

Summary
Objective
Exposure to chemical warfare nerve agents (CWNAs), such as soman (GD), can induce status epilepticus (SE) that becomes refractory to benzodiazepines when treatment is delayed, leading to increased risk of epileptogenesis, severe neuropathology, and long‐term behavioral and cognitive deficits. Rodent models, widely used to evaluate novel medical countermeasures (MCMs) against CWNA exposure, normally express plasma carboxylesterase, an enzyme involved in the metabolism of certain organophosphorus compounds. To better predict the efficacy of novel MCMs against CWNA exposure in human casualties, it is crucial to use appropriate animal models that mirror the human condition. We present a comprehensive characterization of the seizurogenic, epileptogenic, and neuropathologic effects of GD exposure with delayed anticonvulsant treatment in the plasma carboxylesterase knockout (ES1−/−) mouse.
Methods
Electroencephalography (EEG) electrode‐implanted ES1−/− and wild‐type (C57BL/6) mice were exposed to various seizure‐inducing doses of GD, treated with atropine sulfate and the oxime HI‐6 at 1 minute after exposure, and administered midazolam at 15‐30 minutes following the onset of seizure activity. The latency of acute seizure onset and spontaneous recurrent seizures (SRS) was assessed, as were changes in EEG power spectra. At 2 weeks after GD exposure, neurodegeneration and neuroinflammation were assessed.
Results
GD‐exposed ES1−/− mice displayed a dose‐dependent response in seizure severity. Only ES1−/− mice exposed to the highest tested dose of GD developed SE, subchronic alterations in EEG power spectra, and SRS. Degree of neuronal cell loss and neuroinflammation were dose‐dependent; no significant neuropathology was observed in C57BL/6 mice or ES1−/− mice exposed to lower GD doses.
Significance
The US Food and Drug Administration (FDA) animal rule requires the use of relevant animal models for the advancement of MCMs against CWNAs. We present evidence that argues for the use of the ES1−/− mouse model to screen anticonvulsant, antiepileptic, and/or neuroprotective drugs against GD‐induced toxicity, as well as to identify mechanisms of GD‐induced epileptogenesis.
Keywords: chemical warfare nerve agents, electroencephalography, ES1−/− mice, organophosphorus, spontaneous recurrent seizures
Key Points.
Carboxylesterase knockout (ES1−/−) mice are more susceptible than wild‐type C57BL/6 mice to the lethal effects of soman
Spontaneous recurrent seizures develop in ES1−/− mice following soman‐induced status epilepticus when midazolam treatment is delayed
Severe neuronal loss and neuroinflammation are present in ES1−/− mice 2 weeks following soman‐induced status epilepticus
ES1−/− mice show acute and subchronic electroencephalography (EEG) power spectra changes after soman‐induced status epilepticus and midazolam treatment is delayed
1. INTRODUCTION
Exposure to chemical warfare nerve agents (CWNAs) can induce severe and prolonged seizures (ie, status epilepticus; SE) as a consequence of cholinergic hyperactivation in limbic and cortical structures.1 Currently, medical countermeasures (MCMs) against CWNA exposure consist of a muscarinic ACh receptor (mACh) antagonist (eg, atropine sulfate) to reduce the severity of peripheral toxic signs, an oxime to reactivate inhibited acetylcholinesterase (eg, 2‐PAM, HI‐6), and a benzodiazepine (eg, diazepam or midazolam [MDZ]) for the treatment of seizures.2 In rodent models of cholinergic‐induced SE, benzodiazepines are effective at terminating seizures and increasing survival when administered promptly; delayed treatment leads to SE that is refractory to benzodiazepine treatment,3 and results in lasting neuroanatomic changes, development of spontaneous recurrent seizures (SRS), and long‐term cognitive/behavioral impairments.4, 5, 6 Rodent models of treatment‐resistant temporal medial lobe epilepsy, caused by an initial acute hippocampal lesion, also echo the chronic consequences (ie, robust neuroinflammation, neuronal network rewiring, and recurrent spontaneous seizures, among others) of uncontrolled seizure activity.7 In all, pharmacoresistance is an unsolved therapeutic challenge as there is currently no approved treatment for CWNA‐induced refractory SE.
Prolonged seizure activity increases synaptic N‐methyl‐d‐aspartate (NMDA) receptors that increase calcium influx and can induce cell death.3, 8, 9 Release of pro‐inflammatory mediators from activated microglia and astrocytes may promote hyperexcitability and contribute to cell loss and synaptic reorganization,10 although the mechanisms by which changes in neuronal circuitry contribute to SRS development are unknown.
The advancement of MCMs against the toxic effects of CWNAs, such as soman (GD), is highly dependent on the use of an appropriate animal model that closely mirrors the level of toxicity observed in human victims of CWNA exposure. Because delayed anticonvulsant administration is highly plausible in a mass casualty scenario where MCMs against CWNA exposure are not readily accessible, the need for research into more effective treatments to counteract benzodiazepine pharmacoresistance is urgent. Rodents are often used for drug screening against CWNA‐induced toxicity, albeit with a major caveat: mice and rats have plasma carboxylesterase (CaE) activity, which scavenges many organophosphorus compounds, rendering the animal more resistant to GD and other organiphosphates.11 In contrast, humans have undetectable plasma CaE activity. Pretreating mice with the CaE inhibitor cresylbenzodioxaphosphorin oxide (CBDP) prior to exposure potentiates GD toxicity but inhibits other esterases that may confound results.12, 13 ES1 gene knockout (ES1−/−) mice were developed to specifically lack plasma CaE while containing normal CaE activity levels in organs.11 ES1−/− mice exhibit lower median lethal doses (LD50) of parathion, chlorpyrifos14 and the less toxic analogue of GD, soman coumarin, when compared to wild‐type mice.11 The present work focused on the characterization of the seizuregenic, epileptogenic, and neuropathologic effects of GD exposure in ES1−/− mice, which may be a useful model to identify improved MCM against CWNA exposure.
2. METHODS
2.1. Animals
Male ES1−/− and their background strain (C57BL/6) mice were obtained from the United States Army Medical Research Institute of Chemical Defense (USAMRICD) breeding colony at 8‐9 weeks of age (19‐22 g) and housed on a 12 hour:12 hour light‐dark cycle with lights on at 0600. Food and water were available ad libitum. Mice were weighed daily. The experimental protocol was approved by the Institute Animal Care and Use Committee at USAMRICD, and all procedures were conducted in accordance with the principles stated in the Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act of 1966 (P.L. 89‐544), as amended.
2.2. Median lethal dose (LD50) of GD in male ES1−/− mice
To determine the LD50 of GD male, ES1−/− mice were exposed subcutaneously (sc) to GD (0.5 mL/kg) in a stage‐wise manner using the methods described by Feder et al15 GD (pinacolyl methylphosphonofluoridate) was obtained from the Edgewood Chemical Biological Center. The LD50 was calculated by applying a probit regression analysis using the maximum likelihood procedure to the combined data from all stages. The 95% confidence intervals (CIs) for the LD50 were calculated using the delta method.
2.3. Surgeries and EEG recordings
Mice were implanted under 2%‐5% isoflurane with ETA‐F10 telemetry transmitters from Data Sciences International (DSI) as described in Lundt et al16 Transmitters were implanted (sc) with wires wrapped around 2 cortical stainless steel screw electrodes placed at 1.5 mm right of the midline, and 1.5 mm anterior, and 3.0 mm posterior to bregma. Buprenex SR was administered (0.5 mg/kg, sc) to minimize pain. Data were recorded using Dataquest Art Acquisition software (DSI) and digitized at 250 Hz.
2.4. Soman exposures and treatments
After 2 weeks of recovery from surgery, ES1−/− mice (n = 6‐8 per group) were exposed to 23, 46, or 82 μg/kg GD (sc) or saline; C57BL/6 mice were exposed to 82 or 285 μg/kg GD (sc) or saline (n = 3‐7 per group). One minute after exposure, mice received an admix (ip) of atropine sulfate (4 mg/kg; Sigma Aldrich), and the bispyridinium oxime HI‐6 (50 mg/kg; Sigma Aldrich). Midazolam (MDZ; 5 mg/kg, ip) was administered 15 minutes after GD‐induced seizure or 20 minutes after saline exposure.
2.5. Behavioral seizure
Noldus Observer XT was used to input behavioral signs of toxicity. The severity of behavioral seizures was scored using a modified Racine17 scale with the following stages: 0, no abnormality; 1, oral movements; 2, head nodding and/or tremors; 3, forelimb clonus; 4, rearing with convulsions; and 5, rearing and falling with convulsions.
2.6. EEG scoring and power spectral analysis
Electroencephalography data were collected continuously from 3 days before GD exposure to 14 days after exposure. Data analysis was done using customized MATLAB algorithms to assess seizure activity defined as rhythmic high‐amplitude spikes lasting 10 seconds or longer (details in Appendix S1).4, 18
2.7. Brain tissue collection and immunohistochemistry
Two weeks after exposure, mice were injected with sodium pentobarbital (75 mg/kg, ip, Fatal Plus; Patterson Veterinary), and perfused with 0.1 mol/L phosphate buffer (PB), followed by 4% paraformaldehyde in 0.1 mol/L PB (FD NeuroTechnologies). Heparin sodium injection (0.1 mL per L of 1000 Units per mL; Henry Schein Animal Health) was added to the PB used in perfusion. Brains were removed and post‐fixed for 6 hours in the same fixative at 4°C. After cryoprotection in 0.1 mol/L PB containing 20% sucrose for at least 72 hours at 4°C, brains were rapidly frozen and stored in a freezer at −80°C until sectioning. Serial cryostat sections (30 μm) were cut coronally through the cerebrum, approximately from bregma 2.80 mm to −4.84 mm. Every first to eighth section of each series of 8 sections (interval: 260 μm) was collected separately. Sections were stored free‐floating in FD section storage solution (FD NeuroTechnologies) at −20°C before immunostaining (details in Appendix S1).19
2.8. Cell counts
Immunostained slides were scanned using an Olympus BX16IVS microscope with a Pike F‐505 camera (Allied Vision). Surveyor (Objective Imaging Inc.) was used to capture images, and Image‐Pro Plus (Media Cybernetics, Inc.) was used to obtain counts of cells immunoreactive for the markers neuronal nuclear protein (NeuN), glial fibrillary acidic protein (GFAP), and glutamic acid decarboxylase (GAD67) in the thalamus, hippocampus, basolateral amygdala, and/or piriform cortex at bregma −1.94. Brain regions were traced for a specific region of interest using anatomic landmarks and then cell density calculated; 3 replicates were scored and averaged. For analysis of highly dense NeuN‐positive (NeuN+) neurons in the CA1 region of the hippocampus, stereology using Stereo Investigator software (MBF Bioscience) was performed in sections (30 μm each; ∼5 sections per mouse) using the optical fractionator method.20 The counting frame was 40 × 40 μm, and the counting grid was 200 × 200 μm. The dissector height was 20 μm.
2.9. Microglia morphology analysis
Morphologic changes in cells immunoreactive for ionized calcium‐binding adapter molecule 1 (Iba1), indicative of microglia activation, were assessed by estimations of cell body to cell size ratio.21, 22 Using the ImageJ software (National Institutes of Health), a modification of previously described methods was used (details in Appendix S1).21
2.10. Data analysis
A Kruskal‐Wallis analysis of variance (ANOVA) test was performed on Racine scores to determine the main effect of GD on toxicity at each time point, followed by multiple comparisons to control group using the Dunn's method. The effect of GD dose on seizure onset was determined by a one‐way ANOVA, followed by Tukey's test. A 2‐way repeated‐measures ANOVA was used to determine the main effect of GD on body weight and on changes in EEG power spectra across time, followed by a Dunnett's test. A one‐way ANOVA was used to determine main effect of GD on NeuN, GAD67, GFAP, and Iba1 immunoreactivity, followed by a Dunnett's test. Differences were considered statistically significant when P < 0.05.
3. RESULTS
3.1. Median lethal dose of GD in male ES1−/− mice
Data from our dose‐lethality response study show that the estimated LD50 of male ES1−/− mice is 19.2 μg/kg (95% CI 18.0‐20.5 μg/kg) and the slope of the GD dose‐response curve is 74.8 (Figure 1). In comparison, the LD50 of GD in C57BL/6 mice is 83 μg/kg,23 approximately 4‐fold higher than in ES1−/− mice.
Figure 1.
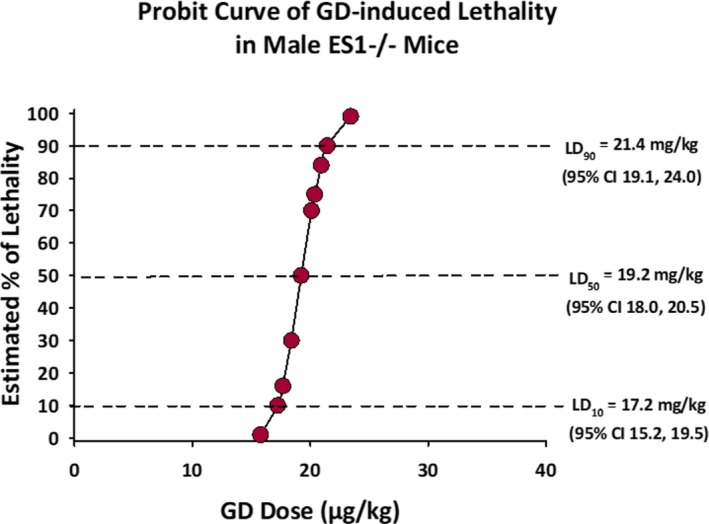
Dose‐response curve for GD‐induced lethality in adult male ES1−/− mice. Each point in the graph represents the average estimated percent of lethality for adult male ES1−/− mice exposed to a particular dose of GD. At 19.2 μg/kg (95% CI 18.0‐20.5) the LD 50 of GD of male ES1−/− mice was 4 times lower than that of C57BL/6 mice reported in the literature. The LD 10 and LD 90 are also shown in the graph. CI, confidence interval; LD, lethal dose
3.2. Acute EEG and behavioral seizures, and development of spontaneous recurrent seizures following subcutaneous GD exposure
Male ES1−/− mice exposed to GD presented with behavioral and EEG seizure activity. Only the 82 μg/kg dose of GD (approximately 4 LD50) resulted in a robust change in EEG amplitude that developed into SE and continued sporadically for at least 48 hours following the initial toxic insult (Figure S1). Exposure to 23 and 46 μg/kg GD (approximately 1 and 2 LD50, respectively) resulted in intermittent seizure activity that did not progress into SE. GD exposure induced dose‐dependent signs of cholinergic toxicity consisting of ataxia, head bobs, and body tremors within 10 minutes of exposure (Figure 2A). Maximum median severity of stage 5 behavioral seizures persisted in mice exposed to 82 μg/kg, whereas mice exposed to 23 or 46 μg/kg GD showed a maximum median of 1.5 and 4, respectively. A main effect of GD dose was detected on the latency to seizure onset [F (1,19) = 12.9, P < 0.001]; mice exposed to 23 μg/kg presented seizure at an average of 9.4 ± 2.1 minutes after GD exposure, which was significantly longer compared to the 46 μg/kg (4.7 ± 1.6 minutes; P = 0.001) and 82 μg/kg (4.4 ± 2.6 minutes; P < 0.001) groups (Figure 2B). Mice that received 82 μg/kg GD presented with SE, with a mean seizure activity duration during the first 24 hours following GD exposure of 555.4 ± 325. 0 minutes (mean ± SD). This dose led to the development of SRSs, characterized by hypersynchronous discharges of high amplitude with duration <1 minute in 5 of 7 ES1−/− mice (Figure 2C). An average number of 6.9 ± 6.6 SRSs were detected during the 14 days following GD‐induced SE with an average latency of 6.1 ± 1.4 days.
Figure 2.
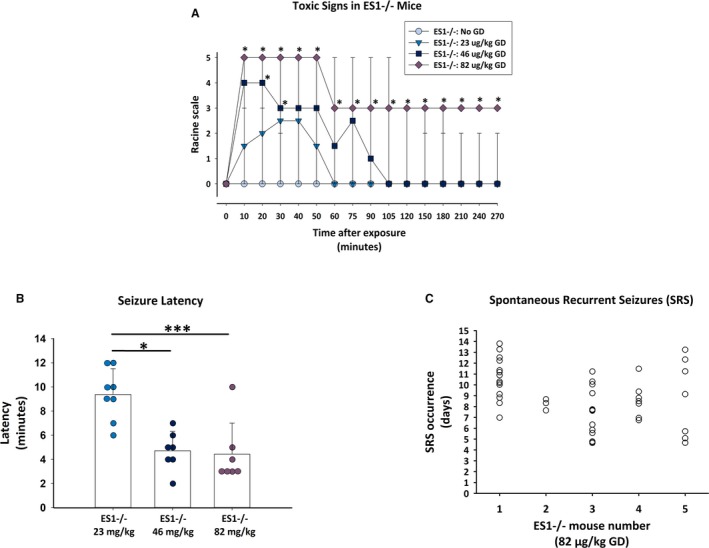
Behavioral and electroencephalography (EEG) seizure activity from male ES1−/− mice exposed to various doses of GD. A, Mice exposed to GD and treated with atropine, HI‐6 at 1 minute after exposure and midazolam (MDZ) 15‐30 minutes after seizure onset showed signs of cholinergic toxicity within minutes; the severity of toxic signs scored according to a modified Racine scale was dose‐dependent, with the 82 μg/kg dose resulting in the most severe signs in ES1−/− mice compared to no GD control; data graphed represents median as well as 90th and 10th percentile. B, In ES1−/− mice, the latency of seizure activity was dependent on the administered dose of GD, with the 82 μg/kg dose resulting in the shortest latency to seizure onset. C, Onset and frequency of spontaneous recurrent seizures (each open circle is an event for one mouse) in ES1−/− mice exposed to ~4 × LD 50 (82 μg/kg GD); a total of 5 of 7 ES1−/− mice exposed to this dose of GD developed chronic seizures during the 2 week period following GD‐induced status epilepticus and delayed MDZ treatment. Onset of spontaneous recurrent seizures (SRS) was an average of 6 days after exposure
In contrast to findings in ES1−/− mice, C57BL/6 mice exposed to 82 μg/kg had intermittent seizure activity, did not develop SE, and showed a maximum Racine score of 3, which was not significantly different from control animals (Figure S2). C57BL/6 mice exposed to 285 μg/kg GD (approximately 4 LD50 in this strain) presented high‐amplitude rhythmic EEG activity characteristic of SE that lasted only an average of 22.5 ± 8.5 minutes, whereas behavioral seizures lasted up to 270 minutes after exposure. Only 1 of 9 C57BL/6 mice exposed to the highest dose of GD and treated with MDZ survived to 24 hours after exposure, and none survived to 48 hours.
3.3. Full‐power spectral analysis during the acute and subchronic periods following exposure to GD
Full‐power spectral analysis was performed on EEG as described in de Araujo Furtado et al.18, 24 There was a main effect of GD exposure on the relative power of delta [F (2,84) = 7.35; P = 0.001], theta [F (2,84) = 9.84; P < 0.001], and gamma [F (2,84) = 5.44, P = 0.005] frequency bands of ES1−/− mice (Figure 3). ES1−/− mice exposed to 82 μg/kg GD show increased relative power of delta for up to 108 minutes after GD exposure (P ≤ 0.05; Figure 3A), theta from time of GD exposure up to euthanasia (P ≤ 0.05; Figure 3B), as well as a decrease in the power of gamma for up to 60 hours after GD exposure (P ≤ 0.05; Figure 3E), compared to no‐GD (control) mice. Conversely, ES1−/− mice exposed to 23 and 46 μg/kg GD showed decreased alpha power up to 36 hours after GD exposure, and increased gamma power for up to 24 hours after GD exposure, compared to control mice. There was no effect of GD exposure on the relative power of the beta band (Figure 3D).
Figure 3.
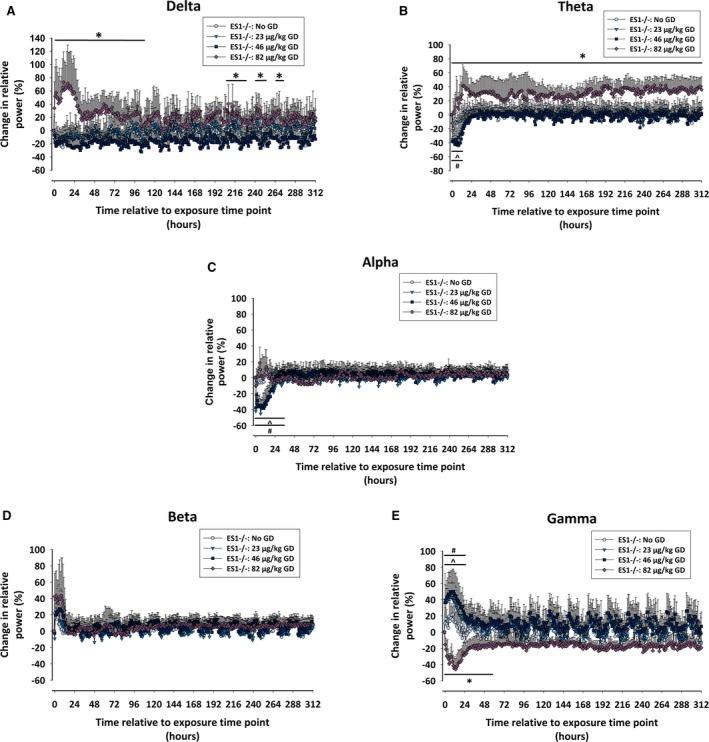
Effects of GD exposure on electroencephalography (EEG) power spectra of male ES1−/− mice. The percent of relative change from baseline in the power of (A) delta (0.1‐4 Hz), (B) theta (4.1‐8 Hz), (C) alpha (8.1‐12 Hz), (D) beta (12.1‐25 Hz), and (E) gamma (25.1‐50 Hz) EEG frequencies was calculated for ES1−/− mice exposed to GD and administered delayed midazolam treatment. Effect of GD exposure on individual power spectra throughout the duration of the study was determined by 2‐way ANOVA analysis, and the change in relative power for mice exposed to 23, 46, and 82 μg/kg GD were compared to values of control (No GD) animals. In ES1−/− mice exposed to 82 μg/kg GD the oscillations in gamma and beta power intensity that coincide with sleep/wake cycles disappear in comparison with the other mouse groups. Statistically significant differences between groups at particular time points are marked by a line drawn above or below the graphed data *P < 0.05, 82 μg/kg GD compared to No GD; ^P < 0.05; 46 μg/kg GD compared to No GD; #P < 0.05, 23 μg/kg GD compared to No GD
In C57BL/6 mice, GD exposure also resulted in changes in EEG power spectra. Because most C57BL/6 mice that were exposed to 4 LD50 of GD did not survive past 24 hours, analysis of the acute effect of GD exposure on EEG power spectra was limited to the first 12 hours following GD‐induced SE. There was a main effect of GD exposure on the relative power of delta [F (2,146) = 5.92; P = 0.020], theta [F (2,146) = 5.38; P = 0.026], alpha [F (2,146) = 4.17, P = 0.048], beta [F (2,146) = 6.86, P = 0.013], and gamma [F (2,146) = 14.48, P = 0.001] frequency bands (Figure S3). C57BL/6 mice exposed to 285 μg/kg GD show increased relative power of delta for up to 450 minutes after GD exposure (P ≤ 0.05; Figure S3A), theta from 140 to 720 minutes after GD exposure (P ≤ 0.05; Figure S3B), alpha from 160 to 600 minutes after GD exposure (P ≤ 0.05; Figure S3C), and beta from 300 to 600 minutes after GD exposure (P ≤ 0.05; Figure S3D) compared to control. Similar to ES1−/− mice exposed to doses of GD that resulted in epileptiform activity without SE, C57BL/6 exposed to 82 μg/kg GD show an increase in the power of gamma from 160 to 720 minutes after exposure (P ≤ 0.05; Figure S3E) compared to control. C57BL/6 mice exposed to the lower dose of GD also present a decrease in theta power from 140 to 550 minutes, and alpha power from 160 to 600 minutes after GD exposure compared to control mice.
Analysis of changes in EEG power spectra in C57BL/6 mice during the 2 weeks after exposure to 82 μg/kg GD showed a main effect of GD exposure on beta [F (2,28) = 49.7; P = 0.002], with GD‐exposed mice showing a decrease from 12 hours after exposure until day of euthanasia (Figure S4D). There was a significant interaction between the effects of GD dose and time on changes in the power of alpha [F (2,28) = 7.19, P < 0.001] and gamma [F (2,28) = 4.89, P < 0.001]. C57BL6/mice exposed to 82 μg/kg GD had decreased power of alpha for up to 24 hours after the time of exposure (P ≤ 0.05, Figure S4C), and an increase in the power of gamma for up to 36 hours after the time of exposure (P ≤ 0.05, Figure S4E). No statistically significant main effects on delta and theta power in C57BL/6 mice were found.
3.4. Body weights
Mice exposed to GD lost body weight during the first 24 hours after exposure but recovered within 1 week of exposure. In ES1−/− mice, there was a main effect of day [F (9,171) = 30.98; P < 0.001] and a significant interaction between group and day [F (27,171) = 2.6, P < 0.001]. ES1−/− mice exposed to 23, 46, and 82 μg/kg GD, respectively, lost on average 7.2%, 4.7%, and 15.9% of body weight during the first 24 hours after exposure. In C57BL/6 mice, a main effect of day [F (9,36) = 14.54; P < 0.001] and a significant interaction between group and day [F (9,36) = 2.97, P = 0.01] was found; mice exposed to 82 μg/kg GD lost 7.7% of their body weight during the first 24 hours after exposure.
3.5. Neuropathologic analysis at 2 weeks following exposure to GD
In ES1−/− mice, a main effect of GD exposure was detected in the CA1 region of the hippocampus [F (1,10) = 9.98, P = 0.010), dorsomedial thalamus [F (1,21) = 3.90, P = 0.023], dorsolateral thalamus [F (1,21) = 29.5, P < 0.001], basolateral amygdala [F (1,21) = 15.0, P < 0.001], and layer 3 of the piriform cortex [F (1,21) = 4.84, P = 0.010]. Although ES1−/− mice exposed to 46 (P = 0.038) and 82 (P < 0.001) μg/kg GD showed reduced NeuN+ cells in the basolateral amygdala, only those exposed to 82 μg/kg GD showed significantly reduced NeuN immunostaining in other brain regions (dorsomedial thalamus, P = 0.023; dorsolateral thalamus, P < 0.001; layer 3 of the piriform cortex, P = 0.018), and CA1 of hippocampus (P = 0.01) compared to control (Figure 4). No main effect of GD exposure on NeuN immunostaining was detected in C57BL/6 mice exposed to 82 μg/kg. A main effect of GD exposure was observed on GAD67 immunostaining [F (1,24) = 5.01, P = 0.008], with mice exposed to 82 μg/kg GD having fewer GABA inhibitory interneurons in layer 3 of the piriform cortex compared to control (P = 0.017; Figure S5). There was no effect in C57BL/6 mice exposed to 82 μg/kg.
Figure 4.
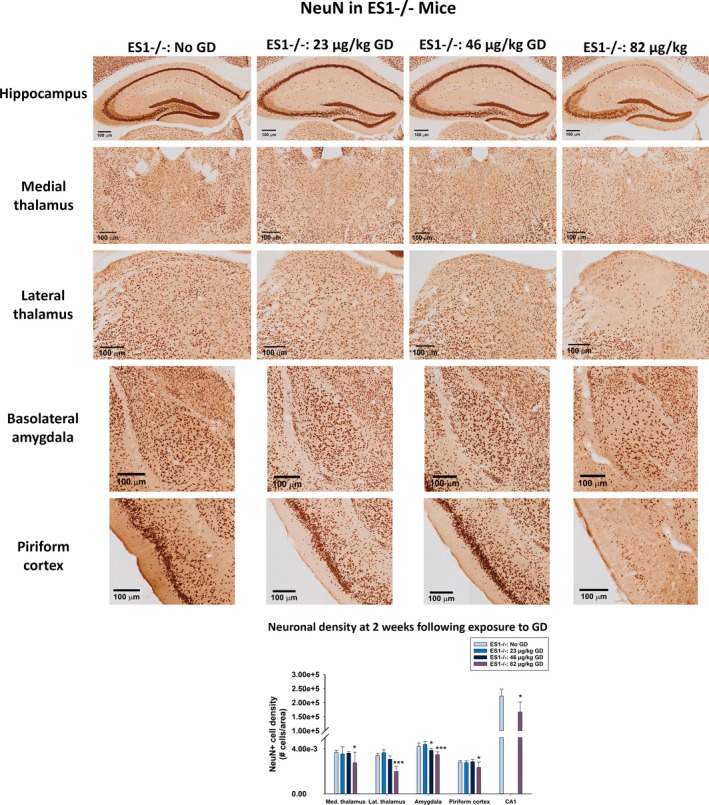
Effect of GD‐induced status epilepticus and delayed midazolam treatment on mature neuronal cell population of male ES1−/− mice. At 2 weeks following exposure to GD and delayed midazolam treatment, ES1−/− mice were perfused and brains collected for immunohistochemistry processing with an antibody against NeuN, a marker for mature neurons. NeuN‐positive (NeuN+) cells were manually quantified in the bregma range of −1.28 to −1.64 mm and cell densities estimated in the dorsomedial thalamus, dorsolateral thalamus, basolateral amygdala, and layer 3 of the piriform cortex. NeuN+ cell density in the CA1 region of the hippocampus was estimated by stereology methods. Cell NeuN+ cell density is shown in graph. *P < 0.05, ***P < 0.001 compared to control (No GD) ES1−/− mice
A robust neuroinflammatory response occurred in ES1−/− at 2 weeks after GD exposure, measured by changes in the morphology and density of Iba1‐positive (Iba1+) microglia, and density of GFAP‐positive (GFAP+) cell density. A main effect of GD was detected in the average cell body‐to‐cell size ratio in the CA1 [F (1,22) = 5.58, P = 0.005], dorsomedial thalamus [F (1,22) = 23.6, P < 0.001], dorsolateral thalamus [F (1,22) = 27.0, P < 0.001], basolateral amygdala [F (1,22) = 34.1, P < 0.001], and layer 3 of the piriform cortex [F (1,22) = 21.7, P < 0.001] (Figure 5). Only ES1−/− mice exposed to 82 μg/kg GD and that developed SE increased the cell body‐to‐cell size ratio in Iba1+ cells in all aforementioned regions (CA1, P = 0.005, dorsomedial thalamus, P < 0.001; dorsolateral thalamus, P < 0.001; basolateral amygdala, P < 0.001; layer 3 of the piriform cortex, P < 0.001) compared to control mice. A main effect of GD was detected in the average Iba1+ cell density in the CA1 [F (1,22) = 4.51, P = 0.013], dorsomedial thalamus [F (1,22) = 11.9, P < 0.001], dorsolateral thalamus [F (1,22) = 17.1, P < 0.001], basolateral amygdala [F (1,22) = 9.32, P < 0.001], and layer 3 of the piriform cortex [F (1,22) = 14.9, P < 0.001]. Only ES1−/− mice exposed to 82 μg/kg GD and that developed SE had increased number of Iba+ cell bodies in CA1 (P = 0.011), dorsomedial thalamus (P < 0.001), dorsolateral thalamus (P < 0.001), basolateral amygdala (P < 0.001), and layer 3 of the piriform cortex (P < 0.001) compared to control. No main effects of GD exposure on either cell body‐to‐cell size ratio or Iba1+ cell bodies were detected in C57BL/6 mice exposed to 82 μg/kg (data not shown).
Figure 5.
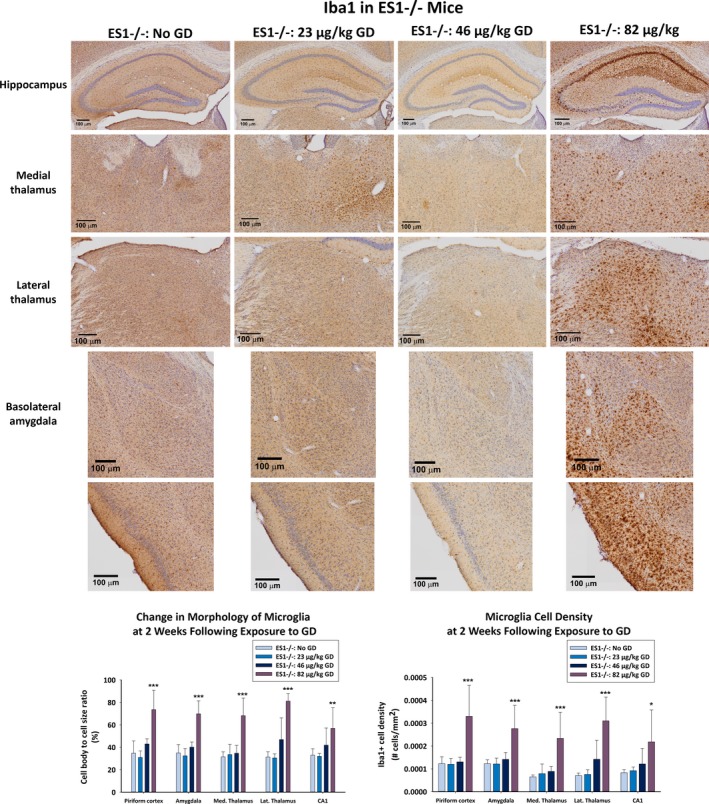
Effect of GD‐induced status epilepticus and delayed midazolam treatment on microglial activation in male ES1−/− mice. At 2 weeks following exposure to GD and delayed midazolam treatment, ES1−/− mice were perfused and brains collected for immunohistochemistry processing with an antibody against Iba1, a marker for microglia. Cresyl violet was used as counterstain for visualization of anatomic landmarks. Measures of cell body‐to‐cell size ratio and cell density of Iba1‐positive (Iba1+) cells were estimated in the bregma range of −1.28 to −1.64 mm in the CA1 region of the hippocampus, dorsomedial thalamus, dorsolateral thalamus, basolateral amygdala, and layer 3 of the piriform cortex; Iba+ cell density is shown in graph. *P < 0.05, **P < 0.01, ***P < 0.001 compared to control (No GD) ES1−/− mice
A main effect of GD exposure on GFAP+ cell density was detected in the dorsomedial thalamus [F (1,21) = 17.4; P < 0.001], dorsolateral thalamus [F (1,21) = 39.5; P < 0.001], basolateral amygdala [F (1,21) = 30.1; P < 0.001], and CA1 region of the hippocampus [F (1,21) = 3.18; P = 0.033] of ES1−/− mice (Figure 6), but not in the piriform cortex. Only ES1−/− mice exposed to 82 μg/kg GD increased GFAP+ cell density in the dorsomedial thalamus (P < 0.001), dorsolateral thalamus, (P < 0.001), basolateral amygdala, (P < 0.001), and CA1 of the hippocampus (P = 0.033), compared to control. There was no effect in C57BL/6 mice exposed to 82 μg/kg GD (data not shown).
Figure 6.
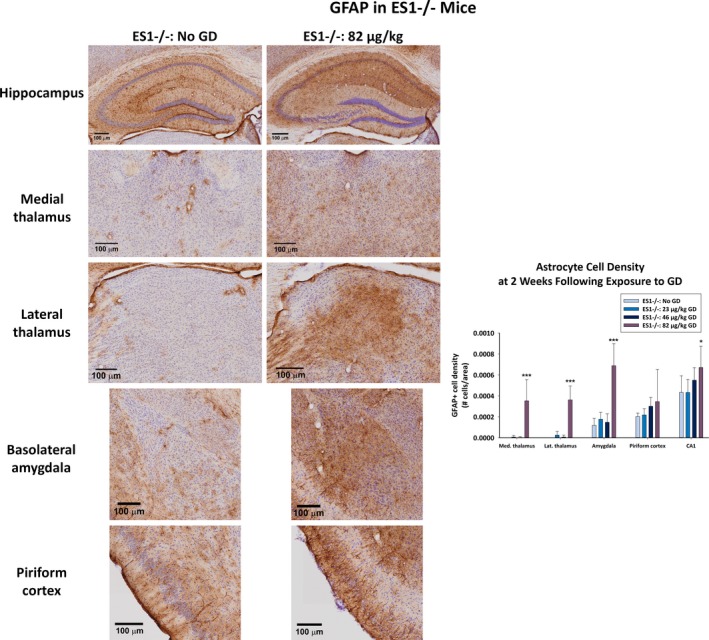
Effect of GD‐induced status epilepticus and delayed midazolam treatment on astrocytic cell response in male ES1−/− mice. At 2 weeks following exposure to GD and delayed midazolam treatment, ES1−/− mice were perfused and brains collected for immunohistochemistry processing with an antibody against GFAP, a marker for astrocytes. Cresyl violet was used as counterstain for visualization of anatomic landmarks. GFAP‐positive (GFAP+) cells were manually counted in the bregma range of −1.28 to −1.64 mm in the dorsomedial thalamus, dorsolateral thalamus, basolateral amygdala, layer 3 of the piriform cortex, and the CA1 region of the hippocampus. GFAP+ cell density is shown in graph. *P < 0.05, ***P < 0.001 compared to control (No GD) ES1−/− mice
4. DISCUSSION
We report on GD‐induced SE and the development of SRSs in ES1−/− mice, which similar to humans lack plasma carboxylesterase. Mice exposed to 4 LD50 developed SE, SRS, and had altered EEG power spectrum when MDZ treatment was delayed to 15 minutes after seizure onset, similar to ours and other's previous findings in GD‐exposed Sprague‐Dawley rats.4, 5, 25 To our knowledge, this is the first characterization of CWNA‐induced SE and epileptogenesis in this novel mouse strain, a more relevant model to human exposure.
GD‐exposed ES1−/− mice that developed SE displayed increases in the delta and theta power bands, the latter being persistently elevated throughout the study. Periods of seizure activity (with frequency components in delta and theta bands), interictal spikes, and an overall increase in sharp wave complexes (with frequency components in delta band) are responsible for the power increases in these bands. It is important to note that the increase in delta activity during the days following GD exposure correlates with the severity of seizure‐induced neuronal damage,26 and delta activity is postulated as a predictive marker of a drug's neuroprotective efficacy.25 The increase in power in the delta band is due to typical high amplitude spike wave activity occurring at 1‐4 Hz, and this type of epileptiform activity can vary in pattern and amplitude following exposure to GD.27 Notably, the increase in power of the delta band also occurs in nonhuman primates displaying SE after CWNA exposure,28 suggesting that this spectral anomaly may be used as a biomarker for imminent brain damage.25
Our observations are extremely relevant, as victims of CWNA exposure can display an increase in sharp wave complexes on EEG during sleep several months after the exposure to the CWNA sarin.29 Similar to our findings, pilocarpine‐induced SE in mice increases theta activity in the preictal and SE phases.30 In addition, case studies of patients diagnosed with idiopathic generalized epilepsy reveal increases in theta and delta power during interictal states.31 Moving forward, it will be interesting to test in the ES1−/− mouse model of CWNA toxicity the hypothesis that drug treatments that reduce the observed increase in power of EEG activity in the 0.1‐8 Hz frequency range would ameliorate chronic epileptic activity that follows prolonged benzodiazepine‐resistant SE. Supporting this idea are data from clinical studies that show that seizure‐free patients diagnosed with idiopathic generalized epilepsy on a daily treatment of valproic acid, a highly effective antiepileptic drug, have reduced power in the delta and theta frequency range.32
Animals exposed to 23 or 46 μg/kg GD and treated with MDZ had transient reduction in the alpha band compared to controls. Paroxysmal high‐voltage rhythmic spike in the alpha band is reduced by diazepam.33 Because MDZ is similarly sedating, a similar effect may occur in mice that did not develop SE.
The power in the gamma band was reduced in parallel to increased power in the delta band. In a model of amygdala kindling,34 increase in delta power and decrease in gamma and beta bands are coupled with epileptogenesis, seizures with behavioral components, and consciousness level. γ‐aminobutyric acid A (GABAA) receptors, ubiquitous in the central nervous system (CNS), might have a critical function in gamma frequency regulation.35, 36, 37 ES1−/− mice exposed to an SE‐inducing dose of GD had loss of GABA interneurons in the piriform cortex. Similarly, rats presenting GD‐induced SE have massive loss of GABAergic neurons38, 39 and downregulation of GABAA receptors after SE,40 which might result in a prolonged reduced power density in the gamma band and other anomalous brain oscillations. Our findings in ES1−/− mice of GD‐induced increased delta and theta power and reduced gamma power agree with findings in rats following exposure to a seizure‐inducing dose of GD.24
Severe and widespread neuronal loss in brain regions associated with seizure propagation was observed in ES1−/− mice 2 weeks following exposure to 4 LD50 GD. ES1−/− mice exposed to lower doses of GD presented with cholinergic signs of toxicity, yet neuronal densities were not different from control, with the exception of neuronal loss in the basolateral amygdala in mice exposed to 2 LD50. In addition, only ES1−/− mice exposed to 4 LD50 GD showed a robust increase in neuroinflammation, demonstrated by increased density of microglia and changes in morphology associated with activation of microglia and increased reactive astrocytes. Chronic neuroinflammation plays a role in the process of epileptogenesis after an initial brain insult.41, 42 Uncontrolled seizure activity in rodents is associated with the activation of a persistent neuroinflammatory response, mediated by microglia and astrocytes, in the days following SE. An increase of inflammatory cytokines [ie, interleukin (IL)‐6, IL‐1β, and tumor necrosis factor alpha (TNF‐α), among other factors] occurs in key brain regions of seizure propagation in animal models of cholinergic‐induced SE.43, 44, 45 Although their role in the clearance of toxic debris is considered to be beneficial in the acute period following brain injury, chronic microglial overactivation creates a neurotoxic environment that worsens neuronal damage46 and contributes to epileptogenesis.
As expected, the LD50 in ES1−/− mice was 4‐fold lower than that reported previously in wild‐type C57BL/6 mice.23 Although in ES1−/− a 4 LD50 GD induced SE, albeit with a high survivability rate, in C57BL/6 mice an equitoxic dose induced <30 minutes of continuous seizure activity with prolonged behavioral toxicity signs and poor survival. It is notable that the C57BL/6 mouse model of temporal lobe epilepsy induced by kainic acid has severe behavioral toxicity that is not always correlated with EEG seizure activity.47 The incongruence in the EEG seizure duration and behavioral convulsions in the C57BL/6 mice strengthens the argument for the necessity of telemetry instrumentation for continuous EEG recording in animal models of seizures.
In summary, this study is the first to show in this innovative animal model CWNA‐induced SE, epileptogenesis, long‐lasting disruptions in EEG power, gliosis, and severe neuropathology. The ES1−/− mouse model, which more accurately mimics the toxicity of CWNA exposure in humans compared to wild‐type mice and rats, may be a useful tool to identify improved MCM against the effects of CWNA exposure, and to identify mechanisms of epileptogenesis.
DISCLOSURE OF CONFLICT OF INTEREST
Authors Marcio de Araujo Furtado and Fu Du were paid under a contract to process and analyze EEG data and to process histopathology, respectively, with both contractors blinded to experimental conditions. All other authors have no conflict of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
ACKNOWLEDGMENTS
This research was supported by the DTRA‐JSTO, Medical S&T Division, the Geneva Foundation, and by NINDs R21 NS103820‐01 to Lucille A. Lange. The authors acknowledge Dr. Douglas Cerasoli, Dr. Linn Cadieux, and Ms. Sandra DeBus for implementation and management of the USAMRICD ES1−/− mouse colony. The authors also acknowledge Dr. Linnzi Wright and Robyn Lee for consultation on the LD50 determinations and the Probit analysis.
Marrero‐Rosado B, de Araujo Furtado M, Schultz CR, et al. Soman‐induced status epilepticus, epileptogenesis, and neuropathology in carboxylesterase knockout mice treated with midazolam. Epilepsia. 2018;59:2206–2218. 10.1111/epi.14582
The views expressed in this manuscript are those of the authors and do not reflect the official policy of the Department of Army, Department of Defense, or the US Government.
REFERENCES
- 1. Myhrer T. Neuronal structures involved in the induction and propagation of seizures caused by nerve agents: implications for medical treatment. Toxicology. 2007;239:1–14. [DOI] [PubMed] [Google Scholar]
- 2. Zilker T. Medical management of incidents with chemical warfare agents. Toxicology. 2005;214:221–31. [DOI] [PubMed] [Google Scholar]
- 3. Niquet J, Baldwin R, Suchomelova L, et al. Benzodiazepine‐refractory status epilepticus: pathophysiology and principles of treatment. Ann NY Acad Sci. 2016;1378:166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Araujo Furtado M, Lumley LA, Robison C, et al. Spontaneous recurrent seizures after status epilepticus induced by soman in Sprague‐Dawley rats. Epilepsia. 2010;51:1503–10. [DOI] [PubMed] [Google Scholar]
- 5. Schultz MK, Wright LK, de Araujo Furtado M, et al. Caramiphen edisylate as adjunct to standard therapy attenuates soman‐induced seizures and cognitive deficits in rats. Neurotoxicol Teratol. 2014;44:89–104. [DOI] [PubMed] [Google Scholar]
- 6. Marrero‐Rosado B, Rossetti F, Rice MW, et al. Age‐related susceptibility to epileptogenesis and neuronal loss in male Fischer rats exposed to soman and treated with medical countermeasures. Toxicol Sci. 2018;164:142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Curia G, Lucchi C, Vinet J, et al. Pathophysiogenesis of mesial temporal lobe epilepsy: is prevention of damage antiepileptogenic? Curr Med Chem. 2014;21:663–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wasterlain CG, Shirasaka Y. Seizures, brain damage and brain development. Brain Dev. 1994;16:279–95. [DOI] [PubMed] [Google Scholar]
- 9. Delorenzo RJ, Sun DA, Deshpande LS. Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintainance of epilepsy. Pharmacol Ther. 2005;105:229–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vezzani A, French J, Bartfai T, et al. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duysen EG, Koentgen F, Williams GR, et al. Production of ES1 plasma carboxylesterase knockout mice for toxicity studies. Chem Res Toxicol. 2011;24:1891–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boskovic B. The influence of 2‐/o‐cresyl/‐4 H‐1: 3: 2‐benzodioxa‐phosphorin‐2‐oxide (CBDP) on organophosphate poisoning and its therapy. Arch Toxicol. 1979;42:207–16. [PubMed] [Google Scholar]
- 13. Clement JG. Importance of aliesterase as a detoxification mechanism for soman (Pinacolyl methylphosphonofluoridate) in mice. Biochem Pharmacol. 1984;33:3807–11. [DOI] [PubMed] [Google Scholar]
- 14. Duysen EG, Cashman JR, Schopfer LM, et al. Differential sensitivity of plasma carboxylesterase‐null mice to parathion, chlorpyrifos and chlorpyrifos oxon, but not to diazinon, dichlorvos, diisopropylfluorophosphate, cresyl saligenin phosphate, cyclosarin thiocholine, tabun thiocholine, and carbofuran. Chem Biol Interact. 2012;195:189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Feder PI, Hobson DW, Olson CT, et al. Stagewise, adaptive dose allocation for quantal response dose‐response studies. Neurosci Biobehav Rev. 1991;15:109–14. [DOI] [PubMed] [Google Scholar]
- 16. Lundt A, Wormuth C, Siwek ME, et al. EEG radiotelemetry in small laboratory rodents: a powerful state‐of‐the art approach in neuropsychiatric, neurodegenerative, and epilepsy research. Neural Plast. 2016;2016:8213878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–94. [DOI] [PubMed] [Google Scholar]
- 18. de Araujo Furtado M, Zheng A, Sedigh‐Sarvestani M, et al. Analyzing large data sets acquired through telemetry from rats exposed to organophosphorous compounds: an EEG study. J Neurosci Methods. 2009;184:176–83. [DOI] [PubMed] [Google Scholar]
- 19. Hsu SM, Raine L, Fanger H. The use of antiavidin antibody and avidin‐biotin‐peroxidase complex in immunoperoxidase technics. Am J Clin Pathol. 1981;75:816–21. [DOI] [PubMed] [Google Scholar]
- 20. Bonthius DJ, McKim R, Koele L, et al. Use of frozen sections to determine neuronal number in the murine hippocampus and neocortex using the optical disector and optical fractionator. Brain Res Brain Res Protoc. 2004;14:45–57. [DOI] [PubMed] [Google Scholar]
- 21. Hovens IB, Nyakas C, Schoemaker RG. A novel method for evaluating microglial activation using ionized calcium‐binding adaptor protein‐1 staining: cell body to cell size ratio. Neuroimmunol Neuroinflamm. 2014;1:82–8. [Google Scholar]
- 22. Tynan RJ, Naicker S, Hinwood M, et al. Chronic stress alters the density and morphology of microglia in a subset of stress‐responsive brain regions. Brain Behav Immun. 2010;24:1058–68. [DOI] [PubMed] [Google Scholar]
- 23. Clement JG, Hand BT, Shiloff JD. Differences in the toxicity of soman in various strains of mice. Fundam Appl Toxicol. 1981;1:419–20. [DOI] [PubMed] [Google Scholar]
- 24. Apland JP, Aroniadou‐Anderjaska V, Figueiredo TH, De Araujo Furtado M, Braga MFM. Full protection against soman‐induced seizures and brain damage by LY293558 and caramiphen combination treatment in adult rats. Neurotox Res. 2018;34:511–24. [DOI] [PubMed] [Google Scholar]
- 25. Carpentier P, Foquin A, Dorandeu F, et al. Delta activity as an early indicator for soman‐induced brain damage: a review. Neurotoxicology. 2001;22:299–315. [DOI] [PubMed] [Google Scholar]
- 26. McDonough JH Jr, Clark TR, Slone TW Jr, et al. Neural lesions in the rat and their relationship to EEG delta activity following seizures induced by the nerve agent soman. Neurotoxicology. 1998;19:381–91. [PubMed] [Google Scholar]
- 27. Rossetti F, de Araujo Furtado M, Pak T, et al. Combined diazepam and HDAC inhibitor treatment protects against seizures and neuronal damage caused by soman exposure. Neurotoxicology. 2012;33:500–11. [DOI] [PubMed] [Google Scholar]
- 28. Lallement G, Clarencon D, Masqueliez C, et al. Nerve agent poisoning in primates: antilethal, anti‐epileptic and neuroprotective effects of GK‐11. Arch Toxicol. 1998;72:84–92. [DOI] [PubMed] [Google Scholar]
- 29. Sekijima Y, Morita H, Shindo M, et al. [A case of severe sarin poisoning in the sarin attack in Matsumoto–one‐year follow‐up of clinical findings, and laboratory data]. Rinsho Shinkeigaku (Clin Neurol). 1995;35:1241–5. [PubMed] [Google Scholar]
- 30. Phelan KD, Shwe UT, Cozart MA, et al. TRPC3 channels play a critical role in the theta component of pilocarpine‐induced status epilepticus in mice. Epilepsia. 2017;58:247–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clemens B. Pathological theta oscillations in idiopathic generalised epilepsy. Clin Neurophysiol. 2004;115:1436–41. [DOI] [PubMed] [Google Scholar]
- 32. Bela C, Monika B, Marton T, et al. Valproate selectively reduces EEG activity in anterior parts of the cortex in patients with idiopathic generalized epilepsy. A low resolution electromagnetic tomography (LORETA) study. Epilepsy Res. 2007;75:186–91. [DOI] [PubMed] [Google Scholar]
- 33. Shaw FZ. 7‐12 Hz high‐voltage rhythmic spike discharges in rats evaluated by antiepileptic drugs and flicker stimulation. J Neurophysiol. 2007;97:238–47. [DOI] [PubMed] [Google Scholar]
- 34. Jalilifar M, Yadollahpour A, Moazedi AA, et al. Classifying amygdala kindling stages using quantitative assessments of extracellular recording of EEG in rats. Brain Res Bull. 2016;127:148–55. [DOI] [PubMed] [Google Scholar]
- 35. Wang XJ, Buzsaki G. Gamma oscillation by synaptic inhibition in a hippocampal interneuronal network model. J Neurosci. 1996;16:6402–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Traub RD, Whittington MA, Stanford IM, et al. A mechanism for generation of long‐range synchronous fast oscillations in the cortex. Nature. 1996;383:621–4. [DOI] [PubMed] [Google Scholar]
- 37. Buzsaki G, Wang XJ. Mechanisms of gamma oscillations. Annu Rev Neurosci. 2012;35:203–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Figueiredo TH, Qashu F, Apland JP, et al. The GluK1 (GluR5) Kainate/{alpha}‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor antagonist LY293558 reduces soman‐induced seizures and neuropathology. J Pharmacol Exp Ther. 2011;336:303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prager EM, Pidoplichko VI, Aroniadou‐Anderjaska V, et al. Pathophysiological mechanisms underlying increased anxiety after soman exposure: reduced GABAergic inhibition in the basolateral amygdala. Neurotoxicology. 2014;44:335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Naylor DE, Liu H, Wasterlain CG. Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci. 2005;25:7724–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koh S. Role of neuroinflammation in evolution of childhood epilepsy. J Child Neurol. 2018;33:64–72. [DOI] [PubMed] [Google Scholar]
- 42. Ravizza T, Gagliardi B, Noe F, et al. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29:142–60. [DOI] [PubMed] [Google Scholar]
- 43. Arisi GM, Foresti ML, Katki K, et al. Increased CCL2, CCL3, CCL5, and IL‐1beta cytokine concentration in piriform cortex, hippocampus, and neocortex after pilocarpine‐induced seizures. J Neuroinflammation. 2015;12:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Spradling KD, Lumley LA, Robison CL, et al. Transcriptional responses of the nerve agent‐sensitive brain regions amygdala, hippocampus, piriform cortex, septum, and thalamus following exposure to the organophosphonate anticholinesterase sarin. J Neuroinflammation. 2011;8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vezzani A, Friedman A, Dingledine RJ. The role of inflammation in epileptogenesis. Neuropharmacology. 2013;69:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Block ML, Zecca L, Hong JS. Microglia‐mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. [DOI] [PubMed] [Google Scholar]
- 47. Puttachary S, Sharma S, Tse K, et al. Immediate epileptogenesis after kainate‐induced status epilepticus in C57BL/6J mice: evidence from long term continuous video‐EEG telemetry. PLoS ONE. 2015;10:e0131705. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials