirtz S., Popp V., Kindermann M., Gerlach K., Weigmann B., Fichtner-Feigl S., Neurath M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017;12:1295–1309. doi: 10.1038/nprot.2017.044. [DOI] [PubMed] [Google Scholar]
- Yang Y., Weng W., Peng J., Hong L., Yang L., Toiyama Y., Gao R., Liu M., Yin M., Pan C. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor-kappaB, and Up-regulating Expression of MicroRNA-21. Gastroenterology. 2017;152:851–866.e24. doi: 10.1053/j.gastro.2016.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq datasets are accessible through GEO: GSE119341 of NCBI’s Gene Expression Omnibus.