

Abstract
The NAD(P)+-hydrolyzing enzyme CD38 is activated in the heart during the process of ischemia and reperfusion, triggering NAD(P)(H) depletion. However, the presence and role of CD38 in the major cell types of the heart are unknown. Therefore, we characterize the presence and function of CD38 in cardiac myocytes, endothelial cells, and fibroblasts. To comprehensively evaluate CD38 in these cells, we measured gene transcription via mRNA, as well as protein expression and enzymatic activity. Endothelial cells strongly expressed CD38, while only low expression was present in cardiac myocytes with intermediate levels in fibroblasts. In view of this high level expression in endothelial cells and the proposed role of CD38 in the pathogenesis of endothelial dysfunction, endothelial cells were subjected to hypoxia-reoxygenation to characterize the effect of this stress on CD38 expression and activity. An activity-based CD38 imaging method and CD38 activity assays were used to characterize CD38 activity in normoxic and hypoxic-reoxygenated endothelial cells, with marked CD38 activation seen following hypoxia-reoxygenation. To test the impact of hypoxia-reoxygenation-induced CD38 activation on endothelial cells, NAD(P)(H) levels and endothelial nitric oxide synthase (eNOS)-derived NO production were measured. Marked NADP(H) depletion with loss of NO and increase in superoxide production occurred following hypoxia-reoxygenation that was prevented by CD38 inhibition or knockdown. Thus, endothelial cells have high expression of CD38 which is activated by hypoxia-reoxygenation triggering CD38-mediated NADP(H) depletion with loss of eNOS-mediated NO generation and increased eNOS uncoupling. This demonstrates the importance of CD38 in the endothelium and explains the basis by which CD38 triggers post-ischemic endothelial dysfunction.
Keywords: endothelium, eNOS, ischemia-reperfusion, NAD(P)(H), nitric oxide, superoxide
INTRODUCTION
The transmembrane enzyme CD38 is a multifunctional protein present in many different cell types (31). Enzymatically, CD38 has both ADP-ribosyl cyclase and NAD(P)+ hydrolase [NAD(P)+ase] activities. As a cyclase, CD38 catalyzes the production of potent Ca2+-mobilizer (phospho)-cyclic ADP-ribose [(P)-cADPR] (25). However, the NAD(P)+ase function, catalyzing the direct conversion of NAD(P)+ to (P)-ADP-ribose, has been shown to be the predominant reaction of CD38 (1, 16). (P)-ADP-ribose may have roles in Ca2+ signaling, as well as apoptosis, through signaling via TRPM2 channels (32).
CD38 was initially discovered as a cell surface molecule involved in antigen recognition on human leukocytes (37). However, over the years CD38 has been discovered to have very diverse roles in a wide variety of cell types. This includes expression on leukocytes such as T lymphocytes where CD38 is involved in activation and proliferation (37), and neutrophils where CD38 regulates the innate immune response through the production of calcium-signaling molecules that signal chemotaxis (33). CD38 has also been shown to be important in nonhematopoietic cells through the production of calcium-signaling second messengers with roles including insulin secretion (41), oxytocin release (26), smooth muscle contraction (3), and neural cell differentiation (49). CD38 has also been shown to be a key regulator of NAD(H) levels (1), which has consequences in the development of metabolic syndrome (5) and aging (9).
Adding to these many roles of CD38, we have recently demonstrated the importance of CD38 enzymatic activation occurring as a result of myocardial ischemia-reperfusion (I/R) injury (7, 39). In rat hearts subjected to 30 min of ischemia and 30 min of reperfusion, levels of NADP(H) in the whole heart were depleted by ~50%, with even greater depletion in the endothelium, >80% (39). This NADP(H) depletion impaired production of nitric oxide (NO) by endothelial nitric oxide synthase (eNOS), which utilizes NADPH as reducing substrate in NO synthesis. Blocking CD38 with flavonoid CD38 inhibitor luteolinidin demonstrated the cardioprotective effect of inhibiting CD38 activation in I/R injury (7). However, complete understanding of CD38 function in the heart is limited by a lack of understanding of its distribution and expression levels in the different cell types of the heart.
The three major primary cell types of the heart are cardiac myocytes (CMs), endothelial cells (ECs), and cardiac fibroblasts (CFs) (46). Cardiac myocytes are the main cell type of which the heart is composed and are critical for the mechanical pump function of the heart. Endothelial cells make up the interior lining of blood vessels and cardiac valves, as well as the endocardium. Fibroblasts provide the critical cellular matrix and connective tissue (4, 34, 46).
To better understand the prior observations made on the whole heart, it is important to consider each individual cell type that may be affected. Therefore, in the current study, we measured CD38 transcription, protein expression, and enzymatic activity in ECs, CFs, and CMs. Much higher expression of CD38 in ECs relative to either CFs or CMs was found. In addition, CD38 in ECs was characterized by immunocytochemistry and CD38 activity-based labeling with a fluorescent small-molecule CD38 substrate. Furthermore, hypoxia-reoxygenation (H/R) was shown to trigger CD38 activation in ECs, resulting in severe depletion of NAD(P)(H) levels that cause eNOS dysfunction and uncoupling with impaired cellular NO production and enhanced superoxide production.
MATERIALS AND METHODS
Materials.
CD38 antibody M-19, actin antibody (I-19), and horseradish peroxidase (HRP)-linked secondary antibodies were purchased from Santa Cruz Biotechnology. CD38 antibody 14.27 was purchased from BioLegend. Rat aortic endothelial cells (ECs) and rat cardiac fibroblasts (CFs) from Sprague-Dawley rats were purchased from Innoprot. ECL immunoblotting detection reagents were purchased from Amersham Biosciences. Rat recombinant CD38 was purchased from Creative Biomart. Glass-bottomed 50-mm dishes for cell imaging were purchased from MatTek Corporation. Cell culture medium was purchased from Cell Applications for CFs. The RNA cleanup and concentration kit was purchased from Norgen Biotek. All other chemicals were purchased from Sigma. CD38 probes SR101-F-araNMN and 6-alkyne-F-araNAD were kindly provided by Dr. Hening Lin (Cornell University, Ithaca, NY).
Animals.
Sprague-Dawley rats were purchased from Envigo and weighed between 250 and 300 g. The CD38−/− mice (12) breeding pairs were obtained from Dr. Eduardo Chini (Mayo Institute) (1). C57Bl/6J mice obtained from Jackson Laboratories were used as controls and are termed wild type (WT). All animal protocols were approved by the Institutional Animal Care and Use Committee of The Ohio State University and conformed to the Guide for the Care and Use of Laboratory Animals published by National Institutes of Health (Bethesda, MD).
Cell culture.
Rat aortic endothelial cells were cultured in our laboratory as previously described (14, 39). ECs were grown in DMEM supplemented with 5% fetal bovine serum, endothelial cell growth factors, nonessential amino acids, and 100 U/ml penicillin and 100 µg/ml streptomycin. Experiments were performed using cells at passages 4–10.
Cardiac fibroblasts (CFs) were cultured in our laboratory according to the manufacturer’s instructions. Cells were grown in cultureware after incubation with attachment factor poly-l-lysine (Sigma) at 2 μg/cm2 for 4 h. Cells were grown in complete rat fibroblast growth medium. Experiments were performed using cells at passages 4–10.
Cardiac myocyte isolation.
Cardiac myocytes were isolated from Sprague-Dawley rat hearts weighing between 250 and 300 g. Rats were heparinized and deeply anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (15 mg/kg). Hearts were then excised and dissected of nonmyocardial tissue in ice-cold Tyrode solution (138 mM NaCl, 4.5 mM KCl, 0.5 mM MgCl2, 0.33 mM Na2HPO4, 5.5 mM glucose, 10 mM HEPES, 15 mM 2,3-butanedione monoxime, and 0.1% bovine serum albumin (BSA) (wt/vol), adjusted to pH 7.4 with NaOH). Hearts were then cannulated via the aorta and perfused with this Tyrode solution for ~2 min to clear the tissue of blood. Then, hearts were perfused in a recirculatory manner with the same Tyrode solution containing 0.6 mg/ml Collagenase II (Worthington Biochemicals) and 50 μM CaCl2. After a 20–25 min digestion period, hearts were removed from the cannula into a dish containing a fresh Collagenase II solution. Hearts were gently torn into small pieces using two blunt end forceps, and incubated at 37°C with gentle agitation for 10–15 min. This cell suspension was then passed through a 200 μm nylon mesh and centrifuged for 3 min at 20 g. Supernatant containing contaminating cells was discarded and the myocyte pellet was resuspended in fresh Tyrode solution. Pelleting and resuspension in fresh Tyrode solution was performed an additional three times to remove contaminating cells.
mRNA extraction and purification.
mRNA was extracted from cells using TRIzol. For ECs and CFs, 3 ml TRIzol was added to cells grown in 60-mm dishes and cells were homogenized by repeated passage through a 25-gauge needle. For cardiac myocytes, 3 ml of TRIzol was added to ~100 mg of cells and this mixture was transferred to a glass tissue homogenizer for homogenization. At this step, chloroform was added to cell lysates at a 1:5 dilution and mixtures were vortexed thoroughly. This mixture was then incubated for 5 min at room temperature and centrifuged at 12,000 g for 15 min at 4°C. The RNA-containing supernatant was removed for RNA purification, performed with an RNA cleanup and concentration kit according to the manufacturer’s instructions. RNA extracts were stored at −80°C until PCR analysis.
Real-time PCR.
Real-time PCR (RT-PCR) analysis was performed by retro-transcribing 250 ng of total RNA using the High Capacity cDNA Reverse Transcription kit (Life Technologies). One microliter of this prepared cDNA was combined with 9 μl of a premade mix that included the TaqMan Fast Advanced Master Mix and the CD38 TaqMan gene expression assay (Rn00565538_m1). The comparative RT-PCR was performed in triplicate, including no-template controls, and analyzed using a 7900HT Fast RT-PCR system. The Ct average of each triplicate was used to perform the relative quantification analysis. RNA input was normalized using rat OAZ1 (Rn00821793_g1) as a reference gene, and the relative expression by cell type has been calculated using the comparative Ct method.
Western blotting.
ECs and CFs grown in 60-mm dishes were washed twice with PBS, then collected by scraping in RIPA buffer consisting of 150 mM NaCl, 10 mM Tris, 1 mM EDTA, 0.1% SDS, 0.1% sodium deoxycholate, and 1% Triton X-100, with freshly added protease inhibitors. In the case of CMs, cells were collected after digestion and washing with a final 20 g centrifugation, then resuspended in RIPA buffer. In all cases, cells were sonicated and allowed to incubate on ice for 30 min. Resulting homogenates were centrifuged at 20,000 g to pellet insoluble material, and supernatants were kept for protein concentration determination using the DC protein assay. Protein totaling 30 μg was neither boiled, nor incubated with reducing agents, and was separated on 4–20% gradient Tris-glycine polyacrylamide gels. In some experiments, rat recombinant CD38 was used as a control. Protein from gels was then semi-dry transferred to PVDF membranes for 75 min and blocked for 60 min at room temperature in 5% milk in Tris-buffered saline with 0.1% Tween 20 (TBST). Membranes were incubated overnight with anti-CD38 antibody diluted at 1:1,000 in 5% milk at 4°C. Membranes were then washed in TBST and incubated for 1 h with HRP-conjugated secondary antibody in TBST at room temperature. Imaging was performed with ECL immunoblotting detection reagents. The intensity of blotting was quantified with ImageJ (National Institutes of Health). Membranes were stripped and similarly immunoblotted with an anti-actin antibody diluted 1:5,000.
CD38 enzymatic activity.
The NAD(P)+ase activity of CD38 was assayed using fluorescence detection. The NAD+ analog nicotinamide 1,N6-ethenoadenine dinucleotide (ε-NAD) was used for NAD(P)+ase measurements (22). For each assay, cell lysate totaling 50 μg of protein was added to a 100 μl reaction mixture containing 200 μM ε-NAD. Reactions were monitored for the conversion of ε-NAD to strongly fluorescent product ε-ADP-ribose (ε-ADPR). Fluorescence was measured at an excitation wavelength of 300 nm and an emission wavelength of 410 nm on a Molecular Devices SpectraMax M2 plate reader.
Hypoxia-reoxygenation.
ECs were washed with PBS and incubated with serum-free DMEM in a hypoxic environment created by placing the flasks containing cells at confluence into a Billups-Rothenberg modular incubator chamber flushed with a 95% N2-5% CO2 gas mixture (14). Cells were kept inside the chamber at 37°C for 0.5, 1, 2, 4, or 24 h followed by reoxygenation for 1 h at 37°C by replacing the hypoxic medium with normoxic HBSS.
Immunocytochemistry of CD38.
ECs were grown in 50-mm dishes containing a fixed, glass size 0 coverslip. Cells were subjected to control or 2, 4, or 24 h hypoxia-reoxygenation treatment. After each, cells were fixed in 4% paraformaldehyde for 10 min and blocked with 1% BSA in PBS containing 0.1% Tween 20 (PBST) and 0.3 M glycine for 1 h at room temperature. Then, cells were incubated with CD38 antibody 14.27 at a 1:200 dilution in PBST overnight at 4°C. After primary antibody incubation, cells were washed 3 × 5 min in PBS, and then incubated with goat anti-mouse Alexa Fluor 594-conjugated secondary antibody (Invitrogen) in PBST at a 1:400 dilution for 1 h at room temperature in the dark. Cells were then washed 3 × 5 min in PBS, and DAPI (1 μM) was added to the cells for 5 min for nuclear staining.
Live cell labeling of CD38.
To image CD38 in cells, a dual labeling approach using small-molecule CD38 substrate SR101-F-araNMN and its blocking substrate, 6-alkyne-F-araNAD, was used (40). SR101-F-araNMN is a cell-permeable and fluorescent CD38 label while 6-alkyne-F-araNAD is a cell-impermeable and nonfluorescent CD38 label, both of which only label catalytically active CD38. Confluent ECs grown in glass-bottomed dishes either underwent control treatment, or 2, 4, or 24 h hypoxia followed by 60 min of reoxygenation. After the reoxygenation period, cells were washed with PBS and were labeled for 10 min with 10 μM SR101-F-araNMN in PBS. After being washed with PBS, cells were fixed with methanol for 10 min at −20°C, and then for 40 additional minutes on ice. In blocking experiments, a 10-min incubation period with 10 μM 6-alkyne-F-araNAD before adding SR101-F-araNMN was performed. DAPI (1 μM) was used as a nuclear stain.
Confocal microscopy of cells.
Cell imaging was performed on a modified-custom fitted Zeiss LSCM 410/REN laser scanning confocal microscope. Cells were imaged through a ×40 oil immersion Apofluor objective (numerical aperture 1.3, working distance = 170 μm). An Ar+-Kr+ laser was used to excite the cells at 488 nm (FITC) or 568 nm (Texas Red) and fluorescence emissions were passed through a FT510 dichroic mirror and detected by a photomultiplier tube equipped with a BP 505–550 filter for FITC or LP590 filter for Texas Red, positioned in front of the pinhole and light path. Scanned optical sections had a thickness of 0.5 μm–0.9 μm. Eight sections were averaged by LSM software for each final image.
Immunohistochemistry of heart tissue.
Wild-type and CD38−/− hearts were perfused with Krebs buffer for 20 min and then embedded in OCT compound. Hearts were then sectioned and used for immunohistochemistry of CD38, CD31 (PECAM-1), and discoidin domain receptor 2 (DDR2). Sections were first thawed at room temperature for 20 min. Then, sections were fixed with 4% paraformaldehyde for 20 min, washed briefly with PBS, and blocked with 10% donkey serum in PBST. After 1 h of blocking at room temperature, CD38 (ab90), CD31, and DDR2 antibodies were added at respective dilutions of 1:50, 1:100, and 1:100 in PBST with 10% donkey serum overnight at 4°C. After overnight incubation, sections were washed 3 × 10 min with PBS, and secondary antibodies (Alexa Fluor 488 for CD38, Alexa Fluor 594 for CD31/DDR2) were added at final dilutions of 1:200 for 2 h. After 3 washes of 10 min each in PBS, slides were mounted and sealed for imaging. Images were taken on an Olympus FV 1000 spectral confocal microscope with a ×60 objective.
siRNA knockdown of CD38.
CD38 knockdown (KD) was achieved using transfection of CD38 siRNA (ThermoFisher 197819) with RNAiMAX transfection reagent. Transfections were performed by separately preparing solutions of CD38 siRNA or scrambled siRNA (ThermoFisher Silencer Negative Control 1) shown to have no effect on cell proliferation, viability, or morphology and RNAiMAX transfection reagent in Opti-MEM medium. These solutions were then mixed together for 20 min at room temperature to allow formation of siRNA-lipid complexes. This mixture was then added to cells incubated in Opti-MEM medium. siRNA transfections were carried out for 12 h, when transfection medium was removed and normal growth medium was added to cells for an additional 12 h, at which point experiments were performed. Western blotting was performed and demonstrated that ~85% decrease in CD38 expression could be achieved.
HPLC analysis of NAD(P)(H).
For assaying cellular NADP(H) content, cells were grown in 60-mm dishes and were subjected to either normoxic treatment or 2, 4, or 24 h of hypoxia followed by 1 h of reoxygenation, with and without CD38 inhibitor luteolinidin. For the CD38 inhibition experiments, luteolinidin was included in the hypoxic and posthypoxic medium at 50 μM. Experiments with siRNA-mediated knockdown (KD) of CD38 were performed under normoxic conditions or after 24 h of hypoxia and 1 h of reoxygenation. After each protocol, cells were harvested by scraping in a derivatization solution of 200 mM potassium cyanide (KCN), 60 mM KOH, and 1 mM diethylenetriaminepentaacetic acid (DTPA). The addition of KCN is for the cyano-derivatization of NAD+ and NADP+, which are not fluorescent otherwise (7, 29, 39). Resulting homogenate was centrifuged to pellet insoluble debris. The supernatant was removed, and chloroform was added 1:1 for protein precipitation by vortexing and centrifugation at 15,000 g for 5 min. After chloroform mixing and centrifugation, three layers result: a lower layer of lipid-containing chloroform, a protein “disc,” and an upper aqueous layer containing polar metabolites. This upper layer was directly injected onto a Supelcosil LC-18-T (25 cm × 4.6 mm × 5 µm) column protected by a C18 guard column. Analytes were eluted with a mobile phase A (MPA) of 200 mM ammonium acetate (pH 5.8) and mobile phase B (MPB) of 200 mM ammonium acetate (pH 5.8) in 50% methanol. Separation was achieved with an initial flow rate of 1.0 ml/min consisting of 8% MPB and a linear methanol gradient (0.4% per minute for 25 min). Analytes were detected via fluorescence spectroscopy (excitation wavelength of 330 nm; emission wavelength of 460 nm). Peaks were assigned by co-elution with analytical standards, and quantitation was performed with use of standard curves prepared from analytical standards.
Nitric oxide measurement by electron paramagnetic resonance.
Spin-trapping measurements of NO from ECs were performed with a Bruker EMX electron paramagnetic resonance (EPR) spectrometer with Fe2+-(N-methyl-d-glucamine dithiocarbamate)2 [Fe2+-(MGD)2] as a NO spin trap (14, 53). The experiments were performed with ~8 × 106 cells grown in 100-mm dishes. Cells were washed with HBSS; then 5 ml of HBSS including CaCl2, MgCl2, the NO spin trap Fe2+-(MGD)2 (0.25 mM Fe2+ and 2.5 mM MGD), and calcium ionophore A23187 (1 μM) were added to each flask, and the cells were incubated for 20 min at 37°C in a humidified environment containing 5% CO2. After incubation, the medium from each flask was collected and concentrated by lyophilization. The trapped NO in the media was quantified by EPR. For NOS inhibition, l-NG-monomethyl arginine (l-NMMA) was used at a concentration of 1 mM to ensure complete inhibition. Spectra recorded from these cellular preparations in a standard quartz flat cell were obtained with the following parameters: microwave power of 10 or 40 mW, modulation amplitude of 4.0 G, and modulation frequency of 100 kHz using an EMX HS resonator.
Superoxide measurement by EPR.
The superoxide spin-trapping studies were performed using the spin trap 5,5′-dimethyl-1-pyrroline-N-oxide (DMPO) at a final concentration of 50 mM. Care was taken to keep the DMPO-containing solutions covered to prevent any light-induced degradation. The DMPO (ultrahigh purity) was purchased from Dojindo (Rockville, MD). EPR spectra were recorded in flat cells at room temperature with a Bruker EMX EPR spectrometer as described above. Measurements were performed at X-band with 100-kHz modulation frequency with 10-mW microwave power, and a modulation amplitude of 1.0 Gauss using an EMX HS resonator.
Statistical analysis.
All values were expressed as means ± SE. Comparisons between two groups were statistically evaluated by Student's t-test. P < 0.05 was considered statistically significant.
RESULTS
CD38 mRNA and expression levels by cell type.
mRNA was measured to determine the level of CD38 transcription occurring in each cell type. RT-PCR showed over 2-fold higher levels of CD38 mRNA in ECs compared with CFs, and ~10-fold higher levels than in CMs (Fig. 1A). CD38 expression was also measured by Western blotting. For each cell type, 30 μg of total protein was loaded to gels, and relative expression of CD38 was tested. Consistent with the RT-PCR results, ECs showed the highest expression of CD38 in the cells tested. Interestingly, despite only 2.5-fold higher mRNA levels, CD38 expression was more than 5-fold higher in ECs than CFs, indicating lower translation of CD38 mRNA to CD38 protein in CFs. Compared with CMs, ECs had greater than 50-fold higher levels of CD38. Overall, CMs contained only very low levels of CD38 mRNA and protein (Fig. 1B).
Fig. 1.
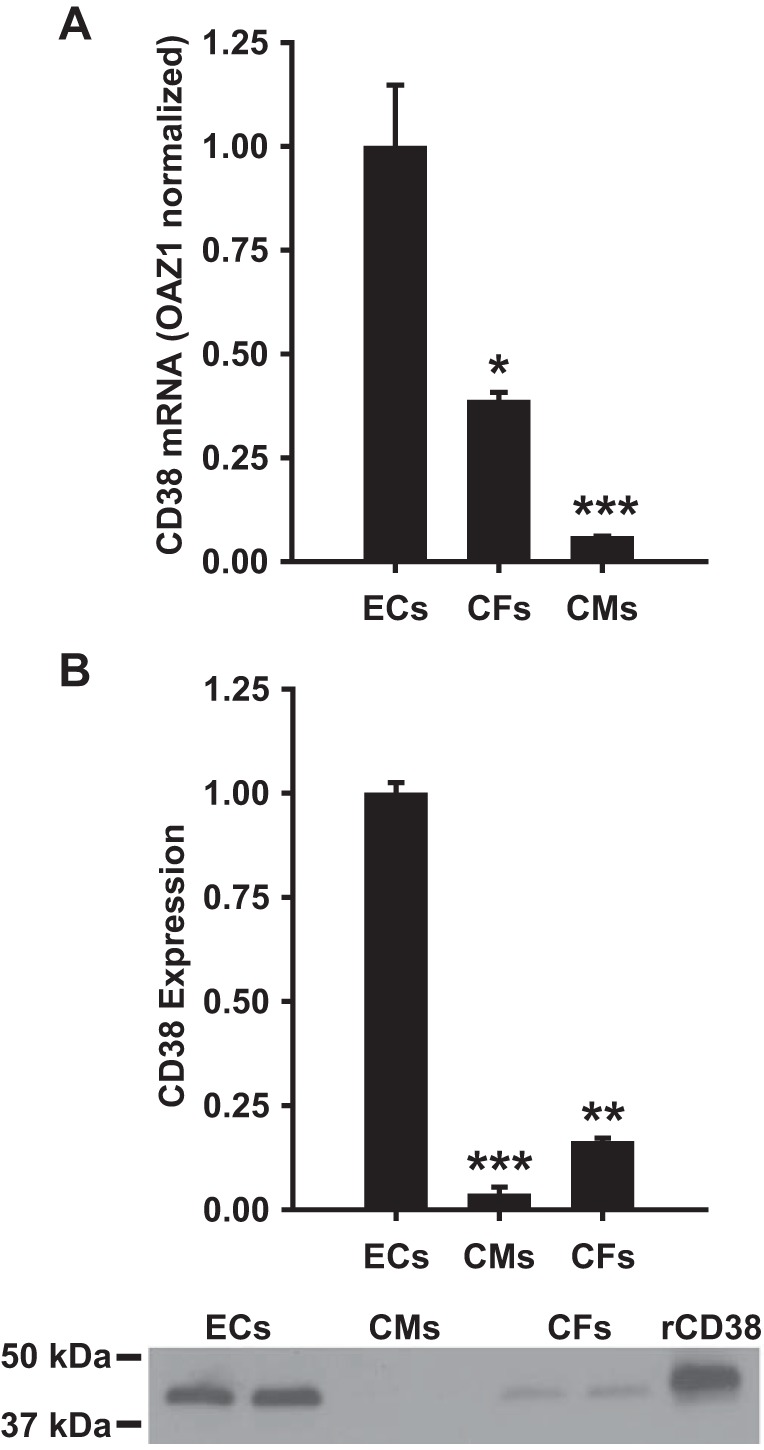
Cellular CD38 mRNA and expression levels. A: CD38 mRNA levels were significantly higher in ECs than in CFs and CMs. Levels were normalized to the housekeeping gene OAZ1, which showed similar levels of mRNA in each cell type. *P < 0.05 vs. ECs,***P < 0.001 vs. ECs (means ± SE; n = 4). B: Western blotting was used to determine the relative expression of CD38 in each cell type. For each cell type, 30 μg of total cellular protein was loaded to gels. The Western blot at the bottom of B shows a typical blot with 2 samples run for each cell type and on the right a standard of recombinant rat CD38 (rCD38) containing 100 ng of CD38. **P < 0.01 vs. ECs, ***P < 0.005 vs. ECs (means ± SE; n = 4).
CD38 activity by cell type.
We also tested the CD38 activity levels in each cell type using an NAD(P)+ase activity assay highly specific to CD38 (22). CD38 has been described as the principle, and perhaps only, NAD(P)+ase in the mammalian system, including the heart, where genetic deletion of CD38 results in complete loss of NAD(P)+ase activity (1). Thus, this highly sensitive assay is CD38-specific. Assays of CD38 activity in ECs and CFs were performed after harvesting cells in normoxic culture conditions. For CMs, CD38 activity was measured immediately after purification of live CMs from whole rat hearts. Consistent with CD38 expression experiments, CD38 activity was higher in ECs than in CFs and CMs, with only trace levels measured in CMs. CD38 activity was measured at 5.00 ± 0.25 nmol·min−1·mg protein−1 in ECs, 1.08 ± 0.18 nmol·min−1·mg protein−1 in CFs, and 0.08 ± 0.01 nmol·min−1·mg protein−1 in CMs (Fig. 2). Thus, CD38 activity is by far the highest in ECs.
Fig. 2.

Cellular CD38 activity levels. A: CD38 NAD(P)+ase activity was measured in ECs, CFs, and CMs based on the increase in fluorescence with the conversion of NAD+ analog ε-NAD to the highly fluorescent product ε-ADPR. The signal is expressed as relative fluorescence units. From this signal the activity is calculated and expressed as nmol·min−1·mg protein−1. For each cell type, 100 μg of total cellular protein was used. B: in ECs, CD38 activity was found to be 5-fold higher than in CFs, and over 50-fold higher than in CMs. *P < 0.05 and **P < 0.01 vs. ECs (means ± SE; n = 4–6).
Immunohistochemistry of CD38, CD31, and DDR2 in the heart.
To confirm that CD38 was strongly expressed in the endothelium of the heart, with lesser expression in CFs and CMs, WT and CD38−/− hearts were stained with a CD38 antibody and antibodies of common endothelial cell (CD31) (35) and cardiac fibroblast (DDR2) (21) markers. As expected, CD38 staining was found in WT but not CD38−/− hearts. CD38 staining in WT hearts largely colocalized with that of CD31, demonstrating strong endothelial expression of CD38, while staining of CD38 on surrounding cardiac myocytes was not seen (Fig. 3A). Some colocalization of CD38 with the cardiac fibroblast marker DDR2 was also found (Fig. 3B). Thus, CD38 in the heart is primarily located in vessels, with some expression on cardiac fibroblasts, and trace or no expression on cardiac myocytes.
Fig. 3.

CD38, CD31, and DDR2 immunostaining in heart tissue. A: CD38 (green) and endothelial cell marker CD31 (red) were stained in WT and CD38−/− (KO) heart sections. Using a differential interference contrast (DIC) image, CD31 is seen to strongly and selectively stain vascular endothelium. CD38 colocalized with CD31 in the WT heart sections, with merging of the two images yielding a strong yellow color. As expected, no CD38 staining is seen in KO. B: CD38 (green) and fibroblast marker DDR2 (red) were stained in WT and CD38−/− heart sections (KO). Some colocalization of CD38 and DDR2 was observed in WT sections, with no CD38 in KO.
Immunocytochemistry of CD38 in hypoxic-reoxygenated endothelial cells.
Based on the high expression level and activity of CD38 in ECs, further studies were performed to assess its baseline activity and localization in ECs. Previously, we observed that CD38 was activated by ischemia-reperfusion (I/R) in hearts (39). Thus, further characterization of CD38 in ECs subjected to hypoxia-reoxygenation (H/R) was also performed. Initially, immunocytochemistry was performed in ECs to determine how cellular localization or expression of CD38 might be affected by H/R. In control ECs, we found significant staining of CD38, which was not seen in control ECs either not receiving primary antibody, or receiving only an irrelevant isotype antibody (mouse IgG2b). Following H/R, the overall magnitude of staining for CD38 was not altered, with a similar intensity of staining as in control cells with 2, 4 and 24 h of hypoxia (Fig. 4). Thus, the total amount of CD38 present in ECs was constant before and after the stress of H/R.
Fig. 4.

Imaging of CD38 in normoxic and hypoxic-reoxygenated endothelial cells with a monoclonal CD38 antibody. A: CD38 in control or hypoxic ECs were immunostained with CD38 antibody 14.27. Staining did not differ between normoxic and hypoxic ECs. Control experiments replacing the CD38 antibody with a control irrelevant primary immunoglobulin (isotype control, isotype) or those lacking primary antibody (− primary) confirmed the specificity of the CD38 staining. B: quantitation of the fluorescence staining demonstrated similar magnitude of staining in normoxic and hypoxic-reoxygenated-treated cells.
Imaging of active CD38 in endothelial cells.
After imaging the expression and localization of CD38 in ECs subjected to normoxia or H/R with a CD38 antibody, we sought to selectively image only catalytically active CD38 using a novel fluorescent small molecule CD38 substrate, SR101-F-araNMN, which covalently binds to the active site (40). To do this, ECs subjected to normoxia or H/R were incubated with SR101-F-araNMN (10 μM). As SR101-F-araNMN only binds active CD38, this labeling method is a novel technique to assess CD38 activation. In normoxic cells, labeling with SR101-F-araNMN showed some staining above background levels, but these levels were relatively low. Following H/R, however, the intensity of SR101-F-araNMN staining increased greatly, particularly in the cells subjected to only 2 h of hypoxia followed by reoxygenation. With use of the nonfluorescent blocking substrate 6-alkyne-F-araNAD, the staining seen in control ECs was totally quenched, and greatly decreased following 2 h of H/R (Fig. 5A). Experiments were then performed to determine the effect of the duration of hypoxia on the magnitude of CD38 activation. With hypoxia durations of 2, 4, or 24 h, followed by 1 h of reoxygenation, similar strong staining indicative of activated CD38 was seen in each (Fig. 5, B and C). Thus, by 2 h of hypoxia, maximum activation of CD38 was seen and remained elevated for up to 24 h of hypoxia.
Fig. 5.
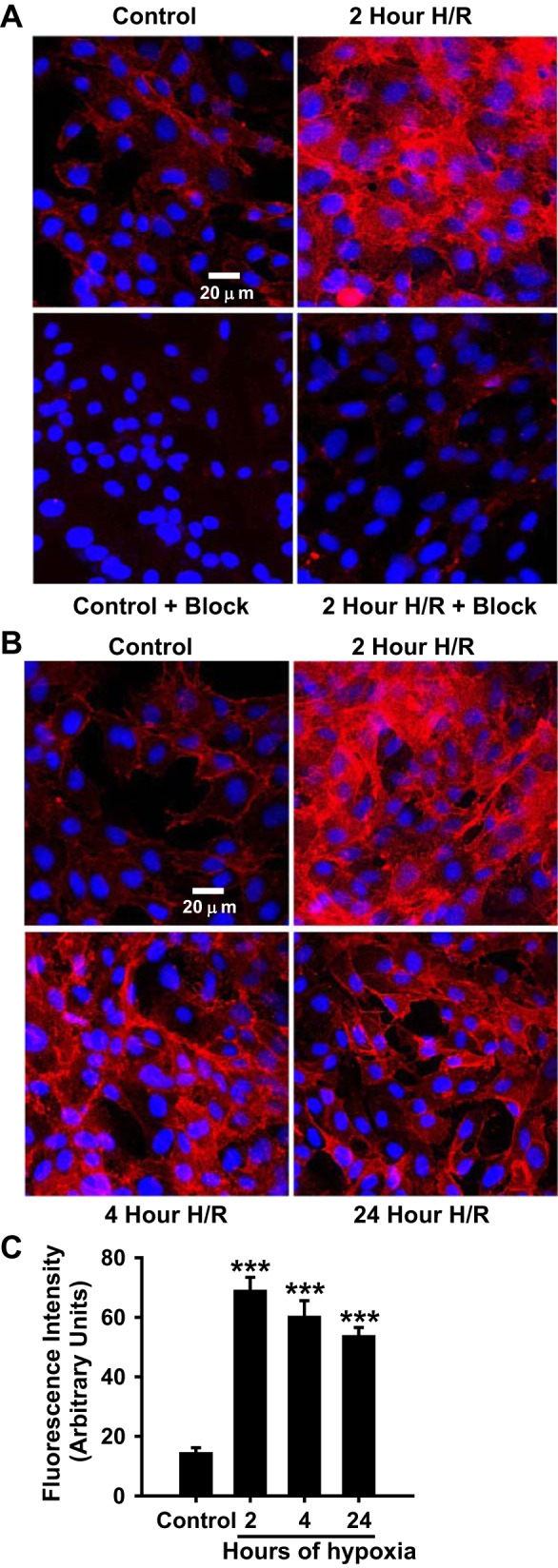
Imaging of active CD38 in endothelial cells. A: ECs subjected to normoxia (Control) or 2 h of hypoxia followed by 1 h of reoxygenation (2 h H/R) were incubated with either 10 μM SR101-F-araNMN or nonfluorescent extracellular CD38 blocking reagent 10 μM 6-alkyne-F-araNAD (+ Block) followed by 10 μM SR101-F-araNMN (+ Block). CD38 labeling increased with 2 h of H/R in unblocked cells, with preincubation of cells with 6-alkyne-F-araNAD blocking the majority of SR101-F-araNMN labeling. B: time course of CD38 activation as measured by incubation with SR101-F-araNMN. C: quantitation of the fluorescence staining obtained from a series of experiments similar to that shown in B (***P < 0.005 vs. Control, means ± SE; n = 4–6). These experiments demonstrate CD38 activation with hypoxia as the SR101-F-araNMN probe is catalysis-based.
CD38 activity and expression in normoxic and H/R-treated ECs.
To further probe posthypoxic CD38 activation in ECs, an activity assay following the conversion of CD38 substrate ε-NAD to fluorescent ε-ADP-ribose was performed in cell homogenates of ECs. Normoxic control ECs had a basal CD38 activity of 5.00 ± 0.25 nmol·min−1·mg protein−1. With 0.5, 1, 2, 4, and 24 h H/R, CD38 activity increased to 6.90 ± 0.67, 8.34 ± 1.21, 14.58 ± 1.82, 10.62 ± 1.39, and 12.44 ± 0.95 nmol·min−1·mg protein−1, respectively, demonstrating clear CD38 activation beginning at 1 h of hypoxia followed by reoxygenation and continuing through 24 h hypoxia followed by reoxygenation (Fig. 6A). Thus, activation seen with increased SR101-F-araNMN labeling was confirmed in vitro with a CD38 activity assay.
Fig. 6.
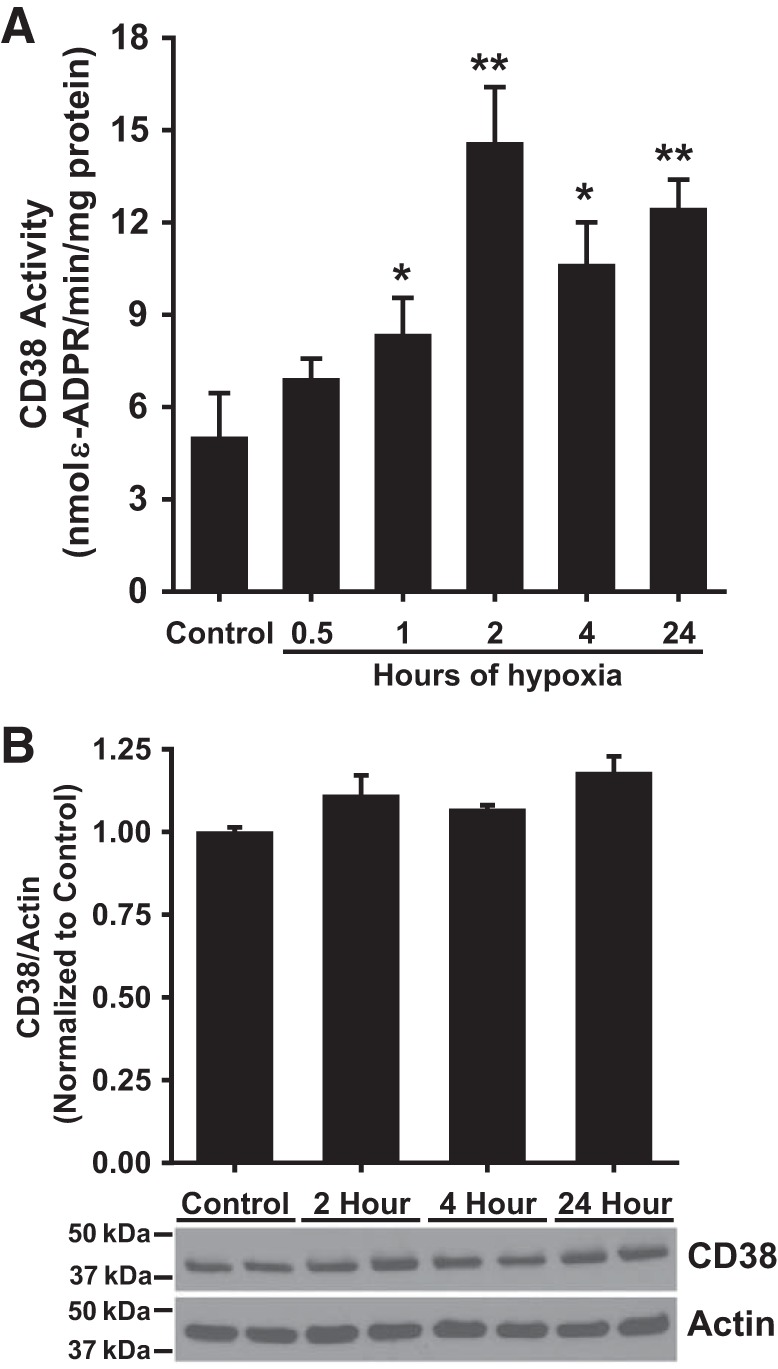
CD38 activity and expression in endothelial cells subjected to varying durations of hypoxia. A: CD38 activity in ECs subjected to H/R increased relative to that of control ECs. CD38 activity significantly increased relative to control beginning with 1 h of hypoxia and peaking at 2 h of hypoxia. *P < 0.05, **P < 0.01 vs. Control (means ± SE; n = 4). B: CD38 expression remained largely unchanged with different durations of hypoxia, indicating activation through another mechanism other than increased enzyme expression (means ± SE; n = 4). The photo blot at the bottom shows a typical Western blot with 2 samples run for control and each duration of hypoxia with data for CD38 and actin.
To determine whether increased CD38 expression was the basis for the increased levels of activated CD38, Western blotting was performed on homogenates of ECs subjected to normoxic control treatment, or 2, 4, or 24 h hypoxia with 1 h of reoxygenation. Western blot experiments revealed little change in CD38 expression occurring with cells subjected to 2, 4, or 24 h H/R compared with control cells (Fig. 6B). Thus, the observed hypoxia-induced CD38 activation is not due to a change in total CD38 expression.
NAD(P)(H) levels in ECs subjected to H/R with and without CD38 inhibition or CD38 knockdown.
To determine how H/R-induced CD38 activation affected the levels of NAD(P)(H) with and without blocking CD38, ECs were subjected to either normoxia or 2, 4, 24 h of H/R with or without CD38 inhibitor luteolinidin (50 μM). H/R resulted in vehicle-treated cells with ~55, 67, and 80% depletion of NADPH, respectively, for the 2, 4, and 24 h hypoxia groups (Fig. 7A). However, with luteolinidin treatment, NADPH depletion at the same time points was largely prevented with depletion of only 23, 30, and 38%. The depletion of CD38 substrate NADP+ was similar to that of NADPH in vehicle-treated ECs undergoing H/R, with its depletion almost entirely blocked by luteolinidin treatment. In these experiments, only the 24 h hypoxia group showed NADP+ depletion (~20%) (Fig. 7A).
Fig. 7.
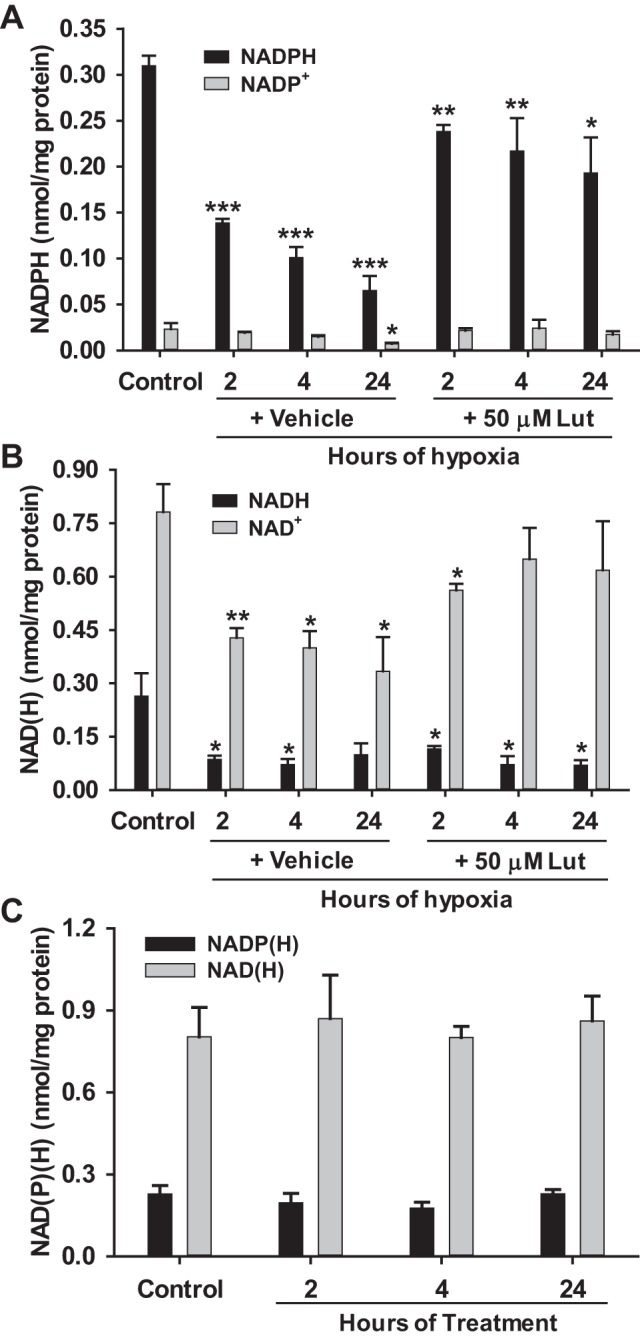
NAD(P)(H) levels in ECs with CD38 inhibition by luteolinidin. A and B: NAD(P)(H) levels were measured by HPLC in normoxic ECs (Control) and hypoxic-reoxygenated ECs either vehicle-treated (+ vehicle) or luteolinidin-treated (+ 50 μM Lut). Levels were preserved in ECs undergoing H/R with 50 μM luteolinidin treatment. *P < 0.05, **P < 0.01, ***P < 0.005 vs. Control (means ± SE; n = 4–6). C: NAD(P)(H) levels were also measured in normoxic ECs with 2, 4, or 24 h of treatment with 50 μM luteolinidin. Luteolinidin treatment in normoxic cells had no effect on cellular NAD(P)(H) levels (means ± SE; n = 3–4).
NAD+ levels, while less affected, were also depleted by H/R (~50% for each vehicle-treated H/R group). CD38 inhibition with luteolinidin protected NAD+ with levels only depleted by ~20% for the 2, 4, and 24 h H/R groups. NADH levels fell between 55 and 75% for each hypoxia group, regardless of treatment (Fig. 7B). Thus, CD38 inhibition with luteolinidin is more effective at preserving cellular NADP(H) than NAD(H) levels after hypoxia.
Lastly, in a parallel group, luteolinidin was incubated with normoxic control cells for 2, 4, or 24 h to determine the effect of luteolinidin alone on cellular NAD(P)(H) levels. In these experiments, luteolinidin had no significant effect on NAD(P)(H), indicating that CD38 does not have a substantial effect on basal NAD(P)(H) levels, and that hypoxia functions as a stimulus for CD38 activation and NAD(P)(H) depletion (Fig. 7C).
NAD(P)(H) measurements were also performed with siRNA-mediated CD38 KD with and without 24 h of hypoxia followed by 1 h reoxygenation. Maximum KD (~85%) of CD38 was optimized with a final siRNA concentration of 10 nM and 18 μl of transfection reagent (RNAiMAX) per ~1 million cells (Fig. 8, A and B). With siRNA-mediated KD of CD38, we were able to more definitively test the effect of blocking CD38 on NAD(P)(H) levels in ECs before and after H/R. With CD38 KD, baseline levels of NADP(H) did not change. However, NAD+ levels were ~25% higher in control cells with CD38 KD, though this failed to reach statistical significance (P = 0.06). With H/R, profound NAD(P)(H) preservation was seen in cells with CD38 KD. While post-H/R NADPH levels were only ~30% of baseline levels in cells receiving scrambled siRNA (scr-siRNA), in cells with CD38 KD, post-H/R NADPH levels were greater than 70% of baseline levels. The magnitude of NAD+ depletion after H/R was less in cells receiving scr-siRNA (50%). However, CD38 KD had a protective effect on this metabolite pool as well, with post-H/R levels that were 80% of baseline (Fig. 8, C and D).
Fig. 8.
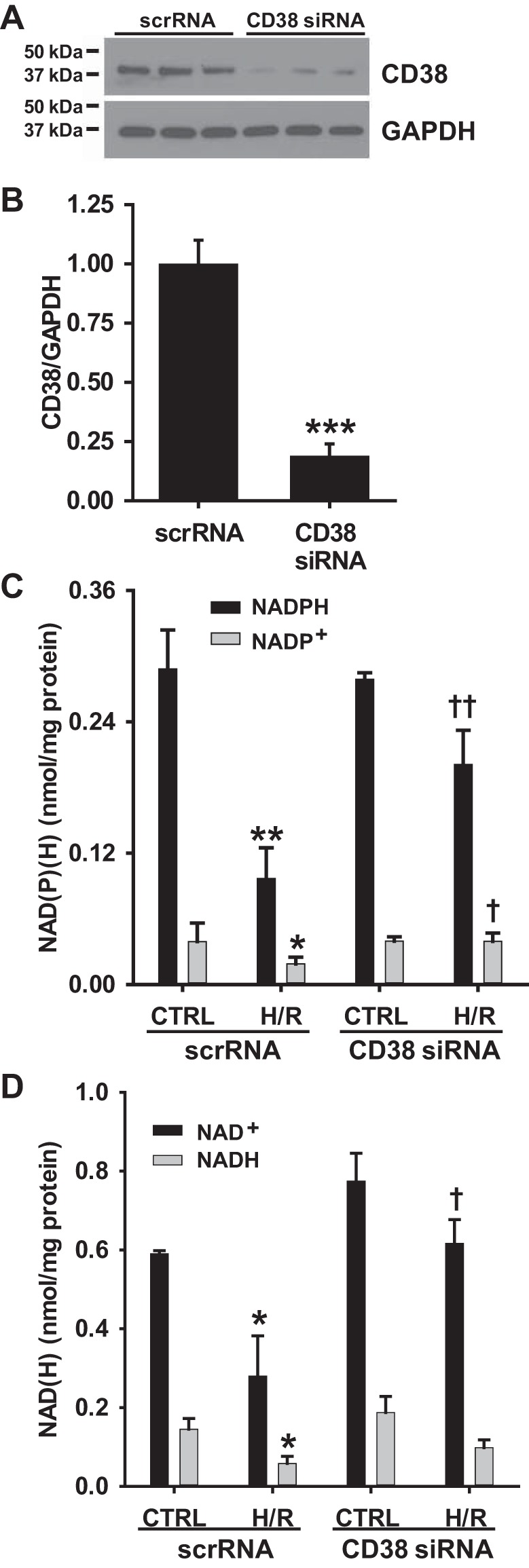
CD38 knockdown and NAD(P)(H) levels after H/R in rat aortic ECs (RAECs). A and B: RAECs were treated with scrambled siRNA (scrRNA) or CD38 siRNA (10 nM) for 24 h and CD38 knockdown (KD) was tested by Western blotting. Compared with scrRNA-treated cells, cells treated with CD38 siRNA had greater than 80% KD of CD38 (***P < 0.005 vs. scrRNA, means ± SE; n = 3). C and D: NAD(P)(H) levels were measured in ECs with and without CD38 KD after either normoxic treatment or 24 h of hypoxia and 1 h of reoxygenation (H/R). With CD38 KD, post-H/R levels of NADP(H) were significantly higher compared with cells receiving scrRNA. *P < 0.05, **P < 0.01 vs. Control scrRNA, †P < 0.05, ††P < 0.01 vs. H/R scrRNA (means ± SE; n = 3).
NO production from ECs undergoing H/R.
To determine how the depleted NAD(P)(H) levels in ECs undergoing H/R affected cellular NO production, NO spin trapping using Fe-MGD was performed with EPR measurements in control and 24 h hypoxia followed by reoxygenation with and without luteolinidin (50 μM) treatment. A prominent triplet signal of trapped NO was measured from normoxic control cells, which was totally blocked by preincubation of cells with NOS inhibitor l-NMMA (1 mM). With 24 h hypoxia followed by 1 h of reoxygenation, cellular production of NO was severely impaired with respect to NO production from control cells. To determine whether hypoxic CD38 activation and resultant NAD(P)(H) depletion contributed to this impairment, the same NO measurements were made in cells undergoing H/R with CD38 inhibition by luteolinidin, which blocks NADPH depletion. In these experiments, NO production remained near control levels, indicating the importance of CD38 in posthypoxic impairment of NO production (Fig. 9, A and B).
Fig. 9.

NO production in normoxic and hypoxic ECs with and without CD38 inhibition (A and B) or knockdown (C and D). A and B: NO was trapped with Fe-MGD and measured using EPR spectroscopy. NO production was greatly impaired after H/R. CD38 inhibition with luteolinidin (50 μM) preserved normal NO production after H/R. NOS inhibition with l-NMMA (1 mM) completely blocked NO production in control cells. ***P < 0.005 vs. CTRL, ++P < 0.01 vs. H/R and ###P < 0.005 vs. (H/R + Lut) (means ± SE; n = 4–6). EPR spectra were measured at X-band, as sum of 20 scans of 60 s each, with 80 ms time constant, using 4.0 Gauss field modulation, and 10 mW of microwave power. C and D: NO production was measured by spin-trapping and EPR in cells undergoing normoxia (CTRL) or H/R with scrambled siRNA (scrRNA) or CD38 siRNA treatment. In control cells, NO production was similar between groups with a prominent NO triplet observed. After H/R, cells receiving scrambled siRNA had greatly diminished NO production, but this was preserved near control levels in cells with CD38 knockdown. ***P < 0.005 vs. CTRL scrRNA, +++P < 0.005 vs. H/R scrRNA (means ± SEM; n = 4–6). EPR spectra were measured at X-band, with 160 ms time constant, 60 s scan time, using 4.0 Gauss field modulation, and 40 mW of microwave power. The higher power provided ~2-fold higher signal amplitude than in A and B.
We also tested the effect of CD38 KD on NO production before and after H/R. Prominent triplet signals of trapped NO were measured in both control cells with scrambled siRNA (scr-siRNA) and with CD38 siRNA. However, while NO production was almost completely lost after H/R in cells receiving scr-siRNA, cells with CD38 KD had largely preserved NO production (Fig. 9, C and D).
Superoxide production in ECs undergoing H/R.
We also tested the effect of H/R on posthypoxic superoxide production with DMPO spin-trapping and EPR. While baseline levels of superoxide were very low, markedly increased superoxide production occurred after H/R (untreated and with scr-siRNA) (Fig. 10). With CD38 KD, some superoxide production remained but levels were ~55% lower than in untreated cells or those treated with scr-siRNA. Next, we tested the effects of various potential inhibitors of superoxide production to determine the mechanisms by which it was generated in these cells. As summarized in Table 1, the observed DMPO-OH signal was near totally quenched by SOD (400 U/ml) or by the xanthine oxidoreductase (XOR) inhibitor oxypurinol (1 mM). The NOS inhibitor NG-nitro-l-arginine methyl ester (l-NAME; 1 mM), which inhibits both NO production from coupled NOS as well as superoxide generation from the oxygenase of uncoupled NOS (11, 17), also decreased the superoxide generation by ~40%. Together this suggests that superoxide generation from XOR is involved in triggering eNOS uncoupling and that eNOS, once uncoupled, produces further superoxide. Interestingly, pretreatment with the specific NADPH oxidase inhibiting TAT-peptide gp91ds-TAT (20 μM) that has been shown to fully inhibit activity (15, 38) had no significant effect on the magnitude of superoxide generation in rat aortic ECs undergoing H/R. This along with the other data suggest that NADPH oxidases have little if any role in this observed superoxide production (see Table 1).
Fig. 10.

Superoxide production in RAECs subjected to H/R with and without CD38 knockdown. Superoxide was spin-trapped with DMPO (50 mM) and measured by EPR. In cells receiving scrambled siRNA (scrRNA), H/R caused marked increases in superoxide as measured by DMPO trapping. Knockdown of CD38 with treatment of cells with CD38 siRNA led to less post-hypoxic formation of superoxide. ***P < 0.005 vs. CTRL scrRNA, +++P < 0.005 vs. (H/R scrRNA) ###P < 0.005 vs. CTRL CD38 siRNA (means ± SE; n = 4–6).). EPR spectra were measured at X-band, with 160 ms time constant, 45 s scan time, using 1.0 Gauss field modulation, and 10 mW of microwave power.
Table 1.
Effect of inhibitors on H/R-induced endothelial radical generation
| Treatment | Radical Generation, % of Untreated |
|---|---|
| Untreated | 100.0 ± 6.5 |
| SOD | 6.7 ± 2.3*** |
| Oxypurinol | 4.8 ± 3.2*** |
| l-NAME | 38.2 ± 7.0*** |
| gp91ds-tat | 97.5 ± 12.5 |
Values are means ± SE; n = 3. Oxygen radical levels were measured with DMPO spin trapping and EPR spectroscopy in RAECs undergoing 24 h of hypoxia/1 h reoxygenation without treatment (untreated), or with SOD (400 U/ml), the XOR inhibitor oxypurinol (1 mM), the NOS inhibitor l-NAME (1 mM), or the Nox2 inhibitor gp91ds-tat (20 μM). While SOD, oxypurinol, and l-NAME all significantly blocked superoxide production from RAECs, the Nox2 inhibitor gp91ds-tat had no significant effect on superoxide production.
P < 0.005 vs. Untreated.
DISCUSSION
With the finding that CD38 is activated in the postischemic heart causing depletion of NAD(P)(H) levels (39), there has been a need to understand how CD38 expression is distributed among the major cell types of the heart. To accomplish this, CD38 mRNA, protein, and activity were measured in endothelial cells (ECs), cardiac myocytes (CMs), and cardiac fibroblasts (CFs). Initially, using pure populations of each cell type, we found that CD38 was highly expressed in endothelial cells compared with cardiac fibroblasts and myocytes (Fig. 1). To further verify this, immunohistology studies were performed mapping CD38 expression in the endothelium, fibroblasts, and cardiac myocytes of wild-type and CD38−/− mouse hearts. CD38 largely colocalized with the endothelial marker CD31, and some colocalization was seen with the fibroblast marker DDR1. In cardiac myocytes; however, no CD38 was seen (Fig. 3). Overall, these findings support the observation that CD38 is highly expressed in cardiac ECs but is absent or expressed only in trace amounts in CMs, as was previously reported based on immunohistochemistry of the rat heart (39).
This cellular localization is important for considering the effects of CD38 activation in the whole heart, where multiple cell types contribute to normal physiological function. In view of this, the protective effects of CD38 inhibitors in ischemia-reperfusion (I/R) must primarily occur through direct effects on cells other than the CMs (7, 39). Rather, these CD38-mediated protective effects, including increased recovery of left ventricle function and decreased infarct size, likely result from protection of endothelial vasodilatory function through preserved NO production, as previously suggested (7, 39). These data also explain the proportionally greater effect of I/R on postischemic levels of endothelial, compared with myocyte, NAD(P)(H) pools (39).
With the finding that CD38 has high expression in endothelial cells, we focused our attention on this cell type, with further studies of hypoxia-reoxygenation designed to simulate that which occurs in ischemic syndromes of the heart. With these experiments, we found hypoxic activation of CD38 with increased labeling of CD38 by catalysis-based substrate SR101-F-araNMN and increased in vitro CD38 hydrolase activity (Fig. 5). This observed increase in CD38 activity occurred in the absence of any detected increase in CD38 expression, suggesting that direct activation of preexisting enzymes occurs following H/R (Fig. 4).
The significance of endothelial CD38 activation was evaluated with measurements of cellular levels of NADP(H) and NAD(H), as well as NO and superoxide production. ECs were subjected to normoxia, or 2, 4, or 24 h H/R with and without CD38 inhibitor luteolinidin or with and without CD38 KD (7, 28). In these experiments, there was a strong duration-dependent effect of hypoxia on cellular NADP(H) and NAD(H) levels, which was blocked by both CD38 inhibition and CD38 KD (Figs. 7 and 8). In these experiments, the NADP(H) pool was more affected than the NAD(H) pool, likely because of the fact that CD38 preferentially catalyzes reactions with NADP+ over NAD+ (~6-fold lower Km for NADP+ than NAD+) (44). Along with depleted NAD(P)(H) after H/R, we also found decreased production of NO. Both CD38 inhibition and CD38 KD were able to preserve posthypoxic NO production (Fig. 9). Thus, protection of the NADP(H) pools by CD38 inhibition or KD is able to preserve normal eNOS-dependent production of NO in ECs subjected to H/R.
Severe oxidative stress with generation of superoxide and secondary oxidants has been shown to occur in nonhuman and human endothelial cells subjected to hypoxia and reoxygenation (14, 52, 54, 55). This could lead to uncoupling of eNOS with loss of NO and gain of superoxide production (45). We have previously observed in bovine aortic endothelial cells (BAECs) subjected to H/R that superoxide-mediated depletion of BH4 occurs and leads to NOS uncoupling (6, 14). Superoxide generation has also been shown to lead to CD38 activation in the postischemic heart (39). Therefore, in the current studies we measured superoxide formation following H/R stress. EPR spin trapping detected a prominent DMPO-OH radical adduct confirmed to be derived from superoxide, since it was quenched by SOD (Table 1). Consistent with the prior studies, H/R-stimulated superoxide generation with observed EPR signals increased by over fivefold compared with control cells in air. Interestingly, with CD38 KD, this radical generation was decreased by ~55%. Since we observed that CD38 inhibition or KD is highly effective in salvaging NADPH, this could differentially affect different pathways of oxidant generation and antioxidant defense. NADPH preservation would support the normal function of eNOS as well as glutathione reductase and related glutathione-mediated antioxidant defense; however, it could also support the generation of superoxide from NADPH oxidases such as activated Nox2.
To better understand the process of H/R-induced radical generation, EPR studies were performed to directly assess the mechanisms responsible for the superoxide generation observed (Table 1). Consistent with prior reports, XOR was shown to be an important source of this endothelial radical generation with >95% quenching of radical generation when this pathway was inhibited (52, 54, 55). Interestingly, the NOS inhibitor l-NAME that inhibits NO and superoxide generation from coupled and uncoupled eNOS, respectively (11, 17), also led to prominent inhibition of superoxide generation, with ~60% decrease seen. This suggests that the oxidative burst caused by XOR in turn triggers NOS uncoupling with further increased and persistent radical generation. This is consistent with the results of a prior study of H/R in BAECs (14). In contrast to the efficacy of inhibiting these pathways, no significant decrease was seen upon specific inhibition of NADPH oxidase assembly/activation using the gp91ds-tat peptide, which has been shown to be a highly specific and potent inhibitor (15, 38). In view of these observations, NADPH oxidase and Noxs would appear to have little if any role in the superoxide radical generation observed. These observations explain why CD38 inhibition through salvage of the NADP(H) pool would serve to protect endothelial function with preserved NO production and decreased superoxide generation.
From this data, the importance of NADPH in modulating eNOS-dependent production of NO and superoxide is clearly demonstrated. It is also clear that with H/R, NADPH levels are sufficiently depleted to adversely affect NADPH-dependent processes in the cell. Relevant to the NO production pathway, NADPH depletion can affect dihydrofolate reductase and sepiapterin reductase, which are NADPH-dependent enzymes required in the recycling pathway and de novo synthesis pathways of BH4, respectively (13, 20). Low postischemic levels of NADPH can contribute to low postischemic levels of BH4, through impairment of these pathways (7, 18, 39). Glutathione reductase is another NADPH-dependent enzyme whose activity is also affected by low cellular NADPH levels. This enzyme, which catalyzes the reduction of oxidized glutathione (GSSG) to reduced glutathione (GSH), has been shown to be vital for regulating normal GSH:GSSG ratios (10, 27). With inhibition of glutathione reductase, S-glutathionylation of eNOS may occur due to an excess of GSSG, resulting in eNOS uncoupling and decreased NO synthesis (11).
Despite clear activation of CD38 with H/R, the molecular mechanism of activation remains unknown. In this study, we observed H/R-mediated CD38 activation in live cells using the catalysis-based CD38 probe SR101-F-araNMN (40), as well as in cell homogenates monitoring conversion of substrate ε-NAD to product ε-ADPR (Figs. 5 and 6). Considering that these assays were performed with total cell homogenate, it is unlikely that the increased activity was due primarily to an internalization step, as has been previously discussed (19, 24, 36, 43, 47, 50). Rather, it may be that there is a molecular alteration to CD38 which confers the enzyme with increased activity. One possibility is modulation of one or more of the six disulfide bonds in the catalytic domain of CD38 (30), which have been shown to be important for CD38 activity (23, 42, 51). In the report of Zhao et al. (51), it was shown that mutation of a cysteine in five of the six disulfide bonds led to near complete inactivation of the enzyme. Thus, CD38 may exist in an inactive state with these critical cysteines in a free thiol form. With the increased oxidative stress seen in the conditions of I/R and H/R (14, 56), free cysteines of CD38 may be driven to their disulfide form, increasing CD38 activity. Another possibility is that CD38 is phosphorylated, leading to its molecular activation (8). With the kinase activation occurring with ischemia (2, 48), phosphorylation of CD38 represents another possible mechanism of CD38 activation.
In summary, we evaluated the levels of CD38 in three cell types of the heart: cardiac myocytes, fibroblasts, and endothelial cells. With relatively low levels in fibroblasts and only trace levels in myocytes, we focused on endothelial cells, which showed high levels of CD38 expression, for further characterization of CD38 with and without H/R. While expression of CD38 was unchanged with H/R, its activity increased in a duration-dependent manner with hypoxia, as determined by the ability of cellular CD38 to bind substrate/label SR101-F-araNMN, and by enzymatic conversion of substrate. This activation was shown to severely deplete cellular levels of NADP(H) and also deplete NAD(H), with CD38 inhibition by luteolinidin or siRNA-mediated KD preventing this depletion. Endothelial depletion of NADP(H) was associated with the onset of eNOS dysfunction and uncoupling that was also mitigated by CD38 inhibition or KD. The present study, combined with our previous work describing the deleterious effects of CD38 in the postischemic heart (7, 39), highlights a need for further delineation of the mechanism of activation of CD38, which is strongly linked to the cellular redox state. Future studies geared toward testing this, as well as inhibition strategies, may benefit from focusing on the endothelium, where CD38 expression was shown to be highest among the cardiac cell types investigated.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-131941 and HL-135648 to J. L. Zweier and National Institute of Diabetes and Digestive and Kidney Diseases Grant R01 DK-113943 to F. L. Christofi.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.B., C.H., and J.L.Z. conceived and designed research; J.B., C.H., and F.L.C. performed experiments; J.B., C.H., F.L.C., and J.L.Z. analyzed data; J.B., C.H., F.L.C., and J.L.Z. interpreted results of experiments; J.B. and C.H. prepared figures; J.B. and J.L.Z. drafted manuscript; J.B., C.H., F.L.C., and J.L.Z. edited and revised manuscript; J.L.Z. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Dr. Hening Lin and Dr. Jonathan Shrimp for the generous gift of the reagents SR101-F-araNMN and 6-alkyne-F-araNAD. We also acknowledge Brian Kemmenoe and Dr. Sara Cole of The Ohio State University Campus Microscopy & Imaging Facility (CMIF) for help with immunohistochemistry experiments.
REFERENCES
- 1.Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun 345: 1386–1392, 2006. doi: 10.1016/j.bbrc.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong SC. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res 61: 427–436, 2004. doi: 10.1016/j.cardiores.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 3.Bai N, Lee HC, Laher I. Emerging role of cyclic ADP-ribose (cADPR) in smooth muscle. Pharmacol Ther 105: 189–207, 2005. doi: 10.1016/j.pharmthera.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol 293: H1883–H1891, 2007. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 5.Barbosa MT, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, Chini EN. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J 21: 3629–3639, 2007. doi: 10.1096/fj.07-8290com. [DOI] [PubMed] [Google Scholar]
- 6.Biondi R, Ambrosio G, De Pascali F, Tritto I, Capodicasa E, Druhan LJ, Hemann C, Zweier JL. HPLC analysis of tetrahydrobiopterin and its pteridine derivatives using sequential electrochemical and fluorimetric detection: application to tetrahydrobiopterin autoxidation and chemical oxidation. Arch Biochem Biophys 520: 7–16, 2012. doi: 10.1016/j.abb.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boslett J, Hemann C, Zhao YJ, Lee HC, Zweier JL. Luteolinidin protects the postischemic heart through CD38 inhibition with preservation of NAD(P)(H). J Pharmacol Exp Ther 361: 99–108, 2017. doi: 10.1124/jpet.116.239459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruzzone S, Moreschi I, Usai C, Guida L, Damonte G, Salis A, Scarfì S, Millo E, De Flora A, Zocchi E. Abscisic acid is an endogenous cytokine in human granulocytes with cyclic ADP-ribose as second messenger. Proc Natl Acad Sci USA 104: 5759–5764, 2007. doi: 10.1073/pnas.0609379104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camacho-Pereira J, Tarragó MG, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, Chini EN. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab 23: 1127–1139, 2016. doi: 10.1016/j.cmet.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cereser C, Boget S, Parvaz P, Revol A. Thiram-induced cytotoxicity is accompanied by a rapid and drastic oxidation of reduced glutathione with consecutive lipid peroxidation and cell death. Toxicology 163: 153–162, 2001. doi: 10.1016/S0300-483X(01)00401-2. [DOI] [PubMed] [Google Scholar]
- 11.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468: 1115–1118, 2010. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cockayne DA, Muchamuel T, Grimaldi JC, Muller-Steffner H, Randall TD, Lund FE, Murray R, Schuber F, Howard MC. Mice deficient for the ecto-nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood 92: 1324–1333, 1998. [PubMed] [Google Scholar]
- 13.Crabtree MJ, Tatham AL, Hale AB, Alp NJ, Channon KM. Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric-oxide synthase coupling: relative importance of the de novo biopterin synthesis versus salvage pathways. J Biol Chem 284: 28128–28136, 2009. doi: 10.1074/jbc.M109.041483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Pascali F, Hemann C, Samons K, Chen CA, Zweier JL. Hypoxia and reoxygenation induce endothelial nitric oxide synthase uncoupling in endothelial cells through tetrahydrobiopterin depletion and S-glutathionylation. Biochemistry 53: 3679–3688, 2014. doi: 10.1021/bi500076r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeLeo FR, Yu L, Burritt JB, Loetterle LR, Bond CW, Jesaitis AJ, Quinn MT. Mapping sites of interaction of p47-phox and flavocytochrome b with random-sequence peptide phage display libraries. Proc Natl Acad Sci USA 92: 7110–7114, 1995. doi: 10.1073/pnas.92.15.7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dousa TP, Chini EN, Beers KW. Adenine nucleotide diphosphates: emerging second messengers acting via intracellular Ca2+ release. Am J Physiol Cell Physiol 271: C1007–C1024, 1996. doi: 10.1152/ajpcell.1996.271.4.C1007. [DOI] [PubMed] [Google Scholar]
- 17.Druhan LJ, Forbes SP, Pope AJ, Chen CA, Zweier JL, Cardounel AJ. Regulation of eNOS-derived superoxide by endogenous methylarginines. Biochemistry 47: 7256–7263, 2008. doi: 10.1021/bi702377a. [DOI] [PubMed] [Google Scholar]
- 18.Dumitrescu C, Biondi R, Xia Y, Cardounel AJ, Druhan LJ, Ambrosio G, Zweier JL. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc Natl Acad Sci USA 104: 15081–15086, 2007. doi: 10.1073/pnas.0702986104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Funaro A, Reinis M, Trubiani O, Santi S, Di Primio R, Malavasi F. CD38 functions are regulated through an internalization step. J Immunol 160: 2238–2247, 1998. [PubMed] [Google Scholar]
- 20.Gao L, Pung YF, Zhang J, Chen P, Wang T, Li M, Meza M, Toro L, Cai H. Sepiapterin reductase regulation of endothelial tetrahydrobiopterin and nitric oxide bioavailability. Am J Physiol Heart Circ Physiol 297: H331–H339, 2009. doi: 10.1152/ajpheart.00007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldsmith EC, Hoffman A, Morales MO, Potts JD, Price RL, McFadden A, Rice M, Borg TK. Organization of fibroblasts in the heart. Dev Dyn 230: 787–794, 2004. doi: 10.1002/dvdy.20095. [DOI] [PubMed] [Google Scholar]
- 22.Graeff RM, Lee HC. Determination of ADP-ribosyl cyclase activity, cyclic ADP-ribose, and nicotinic acid adenine dinucleotide phosphate in tissue extracts. Methods Mol Biol 1016: 39–56, 2013. doi: 10.1007/978-1-62703-441-8_4. [DOI] [PubMed] [Google Scholar]
- 23.Guida L, Franco L, Zocchi E, De Flora A. Structural role of disulfide bridges in the cyclic ADP-ribose related bifunctional ectoenzyme CD38. FEBS Lett 368: 481–484, 1995. doi: 10.1016/0014-5793(95)00715-L. [DOI] [PubMed] [Google Scholar]
- 24.Han MK, Kim SJ, Park YR, Shin YM, Park HJ, Park KJ, Park KH, Kim HK, Jang SI, An NH, Kim UH. Antidiabetic effect of a prodrug of cysteine, l-2-oxothiazolidine-4-carboxylic acid, through CD38 dimerization and internalization. J Biol Chem 277: 5315–5321, 2002. doi: 10.1074/jbc.M106439200. [DOI] [PubMed] [Google Scholar]
- 25.Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, Walseth TF, Lee HC. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 262: 1056–1059, 1993. doi: 10.1126/science.8235624. [DOI] [PubMed] [Google Scholar]
- 26.Jin D, Liu HX, Hirai H, Torashima T, Nagai T, Lopatina O, Shnayder NA, Yamada K, Noda M, Seike T, Fujita K, Takasawa S, Yokoyama S, Koizumi K, Shiraishi Y, Tanaka S, Hashii M, Yoshihara T, Higashida K, Islam MS, Yamada N, Hayashi K, Noguchi N, Kato I, Okamoto H, Matsushima A, Salmina A, Munesue T, Shimizu N, Mochida S, Asano M, Higashida H. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 446: 41–45, 2007. doi: 10.1038/nature05526. [DOI] [PubMed] [Google Scholar]
- 27.Kassahun K, Jochheim CM, Baillie TA. Effect of carbamate thioester derivatives of methyl- and 2-chloroethyl isocyanate on glutathione levels and glutathione reductase activity in isolated rat hepatocytes. Biochem Pharmacol 48: 587–594, 1994. doi: 10.1016/0006-2952(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 28.Kellenberger E, Kuhn I, Schuber F, Muller-Steffner H. Flavonoids as inhibitors of human CD38. Bioorg Med Chem Lett 21: 3939–3942, 2011. doi: 10.1016/j.bmcl.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 29.Klaidman LK, Leung AC, Adams JD Jr. High-performance liquid chromatography analysis of oxidized and reduced pyridine dinucleotides in specific brain regions. Anal Biochem 228: 312–317, 1995. doi: 10.1006/abio.1995.1356. [DOI] [PubMed] [Google Scholar]
- 30.Lee HC. Structure and enzymatic functions of human CD38. Mol Med 12: 317–323, 2006. doi: 10.2119/2006-00086.Lee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev 88: 841–886, 2008. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- 32.Massullo P, Sumoza-Toledo A, Bhagat H, Partida-Sánchez S. TRPM channels, calcium and redox sensors during innate immune responses. Semin Cell Dev Biol 17: 654–666, 2006. doi: 10.1016/j.semcdb.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Partida-Sánchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, Kusser K, Goodrich S, Howard M, Harmsen A, Randall TD, Lund FE. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med 7: 1209–1216, 2001. doi: 10.1038/nm1101-1209. [DOI] [PubMed] [Google Scholar]
- 34.Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD. Revisiting cardiac cellular composition. Circ Res 118: 400–409, 2016. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pusztaszeri MP, Seelentag W, Bosman FT. Immunohistochemical expression of endothelial markers CD31, CD34, von Willebrand factor, and Fli-1 in normal human tissues. J Histochem Cytochem 54: 385–395, 2006. doi: 10.1369/jhc.4A6514.2005. [DOI] [PubMed] [Google Scholar]
- 36.Rah SY, Park KH, Nam TS, Kim SJ, Kim H, Im MJ, Kim UH. Association of CD38 with nonmuscle myosin heavy chain IIA and Lck is essential for the internalization and activation of CD38. J Biol Chem 282: 5653–5660, 2007. doi: 10.1074/jbc.M609478200. [DOI] [PubMed] [Google Scholar]
- 37.Reinherz EL, Kung PC, Goldstein G, Levey RH, Schlossman SF. Discrete stages of human intrathymic differentiation: analysis of normal thymocytes and leukemic lymphoblasts of T-cell lineage. Proc Natl Acad Sci USA 77: 1588–1592, 1980. doi: 10.1073/pnas.77.3.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice. Circ Res 89: 408–414, 2001. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 39.Reyes LA, Boslett J, Varadharaj S, De Pascali F, Hemann C, Druhan LJ, Ambrosio G, El-Mahdy M, Zweier JL. Depletion of NADP(H) due to CD38 activation triggers endothelial dysfunction in the postischemic heart. Proc Natl Acad Sci USA 112: 11648–11653, 2015. doi: 10.1073/pnas.1505556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shrimp JH, Hu J, Dong M, Wang BS, MacDonald R, Jiang H, Hao Q, Yen A, Lin H. Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe. J Am Chem Soc 136: 5656–5663, 2014. doi: 10.1021/ja411046j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takasawa S, Nata K, Yonekura H, Okamoto H. Cyclic ADP-ribose in insulin secretion from pancreatic beta cells. Science 259: 370–373, 1993. doi: 10.1126/science.8420005. [DOI] [PubMed] [Google Scholar]
- 42.Tohgo A, Takasawa S, Noguchi N, Koguma T, Nata K, Sugimoto T, Furuya Y, Yonekura H, Okamoto H. Essential cysteine residues for cyclic ADP-ribose synthesis and hydrolysis by CD38. J Biol Chem 269: 28555–28557, 1994. [PubMed] [Google Scholar]
- 43.Trubiani O, Guarnieri S, Orciani M, Salvolini E, Di Primio R. Sphingolipid microdomains mediate CD38 internalization: topography of the endocytosis. Int J Immunopathol Pharmacol 17: 293–300, 2004. doi: 10.1177/039463200401700309. [DOI] [PubMed] [Google Scholar]
- 44.Vu CQ, Lu PJ, Chen CS, Jacobson MK. 2′-Phospho-cyclic ADP-ribose, a calcium-mobilizing agent derived from NADP. J Biol Chem 271: 4747–4754, 1996. doi: 10.1074/jbc.271.9.4747. [DOI] [PubMed] [Google Scholar]
- 45.Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem 273: 25804–25808, 1998. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- 46.Xin M, Olson EN, Bassel-Duby R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Cell Biol 14: 529–541, 2013. doi: 10.1038/nrm3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu M, Li XX, Ritter JK, Abais JM, Zhang Y, Li PL. Contribution of NADPH oxidase to membrane CD38 internalization and activation in coronary arterial myocytes. PLoS One 8: e71212, 2013. doi: 10.1371/journal.pone.0071212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin T, Sandhu G, Wolfgang CD, Burrier A, Webb RL, Rigel DF, Hai T, Whelan J. Tissue-specific pattern of stress kinase activation in ischemic/reperfused heart and kidney. J Biol Chem 272: 19943–19950, 1997. doi: 10.1074/jbc.272.32.19943. [DOI] [PubMed] [Google Scholar]
- 49.Yue J, Wei W, Lam CM, Zhao YJ, Dong M, Zhang LR, Zhang LH, Lee HC. CD38/cADPR/Ca2+ pathway promotes cell proliferation and delays nerve growth factor-induced differentiation in PC12 cells. J Biol Chem 284: 29335–29342, 2009. doi: 10.1074/jbc.M109.049767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao YJ, Lam CM, Lee HC. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci Signal 5: ra67, 2012. doi: 10.1126/scisignal.2002700. [DOI] [PubMed] [Google Scholar]
- 51.Zhao YJ, Zhang HM, Lam CM, Hao Q, Lee HC. Cytosolic CD38 protein forms intact disulfides and is active in elevating intracellular cyclic ADP-ribose. J Biol Chem 286: 22170–22177, 2011. doi: 10.1074/jbc.M111.228379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zweier JL, Broderick R, Kuppusamy P, Thompson-Gorman S, Lutty GA. Determination of the mechanism of free radical generation in human aortic endothelial cells exposed to anoxia and reoxygenation. J Biol Chem 269: 24156–24162, 1994. [PubMed] [Google Scholar]
- 53.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci USA 84: 1404–1407, 1987. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zweier JL, Kuppusamy P, Lutty GA. Measurement of endothelial cell free radical generation: evidence for a central mechanism of free radical injury in postischemic tissues. Proc Natl Acad Sci USA 85: 4046–4050, 1988. doi: 10.1073/pnas.85.11.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zweier JL, Kuppusamy P, Thompson-Gorman S, Klunk D, Lutty GA. Measurement and characterization of free radical generation in reoxygenated human endothelial cells. Am J Physiol Cell Physiol 266: C700–C708, 1994. doi: 10.1152/ajpcell.1994.266.3.C700. [DOI] [PubMed] [Google Scholar]
- 56.Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res 70: 181–190, 2006. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]