

Summary
Populus alba is widely distributed and cultivated in Europe and Asia. This species has been used for diverse studies. In this study, we assembled a de novo genome sequence of P. alba var. pyramidalis (= P. bolleana) and confirmed its high transformation efficiency and short transformation time by experiments. Through a process of hybrid genome assembly, a total of 464 M of the genome was assembled. Annotation analyses predicted 37 901 protein‐coding genes. This genome is highly collinear to that of P. trichocarpa, with most genes having orthologs in the two species. We found a marked expansion of gene families related to histone and the hormone auxin but loss of disease resistance genes in P. alba if compared with the closely related P. trichocarpa. The genome sequence presented here represents a valuable resource for further molecular functional analyses of this species as a new tree model, poplar breeding practices and comparative genomic analyses across different poplars.
Keywords: Populus alba, Populus bolleana, comparative genomics, transformation efficiency, gene families
Introduction
Poplars have been selected as the model for a range of studies on trees at the molecular level for three reasons (Brunner et al., 2004). First, they were reported to be genetically transformable three decades ago (Fillatti et al., 1987). Second, poplars have a small genome size, short rotation cycle, easy in‐vitro regeneration and rapid vegetative propagation compared with other trees (Bradshaw et al., 2000; Brunner et al., 2004). Third, the genome sequence of one poplar species, P. trichocarpa, was reported more than a decade ago (Tuskan et al., 2006). A genetic transformation system for P. trichocarpa was established just after its genome had been reported (Song et al., 2006), it is still difficult to transform and grow this species in some labs or regions of the North Hemisphere. In numerous molecular studies on poplar have therefore used the P. trichocarpa genome for gene sequence and expression analyses, but for physiological and phenotypic tests other hybrid poplars have been transformed, for example, P. tremula × tremuloides (Ohtani et al., 2011), P. alba × grandidentata (Maloney and Mansfield, 2010), P. alba × P. tremula (Cho et al., 2016) and P. simonii × P. nigra (Zhao et al., 2017). Attributed to the different genetic backgrounds and gene sequences and/or variations in copy number of homologs between different species, such heterogeneous transformation (Han et al., 1997, 2000; Ma et al., 2004) may give rise to numerous unexpected results in phenotypic and molecular analyses. Therefore, it is necessary to sequence the genomes of more species, especially those with widespread distribution and cultivation. These genome resources are useful not only for functional dissection of genes and the genetic optimization of fibre and biomass production and abiotic stress resistance traits in these poplars, but also important for comparative genomic studies across different poplars.
Populus alba, called as the white poplar, is an ecologically and economically important species of the section Populus (Eckenwalder, 1996). This species is widely distributed and cultivated in Europe and Asia (Lazowski, 1997). The natural populations of this species hybridize frequently with other closely related species (for example, P. tremula) producing numerous natural hybrids (Lexer et al., 2005; Van Loo et al., 2008). This species has been widely used in the numerous labs for diverse studies (e.g. Lexer et al., 2005; Van Loo et al., 2008; Wang et al., 2008). The previous studies suggest that P. alba is easily genetically transformed (Soliman et al., 2017; Wang et al., 2008) and one genotype of this species can start to flower very quickly within only 9 months after being regenerated (Meilan et al., 2004). One variety of this species, var. pyramidalis (= P. bolleana) has been widely cultivated for urban afforestation, ecological restoration and wood use from northwest (Xinjiang) to northern China (Beijing) because of its rapid growth, lack of seed catkins, erect stems and high biomass production (Xu, 1988; Xu et al., 2011; Zhang et al., 2008). This variety was selected, domesticated and clonally propagated by means of branch cuttings from one or a very limited number of male individuals of P. alba obtained from its native, dryland distributions in central Asia (Yang et al., 1992). The cutting clones of var. pyramidalis usually start to flower within 5 years. In this study, we firstly sequenced the genome of this variety and compared the genomic differences between it and the closely related species. We then confirmed the high transformation efficiency of P. alba as reported before (Soliman et al., 2017; Wang et al., 2008). We believe that this genome resource will be highly useful for molecular analyses of the gene functions in poplar trees and comparative genomic analyses across different poplars.
Results
Genome sequencing, assembly and annotation
We sequenced the genome of a clonally propagated male individual of P. alba var. pyramidalis using a whole‐genome shotgun strategy. About 320× Illumina data were generated (Table S1) and assembled into an initial genome sequences spanning 406.8 Mb, with a contig N50 of 9.8 kb and a scaffold N50 of 348.9 kb (Figure S1A; Table S2). To overcome challenges posed by the relatively high number of repeats and heterozygosity of this genome (Figure S1B), we also generated about 30× PacBio RS raw data to improve this short‐read assembly. The size of the final assembly after removing scaffolds <1 kb in length comprised 17 797 scaffolds with contig and scaffold N50 size of 26 535 bp and 459 178 bp, respectively (Table S2), representing over 87% of the total genome size as estimated from k‐mer analysis (536 Mb) conducted using KmerGene software (Chikhi and Medvedev, 2013). Our assessment of the quality of the assembly suggested that most of the genome was assembled (Figure 1). A total of 201 Mb (44.61% of the genome) was annotated as consisting of repetitive sequences, similar to the values determined for genomes of other poplar species (Ma et al., 2013; Tuskan et al., 2006; Yang et al., 2017; Table S3). The heterozygosity level of P. alba var. pyramidalis was estimated to be 0.53% on the basis of mapping short library reads to the draft genome (2 394 196 SNPs and 414 130 indels).
Figure 1.
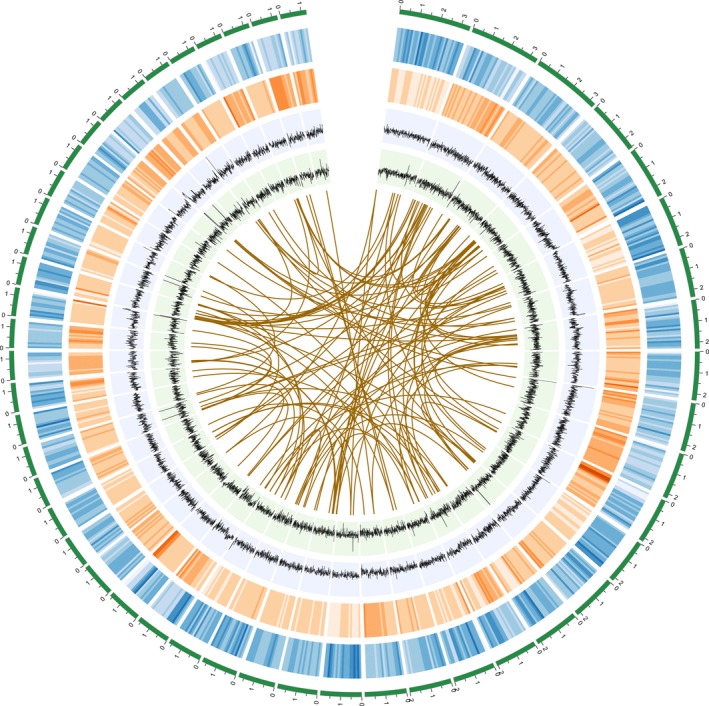
Populus alba var. pyramidalis genome. From the outer edge inward, circles represent the 50 largest DNA sequence scaffolds (green), the genes on each scaffold (blue), repeat density at 10 kb intervals (orange), GC density at 10 kb intervals (green), and the sequenced reads density at 10 kb intervals (grey). Links in the core connect duplicated sets of genes (E‐value threshold of <1e−10 and 85% similarity).
A combination of de novo and homology‐based gene prediction generated a final gene setincluding 37 901 protein‐coding genes (Figure 1; Table S4), with the gene structures being refined using alignments with transcriptomes from four different tissue types (leaf, phloem, xylem and root; Table S5). Of these genes, 4779 were predicted to generate multiple transcript variants due to alternative splicing. The predicted genes were then functionally annotated by a consensus approach, using InterPro (Hunter et al., 2008), Gene Ontology (GO; Ashburner et al., 2000), Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa and Goto, 2000) and Swiss‐Prot (Boeckmann et al., 2003). In total, 32 513 genes (85.8% of the predicted genes) have known homologs in protein databases (Table S6). We further assessed the completeness of the genome assembly, based on comparison with a benchmark of 429 conserved eukaryote genes using the benchmarking sets of universal single‐copy ortholog (BUSCO) v3 method (Simão et al., 2015). The results indicated that our annotation of the P. alba var. pyramidalis genome is nearly complete, with 91.10% of the complete BUSCOs, a value similar to P. trichocarpa and P. euphratica (Table S7). In addition, we also identified 569 ribosomal RNA (rRNA), 940 transfer RNA, 123 small nuclear RNA and 1050 microRNA genes in the assembled genome (Table S8).
Comparative genome analysis
Phylogenetic analysis based on the genomic evidence suggested that P. alba var. pyramidalis is more closely related to P. trichocarpa than to P. euphratica. The divergence between P. alba var. pyramidalis and P. trichocarpa was estimated to have occurred ~13 Mya (Figure S1C). As expected, P. alba var. pyramidalis had the same whole genome duplications (WGD) as P. trichocarpa and P. euphratica (Figure 2A). In addition, we identified a total of 3363 collinear blocks of about 300 Mb in length between P. alba var. pyramidalis and P. trichocarpa (Figure 2B). P. alba var. pyramidalis shared 16 846 gene families (including 28 710 genes) with P. trichocarpa, representing 76% of the total annotated genes (Figure 2C; Figure S1D). We further performed tests for deviations in the Ka/Ks ratio (non‐synonymous substitutions per non‐synonymous site to synonymous substitutions per synonymous site) for these homologous genes and 865 gene pairs were identified to have high diversification ratios (Ka/Ks > 1). GO enrichment indicated these genes were mainly functioned in “primary metabolic process” and “defense response”, including these well‐known defense response genes CPR1 (Kim et al., 2010), LEA (Salleh et al., 2012), and BIR1 (Zhang et al., 2013; Table S9). Besides, we found that there were 869 P. alba var. pyramidalis specific gene families (Figure 2C), which were also enriched in ‘defense response’ (GO:0006952, 76 genes, P = 2.17 × 10−20), including eight families containing ‘salt stress response/antifungal’ domains. Whereas 1427 gene families specific to P. trichocarpa were enriched in ‘photosystem II reaction center’ (GO:0009539, 11 genes, P = 2.67 × 10−13; Figure S2).
Figure 2.
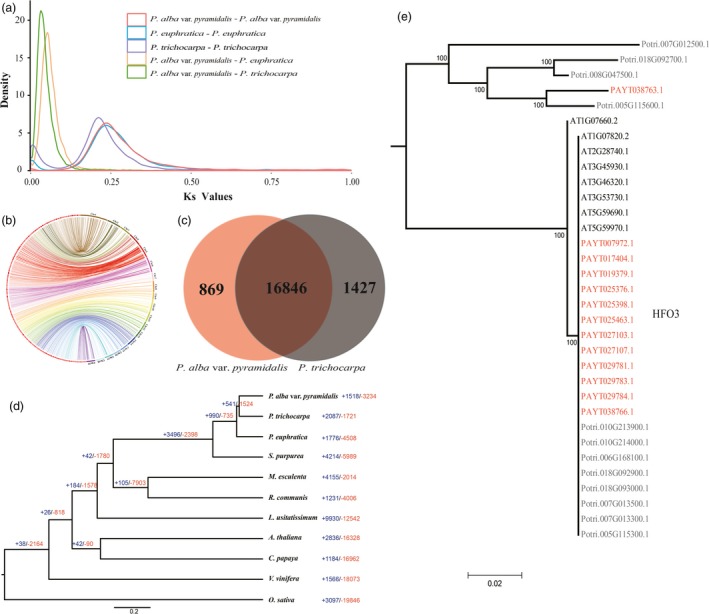
Genome characteristics. (a) Distribution of synonymous nucleotide substitutions (Ks). (b) Circos plots showing synteny between the Populus alba var. pyramidalis (left) and P. trichocarpa (right) genomes. (c) Venn diagram showing the number of gene families shared between P. alba var. pyramidalis and P. trichocarpa. (d) Expansion and contraction of gene families in ten species. (e) Phylogenetic trees of HFO3 genes in Arabidopsis, P. alba var. pyramidalis and P. trichocarpa.
Gene family expansion and contraction
We found that 1518 gene families were expanded in the P. alba var. pyramidalis genome compared to other plant species (Figure 2d). GO enrichment showed that these expanded families were significantly enriched in the terms ‘ADP binding’ (GO:0043531, 103 genes, P = 2.06 × 10−14), ‘defense response’ (GO:0006952, 193 genes, P = 5.47 × 10−12), and ‘secondary metabolic process’ (GO:0019748, 174 genes, P = 1.35 × 10−9; Table S10). Among these families, PUP and auxin/indole‐3‐acetic acid (Aux/IAA) genes related to cytokinin and auxin responses were expanded with a high expression in phloem and xylem (Figure S3), probably related to the fast growth of this variety. In addition, we found that homologs of Arabidopsis HFO3 (Tenea et al., 2009) histone genes, which could increase Agrobacterium‐mediated transformation when over‐expressed (Tenea et al., 2009), were noticeably expanded in P. alba var. pyramidalis (Figure 2E).
In contrast, we found that 3234 gene families, including those containing nucleotide‐binding sites (NBSs) with key roles in plant disease resistance, were very much contracted in P. alba var. pyramidalis genome, with some genes containing the NBS domain being lost altogether (Figure 3; Table S11). For example, only one NBS gene copy containing TIR (Toll⁄interleukin‐1 receptor) domain, which belong to TN and TNL subfamilies, was found in P. alba var. pyramidalis genome (Figure 3b). Sequence alignment of these homologous genes showed that most TIR domains of NBS genes were lost in P. alba var. pyramidalis (Figure S4). The contraction of NBS gene family in P. alba var. pyramidalis genome was also confirmed when compared with other closely related Salicaceae species (Table S12).
Figure 3.
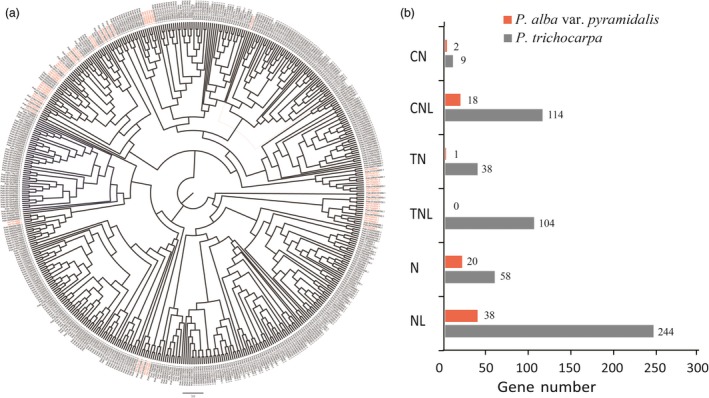
Phylogenetic tree of NBS genes and it's contraction in Populus alba var. pyramidalis. (a) Phylogenetic tree of NBS genes in P. alba var. pyramidalis and P. trichocarpa. (b) Numbers of NBS genes subfamilies in P. alba var. pyramidalis and P. trichocarpa.
Genetic transformation efficiency and gene knock‐out in P. alba var. pyramidalis
We next examined the efficiency of transformation of this variety with the standard Agrobacterium‐mediated system in poplars (Figure 4, Methods S1). We assessed different vectors for transferring the Hyg gene (Table 1). All young leaves subjected to co‐cultivation survived and we checked for the presence of the Hyg gene in at least one callus from each targeted leaf (Figure S5A). The average percentage of transgenic calli was about 80%. We then examined the success rate for inducing sprout regeneration and found that around 35.91% of the transgenic calli could produce shoots (Figure 4C,D). We excised these transgenic shoots from transgenic calli and cultured them in rooting medium. The average rooting efficiency was around 83.05%. The final transformation rates obtained were between 17.59% and 28.51% with an average rate of 23.6% (Table 1; Figure 4E). The average time from co‐cultivation to whole plant regeneration was about 80 days (Table S13).
Figure 4.

The genetic transformation process and results of transient transformation. (a) Simultaneous selection and regeneration on WPM‐reg plates. (b) Multiple green transgenic calli. (c) Transformed shoots regenerated from calli on WPM‐elo media. (d) A regenerated transgenic whole plant on rooting medium. (e) A transgenic plant under greenhouse conditions. (f–i) Transient GFP fusion protein expression assay in nucleus. (f) bright field; (g) DAPI; (h) green fluorescence; (i) overlay. Scale bar = 20 μm.
Table 1.
Summary of transformation efficiency results obtained with different vectors
| Vector | Sprout induction (%) | Root induction (%) | Transgenesis rate (%) | Total efficiency (%) |
|---|---|---|---|---|
| pCAMBIA1305 | 34/94 (36.17) | 77/85 (90.59) | 67/77 (87.01) | 28.51 |
| PYL‐CRISPR‐CAS9‐HD | 25/69 (36.23) | 54/68 (79.41) | 44/54 (81.48) | 23.44 |
| PYL‐CRISPR‐CAS9‐HD | 25/73 (34.25) | 25/37 (67.57) | 19/25 (76.00) | 17.59 |
| PCXSN | 19/56 (33.93) | 68/77 (88.31) | 36/50 (72.00) | 21.57 |
| PCXSN | 24/70 (34.29) | 19/24 (79.17) | 17/19 (89.47) | 24.29 |
| pCAMBIA1302 | 38/101 (37.62) | 95/106 (89.62) | 33/50 (66.00) | 22.25 |
| pCAMBIA1302 | 35/90 (38.89) | 78/90 (86.67) | 41/50 (82.00) | 27.64 |
| Average | 35.91 | 83.05 | 79.14 | 23.60 |
We also carried out a transient transformation assay for this variety, using a rapid transformation system for gene function analysis based on the method described by Takata and Eriksson (2012). We transferred a C2H2‐AZF gene in order to examine its intracellular localization in nucleus. Transient transformation was monitored by the expression of green fluorescence protein (GFP) from the vector (Figure 4F–I). Over 40% cells per leaf were found to show GFP signals in the nucleus under fluorescence stereomicroscope.
Finally, we performed gene knockout experiments in this variety. We followed the method of Fan et al. (2015) to knock out a C2H2‐AZF gene by means of a clustered regularly interspaced short palindromic repeats (CRISPR)‐associated protein (Cas) mediated system. We designed three guide RNAs to target the C2H2‐AZF gene and the knock‐out results were verified by performing qRT‐PCR and sequencing the PCR amplification products of the DNA fragments targeted. We found that the gene had been successfully knocked out in 89% (92 in total) of the samples (Figure S5B). Taken together, our experiments confirmed the high genetic transformation efficiency of P. alba (e.g. Soliman et al., 2017; Wang et al., 2008). Therefore, this species can be used for diverse molecular studies.
Discussion
In this study, we reported the genome sequence of P. alba var. pyramidalis and examined its genomic differences among the closely related species. We found that this variety diverged from P. trichocarpa around 13 million years ago (Figure S1C). Both species had undergone two whole genome duplications and they exhibited extensive collinearity across the gene space (Figure 2B). We annotated 37 901 genes, similar to the total number of genes (41 335) identified in P. trichocarpa. At least 24 278 genes within the collinear regions between these two species are orthologous. We further identified 865 diversified gene pairs and 869 specie‐specific gene families, mainly enriched in abiotic stress response, will help P. alba var. pyramidalis adapted to diverse environment.
We also found a few genes to be species‐specific in P. alba var. pyramidalis due to the expansion of gene families involved in hormone metabolism and response. Hormones (especially auxin) are important factors affecting plant growth, and Aux/IAA proteins play a pivotal role in the perception and signalling of the hormone auxin (Liscum and Reed, 2002; Paponov et al., 2008). Importantly, we found that genes encoding protein containing the NBS domain were greatly contracted in P. alba var. pyramidalis compared with P. trichocarpa. These NBS genes play a critical role in disease resistance (including resistance to both bacteria and viruses) (Dangl and Jones, 2001). A total of 79 NBS gene copies were identified in P. alba var. pyramidalis genome, whereas 567, 251, 150, 419 and 205 copies were identified in P. trichocarpa (Tuskan et al., 2006), P. euphratica (Ma et al., 2013), P. pruinosa (Yang et al., 2017), S. purpurea (https://phytozome.jgi.doe.gov/pz/portal.html) and S. suchowensis (Dai et al., 2014), respectively (Table S12). These NBS genes were further classified into six subfamilies and all these subfamilies were contracted greatly in P. alba var. pyramidalis. It should be noted that gene copies and genomic structures vary not only greatly between species, but also between different genotypes of the same species according to the recent pan‐genome analyses (Pinosio et al., 2016; Zhang et al., 2018). However, how these genomic differences contribute to the species‐ or genotype‐specific traits need further studies for poplars in the future.
In addition, our experiments confirmed that the genetic transformation efficiency for P. alba is high as suggested before on this variety and the other genotype (Soliman et al., 2017; Wang et al., 2008). Therefore, this species may represent a useful new tree model for transformation‐based analyses for three reasons. First, leaves taken from cuttings can be used directly as material for transgenic experiments, which is preferable to the stem internodes used. Second, our final genetic transformation efficiency was on average 23.6%, a high value among poplars. Finally, we found that the entire process from co‐cultivation to whole plant regeneration required an average time of <3 months (80 days; Figure 4A–E), which could save a lot of time. Our subsequent transient transformation assays and gene knock‐out experiments similarly suggest it can be used for other molecular studies. In addition, some genotype of this species could start to flower far earlier than other poplars (Meilan et al., 2004) although it remains tested whether our transformation protocol works well for this genotype. All these findings indicate that P. alba shows high transformation efficiency and is likely to represent a new candidate model for genetic transformation and gene function tests in poplar tree species.
In conclusion, we reported the genome sequence of P. alba for the first time and confirmed its high transformation efficiency. Both the genome sequence and the transformation protocol presented here will accelerate our molecular understanding of this tree species, its breeding program and other diverse studies. Especially, we showed the genomic divergence between P. alba and other closely related species, which indicates that comparative genomic analyses through sequencing more species are necessary to a deep evolutionary understanding of the poplar adaption and diversification.
Materials and methods
Genome sequencing and assembly
Genomic DNA was extracted from leaf tissues of P. alba var. pyramidalis with a standard CTAB (cetyl trimethylammonium bromide) method (Porebski et al., 1997). We carried out whole genome shotgun sequencing with the Illumina Hiseq 2500 platform (Illumina, CA). Seven paired‐end sequencing libraries with insert sizes of approximately 270 bp, 500 bp, 800 bp, 2 kb, 5 kb, 10 kb and 20 kb were constructed, generating a total of 170 Gb of data. RNA samples were prepared from leaves, phloem, xylem, and roots of a 2‐year‐old individual and sequenced on the Illumina Hiseq 2500 platform (Illumina). 15 Gb of PacBio RS reads with an N50 of over 8 kb were sequenced on the PacBio RS II platform (Pacific Biosciences, CA).
We first generated the Illumina‐based de novo genome assembly using Platanus with k‐mer auto‐extension and the option “‐u = 0.2” (Kajitani et al., 2014). Next, all PacBio RS reads were used to fill the gaps by SSPACE‐LongRead v1‐1 (Boetzer et al., 2010) with default parameters after error correction by the Lordec software package v0.6 (Salmela and Rivals, 2014) with all the Hiseq 2500 short reads. Finally, PBJelly v15.8.24 (English et al., 2012) and GapCloser v1.12 (Li et al., 2008) were used with default parameters to improve the genome assembly.
Repeat annotation
For transposable element annotation, RepeatMasker v4.05 (Tarailo‐Graovac and Chen, 2009) and RepeatProteinMasker (Tarailo‐Graovac and Chen, 2009) were used with default parameters against Repbase (Xu and Wang, 2007) to identify known repeats in the P. alba var. pyramidalis genome. In addition, RepeatModeler (Tarailo‐Graovac and Chen, 2009) and LTR_FINDER (Jurka et al., 2005) were used to identify de novo evolved repeats in the assembled genome. Parameters for LTR_FINDER were set to ‘Match = 2, Mismatch = 7, Delta = 7, PM = 80, PI = 10, Minscore = 50, and MaxPeriod = 2000’.
Gene prediction and annotation
Three methods were used to predict protein‐coding genes: transcriptome‐based predictions, de novo predictions, and homology‐based predictions. For transcriptome‐based predictions, RNA from four tissues (leaves, xylem, phloem and root) was isolated and RNA‐seq data (NCBI SRR6003833–SRR6003836), processed by Trinity v2.2 (Grabherr et al., 2011), were used for gene annotation. For de novo predictions, Augustus v3.21 (Stanke et al., 2006), GenScan v1.4 (Burge and Karlin, 1997), glimmerHMM (Majoros et al., 2004), GeneMark v3.47 (Lukashin and Borodovsky, 1998) and SNAP (Korf, 2004) analyses were performed on the repeat‐masked genome, with parameters trained from transcriptome assembly data. Predicted protein sequences from Arabidopsis thaliana, P. trichocarpa, Ricinus communis and Vitis vinifera were used for homology‐based predictions with Phytozome v12 (Goodstein et al., 2011). The homology, de novo and transcriptomic gene sets were merged to form a comprehensive non‐redundant reference gene set using the EVidenceModeler (Haas et al., 2008) and PASA v2.0.2 (Haas et al., 2003) software packages. Functional annotation of the predicted gene models was based on comparison with the Swiss‐Prot (Boeckmann et al., 2003) and KEGG databases (Kanehisa and Goto, 2000) with a minimal e‐value of 1e‐5. GO terms were assigned to the annotated genes using the Blast2GO pipeline (version 3.1.3; Conesa et al., 2005). Protein domains and functions were analyzed using InterProScan (version 5.13–5. 20).
Genome Quality Evaluation and gene clustering analyses
The qualities of the assembly and gene annotation were assessed using BUSCO v3 (Simão et al., 2015). We compared the P. alba var. pyramidalis genome sequence against a set of core eukaryotic genes using BUSCO. Syntenic blocks and gene collinearity were inferred using MCScanX (Wang et al., 2012) and Last software v2.28.2 (http://last.cbrc.jp/) and were visualized using Circos v0.69 (Krzywinski et al., 2009). Synonymous (Ks) and non‐synonymous (Ka) substitution rates for gene pairs were computed using the ‘YN00’ method from the PAML package v4.8 (Yang, 2007). To identify SNPs and indels in the P. alba var. pyramidalis genome, we mapped the sequenced short reads to the draft P. alba var. pyramidalis genome using BWA v0.7.12‐r1039 (Li and Durbin, 2009) and called SNPs and indels using Samtools v0.1.19‐44428 cd (Li et al., 2009).
Ortholog clustering analysis was performed using OrthoMCL v2.0.9 (Li et al., 2003) applied to all the protein‐coding genes of P. alba var. pyramidalis and A. thaliana, Manihot esculenta, Linum usitatissimum, Salix purpurea, V. vinifera, Oryza sativa, Carica papaya, P. trichocarpa, P. euphratica and R. communis. The MCMCTREE program, implemented in the PAML package v4.8 (Yang, 2007), was used to estimate divergence times with calibration times referred to Ma et al. (2013). The phylogenetic tree was constructed from single copy genes by PhyML (Guindon et al., 2010). In order to compare variations of gene copies between P. alba and closely related species, we further downloaded genomes of P. euphratica (Ma et al., 2013), P. pruinosa (Yang et al., 2017), S. purpurea (https://phytozome.jgi.doe.gov/pz/portal.html), S. suchowensis (Dai et al., 2014) and P. trichocarpa (Tuskan et al., 2006). Although genomes of P. tremula and tremuloides are also available through PopGenIE (http://popgenie.org/), the poor assemblies limit their comparisons with P. alba and other species. Species‐specific gene families were identified with the cluster of genes form only one specie. Gene expansion and contraction analysis was conducted using the CAFÉ program (version 3.1; De Bie et al., 2006) with information from the estimated phylogenetic tree. The Hidden Markov Model (Eddy, 1998) profile for domains from the Pfam database (26.0; Finn et al., 2009) and HMMER software (version 3.1; Finn et al., 2011) were used to identify gene families. Resistance genes were identified by the presence of the NBS domain and classified into six groups (CN: CC‐NBS; CNL: CC‐NBS‐LRR; TN: TIR‐NBS; TNL: TIR‐NBS‐LRR; N: NBS; NL: NBS‐LRR).
Genetic transformation process
One‐year‐old P. alba var. pyramidalis clones propagated from cuttings grown in a greenhouse at 25 °C under cycles of 16 h light/8 h darkness (6:30–22:30; 100 μmol/m2/s) and 60% humidity, were used for transformation. After disinfecting with 12% sodium hypochlorite, the leaves of P. alba var. pyramidalis was cut into pieces and put on Woody Plant Medium (with 2 mg/L zeatin, 1 mg/L naphthalene acetic acid and 100 μmol/L acetosyringone) for induction. When the explants had been induced to produce new plants under aseptic conditions, they could be used for the transformation process. This was performed according to the P. alba var. pyramidalis transformation protocol given in Methods S1. The transformation time and success rate were calculated for each step.
Transient transformation process
Sterile rooted cuttings from P. alba var. pyramidalis clones, grown in a greenhouse at 25 °C under cycles of 16 h light/8 h darkness (100 μmol/m2/s), were used for transient transformation. The pCXDG‐based expression vector employed here harbours a GFP gene driven by the Cauliflower Mosaic Virus 35S (CaMV35S) promoter. The expression vectors were transformed into Agrobacterium tumefaciens strain GV3101. Agrobacterium harbouring individual vectors was inoculated into YEP media with appropriate antibiotics. An overnight culture of Agrobacterium was harvested at an OD600 of 0.3, centrifuged at 5000 × g for 10 min, and re‐suspended in 50 mL of infiltration medium (0.5 × MS medium containing 5 mm MES‐KOH (pH 5.6), and 200 μM acetosyringone) to an OD600 of 0.3. The bacterial suspension was incubated at room temperature for three hours with gentle shaking in the dark. Then Agrobacterium infiltration was performed by applying a vacuum three time for three minutes. The cuttings were then put on paper towels to remove excess infiltration medium and transplanted into 0.5 × MS medium (pH 5.6) with 0.6% (w/v) agar and 50 μg/mL cefotaxime. We followed the method of Takata and Eriksson (2012) to conduct a transient transformation assay of the C2H2‐AZF (PAYT023741.1) gene in order to measure expression in the nucleus. The cuttings were returned to the initial growing conditions for 3 days before imaging. Images of whole leaves were monitored using a fluorescence stereomicroscope (Leica TCS SP8, Germany) with excitation at 488 nm to detect GFP fluorescence.
Gene knock‐out experiments
We followed the procedures of Fan et al. (2015) to perform gene knock‐out experiments. One‐year‐old P. alba var. pyramidalis clones propagated from cuttings were used for CRISPR/Cas9‐mediated targeted mutagenesis. The AZF genomic DNA fragment was amplified by PCR with gene‐specific primers (AZF‐F: 5′‐ACCTTTCCTTCTCTCTTCGGAT‐3′; AZF‐R: 5′‐TCCAACAATCTTCCTAATTGAACCT ‐3′). The PCR product was cloned and sequenced, and the sequence was used to select CRISPR/Cas9 target sites. Three output target sites were selected for designing sgRNA sequences based on their locations in the gene and their GC contents. Three target sites were assembled in plasmids designated ATU3b, ATU6‐1 and ATU6‐29 with the specific primers (ATU3b‐F: 5′‐gtcaTCGTAGTGATTCCCCTTCAA‐3′, ATU3b‐R: 5′‐aaacTTGAAGGGGAATCACTACGA‐3′; ATU6‐1F:5′‐ attgTTGAAAGGAGTGGCTGTTGT‐3′, ATU6‐1R: 5′‐ aaacACAACAGCCACTCCTTTCAA‐3′; ATU6‐29F: 5′‐attgCGCCACGAGCGAGCATGATA‐3′, ATU6‐29R: 5′‐ aaacTATCATGCTCGCTCGTGGCG‐3′). The binary pYLCRISPR/Cas9 multiplex genome targeting vector system carrying the CAS9 coding gene and three plasmids with sgRNA cassettes driven by AtU3b, AtU6‐1 and AtU6‐29 were generated. After completion of the transgenic procedure, the plants obtained were selected on 9 mg/L hygromycin and the mutations they contained were identified through Sanger sequencing of individual clones; the mutation rate in transgenic plants was calculated according to the ratio of mutated clonal amplicons to total sequenced clonal amplicons.
Availability
Sequence data from this study can be found at the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov) under SRA accession number SRR5990011 (library with 10 kb insert size), SRR5990012 (20 kb), SRR5990014 (270 bp), SRR5990015 (2 kb), SRR5990016 (500 bp), SRR5990017 (5 kb), SRR5990018 (800 bp) and SRR5990031 (PacBio RS data). The clone sources of P. alba var. pyramidalis can be obtained through the corresponding author. The whole genome sequence data and the annotation file reported in this paper have been deposited in the Genome Warehouse in BIG Data Center (BIG Data Center Members, 2018), Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession number GWHAAEP00000000 that is publicly accessible at http://bigd.big.ac.cn/gwh.
Funding
The research was supported by the National Key Research and Development Program of China (2016YFD0600101), the National High‐Tech Research and Development Program of China (2013AA102605), the National Science Foundation of China (31561123001, 31470620 and 31500502) and the ‘111’ collaboration project.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Methods S1 The protocol of Populus alba var. pyramidalis genetic transformation system.
Figure S1 Genome assembly process and characteristics.
Figure S2 GO enrichment of specie‐specific genes.
Figure S3 Phylogenetic tree and expression conditions of PUP gene family and Aux/IAA proteins.
Figure S4 Homologous NBS R genes in Populus alba var. pyramidalis were lost TIR and NBS domain.
Figure S5 Transformation of Populus alba var. pyramidalis.
Table S1 Sequencing data used for genome assembly.
Table S2 Assemble features of Populus alba var. pyramidalis genome.
Table S3 Summary of transposon content in the genome.
Table S4 Summary of predicted protein‐coding gene annotations and their supporting evidence types.
Table S5 RNA‐seq data of four tissues.
Table S6 Functional annotation of predicted genes.
Table S7 Evaluation of completeness of the genome assembly using BUSCOs.
Table S8 Annotation of non‐coding RNAs.
Table S9 GO enrichment of high diversification gene pairs.
Table S10 GO enrichment of expanded genes families.
Table S11 GO enrichment of contracted genes families.
Table S12 NBS gene numbers identified in different poplars.
Table S13 Summary of culturing time of different transformation stage.
Acknowledgements
We thank the Program Core Facility of School of Life Sciences of Lanzhou University for providing us with fluorescence microscopy facilities.
References
- Ashburner, M. , Ball, C.A. , Blake, J.A. , Botstein, D. , Butler, H. , Cherry, J.M. , Davis, A.P. et al (2000) Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIG Data Center Members (2018) Database resources of the BIG Data Center in 2018. Nucleic Acids Res. 46(Database issue), D14–D20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckmann, B. , Bairoch, A. , Apweiler, R. , Blatter, M.‐C. , Estreicher, A. , Gasteiger, E. , Martin, M.J. et al (2003) The SWISS‐PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer, M. , Henkel, C.V. , Jansen, H.J. , Butler, D. and Pirovano, W. (2010) Scaffolding pre‐assembled contigs using SSPACE. Bioinformatics, 27, 578–579. [DOI] [PubMed] [Google Scholar]
- Bradshaw, H. , Ceulemans, R. , Davis, J. and Stettler, R. (2000) Emerging model systems in plant biology: poplar (Populus) as a model forest tree. J. Plant Growth Regul. 19, 306–313. [Google Scholar]
- Brunner, A.M. , Busov, V.B. and Strauss, S.H. (2004) Poplar genome sequence: functional genomics in an ecologically dominant plant species. Trends Plant Sci. 9, 49–56. [DOI] [PubMed] [Google Scholar]
- Burge, C. and Karlin, S. (1997) Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94. [DOI] [PubMed] [Google Scholar]
- Chikhi, R. and Medvedev, P. (2013) Informed and automated k‐mer size selection for genome assembly. Bioinformatics, 30, 31–37. [DOI] [PubMed] [Google Scholar]
- Cho, J.‐S. , Nguyen, V.P. , Jeon, H.‐W. , Kim, M.‐H. , Eom, S.H. , Lim, Y.J. , Kim, W.‐C. et al (2016) Overexpression of PtrMYB119, a R2R3‐MYB transcription factor from Populus trichocarpa, promotes anthocyanin production in hybrid poplar. Tree Physiol. 36, 1162–1176. [DOI] [PubMed] [Google Scholar]
- Conesa, A. , Götz, S. , García‐Gómez, J.M. , Terol, J. , Talón, M. and Robles, M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21, 3674–3676. [DOI] [PubMed] [Google Scholar]
- Dai, X. , Hu, Q. , Cai, Q. , Feng, K. , Ye, N. , Tuskan, G.A. , Milne, R. et al (2014) The willow genome and divergent evolution from poplar after the common genome duplication. Cell Res. 24, 1274–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangl, J.L. and Jones, J.D. (2001) Plant pathogens and integrated defence responses to infection. Nature, 411, 826. [DOI] [PubMed] [Google Scholar]
- De Bie, T. , Cristianini, N. , Demuth, J.P. and Hahn, M.W. (2006) CAFE: a computational tool for the study of gene family evolution. Bioinformatics, 22, 1269–1271. [DOI] [PubMed] [Google Scholar]
- Eckenwalder, J.E. (1996) Systematics and evolution of Populus In Biology of Populus, and its Implications for Management and Conservation (Stettler R.F., Bradshaw T., Heilman P. and Hinckley T., eds), pp. 542 Ottawa, ON: NRC Research Press. [Google Scholar]
- Eddy, S.R. (1998) Profile hidden Markov models. Bioinformatics, 14, 755–763. [DOI] [PubMed] [Google Scholar]
- English, A.C. , Richards, S. , Han, Y. , Wang, M. , Vee, V. , Qu, J. , Qin, X. et al (2012) Mind the gap: upgrading genomes with Pacific Biosciences RS long‐read sequencing technology. PLoS One, 7, e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, D. , Liu, T. , Li, C. , Jiao, B. , Li, S. , Hou, Y. and Luo, K. (2015) Efficient CRISPR/Cas9‐mediated targeted mutagenesis in Populus in the first generation. Sci. Rep. 5, 12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillatti, J.J. , Sellmer, J. , McCown, B. , Haissig, B. and Comai, L. (1987) Agrobacterium mediated transformation and regeneration of Populus . Mol. Gen. Genet. 206, 192–199. [Google Scholar]
- Finn, R.D. , Mistry, J. , Tate, J. , Coggill, P. , Heger, A. , Pollington, J.E. , Gavin, O.L. et al (2009) The Pfam protein families database. Nucleic Acids Res. 38, D211–D222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R.D. , Clements, J. and Eddy, S.R. (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodstein, D.M. , Shu, S. , Howson, R. , Neupane, R. , Hayes, R.D. , Fazo, J. , Mitros, T. et al (2011) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr, M.G. , Haas, B.J. , Yassour, M. , Levin, J.Z. , Thompson, D.A. , Amit, I. , Adiconis, X. et al (2011) Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon, S. , Dufayard, J.‐F. , Lefort, V. , Anisimova, M. , Hordijk, W. and Gascuel, O. (2010) New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. [DOI] [PubMed] [Google Scholar]
- Haas, B.J. , Delcher, A.L. , Mount, S.M. , Wortman, J.R. , Smith, R.K. Jr , Hannick, L.I. , Maiti, R. et al (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J. , Salzberg, S.L. , Zhu, W. , Pertea, M. , Allen, J.E. , Orvis, J. , White, O. et al (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, K.‐H. , Gordon, M.P. and Strauss, S.H. (1997) High‐frequency transformation of cottonwoods (genus Populus) by Agrobacterium rhizogenes. Can. J. For. Res. 27, 464–470. [Google Scholar]
- Han, K.‐H. , Meilan, R. , Ma, C. and Strauss, S. (2000) An Agrobacterium tumefaciens transformation protocol effective on a variety of cottonwood hybrids (genus Populus). Plant Cell Rep. 19, 315–320. [DOI] [PubMed] [Google Scholar]
- Hunter, S. , Apweiler, R. , Attwood, T.K. , Bairoch, A. , Bateman, A. , Binns, D. , Bork, P. et al (2008) InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka, J. , Kapitonov, V.V. , Pavlicek, A. , Klonowski, P. , Kohany, O. and Walichiewicz, J. (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. [DOI] [PubMed] [Google Scholar]
- Kajitani, R. , Toshimoto, K. , Noguchi, H. , Toyoda, A. , Ogura, Y. , Okuno, M. , Yabana, M. et al (2014) Efficient de novo assembly of highly heterozygous genomes from whole‐genome shotgun short reads. Genome Res. 24, 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa, M. and Goto, S. (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, I.S. , Kim, H.Y. , Shin, S.Y. , Kim, Y.S. , Lee, D.H. , Park, K.M. , Yoon, H.S. (2010) A cyclophilin A CPR1 overexpression enhances stress acquisition in Saccharomyces cerevisiae . Mol. Cells, 29, 567–574. [DOI] [PubMed] [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinformatics, 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski, M. , Schein, J. , Birol, I. , Connors, J. , Gascoyne, R. , Horsman, D. , Jones, S.J. et al (2009) Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazowski, W. (1997). Auen in Österreich – Vegetation, Landschaft und Naturschutz. Monographien Band 81. Vienna, Austria: Federal Environment Agency. [Google Scholar]
- Lexer, C. , Fay, M.F. , Joseph, J.A. , Nica, M. and Heinze, B. (2005) Barrier to gene flow between two ecologically divergent populus species, P. alba (white poplar) and P. tremula (European aspen): the role of ecology and life history in gene introgression. Mol. Ecol. 14, 1045–1157. [DOI] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Stoeckert, C.J. and Roos, D.S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R. , Li, Y. , Kristiansen, K. and Wang, J. (2008) SOAP: short oligonucleotide alignment program. Bioinformatics, 24, 713–714. [DOI] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscum, E. and Reed, J. (2002) Genetics of Aux/IAA and ARF action in plant growth and development. Plant Mol. Biol. 49, 387–400. [PubMed] [Google Scholar]
- Lukashin, A.V. and Borodovsky, M. (1998) GeneMark. hmm: new solutions for gene finding. Nucleic Acids Res. 26, 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, C. , Strauss, S. and Meilan, R. (2004) Agrobacterium‐mediated transformation of the genome‐sequenced poplar clone, Nisqually‐1 (Populus trichocarpa). Plant Mol. Biol. Rep. 22, 311a. [Google Scholar]
- Ma, T. , Wang, J. , Zhou, G. , Yue, Z. , Hu, Q. , Chen, Y. , Liu, B. et al (2013) Genomic insights into salt adaptation in a desert poplar. Nat. Commun. 4, 2797. [DOI] [PubMed] [Google Scholar]
- Majoros, W.H. , Pertea, M. and Salzberg, S.L. (2004) TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene‐finders. Bioinformatics, 20, 2878–2879. [DOI] [PubMed] [Google Scholar]
- Maloney, V.J. and Mansfield, S.D. (2010) Characterization and varied expression of a membrane‐bound endo‐β‐1, 4‐glucanase in hybrid poplar. Plant Biotechnol. J. 8, 294–307. [DOI] [PubMed] [Google Scholar]
- Meilan, R. , Sabatti, M. , Ma, C. and Kuzminsky, E. (2004) An early‐flowering genotype of Populus . J. Plant Biol. 47, 52–56. [Google Scholar]
- Ohtani, M. , Nishikubo, N. , Xu, B. , Yamaguchi, M. , Mitsuda, N. , Goué, N. , Shi, F. et al (2011) A NAC domain protein family contributing to the regulation of wood formation in poplar. Plant J. 67, 499–512. [DOI] [PubMed] [Google Scholar]
- Paponov, I.A. , Paponov, M. , Teale, W. , Menges, M. , Chakrabortee, S. , Murray, J.A. and Palme, K. (2008) Comprehensive transcriptome analysis of auxin responses in Arabidopsis . Mol. Plant, 1, 321–337. [DOI] [PubMed] [Google Scholar]
- Pinosio, S. , Giacomello, S. , Faivre‐Ramapant, P. , Taylor, G. , Jorge, V. , Le Paslier, M.C. , Zaina, G. et al (2016) Characterization of the poplar pan‐genome by genome‐wide identification of structural variation. Mol. Biol. Evol. 33, 2706–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porebski, S. , Bailey, L.G. and Baum, B.R. (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15, 8–15. [Google Scholar]
- Salleh, F.M. , Evans, K. , Goodall, B. , Machin, H. , Mowla, S.B. , Mur, L.A. , Runions, J. et al (2012) A novel function for a redox‐related LEA protein (SAG21/AtLEA5) in root development and biotic stress responses. Plant Cell Environ. 35, 418. [DOI] [PubMed] [Google Scholar]
- Salmela, L. and Rivals, E. (2014) LoRDEC: accurate and efficient long read error correction. Bioinformatics, 30, 3506–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Soliman, M.H. , Hussein, M.H.A. , Gad, M. and Mohamed, A.S. (2017) Genetic transformation of white poplar (Populus alba L.) with glutaredoxin‐2 gene. Biosci. Res. 14, 464–472. [Google Scholar]
- Song, J. , Lu, S. , Chen, Z.‐Z. , Lourenco, R. and Chiang, V.L. (2006) Genetic transformation of Populus trichocarpa genotype Nisqually‐1: a functional genomic tool for woody plants. Plant Cell Physiol. 47, 1582–1589. [DOI] [PubMed] [Google Scholar]
- Stanke, M. , Keller, O. , Gunduz, I. , Hayes, A. , Waack, S. and Morgenstern, B. (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata, N. and Eriksson, M.E. (2012) A simple and efficient transient transformation for hybrid aspen (Populus tremula × P. tremuloides). Plant Methods, 8, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo‐Graovac, M. and Chen, N. (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics, Chapter 4: Unit 4.10. [DOI] [PubMed] [Google Scholar]
- Tenea, G.N. , Spantzel, J. , Lee, L.‐Y. , Zhu, Y. , Lin, K. , Johnson, S.J. and Gelvin, S.B. (2009) Overexpression of several Arabidopsis histone genes increases Agrobacterium‐mediated transformation and transgene expression in plants. Plant Cell, 21, 3350–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan, G.A. , Difazio, S. , Jansson, S. , Bohlmann, J. , Grigoriev, I. , Hellsten, U. , Putnam, N. et al (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science, 313, 1596–1604. [DOI] [PubMed] [Google Scholar]
- Van Loo, M. , Joseph, J.A. , Heinze, B. , Fay, M.F. and Lexer, C. (2008) Clonality and spatial genetic structure in Populus × canescens and its sympatric backcross parent P. alba in a central European hybrid zone. New Phytol. 177, 506–516. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Zhu, M. and Wei, Z. (2008) Cotton laccase gene overexpression in transgenic Populus alba var. pyramidalis and its effects on the lignin biosynthesis in transgenic plants. Fen Zi Xi Bao Sheng Wu Xue Bao, 41, 11–18. [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , DeBarry, J.D. , Tan, X. , Li, J. , Wang, X. , Lee, T.‐H. et al (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W. (1988) Poplar. Harbin: Heilongjiang People's Press. [Google Scholar]
- Xu, Z. and Wang, H. (2007) LTR_FINDER: an efficient tool for the prediction of full‐length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. , Tong, L. , Li, F. , Kang, S. and Qu, Y. (2011) Sap flow of irrigated Populus alba var. pyramidalis and its relationship with environmental factors and leaf area index in an arid region of Northwest China. J. For. Res. 16, 144–152. [Google Scholar]
- Yang, Z. (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang, C.Y. , Shen, K.M. and Mao, Z.M. (1992) Populus L In Flora Xinjiangensis Tomus 1 (Yang C.Y., ed.), pp. 122–158. China: Urumqi Xinjiang Science, Technology & Hygiene Publishing House. [Google Scholar]
- Yang, W. , Wang, K. , Zhang, J. , Ma, J. , Liu, J. and Ma, T. (2017) The draft genome sequence of a desert tree Populus pruinosa . Gigascience, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z.‐H. , Kang, X.‐Y. , Li, D.‐L. and Chen, H.‐W. (2008) Pollen development and multi‐nucleate microspores of Populus bolleana Lauche. For. Stud. China, 10, 107–111. [Google Scholar]
- Zhang, W. , Fraiture, M. , Kolb, D. , Löffelhardt, B. , Desaki, Y. , Boutrot, F.F. , Tör, M. et al (2013) Arabidopsis receptor‐like protein30 and receptor‐like kinase suppressor of BIR1‐1/EVERSHED mediate innate immunity to necrotrophic fungi. Plant Cell, 25, 4227–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Feng, Q. , Lu, H.Y. , Li, Y. , Wang, A.H. , Tian, Q.L. , Zhan, Q.L. et al (2018) Pan‐genome analsyes highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 50, 278–284. [DOI] [PubMed] [Google Scholar]
- Zhao, H. , Jiang, J. , Li, K. and Liu, G. (2017) Populus simonii × Populus nigra WRKY70 is involved in salt stress and leaf blight disease responses. Tree Physiol. 37, 827–844. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods S1 The protocol of Populus alba var. pyramidalis genetic transformation system.
Figure S1 Genome assembly process and characteristics.
Figure S2 GO enrichment of specie‐specific genes.
Figure S3 Phylogenetic tree and expression conditions of PUP gene family and Aux/IAA proteins.
Figure S4 Homologous NBS R genes in Populus alba var. pyramidalis were lost TIR and NBS domain.
Figure S5 Transformation of Populus alba var. pyramidalis.
Table S1 Sequencing data used for genome assembly.
Table S2 Assemble features of Populus alba var. pyramidalis genome.
Table S3 Summary of transposon content in the genome.
Table S4 Summary of predicted protein‐coding gene annotations and their supporting evidence types.
Table S5 RNA‐seq data of four tissues.
Table S6 Functional annotation of predicted genes.
Table S7 Evaluation of completeness of the genome assembly using BUSCOs.
Table S8 Annotation of non‐coding RNAs.
Table S9 GO enrichment of high diversification gene pairs.
Table S10 GO enrichment of expanded genes families.
Table S11 GO enrichment of contracted genes families.
Table S12 NBS gene numbers identified in different poplars.
Table S13 Summary of culturing time of different transformation stage.