

Abstract
Two population-based studies key to advancing knowledge of brain aging are the Honolulu-Asia Aging Study (HAAS) and the Nun Study. Harmonization of their neuropathologic data allows cross comparison, with findings common to both studies likely generalizable, while distinct observations may point to aging brain changes that are dependent on sex, ethnicity, environment, or lifestyle factors. Here, we expanded the neuropathologic evaluation of these 2 studies using revised NIA-Alzheimer’s Association guidelines and compared directly the neuropathologic features of resistance and apparent cognitive resilience. There were significant differences in prevalence of Alzheimer disease neuropathologic change, small vessel vascular brain injury, and Lewy body disease between these 2 studies, suggesting that sex, ethnicity, and lifestyle factors may significantly influence resistance to developing brain injury with age. In contrast, hippocampal sclerosis prevalence was very similar, but skewed to poorer cognitive performance, suggesting that hippocampal sclerosis could act sequentially with other diseases to impair cognitive function. Strikingly, despite these observed differences, the proportion of individuals resistant to all 4 diseases of brain or displaying apparent cognitive resilience was virtually identical between HAAS and Nun Study participants. Future in vivo validation of these results awaits comprehensive biomarkers of these 4 brain diseases.
Keywords: Alzheimer disease neuropathologic change, Cognitive resilience, NIA-AA guidelines, Population-based cohort
INTRODUCTION
Alzheimer disease (AD) is a common, chronic neurodegenerative disease for which advancing age and inheritance of the ε4 allele of the apolipoprotein E gene (APOE) are the major known risk factors. According to recent consensus guidelines (1), AD is considered a pathophysiologic process, or set of processes, characterized by structural changes in brain including amyloid beta (Aβ) deposition, neurofibrillary degeneration, as evidenced by accumulation of neurofibrillary tangles (NFTs), and neuritic plaque formation, which together constitute AD neuropathologic change. This process often, but not always, culminates in clinical expression of AD dementia (1).
Multiple research cohorts have been established for clinico-pathologic correlation of AD. Fewer population-based cohorts have been established, but these have highlighted 3 important neuropathologic features of the dementia syndrome. First, the neuropathologic changes of AD uncommonly exist in isolation in the brains of older individuals, but rather are more often variably combined, or comorbid, with other lesions that underlie the dementia syndrome (2–7). Such lesions include microinfarcts from vascular brain injury (VBI) (8), Lewy bodies (LBs) observed in a variety of clinical contexts that are collectively known pathologically as LB disease (LBD) (9), and hippocampal sclerosis (HS) (10, 11). Second, the neuropathologic changes of AD, VBI, LBD, and HS are varyingly latent, meaning that each can be present in individuals who were shown to be cognitively unimpaired proximate to death (6, 7). Third, the frequency and severity of these neuropathologic changes in cognitively intact older individuals varies, with some showing a lesion burden considered sufficient evidence for dementia (6, 7, 12–20), a situation commonly referred to as apparent cognitive resilience. Together, these repeated findings highlight 2 important subsets among those who maintain cognitive function into old age: those resistant to disease who do not develop neuropathologic lesions, and those apparently resilient to the clinical expression of disease who do develop abundant neuropathologic lesions but fail to succumb to the expected cognitive impairment. Important gaps in our knowledge concerning such resistance and resilience include the extent to which individuals are resistant to one or more of the diseases that commonly afflict aging brain, and the extent to which resistance and apparent cognitive resilience may vary with sex, ethnicity, or lifestyle factors.
Two population-based studies that have been key to advancing our knowledge of brain aging are the Honolulu-Asia Aging Study (HAAS) and the Nun Study (17, 18, 21–24). The HAAS comprises men of Japanese ancestry born on Oahu between 1900 and 1919, while the Nun Study comprises Roman Catholic School Sisters of Notre Dame, who were predominantly Caucasian and born in the United States between 1890 and 1916 (21, 23, 24). Given the broad differences in sex, ethnicity, and lifestyle factors—such as education level, diet, and incidence of smoking (20, 25, 26)—of these 2 groups, we hypothesized that findings common to HAAS and Nun Study participants are likely generalizable to most people in the US, while observations distinct to one or the other study may point to changes in aging brain that are dependent on sex, ethnicity, environment, or lifestyle factors (20). Recently, we harmonized existing neuropathologic data from these 2 iconic studies to allow cross comparison (20). Here, we expanded the neuropathologic evaluation of these 2 studies by applying the revised NIA-AA neuropathologic guidelines (1), and compared directly the neuropathologic features of the 2 cohorts to investigate features of resistance and apparent resilience that are both shared and distinct.
MATERIALS AND METHODS
Cohorts
Standard protocol approvals, registrations, and patient consents were as previously described by us (20). Briefly, the Nun Study was reviewed and approved by Universities of Kentucky and Minnesota institutional review boards (IRBs), and the HAAS was reviewed and approved by the Kuakini Hospital IRB. As previously noted (20), autopsy rates were 25% for HAAS participants, while, prior to death, all participating School Sisters of Notre Dame agreed to autopsy, with final authorizations provided by the Provincial Leader; an autopsy has been completed for 90% of the Nun Study participants.
Cognitive Assessment
Nun Study primary cognitive testing was done annually with the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) neuropsychological battery (27). HAAS participants were screened at each examination with the Cognitive Abilities and Screening Instrument (CASI) (28). Scores were aligned as previously described by us (20).
Neuropathologic Evaluation
We applied the 2012 NIA–AA guidelines to generate “A” (Aβ plaques by Thal stain), “B” (NFTs by Braak stain), and “C” (neuritic plaques from modified CERAD staining) scores (1) for each case. “B” and “C” scores were obtained mostly from previously stained slides. “A” scores were determined using amyloid beta immunohistochemistry exactly as recommended in the guidelines (1) and as previously done by us for other studies (29, 30).
Indices of Disease Burden
We have previously devised and applied indices of the extent of neuropathologic change designated as severe, moderate, or none/negligible (20). Briefly, severe AD pathologic change (index = 1.0) is defined as Braak stage V or VI, moderate (index = 0.4) as Braak stage IV, and none/negligible (index = 0) as Braak stage <IV (31). Severe LBD (index = 1.0) is based on a McKeith score of 7 or higher (9), moderate (index = 0.4) on a McKeith score of 2–6, and none/negligible (index = 0) for no LBs observed. Severe HS (index = 1.0) is based on bilateral occurrence, moderate (index = 0.4) on unilateral occurrence, and none/negligible (index = 0) for no evidence of the abnormality (10, 11). Severe microinfarcts (index = 1.0) requires more than 3 microinfarcts counted in standard screening sections, moderate (index = 0.4) requires 2 or 3 microinfarcts, and none/negligible (index = 0) means one or none are found.
Analysis
Statistical analyses were performed using GraphPad Prism (San Diego, CA). Student t-tests and χ-square tests were used with α = 0.05.
RESULTS
The NIA-AA revised criteria were applied to all eligible HAAS and Nun Study cases (Table 1). In total, we generated 1262 complete NIA-AA ABC Scores in what we have designated the neuropathology (NP) groups. Of these, 747 with ≤2 years from last clinical evaluation to autopsy and APOE genotype were designated clinico-pathologic correlation (CPC) cases (Table 1).
TABLE 1.
Number of Brain Autopsies in the Neuropathology (NP) and Clinico-Pathologic Correlation (CPC) Subsets for the Nun Study and Honolulu-Asia Aging (HAAS) Study
| HAAS (#) | Nun Study (#) | Total (#) | |
|---|---|---|---|
| Total brain autopsies (#) | 852 | 605 | 1457 |
| … with complete NIA-AA ABC Scores (NP) | 762 | 500 | 1262 |
| … and ≤2 years from last clinical evaluation to autopsy | 408 | 388 | 796 |
| … and APOE genotype (CPC) | 392 | 355 | 747 |
We first examined which NP groups were resistant to different aspects of AD neuropathologic change, that is, resistant to Aβ deposition (A score = 0), neurofibrillary degeneration (B score = 0), or neuritic plaque accumulation (C score = 0). Figure 1 shows the distribution of NIA-AA A, B, and C scores for the 2 NP groups. In both cohorts, participants were most resistant to neuritic plaque accumulation and least resistant to neurofibrillary degeneration. HAAS participants were more resistant than Nun Study participants, primarily to Aβ deposition. χ-square tests comparing the distribution of NIA-AA scores among individuals in the 2 NP cohorts were significant for A score (p < 0.0001), B score (p < 0.0001), and C score (p < 0.0001), with the Nun Study participants having a higher median A score (3 compared to 2 for HAAS), but the same median B and C score as HAAS participants (2 in both). These results were the same when analysis was confined to the CPC subsets of each study (not shown).
FIGURE 1.

Heat maps of NIA-AA ABC scores for HAAS-NP and Nun Study–NP groups, ordered from lowest to highest for A, then B, and then C score. Each row presents the results from each of the 762 men in HAAS-NP (A score) or each of the 500 women in Nun Study–NP. χ-square tests comparing the distribution of NIA-AA scores between the HAAS-NP and Nun Study–NP groups were significant for A score (p < 0.0001), B score (p < 0.0001), and C score (p < 0.0001).
We next looked at the extent of AD neuropathologic change, classified as Not, Low, Intermediate, or High, as derived from the ABC scores according to the NIA-AA guidelines (1). As expected from these scores, the HAAS-NP group (Fig. 2A) had more than twice as many participants who were categorized as Not AD neuropathologic change by NIA-AA guidelines than the Nun Study–NP group (χ-square test p < 0.0001). Figure 2B plots the distribution of B scores for those HAAS-NP and Nun Study–NP cases who were Not AD neuropathologic change because their NIA-AA A and C scores were 0, and reflects the relatively common observation of NIA-AA B scores of 1 or 2 (and rarely B score of 3) in older individuals who lack Aβ accumulation. Interestingly, the distribution of B scores in those with A and C scores of 0 was significantly different between the 2 groups (p < 0.001) with HAAS B scores shifted to higher values. Together, these results show that the HAAS-NP group was more resistant to Aβ accumulation than Nun Study–NP group, and for those without Aβ accumulation, the Nun Study–NP group was more resistant to neurofibrillary degeneration.
FIGURE 2.

Percent frequency distribution plots for HAAS-NP and Nun Study–NP. (A) Plots the distribution of the NIA-AA level of AD neuropathologic changes, and (B) plots the distribution of NIA-AA B scores among those with NIA-AA A and C scores of 0.
Next, we focused on the CPC subsets with cognitive data to assess apparent resilience. We further restricted the CPC subsets to those with known APOE genotype (Table 1) to permit assessment of its potential impact on apparent resilience. Table 2 presents descriptive and genetic data for the HAAS-CPC and Nun Study–CPC groups; note the previously described difference in mean level of education between the 2 groups. Based on eligibility criteria for the CPC subsets, expectedly the interval between last clinical evaluation and death was not different between the HAAS-CPC and the Nun Study–CPC. The HAAS used CASI as a global cognitive screen, and the HAAS-CPC subset had a median CASI score of 66 out of 100. The Nun Study used CERAD total score as a global cognitive screen, and the Nun Study–CPC subset had a median result that was approximately one-half of the maximum, viz., median CERAD total score of 51 out of 100 (32).
TABLE 2.
Characteristics of Clinico-Pathologic Correlation (CPC) Subsets of the Nun Study (NS) and Honolulu-Asia Aging Study (HAAS)
| Age at death (y) |
Education (y)* |
APOE ε4
allele |
Last CASI score† |
Last CERAD score† |
|
|---|---|---|---|---|---|
| Mean ± SD | Mean ± SD | % total | Median (25th–75th %tile) | ||
| HAAS-CPC | 88 ± 6 | 11 ± 3 | 11 | 66 (30–82) | — |
| Nun Study–CPC | 91 ± 5 | 16 ± 3 | 13 | — | 51 (22–69) |
p < 0.0001.
Out of total of 100.
Previously, we devised index measures of AD neuropathologic change and commonly observed comorbid lesions in brain of older individuals, including LBD, MVI, and HS (20), to facilitate comparison of total burden of these diseases in brain. Figure 3 shows the distribution of none/negligible, moderate, and severe index measures for individuals in HAAS-CPC and NP-CPC stratified by quartile for performance at last cognitive screen. We have previously described the individual complex interaction among these 4 diseases and the likelihood of cognitive impairment (20). Here we focused on those resistant to all 4 diseases of brain. Indeed, the percentage of individuals resistant to all 4 types of pathologic changes by performance quartile is remarkably similar for these 2 very different cohorts (Fig. 4).
FIGURE 3.

Heat maps of neuropathologic indices for HAAS-CPC and Nun Study–CPC, ordered from lowest to highest for AD index, then VBI index, then LBD index, and then HS index. Each numbered row presents the results from each man in HAAS-CPC or each woman in Nun Study–CPC. Percentages at the top of each column are frequency of moderate or severe index pathologic change for each disease.
FIGURE 4.

Percent of individuals with none/negligible neuropathologic change for AD, VBI, LBD, and HS in HAAS and Nun Study participants stratified by cognitive performance quartile (Q) within 2 years of death. Q1 is the highest and Q4 the lowest performing group.
We also investigated whether APOE genotype might be related to resistance to brain injury from these 4 diseases. Table 3 shows the results of χ-square tests for the distribution of APOE ε2, ε3, and ε4 alleles for those individuals with none/negligible versus moderate or severe neuropathologic index for each disease type. Among instances that yielded a statistically significant result, the moderate or severe neuropathologic index groups had a greater proportion of APOE ε4 alleles, and a lower proportion of APOE ε2 alleles, than the none/negligible neuropathologic index group, with one exception. The exception was LBD in HAAS participants who had lower proportions of APOE ε2 and APOE ε4 in the moderate or severe neuropathologic index groups; for this reason, the combined analysis for LBD is weaker than for the Nun Study alone.
TABLE 3.
Proportion of APOE Alleles and χ-Square Values for the Distribution of APOE Alleles Among Individuals With None/Negligible Neuropathologic Index (20) of Each of the 4 Neurodegenerative Diseases Versus Those With Moderate or Severe Indices For the Combined Clinico-Pathologic Correlation (CPC) Subsets of the Nun Study and the Honolulu-Asia Aging Study (HAAS)
| Neuropathologic Index: | None/ Negligible |
Intermediate/
High |
χ-Square Value | ||||
|---|---|---|---|---|---|---|---|
| APOE (%) | ε2 | ε3 | ε4 | ε2 | ε3 | ε4 | |
| AD | 12 | 82 | 6 | 5 | 72 | 23 | 50**** |
| VBI | 10 | 79 | 11 | 5 | 73 | 22 | 11** |
| LBD | 13 | 76 | 11 | 4 | 81 | 15 | 7* |
| HS | 9 | 78 | 13 | 8 | 78 | 14 | 3 |
AD, Alzheimer disease, VBI, vascular brain injury, LBD, Lewy body disease, HS, hippocampal sclerosis.
p < 0.0001.
p < 0.01.
p < 0.05.
Although, the proportion of individuals resistant to neuropathologic changes from the 4 diseases was very similar in these 2 cohorts, the prevalence of the different brain diseases was dissimilar in HAAS and Nun Study participants. The prevalence of the 4 common brain diseases is noted at the top of columns in Figure 3 and summarized in Figure 5 as the difference in prevalence in the HAAS versus the Nun Study stratified by cognitive performance quartile (with Q1 the highest performance and Q4 the lowest). Small vessel VBI was more common among HAAS (p < 0.001), and LBD was more common among Nun Study participants (p < 0.0001) across all quartiles, but most pronounced in Q3 and Q4. Although overall the HAAS-CPC group was more resistant to AD neuropathologic change than Nun Study–CPC; this relationship was complex and varied by cognitive performance quartile, such that HAAS-CPC participants had greater AD neuropathologic change than Nun Study–CPC in Q1 and Q2, roughly equal prevalence in Q3, and lower prevalence in Q4. The prevalence of HS was very similar in all quartiles in the 2 groups.
FIGURE 5.
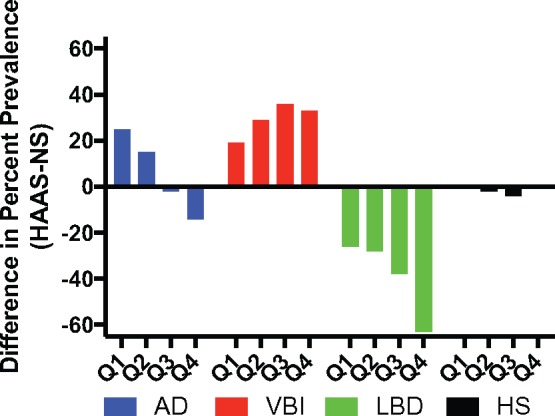
Difference in the percent prevalence of the neuropathologic changes of AD, VBI, LBD, or HS in HAAS and Nun Study participants stratified by quartile (Q) of last cognitive test results. Q1 is the highest and Q4 the lowest performing group.
zFinally, we investigated individuals in Q1 who had high levels of neuropathologic changes for AD, VBI, LBD, or HS, or their combination, within 2 years of death (Fig. 6). High level neuropathologic changes for any one of these diseases is considered sufficient evidence for a clinical diagnosis of dementia, so their presence in individuals in Q1 is a state of apparent cognitive resilience. In HAAS, 21 participants (5%, 4 were heterozygous for APOE ε4) with last cognitive evaluation in Q1 had high levels of neuropathologic changes for 1 or 2 of these diseases; none had high levels of 3 or 4 diseases. The 3 Q1 HAAS individuals with comorbid disease each had high levels of AD and VBI. In the Nun Study, 24 individuals (7%, 1 was homozygous and 2 were heterozygous for APOE ε4) with last cognitive evaluation in Q1 had high levels of neuropathologic changes for 1 or 2 of these diseases; again, none had 3 or 4 comorbid diseases. The 3 Nun Study participants who had high levels of 2 diseases were VBI plus LBD in 2 participants and high level AD plus LBD in another. Using χ-square tests, the distribution of APOE alleles was not significantly different among the Q1 participants with high neuropathologic index of disease (apparent cognitive resilience) versus those in Q1 with none/negligible neuropathologic index of disease for both HAAS and Nun Study. Although the number of observations for apparent cognitive resilience is low, these data do not support the APOE locus as a major influence on apparent cognitive resilience to these 4 diseases.
FIGURE 6.

Percent prevalence of severe neuropathologic index for AD, VBI, LBD, HS, or their combination in HAAS and Nun Study participants who performed in the top quartile (Q1) for cognitive performance within 2 years of death.
DISCUSSION
Large community or population-based autopsy cohorts include older individuals who retain high cognitive function and therefore allow for a different perspective than case-control research cohorts of age-related diseases that threaten cognitive health. High cognitive function may be retained in advanced age either because of resistance to clinically meaningful neuropathologic lesions, or despite heavy disease burden (also known as apparent cognitive resilience). Here we report for the first time NIA-AA neuropathologic evaluations of HAAS and Nun Study, 2 iconic studies of brain aging and dementia. These 2 studies are inherently different in that the HAAS comprises men of Japanese ancestry living on the island of Oahu, while Nun Study participants were women from mainland United States and predominantly Caucasian. It is these broad demographic differences that motivated the direct comparison of these 2 studies; we reasoned that findings common to these 2 very different groups are more likely generalizable to most other populations within the United States, while observations distinct to one or the other cohort may point to changes in aging brain that are dependent on sex, ethnicity, environment, or lifestyle factors. Broadly, the 2 groups were remarkably similar in the proportion of individuals resistant to neuropathologic changes of AD, VBI, LBD, and HS, the proportion of individuals with apparent resilience to clinical expression of overall high levels of neuropathologic changes, increasing comorbidity with increasing cognitive impairment, and the prevalence and distribution of HS. The 2 groups differed in the neuropathologic burden of AD, LBD, and VBI, their distribution relative to cognitive performance, and their relationship of LBD, VBI, and HS to APOE genotype.
We used 2 different subsets of participants in our analyses. The first was the NP subset, defined by those participants who had complete NIA-AA consensus neuropathologic evaluations. We completed ABC scores on all cases where the required tissue blocks were available, resulting in 82% of all brain autopsies from these 2 studies being included in the NP groups. The second was a subset of the NP groups that also had last cognitive evaluation within 2 years of death, and who had APOE genotype determined; these CPC groups were used for clinico-pathologic correlation. Last clinical evaluation proximate to death was a key element in our focus on cognitive resilience in order to limit the interval between screening assessment of cognitive function and neuropathologic evaluation. We have shown previously that clinico-pathologic correlations remain unchanged when this interval is limited to 2 years or less (20). Average intervals from last cognitive screen in the present study were 0.8 and 1.0 years, and we note that the interval between cognitive screening and neuroimaging commonly can be 0.5 years. A greater fraction of Nun Study participants were within the 2-year limit because of differences in the organization of follow up assessments between the 2 studies (19–21, 23).
AD neuropathologic changes were frequent among HAAS-NP and Nun Study–NP. Individuals in both cohorts were most resistant to neuritic plaque accumulation and least resistant to neurofibrillary degeneration. However, there were significant differences between the 2 groups. Indeed, the distribution of A, B, or C Score was significantly greater in Nun Study participants for each score. Although we cannot pinpoint the reasons for apparent lower resistance to AD neuropathologic change in Nun Study participants, it is interesting to note that our results are consistent with reports that indicate a greater risk for AD dementia among women (33–35). Although Nun Study participants were less resistant to accumulation of AD neuropathologic changes than HAAS participants, when focusing on those who lacked Aβ accumulation in brain (A score and C score of 0), HAAS participants were less resistant to neurofibrillary degeneration, perhaps echoing earlier observations of what was then called tangle-predominant or tangle-only dementia being more common in individuals of Asian ancestry. Overall, our results show that in these 2 very different groups, resistance to AD neuropathologic changes was greatest for neuritic plaque formation and least for neurofibrillary degeneration. Nun Study participants were less resistant to AD neuropathologic changes, and HAAS participants were less resistant to neurofibrillary degeneration in the absence of Aβ accumulation. Our results suggest that AD neuropathologic changes may be subject to partial modification by sex, ancestry, and/or lifestyle factors.
An intriguing rare occurrence in both cohorts was isocortical neurofibrillary degeneration in those without Aβ accumulation. Although these cases were few, the distribution of neurofibrillary degeneration was typical for higher Braak stages in both HAAS and NS cases. That such examples are rare reinforces emerging data from experimental models that supports some mechanistic interaction between Aβ accumulation and extension of neurofibrillary degeneration from medial temporal lobe structures to isocortex. However, rare cases of isocortical neurofibrillary degeneration in the absence of Aβ accumulation suggests that Aβ accumulation may not be the exclusive promoter for isocortical expansion of neurofibrillary degeneration. If true, our data suggest that the frequency of hypothesized alternate promoters for isocortical expansion of neurofibrillary degeneration was more common in HAAS participants (36–44).
Both HAAS and Nun Study participants showed significantly increased frequency of all 4 neuropathologic changes with decreasing cognitive performance. While it is not possible to determine longitudinal change from autopsy data, if some combination of these 4 diseases was sequential, or even usually sequential, then one would expect to see this relationship emerge in the cross-sectional data advancing from rare in Q1 to most common in Q4. Only HS met this criterion, raising the possibility that the sequential addition of HS to other types of brain injury may conspire to impair cognitive performance in older individuals. The shared feature of comorbidity among these 2 different cohorts is critically important to consider because it raises the possibility that, for the majority of older individuals at all levels of cognitive performance, therapies directed at just AD, VBI, LBD, or HS might have benefit restricted to the relatively small subset of individuals with only that one disease. Moving forward, comprehensive in vivo biomarker testing for these 4 common diseases will be critical for organization of clinical trials and patient management.
Interestingly, our results show very similar rates of apparent cognitive resilience (5% in the HAAS and 7% in the Nun Study) to high level neuropathologic changes; however, the particular types of neuropathologic change varied between the 2 groups. One interpretation of these results is that like resistance to neuropathologic change, apparent cognitive resilience is not disease-specific but rather resides in a subset of individuals regardless of the disease or combination of diseases present in brain.
Our results support prior studies that demonstrate an association of APOE genotype with resistance to certain diseases; however, it was not a major factor in apparent cognitive resilience (45–48). As expected, inheritance of APOE ε4 allele was strongly associated with higher neuropathologic index for AD in both HAAS and Nun Study, although this association was considerably stronger in the Nun Study. We observed the same pattern for APOE with VBI and more strongly for LBD in the Nun Study but not HAAS. It is worth noting that in Nun Study participants, the association of APOE genotype with LBD was stronger than the association of APOE genotype with AD neuropathologic change in HAAS. In contrast, HS was weakly associated with APOE ε4 in HAAS but not Nun Study participants. Previously, we have associated APOE ε4 with greater risk of LBD in the research cohort of the Parkinson’s Disease Cognitive Genetics Consortium, a mixture of largely Caucasian men and women from across the United States (49). Others have suggested that VBI also may be associated with APOE ε4 (50–52). Our results suggest that, unlike the strong association of APOE genotype with AD neuropathologic change in both studies, the other neuropathologic associations with APOE genotype may be contextual and not observed in all populations.
In summary, there were significant differences in the prevalence of AD neuropathologic changes, small vessel VBI, and LBD between the participants in these 2 studies, suggesting that sex, ethnicity, and/or lifestyle factors may significantly influence the resistance to developing these types of brain injury as we age. In contrast, prevalence of HS was very similar in the 2 groups, but skewed to poorer cognitive performance, suggesting that HS potentially could act sequentially with other diseases to impair cognitive function. Strikingly, despite these differences in brain lesions, the proportion of individuals resistant to all 4 diseases of brain and the proportion of individuals displaying apparent cognitive resilience was virtually identical between HAAS and Nun Study participants. These were unexpected findings because we anticipated resistance and resilience to be disease specific. Although speculative, these data raise the possibility that some individuals are generally resistant or resilient to brain injury with age, rather than resistance or resilience being focused on a particular disease.
ACKNOWLEDGMENTS
The authors would like to thank Allison Beller, Natalie Coleman, Kim Howard, and Aimee Schantz for administrative and technical assistance; the School Sisters of Notre Dame and men of the HAAS for their generous participation in these studies; and the late Dr. William Markesbery for his contributions to both studies and to the field of neuropathology.
REFERENCES
- 1. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 2012;8:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 2012;123:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brayne C, Richardson K, Matthews FE, et al. Neuropathological correlates of dementia in over-80-year-old brain donors from the population-based Cambridge city over-75s cohort (CC75C) study. J Alzheimers Dis 2009;18:645–58 [DOI] [PubMed] [Google Scholar]
- 4. Kovari E, Charidimou A, Herrmann FR, et al. No neuropathological evidence for a direct topographical relation between microbleeds and cerebral amyloid angiopathy. Acta Neuropathol Commun 2015;3:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jellinger KA, Attems J.. Challenges of multimorbidity of the aging brain: a critical update. J Neural Transm (Vienna) 2015;122:505–21 [DOI] [PubMed] [Google Scholar]
- 6. Schneider JA, Aggarwal NT, Barnes L, et al. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis 2009;18:691–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sonnen JA, Santa Cruz K, Hemmy LS, et al. Ecology of the aging human brain. Arch Neurol 2011;68:1049–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sonnen JA, Larson EB, Crane PK, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol 2007;62:406–13 [DOI] [PubMed] [Google Scholar]
- 9. McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 1996;47:1113–24 [DOI] [PubMed] [Google Scholar]
- 10. Zarow C, Weiner MW, Ellis WG, et al. Prevalence, laterality, and comorbidity of hippocampal sclerosis in an autopsy sample. Brain Behav 2012;2:435–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nelson PT, Schmitt FA, Lin Y, et al. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain 2011;134:1506–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bennett DA, Arnold SE, Valenzuela MJ, et al. Cognitive and social lifestyle: links with neuropathology and cognition in late life. Acta Neuropathol 2014;127:137–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boyle PA, Yu L, Wilson RS, et al. Relation of neuropathology with cognitive decline among older persons without dementia. Front Aging Neurosci 2013;5:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jellinger KA, Attems J.. Neuropathological approaches to cerebral aging and neuroplasticity. Dialogues Clin Neurosci 2013;15:29–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Knopman DS, Parisi JE, Salviati A, et al. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 2003;62:1087–95 [DOI] [PubMed] [Google Scholar]
- 16. Mufson EJ, Malek-Ahmadi M, Perez SE, et al. Braak staging, plaque pathology, and APOE status in elderly persons without cognitive impairment. Neurobiol Aging 2016;37:147–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Riley KP, Snowdon DA, Markesbery WR.. Alzheimer’s neurofibrillary pathology and the spectrum of cognitive function: findings from the nun study. Ann Neurol 2002;51:567–77 [DOI] [PubMed] [Google Scholar]
- 18. SantaCruz KS, Sonnen JA, Pezhouh MK, et al. Alzheimer disease pathology in subjects without dementia in 2 studies of aging: the Nun Study and the Adult Changes in Thought Study. J Neuropathol Exp Neurol 2011;70:832–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Snowdon DA. Healthy aging and dementia: findings from the Nun Study. Ann Intern Med 2003;139:450–4 [DOI] [PubMed] [Google Scholar]
- 20. White LR, Edland SD, Hemmy LS, et al. Neuropathologic comorbidity and cognitive impairment in the Nun and Honolulu-Asia Aging Studies. Neurology 2016;86:1000–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Snowdon DA, Greiner LH, Mortimer JA, et al. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997;277:813–7 [PubMed] [Google Scholar]
- 22. Launer LJ, Hughes TM, White LR.. Microinfarcts, brain atrophy, and cognitive function: the Honolulu Asia Aging Study Autopsy Study. Ann Neurol 2011;70:774–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. White L, Petrovitch H, Hardman J, et al. Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann N Y Acad Sci 2002;977:9–23 [DOI] [PubMed] [Google Scholar]
- 24. Gelber RP, Launer LJ, White LR.. The Honolulu-Asia Aging Study: epidemiologic and neuropathologic research on cognitive impairment. Curr Alzheimer Res 2012;9:664–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Butler SM, Snowdon DA.. Trends in mortality in older women: findings from the Nun Study. J Gerontol B Psychol Sci Soc Sci 1996;51:S201–8 [DOI] [PubMed] [Google Scholar]
- 26. Abbott RD, Ross GW, White LR, et al. Environmental, life-style, and physical precursors of clinical Parkinson’s disease: recent findings from the Honolulu-Asia Aging Study. J Neurol 2003;250 Suppl 3:Iii30–9 [DOI] [PubMed] [Google Scholar]
- 27. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991;41:479–86 [DOI] [PubMed] [Google Scholar]
- 28. Teng EL, Hasegawa K, Homma A, et al. The Cognitive Abilities Screening Instrument (CASI): a practical test for cross-cultural epidemiological studies of dementia. Int Psychogeriatr 1994;6:45–58 [DOI] [PubMed] [Google Scholar]
- 29. Montine TJ, Monsell SE, Beach TG, et al. Multisite assessment of NIA-AA guidelines for the neuropathologic evaluation of Alzheimer’s disease. Alzheimer’s Dement 2016;12:164–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Flanagan ME, Marshall DA, Shofer JB, et al. Performance of a condensed protocol that reduces effort and cost of NIA-AA guidelines for neuropathologic assessment of Alzheimer disease. J Neuropathol Exp Neurol 2017;76:39–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Braak H, Braak E.. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–59 [DOI] [PubMed] [Google Scholar]
- 32. Chandler MJ, Lacritz LH, Hynan LS, et al. A total score for the CERAD neuropsychological battery. Neurology 2005;65:102–6 [DOI] [PubMed] [Google Scholar]
- 33. Altmann A, Tian L, Henderson VW, et al. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol 2014;75:563–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barnes LL, Wilson RS, Bienias JL, et al. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry 2005;62:685–91 [DOI] [PubMed] [Google Scholar]
- 35. Filon JR, Intorcia AJ, Sue LI, et al. Gender differences in Alzheimer disease: brain atrophy, histopathology burden, and cognition. J Neuropathol Exp Neurol 2016;75:748–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bright J, Hussain S, Dang V, et al. Human secreted tau increases amyloid-beta production. Neurobiol Aging 2015;36:693–709 [DOI] [PubMed] [Google Scholar]
- 37. Selkoe DJ, Hardy J.. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 2016;8:595–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jack CR Jr, Vemuri P, Wiste HJ, et al. Evidence for ordering of Alzheimer disease biomarkers. Arch Neurol 2011;68:1526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Crary JF. Primary age-related tauopathy and the amyloid cascade hypothesis: the exception that proves the rule?. J Neurol Neuromed 2016;1:53–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128:755–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jellinger KA, Alafuzoff I, Attems J, et al. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol 2015;129:757–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mungas D, Tractenberg R, Schneider JA, et al. A 2-process model for neuropathology of Alzheimer’s disease. Neurobiol Aging 2014;35:301–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Royall DR, Palmer RF.. The temporospatial evolution of neuritic plaque-related and independent tauopathies: implications for dementia staging. J Alzheimer’s Dis 2014;40:541–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Farfel JM, Yu L, De Jager PL, et al. Association of APOE with tau-tangle pathology with and without beta-amyloid. Neurobiol Aging 2016;37:19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 1994;7:180–4 [DOI] [PubMed] [Google Scholar]
- 46. Hsiung GY, Sadovnick AD, Feldman H.. Apolipoprotein E epsilon4 genotype as a risk factor for cognitive decline and dementia: data from the Canadian Study of Health and Aging. CMAJ 2004;171:863–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nicoll JA, Savva GM, Stewart J, et al. Association between APOE genotype, neuropathology and dementia in the older population of England and Wales. Neuropathol Appl Neurobiol 2011;37:285–94 [DOI] [PubMed] [Google Scholar]
- 48. Berge G, Sando SB, Rongve A, et al. Apolipoprotein E epsilon2 genotype delays onset of dementia with Lewy bodies in a Norwegian cohort. J Neurol Neurosurg Psychiatry 2014;85:1227–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tsuang D, Leverenz JB, Lopez OL, et al. APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 2013;70:223–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zito G, Polimanti R, Panetta V, et al. Antioxidant status and APOE genotype as susceptibility factors for neurodegeneration in Alzheimer’s disease and vascular dementia. Rejuvenation Res 2013;16:51–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Souza DR, de Godoy MR, Hotta J, et al. Association of apolipoprotein E polymorphism in late-onset Alzheimer’s disease and vascular dementia in Brazilians. Braz J Med Biol Res 2003;36:919–23 [DOI] [PubMed] [Google Scholar]
- 52. Chen KL, Sun YM, Zhou Y, et al. Associations between APOE polymorphisms and seven diseases with cognitive impairment including Alzheimer’s disease, frontotemporal dementia, and dementia with Lewy bodies in southeast China. Psychiatr Genet 2016;26:124–31 [DOI] [PMC free article] [PubMed] [Google Scholar]