

Abstract
We hypothesized that autophagy and associated lysosome function serve as a critical modulator during Nod-like receptor family pyrin domain containing 3 (Nlrp3) inflammasome activation on proatherogenic stimuli. We first demonstrated that 7-ketocholesterol stimulated Nlrp3 inflammasome formation and activation as shown by increased colocalization of inflammasome components [Nlrp3 versus apoptosis associated speck-like protein (Asc) or caspase-1] and enhanced cleavage of caspase-1 into active caspase-1 to generate IL-1β in coronary artery smooth muscle cells. Deletion of the CD38 gene (CD38−/−) that regulates lysosome function and autophagic flux also led to Nlrp3 inflammasome formation and activation. In the presence of rapamycin, the effects of either 7-ketocholesterol treatment or CD38 gene deletion were abolished. The autophagy inhibitor spautin-1 and the lysosome function blocker bafilomycin A1 also enhanced Nlrp3 inflammasome formation and activation. In animal experiments, we found that increased colocalization of Nlrp3 versus Asc or caspase-1 enhanced IL-1β accumulation and caspase-1 activity in the coronary arterial wall of CD38−/− mice on the Western diet compared with CD38+/+ mice. This increased colocalization was blocked by treatment with rapamycin but enhanced by chloroquine, a water-soluble blocker of autophagic flux. Morphologic examinations confirmed that the media of coronary arteries was significantly thicker in CD38−/− mice on the Western diet than CD38+/+ mice. In conclusion, the deficiency of autophagic flux promotes Nlrp3 inflammasome formation and activation in coronary artery smooth muscle cells on proatherogenic stimulation, leading to medial thickening of the coronary arterial wall.
Nod-like receptor family pyrin domain containing 3 (Nlrp3) belongs to the Nod-like receptor (NLR) family, which can form Nlrp3 complexes with the adaptor protein apoptosis associated speck-like protein (Asc) and pro–caspase-1.1, 2 The formation of this large multiprotein complex, called Nlrp3 inflammasome, leads to the activation of caspase-1, which is required for maturation and production of mature IL-1β and IL-18 by cleavage of their precursor.3, 4, 5 It has been reported that various human inflammatory disorders may result from the dysregulated production of IL-1β derived from Nlrp3 inflammasomes, including atherosclerosis,6 inherited cryopyrin-associated periodic syndromes,7 gouts,8 glomerular sclerosis,9 and type 2 diabetes.3 The formation and activation of Nlrp3 inflammasomes by the adipokine visfatin may be critical for instigating endothelial inflammatory response, leading to endothelial dysfunction and arterial inflammation and thereby initiating atherosclerosis during obesity.10 However, so far it remains unknown how the inflammasome activation is regulated within vascular cells and what mechanism controls inflammasome products to be transported and metabolized given that they are not synthesized through classical protein synthesis pathways through the endoplasmic reticulum.
Autophagy is a cellular process through which cytoplasmic components, such as damaged organelles and protein aggregates, are delivered to lysosomes for degradation and recycled back.11, 12 Basal-level autophagy in various tissues maintains constitutive turnover of cytosolic components. However, starvation autophagy is stimulated to recycle nutrients and generate energy for cell survival under stress (oxidative insult) conditions.11 As an evolutionarily conserved process, autophagy plays an essential role in various cellular processes, such as the clearance of pathogens and cell death, thereby being importantly implicated in human physiology and diseases.13 Because different pharmacologic compounds can modulate autophagic machinery, this process is also a potential therapeutic target for a variety of diseases.14 Given the critical role of autophagy in turnover of organelles or other intracellular molecules via elimination of damaged and aged cellular components, it may also be involved in the control of Nlrp3 inflammasome formation and activation because various organelle stressors activate Nlrp3 inflammasomes, such as damaged mitochondria, phagosome-lysosome, and endoplasmic reticuli if they are not entirely degraded via the autophagic process. In addition, several stimulatory metabolites activate Nlrp3 inflammasomes in vascular tissues, such as cholesterol crystals and monosodium urate crystals, and their action mechanism is associated with lysosome injury, which may change the autophagic process.15, 16 Therefore, autophagy may suppress Nlrp3 inflammasome activation via reducing the activation of Nlrp3 inflammasome induced by organelle stress,17, 18, 19 whereas the loss of autophagy may result in inflammasome activation, leading to different inflammatory disorders, such as atherosclerosis, pneumonia, diabetes, sepsis, and colitis.20 There is also evidence that inducers of autophagy may attenuate the production of mature IL-1β and IL-18 via the Nlrp3 inflammasome pathway.21, 22
Nlrp3 inflammasome activation has been reported to mediate early inflammatory responses after vascular injury and that increased production of IL-1β and IL-18 are associated with human plaque vulnerability and atherogenesis.23, 24 Studies from our group reported that the CD38 signaling pathway is critical for autophagy maturation and deficient CD38 signaling and consequent dysregulation of autophagy may lead to arterial wall thickening as shown in CD38−/− mice.25, 26 Zhang et al27 reported that CD38 gene deletion impaired autophagic flux and caused defective autophagosome trafficking in coronary artery smooth muscle cells (CASMCs), which is attributed to atherogenesis. However, it remains unknown how autophagic flux derangement leads to atherosclerotic pathology in coronary arteries and whether it is associated with Nlrp3 inflammasome activation. This study was designed to answer these essential questions.
Materials and Methods
Mice
CD38 wild-type (CD38+/+) and CD38 knockout (CD38−/−) C57BL/6J male mice (8 to 12 weeks old) were used in this study. All mice were maintained in the environmentally controlled room (25°C and approximately 40% to 50% humidity) with a 12-hour light/dark cycle. Both CD38+/+ and CD38−/− mice were randomly separated into different experimental groups and fed with normal diet (ND) or Western diet (WD) for 8 weeks. All procedures were performed in accordance with the NIH's Guide for the Care and Use of Laboratory Animals28 and were approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University.
Isolation and Culture of Mouse CASMCs
CASMC isolation from mice has been described previously.29 Briefly, 2% isoflurane was used to anesthetize the mice. Then, the carotid arteries were removed and put into phosphate-buffered saline (PBS) on ice. The adventitia was removed from the artery using angled forceps under the microscope. The tissue was washed three times with PBS and was cut into pieces that were approximately 1 to 2 mm using microdissecting scissors in the cell culture hood. The dissected tissues were washed two to three times with cell culture medium and then added into a cell culture dish without medium for 2 hours. Then, fresh medium was added into the tissue culture dish, and tissues were incubated in a humidified 37°C, 5% carbon dioxide incubator. Dulbecco's modified Eagle's medium supplemented with 10% FBS and 2% antibiotics was used to culture tissues. CASMCs were isolated when they spread out from the dissected tissue. After 5 to 10 days, CASMCs were cloned by picking those cells from cell-yielding islands in the dish. The identification and purity of CASMCs in culture were confirmed as described previously.30
Western Blot Analysis
Western blot analysis was performed as described previously.31 Briefly, cell culture dish was placed on ice and the cells were washed with ice-cold PBS. Total protein was extracted using ice-cold lysis buffer with protein inhibitor. The cell suspension was maintained on ice for 30 minutes. After centrifugation at 12,300 × g for 15 minutes at 4°C, the supernatant was transferred to a new tube kept on the ice. Protein concentration was measured and resuspended to 2 μg/μL in loading buffer. The equal amount of protein was loaded into the wells of SDS-PAGE gel along with the molecular weight marker. The gel was run at 100 V for 2 hours at room temperature and then transferred to a polyvinylidene difluoride membrane at 100 V for 1 hour at 4°C. The membrane was blocked with 5% nonfat milk in Tris-buffered saline and Tween 20 buffer at room temperature for 30 minutes. The membrane was incubated with primary antibodies against pro–caspase-1 or cleaved caspase-1 (1:500 dilution, Santa Cruz Biotechnology, Santa Cruz, CA) in blocking buffer overnight at 4°C, followed by incubation with the secondary antibody labeled with horseradish peroxidase for 1 hour at room temperature. The membrane was developed with the Odyssey FC Imaging System after being washed three times with Tris-buffered saline and Tween 20 at room temperature. β-Actin (1:8000 dilution, Santa Cruz) was reported to serve as a loading control. The intensity of the bands was quantified using ImageJ version 6.0 (NIH, Bethesda, MD; https://imagej.nih.gov/ij).
Immunofluorescence Staining
Cells cultured in eight-well plates or coronary arteries on frozen slides were rinsed in PBS for 5 minutes and fixed in 4% paraformaldehyde in PBS (pH 7.4) for 10 minutes at room temperature. The samples were washed three times in PBS for 5 minutes and were incubated for 10 minutes with PBS that contained 0.1% Triton X-100. Then the samples were washed three times for 5 minutes in PBS and incubated with 3% bovine serum albumin (BSA) in PBS for 30 minutes to block unspecific binding of antibodies. The samples were incubated with the diluted primary antibody in 3% BSA overnight at 4°C in a humidified chamber followed by incubation with either Alexa-488– or Alexa-555–labeled secondary antibody in 3% BSA for 1 hour at room temperature in the dark room. The sections were washed three times with PBS for 10 minutes each in the darkroom to decant the secondary antibody solution. Finally, the sections were mounted with a drop of mounting medium with DAPI and sealed with nail polish for taking images using the confocal laser scanning microscope (710 LSM; Zeiss, Dublin, CA). Image ProPlus software version 6.0 (Media Cybernetics, Bethesda, MD) was used to measure the colocalization of Nlrp3 with Asc or caspase-1. Pearson correlation coefficients (PCCs) were expressed to summarize the data of colocalization efficiency as described previously.32, 33
Immunohistochemistry
The mouse hearts were perfused with PBS and fixed in 10% formalin at room temperature. The samples were embedded in paraffin, and 6-μm slides were cut from the embedded blocks. The slides were heated on the hot plate at 65°C for 10 minutes and were put in xylene for 10 minutes to remove the paraffin. Rehydration was performed in graded ethanol (100%, 95%, 75%) and water. Antigen retrieval was performed in sodium citrate buffer (pH 6.0) at 98°C for 15 minutes. Three percent hydrogen peroxide in methanol was used to quench the endogenous peroxidase activity. The samples were blocked with 2.5% horse serum for 1 hour at room temperature. Incubation with the indicated primary antibodies anti–IL-1β (1:100) diluted in PBS was performed overnight at 4°C. After rinsing three times with PBS, the samples were incubated with biotinylated secondary antibodies and a streptavidin-peroxidase complex for 30 minutes at room temperature. Then the samples were sequentially developed with 3,3′-diaminobenzidine solution for 5 minutes. Finally, the sections were counterstained in hematoxylin for 5 minutes, dehydrated in graded ethanol (75%, 95%, 100%), and mounted with Permount. Negative controls were prepared without the primary antibodies. The area percentage of the positive staining was calculated using Image Pro-Plus software version 6.0.34
In Situ Analysis of Caspase-1 Activity
Caspase-1 activity was performed with the similar protocol described previously.35 The FAM-FLICA Caspase-1 Assay Kit (ImmunoChemistry Technologies, LLC, Bloomington, MN) was used to label active caspase-1 enzyme in the frozen sections of coronary artery in the heart. The sections were washed three times in PBS for 5 minutes at room temperature and were fixed in 4% paraformaldehyde in PBS (pH 7.4) for 10 minutes at room temperature. The samples were washed three times in PBS for 5 minutes and were incubated for 10 minutes with PBS that contained 0.1% Triton X-100. Then the samples were washed three times for 5 minutes in PBS and incubated with 3% BSA in PBS for 30 minutes to block unspecific binding of antibodies. The sections were incubated with anti-rabbit α-smooth muscle actin (α-SMA) (1:500; Abcam, Cambridge, UK) overnight at 4°C. After being washed three times in PBS, the sections were co-stained with Alexa-555–labeled anti-rabbit secondary antibody and fluorescent-labeled inhibitor of caspases (FLICA) reagent (1:10) in the kit for 1.5 hours at room temperature. The sections were washed three times with PBS for 10 minutes each in the darkroom to decant the secondary antibody solution and FLICA reagent. Finally, the samples were mounted with a drop of mounting medium with DAPI and sealed with nail polish for taking images using a confocal laser scanning microscope (710 LSM). Image Pro-Plus software version 6.0 was used to measure the colocalization of von Willebrand factor with FLICA. PCCs were expressed to summarize the data of colocalization efficiency as described previously.32
Morphologic Examination and Medial Thickening Analysis
Hematoxlin and eosin staining was performed to observe the morphologic changes as described previously.36 Briefly, hearts were separated from the base of the aorta and transversely cut into two parts. The half that contained the apex was stored at −80°C and the other half was immersed in 10% neutral buffered formalin for >48 hours. The formalin-fixed heart was embedded with paraffin and then cut into 6-μm serial sections for histopathologic evaluation. For hematoxylin and eosin staining, sections were deparaffinized heated on the hot plate at 65°C for 10 minutes and were put in xylene for 10 minutes. Rehydration was performed in graded ethanol (100%, 95%, 75%) and water. After being washed with running water, the sections were immersed in the hematoxylin and hydrochloride alcohol. As soon as the color turned to antiblue, the sections were stained with eosin. After that, sections were washed with running water and dehydrated with water and different graded ethanol (75%, 95%, 100%). Finally, the slides were mounted with Permount (xylene based) and were photographed under the microscope. Medial thickening was measured using Image Pro-Plus software version 6.0. The medial area was determined by subtraction of the lumen area from the outer medial area. The three thickest vessels were chosen to calculate the means of vessel medial thickness and medial thickness-to-lumen radius ratio for each mouse.37
Statistical Analysis
Data are presented as means ± SEM. Significant differences between and within multiple groups were examined using analysis of variance followed by Duncan's multiple-range test. Sigmaplot software version 12.5 (Systat Software, San Jose, CA) was used to perform the statistical analysis. Differences were considered statistically significant at P < 0.05.
Results
Changes in Nlrp3 Inflammasome Formation in CASMCs during Inhibition or Enhancement of Autophagic Flux
Because autophagy and associated lysosome function serve as critical modulators in Nlrp3 inflammasome formation on proatherogenic stimulation, inflammasome formation in 7-ketocholesterol (7-Ket)–treated mouse CASMCs from wild-type (CD38+/+) mice and mice with deletion of CD38 gene (CD38−/−) were first analyzed to determine whether lysosome dysfunction associated with CD38 deficiency is involved in the formation of Nlrp3 inflammasomes. By confocal microscopy, the colocalization (yellow spots within cells) of Nlrp3 with Asc or caspase-1 was found to be increased on 7-Ket stimulation and in cells with CD38 deletion, indicating the aggregation or assembly of these inflammasome molecules (Figure 1, A and C). Figure 1, B and D, shows the PCCs of Nlrp3 with Asc or caspase-1, representing their colocalization efficiency. Both 7-Ket stimulation and CD38 deletion significantly increased Nlrp3 inflammasome formation. However, an autophagy enhancer, rapamycin (Rap), significantly decreased the colocalization of Nlrp3 versus Asc or caspase-1 induced by 7-Ket stimulations in both CD38+/+ and CD38−/− CASMCs. Therefore, the induction of autophagy by Rap may prevent the formation of the Nlrp3 inflammasome, but the deficiency of autophagy induced the formation of the Nlrp3 inflammasome.
Figure 1.
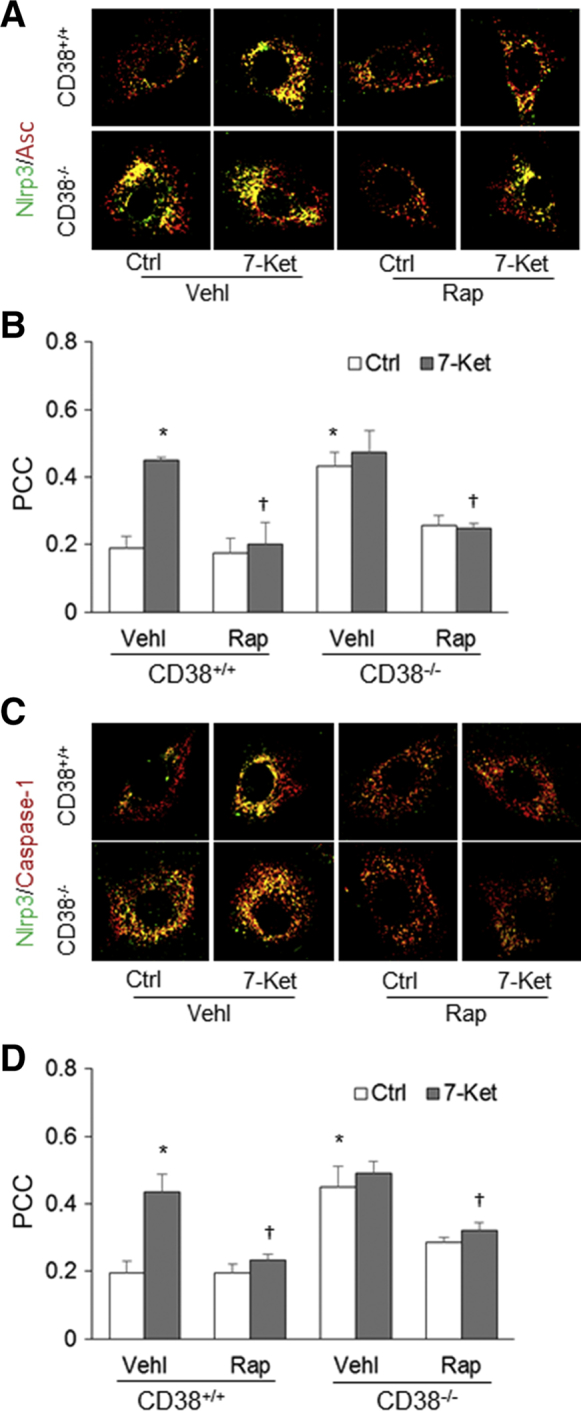
Nod-like receptor family pyrin domain containing 3 (Nlrp3) inflammasome formation in coronary artery smooth muscle cells (CASMCs) during inhibition or enhancement of autophagic flux. A: Representative fluorescent confocal microscope images depict the effects of 7-ketocholesterol (7-Ket) and CD38 gene deletion on the colocalization of Nlrp3 with the apoptosis associated speck-like protein (Asc) in CASMCs from CD38+/+ and CD38−/− mice. B: The fold changes in the Pearson correlation coefficient (PCC) for the colocalization of Nlrp3 with Asc. C: Representative fluorescent confocal microscope images depict the effects of rapamycin (Rap) (10 nmol/L for 24 hours) and CD38 gene deletion on the colocalization of Nlrp3 with caspase-1 stimulated by 7-Ket (10 μg/mL for 24 hours) in CASMCs from CD38+/+ and CD38−/− mice. D: The fold changes in the PCC for the colocalization of Nlrp3 with caspase-1. Data are expressed as means ± SEM. n = 5. ∗P < 0.05 versus the control (Ctrl) vehicle (Vehl) group; †P < 0.05 versus the 7-Ket Vehl group. Original magnification, ×1000.
Changes in Nlrp3 Inflammasome Activation in CASMCs during Inhibition or Enhancement of Autophagic Flux
Next, Nlrp3 inflammasome activation was analyzed by measurement of the active caspase-1 level (19 kDa) by Western blot analysis, spectrometric assay of caspase-1 activity, and enzyme-linked immunosorbent assay quantitation of IL-1β production in CASMCs. 7-Ket significantly increased cleavage of pro–caspase-1 into bioactive caspase-1 in CD38+/+ CASMCs, which was blocked by Rap treatment (Figure 2A). In CD38−/− CASMCs, even under the basal condition, active caspase-1 levels were increased (Figure 2B), suggesting that deletion of the CD38 gene may activate the Nlrp3 inflammasome, thereby causing cleavage of pro–caspase-1 into bioactive caspase-1. 7-Ket had no further effect on the level of active caspase-1 in CD38−/− CASMCs, whereas Rap caused a significant decrease both under control conditions and on 7-Ket stimulation (Figure 2, A and B).
Figure 2.
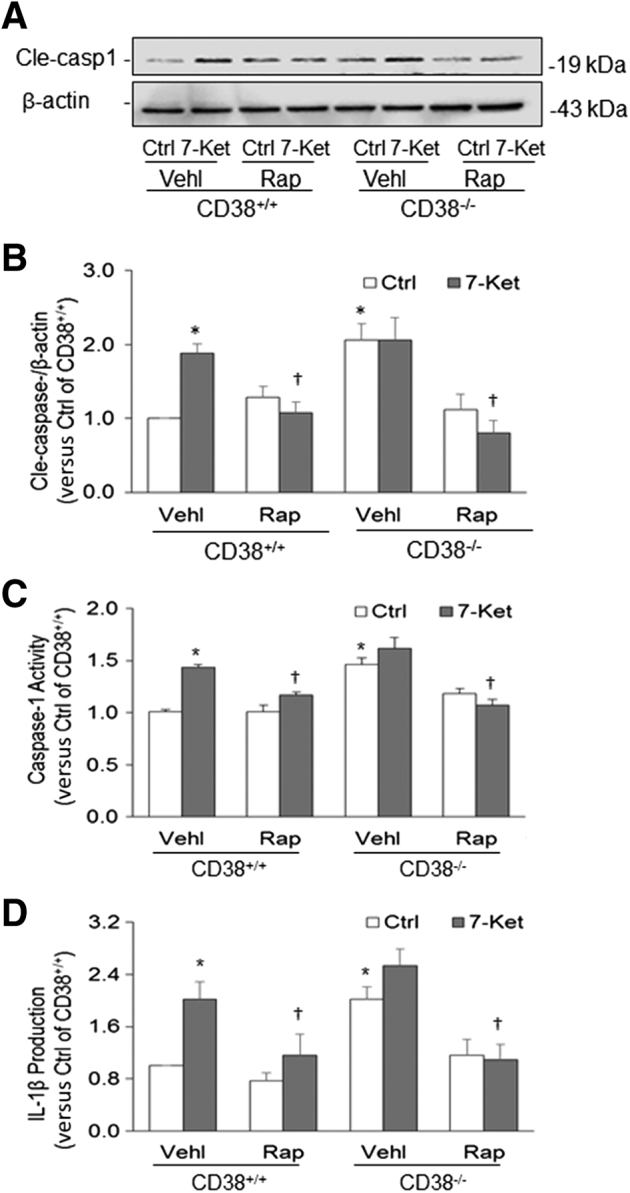
Nod-like receptor family pyrin domain containing 3 (Nlrp3) inflammasome activation in coronary artery smooth muscle cells (CASMCs) during inhibition or enhancement of autophagic flux. A: Representative Western blot gel shows the effects of rapamycin (Rap) (10 nmol/L for 24 hours) and CD38 gene deletion on cleaved caspase-1 (Cle-casp1) level induced by 7-ketocholesterol (7-Ket) in CASMCs from CD38+/+ and CD38−/− mice. B: Quantitative analysis of Western blot. C: Caspase-1 activity in CASMCs detected by chemical analysis of enzyme activity. D: The IL-1β level in cell culture supernatants measured by enzyme-linked immunosorbent assay. Data are expressed as means ± SEM. n = 8 (A and B); n = 5 (C and D). ∗P < 0.05 versus the control (Ctrl) vehicle (Vehl) group; †P < 0.05 versus the 7-Ket Vehl group.
Increased caspase-1 activity and IL-1β production was observed in 7-Ket–stimulated CD38+/+ CASMCs, and increased activation of the Nlrp3 inflammasome was substantially blocked by Rap (Figure 2, C and D). In addition, deletion of the CD38 gene caused a significant increase in caspase-1 activity and IL-1β production, which indicates that the CD38 gene is implicated in Nlrp3 inflammasome activation. In these CD38−/− CASMCs, treatment with 7-Ket did not show a significant increase in caspase-1 activity and IL-1β production, whereas Rap reduced caspase-1 activity and IL-1β production. It seems that improving lysosome function and autophagic flux by Rap treatment antagonized the activating effects of 7-Ket and CD38 gene deletion on Nlrp3 inflammasomes.
Changes in Nlrp3 Inflammasome Formation and Activation during Inhibition of Autophagosome Generation or Suppression of Lysosome Function
Whether inhibition of autophagosome generation and direct suppression of lysosome function are associated with Nlrp3 inflammasome activity was tested. Both inhibition of autophagosome formation by spautin-1 (Spa-1) and suppression of vacuolar H(+)-ATPase in the lysosome by bafilomycin A1 (Baf) markedly increased colocalization of Nlrp3 with Asc or caspase-1 in wild-type CASMCs, whether these cells were treated with 7-Ket or not (Figure 3, A and C). Quantitative analysis showed that the PCC of Nlrp3 with Asc or caspase-1 increased in CASMCs treated with Spa-1 or Baf. This result suggests that normal autophagic processes, including both autophagosome formation and its flux, are important in the regulation of Nlrp3 inflammasome assembly (Figure 3, B and D).
Figure 3.

Effects of spautin-1 (Spa-1) and bafilomycin (Baf) on inflammasome formation in coronary arterial smooth muscle cells (CASMCs) from CD38+/+ and CD38−/− mice. A: Representative confocal fluorescent images show that Spa-1 and Baf treatment increases the colocalization of Nod-like receptor family pyrin domain containing 3 (Nlrp3) and apoptosis associated speck-like protein (Asc) in CASMCs from CD38+/+ mice stimulated with or without 7-ketocholesterol (7-Ket). B: The Pearson colocalization coefficient (PCC) of Nlrp3 with Asc. C: Representative fluorescent confocal microscope images show that Spa-1 and Baf treatment increases the colocalization of Nlrp3 with caspase-1 in CASMCs from CD38+/+ mice stimulated with or without 77-Ket. D: The fold changes in the PCC for the colocalization of Nlrp3 with caspase-1. Data are expressed as means ± SEM. n = 5. ∗P < 0.05 versus the control (Ctrl) vehicle (Vehl) group; †P < 0.05 versus the 7-ket Vehl group. Original magnification, ×1000.
By Western blot analysis, it was found that treatment with Spa-1 and Baf significantly increased the cleavage of pro–caspase-1 into bioactive caspase-1, in particular, when these cells were stimulated by 7-Ket (Figure 4A). Inhibition of autophagosome formation or disruption of lysosome function to suppress autophagic flux significantly increased activation of caspase-1, under both control conditions or on 7-Ket stimulation (Figure 4B). This enhanced activation of the Nlrp3 inflammasome was also confirmed by enhanced caspase-1 activity (Figure 4C) and increased IL-1β levels in culture media (Figure 4D). These findings suggest that autophagosome aggregation with reduced autophagic degradation of ubiquitinated cargoes may result in organelle stress that leads to Nlrp3 inflammasome formation and activation.
Figure 4.

Effects of spautin-1 (Spa-1) and bafilomycin (Baf) on inflammasome activation in coronary arterial smooth muscle cells (CASMCs) from CD38+/+ and CD38−/− mice. A: Representative gel documents of Western blot show the effect of Spa-1 and Baf on cleaved caspase-1 (Cle-casp1) levels in CASMCs from CD38+/+ mice stimulated with or without 7-ketocholesterol (7-Ket). B: Quantitative analysis of Western blot gel bands specific to Cle-casp1 versus β-actin. C: Caspase-1 activity in CASMCs detected by chemical analysis of enzyme activity. D: IL-1β level in cell culture supernatants. Data are expressed as means ± SEM. n = 5. ∗P < 0.05 versus the control (Ctrl) vehicle (Vehl) group; †P < 0.05 versus the 7-Ket Vehl group.
Enhanced Nlrp3 Inflammasome Formation in the Coronary Arterial Wall of CD38−/− Mice
The in vitro findings strongly link loss of autophagosome aggregation attributable to autophagic flux or lysosome dysfunction to Nlrp3 inflammasome action, which is consistent with previous reports about deficient autophagy to various inflammatory disorders, such as sepsis, colitis, pneumonia, atherosclerosis, and diabetes.17, 18, 19, 20 With the use of a mouse model with CD38 gene deletion, which was shown to have lysosome dysfunction and deranged autophagic flux, the relevance of autophagy regulation of Nlrp3 inflammasome formation and activation to vascular injury and inflammation was tested under the WD. Colocalization of Nlrp3 with Asc or caspase-1 was much higher in the arterial wall of CD38−/− mice compared with CD38+/+ mice even though when they were on the ND. The Western diet markedly increased the colocalization of Nlrp3 with Asc or caspase-1 in CD38+/+ mice, but there was no further increase in such aggregation of Nlrp3 inflammasome components in CD38−/− mice. The water-soluble lysosome function inhibitor chloroquine produced similar effects in wild-type mice, which was similar to that produced by CD38 gene deletion. However, Rap treatment reversed the effects of WD plus chloroquine and CD38 deletion (Figure 5, A and C). The PCC analysis showed that the colocalization coefficient of Nlrp3 with Asc or caspase-1 was significantly higher in the arterial wall of CD38+/+ mice receiving the WD with chloroquine treatment and in CD38−/− mice under both ND and WD. Rap treatment reversed this Nlrp3 inflammasome formation almost to the normal level (Figure 5, B and D).
Figure 5.

Nod-like receptor family pyrin domain containing 3 (Nlrp3) inflammasome formation in the coronary arterial wall of CD38+/+ and CD38−/− mice on the normal diet (ND) or Western diet (WD). A: Representative fluorescent confocal microscope images depict the effects of chloroquine (Chl) and rapamycin (Rap) on the colocalization of Nlrp3 with the apoptosis associated speck-like protein (Asc) in the mouse coronary arterial wall. B: The fold changes in the Pearson correlation coefficient (PCC) for the colocalization of Nlrp3 with Asc. C: Representative fluorescent confocal microscope images depict the effects of Chl and Rap on the colocalization of Nlrp3 with caspase-1 in the mouse coronary arterial wall. D: Summarized data show the fold changes in the Pearson correlation coefficient (PCC) for the colocalization of Nlrp3 with caspase-1. Confocal fluorescence microscopy coronary arterial wall images: representative magnified form of the coronary arterial wall located in the corner (insets in A and C). Data are expressed as means ± SEM. n = 5. ∗P < 0.05 versus ND CD38+/+ vehicle (Vehl groups); †P < 0.05 versus Vehl groups. Original magnification: ×400 (A and C, main images); ×300 (insets).
Enhanced Nlrp3 Inflammasome Activation in the Coronary Arterial Wall of CD38−/− Mice
To determine the Nlpr3 inflammasome activation in the arterial wall, FLICA, which is a specific green fluorescent probe for binding the active form of caspase-1, was first detected. Confocal microscopy demonstrated increased caspase-1 activity (green FLICA fluorescence) only in the coronary arterial wall of CD38+/+ mice receiving chloroquine and WD, which was colocalized with α-SMA (Figure 6A). This increased colocalization of FLICA with α-SMA was even observed in the arterial wall of CD38−/− mice on the ND. In these CD38−/− mice, Rap treatment almost completely blocked the colocalization of FLICA with α-SMA regardless of whether they were on the ND and WD or receiving chloroquine. The FLICA staining as shown by the colocalization of FLICA and α-SMA under confocal microscopy is summarized in Figure 6B. FLICA staining in the coronary arterial wall was significantly higher in CD38+/+ mice receiving the WD with chloroquine treatment and in CD38−/− mice under both ND and WD. When these mice were treated with Rap, increased FLICA staining was completely prevented.
Figure 6.
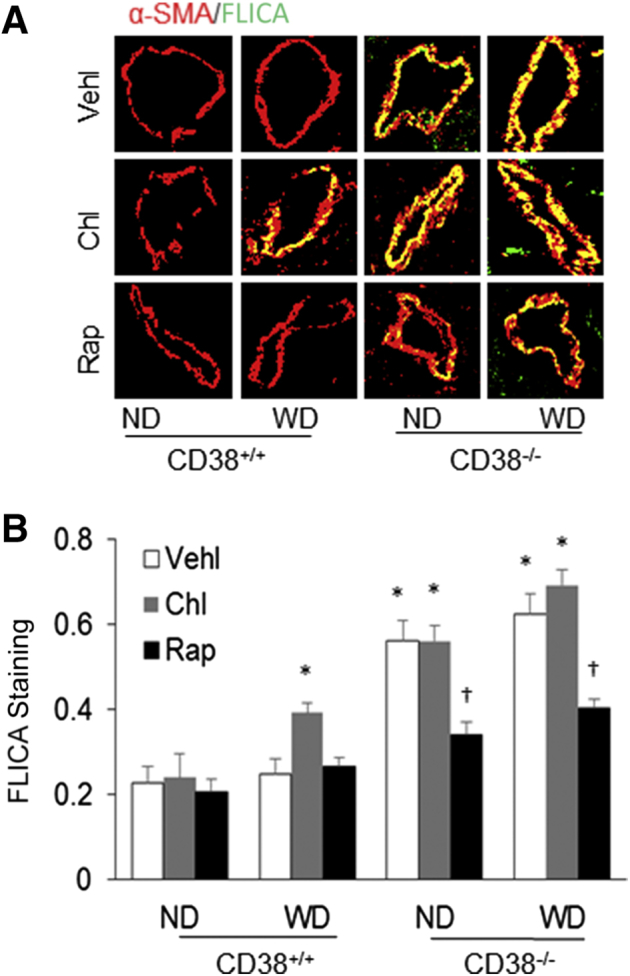
Nod-like receptor family pyrin domain containing 3 (Nlrp3) inflammasome activation in the coronary arterial wall of CD38+/+ and CD38−/− mice on the normal diet (ND) or Western diet (WD). A: Representative confocal fluorescence images show colocalization of active caspase-1 probe fluorescent-labeled inhibitor of caspases (FLICA) (green) with the α-smooth muscle actin (α-SMA) marker (red) in the mouse coronary arterial wall. B: The yellow dots or patches indicating the colocalization of FLICA with α-SMA are quantitated to show the relative caspase-1 activity. Data are expressed as means ± SEM. n = 5. ∗P < 0.05 versus ND CD38+/+ vehicle (Vehl) groups; †P < 0.05 versus Vehl groups. Original magnification, ×300.
By using immunohistochemical analysis, IL-1β levels were also detected in the arterial wall of both mouse strains under different treatments. Corresponding to the changes in FLICA shown above, increased IL-1β was mainly observed in the coronary arterial wall of CD38+/+ mice receiving chloroquine and the WD. The IL-1β levels were also increased in the arterial wall of CD38−/− mice on the ND, but chloroquine treatment did not further increase IL-1β levels in these CD38−/− mice. However, Rap treatment substantially reduced or attenuated IL-1β increase under these conditions (Figure 7A). The quantitation of the positive staining areas of IL-1β in the coronary arterial wall showed that IL-1β in the coronary arterial wall significantly increased in CD38+/+ mice receiving the WD with chloroquine treatment and in CD38−/− mice under both ND and WD or receiving chloroquine. When these mice were treated with Rap, the increase in IL-1β in the coronary arterial wall was significantly blocked (Figure 7B).
Figure 7.
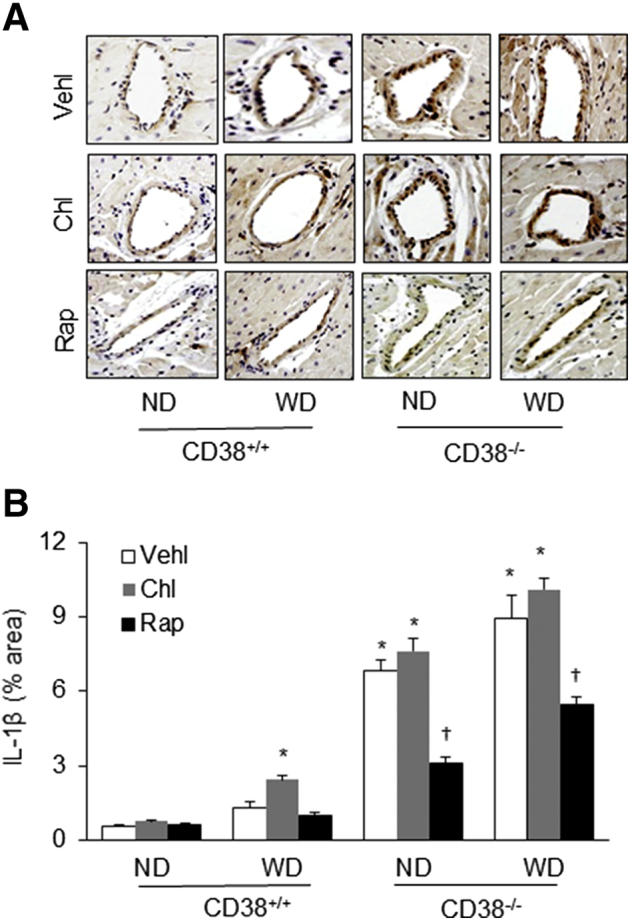
Immunohistochemical analysis of IL-1β levels in the arterial wall of CD3+/+ and CD38−/− mice on the normal diet (ND) or Western diet (WD). A: Representative immunohistochemical staining microphotographs show IL-1β levels in the coronary arterial wall of mice receiving different treatments. B: The area percentage of IL-1β positive staining in the coronary arterial wall. Data are expressed as means ± SEM. n = 5. ∗P < 0.05 versus ND CD38+/+ vehicle (Vehl) groups; †P < 0.05 versus Vehl groups. Original magnification, ×300. Chl, chloroquine; Rap, rapamycin.
Medial Thickening in the Coronary Arterial Wall of CD38−/− Mice
Morphologic examinations confirmed that the media of the coronary arterial wall were markedly thicker in CD38+/+ mice on the WD receiving chloroquine and in CD38−/− mice on the WD or receiving chloroquine treatment. Rap treatment was found to block this medial thickening induced by CD38 gene deletion with WD and by disruption of lysosome function with chloroquine (Figure 8A). The media/lumen ratio calculation in the coronary arterial wall demonstrated that the thickening of the coronary arterial wall was significantly enhanced in CD38+/+ mice with WD receiving chloroquine treatment and in CD38−/− mice under the WD or receiving chloroquine. When these mice were treated with Rap, the coronary arterial medial thickening was almost completely suppressed (Figure 8B).
Figure 8.
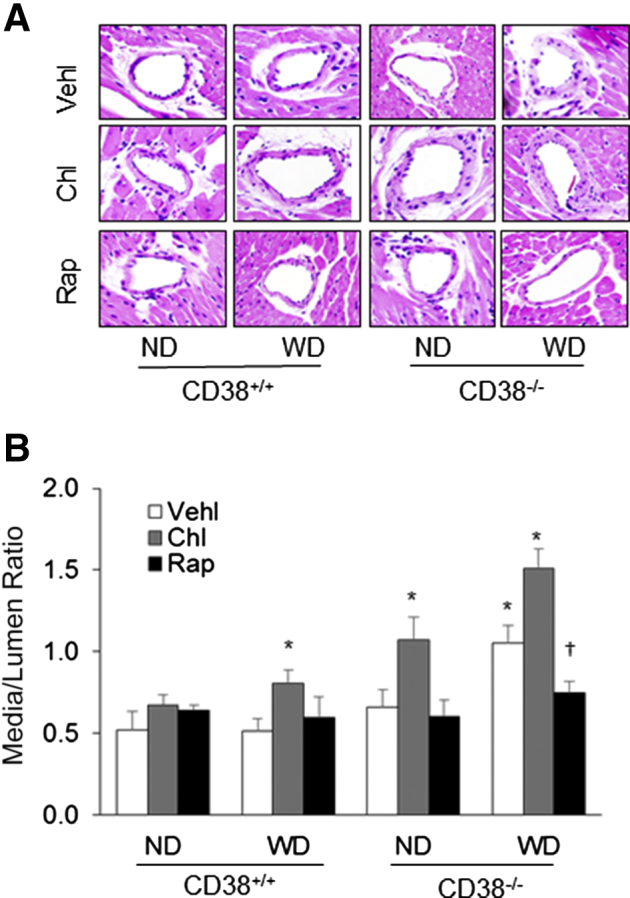
Morphologic examination of the coronary arterial wall in CD38+/+ and CD38−/− mice on the normal diet (ND) or Western diet (WD). A: Representative hematoxylin and eosin staining of the coronary arteries from CD38+/+ and CD38−/− mice showing potential medial thickening. B: Summary data of measured media/lumen ratio of the coronary arteries from mice with different treatments. Data are expressed as means ± SEM. n = 5. ∗P < 0.05 versus ND CD38+/+ vehicle (Vehl) groups; †P < 0.05 versus Vehl groups. Original magnification, ×300. Chl, chloroquine; Rap, rapamycin.
Discussion
The present study demonstrated that on proatherogenic stimuli, autophagy inhibition and associated lysosome dysfunction induced the formation and activation of Nlrp3 inflammasomes both in vitro and in vivo. In cultured CASMCs, a proatherogenic stimulus, 7-Ket, induced the formation and activation of the Nlrp3 inflammasome, which increased caspase-1 activity and IL-1β production. However, Rap, a lysosome function and autophagy enhancer, decreased 7-Ket–induced responses via enhancing activation of autophagy function. Moreover, inhibition of autophagosome formation by Spa-1 and blockade of lysosome function by Baf increased 7-Ket–induced formation and activation of Nlrp3 inflammasomes in CASMCs and the coronary arterial wall from mice. In animal experiments, deletion of CD38 gene along with the WD resulted in the increased inflammasome formation and thereby enhanced caspase-1 activity and IL-1β production in the coronary arterial wall. Similarly, chloroquine, a water-soluble lysosome function inhibitor, also induced the formation and activation of the Nlrp3 inflammasome in the coronary arterial wall. Correspondingly, Rap almost completely blocked the formation and activation of the Nlrp3 inflammasomes induced by the WD. These changes induced by interventions of lysosome function or autophagic process were accompanied by alterations of coronary medial thickness, with enhanced thickening during Nlrp3 inflammasome activation that was suppressed by improvement of lysosome function and reduction of Nlrp3 inflammasome activation. These results strongly suggest that the formation and activation of Nlrp3 inflammasomes are tightly regulated by autophagy maturation and lysosome function in CASMCs and that autophagy deficiency and associated lysosome dysfunction may be contributing factors to coronary arterial inflammation and medial thickening on proatherogenic stimulations.
The literature cites that the activation of Nlrp3 inflammasome may trigger important mechanism in response to atherogenic stimuli, resulting in vascular inflammation that leads to atherosclerosis.32, 35, 36 On the basis of previous reports, 7-Ket was used as a proatherogenic stimulus that was reported to activate this inflammasome in endothelial cells through different mechanisms.38 In particular, 7-Ket as a soluble proatherogenic stimulus may mainly activate the Nlrp3 inflammasome via redox signaling pathway.39 In the present study, it was first determined whether CD38/ADP-ribosylcyclase signaling that regulates autophagy and associated lysosome function are involved in 7-Ket–induced Nlrp3 inflammasome formation and activation in CASMCs. CD38 gene deletion significantly increased the Nlrp3 inflammasome formation and activation, which was enhanced by 7-Ket stimulation. To our knowledge, this is the first report showing that deletion of the CD38 gene, a lysosome function regulator in CASMCs, leads to Nlrp3 inflammasome activation. Previously, our laboratory reported that genetic deficiency of CD38 results in the phenotypic switching of coronary arterial myocytes into atherogenic status, suggesting that the CD38-mediated signaling pathway plays important roles in the regulation of vascular function.40 It has been demonstrated that CD38 gene deletion suppressed Nrf2 activity via NOX4-dependent O2.− production, leading to dedifferentiation of coronary arterial myocytes.40 In another study, our group demonstrated that CD38 deficiency enhanced collagen I deposition and atherosclerosis along with increased LC3B, indicating autophagy dysfunction in mice fed with a fat-enriched WD.26 In regard to this, other studies also provide evidence of CD38 involvement in various inflammatory diseases,41, 42 which may be associated with its regulatory role in intracellular calcium and consequent inflammation.43 All these findings indicate that CD38 plays an important role in inflammation that can lead to vascular inflammatory disease, such as atherosclerosis. The results of the present study extend our understanding of CD38 involvement in vascular inflammation to the formation and activation of Nlrp3 inflammasomes. CD38 deficiency has been suggested to activate the Nlrp3 inflammasome, resulting in inflammatory and sclerotic changes in coronary arteries.
Next, the effects of autophagy and associated lysosome dysfunction attributable to CD38 deficiency and lysosome function suppression on Nlrp3 inflammasome activation were determined. Autophagy or the lysosome function facilitator Rap significantly decreased Nlrp3 inflammasome formation and activation in CASMCs on 7-Ket stimulation or CD38 gene deletion. These data suggest that a defect of lysosome function and impaired autophagy at the maturation step may contribute to the enhancement of Nlrp3 inflammasome activities. Although how the deficiency of autophagic flux induces activation of Nlrp3 inflammasomes was not explored. Previous studies have found that the frustrated lysosome or deranged autophagy may stimulate sterile inflammation by the production of damage-associated molecular patterns.44 It is well known that autophagy promotes the turnover of organelles via elimination of damaged and aged organelles. There are a variety of organelle stressors that activate the formation of Nlrp3 inflammasome complex, such as damaged mitochondria, phagosome-lysosome, and ER, thereby inducing production of IL-1β and IL-18.15, 16 The literature reports that internal or external danger signals cause the formation of Nlrp3 inflammasome complex via recruitment of Asc and caspase-1.4, 45 This inflammasome complex triggers the production of mature IL-1β and IL-18, thereby leading to an elevated inflammatory response. In addition to the production of bioactive ILs, caspase-1 is also considered to be crucial in tissue injury and organ or systemic diseases through its uncanonical effects.9, 46
Recent studies have revealed that the Nlrp3 inflammasome and its products, such as IL-1β and IL-18, are associated with human plaque vulnerability and atherogenesis.23, 24 In the present study, it was confirmed that Nlrp3 inflammasome activation was suppressed by enhancing autophagic flux in both isolated CASMCs and the coronary arterial wall, suggesting that autophagic regulation of the Nlrp3 inflammasome is also important in the control of vascular structural and functional integrity. If this autophagic regulation of the Nlrp3 inflammasome in vascular SMCs failed as shown by lysosome function inhibition by chloroquine or gene deletion of CD38, vascular injury occurs as shown in coronary medial thickening even without proatherogenic stimulations. Furthermore, improving lysosome function and autophagic flux by Rap treatment antagonized this effect. In previous studies, CD38 has been demonstrated to be involved in autophagy regulation by modulating intracellular Ca2+ signaling.25, 26 This CD38-mediated signaling pathway is critical for autophagy maturation, smooth muscle cell phenotype transition, and arterial wall thickening in CD38−/− mice. CD38 gene deletion causes defective autophagosome trafficking, which in turn impairs autophagic flux in CASMCs.27 Inhibition of lysosome function significantly enhanced Nlrp3 inflammasome formation and activation as shown by significantly increased caspase-1 activity and IL-1β levels (a product of inflammasome activation), which was associated with the deficiency of autophagic flux. To our knowledge, this represents the first experimental evidence showing that autophagy and associated lysosome function serve as a critical modulator in Nlrp3 inflammasome activation on proatherogenic stimuli, which importantly protects CASMCs and coronary arteries from the inflammatory injury.
The Nlrp3 inflammasome/IL-1β pathway has been reported to promote atherogenesis in humans and mice,47, 48 which was due to not only an increase in the inflammatory condition of the lesion10, 49 but also vascular remodeling. The present study demonstrated that disruption of autophagy due to pharmacologic inhibition or lysosome dysfunction induced Nlrp3 inflammasome activation and thereby led to coronary medial thickening, one of the typical pathologic changes during atherogenesis. It is assumed that this coronary medial thickening may also be associated with both inflammatory stimulation and uncanonical effects induced by Nlrp3 inflammasome activation. In this regard, the Nlrp3 inflammasome formation and activation have been reported to directly facilitate the activation of tightly controlled myofibroblasts, resulting in myofibroblast proliferation, differentiation, and transformation. Recent studies considered this uncanonical action of the Nlrp3 inflammasome formation together with its inflammatory response as a common pathological process mediating divergent fibrogenic diseases.50 This effect of Nlrp3 inflammasome formation and activation has also been reported in some other organs or tissues, such as its contribution to adverse cardiac remodeling,51 which may be attributed to the phenotype change events of resident cardiac fibroblasts. It has been reported that this phenotype change of smooth muscle cells during inflammasome activation is not dependent on IL-1β, IL-18, or even caspase-1, which is considered an uncanonical action of caspase-1 activity or inflammasome activation.52 It is possible that the coronary medial thickening found in the present study during Nlrp3 inflammasome activation is at least partially associated with arterial smooth muscle cell transdifferentiation or phenotype switching.
Many studies have demonstrated that atherosclerosis starts in early life and its prevalence and extent increase with age. The inflammasome is one of the changes associated with aging.53 Many damage-associated molecular patterns are elevated during aging and thus may trigger Nlrp3 inflammasome activation to produce age-related inflammation.54 The association of inflammasomes and autophagy also has relevance for the field of aging. Salminen et al55 reported that the aging process is associated with a deterioration in autophagic capacity, resulting in Nlrp3 activation. In fact, there is literature suggesting that impaired autophagy and lysosomal dysfunction in neurodegenerative disorders can fail to clear accumulated protein aggregates.56, 57 In the current study, we used the CD38−/− model to further confirm that lysosomal dysfunction is also an important factor in the aging process.
In summary, the present study demonstrated that lysosome dysfunction and autophagy derangement attributable to CD38 gene deletion and lysosomal V–ATPase inhibition resulted in Nlrp3 inflammasome formation and activation and consequent coronary arterial inflammation and remodeling. The results suggest that sufficient autophagy is essential for the control of Nlrp3 inflammasome activation, preventing medial thickening of coronary arteries in response to proatherogenic stimuli.
Acknowledgments
P.-L.L. designed the study; X.Y., O.B., and N.M. performed experiments; H.L. provided input on technical experiments and animal treatment; X.Y. and O.B. interpreted data; all the authors wrote and edited the manuscript and approved the final version.
Supplemental Data
References
- 1.Mariathasan S., Newton K., Monack D.M., Vucic D., French D.M., Lee W.P., Roose-Girma M., Erickson S., Dixit V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 2.Zhou R., Tardivel A., Thorens B., Choi I., Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 3.Dinarello C.A., Donath M.Y., Mandrup-Poulsen T. Role of IL-1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;17:314–321. doi: 10.1097/MED.0b013e32833bf6dc. [DOI] [PubMed] [Google Scholar]
- 4.Martinon F., Mayor A., Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 5.Neven B., Callebaut I., Prieur A.M., Feldmann J., Bodemer C., Lepore L., Derfalvi B., Benjaponpitak S., Vesely R., Sauvain M.J., Oertle S., Allen R., Morgan G., Borkhardt A., Hill C., Gardner-Medwin J., Fischer A., de Saint Basile G. Molecular basis of the spectral expression of CIAS1 mutations associated with phagocytic cell-mediated autoinflammatory disorders CINCA/NOMID, MWS, and FCU. Blood. 2004;103:2809–2815. doi: 10.1182/blood-2003-07-2531. [DOI] [PubMed] [Google Scholar]
- 6.Duewell P., Kono H., Rayner K.J., Sirois C.M., Vladimer G., Bauernfeind F.G., Abela G.S., Franchi L., Nunez G., Schnurr M., Espevik T., Lien E., Fitzgerald K.A., Rock K.L., Moore K.J., Wright S.D., Hornung V., Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mateeva V., Kadurina M. Cryopyrin-associated periodic syndrome caused by a novel mutation in the NLRP3 gene. Exp Dermatol. 2014;23:2. [Google Scholar]
- 8.Martinon F., Petrilli V., Mayor A., Tardivel A., Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 9.Abais J.M., Xia M., Zhang Y., Boini K.M., Li P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal. 2015;22:1111–1129. doi: 10.1089/ars.2014.5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia M., Boini K.M., Abais J.M., Xu M., Zhang Y., Li P.L. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am J Pathol. 2014;184:1617–1628. doi: 10.1016/j.ajpath.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 12.Feng Y., He D., Yao Z., Klionsky D.J. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lavandero S., Troncoso R., Rothermel B.A., Martinet W., Sadoshima J., Hill J.A. Cardiovascular autophagy: concepts, controversies, and perspectives. Autophagy. 2013;9:1455–1466. doi: 10.4161/auto.25969. [DOI] [PubMed] [Google Scholar]
- 14.Rubinsztein D.C., Codogno P., Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elliott E.I., Sutterwala F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 2015;265:35–52. doi: 10.1111/imr.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Latz E., Xiao T.S., Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jabir M.S., Hopkins L., Ritchie N.D., Ullah I., Bayes H.K., Li D., Tourlomousis P., Lupton A., Puleston D., Simon A.K., Bryant C., Evans T.J. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy. 2015;11:166–182. doi: 10.4161/15548627.2014.981915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamkanfi M., Kanneganti T.D., Van Damme P., Vanden Berghe T., Vanoverberghe I., Vandekerckhove J., Vandenabeele P., Gevaert K., Nunez G. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics. 2008;7:2350–2363. doi: 10.1074/mcp.M800132-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 20.Guo W., Sun Y., Liu W., Wu X., Guo L., Cai P., Shen Y., Shu Y., Gu Y., Xu Q. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. 2014;10:972–985. doi: 10.4161/auto.28374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martins J.D., Liberal J., Silva A., Ferreira I., Neves B.M., Cruz M.T. Autophagy and inflammasome interplay. DNA Cell Biol. 2015;34:274–281. doi: 10.1089/dna.2014.2752. [DOI] [PubMed] [Google Scholar]
- 22.Harris J., Hartman M., Roche C., Zeng S.G., O'Shea A., Sharp F.A., Lambe E.M., Creagh E.M., Golenbock D.T., Tschopp J., Kornfeld H., Fitzgerald K.A., Lavelle E.C. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286:9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paramel Varghese G., Folkersen L., Strawbridge R.J., Halvorsen B., Yndestad A., Ranheim T., Krohg-Sorensen K., Skjelland M., Espevik T., Aukrust P., Lengquist M., Hedin U., Jansson J.H., Fransen K., Hansson G.K., Eriksson P., Sirsjo A. NLRP3 inflammasome expression and activation in human atherosclerosis. J Am Heart Assoc. 2016;5:e003031. doi: 10.1161/JAHA.115.003031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi X., Xie W.L., Kong W.W., Chen D., Qu P. Expression of the NLRP3 inflammasome in carotid atherosclerosis. J Stroke Cerebrovasc Dis. 2015;24:2455–2466. doi: 10.1016/j.jstrokecerebrovasdis.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 25.Xiong J., Xia M., Xu M., Zhang Y., Abais J.M., Li G., Riebling C.R., Ritter J.K., Boini K.M., Li P.L. Autophagy maturation associated with CD38-mediated regulation of lysosome function in mouse glomerular podocytes. J Cell Mol Med. 2013;17:1598–1607. doi: 10.1111/jcmm.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bao J.X., Zhang Q.F., Wang M., Xia M., Boini K.M., Gulbins E., Zhang Y., Li P.L. Implication of CD38 gene in autophagic degradation of collagen I in mouse coronary arterial myocytes. Front Biosci (Landmark Ed) 2017;22:558–569. doi: 10.2741/4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y., Xu M., Xia M., Li X., Boini K.M., Wang M., Gulbins E., Ratz P.H., Li P.L. Defective autophagosome trafficking contributes to impaired autophagic flux in coronary arterial myocytes lacking CD38 gene. Cardiovasc Res. 2014;102:68–78. doi: 10.1093/cvr/cvu011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Committee for the Update of the Guide for the Care and Use of Laboratory Animals; National Research Council . National Academies Press; Washington, DC: 2011. Guide for the Care and Use of Laboratory Animals: Eighth Edition. [Google Scholar]
- 29.Adhikari N., Shekar K.C., Staggs R., Win Z., Steucke K., Lin Y.W., Wei L.N., Alford P., Hall J.L. Guidelines for the isolation and characterization of murine vascular smooth muscle cells. A report from the International Society of Cardiovascular Translational Research. J Cardiovasc Transl Res. 2015;8:158–163. doi: 10.1007/s12265-015-9616-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu M., Zhang Y., Xia M., Li X.X., Ritter J.K., Zhang F., Li P.L. NAD(P)H oxidase-dependent intracellular and extracellular O2*- production in coronary arterial myocytes from CD38 knockout mice. Free Radic Biol Med. 2012;52:357–365. doi: 10.1016/j.freeradbiomed.2011.10.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan X., Wang L., Bhat O.M., Lohner H., Li P.L. Differential effects of short chain fatty acids on endothelial Nlrp3 inflammasome activation and neointima formation: antioxidant action of butyrate. Redox Biol. 2018;16:21–31. doi: 10.1016/j.redox.2018.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abraham N.G., Sodhi K., Silvis A.M., Vanella L., Favero G., Rezzani R., Lee C., Zeldin D.C., Schwartzman M.L. CYP2J2 targeting to endothelial cells attenuates adiposity and vascular dysfunction in mice fed a high-fat diet by reprogramming adipocyte phenotype. Hypertension. 2014;64:1352–1361. doi: 10.1161/HYPERTENSIONAHA.114.03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia M., Zhang C., Boini K.M., Thacker A.M., Li P.L. Membrane raft-lysosome redox signalling platforms in coronary endothelial dysfunction induced by adipokine visfatin. Cardiovasc Res. 2011;89:401–409. doi: 10.1093/cvr/cvq286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang C., Yi F., Xia M., Boini K.M., Zhu Q., Laperle L.A., Abais J.M., Brimson C.A., Li P.L. NMDA receptor-mediated activation of NADPH oxidase and glomerulosclerosis in hyperhomocysteinemic rats. Antioxid Redox Signal. 2010;13:975–986. doi: 10.1089/ars.2010.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y., Pitzer A.L., Li X., Li P.L., Wang L., Zhang Y. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial junction disruption: role of HMGB1. J Cell Mol Med. 2015;19:2715–2727. doi: 10.1111/jcmm.12657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang R.Z., Qiu H., Wang N., Long F.L., Mao D.W. Effect of Rheum palmatum L. on NF-kappaB signaling pathway of mice with acute liver failure. Asian Pac J Trop Med. 2015;8:841–847. doi: 10.1016/j.apjtm.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 37.Liu J.L., Bishop S.P., Overbeck H.W. Morphometric evidence for non-pressure-related arterial wall thickening in hypertension. Circ Res. 1988;62:1001–1010. doi: 10.1161/01.res.62.5.1001. [DOI] [PubMed] [Google Scholar]
- 38.Li X., Zhang Y., Xia M., Gulbins E., Boini K.M., Li P.L. Activation of Nlrp3 inflammasomes enhances macrophage lipid-deposition and migration: implication of a novel role of inflammasome in atherogenesis. PLoS One. 2014;9:e87552. doi: 10.1371/journal.pone.0087552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajamaki K., Lappalainen J., Oorni K., Valimaki E., Matikainen S., Kovanen P.T., Eklund K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765. doi: 10.1371/journal.pone.0011765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu M., Li X.X., Wang L., Wang M., Zhang Y., Li P.L. Contribution of Nrf2 to atherogenic phenotype switching of coronary arterial smooth muscle cells lacking CD38 gene. Cell Physiol Biochem. 2015;37:432–444. doi: 10.1159/000430366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guedes A.G., Deshpande D.A., Dileepan M., Walseth T.F., Panettieri R.A., Jr., Subramanian S., Kannan M.S. CD38 and airway hyper-responsiveness: studies on human airway smooth muscle cells and mouse models. Can J Physiol Pharmacol. 2015;93:145–153. doi: 10.1139/cjpp-2014-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider M., Schumacher V., Lischke T., Lucke K., Meyer-Schwesinger C., Velden J., Koch-Nolte F., Mittrucker H.W. CD38 is expressed on inflammatory cells of the intestine and promotes intestinal inflammation. PLoS One. 2015;10:e0126007. doi: 10.1371/journal.pone.0126007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deshpande D.A., Guedes A.G., Lund F.E., Subramanian S., Walseth T.F., Kannan M.S. CD38 in the pathogenesis of allergic airway disease: potential therapeutic targets. Pharmacol Ther. 2017;172:116–126. doi: 10.1016/j.pharmthera.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Álvarez K., Vasquez G. Damage-associated molecular patterns and their role as initiators of inflammatory and auto-immune signals in systemic lupus erythematosus. Int Rev Immunol. 2017;36:259–270. doi: 10.1080/08830185.2017.1365146. [DOI] [PubMed] [Google Scholar]
- 45.Schroder K., Zhou R.B., Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327:296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 46.Denes A., Lopez-Castejon G., Brough D. Caspase-1: is IL-1 just the tip of the ICEberg? Cell Death Dis. 2012;3:e338. doi: 10.1038/cddis.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fearon W.F., Fearon D.T. Inflammation and cardiovascular disease: role of the interleukin-1 receptor antagonist. Circulation. 2008;117:2577–2579. doi: 10.1161/CIRCULATIONAHA.108.772491. [DOI] [PubMed] [Google Scholar]
- 48.Fu X.D., Huang X.W., Li P., Chen W.Y., Xia M. 7-Ketocholesterol inhibits isocitrate dehydrogenase 2 expression and impairs endothelial function via microRNA-144. Free Radic Biol Med. 2014;71:1–15. doi: 10.1016/j.freeradbiomed.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 49.Boini K.M., Xia M., Abais J.M., Li G.B., Pitzer A.L., Gehr T.W.B., Zhang Y., Li P.L. Activation of inflammasomes in podocyte injury of mice on the high fat diet: effects of ASC gene deletion and silencing. Biochim Biophys Acta. 2014;1843:836–845. doi: 10.1016/j.bbamcr.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Artlett C.M., Thacker J.D. Molecular activation of the NLRP3 Inflammasome in fibrosis: common threads linking divergent fibrogenic diseases. Antioxid Redox Signal. 2015;22:1162–1175. doi: 10.1089/ars.2014.6148. [DOI] [PubMed] [Google Scholar]
- 51.Toldo S., Mezzaroma E., Mauro A.G., Salloum F., Van Tassell B.W., Abbate A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid Redox Signal. 2015;22:1146–1161. doi: 10.1089/ars.2014.5989. [DOI] [PubMed] [Google Scholar]
- 52.Bracey N.A., Duff H.J., Muruve D.A. Hierarchical regulation of wound healing by NOD-like receptors in cardiovascular disease. Antioxid Redox Signal. 2015;22:1176–1187. doi: 10.1089/ars.2014.6125. [DOI] [PubMed] [Google Scholar]
- 53.Franceschi C., Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69:S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 54.Cutler R.G., Kelly J., Storie K., Pedersen W.A., Tammara A., Hatanpaa K., Troncoso J.C., Mattson M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salminen A., Kaarniranta K., Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) 2012;4:166–175. doi: 10.18632/aging.100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lynch-Day M.A., Mao K., Wang K., Zhao M.T., Klionsky D.J. The role of autophagy in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009357. doi: 10.1101/cshperspect.a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nixon R.A. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.