

 Radon is a naturally occurring radionuclide, which has a wide environmental distributed.
Radon is a naturally occurring radionuclide, which has a wide environmental distributed.
Abstract
Radon is a naturally occurring radionuclide, which has a wide environmental distributed. It emits multiple high linear energy transfer (LET) alpha particles during radiative decay, and has been regarded as a human carcinogen by the International Agency for Research on Cancer. Currently, residential radon exposure is considered as the second highest cause of lung cancer and the leading cause among nonsmokers. Radon exposure leads to genomic instability, which causes the accumulation of multiple genetic changes and leads to cancer development. However, the molecular basis underlying carcinogenesis, especially the radon-induced changes to mitochondria, has not been fully elucidated. The aim of this study was to explore the dynamic changes in mitochondria along with the cell transformations induced by long-term radon exposure. A malignant transformation model of BEAS-2B cells was established with upto 40 times the usual radon exposure (20 000 Bq m–3, 30 min each time every 3 days). Long-term radon exposure induced EMT-like transformation of epithelial cells in our study, evidenced by decrease in epithelial markers and increase in mesenchymal markers, as well as the loss of cell–cell adhesion and alterations to the morphology of cells from compact shape to a spindle shaped, fibroblast-like morphology. Additionally, the proliferation and migration of cells were increased and apoptosis was decreased with long-term radon exposure. Furthermore, mitochondrial function was up-regulated and the levels of oxidative stress were repressed with long-term radon exposure. Our work explored the dynamic changes of mitochondrial in radon induced malignant transformation of lung bronchial epithelial cells, which could partially elucidate the role of mitochondria in radon induced cell malignancy.
Introduction
Lung cancer is a major cause of cancer deaths worldwide, and there is an estimated 1 825 000 new cases of lung cancer diagnosed and 1 590 000 associated deaths every year, according to GLOBOCAN 2012.1 So far, lung cancer has been confirmed to be associated with tobacco smoke exposure. However, lung cancer in patients without smoke exposure accounts for 10–25% of all cases, ranking it as the 7th most common cause of cancer-related death.2,3 Since the mechanisms contributing to the onset and pathogenesis of lung cancer in non-smokers is different from those induced by tobacco, the study of non-tobacco related carcinogens is fundamental to the better understanding of the biology of lung cancer arising in non-smokers.4–6
Radon is a ubiquitous, colorless, odorless, radioactive gas and is considered a human carcinogen. It derives from the series decay of uranium and thorium, which exist majorly in soil and rocks.7 Numerous studies have reported that the levels of radon and its progeny are at more than 50 Bq m–3, or ever higher than 150 Bq m–3, in many areas.8 International Agency for Research on Cancer (IARC) has already defined radon as a lung cancer carcinogen based on the radon-exposed miners cohort from 1988.9 Nevertheless, a series of studies have reported that long-term radon exposure in residential housing is associated with lung cancer risk, regardless of the patient's smoking status.10–12 Thus, in 2014, IARC concluded that radon and its progeny cause lung cancer in humans.13 The USA Environmental Protection Agency has previously established an action level of radon as 148 Bq m–3, which was re-adjusted by World Health Organization (WHO) to 100 Bq m–3.14,15
The carcinogenic risk of radon exposure is caused by its radioactive decay and the subsequent emission of high energy alpha decay particles (α-decay).16 It is estimated that radon causes approximately 21 000 cases of lung cancer per year.17 Although chemically inert, radon could decay into active progenies that can be inhaled by humans and subsequently, can reach human lung epithelial cells. Deposited radon progeny also decays to generate alpha-particles, which could damage DNA both directly or through generation of free radicals.18 Besides, observable levels of cytokines can be detected in the supernatants of cells exposed to alpha-particle radiation, which indicated a possible role of cytokines in radon-induced carcinogenesis.19 Additionally, radiation also induced mitochondria damage,20–23 which may contribute to the radon-induced carcinogenesis as well. However, previous studies on the mechanism of radon-induced carcinogenesis mostly considered the effect of radiation on nuclei, while the mitochondrial damage caused by radiation exposure in the lung cell transformation was mostly ignored, thereby largely underestimated the harm of radiation to human health.24 Thus, the aim of this study was to examine the dynamic changes in mitochondria induced by radon exposure in human bronchial epithelial cells with epithelial–mesenchymal transition (EMT).
Materials and methods
Cell culture and exposure conditions
BEAS-2B cells (derived from human bronchial epithelial cells) were purchased from American Type Culture Collection (ATCC, USA) and grown in Dulbecco's modification of Eagle's medium (DMEM) (Corning) supplemented with 10% fetal bovine serum (FBS, Hyclone, Logan, UT) and 0.1% gentamycin (Invitrogen, Carlsbad, CA). The cells were maintained at 37 °C in a humidified incubator with 5% CO2. Approximately 1 × 105 cells were seeded on a trans-well membrane in micro-titer plates (Corning, USA). 24 h later, the cells were exposed to radon (Rn), which was produced by the decay of radium.
Cells were placed in a gas inhalation chamber (MED8170, Tianjin Hope Industry & Trade Co. Ltd, Tianjin, China). The gas chamber was connected to a multifunctional radon chamber purchased from Donghua University in China. Radon and its progeny were produced by a radon source using a Changhe pump machine (model BT00-300, China), which were then pumped into the gas chamber. The medium on the upper chamber of transwell plate was removed, and the cells were directly exposed to radon and its progeny at a concentration of 20 000 Bq m–3 for 30 min each time according to our previous preliminary study. After exposure, fresh medium was added and the cells were cultured for 3 days before being subjected with the next exposure.25 ‘Rn-0’ is referred to the group of cells without radon exposure. Other groups of cells were exposed to the indicated times of radon exposure using the same condition as described above [(Rn-5) group of cells with 5 times radon exposure; (Rn-10) group of cells with 10 times radon exposure; (Rn-20) group of cells with 20 times radon exposure; (Rn-30) group of cells with 30 times radon exposure; and (Rn-40) group of cells with 40 times radon exposure.]
Cell morphology observation
Cell morphologies and shapes with the indicated radon exposure times were observed and captured with an inverted microscope (LEICA DM 2500).
Cell cycle analysis
Control and the indicated radon-exposure groups of cells were harvested. Approximately 1 × 106 cells from each group were fixed with 75% chilled ethanol overnight at 4 °C. Subsequently, the cells were stained with propidium iodide (PI) (Beyotime, China) in the dark at room temperature for 30 min. Signals were examined using flow cytometry (Becton Dickinson and Company, USA) with excitation at 493 nm and emission at 630 nm.
mRNA extraction and real-time RT-PCR
To examine the effect of radon on the mtDNA level, total RNA was extracted from each group of cells using TRIzol reagent from CWBIO (Beijing, China), according to the manufacture's protocol. The RNA concentration was determined by a NanoDrop 2000 spectrophotometer (NanoDrop Technology, Wilmington, DE, USA). Equal amounts of mRNA were used to generate cDNA using a HiFiScript cDNA synthesis kit following the manufacturer's instructions (CWBIO, Beijing, China). The RT-PCR procedures were described previously.26 The sequences of primers are as follows:
12S rRNA: forward (AGAACACTACGAGCCACAGC);
reverse (ACTTGCGCTTACTTTGTAGCC);
18S rRNA: forward (GGAGTATGGTTGCAAAGCTC);
reverse (CGCTCCACCAACTAAGAACG);
E-cadherin: forward (GCCAAAGACAGAGCGGAACTAT);
reverse (ATGTGTTCAGCTCAGCCAGC);
N-cadherin: forward (CAACTTGCCAGAAAACTCCAGG);
reverse (ATGAAACCGGGCTATCTGCTC);
Vimentin: forward (TGCCGTTGAAGCTGCTAACTAC);
reverse (TAGGTGGCAATCTCAATGTC);
CK-8: forward (GAGGCATCACCGCAGTTAC);
reverse (TTGCTTCGAGCCGTCTTCT)
GAPDH: forward (CTGACTTCAACAGCGACACC);
reverse (TGCTGTAGCCAAATTCGTTGT).
The conditions used for real-time PCR conditions were: initial denaturation at 95 °C, 10 min, 40 cycles of amplification (95 °C for 10 s; 60 °C for 30 s; 72 °C for 32 s), a melting curve (95 °C for 15 s; 60 °C for 1 min; 95 °C for 15 s), and a cooling cycle (60 °C, 15 s). The mean crossing point (Cp) values and standard deviations (SD) were determined. The Cp values were normalized to the respective Cp values of 18S rRNA reference gene. The data are presented as a fold change in gene expression compared to the control group.
Immunoblot analysis
Cells were collected from each group in RIPA buffer (Beyotime, China) containing 1 mM phenylmethanesulfonyl fluoride (PMSF) (Beyotime, China). Cell lysates were sonicated on ice to allow full digestion, followed by boiling for 10 min. The protein concentration was measured using a BCA protein concentration kit according to the manufacturer's instructions (Beyotime Biotechnology, China). Equal amount of protein from each sample was electrophoresed through an SDS-polyacrylamide gel and subjected to immunoblot analysis with cytochrome C, Vimentin, E-cadiherin, and GAPDH antibodies, which were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
In vitro scratch wound healing assay
The wound healing assay was performed as described elsewhere.27 In brief, UV-sterilized polydimethylsiloxane (PDMS) blocks (1 mm × 2 cm) were placed onto 35 mm glass bottom dishes. Cells from each groups were seeded at a density of 1 × 105 cells per well, and after 48 h, the slab was removed to allow cell migration in DMEM without serum for 36 h. Phase contrast micrograph images were captured at 0 h, 12 h, 24 h, and 36 h. The relative distances traveled by the leading edge within 12 h, 24 h, and 36 h were assessed using ImageJ free software (; https://imagej.nih.gov/ij/download.html).
Cell viability and colony formation assay
Cell viability assay was performed using a CCK-8 assay (Beyotime, China).1500 cells from the various radon exposure groups were seeded and dispensed into 96-well plates at 100 μl per well, followed by 72 h incubation. For cell viability measurement, 10 μl CCK-8 solution was directly added to the cells, followed by incubation at 37 °C for 2 h. The absorbance at 450 nm was measured using a Synergy 2 multi-mode micro-plate reader (Biotek, Seattle, USA). All samples were analyzed in triplicates for each group and the data are presented as the means of three independent experiments. The data are presented as a fold change in cell viability compared to the control group (Rn-0).
For colony formation, 1 × 103 BEAS-2B cells from each radon exposure group were initially seeded into a well of a 6-well plate and maintained in medium containing 10% FBS, which was refreshed every two days. After the cells had incubated for 14 days at 37 °C in 5% CO2, the cells were fixed with methanol and stained with 1% crystal violet for 15 min before being counted. The colony numbers were counted using Image J software and all experiments were repeated three times. The colony-forming efficiency (%) was calculated as the ratio of colony number and cell seeding number.
ATP measurement
The ATP level was determined according to the manufacture's protocol using the ATP detection kit (Beyotime, China). Briefly, cells from each group were harvested and washed twice with phosphate buffer saline (PBS). After the cells were lysed with 200 μl lysis buffer, the supernatants from all samples were harvested for ATP measurements. All samples were run in triplicates for each group and the data represent means of three independent experiments. The data are presented as a fold change in ATP value compared to the control group (Rn-0).
ROS detection
Approximately 1.5 × 106 cells with indicated times of radon exposure were harvested and washed with PBS twice, followed by incubation with fresh medium containing 2,7-dichlorodi-hydrofluorescein diacetate (H2DCFDA, Beyotime, China, 10 μg ml–1 final concentration) for 30 min at 37 °C. Fluorescence was measured using flow cytometry (Becton Dickinson and Company, USA) with excitation at 488 nm and emission at 515–545 nm. All procedures following H2DCFDA incubation were performed in the dark.
Detection of MMP levels
To detect MMP and monitor the levels of apoptosis in cells with radon exposure, a Mitochondrial Membrane Potential Kit was used according to the manufacturer's instructions (Beyotime, China). Briefly, 1 × 106 cells from each group were harvested and incubated with the JC-1 fluorescent probes for 20 min at 37 °C in the dark. After incubation, the cells were washed with PBS and analyzed within 30 min using flow cytometry (Becton Dickinson and Company, USA).
Annexin V-FITC/PI double staining
Levels of cell apoptosis were assessed using the Annexin V-FITC Apoptosis Detection kit (Beyotime, China). Cells were suspended with 195 μl Annexin V-FITC binding buffer. Then, 10 μl PI solution and 5 μl FITC-conjugated Annexin V were added to the cells for incubation at room temperature for 20 min in the dark. The levels of cell apoptosis were detected with flow cytometry (Becton Dickinson and Company, USA).
Cell migration assays
Cell migration was assessed using a Boyden chamber assay. For these experiments, 2.5 × 105 cells from each group were seeded onto the upper well of a Costar Transwell chamber (8 μm; Corning Life Sciences, Tweksbury, MA, USA). 24 hours later, cells that had migrated to the bottom side of the membrane were fixed in 70% ethanol and stained with crystal violet. After being stained, non-migrating cells in the upper chamber were removed using cotton tipped applicators. The membranes were then mounted onto object slides. At least three independent experiments were performed, and the amounts of migrated cells were quantified by Image J software.
Statistics
Results are presented as fold changes to the control groups (Rn-0). The data are shown as mean ± SD of three independent experiments performed in duplicate (real-time RT-PCR) or triplicate (CCK-8 assay, ROS and MMP detection, Annexin V-FITC/PI double staining, and ATP measurement). Statistical tests were performed using SPSS 20.0. Unpaired Student's t-test was applied to compare the means of two groups. One-way ANOVA with Bonferroni's correction was applied to compare the means of three or more groups. A value of p < 0.05 was considered to be significant.
Results
Radon exposure induces mesenchymal-like morphological changes in lung epithelium cells
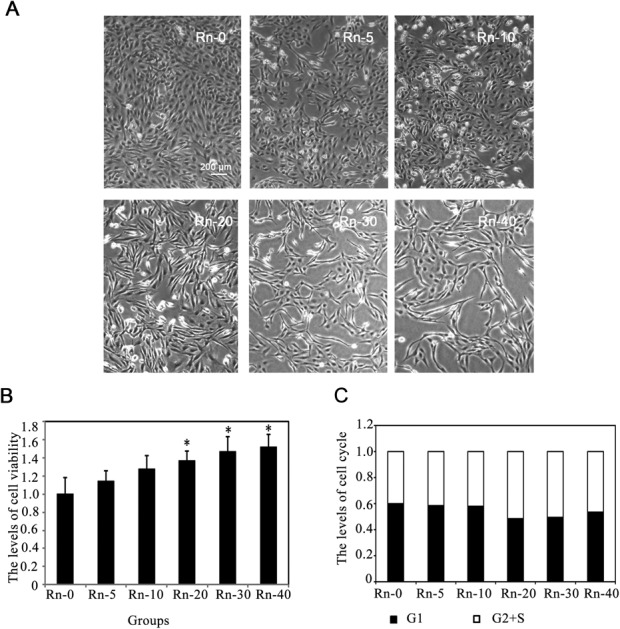
Radon is considered as an environmental toxin, which could increase the possibility of lung cancer development with long-term exposure.28 Thus, we first determined if upon long-term exposure, low dose of radon could induce EMT-like transformation of epithelial cells in our study. BEAS-2B cells from different radon exposure groups were observed under the microscope. After radon exposure, an alteration in BEAS-2B cell morphology was apparent. Fig. 1A shows that exposure to radon induces immortalized lung epithelial cells (BEAS-2B cells) to undergo EMT-like transformation, evidenced by the loss of cell–cell adhesion and the alteration in morphology from compact shape to spindle shaped, fibroblast-like morphology. Then, we detected the cell viability at the indicated times of radon exposure. The results showed that radon exposure could increase the levels of cell viability in a time-dependent manner (Fig. 1B).
Fig. 1. The morphological changes of lung epithelial cells caused by radon exposure. (A) Followed byradon exposure for the indicated times, the morphology of BEAS-2B cells was captured. The cells were divided into control group (Rn-0) and radon exposure groups [(Rn-5) group of cells with 5 times radon exposure; (Rn-10) group of cells with 10 times radon exposure; (Rn-20)group of cells with 20 times radon exposure; (Rn-30) group of cells with 30 times radon exposure; (Rn-40) group of cells with 40 times radon exposure.] Representative cell images from each group are shown. (B) Radon exposure up-regulated the cell proliferation in a time dependent manner. BEAS-2B cells from each groups were harvested. The levels of cell viability were determined by MTT assay for 72 h. Results are expressed as means ± SD (n = 4). (*p < 0.05 radon exposure groups vs. control group). (C) Radon exposure did not affect the cell cycle. BEAS-2B cells from each groups were harvested. The levels of cell cycle were determined.

Next, the markers of epithelial cells and mesenchymal cells were measured by realtime RT-PCR assay (Fig. 2A). The results indicated that long-term radon exposure could cause a decrease in cell epithelial markers (E-cadherin and CK-8) and increase the cell mesenchymal markers (Vimentin and N-cadherin), which is consistent with the above observation of radon induced-cell EMT-like transformation. Besides, the protein expression of E-cadherin and Vimentin were also detected by western blot assay, which also gave the similar results that radon exposure could cause EMT-like transformation in the epithelial cells (Fig. 2B). Furthermore, the migration of an indicated group of cells was evaluated by Boyden chamber assay (Fig. 2C). Results showed that the migration ability of BEAS-2B cells was significantly increased with increasing radon exposure. Overall, long-term radon exposure could dramatically cause EMT-like transformations in the cells in our study.
Fig. 2. Long-term radon exposure caused the epithelial cells EMT-like transformation. (A)The mRNA of BEAS-2B cells from each group were extracted after the indicated times of radon exposure. The relative of mRNA expression of Vimentin, N-cadherin, E-cadherin and CK-8 was measured by realtime RT-PCR assay.(B) The protein levels of E-cadherin and Vimentin were detected by western blot assay and the relative ratios of specific protein expression normalized to GAPDH is shown (lower panel). (C) Cell migration analysis of BEAS-2B cells from each radon exposure group were evaluated by Boyden chamber assay. Results are expressed as means ± SD (n = 4). (*p < 0.05 radon exposure groups vs. control group).

Long-term radon exposure could decrease cell apoptosis
Since it has been reported that various toxins(such as, cigarette smoke extract) could induce cell apoptosis by G2/S arrest in the lung epithelial cells,29 we examined whether long-term exposure to low dose radon could affect the cell cycle progression in human epithelial cells. The proportion of cells in different phases of the cell cycle was determined by flow cytometry. Radon exposure did not arrest the cell cycle in BEAS-2B cells (Fig. 1C), indicating that a low dose radon exposure does not interrupt cell cycle progression.
As shown in Fig. 1, radon exposure could induce morphological changes of EMT-like cells and increase the levels of cell proliferation. To further clarify if the lung epithelial cells (BEAS-2B) were transformed by low dose radon in long-term exposure, we examined the levels of cell apoptosis by flow cytometry analysis with Annexin V/PI staining (Fig. 3A). The results indicated that the levels of cell apoptosis were repressed by long-term radon exposure, which is an outstanding characteristic of cancer cells (Fig. 3B).
Fig. 3. The levels of cell apoptosis were decreased with the long-term radon exposure. The BEAS-2B cells from each group were harvested after the indicated times of radon exposure. The levels of cell apoptosis were analyzed.(A) Apoptotic cells were detected with Annexin V/PI staining by flow cytometry analysis; representative apoptotic images of each group of cells with the indicated times of radon exposure are shown. (B) The levels of cell apoptosis in various radon exposure times were calculated. Results are expressed as means ± SD (n = 4). (*p < 0.05 radon exposure groups vs. control group).

Effects of radon exposure on cell migration
Promoting cancer cell migration is one of the major characteristics of EMT. A scratch assay was carried out to analyze BEAS-2B cells migration in a wounded monolayer in response to radon exposure. Over a period of 36 h, the wounded area remained largely unchanged in the untreated monolayer. In contrast, cells with various times of radon exposure migrated to refill the wounded area in the same time-period (Fig. 4A). Long-term radon exposure induced EMT was associated with increased cells migration as shown in Fig. 4B, as the uncovered wound area was shrunk in radon exposure groups in a time-dependent manner.
Fig. 4. Radon exposure promotes cell migration. (A) A wound healing assay was used to detect the migration ability of BEAS-2B cells after radon exposure. Representative cell images from each indicated times of exposure group in the are shown. (B) Quantification of wound healing is shown. Results are expressed as means ± SD (n = 4). (*p < 0.05 indicated times of radon exposure vs. control group).

Radon exposure increases the cell’ ability of cloning
Similar to our observations from the cellular migration assay, we measured proliferation by a colony formation assay (Fig. 5A). Consistent with our results of MTT assay, the data showed that radon exposure increased colony growth in a time-dependent manner (Fig. 5B).
Fig. 5. Radon exposure increases the cells’ ability of clone-forming. (A) A plate clone formation assay of BEAS-2B cells after exposure to radon is shown. 1 × 103 cells from each group were plated into a new D-100 cell culture dish for clone formation assay in 2 weeks. Representative images from each group are shown. (B) Relative ratio of colony formation is shown. Results are expressed as means ± SD (n = 4). (*p < 0.05 indicated radon exposure times vs. control group).

Long-term radon exposure causes EMT-like cell transformation with up-regulated mitochondrial function and repressed levels of oxidative stress
Since radon exposure could promote cellular proliferation and migration, reduce the cell apoptosis, and induce Mesenchymal-like morphological changes in lung epithelium cells, we decided to explore the mechanisms underlying the EMT-like cell transformation induced by radon exposure. The levels of oxidative stress, MMP and ATP in the cells from the indicated groups of radon exposure were measured (Fig. 6). In the cells exposed to low doses of radon, ROS levels consistently decreased (Fig. 6A). Meanwhile, the protein levels of cytochrome C were up-regulated, which peaked at 5 times radon exposure, and were restored to non-exposure (control) levels in the cells with 40 times radon exposure (Fig. 6B). Next, since radon exposure induced cell proliferation and caused EMT-like cells transformation, we evaluated whether these effects could be involved in the alteration of energetic metabolism. As shown in Fig. 6C, radon increased the cellular ATP levels after 20 times radon exposure (p < 0.05), indicating that the cellular ATP increase is involved in the cell proliferation.
Fig. 6. Long-term radon exposure enhances the number and function of mitochondria, and decreases oxidative stress in the cells. (A) Radon exposure decreased the ROS levels in the cells. (B) Radon exposure induced the protein levels of cytochrome C (upper panel). Relative ratio of Cytochrome C expression normalized to GAPDH is shown (lower panel). (C) Radon exposure time-course dependently increased the levels of ATP. (D) Radon exposure raised the copy number of mtDNA. (E) Radon exposure increased the levels of MMP. Results are expressed as means ± SD (n = 4). (*p < 0.05 indicated radon exposure times vs. control group).

Then, to clarify whether this increase is associated with any mitochondrial alterations, the level of MMP and the copy number of mtDNA were investigated. The levels of MMP increased upon radon exposure, in a similar manner as the mtDNA copy number (Fig. 6D–E). These results indicated that prolonged low dose radon exposure could increase the number and function of mitochondria, which leads to more ATP production and promotes cell proliferation.
Discussion
Although the cause of lung cancer is still not clear, certain risk factors have been proposed. Radon exposure is the second greatest risk factor for the development of lung cancer.17 Radon is a naturally occurring radionuclide in the environment, which emits high linear energy transfer (LET) alpha particles during decay.7,16 In the past few years, several genome-wide screens for the investigation of bio-molecular effects of radon exposure on the human lung epithelium have been reported in the literature.6,30 However, few studies focus on the dysfunction of mitochondria,25 and the mechanism of the development and progression of lung cancer caused by radon exposure remains unexplained. In this study, we established a malignant transformation model of BEAS-2B cells with a time series of radon exposure. We found that long-term radon exposure caused EMT-like cell transformations with up-regulation of mitochondrial function and repression of oxidative stress. Our work explored the dynamic changes in mitochondria in radon induced malignant transformation of lung bronchial epithelial cells, which could partially explain the role of mitochondrial dysfunction in radon induced cell malignancies.
Tumor invasion and metastasis are the leading cause of death in patients with advanced lung cancer. EMT is a crucial event during the tumor progression, and a series of studies found that EMT is activated in various types of cancers.31–33 Currently, EMT has been suggested to play important roles in the initiation of metastasis of lung cancers. Radon has been suggested to cause lung carcinogenesis; and exposure to radon can induce cell malignant transformation.25 Consequently, these evidences suggest a possible link between radon exposure and EMT. Thus, understanding the overall effects of radon exposure on lung cell physiology is very important.
However, it is reported that after treatment with radon for 10 times, cell malignant transformation can be induced in HBE (human bronchial epithelial) cells.25 They also showed alterations to the p53-mediated metabolism upon radon exposure. Since p53 is known to contribute to the balance and the utilization of respiratory and glycolytic pathways in the mitochondria,33 it could be speculated that p53-mediated signaling pathway plays a crucial role in radon-caused malignant cell transformations. However, we established in this study another cell model with BEAS-2B cells, which is also of normal human bronchial epithelial cells. According to our previous study, the p53 gene is of wild type in HBE cells and is mutated in BEAS-2B.34In Liu's study, they reported that after 10 times radon exposure, HBE cells were malignantly transformed. Meanwhile, we found in this study that BEAS-2B cells could also be significantly transformed with up to 10 times radon exposure. Furthermore, BEAS-2B cells did not show more sensitivity than HBE cells to radon-induced cell carcinogenesis. Thus, p53 gene may not be the only pivotal regulator associated with the radon-induced lung epithelial cells’ transformations. In a future study, we will further explore the up-stream targets that lead to the mitochondrial dysfunction, which could further provide explore the anti-lung cancer activities induced by radon exposure and potentially contribute to lung cancer medical therapies.
In Fig. 6, the data demonstrated that the level of ROS was decreased with increased radon exposure. So far, a series of cancer studies have showed that the Nrf2 signaling pathway was induced in cancer tissues.34–36 Moreover, An's group also reported that in arsenite-induced human keratinocyte transformation, the level of ROS was decreased with a significant activation of Nrf2 signaling pathway,37 which is consistent with our results. The Nrf2-mediated antioxidant response represents a critical cellular defense mechanism. Thus, when the Nrf2 signaling pathway is up-regulated, the ROS level will be accordingly depressed.
Funding details
The work was supported by the grants as follows:
Chinese National Nature Science Foundation <number: 81673126, 81703205>;
Youth Foundation of Jiangsu Province <number: BK20160333>;
China Postdoctoral Science Foundation <number: 2016M600440, 2017T100402>;
Natural Science Research in Colleges and Universities of Jiangsu Province <number: 16KJB330008>;
Postdoctoral Science Foundation of Jiangsu Province <number: 1601079C>.
Conflicts of interest
The authors declare no conflict of interest.
References
- http://globocan.iarc.fr/Default.aspx, Globocan, 2012.
- Torre L. A., Bray F., Siegel R. L., Ferlay J., Lortet-Tieulent J., Jemal A. CA-Cancer J. Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- Sun S., Schiller J. H., Gazdar A. F. Nat. Rev. Cancer. 2007;7:778–790. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- Ferlay J., Shin H. R., Bray F., Forman D., Mathers C., Parkin D. M. Int. J. Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- Badar F., Meerza F., Khokhar R. A., Ali F. A., Irfan N., Kamran S., Shahid N., Mahmood S. Asian Pac. J. Cancer Prev. 2006;7:245–248. [PubMed] [Google Scholar]
- Subramanian J., Govindan R. J. Clin. Oncol. 2007;25:561–570. doi: 10.1200/JCO.2006.06.8015. [DOI] [PubMed] [Google Scholar]
- IARC monographs on the evaluation of carcinogenic risks to humans - Ionizing radiation, Part 2: Some internally deposited radionuclides, 2001. [PMC free article] [PubMed]
- Hubaux R., Becker-Santos D. D., Enfield K. S., Lam S., Lam W. L., Martinez V. D. Environ. Health: Global Access Sci. Source. 2012;11:89. doi: 10.1186/1476-069X-11-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Agency for Research on Cancer, Man-made mineral fibres and radon, 1988, vol. 43. [Google Scholar]
- Darby S., Hill D., Deo H., Auvinen A., Barros-Dios J. M., Baysson H., Bochicchio F., Falk R., Farchi S., Figueiras A., Hakama M., Heid I., Hunter N., Kreienbrock L., Kreuzer M., Lagarde F., Makelainen I., Muirhead C., Oberaigner W., Pershagen G., Ruosteenoja E., Rosario A. S., Tirmarche M., Tomasek L., Whitley E., Wichmann H. E., Doll R. Scand. J. Work, Environ. Health. 2006;32(Suppl. 1):1–83. [PubMed] [Google Scholar]
- Krewski D., Lubin J. H., Zielinski J. M., Alavanja M., Catalan V. S., Field R. W., Klotz J. B., Letourneau E. G., Lynch C. F., Lyon J. L., Sandler D. P., Schoenberg J. B., Steck D. J., Stolwijk J. A., Weinberg C., Wilcox H. B. J. Toxicol. Environ. Health, Part A. 2006;69:533–597. doi: 10.1080/15287390500260945. [DOI] [PubMed] [Google Scholar]
- Lubin J. H., Wang Z. Y., Boice Jr. J. D., Xu Z. Y., Blot W. J., De Wang L., Kleinerman R. A. Int. J. Cancer. 2004;109:132–137. doi: 10.1002/ijc.11683. [DOI] [PubMed] [Google Scholar]
- Loomis D., Huang W., Chen G. Chin. J. Cancer. 2014;33:189–196. doi: 10.5732/cjc.014.10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosalorenzo R., Ruanoravina A., Ramis R., Aragonés N., Kelsey K. T., Carballeiraroca C., Fernándezvillar A., Lópezabente G., Barrosdios J. M. Int. J. Radiat. Biol. 2016;93:222–230. [Google Scholar]
- Barbosalorenzo R., Barrosdios J. M., Ruanoravina A. Int. J. Epidemiol. 2017;46(2):767–768. [Google Scholar]
- Darby S., Hill D., Auvinen A., Barros-Dios J. M., Baysson H., Bochicchio F., Deo H., Falk R., Forastiere F., Hakama M., Heid I., Kreienbrock L., Kreuzer M., Lagarde F., Makelainen I., Muirhead C., Oberaigner W., Pershagen G., Ruano-Ravina A., Ruosteenoja E., Rosario A. S., Tirmarche M., Tomasek L., Whitley E., Wichmann H. E., Doll R. Br. Med. J. 2005;330:223. doi: 10.1136/bmj.38308.477650.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez D., Alshanqeety O., Warner K. E., Lantz P. M., Courant P. N. Am. J. Public Health. 2011;101:310–314. doi: 10.2105/AJPH.2009.189225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samet J. M., Avila-Tang E., Boffetta P., Hannan L. M., Olivo-Marston S., Thun M. J., Rudin C. M. Clin. Cancer Res. 2009;15:5626–5645. doi: 10.1158/1078-0432.CCR-09-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan V., Howland M., Kutzner B., McNamee J. P., Bellier P. V., Wilkins R. C. Int. J. Hyg. Environ. Health. 2012;215:339–344. doi: 10.1016/j.ijheh.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Jeng-Jong H., Guan-Lu L., Shyh-Chang S. China's Med. 1999;112:340. [Google Scholar]
- Yamashita S. Biochem. Biophys. Res. Commun. 2011;414:795–800. doi: 10.1016/j.bbrc.2011.10.006. [DOI] [PubMed] [Google Scholar]
- Min K., Kojima J., Saito K., Furusaki S., Sugo T. Cancer Res. 2001;61:3894–3901. [Google Scholar]
- Kulkarni R., Marples B., Balasubramaniam M., Thomas R. A., Tucker J. D. Radiat. Res. 2010;173:635–644. doi: 10.1667/RR1737.1. [DOI] [PubMed] [Google Scholar]
- Kuncic Z., Byrne H. L., Mcnamara A. L., Guatelli S., Domanova W., Incerti S. Comput. Math. Methods Med. 2012;2012:147252. doi: 10.1155/2012/147252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Wang X., Tong J. J. Toxicol. Environ. Health, Part A. 2016;79:436–441. doi: 10.1080/15287394.2016.1176629. [DOI] [PubMed] [Google Scholar]
- Tao S., Rojo de la Vega M., Quijada H., Wondrak G. T., Wang T., Garcia J. G., Zhang D. D. Sci. Rep. 2016;6:18760. doi: 10.1038/srep18760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anon E., Serra-Picamal X., Hersen P., Gauthier N. C., Sheetz M. P., Trepat X., Ladoux B. Proc. Natl. Acad. Sci. U. S. A. 2012;109:10891–10896. doi: 10.1073/pnas.1117814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn E. J., Hooper M., Riker C., Butler K. M., Rademacher K., Wiggins A., Rayens M. K. J. Environ. Health. 2017;79:8. [PMC free article] [PubMed] [Google Scholar]
- D'Anna C., Cigna D., Costanzo G., Ferraro M., Siena L., Vitulo P., Gjomarkaj M., Pace E. Life Sci. 2015;126:10–18. doi: 10.1016/j.lfs.2015.01.017. [DOI] [PubMed] [Google Scholar]
- Hernandez-Boussard T., Rodriguez-Tome P., Montesano R., Hainaut P. Hum. Mutat. 1999;14:1–8. doi: 10.1002/(SICI)1098-1004(1999)14:1<1::AID-HUMU1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Hayashida T., Jinno H., Kitagawa Y., Kitajima M. J. Oncol. 2011;2011:591427. doi: 10.1155/2011/591427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo J. F., Yu C. C., Chiou S. H., Huang C. Y., Jan C. I., Lin S. C., Liu C. J., Hu W. Y., Yu Y. H. Cancer Res. 2011;71:1912–1923. doi: 10.1158/0008-5472.CAN-10-2350. [DOI] [PubMed] [Google Scholar]
- Maitah M. Y., Ali S., Ahmad A., Gadgeel S., Sarkar F. H. PLoS One. 2011;6:e16068. doi: 10.1371/journal.pone.0016068. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Tao S., Wang S., Moghaddam S. J., Ooi A., Chapman E., Wong P. K., Zhang D. D. Cancer Res. 2014;74:7430–7441. doi: 10.1158/0008-5472.CAN-14-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y., Liu Q., He X., Yuan X., Chen Y., Chu Q., Wu K. J. Hematol. Oncol. 2016;9:14. doi: 10.1186/s13045-016-0246-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao S., Rojo de la Vega M., Chapman E., Ooi A., Zhang D. D. Mol. Carcinog. 2018;57(2):182–192. doi: 10.1002/mc.22745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Ma Y., Yang X., Xu X., Zhao Y., Zhu Z., Wang X., Deng H., Li C., Gao F., Tong J., Yamanaka K., An Y. Free Radical Biol. Med. 2015;89:209–219. doi: 10.1016/j.freeradbiomed.2015.07.153. [DOI] [PubMed] [Google Scholar]