

Abstract
The thick ascending limb plays a key role in maintaining water and electrolyte balance. The importance of this segment in regulating blood pressure is evidenced by the effect of loop diuretics or local genetic defects on this parameter. Hormones and factors produced by thick ascending limbs have both autocrine and paracrine effects, which can extend prohypertensive signaling to other structures of the nephron. In this review, we discuss the role of the thick ascending limb in the development of hypertension, not as a sole participant, but one that works within the rich biological context of the renal medulla. We first provide an overview of the basic physiology of the segment and the anatomical considerations necessary to understand its relationship with other renal structures. We explore the physiopathological changes in thick ascending limbs occurring in both genetic and induced animal models of hypertension. We then discuss the racial differences and genetic defects that affect blood pressure in humans through changes in thick ascending limb transport rates. Throughout the text, we scrutinize methodologies and discuss the limitations of research techniques that, when overlooked, can lead investigators to make erroneous conclusions. Thus, in addition to advancing an understanding of the basic mechanisms of physiology, the ultimate goal of this work is to understand our research tools, to make better use of them, and to contextualize research data. Future advances in renal hypertension research will require not only collection of new experimental data, but also integration of our current knowledge.
I. INTRODUCTION
Hypertension is the leading cause of “loss of health” worldwide. In the United States, the incidence has increased from ~18% in the 1960s to nearly 30% today. Similar trends are emerging globally as less developed countries improve their economies and their populations become more sedentary and adopt so-called Western diets. Recent studies have shown that aggressive treatment of blood pressure to a goal of 120/80 mmHg is more beneficial than the previous target of 140/90 mmHg (317). A large contingent of both clinicians and researchers thinks that sustained elevated blood pressure is not possible without a renal defect, as pressure natriuresis would rapidly reestablish a normal blood pressure (240, 241).
The renal nephron can be subdivided into at least 13 different segments, but our purpose is to review and discuss the existing evidence for the role of the thick ascending limb in the regulation of blood pressure, primarily hypertension, and to indicate where further studies are necessary. While this task may seem straightforward, it is, to the contrary, quite complicated. Issues such as the interaction of thick ascending limbs with other structures are only now beginning to be investigated. Furthermore, the thick ascending limb is not truly a single segment but is at least two segments with cortical and medullary thick ascending limbs that possess quite different characteristics. Finally, we have known for more than 4 decades that within each of these segments there are at least two morphologically different cell types (12), and yet there is little evidence of functional implications of such differences (335, 480, 691). As such, this area of research is open to new initiatives.
In assessing the literature, one must be cognizant of the fact that every research technique plays a different role in elucidating complex physiological mechanisms such as blood pressure regulation, and apparent discrepancies in conclusions may be attributable to differences in techniques and their limitations. For instance, molecular biophysical analyses of proteins and patch-clamp studies of individual channels offer very detailed information with few confounding variables but reveal little about how the results of such studies affect an organism as a whole. In contrast, whole animal studies provide information on the organism as a whole but are influenced by a high number of confounding variables, making it difficult to attribute mechanisms (FIGURE 1). Not recognizing the limitations of the experimental procedures used can lead to erroneous conclusions, which may or may not be obvious and create confusion in the literature. For instance, early studies using systemic infusions of the nonselective nitric oxide (NO) synthase (NOS) blocker nitro-l-arginine methyl ester (l-NAME) reported that the increase in renal Na+/H+ exchanger type 3 (NHE3) abundance, as measured by Western blot during aldosterone escape, depends on NO (697). However, the authors did not account for the l-NAME-induced increase in blood pressure, and later studies showed decreased NHE3 abundance due to pressure natriuresis (475). Thus the misinterpretation of the data, and subsequent erroneous conclusions, arose from failure to measure a key parameter.
FIGURE 1.
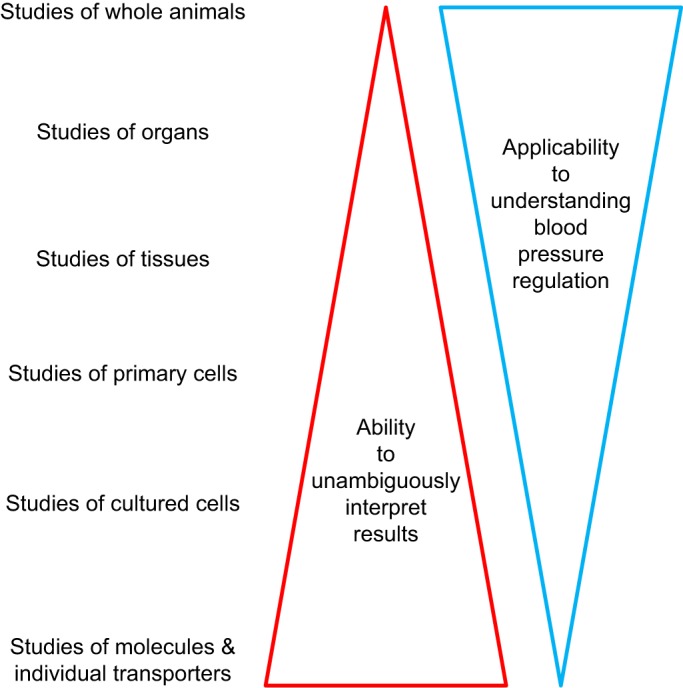
Inverse correlation between certainty of conclusions and applicability to understanding of blood pressure regulation based on experimental settings.
At times such issues are obvious, and at others less so. Originally knockout mice were heralded as the answer for off-target effects of drugs. Knockout technology offers the possibility to study the effects of the deletion of one or more gene products and the influence of heterozygosity in which only one allele of the gene of interest is silenced. Subsequently we learned that not all genes can be studied because their deletion is either lethal or causes unpredicted developmental abnormalities (457); effects in multiple tissues/organs create problems; and compensation by changes in expression of other genes, whether predicted or not, confounds interpretation of the data. As an example, the presence of claudin-16 is necessary to develop a full phenotype in claudin-10 knockout mice (62). Similar problems are likely to affect experiments with the more recently developed knockout rats (196, 197).
Recently, a newer genomic editing technology using clustered regularly interspaced short palindromic repeats (CRISPR) and the CRISPR-associated protein (Cas) 9 has emerged (69, 781). This new system could be useful to more rapidly generate both inducible and tissue-specific knockout animals, or even perform knockout on demand (89) in different tissues, or in nephron segments by packaging CRISPR/Cas9 into adenovirus under the control of specific promoters (26). However, as with all developments in the past, the limitations of this technology will eventually become apparent with widespread use. This review attempts to identify such issues to provide a better understanding of apparently conflicting data.
The following section provides a brief overview of the physiology of thick ascending limbs, required to lay the foundation for a more detailed discussion of how it is changed during hyper- and hypotension, and, in turn, how thick ascending limb NaCl reabsorption contributes to blood pressure regulation.
II. TRANSPORT
A. Transcellular Pathway
The thick ascending limb of the loop of Henle is crucial in maintaining NaCl and water balance. Early micropuncture studies showed that this segment reabsorbs ~25–30% of the NaCl filtered by the glomerulus, while being impermeable to water (44). Thus it dilutes the forming urine (67), explaining why it is frequently referred to as the diluting segment. Studies of isolated, perfused thick ascending limbs in the early 1970s reported lumen positive voltages, which led to the conclusion that NaCl transport consisted of active Cl− transport with Na+ passively following (68). The theory of active Cl− transport was ultimately proven incorrect when subsequent evidence showed that Na+ and Cl− are transported from the lumen into thick ascending limb cells with K+ in a 1:1:2 stoichiometry, suggesting the presence of an apical Na+-K+-2Cl− cotransporter (226). These findings serve as a warning that preconceived ideas can easily lead one to misinterpret even the best data.
In the general model of thick ascending limb, 50–70% of NaCl and NaHCO3 reabsorption occurs via a transcellular process, whereas 30–50% moves through the paracellular pathway. Generally, electroneutral Na+-K+-Cl− cotransport accounts for ~70% of Na+ and 100% of Cl− entry into the cell, whereas Na+/H+ exchange accounts for the remaining Na+ influx and all of the reabsorption, although species differences do exist. Once in the cell, Na+ is extruded in exchange for K+ by basolateral Na+-K+-ATPase. Cl− exits via basolateral KCl cotransport or Cl− channels. Movement of K+ is more complex. About one-half of the K+ taken up by Na+-K+-Cl− cotransport recycles to the lumen via apical K+ channels. The other one-half enters the interstitium via basolateral K+ channels or KCl cotransporters; thus K+ is reabsorbed by thick ascending limbs. Recycling of K+ into the lumen explains the positive lumen potential found by early investigators. Because K+ channels dominate membrane permeability and Cl− exits the cell via electrogenic channels, factors affecting membrane voltage through effects on K+ or Cl− permeability can, in theory, alter NaCl entry via the electroneutral Na+-K+-2Cl− cotransporter type 2 (NKCC2). A representation of transporters thought to be important in blood pressure regulation that are expressed in the thick ascending limb as well as the predominant direction of moving ions is presented in FIGURE 2. Somewhat amazingly, the roles of many thick ascending limb transporters that should relate to net NaCl reabsorption have not been directly tested under normal conditions, and there are only assumptions as to their importance. This becomes more remarkable in models of hypertension, where neither expression nor activity nor regulation of most of these transporters has been studied. These gaps in knowledge will be emphasized in this review. A brief review of the basic physiology/biophysics of the transporters thought to be important in thick ascending limb NaCl reabsorption follows. The level of detail in these sections is proportional to the existing knowledge of their roles in hyper- and hypotension.
FIGURE 2.
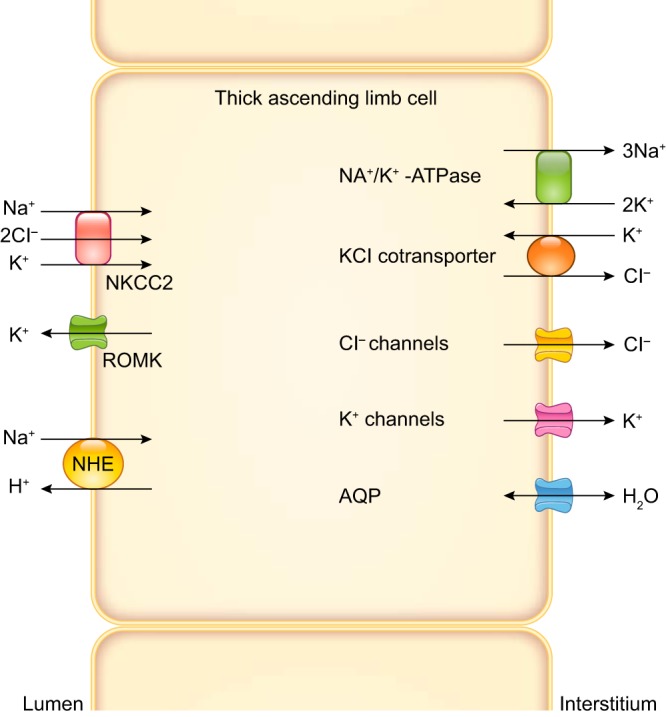
Apical and basolateral transporters expressed by thick ascending limbs thought to be important in blood pressure regulation. Predominant directions of ion movement are depicted by arrows. However, all except the Na+-K+-ATPase can move ions in either direction. The apical transporters are Na+-K+-2Cl− cotransporter type 2 (NKCC2), renal outer medullary K+ channel (ROMK), and Na+/H+ exchanger type 3 (NHE). Multiple ROMK isoforms are expressed by thick ascending limbs. Multiple NHE isoforms are expressed by thick ascending limbs, but NHE3 is the most abundant. The basolateral transporters are Na+-K+-ATPase, KCl cotransporter, Cl− channels, K+ channels, and aquaporin 1 (AQP). The Na+-K+-ATPase ultimately provides the driving force for all NaCl movement. Three subunits comprise the Na+-K+-ATPase. Although there are several α-, β-, and γ-subunits, α1, β1, γA/γB splice variants predominate. At least two different Cl− channels are expressed by thick ascending limbs. At least 2 different K+ channels are expressed in this segment. AQP is expressed by thick ascending limbs, but other water channels may also be present.
1. Apical transport
a) na+-k+-2cl− cotransporter 2.
The bulk of transcellular Na+ reabsorption by thick ascending limbs is mediated by the NKCC2 encoded by the SLC12A1 gene (182). Full-length NKCC2 is composed of ~1,100 amino acids with a predicted molecular mass of 120–121 kDa. Modeling of the cDNA sequence indicates that NKCC2 contains 12 transmembrane helices in the central region (22) with amino- (NH2)- and carboxy- (COOH)-terminal cytoplasmic tails (527, 528). Alternative splicing of exon 4, which encodes the second transmembrane segment, yields three full-length isoforms: A, B, and F. These isoforms differ significantly in their ion affinities (202) and are differentially expressed along the thick ascending limb (306, 527, 758), going from high capacity/low affinity in the medullary portion where NaCl concentration is elevated, to high affinity/lower capacity in distal parts where NaCl concentration is lower. NKCC2-F (Cl−: k½ = 111.3 mM) is expressed in medullary thick ascending limbs; NKCC2-A (Cl−: k½ = 44.7 mM) is expressed in both medullary and cortical thick ascending limbs; and NKCC2-B, which has the highest affinity (Cl−: k½ = 8.9 mM), is primarily expressed in cortical thick ascending limbs and macula densa (22, 463). A Na+-restricted diet causes a shift from NKCC2-A to the higher affinity NKCC2-B in both the cortical and outer medullary portions of the thick ascending limb (611), thereby altering transport capacity (149) to new requirements. Furosemide and its analog bumetanide can inhibit all NKCC2 isoforms. A detailed discussion of the physiology of this transporter can be found in several recent reviews (22, 427, 463, 467, 610).
Regulation of NKCC2 activity is complex, involving several mechanisms, including membrane trafficking, phosphorylation, and protein-protein interactions (22). Current evidence indicates that these mechanisms work in concert. Membrane trafficking regulates NKCC2 activity by modulating the number of transporters in the apical membrane by balancing endocytosis and exocytosis from intracellular vesicles. Biotinylation studies conducted in rat thick ascending limbs indicate that only ~5% of total NKCC2 is located in the apical membrane under basal conditions (499). About 40% of total NKCC2 is found within 0.1 µm of the apical membrane, with the remainder between 0.10 and 1.5 µm away (201). However, even though the number of NKCC2 proteins in the apical membrane may be constant, there is continuous insertion into and retrieval of transporters. The retrieval rate for NKCC2 measured by total internal reflection fluorescence microscopy in thick ascending limb cells is ~1%/min (24, 320).
NKCC2 activity is also regulated by phosphorylation. Phosphoproteomic studies conducted in rat thick ascending limbs showed phosphorylation at several amino acids, including Ser87, Thr96, Thr101, and Ser126, and Ser874 (166, 234). Thr96, Thr101, and Ser126 in the rat sequence appear to be key regulatory amino acids (140, 174, 548).
Regulation of NKCC2 activity by protein-protein interactions has not yet been implicated in controlling blood pressure. We did not consider interactions of transporters with kinases such as SPAK and ORS1 (535, 584) as true protein-protein interactions since these kinases affect activity by phosphorylation rather than by a continued interaction of the proteins themselves. Additional discussion of NKCC2 splicing, phosphorylation, and trafficking can be found elsewhere (22, 27, 427, 467, 584).
b) rectifying k+ channels.
Continuous uptake of K+ by NKCC2 requires a constant efflux of this ion from the cell. About one-half of the reabsorbed K+ recycles back to the lumen via Kir1.1 channels, also known as renal outer medullary K+ channels, or ROMK (261). ROMK channels are coded by the KCNJ1 gene. Alternative splicing produces three different gene products: Kir1.1a (ROMK1), Kir1.1b (ROMK2), and Kir1.1c (ROMK3) (281). Immunofluorescence experiments conducted in rat kidney slices using different antibodies against ROMK showed a strong signal in the apical membrane of most thick ascending limb cells, whereas a minority of cells were negative (745). This heterogeneity may also be reflected in functional differences (691). Only ROMK2 and ROMK3 are expressed in thick ascending limbs (55). ROMK2 and -3 are composed of 372 and 378 amino acids, respectively, with apparent molecular masses of ~45 kDa. ROMK channels are inwardly rectifying K+ channels (312, 388, 599). They have two transmembrane-spanning helices and cytoplasmic NH2-terminal and COOH-terminal domains (318). At least two channels are formed by ROMK subunits with conductances of around 30 and 70 pS (268, 401). The lower conductance channel is constitutively expressed, and the abundance of the 70 pS increases with elevated dietary K+ (401).
Recycling of K+ by ROMK is essential to maintain the characteristic thick ascending limb positive lumen potential of 5–10 mV (130, 423, 559). Compounds that block ROMK, such as glibenclamide or U37883A, reduce Na+ absorption (718), whereas compounds that open ROMK, such as minoxidil, increase NaCl absorption (717). Thus recycling of K+ through ROMK is necessary for maintaining NKCC2 activity. However, when the apical membrane K+ permeability is artificially enhanced with valinomycin, no increase in transport is observed (506), suggesting that the dependency of NKCC2 activity on ROMK goes beyond the maintenance of a positive transmembrane voltage by K+ recycling and could include direct protein-protein interactions between the two transporters. To our knowledge, this possibility has not been investigated. ATP (261, 285) directly activates the channel as cellular acidification inhibits it (440). ROMK activity is also controlled by phosphorylation, although this process has not been studied in great detail (378, 647, 648). A more detailed discussion of this transporter can be found in recent reviews (266, 735).
c) na+/h+ exchangers.
Net reabsorption by isolated, perfused rat thick ascending limbs ranges from 8 to 40 pmol·mm−1·min−1, representing 10–20% of that for Cl−. Reabsorption is mediated by luminal Na+/H+ exchange (218). In vivo, the resultant Na+ reabsorption accounts for >10% of the Na+ reabsorbed by rat thick ascending limbs (623). Physiologically Na+/H+ exchange is important in this segment because it reduces the bicarbonate concentration of the forming urine from ~20 mM at the hairpin turn of the loop of Henle to ~5 mM at the end of the thick ascending limb (64, 144). Additionally, the amount of Na+ reabsorbed by Na+/H+ exchange is of a similar magnitude to that reabsorbed by the entire collecting duct.
Na+/H+ exchangers are electroneutral transporters that exchange Na+ and H+ in a 1:1 stoichiometry. All NHE isoforms have 10–12 putative transmembrane helixes and a long cytoplasmic carboxyl domain (765). There are a handful of renal NHE isoforms (NHE1, NHE2, NHE3, NHE4, and NHE8) (54). NHE2 and -3, expressed by the SLC9A2 and SLC9A3 genes, are expressed in apical membrane of medullary and cortical thick ascending limb in rats and mice (94, 659, 760) and involved in NaHCO3 reabsorption. However, no NHE3 staining is evident in the apical membrane of rabbit thick ascending limbs (52), which is consistent with the lack of reabsorption by this segment in this species. NHE3 is regulated by phosphorylation at Ser552 and Ser605, trafficking, and protein-protein interactions. Although these processes have been studied in some detail in cells and in other nephron segments (104, 110, 257, 291, 354), the information about the regulation of NHE3 in thick ascending limbs remains very limited. A more detailed discussion of this transporter can be found in recent reviews (54, 510).
2. Basolateral transport
a) na+-k+-atpase.
Basolateral Na+-K+-ATPase uses energy from ATP hydrolysis to drive 3 Na+ out of the cell in exchange for 2 K+. This process establishes the electrochemical Na+ gradient used to energize NKCC2 and NHE3. Inhibition of Na+-K+-ATPase using digitalis disrupts the thick ascending limb lumen-positive potential (265), which also affects paracellular transport of other cations. Thus Na+-K+-ATPase plays a pivotal role in driving the reabsorption of Na+, Cl−, and other ions by thick ascending limbs.
Na+-K+-ATPase comprises three subunits: α, β, and γ. α- and β-subunits are in an equimolar ratio in purified preparations of Na+-K+-ATPase (117, 160). The α-subunit has eight transmembrane domains (624) and a molecular mass of 112 kDa (117). It contains the cation and ATP-binding sites and is the catalytic subunit (32, 159). Amino and carboxy termini are cytoplasmic (624). The β-subunit has a total molecular mass of 55 kDa, 35 kDa of which are from the protein component (117). This subunit has one membrane-spanning domain (158, 342) and is required for the maturation of the enzyme and its localization in the plasma membrane (767, 768). The ~10-kDa γ-subunit was characterized in purified preparations of Na+-K+-ATPase (570), and it is thought to modulate the enzyme’s affinity for cations.
Na+-K+-ATPase has multiple isoforms of its α- and β-subunits. The thick ascending limb primarily expresses the α1- and β1-subunits in equal amounts (160, 694), encoded by the ATP1A1 and ATP1B1 genes, respectively.
The γ-subunit of Na+-K+-ATPase is a member of the FXYD family. There are seven members of the FXYD protein family (FXYD1–FXYD7) (194). The FXYD2 gene encodes two splice variants (FXYD2a and FXYD2b) that are both expressed in the thick ascending limb. FXYD2a and FXYD2b proteins correspond to two isoforms of the gamma subunit γa and γb (369). Medullary thick ascending limb cells express both the γa- and γb-isoforms, whereas cortical thick ascending limb cells express only γb (558). The role of the γ-subunit in regulation of Na+-K+-ATPase activity in thick ascending limbs is poorly understood, but likely important in the regulation of NaCl reabsorption.
Na+-K+-ATPase can be regulated by phosphorylation of several residues of the α-subunit, including Ser11, Ser18, Ser23, and Ser938. Phosphorylation of Ser23 in thick ascending limbs has been reported to inhibit activity in contrast to the effect in proximal tubules (47). In thick ascending limbs, cAMP-dependent phosphorylation increases activity, although the residue affected is unknown (348). Trafficking in the proximal tubule also regulates the Na+-K+-ATPase, but this process has not been studied in detail in thick ascending limbs. A more detailed discussion of this transporter can be found in recent reviews (165, 677).
b) kcl cotransporter.
KCl cotransport accounts for about one-half of K+ and Cl− exit across the basolateral membrane in a 1:1 stoichiometry (226, 337). There are four KCC isoforms (KCC1, KCC2, KCC3, KCC4); however, only KCC4 was localized to the thick ascending limb (706). Like other electroneutral cation Cl− cotransporters, KCC proteins contain 12 membrane-spanning domains (464). KCC isoforms share a large central hydrophobic region flanked by cytoplasmic hydrophilic NH2- and COOH-terminal domains (200, 465). KCC4 cDNA predicts a polypeptide of 1,150 amino acids (465) encoded by the SLC12A4 gene. Regulation of KCC is not thoroughly understood. Its role in salt reabsorption and blood pressure regulation are similarly unclear. A more detailed discussion of this transporter can be found in a recent review (463).
c) Cl− channels.
The Cl− channels found in the basolateral membrane of the thick ascending limb belong to the ClC family. They account for about one-half of the required Cl− exit to maintain NaCl reabsorption and contribute to the basolateral membrane potential. Because they are electrogenic transporters, the negative intracellular voltage of −40 to −70 mV drives Cl− exit via these channels. Two family members of ClC are expressed in thick ascending limbs: ClC-K1 and ClC-K2 (710). ClC-K proteins are composed of 697 amino acids with a molecular mass of 75 kDa (344, 698). They have 12 hydrophobic, membrane-spanning domains and intracellular NH2- and COOH-terminals (170, 698). Functional expression requires the Barttin subunit, which facilitates insertion into the plasma membrane (155, 710).
Although both ClC-K1 and -K2 appear to be expressed in thick ascending limbs, whether one or two channels are expressed functionally is open for debate (572). On one hand, immunohistochemistry studies only showed ClC-K2 in the basolateral membrane of thick ascending limbs (352, 761). However, the large degree of homology between ClC-K1 and ClC-K2 sequences may have been a limitation of these studies, as it is not clear whether the antibodies used were in fact able to distinguish between the two channels. On the other hand, patch-clamp studies in mouse thick ascending limbs reported two different Cl− channels with conductances of ~10 and 45 pS, where the larger of the two channels was only found in ~8% of the patches, and the smaller in nearly 50% (233). The low frequency of occurrence of the larger conductance channel may limit the ability of other investigators to detect it. Finally, recent data from experiments using knockout mice indicate that ClC-K2 is the main basolateral Cl− channel in thick ascending limbs, and that it is essential for salt reabsorption by this segment (270). Still the extent of ClC-K1 contribution (if any) to basolateral Cl− permeability remains an open question. A detailed discussion of these channels can be found in a recent review (157).
d) k+ channels.
Basolateral K+ channels transport K+ taken up by Na+-K+-ATPase and NKCC2 out of the cell. Four gene families encode for basolateral K+ channels: KCNK, KCNJ, KCNQ, and SLO. The KCNK gene family encodes several inward rectifying K+ channels that are present in the kidney, with conductances ranging from ~20 to 65 pS (97, 380). These channels contain four transmembrane domains and two pore-forming regions. KCNK1 encodes a 336-amino acid peptide (380); four subunits form the K+-selective pore (414). Thus it is likely that KCNK channels form dimers (380). KCNK1 protein is expressed in the cortical thick ascending limb of rabbits (493), whereas KCNK12 and KCNK13 have been found on the basolateral membrane of the thick ascending limb of mice and rats (676). It is not clear whether these differences are due to variation between species or cortical vs. medullary segments.
The KCNJ family also encodes inward rectifying K+ channels (285). KCNJ proteins contain two membrane-spanning domains separated by an intervening loop that forms part of the pore. The amino terminus and longer carboxy terminus reside in the cytoplasm (281). The conductance of basolateral KCNJ channels is 10–40 pS (245, 400, 770). Four subunits are required to form a functional channel (281). KCNJ10 can form either an homometric channel or an heterometric channel with KCNJ16 (245, 693). Both KCNJ10 and KCNJ16 subunits were found in the basolateral membrane of cortical thick ascending limbs in mice (770). KCNJ13 is located in the basolateral membrane of thick ascending limbs in guinea pigs (134).
The KCNQ family encodes voltage-activated K+ channels. KCNQ1 mRNA was weakly detected in cortical thick ascending limbs, and it was localized to the basolateral membrane by immunostaining (775). Finally, the SLO family encodes a 150-pS K+ channel that is present in thick ascending limb cells. The SLO channel is activated by Na+ and Cl− and is neither pH nor ATP sensitive. The expression of SLO2.2 in mouse thick ascending limb cells has been demonstrated by RT-PCR (523). The details of basolateral K+ channel physiology have been reviewed elsewhere (245).
e) aquaporins.
Aquaporins (AQP) are included in this discussion because they are a special case. Although the thick ascending limb epithelium is water impermeant, the cells of this segment swell and shrink when the basolateral aspect is exposed to hypo- and hypertonic solutions, respectively (267, 585). This observation indicates that thick ascending limbs must express some members of the AQP family or closely related proteins that perform the same function. AQPs all have six strongly hydrophobic regions that span the membrane (556), with intracellular NH2- and COOH-terminals (556). AQP channels exist in membranes as homotetramers in which each subunit functions as an independent water pore (334, 441, 620). Although many AQP are expressed in the kidney, only AQP1 has been definitively identified in thick ascending limbs (76). Western blots from microdissected thick ascending limbs showed the presence of AQP1, and immunofluorescence demonstrated that AQP1 is in the basolateral but not apical membrane, as expected (76). In AQP1 knockout mice, the rate of water flux across the basolateral membrane was reduced by ~50% in comparison to wild-type mice (76). This suggests that other water channels contribute to basolateral water flux in this segment, but these have not been identified. AQP2 mRNA has been reported to be expressed in thick ascending limbs (377), but the physiological value of this finding is questionable. In theory, AQP2 is localized to the apical membrane and stimulated by vasopressin (103). Given that 1) apical AQP2 and basolateral AQP1 would make thick ascending limbs water permeant, which they are clearly not; 2) water permeability of this segment is not stimulated by vasopressin, even though V2 receptors are expressed; and 3) the mRNA report was not corroborated by data demonstrating protein expression (377), it seems unlikely that the presence of AQP2 mRNA in thick ascending limbs would result in any functional expression of the transporter.
AQP1 is discussed in this review because it has been reported to serve as a gas channel, increasing the efflux of NO in cultured cells (276) and native vascular smooth muscle and endothelial cells (275). Thus AQP1 may be involved in transport of NO out of thick ascending limb cells into vasa recta or other cells, as discussed below.
B. Paracellular Pathway
As a result of transcellular transport, there is a lumen-positive potential in thick ascending limbs that drives Na+, Ca2+, and Mg2+ reabsorption via the paracellular pathway, i.e., through the space between adjacent cells (265). Up to 50% of the total Na+ reabsorbed by thick ascending limbs traverses this route (263), which is selective for Na+ over Cl− with a Na+-to-Cl− permeability ratio (PNa+/PCl−) of ~2 (68, 225, 265, 455). An important point to consider is that the estimation of the magnitude of flux through the paracellular route was made based on experiments using perfused tubules with symmetrical solutions. The situation in vivo may be different. Initially, the positive voltage drives Na+ out of the lumen because the concentration gradient is negligible. As Na+ is reabsorbed along the tubule (and the luminal concentration of Na+ reduced), the chemical gradient between the lumen and the interstitium becomes large enough to reverse the paracellular flux of Na+ against the electrical gradient (FIGURE 3). Proof of this phenomenon comes from stop-flow studies showing that net reabsorption of Na+ stops when the luminal concentration of this ion falls below 20 mM (239, 532). This is not likely a cessation of transport, given the affinities of NKCC2 for Na+, K+, and Cl−, but rather the paracellular and transcellular fluxes becoming equal in magnitude but opposite in direction.
FIGURE 3.

Paracellular movement of Na+ in thick ascending limbs. Early in the medullary thick ascending limb, both luminal and interstitial Na+ concentrations are high. There the lumen positive voltage (V+) drives Na+ out of the lumen into the interstitium. However, as Na+ is reabsorbed, the concentration gradient for Na+ becomes large enough to reverse the paracellular flux of Na+ so that it now enters the lumen from the interstitium. The exact point at which the flip in directions occurs is unclear and depends on many factors.
The paracellular route ultimately depends on the proteins connecting the adjacent epithelial cells. Contiguous epithelial cells are connected through a series of specialized intercellular unions in their lateral walls, the tight junctions. The most apical portion of the complex is organized into protein strands that interact with strands on the membrane of the opposing cell (350). These junctions form a barrier with ionic and molecular size selectivity impeding movement of water and solutes (161, 549).
The integral membrane proteins of tight junctions include occludins, claudins, and junction adhesion molecules (451). The claudin family of proteins, which is composed of at least 27 different members (450) that range in weight from 20 to 28 kDa, is thought to confer selectivity to the paracellular pathway. Claudins have four transmembrane domains, two extracellular loops, and cytosolic amino and carboxy termini (373). Selectivity is conferred by acidic, negatively charged amino acids in the first extracellular loop. Replacing these for basic or neutral ones can reverse the preference for cations to anions (108, 109). The selectivity of claudins can be regulated acutely by posttranslational modifications, such as phosphorylation (122, 375, 668, 669, 748), but very few studies have focused on the physiological relevance of these modifications.
Claudins form both cis (with a claudin in the same membrane) and trans (with a claudin in the membrane of the adjacent cell) interactions in homo- or heteromultimeric combinations (15, 362). At least two claudins in opposing cells are needed to form a pore (15). The second extracellular loop participates in the trans interactions between claudins; mutations in the second extracellular loop do not affect cis-interactions, but result in decreased enrichment at membrane contacts between adjacent cells (537), reducing transepithelial resistance (536). In the kidney, the distribution of claudins varies in the different nephron segments; specifically, rat, mouse, and human thick ascending limbs express claudin-3, -10 (more precisely, splice variant claudin-10b), -11, -16, and -19 mRNA (236, 237, 350, 468, 701). In addition, claudin protein expression in mice and rat thick ascending limbs follows a mosaic pattern, with colocalized expression of claudin-3, -16, and -19, but not claudin-10b (448). Claudin-10b was found enriched in the inner stripe of the outer medulla and thick ascending limbs, whereas claudin-3, -16, and -19 were enriched in the outer stripe and the cortex (236, 448). As a consequence of this distribution, the inner stripe of the outer medulla showed a higher PNa+/PCl− than the outer stripe and cortex, and the opposite was true for PMg2+/PNa+. These data suggest a higher preference for monovalent ions in the inner stripe of the outer medulla that decreases toward the outer medulla, whereas the opposite is true for divalent ions (448).
Overall cation reabsorption by thick ascending limbs can be modulated differently by the presence of distinct claudin isoforms. For instance, deletion of claudin-10 reduces Na+ permeability (63). In contrast, claudin-16-deficient mice exhibit decreased Mg2+ and K+ plasma levels, elevated Mg2+ and Ca2+ urine excretion, with no changes in Na+ excretion (299). These animals are hypotensive and have elevated plasma aldosterone levels (+30%) (299). While overall Na+ excretion is unchanged, it could be that Na+ reabsorption is altered in the thick ascending limb specifically, leading to the activation of the renin-angiotensin-aldosterone system, which stimulates aldosterone production and possibly promotes Na+ reabsorption in the collecting duct. This explanation is supported by the increased urinary K+ excretion, which can be explained by the fact that elevated aldosterone promotes its secretion into the lumen in collecting ducts. Isolated, perfused thick ascending limbs from these knockdown mice show a loss of the lumen-positive potential and decreased cation selectivity. This suggests that claudin-16 confers nonselective cation permeability to the tight junctions of thick ascending limbs. Within the mouse nephron, claudin-19 mRNA is primarily expressed in the thick ascending limb (359). Claudin-19 knockdowns resemble the phenotype observed in claudin-16 knockdown mice, which include a reduction in plasma Mg2+ levels, elevated Mg2+ and Ca2+ urine excretion (1.6- and 3-fold higher, respectively), unchanged Na+ excretion, and elevated aldosterone levels (300).
III. HORMONES AND FACTORS
A. Control of the Thick Ascending Limb
To fully understand how the thick ascending limb may affect blood pressure, one needs a basic understanding of the hormones and factors that regulate Na+ reabsorption in this segment. A limited discussion of this topic follows, focused primarily on those factors that have been identified to be relevant in models of hypertension; however, this is by no means an exhaustive list of key factors.
1. Arginine vasopressin
The nonapeptide arginine vasopressin (AVP), also known as antidiuretic hormone, stimulates renal water retention and is a potent vasoconstrictor. The primary function of vasopressin is water homeostasis. This is achieved, in part, by increasing NaCl reabsorption by thick ascending limbs, which creates the osmotic gradient necessary for fluid reabsorption later in the nephron (27, 361). Two groups initially reported (nearly simultaneously) that vasopressin stimulates thick ascending limb transport in isolated, perfused tubules (243, 607). Both demonstrated an increase in transepithelial voltage and lumen to bath Cl− flux after vasopressin treatment. Vasopressin effects on thick ascending limb transport seem to be limited to the medullary portion; as it increases transepithelial voltage and net Cl− reabsorption in medullary but not cortical thick ascending limbs (264, 265). Subsequently, it was shown that the effects of vasopressin were primarily due to changes in NKCC2 activity (454, 658). Given the effects of vasopressin on NKCC2, it may regulate an amount of Na+ reabsorption similar to aldosterone. The increase in NKCC2 activity is due to phosphorylation and insertion of additional transporters into the apical membrane. The vasopressin analog desmopressin stimulates phosphorylation at amino-terminal threonine residues of NKCC2 in vivo in mice, and electron microscopy showed a 1.6-fold increase in the number of NKCC2 cotransporters in the apical membrane in vasopressin-treated animals vs. controls (201).
Similar to the collecting duct, the effects of vasopressin in thick ascending limbs are mediated by the cAMP/protein kinase A signaling cascade. Vasopressin elevates cAMP in thick ascending limbs (307, 308), and cAMP mimics the effects of vasopressin on unidirectional Cl− fluxes (243, 607), transepithelial voltage, and net Cl− reabsorption in medullary thick ascending limbs, while having no effect in cortical segments (264, 265). Finally, cAMP stimulates the number of individual transporters in the apical membrane via exocytotic insertion, and this outcome can be prevented by inhibiting protein kinase A (81). Taken together, these results indicate that the mechanisms by which vasopressin increases thick ascending limb NaCl reabsorption involve binding V2 receptors and activating a G protein/adenylate cyclase/cAMP/PKA cascade, leading to an increase in NKCC2 activity via phosphorylation and insertion of additional cotransporters into the apical membrane (FIGURE 4). However, it is currently unclear whether phosphorylation or insertion of additional transporters is more important or whether one causes the other.
FIGURE 4.

Arginine vasopressin signaling in thick ascending limbs. Arrows indicate stimulation, and T-lines indicate inhibition. Dashed lines indicate that the complete signaling cascade is unknown. AVP, arginine vasopressin; NHE3, Na+/H+ exchanger type 3; NKCC2, Na+-K+-2Cl− cotransporter type 2; PKA, cAMP-dependent protein kinase; ROMK, renal outer medullary K+ channel; V2R, arginine vasopressin type 2 receptor.
Vasopressin increases transepithelial conductance, indicating that it alters an electrogenic transport process (264, 265); therefore, in addition to NKCC2, vasopressin and its downstream signaling molecule cAMP may enhance NaCl reabsorption by increasing K+ recycling across the apical membrane by affecting ROMK. Patch-clamp studies show that this is primarily due to stimulation of 30 pS apical K+ channels, a result mimicked by cAMP (723). Vasopressin also augments the activity of apical 70-pS K+ channels. The latter was reproduced by forskolin and cAMP analogs. A protein kinase A inhibitor completely abolished vasopressin-stimulated K+ channel activity, thus linking vasopressin to cAMP effects (391). These results suggest that vasopressin stimulates ROMK via binding of V2 receptors and activation of the adenylate cyclase/cAMP/PKA signaling cascade (FIGURE 4). Because vasopressin increases NKCC2 activity, the regulation of ROMK activity could be a physiological strategy to balance the net reabsorption of K+. Further studies regarding the actions of cAMP, the downstream effector of vasopressin, on the different thick ascending limb transporters are reviewed elsewhere (81, 348, 498, 571).
Because vasopressin also influences net acid excretion, studies in isolated thick ascending limbs have been conducted to assess the role of this hormone in reabsorption. Adding vasopressin to the bath decreased reabsorption by 50%, and this effect disappeared when vasopressin was removed from the bath. The inhibition of reabsorption was also observed when vasopressin in the bath was added concomitantly to furosemide in the perfusate, showing that this phenomenon was independent of NaCl reabsorption. These effects were mediated by cAMP, as shown by the decrease in reabsorption caused by 8-bromo-cAMP or forskolin. Inhibition of reabsorption by vasopressin could be a mechanism to maintain pH during antidiuretic states and is probably mediated by NHE3 (212) (FIGURE 4). Prostaglandin E2 (PGE2) plays a role in vasopressin’s effects on reabsorption, but these will be discussed in the sect. IIIA6, Arachidonic acid metabolites.
2. Nitric oxide
NO is a free radical commonly classified as a reactive nitrogen species. Although NO is also technically a reactive oxygen species (ROS), it is generally not functionally considered to belong in this category, because the actions of most ROS are assumed to be prohypertensive, whereas NO protects against elevations in blood pressure. As such, NO is a key player in the control of blood pressure, in part by regulating renal Na+ excretion and blood flow (164, 222, 420, 424, 435, 436, 603). The biology of other reactive nitrogen species has been reviewed elsewhere (522).
a) production of nitric oxide and regulation of transcellular transport.
NO is produced by NOS enzymes from l-arginine. The three NOS isoforms, NOS1, NOS2, and NOS3, were formerly known as neuronal NOS, inducible NOS, and endothelial NOS, respectively, based on the tissue or conditions in which they were first characterized. All three NOS isoforms are found in thick ascending limbs (208, 562, 714), where NO regulates net NaCl reabsorption through inhibition of both transcellular and paracellular pathways (455, 456, 502, 506, 543). Given the central role that NO plays in regulating thick ascending limb salt reabsorption, in addition to the fact that its production or actions in several forms of hypertension are disrupted, this topic deserves to be described in some detail.
The first report concerning the effects of NO on thick ascending limb NaCl reabsorption showed that a NO donor reduced net Cl− flux in isolated, perfused thick ascending limbs. In the same study, a similar response was observed when net Cl− flux was measured in the presence of the NOS substrate, 0.5 mM l-arginine, which was attenuated by the nonselective NOS inhibitor l-NAME (543). At that time, the effect of l-arginine seemed curious. NOSs were thought to be allosterically regulated and simply adding substrate should not have activated the enzyme. To explain l-arginine’s effects, we argued that basal intracellular Ca2+ was high enough to partially activate NOS because the k½ values of NOS1 and -3 were ~200 nM, whereas intracellular Ca2+ was ~100 nM. Thus basal intracellular Ca2+ was great enough to support ~30% of maximum activity. This instance serves as a cautionary tale because, whereas this explanation satisfied the reviewers, it was later shown to be incorrect, as explained below in the sect. IIIA2C, regulation of nitric oxide by luminal flow.
Any, or a combination of, NOS isoforms could, in theory, produce the NO responsible for inhibiting transport. Studies measuring net Cl− flux in the presence of l-arginine or NO donors in isolated thick ascending limb tubules from NOS1, NOS2, and NOS3 knockout mice were done to identify the NOS isoform responsible for this inhibition. l-Arginine reduced net Cl− flux in tubules from NOS1 and NOS2 knockout mice and wild-type mice, but not in those from NOS3 knockout mice. When thick ascending limbs from this last group were incubated with a NO donor, net Cl− flux was reduced, showing that these tubules could still respond in an appropriate way to NO (542). In addition, rescuing NOS3 expression using adenovirus-mediated gene transfer in thick ascending limbs from NOS3 knockout mice restored both l-arginine-stimulated NO production and inhibition of Cl− reabsorption (507). These data indicate that NOS3 was the isoform mediating the effects on net Cl− transport in this segment under normal physiological conditions. The data do not speak to whether or not the other isoforms become important under pathophysiological circumstances.
In vivo studies showed the importance of renal and thick ascending limb NOS3 in the regulation of urinary volume and Na+ excretion (530). NOS3 knockout mice exhibited decreased urinary volume and urinary Na+ excretion without changes in blood pressure, vasopressin levels, plasma renin concentration, or glomerular filtration rate (GFR) in response to a volume load. However, when animals were given bumetanide, differences in urinary volume between NOS3 knockouts and wild-type mice were abolished, supporting the idea that NO produced by NOS3 exerts an effect on the transport mechanisms of thick ascending limbs and possibly other tissues, as discussed below. It is not known, however, why only NOS3 produces NO in this segment, even though NOS1 and -2 are also present. It may be that, under pathological conditions, NOS1 and NOS2 play a role in regulating thick ascending limb transport, or they may be involved in regulating other processes, as suggested by others (129). Alternatively, distinct NOS isoforms could be localized in different compartments as a mean of gaining specificity of their function.
A decrease in net Cl− flux could be explained by either a decrease in luminal Cl− entry or an increase in basolateral Cl− exit. Measurements of intracellular Na+ and Cl− in the presence or absence of NO donors in isolated, perfused rat thick ascending limbs show that NO decreases both (506), and that these effects were due to a reduction in NKCC2 activity. Because the activity of NKCC2 also depends on K+, we performed additional experiments to rule out the possibility that the NO donor was exerting its effects on the luminal K+ channels and indirectly affecting NKCC2. NO did not change the depolarization of the apical membrane caused by increasing luminal K+ to 25 mM. Furthermore, inhibition of net Cl− reabsorption caused by an NO donor remained in the presence or absence of valinomycin, a K+ ionophore that increases K+ permeability. These findings indicate that NO inhibited net Cl− reabsorption independently of the increase in K+ permeability. Finally, we found no acute effects of NO on basolateral Na+-K+-ATPase (506).
The signaling cascade by which NO inhibits NKCC2 activity involves both cGMP and cAMP (FIGURE 5). The soluble guanylate cyclase inhibitor LY-83583 blocked the decrease in net Cl− reabsorption by isolated, perfused thick ascending limbs caused by l-arginine (502), while dibutyryl cGMP mimics this effect (21). A role for a reduction in cAMP was supported by the fact that phosphodiesterase-2 (PDE2) inhibitors greatly diminished the ability of l-arginine (502) and dibutyryl cGMP to reduce Cl− reabsorption (21). This phosphodiesterase is activated by cGMP and selectively degrades cAMP. The cGMP-dependent protein kinase inhibitor KT-5823 failed to prevent the inhibition of net Cl− reabsorption induced by l-arginine. Finally, when tubules were treated with dibutyryl-cAMP (a membrane-permeable analog of cAMP resistant to hydrolysis by phosphodiesterase), l-arginine did not exert an inhibitory effect on net Cl− reabsorption.
FIGURE 5.

Nitric oxide (NO) signaling and known effects on individual transporters involved in transcellular NaCl reabsorption in thick ascending limbs. Arrows indicate stimulation, and T-lines indicate inhibition. Dashed lines indicate that the complete signaling cascade is unknown. AMP, adenosine monophosphate; ClC-K, Cl− channel; CLD19, claudin 19; NHE3, Na+/H+ exchanger type 3; NKCC2, Na+-K+-2Cl− cotransporter type 2; NOS3, NO synthase type 3; PDE2, cGMP-stimulated phosphodiesterase; PKA, cAMP-dependent protein kinase; PKG, cGMP-dependent protein kinase; ROMK, renal outer medullary K+ channel; sGC, soluble guanylyl cyclase.
The short-term mechanism by which cGMP, and presumably NO, regulates NKCC2 activity is via changes in protein trafficking (21). Incubation with cGMP analogs decreased surface NKCC2 levels, but not the total pool of NKCC2. When the PDE2 inhibitor BAY 60-7550 (100 nM) was present, addition of cGMP did not alter apical membrane surface NKCC2 expression, consistent with net flux experiments (21). The mechanism(s) by which cGMP acts to regulate trafficking of the transporter is not yet completely clear, but it likely involves a decrease in the insertion of transporters into the membrane. Individual cotransporters are continuously inserted into and taken up from the luminal membrane (22). It appears that NO acts by increasing cGMP, which in turn decreases cAMP (21). The decrease in cAMP then leads to a reduction in exocytotic insertion of NKCC2 into the apical membrane, thereby diminishing the number of transporters (81). This process results in lower NKCC2 activity and blunted net NaCl reabsorption. However, further studies are necessary to characterize this mechanism.
NO reduces not only NaCl reabsorption via NKCC2, but also NaHCO3 reabsorption, via actions on NHEs. NO donors reduced intracellular pH recovery, a measure of NHE activity, after an acid load in isolated, perfused thick ascending limbs (191). Both apical and basolateral NHEs were affected. In line with these results, reabsorption was reduced by endogenously produced NO (500). Incubation of tubules with cGMP analogs decreased reabsorption, and KT-5823, an inhibitor of cGMP-dependent protein kinase, abolished the inhibitory effect of l-arginine on reabsorption. These data indicate that cGMP and cGMP-dependent protein kinase mediated the inhibitory actions of NO on NHE. Thus, although NO inhibits NKCC2 and NHEs, its actions are mediated by different signaling cascades.
Currently, it is unclear how cGMP-dependent protein kinase blunts NHE activity. However, NHE3 activity can be regulated by trafficking (143, 389, 749) and phosphorylation (118) in the proximal tubule. While these possibilities have not yet been explored in the thick ascending limb, the involvement of cGMP-dependent protein kinase suggests direct phosphorylation of the transporter. Alternatively, the cascade could be more complex, involving a mediator such as the dopamine- and cAMP- regulated phosphoprotein (DARPP-32) (442). DARPP-32 regulates transport in thick ascending limbs where it is highly expressed (53, 175). Both cGMP- and cAMP-dependent protein kinases can phosphorylate DARPP-32, leading to the inhibition of phosphatase-1 (16, 53, 176).
In addition to NKCC2 and NHE, NO affects other transporters important for thick ascending limb NaCl reabsorption, but these have not been studied thoroughly. NO can inhibit conductance of a basolateral 10-pS Cl− channel in mouse thick ascending limbs through cGMP and cGMP-dependent protein kinase (741). The authors suggested that these channels are the most abundant Cl− channels in the basolateral membrane; however, their total conductance is only slightly greater than that of the 45-pS channels, because the latter has more than four times that of the single channel conductance. It is not clear whether this finding affects the interpretation of other results discussed previously regarding NKCC2 or the effects of NO on paracellular resistance discussed in the next section.
Additionally, patch-clamp data show that NO stimulates apical 70-pS ROMK channels in this segment (402, 731). In theory, the actions of NO on ROMK would be expected to increase NaCl reabsorption, both by augmenting the lumen positive potential and by enhancing NKCC2 activity. It could possibly also compensate for the inhibition of net NaCl reabsorption predicted to result from a NO-induced reduction in basolateral Cl− channel activity. However, as noted above, NO reduces net salt reabsorption in this segment. This demonstrates the danger of extrapolating results from the data without regard to other important parameters, as discussed in the introduction of this review. Additional studies of these and other transporters are clearly necessary to generate a complete picture of the regulation of transcellular NaCl and NaHCO3 reabsorption by NO in this segment.
b) effects of nitric oxide on paracellular resistance.
In addition to regulating specific transport proteins involved in transcellular movement of NaCl and NaHCO3, recent publications show that NO affects the paracellular pathway. At present, there are only two reports on native thick ascending limbs (455, 456). By measuring dilution potentials in isolated, perfused thick ascending limbs, we found that NO donors and endogenously produced NO decreased the Na+ to Cl− permeability ratio (PNa+/PCl−), a measure of the characteristics of the paracellular pathway (455) and paracellular resistance (456). The latter parameter was calculated by cable analysis from experimental data where voltage deflections after a current injection were measured with or without l-arginine. The effect of l-arginine was abolished by l-NAME, indicating that it was due to NO. With both PNa+/PCl− and paracellular resistance, absolute permeabilities of Na+ and Cl− were calculated. Surprisingly, NO increased both PNa+ and PCl−, but the increase in PCl− was greater. Treatment of tubules with a cGMP membrane-permeable analog mimicked the effects of NO. The cGMP-dependent protein kinase inhibitor, KT-5823, blocked the effects of NO, whereas the PDE2 inhibitor BAY 60-7550 did not. These data show that NO increases PNa+ and PCl− via activation of soluble guanylate cyclase, elevation of cGMP, and stimulation of cGMP-dependent protein kinase. However, it is not yet known how this cascade triggers a change in the selectivity of the paracellular pathway. It is possible that the actions of NO are ultimately mediated by the claudins present in the tight junctions of the thick ascending limb. Directly or indirectly, the members of the signaling cascade could be inducing posttranslational modifications, such as phosphorylation, or a change in membrane expression.
Mathematical modeling allowed us to predict how the actions of NO affected the luminal Na+ concentration along the thick ascending limb (456). At the end of the tubule, NO treatment resulted in a higher luminal concentration of Na+ compared with the absence of NO treatment. The model also predicted that the magnitude of the inhibitory effect on net Na+ reabsorption through the paracellular route is similar to that of the transcellular pathway. This conclusion emphasizes the importance of the paracellular pathway in the anti-hypertensive effects of NO, an area that is just beginning to be explored.
c) regulation of nitric oxide production by luminal flow.
Luminal flow varies in the thick ascending limb under physiological conditions. A high-salt diet increases luminal flow through thick ascending limbs. Given the importance of dietary salt, NO, and thick ascending limbs to blood pressure regulation, it is important to discuss the ability of flow to stimulate NO production.
As described in the previous section, the original experiments in which knockout mice were used to determine the source of NO responsible for inhibiting transport were performed in the presence of luminal flow. At that point, we were completely unaware that flow itself could stimulate NO production (542). We eventually showed that increasing luminal flow from no-flow to a high physiological flow rate stimulated NO production in isolated, perfused thick ascending limbs (504). The NO response to luminal flow was blunted by l-NAME, suggesting that flow activates NOS (504). Subsequently, we showed that the NOS isoform responsible for flow-stimulated NO production was NOS3 using knockout mice (78).
In light of these findings, we can now better explain our early observations where the simple addition of l-arginine to the bathing solution inhibited Cl− reabsorption by thick ascending limbs. NOS3 was being activated by a flow-induced phosphorylation that allowed basal intracellular Ca2+ to fully activate the enzyme. Addition of l-arginine just provided the necessary substrate to support NO production. Thus our argument based solely on intracellular Ca2+, which we had so effectively made to reviewers, was ultimately proven wrong! In the end, the nature of the experiment, i.e., the method used to measure transport, provided the stimulus for NOS activation. No one had ever thought to consider such a basic component of the experimental design to be an important experimental parameter to control. This is a prime example of the issues raised in the opening paragraphs of this review: investigators, beware!
Luminal flow increases ion delivery, stretch, pressure, and shear stress. Studies in which each variable was changed, either independently or in an opposite direction to the others, demonstrated that flow-stimulated shear stress was the parameter responsible for enhancing NO production (78). Studies have resolved at least part of the question as to how the mechanical stimulus was transduced into a chemical one by thick ascending limbs, although significant issues remain. This segment expresses at least two mechano-sensitive channels: transient receptor potential vanilloid type 4 (TRPV4) (680) and transient receptor potential polycystic type 2 (TRPP2) channels (173). Both are slightly selective ion channels that essentially become Ca2+ channels because of the large electrochemical gradient across the cell membrane for Ca2+. In experiments carried out in our laboratory, flow increased intracellular Ca2+ approximately fivefold compared with baseline, and no increase in Ca2+ was seen in the absence of extracellular Ca2+ (74). Two different TRPV4 inhibitors, ruthenium red and RN-1734, reduced peak Ca2+. Additionally, the flow-induced rise in intracellular Ca2+ was blunted in tubules transfected with TRPV4-shRNA in proportion to the knockdown of TRPV4 (74). These data indicate that TRPV4 is necessary for flow-induced increases in Ca2+ but not necessarily flow stimulation of NO production. One study proved this point by showing that TRPV4 antagonists ruthenium red and RN-1734 blocked flow-induced NO production in isolated, perfused thick ascending limb tubules (75). Thick ascending limbs that underwent in vivo adenoviral transduction with shRNA to knockdown TRPV4 exhibited blunted flow-induced NO production in proportion with the reduction in TRPV4 protein. The TRPV4 shRNA did not change NOS3 protein expression. Furthermore, two different TRPV4 agonists, 4α-phorbol-12,13-didecanoate and GSK1016790A stimulated NO production in the absence of flow in intact tubules, while they failed to stimulate NO production in transduced tubules (75). Depletion of extracellular Ca2+ prevented flow-induced NO production, providing the final link between TRPV4, intracellular Ca2+, and NO (75). This result provides solid evidence that TRPV4 is acting as a mechanosensor in response to flow that triggers the NO signaling cascade (FIGURE 6).
FIGURE 6.

Flow-induced nitric oxide (NO) production via the mechanosensitive transient receptor potential vanilloid type 4 (TRPV4) channel requires the activation of both basolateral P2X and luminal P2Y purinergic receptors. These receptors activate the phosphatidylinositol 3-kinase (PI3K), which in turn phosphorylates protein kinase B (Akt), which phosphorylates NO synthase type 3 (NOS3), increasing NO production.
Although considerable data support a role for TRPV4, they do not rule out the possibility that TRPP2 is also involved. TRPV4 and TRPP2 function as tetramers. Functional channels could contain tetramers with TRPV4-to-TRPP2 ratios of 4:0, 3:1, 2:2, and/or 1:3 (650, 772). Physiological conditions could change channel activity by altering the ratio in addition to the posttranslational modifications. Flow-stimulated NO likely plays a large role in regulating thick ascending limb NaCl reabsorption, and thus blood pressure, making this area an important focus of future research.
The next step in the cascade appears to be ATP release. Luminal flow stimulated ATP release in isolated, perfused thick ascending limbs (77, 322). In isolated, perfused thick ascending limbs, the purinergic type 2 (P2)-receptor antagonist suramin reduced NO production, indicating that the P2 receptors are responsible for the NO response to luminal flow (77, 322). In addition, quenching ATP with hexokinase, added to either the luminal or basolateral bath, blunted flow-stimulated NO production (77, 322). Given that hexokinase is a large enzyme, the likelihood of it crossing the tight junctions is very low, which strongly suggests that both luminal and basolateral release of ATP are required for flow-induced NO production (77, 322). Similar results were obtained with apyrase (77, 322). In addition, the P2X-selective antagonist NF023 prevented flow-enhanced NO production when added to the basolateral, but not the luminal side, which suggests that flow-induced NO production requires the activation of basolateral P2X receptors and luminal P2Y (77), respectively (FIGURE 6). Flow-induced increases in intracellular Ca2+ also appear to depend on both luminal P2Y and basolateral P2X receptors in mouse medullary thick ascending limbs (322). It is unknown why occupancy of both luminal P2Y and basolateral P2X receptors is required, and this question deserves further study.
The role of P2 receptors in flow-induced signaling and the relationship to TRPV4 channels is not without controversy. First, our laboratory had previously reported that the P2X-selective agonist β-γ-Me-ATP decreased oxygen consumption (a measure of active Na+ transport) in thick ascending limb suspensions, and that the P2X-selective antagonist NF023 prevented ATP-induced inhibition of oxygen consumption. However, the P2Y-selective agonist UTP caused only a small decrease in oxygen consumption (630). Taken at face value, these data would seem to indicate that the effects of ATP are only mediated by P2X receptors. However, β-γ-Me-ATP is selective for P2X receptors, but it does not exclusively activate this subclass of P2 receptors, and its selectivity is dose dependent. Thus the most likely explanation for the apparent discrepancy between our two results is that the agents used were not 100% selective. A second controversy is whether TRPV4 is activated by flow and then ATP is released, or that ATP is released directly by flow, increasing intracellular Ca2+. We found that scavenging ATP only modestly reduced flow-induced changes in intracellular Ca2+ in rat medullary thick ascending limbs. These data show that TRPV4 is activated before ATP is released (74). In contrast, in mouse tubules the flow-induced increase in intracellular Ca2+ was completely abrogated by ATP scavenging, leaving no role for TRPV4 (322). It is unclear whether this is a true species difference or due to differences in experimental design and/or environmental factors such as diet. In any case, additional studies are needed to resolve this issue and to understand how ATP gets out of the cell.
After ATP release, phosphatidylinositol 3-kinase (PI3-kinase) is activated and then phosphorylates and activates Akt and NOS3. PI3-kinase inhibitors prevented flow-stimulated and ATP-augmented NO production (628). PI3-kinase, in turn, activates Akt. There are three Akt isoforms. Akt1 mediates ATP-enhanced NO based on data from a FRET Akt activity reporter, measurements of phosphorylation, and dominant-negative mutants (628). The flow/PI3-kinase/Akt cascade ultimately caused phosphorylation of NOS3 at Ser1179, a stimulatory site (504). A representation of the mechanism by which luminal flow activates NO production is shown in FIGURE 6.
When activated by luminal flow, NOS3 translocates to different regions of thick ascending limb cells. Subcellular localization using immunostaining showed that, in the absence of flow, NOS3 was distributed relatively evenly throughout the cytoplasm and subapical and basolateral spaces. In response to luminal flow (20 nl/min), NOS3 translocated mostly to the apical membrane. When this protocol was repeated in the presence of cytochalasin D, a disruptor of the actin cytoskeleton, the translocation of NOS3 did not take place, and the increase in NO production was markedly reduced. These data indicate that an intact cytoskeleton is necessary for NOS3 to be able to translocate and be activated by luminal flow (504). Additional experiments showed that PI3-kinase mediates flow-induced NOS3 translocation and demonstrated the necessity of heat shock protein 90 (505). Whether NOS3 is translocated and then activated or vice versa is unknown. Interestingly, NOS3 translocates from the membrane to cytosolic structures when endothelial cells are stimulated with bradykinin (550), a seemingly opposite process to that occurring in thick ascending limbs.
The signaling cascade activated by luminal flow appears to be unnecessarily complicated. NOS3 can be activated directly by an increase in intracellular Ca2+. This observation raises the question as to why, or even more appropriately why doesn’t, the increase in Ca2+ caused by TRPV4 activation activate NOS3 by itself. Scavenging ATP or blocking PI3-kinase prevents the flow-induced increase in NO production, which shows that Ca2+ does not have an alternative pathway to modulate NO production. An additional question is why doesn’t the increase in Ca2+ activate NOS1, which can also be stimulated by an elevation in Ca2+/calmodulin (3, 205). Clearly, there must be subcellular compartmentalization of the Ca2+ signal, but this finding has not been reported to date.
d) regulation of nitric oxide synthases by dietary sodium.
High-salt diets alter NOS3 levels in the outer medulla (274, 278, 433, 497). This increase in protein abundance was generally assumed to be primarily due to expression changes by thick ascending limbs, but it was not directly demonstrated until Western blots were performed on freshly dissected medullary segments (497). Seven to ten days of high salt increased NOS3 protein by approximately fourfold; the ability of an NO donor to inhibit transport was also dramatically enhanced. However, NO production, as measured by either the NO-sensitive dye DAF-2DA or NO electrode, did not increase (497), which seemed curious. The lack of an increase in NO production even in the context of elevated NOS3 expression appeared to be due to changes in phosphorylation of NOS3 caused by salt. Feeding Sprague-Dawley rats high salt had a biphasic effect on NOS3 protein expression. Between 1 and 7 days, high salt increased NOS3 protein, but by 28 days they had returned to control levels (278). NO release measured with an NO-selective electrode peaked at day 1, but it showed no differences between 3 and 7 days. This temporal dissociation of expression and production was accompanied by ~40% decrease in phosphorylation of threonine 495 (Thr495), an inhibitory site, at day 1 and a >200% increase after day 3 (278). At present, the cause of the elevation of phosphorylation at Thr495, and therefore NO production inhibition by a high-salt diet, is unknown but may be due to flow-induced -induced activation of protein kinase C (293). We have proposed that acutely high salt enhances primarily NO production over , because flow-induced NO (as would be caused by high salt) inhibits flow-stimulated production (discussed below). This inhibition is not complete, however, and activates protein kinase C, which can phosphorylate NOS3 at Thr495 (171). We have also hypothesized that the decrease in NOS3 protein levels is due to salt stimulating peroxynitrite production from NO and because peroxynitrite diminishes NOS3 expression (561). At present, this idea remains mostly speculative but is important to our understanding of salt-sensitive hypertension and should be explored further.
3. Endothelin
The endothelin family of peptides comprises three members, all three of them composed of 21 amino acids and encoded by three different genes: ET-1, ET-2, and ET-3 (309). ET-1 was the first of them to be identified as a potent vasoconstrictor produced by vascular endothelial cells (314, 750). There are two endothelin receptors in humans: ETA, predominantly in vascular smooth muscle and myocytes, and ETB, most abundant in endothelial cells and renal tubules. The ET-1/ETA binding can last up to 2 h, whereas the ET-1/ETB binding is more labile (544). These receptors often have opposing effects. In the kidney, every cell expresses ET receptors. They are more abundantly located on the basolateral membrane, where they are thought to have autocrine/paracrine effects rather than an endocrine function (357).
Early studies showed that endothelin caused natriuresis without altering GFR or renal blood flow (531). It was later reported that ET-1 has an overall inhibitory effect of Na+ transport in the nephron, primarily via activation of the ETB receptor and, to a lesser extent, the ETA receptor (355). The role of ETB in Na+ transport and reabsorption is best evidenced by the fact that defects or chronic blockade of ETB receptor function results in salt-sensitive hypertension (188, 547).
The thick ascending limb is the second biggest tubular source of ET-1 (355). The first evidence of an inhibitory effect of endothelin on thick ascending limbs comes from experiments in isolated, perfused mouse thick ascending limbs, where ET-1 and ET-3 applied either in the bath or in the lumen inhibited Cl− reabsorption (128). Our group later showed that the NOS inhibitor l-NAME blocked the ET-1-mediated decrease in Cl− reabsorption in isolated, perfused rat thick ascending limbs. These results were supported by data showing that removing l-arginine from the experimental solutions prevented the inhibition of Cl− reabsorption after adding ET-1 (541). A subsequent study showed that the inhibition mediated by ET-1 was the result of reduced NKCC2 activity caused by activation of NOS3 (277). In both mice and rats, the inhibitory actions of ET-1 on thick ascending limb transport are due to activation of ETB receptors. The ET-1-induced decrease of Cl− reabsorption was blocked by adding a selective ETB antagonist BQ-788 (128, 541) and reproduced in the presence of the ETB-receptor agonist sarafotoxin S6c (128). Additionally, the selective ETA-receptor antagonist failed to block the inhibitory effects of ET-1 (541).
The signaling cascades affected after ETB-receptor activation appear to differ in rats and mice. In rat thick ascending limbs, the PI3-kinase inhibitor wortmannin blocked ET-1-stimulated NO production. Using a FRET activity reporter, we showed that ET-1 increased Akt activity, and wortmannin blunted this effect. Inhibition of Akt also prevented ET-1 from increasing NO production (FIGURE 7). Akt activation ultimately caused phosphorylation of NOS3 at Ser1177, thereby activating it (277). In contrast, in mice, protein kinase C inhibitors have been reported to block ET-1-induced inhibition of Cl− reabsorption (128). These data suggest that protein kinase C mediates the effects of ET-1; however, no increase in intracellular Ca2+ was found. The lack of an increase in Ca2+ seems to be at odds with the conclusion that protein kinase C mediated the inhibition by ET-1, unless it was due to activation one of the Ca2+-independent protein kinase C isoforms. It is important to note that the mouse experiments were performed in the absence of l-arginine, and these authors did not test whether NO was involved (128).
FIGURE 7.

Endothelin signaling in rat thick ascending limbs. Arrows indicate stimulation, and T-lines indicate inhibition. Phosphoinositol-dependent kinase (PDK) is known to be an intermediary in other systems but has not been directly demonstrated in mediating the effects of endothelin in thick ascending limbs and so is in gray. Akt, protein kinase B; ET-1, endothelin 1; ETB, endothelin type B receptor; NKCC2, Na+-K+-2Cl− cotransporter type 2; NO, nitric oxide; NOS3, NO synthase type 3; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol (4,5)-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate.
It is interesting to note that only 4 µM l-arginine was required to support NO-dependent ET-1-induced inhibition of net Cl reabsorption (541), whereas more than 100 µM l-arginine are required for flow-induced NO production (501, 504, 505). Given that the k½ of NOS3 for l-arginine is ~4 µM, whereas the k½ of the Y+ amino acid transporter that facilitates l-arginine entry into thick ascending limb cells is ~200 nM (106), these data likely indicate that ET-1 regulates l-arginine transport in addition to NOS3 activity, whereas luminal flow does not. This issue is extremely important in the larger unanswered question of how thick ascending limb NaCl reabsorption contributes to blood pressure regulation.
ET-1 not only acutely stimulates NOS3, thereby inhibiting thick ascending limb NaCl reabsorption, but it also enhances NOS3 protein levels (273, 274). Our group showed that 7 days of high-salt diet increased outer medullary osmolality. Mimicking the increase in osmolality in primary cultures of thick ascending limbs augmented ET-1 release. The elevated osmolality resulting from a high-salt diet increased NOS3 protein in primary culture of thick ascending limbs, and this effect was prevented by BQ-788, indicating ETB receptors mediated this effect (274). Dietary salt induces increases in luminal flow in the distal nephron and the outer medulla, and it may be the stimulus for ET-1 production, which is essential to maintain a normal blood pressure in the context of high salt (193, 305, 411, 613). Interestingly, elevated luminal flow (as would be caused by a high-salt diet) stimulates ET-1 production in cortical collecting ducts (411). This subject has not been studied in thick ascending limbs to date.
4. Catecholamines
Thick ascending limbs have among the highest innervation of all nephron segments (31). Offering the first evidence of adrenergic modulation of thick ascending limb transport, Bailly et al. (29) showed that the β-adrenergic agonist isoproterenol increased electrolyte transport in isolated, perfused mouse thick ascending limbs. Net fluxes of Ca2+, Mg2+, Na+, and Cl− were increased in isoproterenol-treated cortical thick ascending limbs vs. nontreated tubules, with no changes in the net flux of K+. Incubation of these tubules with the β-blocker propranolol prevented the increase. In contrast, isoproterenol stimulated net fluxes of Na+ and Cl− in medullary thick ascending limbs, but not those of K+, Ca2+, and Mg2+. Such data again emphasize some of the functional differences between these segments. These effects were likely mediated by cAMP because incubation of microdissected tubules with isoproterenol stimulated adenylate cyclase activity (29) (FIGURE 8). In contrast to β-adrenergic agonists, the selective α2-adrenergic-receptor agonist clonidine (an α2-adrenergic agonist used as an antihypertensive drug) inhibited net Cl− flux in isolated, perfused rat medullary thick ascending limbs (539), and this inhibition was blunted in the presence of the α2-adrenergic antagonist rauwolscine. When clonidine was added to tubules previously incubated with l-NAME, or in the absence of l-arginine, the reduction in net Cl− flux was similarly blocked. These data indicate that NO mediates the effects of α2-adrenergic regulation of Cl− transport in thick ascending limbs. Pretreatment with the guanylate cyclase inhibitor LY-83583 or the PI3-kinase inhibitor wortmannin prevented the clonidine-induced decrease in Cl− flux, providing a more detailed description of the signaling cascade of the actions resulting from α2-adrenergic-receptor activation (539) (FIGURE 8).
FIGURE 8.

Adrenergic signaling and known effects on transporters involved in NaCl reabsorption in thick ascending limbs. Arrows indicate stimulation, and T-lines indicate inhibition. Dashed lines indicate that the complete signaling cascade is unknown. Relatively lower concentrations of norepinephrine (NE) are needed to stimulate α2-receptors compared with β-receptors. Thus the red triangle representing NE is smaller. It is currently unclear how β-receptors activate mitogen-activated protein kinase (MAPK) in thick ascending limbs. It is also unclear how they activate Na+-K+-2Cl− cotransporter type 2 (NKCC2). α2R: α2-adrenergic receptor; βR, β-adrenergic receptor; NKCC2, Na+-K+-2Cl− cotransporter type 2; NO, nitric oxide; NOS3, NO synthase type 3; PI3K, phosphatidylinositol 3-kinase; PKA, cAMP-dependent protein kinase.
Experiments measuring the effect of norepinephrine (NE) on Cl− flux in isolated, perfused rat medullary thick ascending limbs found a biphasic effect, in which 10 and 100 pM inhibited, and 1 and 10 µM stimulated, Cl− reabsorption (538). In this same report, experiments were conducted using a series of α1-, α2-, and β-adrenergic receptor-specific agonists or adding NE in the presence of α2- and β-adrenergic receptor antagonists to show that 1) α1-adrenergic receptors do not participate in the effects of NE; 2) α2-adrenergic receptor activation by NE decreases transport; and 3) β-adrenergic receptor activation by NE stimulates NaCl reabsorption (538). Similar results were reported in other studies (29). As might be anticipated, stimulation of NKCC2 activity by isoproterenol is a result of enhanced exocytic insertion of NKCC2 in the apical membrane through a cAMP-dependent process (252). Adrenergic regulation of NaCl reabsorption is not limited to acute effects. Studies in cultured mouse thick ascending limb cells show that β-adrenergic-receptor stimulation by NE increases NKCC2 protein expression via a signaling cascade involving cAMP, protein kinase A, and mitogen-activated protein kinase (638). Treatment with the α-receptor blocker phentolamine further increased NKCC2 protein by 30% (638). The chronic effects of systemic and intrarenal elevations of NE will be discussed in sect. IVB, Induced Models of Hypertension.
5. Superoxide and hydrogen peroxide
The family of ROS includes several molecules, such as singlet oxygen, hydroxyl radical, and hypochloride anion; however, the best studied ROS in the kidney are superoxide () and hydrogen peroxide (H2O2) (192, 208). Although is produced in all regions of the kidney, the outer medulla is the main renal source of (779) and H2O2 (99).
Studies of and H2O2 are fraught with problems; the half-life of is extremely short, and it can react with nearly any molecule that contains a double bond (258, 609, 739). In general, both and H2O2 are difficult to measure in intact cells, and great care is required to get reproducible results. One possible explanation is that the indicators used to measure may not be as selective as advertised and measure other reactive oxygen containing molecules such as OH·, , or singlet oxygen. In fact, the selectivity of many of the reagents used in the study of ROS can change, depending on environmental conditions, and they may react with tubing or other plastics routinely used in experiments (unpublished observations; personal communications D. Kohan, University of Utah). The issues and scientific guidelines regarding measurements of ROS and redox-dependent signaling have been recently reviewed in a Scientific Statement of the American Heart Association (227).
production in the renal medulla is linked to the development of hypertension, as proven by the increase in blood pressure elicited by the chronic inhibition of superoxide dismutase (SOD), which catalyzes the formation of H2O2 from . These animals showed decreased medullary flow but an intact cortical flow, pointing to the central role of oxidative stress in the renal medulla in the development of hypertension (422). Acute infusion of diethyldithiocarbamate in dogs increased renal vascular resistances, and it led to decreases in renal blood flow and decreases in diuresis and natriuresis without changes in GFR (419). Because SOD is responsible for degradation, it was reasonable to hypothesize that administering the scavenger tempol would lead to decreases in blood pressure in the context of increased oxidative stress. However, medullary infusion of tempol failed to decrease mean arterial pressure, unless catalase was concomitantly infused, and medullary infusion of H2O2 by itself raised blood pressure in the absence of diethyldithiocarbamate (421). Tempol infusion alone led to an increase in UNaV and UV, but, when coinfused with catalase, the natriuretic and diuretic effects were larger. These results suggest that, even when tempol suppresses and its effects, those of H2O2 remain present (99). No matter whether medullary H2O2 or is the ROS responsible for increases in blood pressure, the absence of changes in cortical blood flow and GFR point to thick ascending limbs and/or vasa recta as the targets of oxidative stress.
Intrarenal ROS can be generated by xanthine oxidase (371), cyclooxygenases (743), uncoupled NOS (210), mitochondria (46), and NADPH oxidase (430). Three different isoforms of the catalytic subunit of NADPH oxidase, Nox1, Nox2, and Nox4, are present in thick ascending limbs (295, 383, 430). Increasing luminal NaCl concentration while keeping flow fixed at 15 nl/min, or increasing flow from 5 to 20 nl/min while keeping luminal NaCl fixed, augmented production in isolated, perfused thick ascending limbs (2). Given that apocynin prevents the ability of flow to stimulate , activation of NADPH oxidase likely mediates flow-induced production (294).
In isolated rat thick ascending limbs perfused with physiological saline, furosemide reduced flow-stimulated production by ~50% (294). Similarly, generation of was reduced by about one-half when tubules were perfused with a solution that contained no NaCl (294). Thus the ability of flow to enhance production owes equally to increased NaCl delivery and reabsorption, and flow itself (294). In addition, and because flow could stimulate production in the absence of NaCl, the authors concluded that the stretch of the epithelial cells, shear stress, or the increase in pressure should be sensed to initiate the response to elevated luminal flow (294).
Subsequent studies indicated that wall stretch was the physical parameter that caused flow-stimulated production. Stretching tubules with minimal shear stress by pinching the distal end closed increased generation, and pinching the distal end closed after tubules had been subjected to free luminal flow maintained production. Furthermore, treating perfused thick ascending limbs for a brief period with collagenase (to allow them to stretch more at a constant flow) augmented generation (190). Protein kinase C mediated flow-stimulated production. In the presence of the protein kinase Cα/β1-selective inhibitor Gö6976, flow-induced production was suppressed. A similar effect was observed in tubules transduced to express dominant-negative protein kinase Cα, but not with those expressing dominant-negative protein kinase Cβ1 (297).
Of the members of the NADPH oxidase family, only Nox4 appeared to be activated in response to luminal flow and Na+ delivery. Flow-induced production was blunted in isolated, perfused tubules from animals transfected with a Nox4 siRNA, but not in those transfected with Nox1 siRNA or in Nox2 knockout mice (295).
The prototypical NADPH oxidase is Nox2. The catalytic subunit of Nox2 is a transmembrane protein of 91 kDa. p22phox is a 22-kDa subunit that is normally associated with the catalytic subunit. Other subunits necessary for Nox2 activation are soluble proteins, including p47phox (6), p67phox, and Rac1 (95). p47phox is thought to be a chaperone for p67phox (615). Upon activation, these proteins are thought to translocate to the membrane and bind the catalytic subunit (229). Apocynin is thought to inhibit NADPH oxidase by preventing this assembly (326, 567, 651). The finding that Nox4 is responsible for flow-induced production in thick ascending limbs raises a myriad of problems and potential controversy. First, the catalytic subunit of Nox4 has a molecular mass of ~65 kDa (622), and it lacks the binding site for p47phox/p67phox (429). Thus Nox4 has been reported to be constitutively active (429, 674). If so, apocynin should not inhibit the activity of Nox4, and Rac1 should not be required for activation by NaCl (631). Because apocynin does inhibit Nox4 activity (430), there must be other, unidentified subunits required for Nox4 activation, or the current paradigm is wrong.
The theory that Nox4 activity requires other subunits is supported by the fact that Nox4 is stimulated by protein kinase C; however, no protein kinase C consensus sequences have been identified on the catalytic subunit or p22phox. Furthermore, p47phox is a chaperone molecule likely involved in assembly of other proteins. If p47phox were to guide such a putative subunit, it could explain why elimination of p47phox prevents flow-induced Nox4 activation (297, 430). The DNA polymerase delta interacting protein 2 (Poldip-2) interacts with Nox4 and is necessary for its activity (409). It colocalizes with Nox4 in thick ascending limbs (600), and knocking Poldip-2 down led to decreased H2O2 production in renal vasculature (660). This protein could, therefore, be a candidate for the putative subunit that requires translocation in conjunction with p47phox. However, it is not clear how protein kinase C and apocynin affect the way Poldip-2 interacts with Nox4 and requires further research. Finally, the current interpretation of all aforementioned data assumes that apocynin, knockout of p47phox, and protein kinase C all act at the same step of the cascade, serving as the source of production. It is possible, if not highly likely, that they are involved in different steps of the signaling cascade, which remain to be identified.
In addition to questions about assembly, there is disagreement about whether Nox4 produces or H2O2. The literature contains many reports of both molecules being produced. Some studies have suggested that Nox4 is capable of producing both, but that it generates more H2O2 than (290, 529, 666). Given the fact that is more reactive than H2O2, H2O2 is a product of reduction, and that most cells have an abundance of SOD, it would seem easier to explain reports that Nox4 produces H2O2 than those indicating that it generates . However, the structure of Nox4, specifically the E-loop, causes the enzyme to produce mostly H2O2 without the intermediate . Mutations in this loop can reverse this process so that Nox4 produces more than H2O2 (666). Although the results of this study were robust, the conclusions were based on in vitro studies using chimeras and, therefore, have limitations.
Adding to the debate, it was suggested that generation by Nox4 may be initiated by H2O2 generated by mitochondria in response to elevated Na+ delivery (491). Experiments using a novel mitochondrial-selective H2O2 sensor showed that, in isolated, perfused thick ascending limbs, increased Na+ delivery enhanced H2O2 production. The H2O2 response to Na+ delivery was blunted by the electron transport chain inhibitors rotenone and antimycin A, but not by apocynin. Taken together, these results suggest that H2O2 response to Na+ delivery was mediated by a mechanism depending on mitochondrial respiration rather than activation of an NADPH-oxidase family member (491).
The relationship between , H2O2, and Na+ reabsorption has been extensively studied in thick ascending limbs (293, 330, 503, 634). Exogenous generated by hypoxanthine/xanthine oxidase increased Cl− reabsorption in isolated, perfused tubules, an effect that was blocked in the presence of SOD. Addition of the scavenger tempol reduced transport by about the same amount as SOD; this effect was reversible as removal of tempol restored Cl− reabsorption (503). Together these data suggest that both extracellular and intracellular enhance Cl− reabsorption. The results were not likely due to H2O2 because they were blocked by the addition of SOD, unless the subsequent generation of H2O2 inhibited transport by this segment. Such an inhibitory effect seemed unlikely as treatment of perfused thick ascending limbs with 200 nM H2O2 did not alter net Cl− transport (503). It is possible though that the concentration of H2O2 used may have simply been too low to have an effect. Although mitochondria produce H2O2, there have been no direct reports of its effects on NaCl reabsorption, or transport activity by thick ascending limbs.
-induced stimulation of Cl− reabsorption was due to activation of NKCC2. Hypoxanthine/xanthine oxidase increased directly measured NKCC2 activity and exogenously added elevated steady-state intracellular Na+ and Cl−, as would be expected with enhanced NKCC2 activity (330). However, it is not yet known whether ROS increase the number of transporters in the apical membrane, their phosphorylation, or both.
Exogenous can also stimulate luminal NHE activity and these effects are blocked by tempol, suggesting that rather than H2O2 stimulates NHE3 activity (331). On the contrary, basolateral NHE activity is reduced by exogenous (331), while Na+-K+-ATPase activity appears not to be acutely affected (330). ROMK activity is inhibited by ROS in the cortical collecting duct, but this has not been studied in thick ascending limbs (722).
ROS not only increase thick ascending limb NaCl reabsorption on their own, but also does so via interactions with NO. Removing by adding tempol and SOD increased l-arginine-dependent NO production in isolated, perfused rat thick ascending limb tubules and also enhanced the inhibitory effect of endogenously produced NO on Cl− reabsorption (501).
The signaling cascade whereby enhances both NKCC2 and NHE activity involves protein kinase C. The nonselective protein kinase C inhibitor staurosporine prevented hypoxanthine/xanthine oxidase from stimulating Cl− reabsorption, as did the protein kinase Cα/β1 inhibitor Gö6976. Exogenously added enhanced the activity of protein kinase Cα and Cδ. Because Gö6976 blocked the effect, the authors concluded that the stimulation was mediated by protein kinase Cα (634). In the NHE experiments, the stimulatory effect of on activity was blocked by staurosporine and Gö6976 (293). Given that protein kinase Cα mediates flow-induced production and also appears to mediate the ability of to enhance transport, there must be subcellular compartmentalization of different pools of protein kinase Cα; however, this has never been demonstrated and calls for further exploration. A representation of the signaling in thick ascending limbs can be found in FIGURE 9.
FIGURE 9.
Superoxide () signaling in thick ascending limbs. Arrows indicate stimulation, and T-lines indicate inhibition. Dashed lines indicate that the complete signaling cascade is unknown. NADPH oxidase (NOX) is likely the primary source of in thick ascending limbs but the isoform activated by a given stimulus may vary. NHE, Na+/H+ exchanger; NKCC2, Na+-K+-2Cl− cotransporter type 2; PKCα, protein kinase Cα.
6. Arachidonic acid metabolites
Arachidonic acid is a polyunsaturated omega-6 fatty acid that can be metabolized by various enzymes, leading to the generation of metabolites with important autacoid functions in the kidney. In the next few paragraphs, we convey the updated effect of a few of these compounds. It is likely that other arachidonic acid derivatives have a role in the regulation of transport in thick ascending limbs; however, those possibilities remain to be explored.
a) prostaglandin e2.
Prostaglandins are a family of compounds that arise from the sequential conversion of arachidonic acid to 1) prostaglandin G2 by cyclooxygenase 1 and 2 (COX1 and COX2); 2) prostaglandin H2 by COX1 and COX2; and 3) chemically distinct prostaglandins by specific terminal prostaglandin synthases (519). Multiple enzymes may catalyze this last step. For instance, PGE2 is formed by at least three different PGE2 synthases (519).
The medullary thick ascending limb is one of the principal targets for PGE2 and expresses the highest number of PGE2 receptors (152). In cultured mouse medullary thick ascending limbs, PGE2 inhibited NKCC2 transport measured as bumetanide-sensitive K+ flux. Vmax decreased, but not the affinity of the transporter for its substrates. Bumetanide binding was also reduced. These data suggest that PGE2 inhibits NKCC2 activity via a reduction in the number of functional units in the membrane (338). PGE2 has also been reported to reduce Na+-K+-ATPase activity in this segment (709). The signaling cascade involved in NKCC2 and pump inhibition by PGE2 is still unclear. Although PGE2 increased cAMP levels, cAMP analogs did not inhibit NKCC2 cotransport, but they stimulated it when cells were grown on permeable supports. Thus the authors concluded that cAMP was not the signaling molecule by which PGE2 inhibited transport (338). The explanation for these results seems simple. High concentrations of PGE2 elevate cAMP levels, whereas low concentrations inhibit it via inhibitory G proteins (470). Such data suggested that multiple PGE receptors are expressed by thick ascending limbs. Given that cAMP stimulates NKCC2 by increasing its exocytic insertion into the plasma membrane (81), it would be unlikely for PGE2 to inhibit this transporter via cAMP. This example serves as a cautionary tale: just because two things are correlated does not necessarily indicate cause and effect.
On the other hand, PGE2 stimulated NHE activity (56) and reversed vasopressin-induced reductions in reabsorption (212, 214), likely because PGE2 inhibits vasopressin-dependent cAMP production, and these effects are mediated by protein kinase C (212).
PGE2 inhibited 50-pS K+ channels on the basolateral membrane in patch-clamp studies performed on medullary thick ascending limb cells (230), and it also reduced apical ROMK activity (391). This is in line with the overall inhibitory effect that this prostaglandin exerts on transport in this segment. The downstream mediators of this effect involved activation of protein kinase C and concerted phosphorylation of the mitogen-activated protein kinases, p38, and ERK (230, 391).
b) 8-iso-prostaglandin f2α.
Isoprostane 8-iso-PGF2α is a modified prostaglandin produced by the nonenzymatic oxidation of PGF2α by ROS (551, 617). When added to either the luminal or basolateral bath, it increases Cl− flux in isolated, perfused rat cortical thick ascending limbs. This effect is reversed by blocking NKCC2 activity with furosemide. The mechanism of action of the stimulatory effect of 8-iso-prostaglandin-F2α involves the activation of cAMP/protein kinase A (80). Increased 8-iso-prostaglandin-F2α plasma and urine levels are found in many hypertension models and are sometimes used as a marker for oxidative stress (612, 759). Given this evidence, the effects of PGF2α on thick ascending limb transport, as well as the role these effects play in the development of hypertension, deserve additional investigation.
c) 20-hydroxyeicosatetraenoic acid.
20-Hydroxyeicosatetraenoic acid (20-HETE) is synthesized from arachidonic acid by cytochrome P-450 omega hydroxylases in the kidney. In the thick ascending limb, 20-HETE inhibits transport through different mechanisms. Early studies showed that 20-HETE decreased 86Rb+ uptake in rabbit medullary thick ascending limb suspensions (154), and this decrease was completely abolished when tubules were pretreated with furosemide (153). Intracellular Na+ and K+ were also reduced, indicating that 20-HETE inhibits NKCC2 without affecting Na+-K+-ATPase activity (154). In contrast, other investigators reported that 20-HETE secondarily inhibits thick ascending limb Na+ uptake by reducing Na+-K+-ATPase activity in rat renal medulla suspensions (763). These studies found that 20-HETE increased intracellular Na+ levels and inhibited 86Rb+ uptake in the presence of furosemide. A fundamental difference between these two studies that could explain the disparate data is that the latter experiments (763) were performed in the presence of the 20-HETE synthesis inhibitor 17-ODYA and indomethacin to prevent endogenous production and further metabolism of 20-HETE by cyclooxygenases, while the former was not (154). 20-HETE inhibited reabsorption in isolated, perfused thick ascending limb tubules, suggesting that it reduces luminal NHE activity (215). Cell-attached patch-clamp experiments performed in rat medullary thick ascending limbs showed that 20-HETE also inhibited the apical 70-pS (719) and basolateral 50-pS K+ channels (231, 724), and 10-pS Cl channels (232) in this segment.
Together these reports suggest a regulatory role for 20-HETE in thick ascending limb transport that is consistent with an integrated mechanism to prevent excess reabsorption of NaCl. However, it is unclear what signaling cascade is activated by 20-HETE or whether it binds directly to specific transport proteins. Further studies are necessary to complement our understanding of 20-HETE in the pathogenesis of hypertension.
7. Angiotensin II
Angiotensin II (ANG II), an octapeptide with important bioactive functions, is produced by the sequential proteolytic cleavage of angiotensinogen to ANG I by renin, and then ANG I to ANG II by angiotensin-converting enzyme. ANG II can activate angiotensin receptors type 1 (AT1) or type 2 (AT2). Surprisingly, given the importance of both ANG II and thick ascending limbs to blood pressure regulation, there have been relatively few studies of the effects of ANG II on NaCl or NaHCO3 reabsorption by this segment.
At physiological concentrations, ANG II inhibits Cl− reabsorption by thick ascending limbs (34, 35, 379). These results are at odds with the generally held view that this hormone stimulates salt retention by the kidney. However, in vivo, ANG II may in fact stimulate salt reabsorption by this segment. Alone, 10−9 M ANG II inhibits Cl− reabsorption, but this same concentration stimulates transport if tubules have been pretreated with either NE (34, 35) or cAMP (35). Similarly, ANG II was found to elevate oxygen consumption, a measure of active transport, in a sustained manner after a transient inhibition in thick ascending limb suspensions treated with vasopressin, which enhances intracellular cAMP (632). It is likely that both the elevation and reduction in Cl− reabsorption are due to changes in NKCC2 activity because ANG II has a biphasic effect on this transporter with 10−6 M stimulating and inhibiting activity (14). Given that the biphasic effect of ANG II on transport in the thick ascending limbs is well established with only minor differences in the zenith and nadir in terms of concentration, the real question is: What is the in vivo concentration of ANG II in the outer medulla?
The proposed signaling cascades activated by ANG II, which mediate its effects on Cl− reabsorption and/or NKCC2 activity, are contradictory and controversial. AT1-receptor activation has been reported to be required for inhibition of transport (14, 379, 632) and its stimulation (14, 632). AT2 receptors have been reported to either have no effect (379) or mediate inhibition of NKCC2 activity (292). Inhibition of transport was reported to 1) be mediated by 20-HETE and not cAMP (14); 2) involve cAMP because a protein kinase A inhibitor could prevent the reduction (35); 3) be due to a decrease in cAMP (632); and 4) be mediated by NO (292). In contrast, the stimulation has been reported to 1) be mediated by protein kinase C (14); 2) somehow involve cAMP and protein kinase A (35); and 3) be mediated by elevated (632).
There is no clear way to explain these disparate data. However, it may be that increasing concentrations of ANG II stimulate different signaling cascades. For instance, endothelin has been shown to both stimulate and inhibit proximal tubule transport and also depend on activation of protein kinase C (185). The relative importance of the various inhibitory pathways (20-HETE vs. NO) may depend on the presence of l-arginine. Reports showing that 20-HETE is responsible for ANG II-induced inhibition of transport are based on experiments lacking l-arginine. It may be easier to reconcile the different signaling cascades involved in stimulation of transport. ANG II enhances protein kinase C activity, and protein kinase C may activate production (279), which elevates transport.
In addition to its effects on NKCC2, ANG II reduces reabsorption. Good et al. (215) demonstrated that 10−8 mM ANG II acutely inhibits reabsorption in isolated, perfused tubules in a reversible manner. These authors did not find evidence of a biphasic effect of ANG II on transport. The effect on reabsorption involved production of 20-HETE and activation of the tyrosine kinase pathway. Further studies are necessary to clarify whether ANG II affects apical or basolateral pathways of reabsorption (215).
Finally, ANG II also acutely stimulates basolateral Cl− channels via AT1 receptors. This effect relies on activation of protein kinase C and consequent increases in NADPH oxidase activity and ROS generation (742). This signaling cascade would, therefore, be similar to the one that enhances NKCC2 activity, as proposed above. A representation of ANG II signaling in thick ascending limbs can be found in FIGURE 10.
FIGURE 10.

Some of the proposed signaling cascades activated by angiotensin II (ANG II) in thick ascending limbs and reported effects on transporters involved in NaCl reabsorption. ANG II effects and signaling in the thick ascending limb are subjects of considerable controversy, and signaling is quite complex. It is likely that both are affected by environmental factors. AT1R, angiotensin II type 1 receptor; AT2R, angiotensin II type 2 receptor; 20-HETE, 20-hydroxyeicosatetraenoic acid; NHE, Na+/H+ exchanger; NKCC2, Na+-K+-2Cl− cotransporter type 2; NO, nitric oxide; NOS3, NO synthase type 3; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; PLA, phospholipase A; PLC, phospholipase C.
The interaction between ANG II, NO, and ROS plays a central role in regulating transport in the thick ascending limb. ANG II increases NO production in isolated medullary rat thick ascending limbs (139, 271) via AT2 receptors (271). Preincubation of the tubules with an Akt inhibitor prevented the effect of ANG II on NO synthesis. Akt1 appeared to mediate these actions because ANG II rapidly increased phosphorylation of Akt1 at Ser473 (at 5 min), and a dominant-negative Akt1 reduced the effect of ANG II on NO production (271). Finally, we demonstrated that ANG II led to the phosphorylation of NOS3 at Ser1177 and Ser663, and that this effect was inhibited in the presence of an Akt inhibitor, revealing the last step in the cascade (271).
Unlike NO, ANG II stimulates production in thick ascending limbs via AT1 receptors (279, 460). This effect was not altered in the presence of the AT2-receptor antagonist PD 123319. In contrast, ANG II failed to stimulate production in the presence of the AT1-receptor antagonist losartan (279). The source of ANG II-stimulated is NADPH oxidase because ANG II failed to increase in the presence of an NADPH oxidase inhibitor or in tubules from p47phox knockout mice. Furthermore, ANG II was not able to induce production in tubules in which Nox4 was knocked down, but it was able to generate in tubules from Nox2 knockout mice (430). These data are somewhat contradictory, because, in theory, Nox4 does not require the assembly of cytoplasmic units to be active (429, 674). However, our current understanding of Nox4 activation is limited and requires further study. The effects of ANG II on production are mediated by protein kinase Cα. The protein kinase Cα/β1 inhibitor Gö6976 blocked the effect, as did transduction of tubules with dominant-negative protein kinase Cα. In contrast, dominant-negative protein kinase Cβ1 did not (279).
It is interesting that ANG II stimulates both NO and at the same concentrations (271, 383). One would expect that, if NO and were generated at the same time, they would destroy each other in the process. One possibility is that NO predominates over , as NO is always measured with generation intact, but is usually assessed in the absence of l-arginine, and, therefore, NO is absent or at negligible concentrations. However, this hypothesis has not been directly tested. Unlike flow-induced NO, which prevents flow from stimulating production (296), ANG II-enhanced NO does not appear to markedly affect ANG II-stimulated synthesis. Rather, ANG II causes NOS to uncouple and become a source of (210). However, if this explanation was complete, one would not be able to measure any ANG II-stimulated NO. Clearly, the effects of ANG II on thick ascending limb transport are poorly understood, as are the signaling cascades that mediate its effects.
8. Aldosterone
Aldosterone is the main mineralocorticoid hormone in humans. In the kidney, aldosterone regulates Na+ reabsorption and K+ secretion and, therefore, plays an important role in electrolyte and water balance. Little is known, however, about the role aldosterone plays in regulating thick ascending limb transport. Two studies have shown that aldosterone decreases reabsorption acutely via an inhibitory effect on NHE3 activity in isolated, perfused rat medullary thick ascending limbs (216, 217), and that these effects are mediated by ERK (216, 217, 729). However, to date, no study has reported aldosterone regulating NKCC2 activity. Although this segment does express both glucocorticoid and mineralocorticoid receptors, the fact that it shows low levels of 11β-hydroxysteroid dehydrogenase type 2, which inactivates glucocorticoids and provides selectivity to mineralocorticoids receptors, suggests that, in the thick ascending limb, aldosterone signaling is not favored (4, 57). Furthermore, aldosterone does not activate the downstream signaling cascades stimulated in the collecting duct, which increases Na+ reabsorption in that segment. This finding then begs the question of which receptors are responsible for inhibition of reabsorption. One possible explanation is that aldosterone-induced effects on transport are due to activation of plasma membrane G protein-coupled, rather than cytoplasmic steroid hormone, receptors. This also raises the question of why thick ascending limbs express mineralocorticoid receptors.
9. Calcium and tumor necrosis factor-α
Thick ascending limbs reabsorb 25–30% of the Ca2+ filtered at the glomerulus (242). Ca2+ reabsorption in medullary segments is primarily due to passive transport driven by the lumen-positive voltage (60, 242). In the cortical segments, Ca2+ transport may be active because net reabsorption exceeds that predicted by the transepithelial potential. Furthermore, it is not inhibited by furosemide, which eliminates the lumen-positive voltage (655). Thus thick ascending limb Ca2+ and Mg2+ reabsorption has a large effect on interstitial concentrations of these ions. In turn, they can then activate the Ca2+-sensing receptor (CaSR), which is ubiquitously expressed along the nephron (495, 583, 757). CaSR is a G protein-coupled receptor whose primary function is to sense the extracellular Ca2+ concentration, as its name implies (169, 247).
Stimulation of the CaSR in thick ascending limbs leads to the activation of the nuclear factor of activated T cells, member 5 (NFAT5) (249, 479), a transcription factor involved in osmotic regulation and hypertonic stress response (17), which is also activated by apical transport (248). Activation of NFAT5 leads to the synthesis and release of tumor necrosis factor-α (TNF-α) (248, 249) by a mechanism yet to be elucidated (FIGURE 11A).
FIGURE 11.

A: synthesis and release of tumor necrosis factor-α (TNF-α) by thick ascending limbs. B: auto- and paracrine functions of TNF-α in thick ascending limbs. AT1R, angiotensin II (ANG II) type 1 receptor; CaSR, calcium (Ca2+) sensing receptor; COX2, cyclooxygenase 2; LPS, lipopolysaccharide; NFAT5, nuclear factor of activated T cells 5; NKCC2, Na+-K+-2Cl− cotransporter type 2; PGE2, prostaglandin E2; ROMK, renal outer medullary K+ channel; TLR4, Toll-like receptor 4; TNFR, TNF-α receptor. See main text for details.
TNF-α activates COX2 (713), thus explaining the finding of earlier studies showing that stimulation of the CaSR in primary cultures of medullary thick ascending limb cells increased PGE2 synthesis (712). Rat thick ascending limb suspensions also produce TNF-α after treatment with lipopolysaccharides (413) or high (0.1 mM) doses of ANG II (168), resulting in inhibition of transport (33, 168) (FIGURE 11, A AND B). Taken together, this information indicates that, once secreted, TNF-α acts as an autacoid to inhibit transport by thick ascending limbs via PGE2.
Finally, activation of the CaSR also has been reported to increase 20-HETE production in isolated rat thick ascending limbs (720), which also results in transport inhibition (724), as discussed previously.
B. Paracrine Function of the Thick Ascending Limb
The three-dimensional structure of the kidney is critical to its overall function. Nowhere is this more apparent than when considering the regulatory role of the thick ascending limb in processes of other cell types. Unfortunately, we know little about the singular cross-talk processes between thick ascending limbs and other cells, let alone their interactions. Worse, modelers have very little information on which to base their mathematical models, even if they could account for the extreme complexity. Still, the field is wide open and offers an extraordinary opportunity for scientists to make substantial contributions.
In addition to its direct ability to alter urinary Na+ excretion and water reabsorption, thick ascending limbs may alter the kidney’s ability to excrete salt and water by affecting other structures that are in close proximity, and even some at considerable distance. These include those that lie downstream of the thick ascending limb that can be affected by the forming urine and those in close proximity to thick ascending limbs via their serosal aspects. The former includes macula densa, distal convoluted tubules, connecting tubules, and cortical collecting ducts, whereas the latter includes proximal straight tubules (both S2 and S3 segments), vasa recta, descending thin limbs, cortical/outer medullary collecting ducts, and interstitial cells.
In the renal cortex, cortical thick ascending limbs are arranged in medullary rays, also known as Ferrein’s pyramids, along with proximal straight tubules and collecting ducts. At the distal end of the cortical thick ascending limb lies the macula densa, a plaque of specialized cells. Immediately afterwards, the distal convoluted tubule begins. Multiple cortical thick ascending limbs surround a single cortical collecting duct. There is an average of seven cortical thick ascending limbs for every cortical collecting duct in rats. The cortical thick ascending limbs are adjacent and equal in number to proximal straight tubules (574).
The organization differs between the outer and inner stripe of the outer medulla. In the inner stripe of the outer medulla, few medullary thick ascending limbs of long loops of Henle are interspersed with descending thin limbs of short-looped nephrons, descending vasa recta, and ascending vasa recta in bundles surrounding a core of descending and ascending vasa recta. The periphery of this vascular bundle features a layer of long-looped medullary thick ascending limbs and a few medullary thick ascending limbs from short-looped nephrons. Finally, the interbundle spaces contain interspersed medullary thick ascending limbs from short-looped nephrons, collecting ducts, descending thin limbs of long-looped nephrons, and capillaries (574) (FIGURE 12). The organization of cortical medullary rays and the outer medulla is critical to the ability of thick ascending limbs to affect the function of other nephron segments and the medullary vasculature. The cross talk between thick ascending limbs and other nephron segments is likely important for the integration of Na+ reabsorption along the nephron and between nephrons. One can easily imagine such integration, given the close localization of cortical thick ascending limbs, proximal tubules, and cortical collecting ducts in the cortex, and descending thin limbs, medullary thick ascending limbs, and outer medullary collecting ducts in the outer medulla, not to mention interstitial cells and medullary blood flow through vasa recta.
FIGURE 12.

Diagram representation of the tubulovascular arrangement at the largest transverse level in and around a vascular bundle (VB). The red dashed circle indicates the VB core in which the descending vasa recta (DVR) and ascending vasa recta (AVR) are marked in solid red and blue, respectively. I–III represent the three types of short-looped nephrons (SLN) to show their locations. The yellow dashed curve outlines the long-looped nephron (LLN) thick ascending limbs (TAL) at the margin of the VB. The proportions of the various structures are based on the tracing data. DTL, descending thin limb. [From Ren et al. (574) with permission.]
The sections below focus on situations in which paracrine factors released by thick ascending limbs alter the function in a second structure. If there is direct data showing that the compound alters function in a second structure, we discuss the potential effects in some detail. However, we only briefly discuss the possibilities in cases where the evidence of a secondary effect is not backed by data directly from thick ascending limbs.
Thick ascending limbs have been shown to alter the function of vasa recta and macula densa through the release of NO and oxidants; however, there is sufficient data to theorize that thick ascending limbs may also regulate these and other tissues via a variety of other compounds, by altering the composition of the forming urine and by regulating interstitial Ca2+.
1. Nitric oxide
The most studied paracrine factor released by thick ascending limbs is NO, a notable fact, as NO is relatively unstable with a half-life in aqueous solutions in the order of seconds (172, 678). The fact that thick ascending limb-derived NO can alter the function of other renal tissues lends credence to the idea that other, more long-lived compounds do as well.
Regulation of vasa recta tone by NO released by thick ascending limbs is perhaps the best studied cross talk between thick ascending limbs and other structures. Descending and ascending vasa recta are vessels that derive primarily from efferent arterioles, but ~10% arise directly from preglomerular afferent arterioles. They form a network of blood vessels in the outer and inner medulla. Originally, it was thought that vasa recta were just passive capillaries that lacked the ability to constrict. However, the seminal work by Pallone et al. (148, 513) showed that the pericytes surrounding the endothelial cells act as modified smooth muscle cells with the capacity to regulate vasa recta diameter, and thus medullary blood flow. As discussed in other sections of this review, decreases in medullary blood flow can contribute to elevation of blood pressure (111, 469), and NO-dependent cross talk between thick ascending limbs and vasa recta is altered in Dahl rats (461).
NO generated by thick ascending limbs is released into the interstitium, as evidenced by the fact that flow-induced NO can be measured using an extracellular NO-sensitive electrode (504). Once outside of thick ascending limb cells, NO is free to diffuse to surrounding tissues (FIGURE 13). Mori and Cowley (460) have shown that, when is reduced by scavenging with tempol, the measured amount of ANG II-stimulated NO in pericytes increases, indicating that NO diffuses from thick ascending limbs to vasa recta, and this is buffered by thick ascending limb . However, these experiments were performed in endothelial cell-denuded vasa recta (460). Consequently, it is unclear how much influence thick ascending limb NO would have in regulating vasa recta tone when the endothelium, another source of NO closer to the pericytes, is intact and capable of producing it.
FIGURE 13.

Paracrine factors released into the lumen or interstitium by thick ascending limbs that could alter vasa recta and collecting duct function. ET-1, endothelin 1; HETE, 5-hydroxyeicosatetraenoic acid; NO, nitric oxide; PGs, prostaglandins.
The macula densa function is also affected by NO. The macula densa, found in the transition between the thick ascending limb and the distal convoluted tubule, is a specialized plaque of cells that contacts the juxtaglomerular cells present on the afferent glomerular arteriole to form the juxtaglomerular apparatus. This structure senses the NaCl concentration, and possibly the flow rate of the forming urine, and sends signals to both afferent and efferent arterioles to either constrict or dilate in a process called tubuloglomerular feedback and efferent tubuloglomerular feedback, respectively. This signal appears to be driven by adenosine and/or ATP and may vary between superficial cortical and juxtamedullary nephrons. NO produced by NOS1 blunts tubuloglomerular feedback presumably by reducing NKCC2 cotransport activity, the initiating step (208, 392, 577, 715). NaCl transport into the macula densa via NKCC2 cotransport is also important in the regulation of renin release from juxtaglomerular cells (398, 533, 614).
Macula densa function can be affected by NO released by thick ascending limbs into the forming urine, which is swept downstream (FIGURE 14). Evidence for this process comes from in vitro studies of tubuloglomerular feedback. Afferent arterioles and attached macula densa were simultaneously microperfused in vitro, and a tubuloglomerular response elicited; as expected, addition of the NOS1 inhibitor 7-nitroindazole enhanced this response. In the presence of 7-nitroindazole, the nonselective NOS inhibitor Nω-nitro-l-arginine methyl ester augmented tubuloglomerular feedback further in macula densa perfused orthograde via thick ascending limbs, but not in those perfused retrograde via the distal tubule. Inhibition of macula densa soluble guanylate cyclase blocked the effect of NO produced by thick ascending limb NOS when macula densas were perfused via thick ascending limbs. Such data indicate that NO produced by thick ascending limbs can enter the forming urine and affect segments downstream (575).
FIGURE 14.
Paracrine factors released into the lumen of thick ascending limbs that have been shown or could alter macula densa and distal convoluted tubule function. ET-1, endothelin 1; HETE, 5-hydroxyeicosatetraenoic acid; NO, nitric oxide; PGs, prostaglandins.
Elevated tubuloglomerular feedback caused by a reduction in macula densa NO production has been shown to cause salt-sensitive hypertension (404). Therefore, we would expect that any decrement in NO reaching the macula densa from the thick ascending limb would have a similar effect. However, although thick ascending limb NO bioavailability is known to be reduced in several models of hypertension, including ANG II-induced hypertension and the Dahl salt-sensitive rat, whether and how crosstalk between thick ascending limbs and macula densa contributes to these forms of high blood pressure have not yet been resolved.
NO produced by the loop of Henle reaches the macula densa and presumably the distal convoluted tubule (FIGURE 14). Factors that increase luminal flow augment NO delivery without affecting its luminal concentration. Perfusing the loop of Henle with furosemide increases NO delivery and concentration (381). This is expected as NaCl reabsorption by thick ascending limbs stimulates production (294), which could reduce NO bioavailability. Whether NO regulates NaCl reabsorption by the distal convoluted tubule is currently unknown.
Collecting ducts lie in close proximity to thick ascending limbs in bundles in the outer medulla that extend into the cortex in medullary rays. The average distance between a thick ascending limb and a collecting duct can be calculated to <100 µm (574). We first showed that NO produced by endothelial cells inhibits cortical collecting duct Na+ reabsorption (653), and that NO blunts vasopressin-stimulated water reabsorption (187). We calculated that NO could diffuse as far as 100 µm from endothelial cells to collecting ducts (652). At that time, it was unknown that thick ascending limbs could produce NO, so we did not take into consideration the possibility that thick ascending limbs could also regulate collecting duct transport by releasing NO. Given their close proximity, it is likely that NO released across thick ascending limb basolateral membranes, possibly mediated by AQP-1 (272, 276), diffuses through the interstitium to the collecting ducts. It seems less likely that NO released into the forming urine by thick ascending limbs would have much influence, given the distance it would have to travel and its half-life. To date, this field remains undeveloped, despite the potentially important fact that the collecting duct is the final site of salt and water reabsorption along the nephron and, therefore, responsible for the fine tuning of urinary volume and urinary Na+ excretion.
Finally, NO has been shown to reduce proximal tubule transport. It is possible, given their location, that thick ascending limb NO could also affect S1 and early S2 segments of the proximal tubule. As described above, late S2 and S3 segments are arranged within medullary rays in close proximity to thick ascending limbs. Thus it is likely that NO from the latter could influence proximal nephron salt and water reabsorption. However, studies have yet to address this area of physiology (FIGURE 15).
FIGURE 15.

Paracrine factors released into the interstitium by thick ascending limbs that could alter proximal tubule and thin descending limb function. ET-1, endothelin 1; HETE, 5-hydroxyeicosatetraenoic acid; NO, nitric oxide; PGs, prostaglandins.
2. Superoxide and hydrogen peroxide
The oxidants and H2O2 are produced by thick ascending limbs (190, 208, 210, 491). The study of in particular demands caution because of the complex chemistry and high reactivity (a half-life in water of ~1 s) of this species. Moreover, the tools to study present several problems (227). Under normal circumstances, produced in thick ascending limbs does not reach the vasa recta (460) because of either scavenging by NO or actual inhibition of production by NO (296). In contrast, when NO bioavailability in thick ascending limbs is chemically reduced, it appears that stimulating the produced by this segment leads to an increase in in the vasa recta, where it may cause vasoconstriction. From such data, we can conclude that produced by thick ascending limbs regulates vasa recta tone in a paracrine manner. Thus, under conditions in which NO is reduced, such as hypertension or reduced luminal flow, thick ascending limb likely constricts vasa recta, reducing medullary blood flow. While the results from the aforementioned study seem straightforward, we find them difficult to reconcile with the short known half-life of in aqueous solutions, its high tendency to react with double bonds of phospholipid side chains, and the unlikelihood that a molecule carrying a net negative charge diffuses across the lipid portion of membranes. Thus an alternative explanation of these results is that thick ascending limb SOD in the intercellular space may form H2O2, and that this less reactive compound diffuses to and induces production by vasa recta pericytes.
Based on the fact that thick ascending limb NO reaches the macula densa, and data showing other oxygen-containing reactive compounds generated in thick ascending limbs modulate vasa recta function, we must assume that the latter species also alter macula densa-mediated processes. In the macula densa, augments tubuloglomerular feedback (177, 641, 734, 771). Originally, this process was thought to be due to reducing NO (576) and also affecting the afferent arteriole (394). More likely, stimulates NKCC2 activity in the macula densa via a protein kinase C-dependent mechanism, similar to what occurs in thick ascending limbs (634).
Thick ascending limbs may also modulate macula densa production. Increases in luminal pH in isolated, perfused macula densa promote production by NADPH oxidase (393). Because thick ascending limbs reabsorb in several species, this segment regulates the pH of the forming urine just before it reaches the macula densa. Therefore, one would expect that factors that reduce thick ascending limb reabsorption would tend to increase production in the macula densa, thereby enhancing tubuloglomerular feedback. At present, this and other important issues in this area have not been addressed.
Because or similar species from thick ascending limbs can reach the vasa recta, one must assume that these compounds would also be able to diffuse to the collecting duct. In A6 cells, a model of this segment, aldosterone has been shown to increase production. In addition, as blunts the ability of NO to inhibit epithelial sodium channel (ENaC) activity, scavenging reduces the activity of this transporter (762). Similarly, ANG II increased ENaC activity in native collecting ducts, as measured by patch clamp. Protein kinase C and NADPH oxidase inhibition prevented the stimulation, suggesting that /H2O2 mediated the effects of ANG II (660). In mpkCCD cells, prorenin had similar effects as those of ANG II, and these were blocked by a NADPH oxidase-1 and -4 inhibitor and by NADPH oxidase-4 siRNA (403). Together, these data indicate a high likelihood that thick ascending limb , H2O2, or both can diffuse to the collecting duct where they increase salt reabsorption. This mechanism is undoubtedly important in the many forms of hypertension that involve enhanced thick ascending limb oxidative stress. However, at present, this field is remarkably understudied.
3. Endothelin
Medullary thick ascending limbs are the second greatest source of renal ET-1 after inner medullary collecting ducts (355). Unlike NO and , ET-1 is a peptide and thus long-lived. As such, it is more likely to diffuse from thick ascending limbs to surrounding structures and to reach downstream tubules via the forming urine. Currently, there is no direct evidence that thick ascending limb ET-1 alters the function of other renal tissues; however, this possibility is extremely likely and offers a promising research opportunity.
Descending vasa recta lie in close proximity to medullary thick ascending limbs. Silldorff and colleagues (627) have shown that ET-1, -2, and -3 all induce vasoconstriction, with ET-1 being most potent. Vasoconstriction elicited by ET-1 was abrogated by an ETA-receptor antagonist, whereas vasoconstriction elicited by ET-3 was blocked by an ETB-receptor agonist. ET-1 causes vasoconstriction in this vascular segment by inhibiting KATP channels (83). The authors speculated that their data were consistent with the theory of localized control of medullary blood flow.
We think there is sufficient evidence to make a case that luminally released ET-1 from thick ascending limbs alters tubuloglomerular feedback via the macula densa (FIGURE 14). Single nephron (SN) GFR increases when ET-1 is infused into the proximal tubule in micropuncture experiments. More proximal luminal exposure of the macula densa itself to endothelin causes a similar increase in SN-GFR without changes in systemic and renal hemodynamics (592). These data suggest that the effects of luminal ET-1 on the macula densa are a physiological mechanism to inactivate the tubuloglomerular feedback that counteracts the effects of systemic ET-1. However, other investigators have reported that ET-1 has no effect on tubuloglomerular feedback (665).
Thick ascending limb ET-1 is likely to reach both cortical and medullary segments of the collecting duct (FIGURE 13). Endothelin was first shown to inhibit vasopressin-stimulated water and Cl− reabsorption, and transepithelial voltage in cortical collecting ducts. However, the receptors and signaling cascades involved were not investigated (684). Subsequently, it was shown that endothelin reduced Na+ reabsorption via both ENaC-dependent and ENaC-independent mechanisms via activation of both ETA and ETB receptors (410). The inhibitory effect on ENaC appears to be due to ubiquitination via the Nedd4-2 ligase (524).Thick ascending limb ET-1 also may inhibit vasopressin-stimulated cortical collecting duct water permeability. It has been shown to reduce vasopressin-enhanced fluid reabsorption in this segment (684). However, the effects of thick ascending limb endothelin on cortical collecting transport may be overridden by endothelin released by the collecting duct itself acting in an autocrine fashion as collecting ducts produce endothelin (516). Similarly, even though ET-1 inhibits Na+-K+-ATPase activity in medullary collecting ducts (769), ET-1 from the thick ascending limb is not likely to influence transport by this segment. Inner medullary collecting ducts are the greatest source of ET-1 in the kidney, and they are not in close proximity to thick ascending limbs.
Thick ascending limb ET-1 may also reach the S2 and S3 segments of the proximal tubule (FIGURE 15). In this segment, ET-1 has a biphasic effect on Na+ and water reabsorption. ET-1 at 10−13 M stimulates transport while 10−9 M inhibits it (185). The augmentation of fluid and Na+ reabsorption is due to both elevated Na+/H+ exchange and Na+/ cotransport (235). Stimulation is protein kinase C dependent (185, 235), whereas inhibition depends on arachidonic acid metabolites (185). Big ET-1, the precursor to ET-1, may have similar effects, as it increases diacylglycerols, activators of protein kinase C in proximal tubules (37). Whether thick ascending limb ET-1 alters distal convoluted tubule function is unknown.
4. Adenosine and adenosine triphosphate
Thick ascending limbs release ATP in response to mechanical stimulation (77, 489) as does nearly every cell type. At present, the field lacks data showing that thick ascending limb ATP or adenosine alters the function of other renal structures; although a study in vascular crosstalk in Dahl salt-sensitive rats showed that treating medullary tissue strips from SS.BN.13 with the P2 receptor antagonist suramin abolished the buffering in vasa recta constriction mediated by thick ascending limbs (486). In opposition to these findings, another study showed that ATP plays a role in regulating medullary blood flow by constricting vasa recta (119). How different ATP receptors play distinct roles in regulating vasa recta tone is, therefore, a subject worthy of exploration. Release of ATP by the thick ascending limb could also augment tubuloglomerular feedback by the macula densa (FIGURE 14). In contrast, ATP may increase (673) or decrease (65) ENaC activity, depending on whether it was released into the interstitium or forming urine (FIGURE 13). Current data suggest that basolateral ATP stimulates transport in this segment (137, 736).
5. Prostaglandins and metabolites of arachidonic acid
Thick ascending limbs produce a variety of prostaglandins and other arachidonic acid metabolites. Currently, nothing is known about the role of these compounds in regulating the function of other renal structures, although a significant body of evidence exists for such factors regulating proximal tubule, thin descending limb, macula densa, collecting duct (525), and vasa recta function (343) (FIGURES 13–15).
6. Luminal concentration of sodium chloride
The most obvious way in which thick ascending limbs may affect downstream salt and water reabsorption is through changes in luminal NaCl concentration. Elevated thick ascending limb salt reabsorption will reduce luminal NaCl concentration, which blunts macula densa-mediated tubuloglomerular feedback. Reduced luminal NaCl will also stimulate renin release from juxtaglomerular cells via a macula densa-mediated mechanism. Given that thick ascending limb NaCl reabsorption is abnormally high in Dahl salt-sensitive and spontaneously hypertensive rat (SHR), the effect of the thick ascending limb on the macula densa may partially explain why plasma renin activity (PRA) remains normal in these models when it should be low because of baroreceptor inhibition of renin release.
Elevated thick ascending limb NaCl reabsorption will also enhance vasopressin-stimulated water reabsorption in the collecting duct by increasing the osmotic gradient from lumen to bath. In contrast, inhibition of salt reabsorption by thick ascending limbs will have the opposite effect.
7. Calcium
Reabsorption of Ca2+ by thick ascending limbs can cause large changes in interstitial concentrations of this ion. Since the CaSR is expressed in many cellular types within the kidney (495, 583, 757), Ca2+ reabsorption by thick ascending limbs can potentially affect the function of surrounding structures. Still, at the present time, there are no reports of changes in interstitial Ca2+ concentrations in hypertensive models, or reports of the role of augmented thick ascending limb transport in such an increase. That said, we can speculate what would happen to the function of a large variety of cells based on current knowledge.
It seems unlikely that Ca2+ reabsorbed by thick ascending limbs would have a significant effect on proximal convoluted tubule function. More than one-half (65%) of filtered Ca2+ is reabsorbed by proximal tubules, and the total length of proximal convoluted tubules in the cortex exceeds that of cortical thick ascending limbs. Additionally, the proximal convoluted tubules are far removed from cortical thick ascending limbs. In contrast, the S2 and S3 segments of the proximal tubule are in close juxtaposition to cortical thick ascending limbs, and most Ca2+ is absorbed early in the proximal tubule. Thus it is likely that divalent ion reabsorption by cortical thick ascending limbs would have a profound effect on S2 and S3 function. However, if it did, it would likely stimulate fluid and salt reabsorption, further contributing to salt retention and hypertension, as shown by studies on the role of the CaSR and increased fluid reabsorption in the proximal tubule (84).
Unlike the proximal tubule, Ca2+ reabsorbed by the thick ascending limb would be expected to reduce vasopressin-stimulated fluid reabsorption by the collecting duct, as shown by studies where the CaSR downregulates expression and trafficking of AQP-2 (72, 557, 564). The effects of Ca2+ reabsorbed by thick ascending limbs would, therefore, tend to have an anti-hypertensive effect on collecting duct transport. The fact that activation of the CaSR reduces vasopressin-stimulated water permeability via a reduction in cAMP explains why proximal tubule transport is stimulated, as Na/H exchange activity is blunted by cAMP and protein kinase A activation.
As described above, thin descending limbs passively reabsorb water. Currently, there is no direct evidence showing that changes in interstitial Ca2+ alter this process. In contrast, increasing either luminal or basolateral Ca2+ reduces the Na+-to-Cl− permeability ratio in thin ascending limbs. This effect is mimicked by neomycin and gentamycin, which are known activators of the CaSR (654). The authors concluded from these data that activation of the CaSR decreases NaCl reabsorption (654). However, a decrease in the Na+-to-Cl− permeability ratio could be caused by a decrease in Na+ permeability, an increase in Cl− permeability, an increase in both parameters with the magnitude of the effect on Cl− being greater, or a decrease in both parameters with the effect on Na+ being of greater magnitude. The actual change in parameters and the NaCl concentration gradient will determine whether NaCl reabsorption is increased or decreased. For instance, we recently showed that NO reduces the Na+-to-Cl− permeability ratio in thick ascending limbs and may reduce Na+ reabsorption (456). Whether or not Ca2+ reabsorption by thick ascending limbs actually affects thin ascending limbs under physiological conditions or in hypertension is not known.
Currently, there is no published evidence that vasa recta tone can be altered by small changes in interstitial Ca2+ via the CaSR. Given that dietary and thus interstitial Ca2+ affects blood pressure like vasa recta tone does, this is likely to be an important area for future research.
IV. ANIMAL MODELS OF HYPERTENSION
The study of renal function in models of hypertension is fraught with problems. A number of factors are frequently overlooked by investigators. First, perhaps most importantly, urinary Na+ excretion alone is not a measure of Na+ balance. Fecal and grooming losses can be considerable, but are often ignored. Many studies report differences in urinary Na+ excretion between hypertensive and normotensive animals at 2 wk or more, assuming these reflect differences in Na+ balance. This is not the case. Both rats and mice quickly come into measurable Na+ balance within a few days after the prohypertensive insult. Differences in urinary Na+ excretion at such time points more likely reflect changes in intake, assuming proper sample collection and handling. Sustained differences in Na+ balance over weeks are clearly impossible. The calculations to prove so are easily performed.
Small changes on the order of ±10% in renal parameters, including urinary Na+ excretion, GFR, and renal blood flow that are frequently labeled “not significant,” can and do affect blood pressure. The explanations for why these changes are declared “not significant” are multifold. They include poor experimental design, lack of statistical power, biological variability, and inability to precisely measure the parameter of interest. For instance, even though we have the analytic power to measure femtomoles (10−15) of Na+ (amounts many orders of magnitude below what a usual biological sample would contain), it is quite another thing to measure Na+ in an aliquot of urine to this level of accuracy. Inevitable contamination of urine by food and/or feces, etc., prevents us from doing so.
The composition of the experimental diet is also frequently overlooked (147). The origin of dietary protein and other components influence the development of hypertension (198, 434). Few investigators bother to use verified, matched low- and high-Na+ diets. However, this may change, albeit slowly, with the National Institutes of Health’s new standards on scientific rigor and authentication of key biological variables.
Blood pressure measurements themselves are subject to variables rarely considered. While most investigators understand that tailcuff measurements are stressful for the animal, most do not consider the fact that rats are social animals, and the isolation necessary for telemetric measurements of blood pressure is also stressful.
Last but not least, problems have arisen in the study of individual signaling cascades in isolation. The information produced in these studies, albeit of great detail, is sometimes disconnected from the existing body of knowledge. This creates difficulties for the functional interpretation and contextualization of the results. This problem is particularly evident in hypertension-focused studies because of the multifactorial nature of this condition.
A. Genetic Models
1. Dahl rat
The Dahl rat has been extensively used in hypertension research as an experimental model that shares many similarities with the development of salt-sensitive hypertension in humans, especially in the African American population, in which this type of hypertension accounts for 75% of the cases (406, 732). The Dahl rat is the best example of the role of the thick ascending limb in an animal model of hypertension, even though the ultimate causes of salt sensitivity in this strain remain unknown. In fact, several studies have pointed to different molecular, biochemical, and physiological mechanisms derived from multiple genetic loci. Part of the complexity of interpreting results from this model owes to the fact that different lines of the Dahl salt-sensitive rat that have gone through genetic bottlenecks are available as explained below.
a) development of different strains of dahl rats.
The Dahl rat was originated in the 1960’s by Lewis K. Dahl et al. (124, 125) at the Brookhaven National Laboratory by breeding Sprague-Dawley rats prone to hypertension when placed on a high-salt diet. Their control strain was also bred from outbred Sprague-Dawley rats, but selected to be resistant to salt-induced hypertension (124, 125). These two strains were designated S and R, respectively (here we will use the notation S/Br and R/Br, but this was not used in publications at the time), but they were not initially pure inbred strains. Rapp and Dene (565) developed the inbred strains using rats coming from the Brookhaven colonies. These rats were designated S/JR and R/JR to distinguish them from the original strains. They were subsequently designated SS/JrHsd and SR/JrHsd (that will be simplified as S/Jr and R/Jr for the purpose of this review) when they were transferred to Harlan Sprague-Dawley (643) (now Envigo). Where possible, we will identify the specific strain of rats used in the studies reported in the following sections. In response to increased dietary NaCl, both S/Br and S/JR rapidly developed high blood pressure and renal damage (124, 565). When kidneys from S/Br were transplanted to R/Br, the recipients became hypertensive when fed a high-salt diet. These data indicated that a renal defect was the cause of salt sensitivity in the original strain of animals (123, 126).
Two decades later, cosegregation studies crossing S/Jr and R/Jr, showed that the elevated blood pressure phenotype cosegregates with a restriction fragment length polymorphism of the renin gene (566). Based on these findings, investigators from the Medical College of Wisconsin in Milwaukee bred a congenic line homozygous for the salt-sensitive renin allele, by selection and backcrossing of rats from the Harlan colony (323). The original purpose of this colony was to identify the genes responsible for the sensitivity to salt in Dahl rats using phenotyping, genetic mapping, consomic lines, and zinc-finger nuclease technology (199, 366, 432, 458); but an additional advantage was that the Milwaukee colony avoided the genetic contamination reported for S/Jr rats in the early 1990’s (643). These animals were later inbred, creating the SS/JrHsD/Mcwi strain, currently available at Charles River Laboratories as SS/JrHsdMcwiCrl. Harlan Sprague-Dawley, on the other hand, eliminated the contaminated colonies and bred a new inbred strain using the noncontaminated foundation colony (711).
An interesting consideration is that the severity of hypertension in the SS/JrHsdMcwi (that will be simplified to S/Mcw) depends on the composition of the diet (198, 434). These animals develop a lower degree of damage and hypertension when fed a grain-based diet than when fed a purified diet; moreover, the source of dietary protein administered to dams influences blood pressure and renal damage in the offspring (198). This implies that new genetic abnormalities or epigenetic programs were bred into the S/Mcw that were not in the original S/Br or S/Jr strains. Thus, because of the breeding strategies and original sources of the rats, Dahl salt-sensitive rats offered by Charles River Laboratories are different from those sold by Envigo. Investigators should have considered this while making comparisons or extrapolating experimental results.
It is evident by now that the genetic heterogeneity of Dahl salt-sensitive rats raises the problem of finding a proper control strain. There is not a simple answer here, and the proper control would largely depend on what experimental problem the investigator is trying to address. Although there is great resistance in the hypertension community to the use of Sprague-Dawley rats as the control for any of the Dahl salt-sensitive strains, from a genetic point of view it may make more sense than using any of the originally derived R stains. The reasons are simple. The salt-sensitive rats, as generated by Dahl and inbred by Rapp, were derived from outbred Sprague-Dawley rats. The salt-resistant strains were also derived from outbred Sprague-Dawley rats. Thus it is highly unlikely that the gene alleles that promote salt sensitivity in the S/Br and S/Jr are simply missing in R/Br and R/Jr rats. Given the multitude of genes that potentially regulate blood pressure, it is far more probable that the R/Br and R/Jr strains have gene alleles that resist the effects of salt, and these may or may not be expressed in S/Br and S/Jr. Genetically, this likely puts the Sprague-Dawley rat in the middle of these strains. This issue was recognized and addressed by choosing the Brown Norway rat, an outbred original parental strain of Sprague-Dawley rats, to generate consomic control strains, as discussed in the following paragraph.
In an attempt to develop a model that would be more useful in genetic studies, and to address the problem of the control, a consomic strain of the S/Mcw presenting less sensitivity to salt was developed. The SS.BN.13 is a S/Mcw in which chromosome 13 was replaced with that from the Brown Norway rat (113). When SS.BN.13 are placed on a high-salt diet, their blood pressure increases to a lesser degree than in S/Mcw (113). The increase in blood pressure caused by high salt in the SS.BN.13 ranges from <20% (113) to near 50% (459) of the S/Mcw. Thus, as they are not completely resistant to salt, the SS.BN.13 does not represent an ideal genetic control either. The development of a moderate condition suggests either that uncontrolled environmental factors play an important role in salt sensitivity, that genes causing salt sensitivity are not restricted to chromosome 13, or both. To investigate these issues, a full range of consomic strains were generated using other chromosomes from the Brown Norway rat (116, 432). Not surprisingly, the consomic strains present a wide range of sensitivities to salt and renovascular abnormalities (116, 366, 408, 432, 434). Some of these consomic strains are relevant to this review and will be discussed in later sections.
b) renal function and thick ascending limbs.
In prehypertensive S/Br and R/Br, renal blood flow and GFR were not different (43, 593). In contrast, pressure natriuresis was blunted in S/Br compared with R/Br because of a reduction in slope (586) (FIGURE 16). The natriuretic response to a Na+ load largely depended on how the load was administered. When on a basal diet containing 0.4% Na+, the S/Br rats showed exaggerated natriuresis and urinary volume (with diluted urine) in response to an isotonic Na+ load given by gavage. However, no differences between S/Br and R/Br were found with respect to Na+ or water excretion when the given load was hypertonic. The authors argued that the isotonic load was unmasking a defect in a concentrating mechanism in the thick ascending limb (42). While the more dilute urine supports such a conclusion, the elevated Na+ excretion and urinary volume does not. One consideration, based on our personal observations, is that S rats are hyperexcitable. Thus the stress caused by manipulation and the gavage could have caused blood pressure to increase more in S/Br than R/Br rats, explaining the differences in excretion rates. We would expect this state to be masked by the hypertonic load, owing to the amount of salt given.
FIGURE 16.
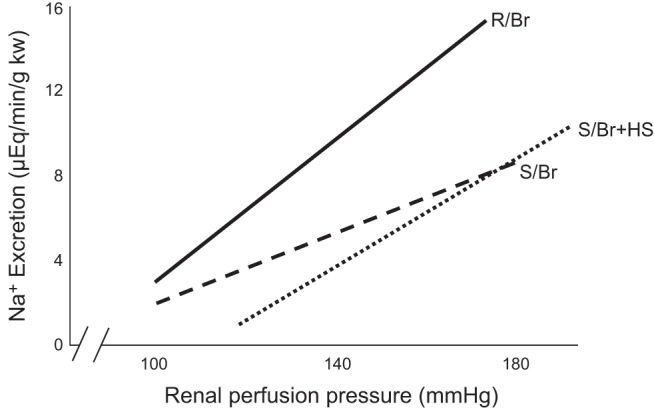
Pressure natriuresis relationship in Dahl salt-resistant rats fed normal salt and salt-sensitive rats fed normal and high-salt diets. kw, kidney weight; R/Br, Dahl salt-resistant rats, Brookhaven strain, fed normal salt; S/Br+HS, Dahl salt-sensitive rats, Brookhaven strain, fed high salt; S/Br, Dahl salt-sensitive rats, Brookhaven strain, fed normal salt. Pressure natriuresis was blunted in S/Br by a decrease in slope, while S/Br+HS additionally showed a rightward shift. This indicates that, when hypertension is established after dietary manipulations, renal changes lead to increases in blood pressure that are no longer salt-sensitive. [Adapted from Roman (586).]
Conscious prehypertensive S/Br rats on a 0.3% Na+ diet had decreased urinary Na+ excretion in response to an intravenous NaCl load (591). Experiments with anesthetized animals also showed that S/Br rats maintained on a salt-restricted diet with blood pressure similar to those in R/Br had decreased Na+ excretion in response to volume expansion (593). Decreases in Na+ excretion in prehypertensive S/Jr rats were also reported during renal function protocols where rats were maintained in a mild diuretic state (589, 778). Together these studies pointed to abnormally high Na+ reabsorption by a tubular segment.
When S/Br and R/Br rats were placed on a 8% salt diet for 2 wk, they showed no differences in Na+ and water balance, although extracellular fluid volume increased in S/Br animals after 1 day (591). The likely explanation for these data is the rapid development of hypertension in the S/Br animals, which began at day 1 and continued for the duration of the study. Greene et al. (224) showed that blood pressure did not rise when S/Br were infused with 20 mEq Na+/day, while volume expansion was prevented by servo-control, indicating that fluid retention causes hypertension. As represented in FIGURE 16, in the salt-sensitive rat with established hypertension, the pressure natriuresis curve not only decreased in slope, but also shifted to the right. This result probably indicates that the loss of renal mass elicited by high perfusion pressures in this model is enough to turn this condition into essential hypertension (not salt sensitive). Supporting this idea, different studies have shown that, when a hypertensive S rat is switched back from a high- to a normal-salt diet, the blood pressure remains elevated in several animals for at least 2 days (114, 452). In fact, the time course of salt-induced hypertension in the Dahl rat seems to have one initial component of <1 wk, where animals exhibit strong salt sensitivity, and a second phase where there is an irreversible resetting of blood pressure to higher values (703, 704).
Several micropuncture studies supported the theory that the primary defect in the S/Br rat resided in the loop of Henle. S/Br fed a diet containing 8% NaCl for 4 wk excreted NaCl slower than R/Br in response to setting renal perfusion levels to those in R/Br. This result correlated with increased loop Cl− reabsorption but not proximal or more distal segments of the nephron (345). In addition, when renal perfusion pressure is set high, mimicking hypertension, loops of Henle of S/Jr rats reabsorbed more Cl− than those of R/Jr, and proximal and distal tubules reabsorbed less. This same pattern was reported for S/Br and R/Br rats, although the blood pressure of the S/Br strain was ~20 mmHg lower than the S/Jr strain when on the 0.3% NaCl diet (589). Further studies extended these observations using either euvolemic or volume-expanded rats on a salt-restricted (0.06% NaCl) diet. After volume expansion, urinary NaCl excretion was decreased in S/Jr, and loop Cl− reabsorption was greater. In euvolemic animals, urinary NaCl excretion and loop Cl− reabsorption were not different, as would be expected for animals in Na+ balance. However, when distal nephron NaCl delivery was increased by perfusing the loop of Henle, as would be caused by a high-salt diet, greater loop Cl− reabsorption was also present in euvolemic S/Jr rats (346).
Because micropuncture experiments showed that loop of Henle Cl− reabsorption was elevated in the salt-sensitive rats, several investigators examined whether this was true for isolated, perfused thick ascending limbs. Under basal conditions, with no hormones or factors present that could alter NaCl reabsorption, S/Jr thick ascending limbs reabsorbed more Cl− than tubules from R/Jr (313). Similarly, we found that thick ascending limbs from S/Jr rats reabsorbed Cl− at rates 10–15% greater than those from R/Jr; however, our experiment lacked the power to resolve this difference (186). Whether this was a result of the small number of animals we used, or due to differences in the source of the rats between experiments remains unclear. However, it is important to note that, even though some experimental techniques cannot resolve a 10–15% increase in NaCl reabsorption, such difference would be expected to have a huge impact in salt retention and be sufficient to cause salt-sensitive hypertension.
Another topic of study important to our understanding of the role of the thick ascending limb in salt-sensitive hypertension is the expression and activity of the transporters involved in NaCl reabsorption.
Genetic studies have shown that the NKCC2 locus in the S/Jr is linked to the development of salt-induced hypertension (280). Studies in S/Jr and R/Jr rats given a low-salt diet (0.2% NaCl) showed that, while NKCC2 activity was increased in the S/Jr, its protein abundance was decreased (13). It is important to mention that, even though animals were kept on a Na+-restricted diet, the S/Jr presented blood pressure values 20 mmHg higher than the R/Jr, which can probably explain the difference in NKCC2 protein expression (13). Adding to the confusion, a subsequent study indicated that total NKCC2 protein was increased in the renal medulla of S/Mcw rats compared with SS.BN.13 and Brown Norway, regardless of the Na+ content of the diet (286). These disparate results may have been due to the existence of multiple isoforms of NKCC2 and the fact that researchers used different NKCC2 antibodies. Alternatively, this can be interpreted in terms of the genetic differences of the rats used.
NKCC2 activity depends not directly on total protein levels, but rather on the number of transporters in the apical membrane and their phosphorylation. When S/Jr were fed a low-salt diet (0.22% NaCl), total NKCC2 protein in thick ascending limb was lower than in R/Jr, but the fraction located in the apical membrane was higher. This change was accompanied by higher levels of NKCC2 phosphorylation in S/Jr, which correlated with greater SPAK/OSR1 activity, as measured by their phosphorylation. Total protein levels of SPAK and OSR1 were similar between strains (23). The opposing effects of lower total, but greater apical, expression and phosphorylation appear to result in no difference in NKCC2 activity (measured as furosemide-sensitive oxygen consumption) when rats were fed a 0.22% NaCl diet (250). A high-salt diet, consisting of 1% NaCl in drinking water, decreased NKCC2 activity by lowering apical NKCC2 expression in R/Jr thick ascending limbs. In contrast, NKCC2 activity did not decrease in response to dietary salt in thick ascending limbs from S/Jr, and, in fact, apical NKCC2 expression increased (250). This demonstrates a lack of NKCC2 regulation in salt sensitivity, perhaps due to NO, 20-HETE, or some other compound.
It has been long known that renin is involved in salt sensitivity of Dahl rats (323, 566, 644). Surprisingly, knocking out the renin gene in S/Mcw (Ren −/−) did not affect NKCC2 protein levels in whole kidney homogenates (526). If one assumes that ANG II upregulates NKCC2 protein expression (370), then knocking out the renin gene should have negatively impacted NKCC2 protein abundance. The lack of an effect on NKCC2 may have been due to the dramatic reduction in blood pressure (~50 mmHg) or the renal abnormalities (457). Alternatively, as discussed earlier, total protein abundance may not reflect NKCC2 activity, and the authors did not directly measure transport (526).
NKCC2 requires intact ROMK activity to recirculate K+ from the cell to the lumen. Total ROMK protein expression was reported to be elevated in the renal medulla of S/Mcw rats compared with SS.BN.13 and Brown Norway, regardless of dietary Na+ (286). The role of ROMK in salt-sensitive hypertension was further explored by developing a ROMK knockout rat in S/Jr background. ROMK heterozygosis did not prevent hypertension but attenuated the increase in blood pressure compared with wild-type S/Jr. This attenuation was more evident when rats were fed an 8% compared with a 4% salt diet (777). A role for thick ascending limb ROMK is also supported by data that show a ROMK inhibitor, ROMKi B, was able to prevent and reverse the rise in blood pressure in S/Jr fed a high-salt diet (776). Together these data all support the necessity of functional ROMK for the development of the hypertensive phenotype in S rats. However, it remains to be determined whether there is a primary elevation in ROMK activity, or if it just follows the elevated activity of NKCC2. As any treatment reducing NKCC2 activity would be expected to blunt salt sensitivity in this model, one should not assume direct cause and effect.
Finally, as apical and basolateral transport of Na+ are tightly coupled, the elevated NKCC2 activity occurring in the S rats is expected to positively impact the basolateral Na+-K+-ATPase. Reinforcing this idea, cosegregation analysis on the F2 of S × R has shown that the NKCC2 and the α1-subunit of Na+-K+-ATPase loci are linked to the development of salt-induced hypertension, and that their interaction defines the level of blood pressure (280).
Studies in microdissected tubules showed that in prehypertensive S/Jr, medullary thick ascending limb Na+-K+-ATPase activity under unstimulated conditions was similar to that found in R/Jr (482). However, when the dopamine 1 receptor agonist fenoldopam was added, it only inhibited the Na+-K+-ATPase in R/Jr tubules (482). Treatment with fenoldopam also failed to increase the levels of cAMP in S/Jr medullary cells. Because this elevation was present when cAMP was increased by forskolin treatment, the authors concluded that there was a dopamine unresponsiveness in S/Jr (482). This lack of response to dopamine in S/Jr was also reported in proximal tubules (481, 482), and confirmed in whole-animal studies where administration of dopamine increased fractional Na+ excretion in R/Jr but not in S/Jr (482). Still, the available information regarding the role of dopamine in this model of hypertension is very limited, and further studies are required.
Finally, sex differences in Dahl rats have been reported (204, 437, 754); however, none of these addressed thick ascending limb function.
c) nitric oxide.
NO is an important regulator of blood pressure, and its deficiency predisposes one to salt-sensitive hypertension (602). As l-arginine is the substrate that supports NO production by all NOS isoforms, l-arginine supplementations were studied in several models of salt-sensitive hypertension as a means to prevent or blunt the elevations in blood pressure. The fact that S/Jr rats have deficiencies in renal production of NO, as evidenced by reduced urinary nitrate and nitrite excretion (301), provides the rationale to explore the effects of l-arginine supplementation in this model.
In the S/Jr rat, both oral and intraperitoneal administration of l-arginine for 5 days blunted the increase in blood pressure caused by an 8% NaCl diet (98). Moreover, in hypertensive S/Jr fed high-salt diets, administration of l-arginine led to a normalization of the pressure natriuresis curve and increased natriuresis (521). Systemic infusion of a high dose of l-arginine normalized pressure natriuresis in S/Jr rats (301). These effects were not due to an improvement in renal structure, as the increase in blood pressure was blunted by administering l-arginine for 3 days (301). Studies designed to identify the location of defective NO synthesis showed that infusing l-arginine only into the renal medulla of S/Jr was sufficient to blunt the increase in blood pressure elicited by 7 days of 4% NaCl intake. The dose of l-arginine was low enough to prevent changes in cortical blood flow or systemic effects when administered intravenously (452). These studies led to the conclusion that NO production was reduced in the renal medulla of the salt-sensitive rat.
To start investigating the cause of the NO deficiency, many studies addressed the expression levels of one or more NOSs. It was found that S/Mcw presented reduced NOS3 and NOS1 mRNA levels in the renal medulla, compared with Brown Norway rats (764). In addition, a significant reduction in NOS1 and NOS3 mRNA levels was reported in the outer medulla of S/Jr compared with R/Jr, when fed either 0.6 or 8% NaCl diets (90). Interestingly, this latter study also found that NOS1 transcription levels in R/Jr rats were higher than in Sprague-Dawley rats, which presented NOS1 mRNA levels similar to those found in S/Jr (90). This fits well with the protection to salt sensitivity in the R/Jr compared with the strain that originated and emphasizes the need of a better control for the S/Jr.
Studies addressing the NO production capacity by measuring conversion of l-arginine by outer medullary preparations showed a reduction in this parameter in the S/Mcw compared with Brown Norway (664). Confirmatory experiments showed that all three isoforms of NOS were reduced in S/Mcw (664). Other reports indicate that renal NOS3 protein expression was not different between S/Jr and R/Jr on a 0.2% NaCl diet; and that switching both S/Jr and R/Jr to a high-salt diet (8% NaCl) did not affect renal NOS3 protein. Finally, it was also reported that S/Jr presented lower levels of NOS2 protein in kidneys, aorta, and heart in both low- and high-salt diets (478). The disparities in these results might also come from the use of different strains and controls. Discrepancies between transcription and protein expression might indicate differences in translational efficiency of the NOSs. However, differences in NOS3 phosphorylation, which also regulates function, have not yet been explored.
One mechanism by which NO deficiency could be mediating salt-sensitive hypertension is by sensitization of the renal medulla to small elevations of hormones that affect thick ascending limbs. Intravenous infusion of a subpressor dose of vasopressin for 2 wk in rats fed a normal-salt diet increased blood pressure in S/MCw, but not in SS.BN.13. However, Na+ balance was not measured, and the systemic infusion could have caused systemic effects, which were not explored (764). Infusion of non-pressor doses of ANG II intravenously for 7 days increased blood pressure in S/Mcw, and this increase was prevented by simultaneous medullary infusion of l-arginine (664). Moreover, our group reported that NO production by thick ascending limbs due to AT2 receptor activation was blunted in tubules from S/Jr, leading to increased NKCC2 activity (292) (FIGURE 17).
FIGURE 17.

Angiotensin (ANG) II type 2 receptor (AT2R)-mediated nitric oxide (NO) production in Dahl rats. A: the signaling cascade whereby AT2R-mediated NO inhibits Na+-K+-2Cl− cotransporter type 2 (NKCC2) activity in the R phenotype. B: the changes present in the S phenotype leading to a reduced inhibition of NKCC2 by NO. Arrows indicate stimulation, and T-lines indicate inhibition. , superoxide; PDE2, cGMP-stimulated phosphodiesterase; PKG, cGMP-dependent protein kinase.
In addition to blunted NO production in the Dahl salt-sensitive rat, the response to NO may also be reduced. In isolated, perfused thick ascending limbs, the NO donors spermine NONOate and nitroglycerin added to the bath in low concentrations decreased Cl− reabsorption to a larger extent in R/Jr than in S/Jr when both strains were fed a 0.2% NaCl diet. Cortical and inner medullary collecting duct transport was reduced by the same extent in both strains (186). This study showed for the first time a defect in NO-mediated inhibition of Cl− transport in the thick ascending limb in these rats, although it did not report a difference in basal rates of Cl− reabsorption between tubules from S/Jr and R/Jr. The lack of a difference in basal transport was probably attributable to the small sample size and not a relevant finding. This publication also raised the possibility that, not only the production of NO, but also the response to it, was altered in the salt-sensitive rat. The second messenger of NO is cGMP. Phosphodiesterase-5 (PDE5) decreases NO activity by decreasing cGMP levels. In S/Jr rats, PDE5 protein expression in isolated thick ascending limbs was increased compared with R/Jr, and the presence of verdanafil was able to restore the NO-mediated inhibition in transport. Blunted inhibition of NO in tubules from S/Jr rats was not due to differences in cGMP levels because cGMP production was elevated in the presence of the general phosphodiesterase inhibitor isobutylmethylxanthine (IBMX). Together these data suggest that decreased NO effects in thick ascending limbs of S/Jr rats are due to increased PDE5 activity and cGMP catabolism and not necessarily to a NO deficiency (251).
In micropuncture experiments, l-arginine was unable to blunt the elevated fractional Cl− reabsorption in thick ascending limbs of S/Jr rats, even though it increased urinary Na+ excretion. It also had no effect on proximal or distal tubule fractional Cl− reabsorption. The authors concluded that the effect of l-arginine must have been preferentially on juxtamedullary nephrons or the collecting system, parts of the kidney not accessible to micropuncture (347). However, the results could also mean that the effect of NO, rather than its production, are defective in the salt-sensitive rat. Another explanation could be that, under these experimental conditions (these animals were undergoing mild diuresis throughout the experiment), other factors, such as production, could be at a maximum and, therefore, inhibiting NO’s actions, as discussed in previous sections. Furthermore, absolute distal Cl− delivery in S/Jr rats given l-arginine was not significantly different from the value in R/Jr rats, whereas it was different in the S/Jr rats not given l-arginine. Because neither SN GFR nor fractional proximal tubule Cl− reabsorption differed, it is unclear whether the failure to detect a difference in fractional Cl− reabsorption was a statistical fluke. This issue relates back to the issues discussed at the beginning of this section.
Finally, thick ascending limb-derived NO also diffuses to the vasa recta, where it is able to buffer ANG II-mediated vasoconstriction in a process known as “tubulo-vascular cross talk” (139). If NO activity is decreased, or if large amounts of diffuse from thick ascending limb to vasa recta, can blunt the buffering actions of NO in vasa recta (460). In prehypertensive S/Mcw, tubulovascular cross talk is defective. ANG II increased NO in thick ascending limbs that diffused to vasa recta in SS.BN.13, but not in S/Mcw, which is in agreement with our laboratory’s results showing the defective signaling from AT2 receptors in thick ascending limbs of S/Jr rats (292). NO buffering was restored when strips of tissue were incubated with the scavenger TIRON, indicating that the excess coming from the thick ascending limb was buffering the effects of NO in vasa recta (461). However, in SS.BN.13, adding l-NAME did not inhibit the buffering actions of NO derived from thick ascending limbs. Interestingly, that inhibition was achieved by incubation with the P2 receptors inhibitor suramin (486). Because ATP release is stimulated by flow in thick ascending limbs and this ATP stimulates NO production (77), this finding opens the door to new research into the importance of purinergic signaling between thick ascending limbs and vasa recta mediated by luminal flow (FIGURE 18).
FIGURE 18.

Thick ascending limb-vasa recta crosstalk after angiotensin II (ANG II) treatment in Dahl rats. Left: the R phenotype, where nitric oxide (NO) production in response to incubation with ANG II blunts ANG II-induced superoxide () production. The increase of NO in the thick ascending limb concomitantly raises NO levels in adjacent vasa recta pericytes and buffers vasoconstriction induced by ANG II-dependent Ca2+ increases. A large part of this buffering effect is mediated by vasa recta purinergic receptors instead of just diffusing NO. Right: the S phenotype, where excess production blunts NO increases in the thick ascending limb and vasa recta. Moreover, increases in are also observed in adjacent vasa recta in response to ANG II. Vasoconstriction buffering is blunted in these animals. AT1R, angiotensin II type I receptor; AT2R, angiotensin II type II receptor; P2, purinergic type 2 receptors.
These results imply that, in the renal medulla of salt-sensitive strains, enhanced production blunts the paracrine actions of the NO derived from the thick ascending limb in vasa recta, which increases vascular resistance and therefore leads to lower perfusion of the renal medulla. This process is one of the mechanisms by which excess may promote the development of salt sensitivity in this model. actions in the renal medulla of the Dahl rat will be discussed in detail in the next section.
d) endothelin and luminal flow.
Because of its ability to increase NO production in the renal medulla, endothelin production by thick ascending limbs plays an important role in hypertension (1, 189, 356, 358, 545). Medullary endothelin concentrations in S/Jr were lower than in R/Jr rats either on a low- (0.3% NaCl) or high-salt diet (8% NaCl). When placed on a high-salt diet, S/Jr were unable to increase renal medullary ET-1, and infusing endothelin in S/Jr fed a high-salt diet delayed the development of hypertension. Finally, S/Jr rats fed a high-salt diet also had a significant reduction in medullary endothelin B receptor expression compared with R/Jr, suggesting that both endothelin deficiency and lack of receptors could contribute to decreased NO in this model (642).
It is not clear why endothelin concentrations are reduced in the Dahl salt-sensitive rat. Endothelin production by thick ascending limbs and medullary collecting ducts is stimulated by hyperosmolality (274, 514, 515) and luminal flow (514, 515), respectively. It is likely that luminal flow stimulates endothelin production in thick ascending limbs as well, although this possibility has not been tested. ATP release is likely a mediator of these effects because of the following: 1) luminal flow stimulates ATP release by inner medullary collecting ducts; 2) the endothelin increase in response to flow is mediated by purinergic signaling (515); and 3) we have unpublished data showing that hypertonic solutions stimulate ATP release from thick ascending limbs. Thus the reduced levels of endothelin in the medulla of salt-sensitive rats may be related to a blunted ability to sense either flow of the forming urine or hypertonicity. It has been shown that mutations that affect blood pressure in the S/Br rat also cause elongation of the cilia in epithelial cells (219). Alternatively, malfunction of one of the mechanosensitive channels, such as TRPV4 (183) or TRPP2 (382, 725) and/or signaling downstream of the ATP release, could mediate abnormal flow-sensing mechanisms in this strain.
e) reactive oxygen species.
There is considerable evidence that ROS play a role in the salt sensitivity of the Dahl rat. For instance, urinary F2-isoprostane excretion (a marker of oxidative stress) was elevated after 1 wk of 8% NaCl in S/Jr compared with R/Jr (447). When S/Jr were infused for 3 wk with tempol concomitantly with 20.6 meq of Na+/day, the increase in blood pressure was blunted, and there was a decrease in production in both the cortex and medulla (446). Administering the antioxidants vitamin C and E to S/Jr at the same time they were fed a high-salt diet for 3 wk decreased renal levels to those found in S/Jr fed a low-salt diet and blunted the salt-induced increase in blood pressure (679). As stated in previous sections, the study of ROS is a complex issue, and the Dahl rat is no exception. This could explain the controversies over the source(s) of ROS, the exact species involved, as well as whether the effects are direct or indirect. In an attempt to bring order to this subject, we discuss each topic individually.
The foundation for elevated thick ascending limb ROS initially came from studies that found increased levels in the outer medulla. Medullary is higher in S/Jr, regardless of the NaCl content of the diet, whereas cortical levels of are similar in S/Jr and R/Jr. Medullary Cu2+/Zn2+ and Mn2+ SOD were reduced in S/Jr on a low-salt diet compared with R/Jr, and medullary Mn2+ SOD was further decreased by high salt (447). Similarly, medullary levels in S/Mcw were double compared with SS.BN.13, even when they were fed a 0.4% NaCl diet (671). Medullary H2O2 concentrations have also been reported to be higher in S/Mcw than in SS.BN.13 when rats were fed a normal diet (670). The elevated H2O2 likely contributed to salt sensitivity because of the following: 1) a high-salt diet (4% NaCl) increased blood pressure and medullary H2O2; 2) these were blunted by medullary infusion of catalase; and 3) medullary H2O2 infusion alone was sufficient to increase blood pressure in SS.BN.13 (670).
Surprisingly, no full publications to date directly demonstrate that elevated ROS in thick ascending limbs leads to enhanced NaCl reabsorption or NKCC2 activity in the Dahl salt-sensitive rat. As such, this section will primarily deal with reports showing that ROS production is increased in salt-sensitive rats. It will be up to the reader to judge whether the circumstantial evidence is sufficient to conclude that abnormal regulation of thick ascending limb salt transport in this model is, at least in part, due to elevated ROS. This obvious critical issue deserves further investigation.
In this section, we will use the generic term ROS to mean either or H2O2 in an attempt to make some sense of the literature. The distinction between the two will be addressed in a separate section. Trying to discuss both ROS sources and the question of /H2O2 simultaneously would only lead to confusion. There are many possible sources of ROS in thick ascending limbs, including NADPH oxidase, xanthine oxidase, uncoupled NOS, cyclooxygenase, and the mitochondria, among others. S/Mcw thick ascending limbs produced more ROS than SS.BN.13. The NADPH oxidase inhibitor diphenylene iodonium blunted most production, whereas the remainder was eliminated by a mitochondrial uncoupling agent. The activities of other potential contributors to ROS levels, including NOS, SOD, catalase, and glutathione peroxidase activities, did not differ between S/Mcw and SS-13BN rats. Infusing apocynin, a second NADPH oxidase inhibitor, into the renal medulla of rats fed a 4% NaCl diet decreased production and led to an accompanying 20-mmHg reduction in blood pressure (671). Together these data strongly suggest that one (or more) of the seven known NADPH oxidases (38) is (are) the primary source(s) of ROS in the Dahl salt-sensitive rat thick ascending limb.
Thick ascending limbs express three NADPH oxidase catalytic subunits: Nox1, Nox2, and Nox4 (295, 430); however, current evidence indicates that only NADPH oxidases 2 and 4 are functional in this segment. As explained previously, current dogma holds that Nox2, but not Nox4, requires assembly with p67phox to produce an active enzyme (429). When S/Mcw were fed an 8% NaCl diet, expression of p67phox protein increased (162). This study suggested that Nox2 rather than Nox4 is the isoform of NADPH oxidase responsible for oxidative stress in the outer medulla. When expression of p67phox was disrupted with zinc-finger nucleases, high-salt-fed S/Mcw-p67phox knockouts exhibited lower blood pressure and reduced medullary oxidative stress (156). These data raise some questions. There were no differences between S/Mcw and SS.13BN in p67phox protein expression when rats were fed a low-salt diet (162), but the same group reported that prehypertensive S/Mcw had double the oxidative stress compared with SS.BN.13 (671). One may well ask: where are the ROS coming from?
Later studies using S/Mcw-Nox4 knockout rats showed these animals had lower blood pressure compared with S/Mcw rats in response to 3 wk of high-salt diet (115). Tissue redox ratio, as measured by the NADH-to-FAD ratio, indicated less oxidative stress in the knockout. However, these rats displayed reduced RNA levels of p67phox in the outer medulla, indicating that knocking out Nox4 affected Nox2 activity. Therefore, the effects observed could not have been solely attributed to knockdown of Nox4 and were most likely due to actions in multiple pathways (115). Further studies are required to evaluate the degree of contribution of different NADPH oxidase isoforms to salt-sensitive hypertension in this model.
Recently, a new regulator of thick ascending limb NADPH oxidase in Dahl salt-sensitive rats has been described. Increasing NaCl delivery to the thick ascending limb led to enhanced ROS production in S/Mcw compared with SS.BN.13. ROS production was amiloride-sensitive, mediated by NADPH oxidase (488), and exacerbated by a high-salt diet. The increase in ROS did not depend on Na+/H+ exchange, but on an H+ channel (487). This channel was identified as HV1. HV1 contributes to NADPH oxidase activity in macrophages and is pH and voltage dependent (133). Therefore, this channel would be especially relevant when animals are fed a high-salt diet, because cells in the thick ascending limb will be depolarized. In S/Mcw-HV1 total knockouts, the salt-induced increase in blood pressure was blunted along with ROS production in the renal medulla. Interestingly, ROS production activated by this proton channel was enhanced by low intracellular Na+ (<10 mM). The explanation given was that, when rats are fed a high-salt diet, there would be increased NKCC2 activity. However, intracellular Na+ would not increase because of Na+-K+-ATPase activation, leading to cell depolarization. This process, together with an acidic environment due to NH4+ secreted by the proximal tubule, would create the conditions for increased H1V activity, production, and hypertension (324). Unfortunately, this explanation has no data to support it at present.
ROS can be also generated by uncoupled NOS. As mentioned before, NOS3 uncoupling may be due to low availability of the substrate (l-arginine) or the cofactor R-tetrahydrobiopterin (BH4) (705). Increased production of ROS leads to the oxidation of BH4 to BH2, decreasing the ratio of BH4 to BH2 (374). In vitro experiments designed to evaluate the role of NOS uncoupling in ROS production showed that l-NAME did not reduce ROS levels in S/Mcw outer medullary tissue when rats had been kept on a normal-salt diet. However, when S/Mcw were fed a 4% NaCl diet, l-NAME decreased ROS to the levels found in normal-salt-fed animals. In contrast, high salt increased both NOS activity and NO production in the control strain SS.BN.13, as indicated by an increase in ROS production in response to incubation with l-NAME (672). Therefore, NOS uncoupling is likely an important source of ROS in the Dahl salt-sensitive rat when on high salt; however, the mechanisms that cause NOS uncoupling are unclear and need further investigation.
In the Dahl salt-sensitive rat, mitochondria have also been proposed as an important source of ROS (773). Fumarate is converted into L-malate by the enzyme fumarase in the mitochondria. The fumarase gene is encoded in chromosome 13. S/Mcw showed a lack of fumarase enzyme activity and fumarate excess when kept on a 0.4% NaCl diet. Intravenous infusion of a fumarate precursor in SS.13BN rats led to salt-induced hypertension and increased ROS production in the renal medulla (681). Therefore, fumarate excess could be contributing to salt-sensitive hypertension by increasing ROS production in the renal medulla.
in the thick ascending limb is produced mainly by Nox2 and Nox4 (295). Nox4 is responsible for flow-induced production in the thick ascending limb of Sprague-Dawley rats (295). While Nox2 needs p47phox and the assembly of cytoplasmic units to be active (472), Nox4 is believed to be constitutive and to produce exclusively H2O2 instead of (429, 484, 619, 666). However, as discussed in a previous section, many publications have shown otherwise (295, 367, 368, 430). These disparate results may be explained by the rapid dismutation of into H2O2 by SOD or the problems that arise from the use of ROS-sensitive dyes. ROS-sensitive dyes have limited specificity, and their performance is highly dependent on the conditions of the experiment (582). Most studies in the Dahl rat have focused on the role of Nox2-induced oxidative stress in the renal medulla, leaving Nox4 studied less thoroughly. The current dogma states that Nox2 produces , and Nox4 produces H2O2. Mounting evidence implies more than one isoform is involved. For instance, the levels of p67phox protein, used as a surrogate of Nox2 activity, was only increased when S/Mcw were on a high-salt diet (162), while oxidative stress levels were already increased in prehypertensive S/Mcw salt-sensitive rats fed a normal-salt diet (671). Furthermore, null mutation of p67phox decreased but did not completely prevent the increase in blood pressure caused by salt, and differences in blood pressure between knockout and wild type were only evident when rats were kept on a high-salt diet (156). These results conflict with those showing that Nox2 is already an important source of oxidative stress in prehypertensive Dahl salt-sensitive rats.
Recently, S/Mcw-p67phox and S/Mcw-Nox4 total knockouts were used to study the contribution of Nox2 and Nox4 to flow-induced and H2O2 production by thick ascending limbs. After 3 days on a 4% NaCl diet, thick ascending limbs were manually dissected and perfused at variable flow rates, ranging from 5 to 20 nl/min, while monitoring ROS production with fluorophores. In these experiments, perfusing tubules at 20 nl/min significantly increased production in both S/Mcw-p67phox and S/Mcw-Nox4 knockouts, as well as in wild type, compared with a nonperfused tubule. However, at 5 nl/min, only wild-type and S/Mcw-Nox4 knockouts increased production above baseline values, suggesting that at low flow rates Nox2 prevails (773). In contrast, cellular production of H2O2 at 20 nl/min was completely abolished in S/Mcw-Nox4 knockouts and reduced by one-half in thick ascending limbs from S/Mcw-p67phox knockouts compared with wild-type S/Mcw. The straightforward interpretation of these data is that Nox4 mainly produces H2O2, whereas Nox2 produces ; however, the results of this paper are puzzling. In the case of intracellular H2O2, at least part of it is coming from the dismutation of ; therefore, H2O2 levels should be elevated in S/Mcw-Nox4 knockouts simply because Nox2 is still active. In this regard, one could argue that the S/Mcw-Nox4 knockout rats present decreased p67phox mRNA in the outer medulla (115); however, that also weakens the conclusion that flow-induced in the S/Mcw-Nox4 knockouts is due to p67phox-dependent Nox2. Because of the link between ROS production, luminal flow of the forming urine, and Na+ reabsorption by the thick ascending limb, increased medullary oxidative stress is considered one of the driving forces for the development of salt-sensitive hypertension in the Dahl rat. A model to explain the consequences of flow-induced ROS in the development of hypertension is shown in FIGURE 19.
FIGURE 19.

Proposed mechanisms whereby increased reactive oxygen species (ROS) cause hypertension and renal injury in the Dahl salt-sensitive rat. An imbalance favoring renal medullary ROS in the Dahl salt-sensitive rat would increase Na+-K+-2Cl− cotransporter type 2 (NKCC2) activity by both direct stimulation and by inhibition of NO synthase type 3 (NOS3). A more active thick ascending limb would decrease Na+ delivery to the macula, thereby reducing tubuloglomerular feedback (TGF). These changes would increase glomerular perfusion pressure and filtration rate, which over time would damage the glomerulus, thereby reducing the filtration surface. In addition, because ROS production is regulated by luminal flow in thick ascending limbs, an increase in filtration rate would augment fluid delivery to this segment, perpetuating the injury. Given the link between ROS production, luminal flow, and Na+ reabsorption by the thick ascending limb, increased medullary oxidative stress is considered one of the driving forces for the development of salt-sensitive hypertension in the Dahl rat.
f) 20-hydroxyeicosatetraenoic acid.
Inhibiting 20-HETE formation led to salt-sensitive hypertension in Sprague-Dawley rats (287). Genetic studies showed that the CYP4504A2 locus in chromosome 5 cosegregates with blood pressure in S/Jr (646). In these animals, 20-HETE activity was found decreased in the renal medulla (412) due to a reduction in cytochrome P-450 protein and activity (646). Thus alterations in renal medullary production of 20-HETE could contribute to salt sensitivity in the Dahl rat.
Microperfusion studies showed that when animals were kept on a low-salt diet, addition of a 20-HETE production inhibitor increased loop Cl− transport in R/Jr and Sprague-Dawley, but not in S/Jr rats (313). These data suggest that basal CYP450 metabolites in S/Jr are low. Consistently, addition of 20-HETE to the perfusate lowered loop Cl− transport in S/Jr but had no effect in R/Jr (313). Together, these data indicate that a deficiency in 20-HETE could be mediating basal elevations in NaCl reabsorption in the S/Jr. Reductions in 20-HETE production by S/Jr thick ascending limbs could be explained by reduced CYP4504A2 protein expression and activity (313, 646). Alternatively, elevated ROS production in the Dahl salt-sensitive rat can enhance 20-HETE degradation, thereby interfering with its actions. For instance, treating rats with tempol in the drinking water for 2 wk led to increased 20-HETE excretion, suggesting that the high oxidative stress in S/Mcw was the cause of reduced 20-HETE (288).
The role of 20-HETE was further supported by chromosome substitution experiments. When a region that includes the CYP4A1, 2, 3, and 8 genes from the Lewis rat in chromosome 5 was transferred to S/Mcw rats to augment 20-HETE production, salt-induced hypertension was blunted, and pressure natriuresis response improved (738). These effects were reversed by administration of an inhibitor of 20-HETE synthesis (738). Although the role of 20-HETE in the development of salt-sensitivity in the Dahl rat (FIGURE 20) has a strong foundation, many questions remain unanswered. These questions include the exact signaling cascade involved and the interactions between gene products on chromosome 5 and chromosome 13 that allow replacing the latter in S/JrHsdMcw with the Brown Norway, which has such a marked effect on blood pressure.
FIGURE 20.

Proposed effects of 20-hydroxyeicosatetraenoic acid (20-HETE) signaling on transport in the Dahl rat. A: the 20-HETE physiology in R. B: the ultimate cause for decreased 20-HETE levels in S remains unknown. However, the reported decreased in cytochrome P-450 A2 (CYP450A2) protein expression and defective angiotensin II type 2 receptor (AT2R) signaling are expected to reduce the formation of 20-HETE. Concomitantly, excess superoxide () may further lower 20-HETE bioavailability. Together, these effects could increase thick ascending limb NaCl reabsorption on S, by lack of inhibitory signals on Na+-K+-2Cl− cotransporter (NKCC2), Na+/H+ exchanger type 3 (NHE3), and renal outer medullary K+ channel (ROMK).
g) transcriptome and mitochondrial studies.
Transcriptome studies showed a differential expression of ~50 genes in response to a 4% NaCl diet administered for either 16 h, 3 days, or 2 wk, in renal medullas of S/Mcw compared with SS.BN.13. Out of the 50 genes showing differential expression patterns, 60% were linked to blood pressure regulation and the development of renal injury. However, only three of these genes were present on chromosome 13 (386).
Most of the differentially expressed genes found in the aforementioned study map to chromosomes other than 13. This shows that SS.BN.13 is not an ideal control. This work laid the foundation for investigation of these newly identified genes as participants in the development of salt-sensitive hypertension. For instance, targeting one of these genes, the 11β-hydroxysteroid dehydrogenase type 1 with small hairpin RNA delivered into the renal medulla of S/Mcw, attenuated salt-sensitive hypertension (395). The other two genes located in chromosome 13 were NGF-inducible antiproliferative putative secreted protein (an anti-proliferative protein that may have a role in extracellular protein kinase C-dependent signal transduction) and the antioxidant enzyme thiol-specific antioxidant protein (93), and their effects remain to be investigated.
Transcriptome studies are a valuable tool; however, changes in mRNA expression in the salt-hypertensive animals could reflect either the primary response to salt, or higher order responses to the elevated blood pressure once hypertension is established. Thus dissecting the cause and consequences of a high-salt diet is not always a straightforward task.
Analysis of mitochondrial function in S/Mcw and SS.BN.13 thick ascending limbs fed either 0.4 or 8% NaCl diet for 1 wk yielded differences in oxygen utilization (774). Proteomics analysis of the mitochondria showed that no proteins were differentially expressed under the basal 0.4% salt diet. However, feeding the animals with the 8% NaCl diet altered the levels of seven proteins (774). One of these proteins was the NADP-dependent isocitrate dehydrogenase, which was found decreased in S/Jr compared with SS.BN.13. Given that this enzyme protects against oxidative damage (325), and oxidative stress is thought to contribute to the salt sensitivity of this model (162, 446, 447, 671), the authors proposed that deficient oxygen utilization in the renal medulla could be contributing to renal damage (774). However, how this process relates to inappropriately elevated NaCl reabsorption remains unclear. One speculation is that increased H2O2 or activates protein kinase C, thereby stimulating transport, as discussed above.
Recent electron microscopy studies of the thick ascending limb showed singularities in the S/Mcw compared with either Sprague-Dawley or SS.BN.13. Although tubules from the three strains appeared similar under light microscopy, medullary thick ascending limbs from salt-sensitive rats fed for 1 wk with a 4% NaCl diet showed a reduced number of long mitochondria (a sign of medullary insufficiency), and a widening of the endoplasmic reticulum (a reticular stress marker). Moreover, S/Mcw exhibited mild renal injury before the development of hypertension (259). Unfortunately, this study only explored the ultrastructure of mitochondria and endoplasmic reticulum in rats fed a 4% NaCl diet and leaves unanswered the question of whether that potential insufficiency is a cause or consequence of the high blood pressure. Finally, using transcriptomic profiles, Yang et al. (755) reported that a high-salt diet (8% NaCl) leads to changes consistent with cell proliferation and cell cycle regulation in S/Mcw, data supported by evidence of an increase in proliferative cells in epithelial thick ascending limbs of S/Mcw fed a high-salt diet. Most of the genes differentially regulated by both a 0.4% and an 8% NaCl diet were present on chromosome 13.
2. Spontaneously hypertensive rat
The SHR was originated at the University of Kyoto by selective inbreeding of Wistar rats. At each generation, rats with blood pressures above average were selected for breeding (327, 492). Wistar Kyoto rats (WKY) were bred from Wistar rats at the University of Kyoto (327) as the normotensive control strain for the SHR. However, the fact that WKYs were developed later than SHRs, together with the high degree of genetic divergence between strains, limits usefulness of WKY as a genetic control (142, 327, 399). In fact, some investigators prefer to compare SHR to other strains (416, 417).
SHRs develop hypertension as early as 3–6 wk of life (587), even in the absence of environmental manipulations (492). Based on this timeline, the natural course of the SHR model can be divided into the following categories: prehypertensive (<6 wk of age), developing hypertension (7–12 wk of age), and established hypertension (>12 wk old) (71, 639). The characteristics of this model resemble essential hypertension in humans, and the SHR model has been extensively used in studies involving stroke (9, 256, 621), myocardial hypertrophy (319, 390), changes in vascular resistances (45, 184, 728), and hypertension and sex hormones (425, 426, 751, 753), among many others.
Several lines of evidence support the role of the kidney in the development of hypertension in SHR. Transplantation of a WKY kidney into a SHR recipient ameliorates hypertension and vice versa (578, 579). Providing further support for this concept, several classical studies showed that young SHRs presented a shift in pressure natriuresis (587) accompanied by an increase in the fractional tubular reabsorption of Na+ and reduced renal medullary blood flow (590). Additionally, SHRs needed higher renal perfusion pressures to maintain urinary Na+ and water excretion, even in the absence of changes in GFR and renal plasma flow (18). Autoregulation studies showed that the natriuretic response in SHRs upon increasing central blood pressure by 50 mmHg was about one-half of that in WKY (588). Interestingly, even though there is evidence of a defect in salt handling by the kidneys, Na+ content reduction in the diet from 140 to 0.5 meq/kg was able to delay the elevation of blood pressure but failed to prevent hypertension in the SHR (40). The reduction in Na+ intake in these experiment was significant, as it falls below the amount of Na+ a rat would take if offered separately from the food (428).
The role of the thick ascending limb in the development of hypertension in the SHR has not been extensively studied. However, based on other animal models of hypertension, one would expect that thick ascending limb salt reabsorption to be either normal or elevated in SHR <14 wk of age and reduced in older animals because of pressure natriuresis. Support of the hypothesis that in SHR there is a blunted tubular reabsorption of Na+ as a consequence of the elevated blood pressure comes from some classic studies showing that salt and water-loaded SHR exhibited higher excretion of Na+ than WKY controls in both the proximal tubule and loop of Henle (138, 289). Other relevant changes in these animals are increased vasopressin and PRA at 10 wk that decreased after the establishment of hypertension (71).
Adult (12-wk-old) SHR kidney showed moderately elevated protein expression of several transporters, including ROMK, NHE3, Na+/ cotransporter 1, and Na+-K+-ATPase (640). However, the biggest changes were in NKCC2. NKCC2 protein expression in the outer medulla of SHRs was sixfold greater than in WKY. These changes were not accompanied by changes in mRNA, suggesting a posttranslational mechanism of protein overexpression taking place in SHR (640). Furthermore, cellular fractionation studies conducted in the inner stripe of the outer medulla of prehypertensive (97 mmHg) SHR and animals with developed hypertension at 8 (137 mmHg) and 14 wk (195 mmHg) of age showed that the progression of blood pressure follows changes in NCKK2 protein expression (639). In these experiments, total NKCC2 increased by threefold at 8 wk and fivefold at 14 wk. In addition, the ratio between membrane and vesicular fractions was 4:1 in SHR compared with <2:1 in WKY at 8 wk and 6:1 vs. 2:1 at 14 wk. Although enhanced NKCC2 protein expression and membrane localization suggest a larger transport capacity by thick ascending limbs of SHR, the membrane fraction of NKCC2 reported for both strains was considerably different than the values in Sprague-Dawley rat (21, 498, 516). The difference in NKCC2 membrane expression is supported by an elevated fractional excretion of Na+ in response to furosemide in SHR compared with WKY (640). These changes likely contribute to elevated blood pressure in this strain. Along the same lines, studies of Na+-K+-ATPase in inner medullary lysates of prehypertensive (4-wk-old) SHR compared with hypertensive mates (12-wk-old) showed that both expression and activity are elevated during hypertension in these animals (416). Unfortunately, these studies used Sprague-Dawley rats as a reference, which makes it difficult to interpret the results. Together, the increases in Na+-K+-ATPase and NKCC2 discussed previously suggest elevated transport rates in SHR thick ascending limbs.
Finally, several studies, some involving the kidney (283, 568, 656, 657), have reported a sexual dimorphism of blood pressure in the SHR strain. However, renal transplantation between female and male SHR showed that the aforementioned dimorphism does not depend on intrinsic renal differences between sexes (253).
a) vasopressin and prostaglandin e2.
Studies using isolated, perfused thick ascending limbs showed that basal rates of Cl− and are similar in SHR and WKY tubules. They also revealed that addition of 10−10 M vasopressin to the bath decreased reabsorption by nearly 50% in both groups. These data indicate that vasopressin signaling is preserved in SHR thick ascending limbs. However, the ability of PGE2 to regulate transport in SHR tubules was blunted compared with WKY. PGE2 did not stimulate reabsorption in SHR thick ascending limbs, but did in those from WKY. Removal of PGE2 from the bath stimulated Cl− reabsorption in WKY tubules, but not in those from SHR. These effects are likely due to PGE2 not being able to activate protein kinase C because phorbol esters (which activate protein kinase C) blunt the differences in transport between strains caused by the prostaglandin (213). Taken together, these results suggest that the lack of PGE2 inhibition of NaCl absorption in SHR thick ascending limbs may contribute to renal salt retention and the development of hypertension.
Silva et al. (633) further studied the effects of AVP on SHR by measuring O2 consumption in thick ascending limb suspensions. AVP increased transport compared with basal conditions, whereas AVP combined with ANG II increased transport vs. AVP. These synergistic effects were blocked by the AT1-receptor blocker losartan. They then hypothesized that these changes could be due to NOS uncoupling, lack of ANG II-mediated cAMP inhibition, or increased . Addition of l-NAME did not change O2 consumption rates. Incubation with ANG II blocked AVP-stimulated cAMP. However, concomitant addition of AVP and ANG II dramatically increased production and O2 consumption. The combined effects of AVP and ANG II on O2 consumption were blunted by inhibiting NADPH oxidase with apocynin or scavenging with tempol (633). Taken together, these results suggest synergistic effects of the AVP/cAMP and ANG II/AT1/ signaling pathways.
b) endothelin.
ET-1 is a potent regulator of Na+ reabsorption, and renal defects in ET-1 synthesis or signaling contribute to the development of hypertension in SHR (1, 303, 358). However, the information specific to the thick ascending limb is limited. Studies in microdissected renal tubules showed that endothelin converting enzyme-1 (ECE-1) mRNA expression was increased in proximal tubules, thick ascending limbs, and inner medullary collecting ducts in prehypertensive (4-wk-old) SHR compared with WKY. These findings were confirmed by Western blots of the renal cortex and medulla. Interestingly, in hypertensive (12-wk-old) SHRs, no differences in ECE-1 protein expression were found, even though mRNA levels remain elevated (141). Although not the only source, ECE-1 is the main source of ET-1. Thus these data suggest that elevated nephron (including thick ascending limbs) ET-1 synthesis in SHR may be contributing to the increase in blood pressure in these animals, downplaying its role once hypertension is established.
c) angiotensin ii.
Many studies have implicated defective ANG II signaling as a cause of hypertension in the SHR (101, 145, 597). Thus it is possible to speculate that the thick ascending limb plays a role in the generation of hypertension in the SHR, as it does in ANG II-induced hypertension. Interestingly, sex studies involving SHRs have reported systemic and renal differences in the renin angiotensin system (100, 146, 283, 568, 752). Still, we know of no direct studies concerning the regulation of thick ascending limb NaCl reabsorption by ANG II in SHR thick ascending limbs. The participation of the thick ascending limb in ANG II-dependent hypertension is discussed in a later section.
3. Milan hypertensive rat
Milan hypertensive strain (MHS) and its control, the Milan normotensive strain (MNS), were originally bred from a Wistar colony at the University of Milan and selected depending on their blood pressure (160–180 mmHg for MHS and 120–125 mmHg for MNS). These animals became hypertensive in the absence of dietary manipulations (50). Differences in blood pressure appeared around 5 wk of age and plateaued somewhere between 7 and 8 wk with values of ~40 mmHg higher in MHS compared with MNS. Similar to that found in studies of Dahl salt-sensitive and SHR rats, kidney transplantation studies showed that a renal defect was most probably the leading cause of hypertension in this strain (49). Glomerular filtration in young prehypertensive MHS is elevated compared with MNS (85). After weaning, MHS retain Na+ in parallel to the increase in blood pressure, reaching a stable level 4 wk later. These changes cannot be attributed to PRA, as it was already decreased at weaning in MHS and kept decreasing as blood pressure increased (48). Thus, given that the prehypertensive phase occurs with a positive Na+ balance and without elevations in PRA, a tubular defect seemed likely (48).
Young MHS (normotensive) express more NKCC2 mRNA and protein in medullary thick ascending limbs compared with age-matched MNS (85). While NHE3 expression in proximal tubules was not different between strains, the Na+-Cl− symporter (NCC) and ENaCα mRNA and protein in distal segments of the nephron were lower in MHS compared with MNS (85). Interestingly, as hypertension developed, this pattern changed, and adult MHS (hypertensive) presented no differences in NKCC2 protein in medullary thick ascending limbs but an increase in NCC in distal convoluted tubules (86). These data suggest that the thick ascending limb may be important in the Na+-retaining phase of the development of high blood pressure in MSH but plays less of a, or no, role once hypertension is established.
The theory that thick ascending limb transport plays no role in the hypertension of adult MHS is controversial. Membrane vesicles from the outer medulla of adult MHS showed NKCC2 activity as measured by furosemide-sensitive 86Rb+ uptake was greater than in those from MNS under nonsaturating conditions (167). Because the difference diminished at higher 86Rb+ concentrations, the data suggested an increase in affinity of the transporter for K+. The significance of this report is questionable, however, given that luminal K+ concentrations are usually at saturating levels in vivo.
Western blots conducted on whole kidney homogenates showed an increase in NKCC2 phosphorylation in adult MSH compared with normotensive MNS (88). These findings were confirmed by immunofluorescence in kidney slices. This study also showed that MHS presented a higher phosphorylation fraction of SPAK, possibly explaining the increase in NKCC2 phosphorylation (88).
Studies conducted in isolated nephron segments showed that MHS had higher Na+-K+-ATPase activity than MNS in both proximal tubules and medullary thick ascending limbs (443). The increase in thick ascending limb Na+-K+-ATPase activity preceded the elevation of blood pressure (444). These findings are consistent with other reports showing that Na+-K+-ATPase protein and mRNA levels are elevated in the outer medulla of prehypertensive MHS compared with MNS, changes that remain even after hypertension has been established (443). The primary reason for the increase in Na+-K+-ATPase activity remains elusive, but could be associated with genetic variants of the actin-binding protein adducin (601, 688, 689). Alternatively, a primary increase in NKCC2 activity could lead to a concomitant increase in activity of Na+-K+-ATPase.
Thus, even though hypertension in MHS has been described in some detail (687), the information related to the physiology of the thick ascending limb in this model is limited to expression studies or measurements of transport in tissue preparations. Complementary measurements of tubular transport in isolated, perfused tubules or by in vivo microperfusion would greatly increase our understanding of this model.
B. Induced Models of Hypertension
1. Angiotensin II-induced hypertension
The renin-angiotensin system (RAS) is an important regulator of blood pressure by its actions on the cardiovascular, renal, and central nervous systems. ANG II is the main effector of the RAS, and its systemic and intrarenal levels are tightly regulated to achieve water and Na+ balance. Abnormal increases in ANG II lead to salt retention and increased blood pressure (207, 598), whereas reductions in ANG II activity help to eliminate excess salt and reduce blood pressure (466, 747). There are several strategies to study the effects of ANG II. Strategies to downregulate the RAS include the knockdown or knockout of ANG II receptors, the use of ANG II receptor blockers that interfere with ANG II signaling, and the use of angiotensin-converting enzyme and renin inhibitors, which reduce ANG II levels by inhibiting its formation. Pharmacological tools are also effective in the treatment of hypertensive patients (70, 667, 702). Furthermore, ANG II-induced hypertension models can be divided into renin-dependent and low-renin models. The first group includes models in which PRA is elevated by the use of a clamp on a renal artery, which stimulates renin secretion and thereby ANG II formation. These models are usually accompanied by a partial nephrectomy to further limit salt excretion capacity. The low-renin group uses the aid of injections or infusion pumps to increase circulating ANG II. This approach permits fine-tuning of ANG II at set levels. Both types of models of ANG II-induced hypertension have been widely used to study the causes (5, 8, 102, 150, 223, 360) and consequences (92, 132, 244, 254) of high blood pressure.
In ANG II-induced hypertension models, the magnitude of the increase in blood pressure, as well as the physiological adaptation to it, depend on the amount of ANG II either produced or infused. In vivo chronic administration of slow-pressor doses of ANG II (<400 ng·kg−1·min−1 for most species) induces a gradual rise in blood pressure. These doses have no measurable effects on blood pressure, urinary volume, or urinary Na+ excretion acutely, but they severely affect these parameters in the mid- or long term (132, 223, 438, 520). Thus the progression of ANG II-induced hypertension in these models resembles essential hypertension. On the other end of the spectrum, infusion of pressor doses of ANG II (400–3,000 ng·min−1·kg−1, depending on the species) causes an immediate increase in blood pressure mimicking malignant hypertension.
Experimental evidence of the importance of the kidney in ANG II-induced hypertension comes from studies transplanting a kidney from a total AT1-receptor knockout donor to a binephrectomized wild-type mouse. This maneuver dramatically blunts the increase in blood pressure caused by chronic infusion of ANG II (120).
Studies of ANG II-induced hypertension models at early time points (<1 wk) predominantly examine the causes of hypertension, whereas those performed after hypertension has fully developed likely report the consequences and adaptation to the increase in blood pressure. In the long run, both pressor and slow-pressor models lead to renal, heart, and vascular damage with high oxidative stress, and serious reduction in the overall health of the animals.
Within certain limits, the increase in blood pressure in ANG II-induced hypertension models can be summarized as follows:
Where [ANG II] is the amount of endogenous or administered ANG II, [Salt] is the dietary salt intake, and Time is the length of the experiment. Given that the maximal blood pressure achievable in chronic models of hypertension is capped at around 200 mmHg (135, 167, 385, 453), it follows that the contribution of dietary salt to the increase in blood pressure is reduced as the amount of ANG II increases and that, over time, the magnitude of the elevation in blood pressure is less sensitive to these parameters. A representation of the interaction between dietary salt and the levels of ANG II on the increase of blood pressure during the first week of infusion can be seen in FIGURE 21. Even in animals infused with slow-pressor doses of ANG II, and after a short period of positive Na+ balance, the sensitivity of blood pressure to salt diminishes with time (211, 598). On the contrary, the fast-paced elevation in blood pressure caused by the infusion of pressor doses of ANG II triggers an immediate pressure natriuretic response, and the animals go to a negative Na+ balance (FIGURE 22). The corollary that can be drawn from these findings is that doses of ANG II in the lower range are more appropriate to study primary changes in renal function and the sensitivity of blood pressure to salt.
FIGURE 21.

Combined effects of dietary salt [Salt] and the levels of angiotensin II [ANG II] on the blood pressure increase (ΔBP) during the first week of ANG II infusion. The partial contributions of [Salt] and [ANG II] to ΔBP are represented on the right.
FIGURE 22.
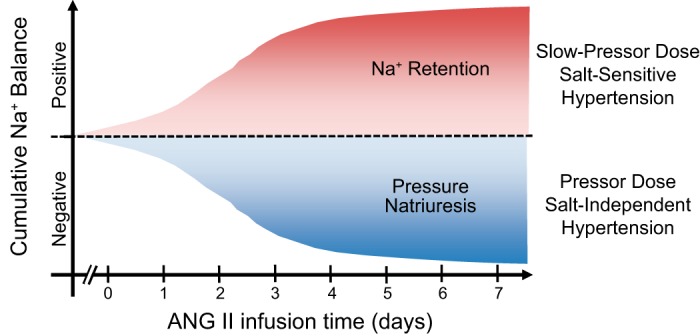
Effect of infusing either pressor or slow-pressor doses of angiotensin II (ANG II) on the cumulative Na+ balance during the first week of treatment.
To discuss the effects of ANG II infusions in the thick ascending limb, we are going to classify studies of up to 1 wk as the initial phase and studies of >1 wk as the adaptive phase. That said, the thick ascending limb in ANG II-induced hypertension cannot be considered as a sole participant. As ANG II stimulates Na+ transport in proximal tubules (107), thick ascending limbs (207, 629, 716), and collecting ducts (349, 534), the contribution of the thick ascending limb to the model should be thought of as a part of a larger process of change occurring all along the nephron (474). In the discussion below, one must keep in mind that ANG II profoundly affects renal hemodynamics (127, 449, 686), which makes it more difficult to dissect direct tubular actions from those that are secondary to changes in hemodynamics.
The initial phase of ANG II-induced hypertension is characterized by an increase in transport rates by thick ascending limbs (207), which is accompanied by a positive Na+ balance and a sustained increase in blood pressure (598). The increase in Na+ reabsorption by thick ascending limbs is likely mediated by increases in NKCC2 protein and membrane expression levels (370), leading to an increase in NKCC2-mediated apical entry of Na+ and secondary elevations in Na+-K+-ATPase activity (207). Part of these changes is due to an increase in ROS and a reduction in NO levels within the renal medulla (279, 562). In fact, tempol administration prevents the development of ANG II-induced hypertension (496, 733). In addition, NO production and signaling is impaired in thick ascending limbs of rats infused with ANG II by the following mechanisms: 1) reduction in NOS3 protein (562); 2) increased T495/S1177 phosphorylation (562); and 3) enhanced degradation of cGMP by PDE5 (563). Together, these alterations are expected to reduce the normal inhibitory signaling on NKCC2, thereby increasing its activity (563). The alterations in NO signaling during ANG II-induced hypertension are presented in FIGURE 23.
FIGURE 23.

Changes in nitric oxide (NO) signaling in thick ascending limbs during angiotensin II (ANG II)-induced hypertension. Black arrows and T-lines indicate normal physiological stimulation and inhibition, respectively. Arrows in red indicate the direction (up: ↑ or down: ↓) of pathological changes occurring during ANG II-induced hypertension. These changes result in altered cGMP levels and reduced inhibition of Na+-K+-2Cl− cotransporter (NKCC2). NOS3, NO synthase type 3; PDE5, phosphodiesterase 5.
After the first week of ANG II infusion, the model progressively transitions to the second phase, in which the adaptive response reestablishes Na+ balance (439, 475). In this phase, the animals reach Na+ balance at the cost of elevated blood pressure to achieve natriuresis, and the rate at which blood pressure increases is dramatically reduced (475, 598). Importantly, after 14 days of ANG II infusion, renal damage, in addition to extrarenal changes, such as increased systemic oxidative stress, vascular stiffness, and cardiac collagen deposition, worsens and likely reduces the overall contribution of the kidney to the pathology (282, 284, 339, 387). In this context, we expect the contributions of the thick ascending limb to salt retention and the maintenance of blood pressure to be reduced.
a) studies in rats.
Infusion of ANG II for 1 wk, at doses ranging from 200 to 400 ng·kg−1·min−1, increases net transport by rat thick ascending limbs (207, 629). These doses also cause NKCC2 activation markers, such as furosemide-induced diuresis and natriuresis, NKCC2 phosphorylation ratio, and NKCC2 apical localization, to be elevated as early as 3 days after the initiation of ANG II infusion and remain elevated up to day 14 (370, 474). Similarly, Na+-K+-ATPase activity is enhanced in thick ascending limbs after 1 wk of infusion of slow-pressor doses of ANG II (207), independent of its protein abundance (207, 370). The direct and relatively easily understood early actions of ANG II progressively become complex as its Na+-retaining effects are counteracted by natriuretic responses caused by elevated blood pressure. After 2 wk of ANG II infusion, a reduction in Na+-K+-ATPase protein is seen in the inner stripe of the outer medulla (475), accompanied by a reduction in NKCC2 protein abundance and phosphorylation (351, 475). These changes are not seen in cortical thick ascending limbs, as the positive effects of ANG II on Na+ transporters remain from the cortical thick ascending limb through medullary collecting duct at this time point (475). Importantly, the reduction in Na+ transporters in the renal medulla seems to be a direct consequence of hypertension, as the elevation of blood pressure by hormones other than ANG II exerts similar effects (351).
To sum up, during the initial phase of ANG II-induced hypertension using slow-pressor doses, the abundance of transporters is stimulated not only in thick ascending limbs (370, 474) but also all along the nephron (474). The Na+-retaining effects of ANG II lead to salt retention, as evidenced by the fact that animals have difficulties excreting a salt bolus (474). Ultimately, this condition causes salt-sensitive hypertension (598). As blood pressure increases, the stimulatory effects of ANG II on thick ascending limb transport are partially overridden by pressure natriuresis (475). A comprehensive review of transporter abundance and phosphorylation profiles in the initial and adaptive phases of ANG II-induced hypertension can be found elsewhere (439).
b) studies in mice.
Working with ANG II-induced hypertension models in mice has the great advantage of available genetically manipulated animals. However, mice have the disadvantage of being resistant to ANG II, requiring much larger doses that make results difficult to compare with those obtained in rats. As an example, mice infused with 400 ng·kg−1·min−1 develop a slow increase in blood pressure (211), similar to that produced by 200 ng·kg−1·min−1 in rats (598). Moreover, even though new technologies are emerging (781), the most widely used methods to generate mutant mice require stem cells from the SV129 strain (a strain with 2 renin genes) to be inserted into a host blastocyst from a C57 strain (a strain with 1 renin gene) (397). This requirement creates problems because investigators either have to devote considerable time and resources to backcrossing mice to obtain an inbred strain on the C57 background or use mice with mixed backgrounds. The latter strategy raises serious issues as to which mice should be used as controls. Further complicating the problem, the C57 strain shows modest salt sensitivity of blood pressure (87). In the end, however, this technology was proved successful to demonstrated that renal AT1 receptors, as well as intrarenal ANG II formation, are essential to fully develop ANG II-induced hypertension (120, 211).
C57 mice infused with 400 ng·kg−1·min−1 ANG II for 2 wk undergo a slow increase in blood pressure of ~30 mmHg (211), comparable to that caused by slow-pressor doses in rats. These animals also have an elevated NKCC2 phosphorylation and phosphorylation ratio, regardless of a slight decrease in protein abundance (211). These changes in protein expression and phosphorylation were accompanied by increased NKCC2 apical localization and an enhanced diuretic response to furosemide (211), evidence of activation of this transporter by ANG II. In contrast, mice infused with 1,000 ng·kg−1·min−1 ANG II for 2 wk presented a steeper elevation in blood pressure and a considerable reduction in NKCC2 abundance in whole kidney homogenates of ~25% (238), and infusion of 2,000 ng·kg−1·min−1 for 10 days also decreased NKCC2 phosphorylation (708). These changes are consistent with either a negative effect of ANG II in thick ascending limb transport or a counterregulatory mechanism. Given that slow-pressor doses increase NKCC2 activity at early time points, it is more probable that the results with ≥1,000 ng·kg−1·min−1 are due to pressure natriuresis rather than direct effects of ANG II itself. This conclusion is supported by sex-based studies where both male and female mice were infused with 800 ng·kg−1·min−1 of ANG II for 1 wk. Infusion of ANG II into young females caused an ~25% increase in NKCC2 protein levels in whole kidney homogenates, accompanied by a modest increase in blood pressure of 20 mmHg (682). However, NKCC2 was reduced by ~40% in young males, which presented an increase in blood pressure of 40 mmHg (682). Interestingly, the increase in NKCC2 protein expression was absent in older females, which responded to ANG II infusion with a steeper rise in blood pressure and a slightly higher final value than that in younger animals (682). These results, analyzed in light of our previous discussion in rat models, build on the idea that short-term infusion of ANG II enhances salt reabsorption by NKCC2, and later on, as hypertension develops, NKCC2 is downregulated by pressure natriuresis.
c) intrarenal renin-angiotensin system.
Cross-transplant experiments in mice lacking the AT1A receptor demonstrated that mice lacking ANG II signaling only in the kidney presented a reduction of ~35% in their basal blood pressures, a reduction comparable to the contribution of all other extrarenal AT1 receptors (121). Moreover, the kidney possesses the complete enzymatic machinery to produce ANG II from locally synthesized angiotensinogen, a system called the intrarenal RAS (473). The activation of the intrarenal RAS is characterized by increased intrarenal content of angiotensinogen, angiotensin-converting enzyme, and ANG II (473). During infusion of slow-pressor doses of ANG II, there is an increase in circulating ANG II level by day 3 and an accumulation of ANG II in the kidney evident at day 10 (780); the increase in blood pressure follows the intrarenal accumulation of the peptide (598, 780). Whereas short-term infusion of ANG II has no effect on renal angiotensinogen abundance (474), chronic infusion elevates cortical angiotensinogen production and urinary excretion (353, 475, 780). In addition, even though liver angiotensinogen is the primary source of renal ANG II (431), production of intrarenal ANG II is essential to the full development of the ANG II-induced hypertensive model. In fact, blood pressure only increases modestly with ANG II infusion in mice lacking intrarenal angiotensin-converting enzyme (211). The increases in blood pressure in these mice resemble the initial phase of the model, before intrarenal RAS is activated.
2. l-NAME-induced hypertension
l-NAME-induced hypertension is a model consisting of the systemic inhibition of NO synthesis by orally or intravenously administered l-NAME. This model displays cardiovascular (298, 445, 595, 661) and renal (61, 178, 469, 662, 685) alterations, as well as alteration to the nervous system (221, 606). Renal NO regulates GFR (372, 396), renal blood flow (372, 740), medullary blood flow (112, 435), pressure natriuresis (418, 604), transport processes (186, 543), and renin release (39, 675). Thus the effects of NOS inhibition on the kidney are considerable.
The bulk of l-NAME-induced hypertension studies was conducted in rats using doses of l-NAME in the drinking water ranging from <5 (36) to >60 mg·kg−1·day−1 (581). In general, the magnitude of the hypertension increases with the dose of l-NAME (726). NOS inhibition alters the capacity of the kidneys to handle Na+. Salt handling during l-NAME-induced hypertension may follow one of two patterns based on the extent of NOS inhibition. Low doses of l-NAME (<5 mg·kg−1·day−1) that partially inhibit NOS cause a reduction in the slope of the pressure natriuresis response (746). This finding indicates that low doses of l-NAME limit the ability of the kidneys to maintain blood pressure within a narrow range when Na+ intake increases, resulting in salt-sensitive hypertension (36, 746). In contrast, doses of l-NAME causing near total NOS inhibition (>10 mg·kg−1·day−1) cause a right shift in the pressure natriuresis response in addition to changing the slope. Therefore, a significant portion of the increase in blood pressure is salt independent and does not respond to salt restriction (746). Animals in this regime rapidly develop hypertension and kidney damage, both of which are worsened by a high-salt diet (179, 683). The inability to excrete salt is such that 20% of the animals die within 6 wk of treatment (746). Discontinuation of the l-NAME treatment partially ameliorates the hypertension, but blood pressure remains elevated for at least 2 wk compared with controls (462, 581).
Studies in conscious dogs showed that 3-day intravenous infusion of very low doses (50 ng·kg−1·min−1) of l-NAME led to reductions in GFR, urinary volume, and urinary Na+ excretion, and increases in cumulative Na+ balance (610) when animals were kept on a normal salt intake. The treatment was stopped before producing significant changes in arterial pressure. Over time, Na+ positive balances would be expected to lead to volume expansion and pressure natriuresis. Thus these findings indicate that l-NAME-induced changes in renal function precede hypertension in this model. Using a similar dose (100 ng·kg−1·min−1), coinfusion of excess NaCl 1 day after the beginning of the l-NAME treatment caused immediate increases in blood pressure (602). Together, these two studies indicated that l-NAME infusion affects the ability of the kidney to handle excess salt and that, at least in dogs, kidneys are more sensitive to NOS inhibition than the vasculature. Otherwise, systemic vasoconstriction and elevation in blood pressure would precede changes in renal function.
Systemic infusions of larger doses of l-NAME (9 mg·kg−1·min−1) in rats caused a sudden increase in blood pressure even when Na+ intake was clamped at a normal level (469). These animals retained Na+ (positive Na+ balance), and their weight increased because of water retention during the 5 days of treatment. After the l-NAME treatment stopped, the rats entered a negative Na+ balance phase, and blood pressure and weight returned to normal within 3 days. These data indicate that the early increase in blood pressure is not due to structural damage, but rather to alterations in Na+ handling by the kidneys. In fact, laser-Doppler measurements of renal blood flow showed that systemic infusion of (9 mg·kg−1·min−1) l-NAME in rats caused a reduction in renal medullary blood flow of 22% with no changes in cortical flow (469). Alterations of renal medullary flow and NO have been described in other models of salt-sensitive hypertension, such the Dahl salt-sensitive rat (98). Together, these findings suggest that renal medullary structures, including the thick ascending limb, participate in the l-NAME-induced model of hypertension.
A schematic chart of the increase in blood pressure as a function of the dose of l-NAME for different dietary Na+ isopleths can be found in FIGURE 24.
FIGURE 24.
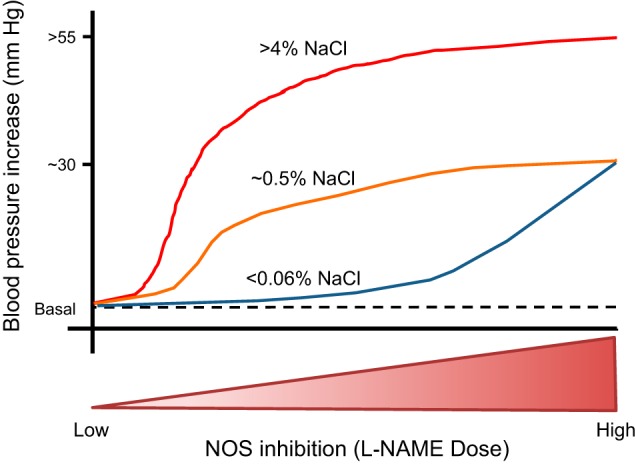
Chart of the increase in blood pressure as a function of the dose of NG-nitro-l-arginine methyl ester (l-NAME) under different dietary Na+ isopleths. NOS, nitric oxide synthase.
The thick ascending limb seems to be an important contributor to l-NAME-induced hypertension. Animals given l-NAME in the drinking water at doses ranging from 10 to 100 mg·kg−1·day −1 presented a dose-dependent increase in NKCC2 activity in suspensions of medullary thick ascending limbs (726). Moreover, at 4 wk of l-NAME treatment, the increase in NKCC2 activity and protein levels correlated with the increase in blood pressure (726). No changes in NKCC2 protein were seen at 4 days of treatment (697), emphasizing the long-term nature of the model.
The involvement of NKCC2 in NO deprivation hypertensive models was further investigated using NOS3 knockout mice. These animals have mildly elevated blood pressure (~10 mmHg), but unaltered glomerular filtration when on normal salt (530, 721). When given an acute water load, NOS3 knockout mice show a 30% reduction in the ability to excrete the load compared with wild types in the first 3 h. Volume-expanded NOS3 knockout mice also retain Na+. These changes are blunted by the NKCC2 inhibitor bumetanide (530), indicating that Na+ retention in thick ascending limbs is involved in producing these effects.
The obvious question that arises from such studies is why NOS3 knockout mice are not salt sensitive, meaning that increasing dietary salt does not dramatically increase blood pressure. The likely explanation for this is that multiple compensatory mechanisms counteract the loss of NOS3 in both the vasculature and kidney. This theory is supported by the fact that NOS3 knockout mice only show an ~10-mmHg difference in blood pressure from wild types, but acute l-NAME treatment increases blood pressure by ≥40 mmHg. This fact also serves to warn against assuming that the effect after knocking out a single gene is actually due to that knockout. There is a strong argument to be made for inducible, tissue-specific knockouts using either cross transplants or tissue-specific viral delivery of the CRISPR/Cas 9 technology.
Clearly, our understanding of the role of thick ascending limbs in l-NAME-induced hypertension is limited. This area is of considerable importance and warrants further study.
3. DOCA-salt hypertension
In the deoxycorticosterone acetate (DOCA)-salt model, the synthetic mineralocorticoid DOCA is administered in combination with a high-salt diet, usually 1% NaCl in the drinking water. The model also frequently includes unilateral nephrectomy (494). Mineralocorticoid excess leads to increased Na+ reabsorption and K+ wasting by collecting ducts, accompanied by fluid retention. These effects are exacerbated by increased Na+ intake and a reduction in renal mass. Thus this model resembles hyperaldosteronism in humans and volume-overload induced hypertension. In the DOCA-salt model, there is cardiovascular remodeling with fibrosis, endothelial dysfunction, and heart conduction abnormalities (476, 608, 744). These effects and the elevated blood pressure are ameliorated by the administration of l-arginine, antioxidants, and substances that increase NO production or bioavailability (96, 163).
Changes in thick ascending limb function in this model are secondary to the effects of DOCA primarily on collecting ducts and distal convoluted tubules. In DOCA-salt hypertension, plasma levels of ET-1 are elevated (11). ETB-receptor number increases in the renal medulla of DOCA-salt animals, and specific ETB antagonism significantly reduces Na+ and water excretion in the DOCA-salt rat (546). On the other hand, administration of an ETA antagonist to DOCA-salt rats reverses the increase in blood pressure primarily through systemic actions (11). Together these data suggest that ETB signaling in renal medulla counteracts fluid- and Na+-retaining effects of DOCA.
As discussed in a previous section, ET-1 reduces transport in thick ascending limbs via NO (560, 562). Renal immunohistochemical analysis of DOCA-salt hypertensive animals revealed large increases in NOS3 protein in thick ascending limbs and other segments (10). This change was accompanied by increased excretion of nitrates and nitrites in the urine, a measure of renal NO production (10). Correlation between renal excretion of NO metabolites and tubular NOS3 protein levels suggests that NO production was enhanced in thick ascending limbs, possibly as a countermeasure of increased blood pressure in response to the expected increase in fluid flow of the forming urine or interstitial osmolality. However, the field lacks studies that focus on segment-specific responses, and the contribution of the thick ascending limb is only theoretical at this time.
4. Norepinephrine-induced hypertension
Kidneys are highly innervated, and sympathetic nerve activity affects renal endocrine and hemodynamic functions. Elevated sympathetic activity in the kidneys has been reported in both human essential hypertension (341, 364) and animal models such as the SHR (328, 329, 365). Even though chronic electrical stimulation of renal nerves or systemic infusions of NE cause hypertension in a dose-dependent manner, the effects of NE in the kidney cannot be clearly categorized as prohypertensive or antihypertensive because of the different nature of the receptors involved.
On one hand, activation of α1- and β-adrenergic receptors, which regulate tubular Na+ reabsorption, cause salt retention (34, 41, 260) and the release of renin (508). On the other hand, activation of α2-adrenergic receptors has been linked to reductions in renin release (573), increased NO production, and inhibition of renal tubular ion transport activity (310, 539, 596, 699).
Thick ascending limbs present a biphasic response to NE, with α2-adrenergic-receptor-mediated inhibition of transport at low doses of NE (<10−7 M) and β-adrenergic-receptor-mediated stimulation of transport at high NE doses (>10−7 M) (538). Thus, in experimental settings where high concentrations of NE are used, the effects of α2-adrenergic receptors are only seen upon the use of a β-blocker, indicating that the Na+-retaining actions of NE likely prevail in these models (41, 538, 596).
The generation of NE-induced hypertensive models as a means to study renal tubular function has its most serious limitation in the strong renal medullary vasoconstriction caused by NE (28, 649), which reduces GFR and, therefore, reduces the capacity to excrete salt and water, regardless of changes in tubular transport. To overcome this issue, Plato and Osborn (540) evaluated the effects of intrarenal infusions of NE using a servo-controlled infusion system in unilaterally nephrectomized and renal denervated dogs. The servo system infused NE into the renal artery at a dose that keeps GFR constant. In this model, hypertension developed over 28 days, and blood pressure significantly decreased after cessation of NE infusion. During the infusion period, circulating concentrations of NE, epinephrine, and AVP remained unchanged, indicating a negligible systemic spillover of NE and strongly suggesting that the elevations in blood pressure were of renal origin.
An interesting aspect of this model is that, after a short period of Na+ retention, the servo-controlled NE infusions were associated with persistent natriuresis and diuresis, accompanied by contractions of vascular volume. Whether these changes were a consequence of intrarenal α2-adrenergic-receptor activation or a manifestation of pressure natriuresis remains unclear from this study. However, the intravenous infusion of a subpressor dose of NE (8 µg·kg−1·h−1) to conscious Sprague-Dawley rats provided evidence that renal α2-adrenergic receptors stimulate NO production, and it showed that the increase in NOS activity within the renal medulla buffers the hypertensive effects of NE (663). In this study, selective blockade of α2-adrenergic receptors by injection of rauwolscine into the renal medulla reduced the renal conversion of l-arginine to l-citrulline and caused subpressor doses of NE to elevate blood pressure (663). The response to rauwolscine infusions was mimicked by infusing l-NAME (663), suggesting that renal medullary α2-adrenergic receptors act through stimulation of NO production. Similarly, inhibition of NOS accentuates the acute hemodynamic responses to renal nerve stimulation and NE infusion in kidneys (7, 518, 573) and the NE-induced constriction in isolated, perfused outer medullary vasa recta (756).
Finally, subcutaneous infusion of pressor doses of NE (1,000 µg·kg−1·h−1) for 7 days in rats caused the systolic blood pressure to rise to ~220 mmHg (351). In these experiments, the animals presented elevated urine volume and natriuresis, and a reduction in NKCC2 in the renal medulla consistent with the pressure natriuresis mechanisms taking place (351), suggesting a prevalence of the α2-adrenergic action (638). Given the importance of circulating epinephrine released by the adrenal gland and sympathetic nerves in regulating Na+ excretion, the role of the thick ascending limb in NE-induced hypertensive models needs to be further investigated.
5. Macronutrients imbalance-induced hypertension
Proteins, fats, and carbohydrates constitute the main macronutrients in diets. A balanced human diet is one in which the macronutrient composition ranges are, expressed as a percentage of caloric demand, as follows: 45–65% carbohydrates, 20–35% fat, and 15–25% proteins. Increases or reductions in the proportion of one particular macronutrient would impact the proportion of the others, creating an imbalance in the diet. This concept is particularly true for fats, owing to their specific caloric content, which causes small deviations in the percentage of fats by weight to produce a large deviation in the contribution percentage to total calories.
Another important consideration in a healthy diet is the nature of the macronutrients. Because of substantial differences in metabolism, i.e., starch is not assimilated in the same way (or speed) as free glucose, different monosaccharides have different systemic effects. For instance, fructose metabolism bypasses the need of insulin and the entry checkpoint to glycolysis, which can increase AMP and activate starvation signaling, even though the cells have excess fuel (66, 269, 376, 700).
During the past 5 decades, significant modifications in the composition of human diet have occurred, mainly driven by industrial food processing. On one hand, industrial food processing allows for longer periods of food preservation and facilitates distribution at a lower price. Indeed, feeding large urban areas with only raw unprocessed foods would not only increase the price, but also reduce the offerings to a few locally produced seasonal products. On the other hand, industrial food processing is the major source of imbalanced macronutrients, favoring fats, simple carbohydrates, and excess salt. A large body of evidence shows that macronutrient imbalances can lead to metabolic diseases; however, in this review, we will focus on animal models that affect renal function, in particular the thick ascending limb, and cause salt-sensitive hypertension. We will discuss the effects of three diets: high-protein, high-fat, and abnormal carbohydrate compositions, such as high fructose in detriment of glucose or starch content.
a) high-protein diets.
Early studies in rats showed that switching animals from a 10% to a 30% casein diet caused an increase in kidney mass, preferably in the outer stripe of the inner medulla and thick ascending limbs (59). In addition, young rats (16 days old) receiving an isocaloric diet that contained either normal- (21% calories) or high-protein levels (50% calories) presented an increase in Na+-K+-ATPase activity in thick ascending limbs within 4 days of treatment, which lasted to the end of the study at 4 wk (316). Similar results were found in adult rats, in which the increase in Na+-K+-ATPase activity was not due to an elevated density of this transporter, but to the hypertrophy of the tubules (58). In these animals, increasing casein content of the diets from 10 to 30% by weight elevated GFR, free water clearance, and urine-concentrating ability (59), and elevated urea excretion from 2.6 to 22.3 mmol·animal−1·day−1 (699). Given that the thick ascending limb is fundamental in generating the hyperosmotic environment necessary to excrete urea, the hypertrophy may be related to these effects.
Human studies to test the effects of partial replacement of carbohydrate with protein on kidney function showed that such an intervention elevated estimated GFR by 5%. However, during the 6 wk of the trial, there was no correlation between changes in renal function and blood pressure (336).
Given that dietary protein supplements that could create an imbalance are commonly used by athletes and groups with certain food preferences, the impact of high-protein diets in renal physiology deserves further investigation.
b) high-fat diets.
High-fat feeding in rats has been used as a model of metabolic syndrome, which is associated with obesity, insulin resistance, elevated blood pressure, and renal damage (73, 203, 485). The shortcoming of using this model to study renal function is that the deleterious effects are additive and systemic, making it difficult to establish a specific cause-effect relationship. In addition, the swiftness of the development of these alterations in experimental models overwhelm small changes that may be of great significance for the slow progression of human disease.
The expression of thick ascending limb transporters was studied by Western blots in Brown Norway rats fed 36% (by weight) fat diet compared with a 4% fat diet used as control for 8 wk (580). The high-fat group presented impaired glucose tolerance, high oxidative stress, and elevated blood pressure (580), and both NKCC2 and ROMK protein expressions were elevated in outer medullary tissue (580). In addition, the high-fat diet increased the natriuretic response to furosemide (580). These findings suggest that the thick ascending limb contributes to Na+ retention and development of hypertension in this model. However, these findings cannot be extrapolated to other obesity and hypertension models. For instance in the Zucker rat, a genetic model, pre-macula densa transporters, including NKCC2 and ROMK, were reduced (51), possibly a secondary effect of a blood pressure increase.
As a corollary, the thick ascending limb very likely contributes to Na+ retention in high-fat hypertensive models, but the fast pace of progression makes these models inappropriate for the study of mild changes in Na+ reabsorption.
c) fructose.
Today average Americans consume at least 11% of their calories as fructose, whereas ≥5% ingest ≥20% of their calories from it (490, 707). This latter group contains more than 16 million people. Over the last 3 decades, the food and beverage industry has driven the switch from sucrose to fructose as a sweetener through the introduction of high-fructose corn syrup. High-fructose corn syrup has lower production and transportation costs, is easier to handle, and is a more potent sweetener than solid, granulated sucrose. Consuming large quantities of fructose has been implicated in metabolic disorders (405, 616), elevated blood pressure (304, 477), salt-sensitive hypertension (483, 618), and renal failure (195, 471, 605).
Salt sensitivity of blood pressure in rodents consuming fructose-enriched diets is well documented (79, 91, 302, 483, 569). Restricting salt prevented hypertension in rats given diets containing fructose ranging from 20% in the drinking water to 66% in the chow (79, 91, 483). However, a serious limitation of most of these studies is that they used amounts of fructose not comparable to those consumed by humans. Thus, even though they have been successful in rapidly mimicking metabolic syndrome-like symptoms, most of these studies are not adequate to serve as the basis of assumptions about the renal effects of moderately fructose-enriched diets, such those consumed by over 16 million Americans.
To circumvent this problem and study renal function before major metabolic alterations and the onset of renal damage occur, our laboratory has been using a moderately enriched fructose diet for 1 wk. We found that rats consuming 20% fructose in their drinking water remain normotensive while on normal salt, but addition of 4% NaCl to the diet produces an increase in blood pressure within 24 h (79). Salt sensitivity of blood pressure was accompanied by an increased sensitivity of renal tubules to ANG II (79, 206); however, the mechanisms by which fructose affects tubular transport and how these impact blood pressure are not yet fully understood.
As of today, there are no full publications about the role of thick ascending limbs in fructose-induced salt-sensitive hypertension. However, preliminary studies in rats show that 20% fructose, but not glucose, diets increase transport by this segment (25). Thick ascending limbs lack the metabolic machinery necessary to reabsorb and metabolize fructose; but they express G protein-coupled sweet taste receptors (T1R2/T1R3) (19). Animals receiving fructose show enhanced membrane expression of NKCC2 (25) and increased phosphorylation at the activating residues T96 and T101 (20), an effect absent in T1R2/T1R3 knockout mice (340). In addition, stimulation of the sweet-taste receptors by fructose in thick ascending limb suspensions of either rats or mice enhance surface NKCC2 levels (19). Taken together, these data suggest that fructose could be enhancing Na+ reabsorption by the thick ascending limb, thereby contributing to salt-sensitivity during moderately enriched fructose feeding.
V. HUMAN HYPERTENSION
The human thick ascending limb is an important regulator of volume homeostasis. The effectiveness of loop diuretics in clinical practice in the treatment of volume expansion diseases, edema, elevated blood pressure, and heart failure is well known. In this regard, even though there are sex differences in the treatment of hypertension in general (136) and in the use of loop diuretics in particular (131), there is only a handful of studies addressing sexual differences in thick ascending limb physiology in humans (105, 246).
In the first part of this section, we will discuss racial differences in Na+ handling by thick ascending limbs, whereas in the second part we will discuss a wide spectrum of mutations in thick ascending limb genes causing a multitude of symptoms, including hypertension, hypotension, and salt waste (321, 415, 626, 635, 637, 645, 766).
A. Racial Differences
The most intriguing data regarding the role of thick ascending limbs in human hypertension comes from racial differences in pressure natriuresis. By definition, salt-sensitive hypertension is the elevation in blood pressure required to eliminate salt due to a shifted pressure natriuresis response. In this regard, African American individuals present higher blood pressures as well as a ~30% reduction in K+ excretion after administration of a salt load than Caucasians (407), and they take longer to eliminate Na+ and return to balance (730). Given that Na+ and K+ are reabsorbed together only in the thick ascending limb, these data suggest that the salt sensitivity seen in African Americans is due to an enhanced Na+ reabsorption by NKCC2. The role of the thick ascending limb in controlling blood pressure from a clinical perspective has been extensively reviewed (332). Even though this knowledge is generally accepted, the exact etiology remains elusive, and several factors, such as aldosterone (553), vasopressin (30), ANG II (228), or even differences in K+ intake, may contribute, as discussed below.
African Americans excrete less urinary K+ than Caucasians (105, 228, 553, 696), which can be explained by nutritional surveys and reference data indicating that African Americans consume less K+ than other US and Canadian populations (517). This situation may also explain the greater rates of salt-sensitive hypertension, as a K+-poor diet is associated with an increased propensity to develop high blood pressure in all races tested thus far (228, 363). Further support of the involvement of a common Na+-K+ transporter such as NKCC2 comes from studies showing that Na+ restriction in the diet blunts racial differences in K+ excretion (696).
The possibility of differences in K+ handling has also been proposed, but the results are inconclusive. Urinary K+ excretion was lower in African Americans compared with Caucasians when they were kept for 3 wk on a fixed K+ intake. This study did not account for other routes of K+ excretion (695). In another 3-wk-long study that fixed K+ intake and accounted for losses in both urine and feces, African Americans presented decreased urinary K+ excretion with no differences in fecal K+ (511). These results are difficult to explain, since, after 3 wk, the participants of these studies must be in K+ balance, and, therefore, rates of K+ excretion should be similar among all participants. This is probably due to the lack of supervision of the diet when participants had their meals off-site, one of the pitfalls of these types of studies. Cumulative K+ gastrointestinal excretion was also not different after 9 days of controlled intake, and this result was accompanied by lower rates of excretion in African Americans (594). Because the proximal tubule and thick ascending limb reabsorb 85% of the filtered K+ and only the thick ascending limb reabsorbs it actively, differences in K+ excretion before individuals reach balance could be due to a defect increasing the activity of the thick ascending limb.
African Americans have lower aldosterone levels before the development of hypertension (552–554) and even after normalizing by K+ intake (407). These lower levels of aldosterone are accompanied by lower levels of PRA (553, 692), similar to that found in the Dahl rat model of salt-sensitive hypertension (315). Also, amiloride reduced blood pressure in Caucasians, but failed to decrease this parameter in African Americans (552), suggesting that ENaC activity is reduced in African Americans with respect to Caucasians, in good agreement with the hypothesis of a higher Na+ uptake by proximal/early distal segments in blacks.
A more active thick ascending limb would lead to an increased positive voltage in the lumen that would promote the uptake of other cations by the paracellular pathway. In agreement with this hypothesis, African American subjects also excreted less Ca2+ (512, 555, 737) and Mg2+ (517). In response to an acute intravenous dose of furosemide, differences in Ca2+ and Mg2+ excretion between African Americans and Caucasians attenuated, going from an ~20 to 10% difference. During the recovery period, Ca2+ and Mg2+ urinary excretions increased more slowly in African American participants. In addition, African Americans concentrate urine to a greater extent than Caucasians with similar levels of vasopressin (105, 246). Taken together, these data support the hypothesis of increased NKCC2 activity in the loop of Henle.
B. Human Genetic Defects
Genetic variations boosting NKCC2 activity would be expected to enhance salt retention and could be an evolutionary adaptation to arid environments with limited water availability. However, when water availability is no longer restrictive and NaCl is in excess, such variations are expected to predispose to salt-sensitive hypertension. As such, different groups have focused their efforts on studying the genetic heterogeneity of thick ascending limb transporters.
To maintain an apical NaCl reabsorption by NKCC2, Cl− is extruded out of the cell by the basolateral ClC-Kb/Barttin channel. Thus increased ClC-Kb/Barttin activity would be expected to stimulate net NaCl reabsorption by thick ascending limbs. Consistent with this idea, a gain of function point mutation in ClC-Kb, ClC-Kb (T481S), was linked to essential hypertension in both black and white men (321, 626). Compared with Asian and Hispanic Americans, this mutation was found to be more prevalent in African and Caucasian Americans (626), and higher in African blacks, with a frequency of 22%, than European whites by ~50% (321). Further characterization of the ClC-Kb(T481S) substitution using two-electrode voltage-clamp in Xenopus oocytes showed that the mutation caused a sevenfold increase in conductance compared with wild-type channels (321).
Mutations of the Barttin subunit of the ClC-Kb/Barttin channel were also studied in African Americans, Asians, Hispanics, Caucasians, and a cohort of hypertensive and normotensive subjects in Ghana (625). Three polymorphisms were identified, V43I, E255Q, and G284D. Two of these mutations, V43I and G284D, conferred a partial loss of function, as shown by channel activity measurements that revealed decreased conductance. However, in population studies, no relationship between these mutations and hypertension was found (625).
A lack of the ability of the CaSR to inhibit NKCC2 activity is expected to cause a cycle of elevated NaCl and Ca2+ reabsorption by thick ascending limbs (151, 181), a condition that can lead to hypertension. In fact, population-based studies conducted in normotensive individuals indicate that common variations (single-nucleotide polymorphisms) in the CaSR showed significant association with blood pressure. In addition, the alleles associated with higher blood pressures associate also with lower urinary Ca2+ excretion in a cross-sectional study (333).
Finally, single nucleotide polymorphisms in the CYP4A11 gene have been related to hypertension in African Americans (180). This gene encodes the enzymes responsible for synthesizing 20-HETE, an important regulator of Na+ transport in the thick ascending limb. These are population-based studies, and no further experimental approaches have been used to determine the importance of this mutation in Na+ handling. A summary of genetic defects leading to elevated blood pressure with origin in the thick ascending limb can be found in FIGURE 25.
FIGURE 25.
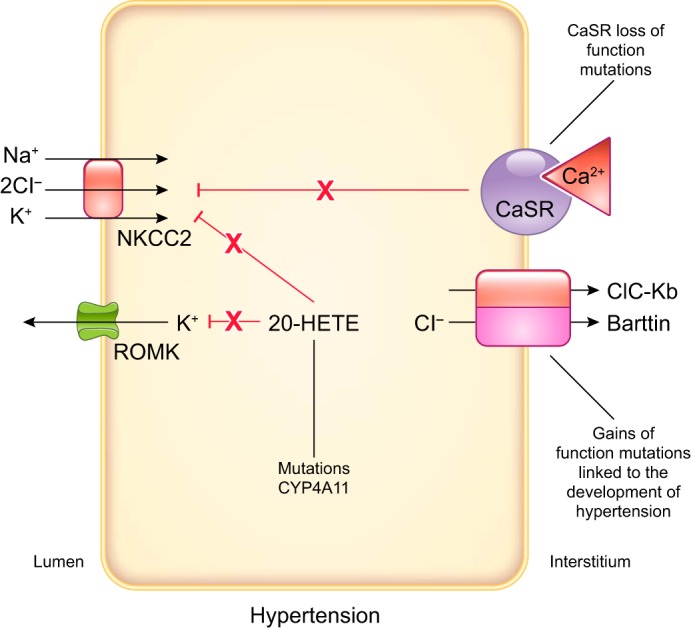
Mutations in proteins expressed by thick ascending limbs that cause hypertension. Loss of function mutations in the basolateral Ca2+-sensing receptor (CaSR) would decrease the inhibitory effect of this receptor on Na+-K+-2Cl− cotransporter (NKCC2), while gain of function mutations in ClC-Kb channels would also lead to increases in NKCC2 activity. Mutations in the CYP4A11 gene may decrease 20-hydroxyeicosatetraenoic acid (20-HETE) levels and, therefore, its inhibitory effects on NaCl reabsorption in the thick ascending limb.
On the other end of the spectrum, a group of mutations that cause salt waste by the thick ascending limb are collectively known as Bartter’s Syndrome. In general, any mutation leading to decreased NKCC2 activity would be expected to lead to a lower voltage in the lumen, K+ loss, and excessive Ca2+ and Mg2+ excretion through the paracellular route. Thus Bartter’s Syndromes of different genetic origins are all associated with hypotension, hypokalemic alkalosis, severe volume loss, and hypercalciuria.
Depending on the gene affected, Bartter’s Syndrome can be differentiated into five types. Mutations affecting the NKCC2 gene lead to neonatal Bartter’s Syndrome type 1, which, besides hypokalemic alkalosis and hypercalciuria, also presents with oligohydramnios, restricted fetal growth, and increased renal prostaglandins (636). Six independent loss-of-function mutations may elicit these severe clinical manifestations in homozygous individuals (262, 636). In the neonatal Bartter’s Syndrome type 2, ROMK is affected by loss-of-function mutations. In this case, NKCC2 activity is reduced by defective recycling of K+ back to the lumen, causing depolarization and a reduction in Ca2+ and Mg2+ reabsorption (262). Loss-of-function mutations in the ClC-Kb transporter lead to the Classic Bartter’s Syndrome type 3 (262, 635), whereas mutations in the Barttin subunit cause the severe Infantile Bartter’s Syndrome type 4 (262). Bartter’s Syndrome type 5 is the only form of the disease due to a gain-of-function mutation in the CaSR gene. When it binds Ca2+, a signaling cascade is initiated that eventually leads to NKCC2 inhibition, promoting excretion of Ca2+ (262, 727) (FIGURE 26).
FIGURE 26.
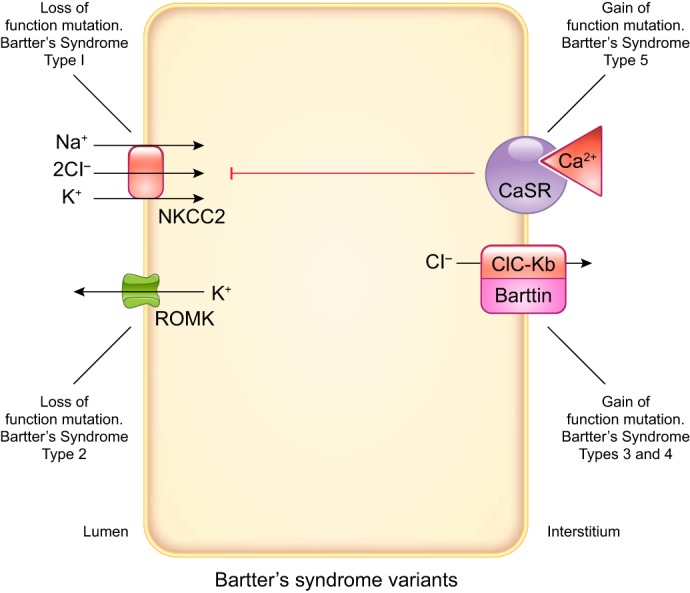
Mutations in proteins expressed by thick ascending limbs that cause Bartter’s Syndrome. This condition is characterized by salt waste and excessive Ca2+ and Mg2+ excretion and may be due to any loss of function mutation in transporters/channels of the thick ascending limb that decreases Na+-K+-2Cl− cotransporter (NKCC2) activity, except for the Ca2+-sensing receptor (CaSR), which leads to NKCC2 inhibition by a gain of function mutation.
Finally, some studies link uromodulin expression to differences in blood pressure. Uromodulin is the most common protein present in urine. Primarily secreted by the thick ascending limb, uromodulin is thought to protect against infectious agents and prevent the formation of renal stones. Genome-wide association studies (GWAS) conducted in Nordic people found a promoter region variant near the uromodulin gene associated with lower risk of hypertension (509). This single nucleotide polymorphism, called the minor G allele, is also associated with lower urinary excretion of uromodulin and is suggested to play a role in Na+ homeostasis (509). Supporting these finding, studies with uromodulin knockout mice have shown that this protein regulates Na+ uptake by the thick ascending limb, and that knockout mice display lower blood pressures than wild type (220). Uromodulin overexpression in mice leads to salt-sensitive hypertension and renal damage (690), and uromodulin mutations have been linked to the development of medullary cystic disease and nephropathy (255). Uromodulin is therefore an important candidate for further investigation and a possible drug target.
The uromodulin studies are a source of great examples of the heritability of blood pressure. As much as an estimated 50% of population variation in blood pressure can be attributed to genetic factors; however, all of the genetic variants identified so far explain at best 2% of this variation (311). Therefore, further studies are required to identify other genes related but not limited to the thick ascending limb. As the limitations of GWAS (82) and traditional genetic studies have made the discovery process very slow, other exploratory tools, such as renal genomics and proteomics, may be more effective and, therefore, need to be further developed (209) and explored.
This section tells the cautionary tale that, even though we understand and are able to make predictions about the outcome of genetically defective or strongly inhibited thick ascending limb transporters, our understanding of their regulatory role in normal human physiology is very limited. Little information is available from clinical studies about the endocrine/paracrine signals governing their function. Thus much of our understanding of physiology fits into a binary model, where the activity of a transporter is unusually high or low, missing the normal physiological range.
VI. CONCLUDING REMARKS: A CALL TO ARMS
A vast array of hormones and paracrine and autocrine factors regulate thick ascending limb transport. The mechanisms by which a limited number of these regulate salt reabsorption in this segment have been discussed in this review. As can be readily seen, despite decades of investigation, we still know little about how many of these affect transporters involved in NaCl reabsorption save NKCC2, and possibly ROMK.
Given that the thick ascending limb reabsorbs ~30% of the filtered load of NaCl and is responsible for generating the osmotic gradient for water reabsorption later in the nephron, it is not surprising that it plays a significant role in the development, and at times mitigation, of both genetic and induced forms of hypertension in animal models, and in humans. However, our understanding of these processes, similar to the regulation of individual transporters, is quite limited. One could easily and understandably take the pessimistic view that we are blind men describing an elephant. As a result, we are able to come to only few useful conclusions short of this or that form of hypertension involves this segment.
The reasons for this are many. First and foremost is that our reductionist view of the regulation of transport processes in the thick ascending limb, and how they are altered in hypertension may have led us into a boxed canyon with no way out. For instance, many in vitro studies fail to consider changes in blood pressure itself to be a key parameter that must be accounted for. Therefore, the effects of experimental variables from those secondary to elevations in blood pressure are largely omitted. Related to this is the element of time. Changes in thick ascending limb transport that contribute to the development of hypertension must, by definition, precede increases in blood pressure. Given that rats and mice come into Na+ balance within a few days, and initial rapid increases in blood pressure occur in a similar time frame in most animal models, the results of studies conducted within this time frame likely reflect primary changes in renal function, while studies of ≥2 wk out investigate the consequences of hypertension rather than its causes.
Furthermore, many of the variables likely to be important in understanding the role of the thick ascending limb in the development of hypertension have not been adequately controlled. It is clear from the elegant genetic studies of the Dahl rat that, even in the most carefully controlled experiment, environmental variables come into play. Perhaps primary among these are genetic backgrounds, the stress on the animals, and dietary factors. Taking the latter two as examples, it is easy to understand how reports of blood pressure and thick ascending limb function can vary. We now are at least partially aware that Dahl salt-sensitive rats are far more prone to stress-induced increases in blood pressure and that the sex of the investigator matters when performing any in vivo study in mice: female scientists cause less stress in the mice than males. As salt intake is a key parameter in the determination of blood pressure, it is not unexpected that different laboratories would obtain varying results when using diets from a range of vendors. At last check, “standard” rat chow had a Na+ content ranging from 0.12 to 0.49%, depending on its source. Similarly, high-salt diets range from 1% NaCl in the drinking water to 8% in the food.
At a minimum, we call on the renal and hypertension communities to describe their experiments in more detail, and not to make the assumption that “it doesn’t matter.” In our own laboratory, we live by the motto that, unless you can demonstrate that it doesn’t matter…it matters!! (Although we are as guilty as anyone of not adequately reporting the details of our experimental design.) Perhaps a select panel should be convened to set a single, specific standard for dietary salt as we did for nomenclature for the RAS. This alone would go a long way to reducing variability in the literature. The National Institutes of Health’s new rules on scientific rigor are an attempt to address such issues, but as long as we, as a community, only pay lip service to these requirements, it will remain difficult to compare results from different laboratories.
As it stands, one could easily be disillusioned by the current state of affairs; however, we take the optimistic view that we are rapidly learning what we do not know, and many fundamental questions remain for imaginative, energetic investigators to tackle.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
ACKNOWLEDGMENTS
Address for reprint requests and other correspondence: A. Gonzalez-Vicente, Dept. of Physiology and Biophysics, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH 44106 (e-mail: agustin.gonzalezvicente@case.edu).
A. Gonzalez-Vicente and F. Saez contributed equally to this work.
REFERENCES
- 1.Abassi ZA, Ellahham S, Winaver J, Hoffman A. The intrarenal endothelin system and hypertension. News Physiol Sci 16: 152–156, 2001. [DOI] [PubMed] [Google Scholar]
- 2.Abe M, O’Connor P, Kaldunski M, Liang M, Roman RJ, Cowley AW Jr. Effect of sodium delivery on superoxide and nitric oxide in the medullary thick ascending limb. Am J Physiol Renal Physiol 291: F350–F357, 2006. doi: 10.1152/ajprenal.00407.2005. [DOI] [PubMed] [Google Scholar]
- 3.Abu-Soud HM, Yoho LL, Stuehr DJ. Calmodulin controls neuronal nitric-oxide synthase by a dual mechanism. Activation of intra- and interdomain electron transfer. J Biol Chem 269: 32047–32050, 1994. [PubMed] [Google Scholar]
- 4.Ackermann D, Gresko N, Carrel M, Loffing-Cueni D, Habermehl D, Gomez-Sanchez C, Rossier BC, Loffing J. In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: differential effect of corticosteroids along the distal tubule. Am J Physiol Renal Physiol 299: F1473–F1485, 2010. doi: 10.1152/ajprenal.00437.2010. [DOI] [PubMed] [Google Scholar]
- 5.Agarwal R, Campbell RC, Warnock DG. Oxidative stress in hypertension and chronic kidney disease: role of angiotensin II. Semin Nephrol 24: 101–114, 2004. doi: 10.1016/j.semnephrol.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Ago T, Nunoi H, Ito T, Sumimoto H. Mechanism for phosphorylation-induced activation of the phagocyte NADPH oxidase protein p47(phox). Triple replacement of serines 303, 304, and 328 with aspartates disrupts the SH3 domain-mediated intramolecular interaction in p47(phox), thereby activating the oxidase. J Biol Chem 274: 33644–33653, 1999. doi: 10.1074/jbc.274.47.33644. [DOI] [PubMed] [Google Scholar]
- 7.Ajikobi DO, Cupples WA. Alpha 2-adrenergic mediation of the effects of angiotensin II on rat renal artery in vitro. Can J Physiol Pharmacol 72: 1019–1024, 1994. doi: 10.1139/y94-142. [DOI] [PubMed] [Google Scholar]
- 8.Alexander BT, Cockrell KL, Rinewalt AN, Herrington JN, Granger JP. Enhanced renal expression of preproendothelin mRNA during chronic angiotensin II hypertension. Am J Physiol Regul Integr Comp Physiol 280: R1388–R1392, 2001. doi: 10.1152/ajpregu.2001.280.5.R1388. [DOI] [PubMed] [Google Scholar]
- 9.Alhusban A, Kozak A, Pillai B, Ahmed H, Sayed MA, Johnson MH, Ishrat T, Ergul A, Fagan SC. Mechanisms of acute neurovascular protection with AT1 blockade after stroke: Effect of prestroke hypertension. PLoS One 12: e0178867, 2017. doi: 10.1371/journal.pone.0178867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allcock GH, Hukkanen M, Polak JM, Pollock JS, Pollock DM. Increased nitric oxide synthase-3 expression in kidneys of deoxycorticosterone acetate-salt hypertensive rats. J Am Soc Nephrol 10: 2283–2289, 1999. [DOI] [PubMed] [Google Scholar]
- 11.Allcock GH, Venema RC, Pollock DM. ETA receptor blockade attenuates the hypertension but not renal dysfunction in DOCA-salt rats. Am J Physiol Renal Physiol 275: R245–R252, 1998. doi: 10.1152/ajpregu.1998.275.1.R245. [DOI] [PubMed] [Google Scholar]
- 12.Allen F, Tisher CC. Morphology of the ascending thick limb of Henle. Kidney Int 9: 8–22, 1976. doi: 10.1038/ki.1976.2. [DOI] [PubMed] [Google Scholar]
- 13.Alvarez-Guerra M, Garay RP. Renal Na-K-Cl cotransporter NKCC2 in Dahl salt-sensitive rats. J Hypertens 20: 721–727, 2002. doi: 10.1097/00004872-200204000-00031. [DOI] [PubMed] [Google Scholar]
- 14.Amlal H, LeGoff C, Vernimmen C, Soleimani M, Paillard M, Bichara M. ANG II controls Na+-K+(NH4+)-2Cl− cotransport via 20-HETE and PKC in medullary thick ascending limb. Am J Physiol Cell Physiol 274: C1047–C1056, 1998. doi: 10.1152/ajpcell.1998.274.4.C1047. [DOI] [PubMed] [Google Scholar]
- 15.Angelow S, Ahlstrom R, Yu AS. Biology of claudins. Am J Physiol Renal Physiol 295: F867–F876, 2008. doi: 10.1152/ajprenal.90264.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aperia A, Hökfelt T, Meister B, Bertorello A, Fryckstedt J, Holtbäck U, Seri I. The significance of L-amino acid decarboxylase and DARPP-32 in the kidney. Am J Hypertens 3: 11S–13S, 1990. doi: 10.1093/ajh/3.6.11S. [DOI] [PubMed] [Google Scholar]
- 17.Aramburu J, Drews-Elger K, Estrada-Gelonch A, Minguillón J, Morancho B, Santiago V, López-Rodríguez C. Regulation of the hypertonic stress response and other cellular functions by the Rel-like transcription factor NFAT5. Biochem Pharmacol 72: 1597–1604, 2006. doi: 10.1016/j.bcp.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 18.Arendshorst WJ, Beierwaltes WH. Renal tubular reabsorption in spontaneously hypertensive rats. Am J Physiol Renal Physiol 237: F38–F47, 1979. doi: 10.1152/ajprenal.1979.237.1.F38. [DOI] [PubMed] [Google Scholar]
- 19.Ares G, Caceres P, Kassem KM, Ortiz PA. Stimulation of sweet taste receptors expressed in the kidney enhance surface NKCC2 levels in thick ascending limbs (TALs) (Abstract). FASEB J 31, Suppl 1: 856.816, 2017. [Google Scholar]
- 20.Ares G, Haque M, Henson E, Ortiz P. A fructose-enriched diet induces salt-sensitive hypertension and enhances NKCC2 and SPAK/OSR1 phosphorylation in thick ascending limbs (TALs) (Abstract). FASEB J 29, Suppl 1: 811.3, 2015. [Google Scholar]
- 21.Ares GR, Caceres P, Alvarez-Leefmans FJ, Ortiz PA. cGMP decreases surface NKCC2 levels in the thick ascending limb: role of phosphodiesterase 2 (PDE2). Am J Physiol Renal Physiol 295: F877–F887, 2008. doi: 10.1152/ajprenal.00449.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ares GR, Caceres PS, Ortiz PA. Molecular regulation of NKCC2 in the thick ascending limb. Am J Physiol Renal Physiol 301: F1143–F1159, 2011. doi: 10.1152/ajprenal.00396.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ares GR, Haque MZ, Delpire E, Ortiz PA. Hyperphosphorylation of Na-K-2Cl cotransporter in thick ascending limbs of Dahl salt-sensitive rats. Hypertension 60: 1464–1470, 2012. doi: 10.1161/HYPERTENSIONAHA.112.202101. [DOI] [PubMed] [Google Scholar]
- 24.Ares GR, Ortiz PA. Constitutive endocytosis and recycling of NKCC2 in rat thick ascending limbs. Am J Physiol Renal Physiol 299: F1193–F1202, 2010. doi: 10.1152/ajprenal.00307.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ares GR, Ortiz PA. Direct renal effects of a fructose-enriched diet: interaction with high salt intake. Am J Physiol Regul Integr Comp Physiol 309: R1078–R1081, 2015. doi: 10.1152/ajpregu.00156.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asico LD, Cuevas S, Ma X, Jose PA, Armando I, Konkalmatt PR. Nephron segment-specific gene expression using AAV vectors. Biochem Biophys Res Commun 497: 19–24, 2018. doi: 10.1016/j.bbrc.2018.01.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachmann S, Mutig K. Regulation of renal Na-(K)-Cl cotransporters by vasopressin. Pflugers Arch 469: 889–897, 2017. doi: 10.1007/s00424-017-2002-2. [DOI] [PubMed] [Google Scholar]
- 28.Badzyńska B, Sadowski J. Moderate intrarenal vasoconstriction after high pressor doses of norepinephrine in the rat: comparison with effects of angiotensin II. Kidney Blood Press Res 34: 307–310, 2011. doi: 10.1159/000328328. [DOI] [PubMed] [Google Scholar]
- 29.Bailly C, Imbert-Teboul M, Roinel N, Amiel C. Isoproterenol increases Ca, Mg, and NaCl reabsorption in mouse thick ascending limb. Am J Physiol Renal Physiol 258: F1224–F1231, 1990. doi: 10.1152/ajprenal.1990.258.5.F1224. [DOI] [PubMed] [Google Scholar]
- 30.Bankir L, Perucca J, Weinberger MH. Ethnic differences in urine concentration: possible relationship to blood pressure. Clin J Am Soc Nephrol 2: 304–312, 2007. doi: 10.2215/CJN.03401006. [DOI] [PubMed] [Google Scholar]
- 31.Barajas L, Powers K, Wang P. Innervation of the renal cortical tubules: a quantitative study. Am J Physiol Renal Physiol 247: F50–F60, 1984. doi: 10.1152/ajprenal.1984.247.1.F50. [DOI] [PubMed] [Google Scholar]
- 32.Bastide F, Meissner G, Fleischer S, Post RL. Similarity of the active site of phosphorylation of the adenosine triphosphatase from transport of sodium and potassium ions in kidney to that for transport of calcium ions in the sarcoplasmic reticulum of muscle. J Biol Chem 248: 8385–8391, 1973. [PubMed] [Google Scholar]
- 33.Battula S, Hao S, Pedraza PL, Stier CT, Ferreri NR. Tumor necrosis factor-alpha is an endogenous inhibitor of Na+-K+-2Cl− cotransporter (NKCC2) isoform A in the thick ascending limb. Am J Physiol Renal Physiol 301: F94–F100, 2011. doi: 10.1152/ajprenal.00650.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baum M. Effect of catecholamines on rat medullary thick ascending limb chloride transport: interaction with angiotensin II. Am J Physiol Regul Integr Comp Physiol 298: R954–R958, 2010. doi: 10.1152/ajpregu.00758.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baum M. Luminal angiotensin II stimulates rat medullary thick ascending limb chloride transport in the presence of basolateral norepinephrine. Am J Physiol Renal Physiol 310: F294–F299, 2016. doi: 10.1152/ajprenal.00447.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baylis C, Mitruka B, Deng A. Chronic blockade of nitric oxide synthesis in the rat produces systemic hypertension and glomerular damage. J Clin Invest 90: 278–281, 1992. doi: 10.1172/JCI115849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beara-Lasić L, Knotek M, Cejvan K, Jaksić O, Lasić Z, Skorić B, Brkljacić V, Banfić H. The effect of big endothelin-1 in the proximal tubule of the rat kidney. Br J Pharmacol 120: 625–630, 1997. doi: 10.1038/sj.bjp.0700956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 39.Beierwaltes WH. Macula densa stimulation of renin is reversed by selective inhibition of neuronal nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol 272: R1359–R1364, 1997. doi: 10.1152/ajpregu.1997.272.5.R1359. [DOI] [PubMed] [Google Scholar]
- 40.Beierwaltes WH, Arendshorst WJ, Klemmer PJ. Electrolyte and water balance in young spontaneously hypertensive rats. Hypertension 4: 908–915, 1982. doi: 10.1161/01.HYP.4.6.908. [DOI] [PubMed] [Google Scholar]
- 41.Bello-Reuss E. Effect of catecholamines on fluid reabsorption by the isolated proximal convoluted tubule. Am J Physiol Renal Physiol 238: F347–F352, 1980. doi: 10.1152/ajprenal.1980.238.5.F347. [DOI] [PubMed] [Google Scholar]
- 42.Ben-Ishay D, Knudsen KD, Dahl LK. Exaggerated response to isotonic saline loading in genetically hypertension-prone rats. J Lab Clin Med 82: 597–604, 1973. [PubMed] [Google Scholar]
- 43.Ben-Ishay D, Knudsen KD, Dahl LK. Renal function studies in the early stage of salt hypertension in rats. Proc Soc Exp Biol Med 125: 515–518, 1967. doi: 10.3181/00379727-125-32135. [DOI] [PubMed] [Google Scholar]
- 44.Bennett CM, Brenner BM, Berliner RW. Micropuncture study of nephron function in the rhesus monkey. J Clin Invest 47: 203–216, 1968. doi: 10.1172/JCI105710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bennett MA, Hillier C, Thurston H. Endothelium-dependent relaxation in resistance arteries from spontaneously hypertensive rats: effect of long-term treatment with perindopril, quinapril, hydralazine or amlodipine. J Hypertens 14: 389–397, 1996. doi: 10.1097/00004872-199603000-00017. [DOI] [PubMed] [Google Scholar]
- 46.Bernal-Mizrachi C, Gates AC, Weng S, Imamura T, Knutsen RH, DeSantis P, Coleman T, Townsend RR, Muglia LJ, Semenkovich CF. Vascular respiratory uncoupling increases blood pressure and atherosclerosis. Nature 435: 502–506, 2005. doi: 10.1038/nature03527. [DOI] [PubMed] [Google Scholar]
- 47.Bertuccio CA, Cheng SX, Arrizurieta EE, Martín RS, Ibarra FR. Mechanisms of Na+-K+-ATPase phosphorylation by PKC in the medullary thick ascending limb of Henle in the rat. Pflugers Arch 447: 87–96, 2003. doi: 10.1007/s00424-003-1144-6. [DOI] [PubMed] [Google Scholar]
- 48.Bianchi G, Baer PG, Fox U, Duzzi L, Pagetti D, Giovannetti AM. Changes in renin, water balance, and sodium balance during development of high blood pressure in genetically hypertensive rats. Circ Res 36, Suppl 1: 153–161, 1975. doi: 10.1161/01.RES.36.6.153. [DOI] [PubMed] [Google Scholar]
- 49.Bianchi G, Fox U, Di Francesco GF, Giovanetti AM, Pagetti D. Blood pressure changes produced by kidney cross-transplantation between spontaneously hypertensive rats and normotensive rats. Clin Sci Mol Med 47: 435–448, 1974. [DOI] [PubMed] [Google Scholar]
- 50.Bianchi G, Fox U, Imbasciati E. The development of a new strain of spontaneously hypertensive rats. Life Sci 14: 339–347, 1974. doi: 10.1016/0024-3205(74)90064-2. [DOI] [PubMed] [Google Scholar]
- 51.Bickel CA, Knepper MA, Verbalis JG, Ecelbarger CA. Dysregulation of renal salt and water transport proteins in diabetic Zucker rats. Kidney Int 61: 2099–2110, 2002. doi: 10.1046/j.1523-1755.2002.00353.x. [DOI] [PubMed] [Google Scholar]
- 52.Biemesderfer D, Pizzonia J, Abu-Alfa A, Exner M, Reilly R, Igarashi P, Aronson PS. NHE3: a Na+/H+ exchanger isoform of renal brush border. Am J Physiol Renal Physiol 265: F736–F742, 1993. doi: 10.1152/ajprenal.1993.265.5.F736. [DOI] [PubMed] [Google Scholar]
- 53.Blau S, Daly L, Fienberg A, Teitelman G, Ehrlich ME. DARPP-32 promoter directs transgene expression to renal thick ascending limb of loop of Henle. Am J Physiol Renal Physiol 269: F564–F570, 1995. doi: 10.1152/ajprenal.1995.269.4.F564. [DOI] [PubMed] [Google Scholar]
- 54.Bobulescu IA, Moe OW. Na+/H+ exchangers in renal regulation of acid-base balance. Semin Nephrol 26: 334–344, 2006. doi: 10.1016/j.semnephrol.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boim MA, Ho K, Shuck ME, Bienkowski MJ, Block JH, Slightom JL, Yang Y, Brenner BM, Hebert SC. ROMK inwardly rectifying ATP-sensitive K+ channel. II. Cloning and distribution of alternative forms. Am J Physiol Renal Physiol 268: F1132–F1140, 1995. doi: 10.1152/ajprenal.1995.268.6.F1132. [DOI] [PubMed] [Google Scholar]
- 56.Borensztein P, Juvin P, Vernimmen C, Poggioli J, Paillard M, Bichara M. cAMP-dependent control of Na+/H+ antiport by AVP, PTH, and PGE2 in rat medullary thick ascending limb cells. Am J Physiol Renal Physiol 264: F354–F364, 1993. doi: 10.1152/ajprenal.1993.264.2.F354. [DOI] [PubMed] [Google Scholar]
- 57.Bostanjoglo M, Reeves WB, Reilly RF, Velázquez H, Robertson N, Litwack G, Morsing P, Dørup J, Bachmann S, Ellison DH, Bostanjoglo M. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules [Erratum in J Am Soc Nephrol 9: 2179, 1998]. J Am Soc Nephrol 9: 1347–1358, 1998. [DOI] [PubMed] [Google Scholar]
- 58.Bouby N, Bankir L. Effect of high protein intake on sodium, potassium-dependent adenosine triphosphatase activity in the thick ascending limb of Henle’s loop in the rat. Clin Sci (Lond) 74: 319–329, 1988. doi: 10.1042/cs0740319. [DOI] [PubMed] [Google Scholar]
- 59.Bouby N, Trinh-Trang-Tan MM, Laouari D, Kleinknecht C, Grünfeld JP, Kriz W, Bankir L, Douté M, Hähnel B, Coutaud C. Role of the urinary concentrating process in the renal effects of high protein intake. Kidney Int 34: 4–12, 1988. doi: 10.1038/ki.1988.138. [DOI] [PubMed] [Google Scholar]
- 60.Bourdeau JE, Burg MB. Voltage dependence of calcium transport in the thick ascending limb of Henle’s loop. Am J Physiol Renal Physiol 236: F357–F364, 1979. doi: 10.1152/ajprenal.1979.236.4.F357. [DOI] [PubMed] [Google Scholar]
- 61.Bouriquet N, Casellas D. Chronic l-NAME hypertension in rats and autoregulation of juxtamedullary preglomerular vessels. Am J Physiol Renal Physiol 269: F190–F197, 1995. doi: 10.1152/ajprenal.1995.269.2.F190. [DOI] [PubMed] [Google Scholar]
- 62.Breiderhoff T, Himmerkus N, Drewell H, Plain A, Günzel D, Mutig K, Willnow TE, Müller D, Bleich M. Deletion of claudin-10 rescues claudin-16-deficient mice from hypomagnesemia and hypercalciuria. Kidney Int 93: 580–588, 2018. doi: 10.1016/j.kint.2017.08.029. [DOI] [PubMed] [Google Scholar]
- 63.Breiderhoff T, Himmerkus N, Stuiver M, Mutig K, Will C, Meij IC, Bachmann S, Bleich M, Willnow TE, Müller D. Deletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosis. Proc Natl Acad Sci USA 109: 14241–14246, 2012. doi: 10.1073/pnas.1203834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buerkert J, Martin D, Trigg D. Segmental analysis of the renal tubule in buffer production and net acid formation. Am J Physiol Renal Physiol 244: F442–F454, 1983. doi: 10.1152/ajprenal.1983.244.4.F442. [DOI] [PubMed] [Google Scholar]
- 65.Bugaj V, Sansom SC, Wen D, Hatcher LI, Stockand JD, Mironova E. Flow-sensitive K+-coupled ATP secretion modulates activity of the epithelial Na+ channel in the distal nephron. J Biol Chem 287: 38552–38558, 2012. doi: 10.1074/jbc.M112.408476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burch HB, Choi S, Dence CN, Alvey TR, Cole BR, Lowry OH. Metabolic effects of large fructose loads in different parts of the rat nephron. J Biol Chem 255: 8239–8244, 1980. [PubMed] [Google Scholar]
- 67.Burg M, Stoner L. Renal tubular chloride transport and the mode of action of some diuretics. Annu Rev Physiol 38: 37–45, 1976. doi: 10.1146/annurev.ph.38.030176.000345. [DOI] [PubMed] [Google Scholar]
- 68.Burg MB, Green N. Function of the thick ascending limb of Henle’s loop. Am J Physiol 224: 659–668, 1973. doi: 10.1152/ajplegacy.1973.224.3.659. [DOI] [PubMed] [Google Scholar]
- 69.Burgio G. Redefining mouse transgenesis with CRISPR/Cas9 genome editing technology. Genome Biol 19: 27, 2018. doi: 10.1186/s13059-018-1409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Burnier M. Angiotensin II type 1 receptor blockers. Circulation 103: 904–912, 2001. doi: 10.1161/01.CIR.103.6.904. [DOI] [PubMed] [Google Scholar]
- 71.Burrell LM, Risvanis J, Dean RG, Patel SK, Velkoska E, Johnston CI. Age-dependent regulation of renal vasopressin V(1A) and V2 receptors in rats with genetic hypertension: implications for the treatment of hypertension. J Am Soc Hypertens 7: 3–13, 2013. doi: 10.1016/j.jash.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 72.Bustamante M, Hasler U, Leroy V, de Seigneux S, Dimitrov M, Mordasini D, Rousselot M, Martin PY, Féraille E. Calcium-sensing receptor attenuates AVP-induced aquaporin-2 expression via a calmodulin-dependent mechanism. J Am Soc Nephrol 19: 109–116, 2008. doi: 10.1681/ASN.2007010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cabandugama PK, Gardner MJ, Sowers JR. The renin angiotensin aldosterone system in obesity and hypertension: roles in the cardiorenal metabolic syndrome. Med Clin North Am 101: 129–137, 2017. doi: 10.1016/j.mcna.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cabral PD, Capurro C, Garvin JL. TRPV4 mediates flow-induced increases in intracellular Ca in medullary thick ascending limbs. Acta Physiol (Oxf) 214: 319–328, 2015. doi: 10.1111/apha.12528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cabral PD, Garvin JL. TRPV4 activation mediates flow-induced nitric oxide production in the rat thick ascending limb. Am J Physiol Renal Physiol 307: F666–F672, 2014. doi: 10.1152/ajprenal.00619.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cabral PD, Herrera M. Membrane-associated aquaporin-1 facilitates osmotically driven water flux across the basolateral membrane of the thick ascending limb. Am J Physiol Renal Physiol 303: F621–F629, 2012. doi: 10.1152/ajprenal.00268.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cabral PD, Hong NJ, Garvin JL. ATP mediates flow-induced NO production in thick ascending limbs. Am J Physiol Renal Physiol 303: F194–F200, 2012. doi: 10.1152/ajprenal.00504.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cabral PD, Hong NJ, Garvin JL. Shear stress increases nitric oxide production in thick ascending limbs. Am J Physiol Renal Physiol 299: F1185–F1192, 2010. doi: 10.1152/ajprenal.00112.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cabral PD, Hong NJ, Hye Khan MA, Ortiz PA, Beierwaltes WH, Imig JD, Garvin JL. Fructose stimulates Na/H exchange activity and sensitizes the proximal tubule to angiotensin II. Hypertension 63: e68–e73, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02564. [DOI] [PubMed] [Google Scholar]
- 80.Cabral PD, Silva GB, Baigorria ST, Juncos LA, Juncos LI, García NH. 8-iso-prostaglandin-F2α stimulates chloride transport in thick ascending limbs: role of cAMP and protein kinase A. Am J Physiol Renal Physiol 299: F1396–F1400, 2010. doi: 10.1152/ajprenal.00225.2010. [DOI] [PubMed] [Google Scholar]
- 81.Caceres PS, Ares GR, Ortiz PA. cAMP stimulates apical exocytosis of the renal Na+-K+-2Cl− cotransporter NKCC2 in the thick ascending limb: role of protein kinase A. J Biol Chem 284: 24965–24971, 2009. doi: 10.1074/jbc.M109.037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Callaway E. New concerns raised over value of genome-wide disease studies. Nature 546: 463, 2017. doi: 10.1038/nature.2017.22152. [DOI] [Google Scholar]
- 83.Cao C, Lee-Kwon W, Silldorff EP, Pallone TL. KATP channel conductance of descending vasa recta pericytes. Am J Physiol Renal Physiol 289: F1235–F1245, 2005. doi: 10.1152/ajprenal.00111.2005. [DOI] [PubMed] [Google Scholar]
- 84.Capasso G, Geibel PJ, Damiano S, Jaeger P, Richards WG, Geibel JP. The calcium sensing receptor modulates fluid reabsorption and acid secretion in the proximal tubule. Kidney Int 84: 277–284, 2013. doi: 10.1038/ki.2013.137. [DOI] [PubMed] [Google Scholar]
- 85.Capasso G, Rizzo M, Evangelista C, Ferrari P, Geelen G, Lang F, Bianchi G. Altered expression of renal apical plasma membrane Na+ transporters in the early phase of genetic hypertension. Am J Physiol Renal Physiol 288: F1173–F1182, 2005. doi: 10.1152/ajprenal.00228.2004. [DOI] [PubMed] [Google Scholar]
- 86.Capasso G, Rizzo M, Garavaglia ML, Trepiccione F, Zacchia M, Mugione A, Ferrari P, Paulmichl M, Lang F, Loffing J, Carrel M, Damiano S, Wagner CA, Bianchi G, Meyer G. Upregulation of apical sodium-chloride cotransporter and basolateral chloride channels is responsible for the maintenance of salt-sensitive hypertension. Am J Physiol Renal Physiol 295: F556–F567, 2008. doi: 10.1152/ajprenal.00340.2007. [DOI] [PubMed] [Google Scholar]
- 87.Carlson SH, Wyss JM. Long-term telemetric recording of arterial pressure and heart rate in mice fed basal and high NaCl diets. Hypertension 35: E1–E5, 2000. doi: 10.1161/01.HYP.35.2.e1. [DOI] [PubMed] [Google Scholar]
- 88.Carmosino M, Rizzo F, Ferrari P, Torielli L, Ferrandi M, Bianchi G, Svelto M, Valenti G. NKCC2 is activated in Milan hypertensive rats contributing to the maintenance of salt-sensitive hypertension. Pflugers Arch 462: 281–291, 2011. doi: 10.1007/s00424-011-0967-9. [DOI] [PubMed] [Google Scholar]
- 89.Carroll KJ, Makarewich CA, McAnally J, Anderson DM, Zentilin L, Liu N, Giacca M, Bassel-Duby R, Olson EN. A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc Natl Acad Sci USA 113: 338–343, 2016. doi: 10.1073/pnas.1523918113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Castrop H, Kurtz A. Differential nNOS gene expression in salt-sensitive and salt-resistant Dahl rats. J Hypertens 19: 1223–1231, 2001. doi: 10.1097/00004872-200107000-00007. [DOI] [PubMed] [Google Scholar]
- 91.Catena C, Cavarape A, Novello M, Giacchetti G, Sechi LA. Insulin receptors and renal sodium handling in hypertensive fructose-fed rats. Kidney Int 64: 2163–2171, 2003. doi: 10.1046/j.1523-1755.2003.00313.x. [DOI] [PubMed] [Google Scholar]
- 92.Cervenka L, Kramer HJ, Malý J, Heller J. Role of nNOS in regulation of renal function in angiotensin II-induced hypertension. Hypertension 38: 280–285, 2001. doi: 10.1161/01.HYP.38.2.280. [DOI] [PubMed] [Google Scholar]
- 93.Chae HZ, Robison K, Poole LB, Church G, Storz G, Rhee SG. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc Natl Acad Sci USA 91: 7017–7021, 1994. doi: 10.1073/pnas.91.15.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chambrey R, Warnock DG, Podevin RA, Bruneval P, Mandet C, Bélair MF, Bariéty J, Paillard M. Immunolocalization of the Na+/H+ exchanger isoform NHE2 in rat kidney. Am J Physiol Renal Physiol 275: F379–F386, 1998. doi: 10.1152/ajprenal.1998.275.3.F379. [DOI] [PubMed] [Google Scholar]
- 95.Chan EC, Jiang F, Peshavariya HM, Dusting GJ. Regulation of cell proliferation by NADPH oxidase-mediated signaling: potential roles in tissue repair, regenerative medicine and tissue engineering. Pharmacol Ther 122: 97–108, 2009. doi: 10.1016/j.pharmthera.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 96.Chan V, Fenning A, Iyer A, Hoey A, Brown L. Resveratrol improves cardiovascular function in DOCA-salt hypertensive rats. Curr Pharm Biotechnol 12: 429–436, 2011. doi: 10.2174/138920111794480552. [DOI] [PubMed] [Google Scholar]
- 97.Chemin J, Patel AJ, Duprat F, Lauritzen I, Lazdunski M, Honoré E. A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J 24: 44–53, 2005. doi: 10.1038/sj.emboj.7600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen PY, Sanders PW. L-arginine abrogates salt-sensitive hypertension in Dahl/Rapp rats. J Clin Invest 88: 1559–1567, 1991. doi: 10.1172/JCI115467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen YF, Cowley AW Jr, Zou AP. Increased H2O2 counteracts the vasodilator and natriuretic effects of superoxide dismutation by tempol in renal medulla. Am J Physiol Regul Integr Comp Physiol 285: R827–R833, 2003. doi: 10.1152/ajpregu.00636.2002. [DOI] [PubMed] [Google Scholar]
- 100.Chen YF, Naftilan AJ, Oparil S. Androgen-dependent angiotensinogen and renin messenger RNA expression in hypertensive rats. Hypertension 19: 456–463, 1992. doi: 10.1161/01.HYP.19.5.456. [DOI] [PubMed] [Google Scholar]
- 101.Cheng HF, Wang JL, Vinson GP, Harris RC. Young SHR express increased type 1 angiotensin II receptors in renal proximal tubule. Am J Physiol Renal Physiol 274: F10–F17, 1998. doi: 10.1152/ajprenal.1998.274.1.F10. [DOI] [PubMed] [Google Scholar]
- 102.Chin SY, Wang CT, Majid DS, Navar LG. Renoprotective effects of nitric oxide in angiotensin II-induced hypertension in the rat. Am J Physiol Renal Physiol 274: F876–F882, 1998. doi: 10.1152/ajprenal.1998.274.5.F876. [DOI] [PubMed] [Google Scholar]
- 103.Christensen BM, Zelenina M, Aperia A, Nielsen S. Localization and regulation of PKA-phosphorylated AQP2 in response to V(2)-receptor agonist/antagonist treatment. Am J Physiol Renal Physiol 278: F29–F42, 2000. doi: 10.1152/ajprenal.2000.278.1.F29. [DOI] [PubMed] [Google Scholar]
- 104.Chu TS, Tsuganezawa H, Peng Y, Cano A, Yanagisawa M, Alpern RJ. Role of tyrosine kinase pathways in ETB receptor activation of NHE3. Am J Physiol Cell Physiol 271: C763–C771, 1996. doi: 10.1152/ajpcell.1996.271.3.C763. [DOI] [PubMed] [Google Scholar]
- 105.Chun TY, Bankir L, Eckert GJ, Bichet DG, Saha C, Zaidi SA, Wagner MA, Pratt JH. Ethnic differences in renal responses to furosemide. Hypertension 52: 241–248, 2008. doi: 10.1161/HYPERTENSIONAHA.108.109801. [DOI] [PubMed] [Google Scholar]
- 106.Closs EI. CATs, a family of three distinct mammalian cationic amino acid transporters. Amino Acids 11: 193–208, 1996. doi: 10.1007/BF00813860. [DOI] [PubMed] [Google Scholar]
- 107.Cogan MG. Angiotensin II: a powerful controller of sodium transport in the early proximal tubule. Hypertension 15: 451–458, 1990. doi: 10.1161/01.HYP.15.5.451. [DOI] [PubMed] [Google Scholar]
- 108.Colegio OR, Van Itallie C, Rahner C, Anderson JM. Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am J Physiol Cell Physiol 284: C1346–C1354, 2003. doi: 10.1152/ajpcell.00547.2002. [DOI] [PubMed] [Google Scholar]
- 109.Colegio OR, Van Itallie CM, McCrea HJ, Rahner C, Anderson JM. Claudins create charge-selective channels in the paracellular pathway between epithelial cells. Am J Physiol Cell Physiol 283: C142–C147, 2002. doi: 10.1152/ajpcell.00038.2002. [DOI] [PubMed] [Google Scholar]
- 110.Collazo R, Fan L, Hu MC, Zhao H, Wiederkehr MR, Moe OW. Acute regulation of Na+/H+ exchanger NHE3 by parathyroid hormone via NHE3 phosphorylation and dynamin-dependent endocytosis. J Biol Chem 275: 31601–31608, 2000. doi: 10.1074/jbc.M000600200. [DOI] [PubMed] [Google Scholar]
- 111.Cowley AW Jr, Mattson DL, Lu S, Roman RJ. The renal medulla and hypertension. Hypertension 25: 663–673, 1995. doi: 10.1161/01.HYP.25.4.663. [DOI] [PubMed] [Google Scholar]
- 112.Cowley AW Jr, Mori T, Mattson D, Zou AP. Role of renal NO production in the regulation of medullary blood flow. Am J Physiol Regul Integr Comp Physiol 284: R1355–R1369, 2003. doi: 10.1152/ajpregu.00701.2002. [DOI] [PubMed] [Google Scholar]
- 113.Cowley AW Jr, Roman RJ, Kaldunski ML, Dumas P, Dickhout JG, Greene AS, Jacob HJ. Brown Norway chromosome 13 confers protection from high salt to consomic Dahl S rat. Hypertension 37: 456–461, 2001. doi: 10.1161/01.HYP.37.2.456. [DOI] [PubMed] [Google Scholar]
- 114.Cowley AW Jr, Stoll M, Greene AS, Kaldunski ML, Roman RJ, Tonellato PJ, Schork NJ, Dumas P, Jacob HJ. Genetically defined risk of salt sensitivity in an intercross of Brown Norway and Dahl S rats. Physiol Genomics 2: 107–115, 2000. doi: 10.1152/physiolgenomics.2000.2.3.107. [DOI] [PubMed] [Google Scholar]
- 115.Cowley AW Jr, Yang C, Zheleznova NN, Staruschenko A, Kurth T, Rein L, Kumar V, Sadovnikov K, Dayton A, Hoffman M, Ryan RP, Skelton MM, Salehpour F, Ranji M, Geurts A. Evidence of the importance of Nox4 in production of hypertension in Dahl salt-sensitive rats. Hypertension 67: 440–450, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cowley AW Jr, Liang M, Roman RJ, Greene AS, Jacob HJ. Consomic rat model systems for physiological genomics. Acta Physiol Scand 181: 585–592, 2004. doi: 10.1111/j.1365-201X.2004.01334.x. [DOI] [PubMed] [Google Scholar]
- 117.Craig WS, Kyte J. Stoichiometry and molecular weight of the minimum asymmetric unit of canine renal sodium and potassium ion-activated adenosine triphosphatase. J Biol Chem 255: 6262–6269, 1980. [PubMed] [Google Scholar]
- 118.Crajoinas RO, Polidoro JZ, Carneiro de Morais CP, Castelo-Branco RC, Girardi AC. Angiotensin II counteracts the effects of cAMP/PKA on NHE3 activity and phosphorylation in proximal tubule cells. Am J Physiol Cell Physiol 311: C768–C776, 2016. doi: 10.1152/ajpcell.00191.2016. [DOI] [PubMed] [Google Scholar]
- 119.Crawford C, Wildman SS, Kelly MC, Kennedy-Lydon TM, Peppiatt-Wildman CM. Sympathetic nerve-derived ATP regulates renal medullary vasa recta diameter via pericyte cells: a role for regulating medullary blood flow? Front Physiol 4: 307, 2013. doi: 10.3389/fphys.2013.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA 103: 17985–17990, 2006. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, Flannery PJ, Spurney RF, Kim HS, Smithies O, Le TH, Coffman TM. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest 115: 1092–1099, 2005. doi: 10.1172/JCI23378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.D’Souza T, Agarwal R, Morin PJ. Phosphorylation of claudin-3 at threonine 192 by cAMP-dependent protein kinase regulates tight junction barrier function in ovarian cancer cells. J Biol Chem 280: 26233–26240, 2005. doi: 10.1074/jbc.M502003200. [DOI] [PubMed] [Google Scholar]
- 123.Dahl LK, Heine M. Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ Res 36: 692–696, 1975. doi: 10.1161/01.RES.36.6.692. [DOI] [PubMed] [Google Scholar]
- 124.Dahl LK, Heine M, Tassinari L. Effects of chronia excess salt ingestion. Evidence that genetic factors play an important role in susceptibility to experimental hypertension. J Exp Med 115: 1173–1190, 1962. doi: 10.1084/jem.115.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dahl LK, Heine M, Tassinari L. Role of genetic factors in susceptibility to experimental hypertension due to chronic excess salt ingestion. Nature 194: 480–482, 1962. doi: 10.1038/194480b0. [DOI] [PubMed] [Google Scholar]
- 126.Dahl LK, Heine M, Thompson K. Genetic influence of the kidneys on blood pressure. Evidence from chronic renal homografts in rats with opposite predispositions to hypertension. Circ Res 34: 94–101, 1974. doi: 10.1161/01.RES.34.1.94. [DOI] [PubMed] [Google Scholar]
- 127.Das S, Mattson DL. Exogenous L-arginine attenuates the effects of angiotensin II on renal hemodynamics and the pressure natriuresis-diuresis relationship. Clin Exp Pharmacol Physiol 41: 270–278, 2014. doi: 10.1111/1440-1681.12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.de Jesus Ferreira MC, Bailly C. Luminal and basolateral endothelin inhibit chloride reabsorption in the mouse thick ascending limb via a Ca2+-independent pathway. J Physiol 505: 749–758, 1997. doi: 10.1111/j.1469-7793.1997.749ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.De Miguel C, Foster JM, Carmines PK, Pollock JS. Interaction between NO synthase and NADPH oxidase in control of sodium transport by the renal thick ascending limb during diabetes. Acta Physiol (Oxf) 209: 148–155, 2013. doi: 10.1111/apha.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.de Rouffignac C, Quamme G. Renal magnesium handling and its hormonal control. Physiol Rev 74: 305–322, 1994. doi: 10.1152/physrev.1994.74.2.305. [DOI] [PubMed] [Google Scholar]
- 131.Deborde T, Amar L, Bobrie G, Postel-Vinay N, Battaglia C, Tache A, Chedid A, Dhib MM, Chatellier G, Plouin PF, Burgun A, Azizi M, Jannot AS. Sex differences in antihypertensive treatment in France among 17 856 patients in a tertiary hypertension unit. J Hypertens 36: 939–946, 2018. doi: 10.1097/HJH.0000000000001607. [DOI] [PubMed] [Google Scholar]
- 132.DeClue JW, Guyton AC, Cowley AW Jr, Coleman TG, Norman RA Jr, McCaa RE. Subpressor angiotensin infusion, renal sodium handling, and salt-induced hypertension in the dog. Circ Res 43: 503–512, 1978. doi: 10.1161/01.RES.43.4.503. [DOI] [PubMed] [Google Scholar]
- 133.DeCoursey TE. Voltage-gated proton channels: molecular biology, physiology, and pathophysiology of the H(V) family. Physiol Rev 93: 599–652, 2013. doi: 10.1152/physrev.00011.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Derst C, Hirsch JR, Preisig-Müller R, Wischmeyer E, Karschin A, Döring F, Thomzig A, Veh RW, Schlatter E, Kummer W, Daut J. Cellular localization of the potassium channel Kir7.1 in guinea pig and human kidney. Kidney Int 59: 2197–2205, 2001. doi: 10.1046/j.1523-1755.2001.00735.x. [DOI] [PubMed] [Google Scholar]
- 135.Dhande IS, Zhu Y, Braun MC, Hicks MJ, Wenderfer SE, Doris PA. Mycophenolate mofetil prevents cerebrovascular injury in stroke-prone spontaneously hypertensive rats. Physiol Genomics 49: 132–140, 2017. doi: 10.1152/physiolgenomics.00110.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Di Giosia P, Giorgini P, Stamerra CA, Petrarca M, Ferri C, Sahebkar A. Gender Differences in Epidemiology, Pathophysiology, and Treatment of Hypertension. Curr Atheroscler Rep 20: 13, 2018. doi: 10.1007/s11883-018-0716-z. [DOI] [PubMed] [Google Scholar]
- 137.Díaz-Sylvester P, Mac Laughlin M, Amorena C. Peritubular fluid viscosity modulates H+ flux in proximal tubules through NO release. Am J Physiol Renal Physiol 280: F239–F243, 2001. doi: 10.1152/ajprenal.2001.280.2.F239. [DOI] [PubMed] [Google Scholar]
- 138.DiBona GF, Rios LL. Mechanism of exaggerated diuresis in spontaneously hypertensive rats. Am J Physiol Renal Physiol 235: 409–416, 1978. doi: 10.1152/ajprenal.1978.235.5.F409. [DOI] [PubMed] [Google Scholar]
- 139.Dickhout JG, Mori T, Cowley AW Jr. Tubulovascular nitric oxide crosstalk: buffering of angiotensin II-induced medullary vasoconstriction. Circ Res 91: 487–493, 2002. doi: 10.1161/01.RES.0000035243.66189.92. [DOI] [PubMed] [Google Scholar]
- 140.Dimke H, Flyvbjerg A, Bourgeois S, Thomsen K, Frøkiaer J, Houillier P, Nielsen S, Frische S. Acute growth hormone administration induces antidiuretic and antinatriuretic effects and increases phosphorylation of NKCC2. Am J Physiol Renal Physiol 292: F723–F735, 2007. doi: 10.1152/ajprenal.00276.2006. [DOI] [PubMed] [Google Scholar]
- 141.Disashi T, Nonoguchi H, Iwaoka T, Naomi S, Nakayama Y, Shimada K, Tanzawa K, Tomita K. Endothelin converting enzyme-1 gene expression in the kidney of spontaneously hypertensive rats. Hypertension 30: 1591–1597, 1997. doi: 10.1161/01.HYP.30.6.1591. [DOI] [PubMed] [Google Scholar]
- 142.Doris PA. Genetics of hypertension: an assessment of progress in the spontaneously hypertensive rat. Physiol Genomics 49: 601–617, 2017. doi: 10.1152/physiolgenomics.00065.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.du Cheyron D, Paillard M, Poggioli J. [Regulation of the luminal Na+/H+ exchanger NHE3 by intracellular protein trafficking]. Nephrologie 23: 219–224, 2002. [PubMed] [Google Scholar]
- 144.DuBose TD Jr, Lucci MS. Effect of carbonic anhydrase inhibition on superficial and deep nephron bicarbonate reabsorption in the rat. J Clin Invest 71: 55–65, 1983. doi: 10.1172/JCI110751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dukacz SA, Feng MG, Yang LF, Lee RM, Kline RL. Abnormal renal medullary response to angiotensin II in SHR is corrected by long-term enalapril treatment. Am J Physiol Regul Integr Comp Physiol 280: R1076–R1084, 2001. doi: 10.1152/ajpregu.2001.280.4.R1076. [DOI] [PubMed] [Google Scholar]
- 146.Eatman D, Wang M, Socci RR, Thierry-Palmer M, Emmett N, Bayorh MA. Gender differences in the attenuation of salt-induced hypertension by angiotensin (1-7). Peptides 22: 927–933, 2001. doi: 10.1016/S0196-9781(01)00404-1. [DOI] [PubMed] [Google Scholar]
- 147.Editorial Team Chow down. Nature 530: 254, 2016. doi: 10.1038/530254a. [DOI] [PubMed] [Google Scholar]
- 148.Edwards A, Cao C, Pallone TL. Cellular mechanisms underlying nitric oxide-induced vasodilation of descending vasa recta. Am J Physiol Renal Physiol 300: F441–F456, 2011. doi: 10.1152/ajprenal.00499.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Edwards A, Castrop H, Laghmani K, Vallon V, Layton AT. Effects of NKCC2 isoform regulation on NaCl transport in thick ascending limb and macula densa: a modeling study. Am J Physiol Renal Physiol 307: F137–F146, 2014. doi: 10.1152/ajprenal.00158.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Edwards BS, Schwab TR, Zimmerman RS, Heublein DM, Jiang NS, Burnett JC Jr. Cardiovascular, renal, and endocrine response to atrial natriuretic peptide in angiotensin II mediated hypertension. Circ Res 59: 663–667, 1986. doi: 10.1161/01.RES.59.6.663. [DOI] [PubMed] [Google Scholar]
- 151.el-Hajj Fuleihan G, Seifter J, Scott J, Brown EM. Calcium-regulated renal calcium handling in healthy men: relationship to sodium handling. J Clin Endocrinol Metab 83: 2366–2372, 1998. doi: 10.1210/jcem.83.7.4915. [DOI] [PubMed] [Google Scholar]
- 152.Eriksen EF, Richelsen B, Gesser BP, Jacobsen NO, Stengaard-Pedersen K. Prostaglandin-E2 receptors in the rat kidney: biochemical characterization and localization. Kidney Int 32: 181–186, 1987. doi: 10.1038/ki.1987.190. [DOI] [PubMed] [Google Scholar]
- 153.Escalante B, Erlij D, Falck JR, McGiff JC. Cytochrome P-450 arachidonate metabolites affect ion fluxes in rabbit medullary thick ascending limb. Am J Physiol Cell Physiol 266: C1775–C1782, 1994. doi: 10.1152/ajpcell.1994.266.6.C1775. [DOI] [PubMed] [Google Scholar]
- 154.Escalante B, Erlij D, Falck JR, McGiff JC. Effect of cytochrome P450 arachidonate metabolites on ion transport in rabbit kidney loop of Henle. Science 251: 799–802, 1991. doi: 10.1126/science.1846705. [DOI] [PubMed] [Google Scholar]
- 155.Estévez R, Boettger T, Stein V, Birkenhäger R, Otto E, Hildebrandt F, Jentsch TJ. Barttin is a Cl− channel beta-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature 414: 558–561, 2001. doi: 10.1038/35107099. [DOI] [PubMed] [Google Scholar]
- 156.Evans LC, Ryan RP, Broadway E, Skelton MM, Kurth T, Cowley AW Jr. Null mutation of the nicotinamide adenine dinucleotide phosphate-oxidase subunit p67phox protects the Dahl-S rat from salt-induced reductions in medullary blood flow and glomerular filtration rate. Hypertension 65: 561–568, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Fahlke C, Fischer M. Physiology and pathophysiology of ClC-K/barttin channels. Front Physiol 1: 155, 2010. doi: 10.3389/fphys.2010.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Farley RA, Miller RP, Kudrow A. Orientation of the beta subunit polypeptide of (Na+ + K+)ATPase in the cell membrane. Biochim Biophys Acta 873: 136–142, 1986. doi: 10.1016/0167-4838(86)90199-8. [DOI] [PubMed] [Google Scholar]
- 159.Farley RA, Tran CM, Carilli CT, Hawke D, Shively JE. The amino acid sequence of a fluorescein-labeled peptide from the active site of (Na,K)-ATPase. J Biol Chem 259: 9532–9535, 1984. [PubMed] [Google Scholar]
- 160.Farman N, Coutry N, Logvinenko N, Blot-Chabaud M, Bourbouze R, Bonvalet JP. Adrenalectomy reduces alpha 1 and not beta 1 Na+-K+-ATPase mRNA expression in rat distal nephron. Am J Physiol Cell Physiol 263: C810–C817, 1992. doi: 10.1152/ajpcell.1992.263.4.C810. [DOI] [PubMed] [Google Scholar]
- 161.Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol 17: 375–412, 1963. doi: 10.1083/jcb.17.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Feng D, Yang C, Geurts AM, Kurth T, Liang M, Lazar J, Mattson DL, O’Connor PM, Cowley AW Jr. Increased expression of NAD(P)H oxidase subunit p67(phox) in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension. Cell Metab 15: 201–208, 2012. doi: 10.1016/j.cmet.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fenning A, Harrison G, Rose’meyer R, Hoey A, Brown L. l-Arginine attenuates cardiovascular impairment in DOCA-salt hypertensive rats. Am J Physiol Heart Circ Physiol 289: H1408–H1416, 2005. doi: 10.1152/ajpheart.00140.2005. [DOI] [PubMed] [Google Scholar]
- 164.Fenoy FJ, Ferrer P, Carbonell L, García-Salom M. Role of nitric oxide on papillary blood flow and pressure natriuresis. Hypertension 25: 408–414, 1995. doi: 10.1161/01.HYP.25.3.408. [DOI] [PubMed] [Google Scholar]
- 165.Féraille E, Doucet A. Sodium-potassium-adenosinetriphosphatase-dependent sodium transport in the kidney: hormonal control. Physiol Rev 81: 345–418, 2001. doi: 10.1152/physrev.2001.81.1.345. [DOI] [PubMed] [Google Scholar]
- 166.Feric M, Zhao B, Hoffert JD, Pisitkun T, Knepper MA. Large-scale phosphoproteomic analysis of membrane proteins in renal proximal and distal tubule. Am J Physiol Cell Physiol 300: C755–C770, 2011. doi: 10.1152/ajpcell.00360.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ferrandi M, Salardi S, Parenti P, Ferrari P, Bianchi G, Braw R, Karlish SJ. Na+/K+/Cl−-cotransporter mediated Rb+ fluxes in membrane vesicles from kidneys of normotensive and hypertensive rats. Biochim Biophys Acta 1021: 13–20, 1990. doi: 10.1016/0005-2736(90)90377-Z. [DOI] [PubMed] [Google Scholar]
- 168.Ferreri NR, Escalante BA, Zhao Y, An SJ, McGiff JC. Angiotensin II induces TNF production by the thick ascending limb: functional implications. Am J Physiol Renal Physiol 274: F148–F155, 1998. doi: 10.1152/ajprenal.1998.274.1.F148. [DOI] [PubMed] [Google Scholar]
- 169.Filopanti M, Corbetta S, Barbieri AM, Spada A. Pharmacology of the calcium sensing receptor. Clin Cases Miner Bone Metab 10: 162–165, 2013. [PMC free article] [PubMed] [Google Scholar]
- 170.Fisher SE, van Bakel I, Lloyd SE, Pearce SH, Thakker RV, Craig IW. Cloning and characterization of CLCN5, the human kidney chloride channel gene implicated in Dent disease (an X-linked hereditary nephrolithiasis). Genomics 29: 598–606, 1995. doi: 10.1006/geno.1995.9960. [DOI] [PubMed] [Google Scholar]
- 171.Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res 88: E68–E75, 2001. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- 172.Flores-Santana W, Switzer C, Ridnour LA, Basudhar D, Mancardi D, Donzelli S, Thomas DD, Miranda KM, Fukuto JM, Wink DA. Comparing the chemical biology of NO and HNO. Arch Pharm Res 32: 1139–1153, 2009. doi: 10.1007/s12272-009-1805-x. [DOI] [PubMed] [Google Scholar]
- 173.Foggensteiner L, Bevan AP, Thomas R, Coleman N, Boulter C, Bradley J, Ibraghimov-Beskrovnaya O, Klinger K, Sandford R. Cellular and subcellular distribution of polycystin-2, the protein product of the PKD2 gene. J Am Soc Nephrol 11: 814–827, 2000. [DOI] [PubMed] [Google Scholar]
- 174.Fraser SA, Gimenez I, Cook N, Jennings I, Katerelos M, Katsis F, Levidiotis V, Kemp BE, Power DA. Regulation of the renal-specific Na+-K+-2Cl− co-transporter NKCC2 by AMP-activated protein kinase (AMPK). Biochem J 405: 85–93, 2007. doi: 10.1042/BJ20061850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fryckstedt J, Aperia A, Snyder G, Meister B. Distribution of dopamine- and cAMP-dependent phosphoprotein (DARPP-32) in the developing and mature kidney. Kidney Int 44: 495–502, 1993. doi: 10.1038/ki.1993.273. [DOI] [PubMed] [Google Scholar]
- 176.Fryckstedt J, Meister B, Aperia A. Control of electrolyte transport in the kidney through a dopamine- and cAMP-regulated phosphoprotein, DARPP-32. J Auton Pharmacol 12: 183–189, 1992. doi: 10.1111/j.1474-8673.1992.tb00376.x. [DOI] [PubMed] [Google Scholar]
- 177.Fu Y, Lu Y, Liu EY, Zhu X, Mahajan GJ, Lu D, Roman RJ, Liu R. Testosterone enhances tubuloglomerular feedback by increasing superoxide production in the macula densa. Am J Physiol Regul Integr Comp Physiol 304: R726–R733, 2013. doi: 10.1152/ajpregu.00341.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Fujihara CK, De Nucci G, Zatz R. Chronic nitric oxide synthase inhibition aggravates glomerular injury in rats with subtotal nephrectomy. J Am Soc Nephrol 5: 1498–1507, 1995. [DOI] [PubMed] [Google Scholar]
- 179.Fujihara CK, Michellazzo SM, de Nucci G, Zatz R. Sodium excess aggravates hypertension and renal parenchymal injury in rats with chronic NO inhibition. Am J Physiol Renal Physiol 266: F697–F705, 1994. doi: 10.1152/ajprenal.1994.266.5.F697. [DOI] [PubMed] [Google Scholar]
- 180.Gainer JV, Lipkowitz MS, Yu C, Waterman MR, Dawson EP, Capdevila JH, Brown NJ, Group AS; AASK Study Group . Association of a CYP4A11 variant and blood pressure in black men. J Am Soc Nephrol 19: 1606–1612, 2008. doi: 10.1681/ASN.2008010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gamba G, Friedman PA. Thick ascending limb: the Na+:K+:2Cl− co-transporter, NKCC2, and the calcium-sensing receptor, CaSR. Pflugers Arch 458: 61–76, 2009. doi: 10.1007/s00424-008-0607-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Gamba G, Miyanoshita A, Lombardi M, Lytton J, Lee WS, Hediger MA, Hebert SC. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J Biol Chem 269: 17713–17722, 1994. [PubMed] [Google Scholar]
- 183.Gao F, Wang DH. Impairment in function and expression of transient receptor potential vanilloid type 4 in Dahl salt-sensitive rats: significance and mechanism. Hypertension 55: 1018–1025, 2010. doi: 10.1161/HYPERTENSIONAHA.109.147710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.García-Tejedor A, Manzanares P, Castelló-Ruiz M, Moscardó A, Marcos JF, Salom JB. Vasoactive properties of antihypertensive lactoferrin-derived peptides in resistance vessels: Effects in small mesenteric arteries from SHR rats. Life Sci 186: 118–124, 2017. doi: 10.1016/j.lfs.2017.07.036. [DOI] [PubMed] [Google Scholar]
- 185.Garcia NH, Garvin JL. Endothelin’s biphasic effect on fluid absorption in the proximal straight tubule and its inhibitory cascade. J Clin Invest 93: 2572–2577, 1994. doi: 10.1172/JCI117268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.García NH, Plato CF, Stoos BA, Garvin JL. Nitric oxide-induced inhibition of transport by thick ascending limbs from Dahl salt-sensitive rats. Hypertension 34: 508–513, 1999. doi: 10.1161/01.HYP.34.3.508. [DOI] [PubMed] [Google Scholar]
- 187.García NH, Pomposiello SI, Garvin JL. Nitric oxide inhibits ADH-stimulated osmotic water permeability in cortical collecting ducts. Am J Physiol Renal Physiol 270: F206–F210, 1996. doi: 10.1152/ajprenal.1996.270.1.F206. [DOI] [PubMed] [Google Scholar]
- 188.Gariepy CE, Ohuchi T, Williams SC, Richardson JA, Yanagisawa M. Salt-sensitive hypertension in endothelin-B receptor-deficient rats. J Clin Invest 105: 925–933, 2000. doi: 10.1172/JCI8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Garvin JL, Herrera M, Ortiz PA. Regulation of renal NaCl transport by nitric oxide, endothelin, and ATP: clinical implications. Annu Rev Physiol 73: 359–376, 2011. doi: 10.1146/annurev-physiol-012110-142247. [DOI] [PubMed] [Google Scholar]
- 190.Garvin JL, Hong NJ. Cellular stretch increases superoxide production in the thick ascending limb. Hypertension 51: 488–493, 2008. doi: 10.1161/HYPERTENSIONAHA.107.102228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Garvin JL, Hong NJ. Nitric oxide inhibits sodium/hydrogen exchange activity in the thick ascending limb. Am J Physiol Renal Physiol 277: F377–F382, 1999. doi: 10.1152/ajprenal.1999.277.3.F377. [DOI] [PubMed] [Google Scholar]
- 192.Garvin JL, Ortiz PA. The role of reactive oxygen species in the regulation of tubular function. Acta Physiol Scand 179: 225–232, 2003. doi: 10.1046/j.0001-6772.2003.01203.x. [DOI] [PubMed] [Google Scholar]
- 193.Ge Y, Bagnall A, Stricklett PK, Strait K, Webb DJ, Kotelevtsev Y, Kohan DE. Collecting duct-specific knockout of the endothelin B receptor causes hypertension and sodium retention. Am J Physiol Renal Physiol 291: F1274–F1280, 2006. doi: 10.1152/ajprenal.00190.2006. [DOI] [PubMed] [Google Scholar]
- 194.Geering K. FXYD proteins: new regulators of Na-K-ATPase. Am J Physiol Renal Physiol 290: F241–F250, 2006. doi: 10.1152/ajprenal.00126.2005. [DOI] [PubMed] [Google Scholar]
- 195.Gersch MS, Mu W, Cirillo P, Reungjui S, Zhang L, Roncal C, Sautin YY, Johnson RJ, Nakagawa T. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. Am J Physiol Renal Physiol 293: F1256–F1261, 2007. doi: 10.1152/ajprenal.00181.2007. [DOI] [PubMed] [Google Scholar]
- 196.Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, Vincent A, Lam S, Michalkiewicz M, Schilling R, Foeckler J, Kalloway S, Weiler H, Ménoret S, Anegon I, Davis GD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jacob HJ, Buelow R. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325: 433, 2009. doi: 10.1126/science.1172447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Geurts AM, Cost GJ, Rémy S, Cui X, Tesson L, Usal C, Ménoret S, Jacob HJ, Anegon I, Buelow R. Generation of gene-specific mutated rats using zinc-finger nucleases. Methods Mol Biol 597: 211–225, 2010. doi: 10.1007/978-1-60327-389-3_15. [DOI] [PubMed] [Google Scholar]
- 198.Geurts AM, Mattson DL, Liu P, Cabacungan E, Skelton MM, Kurth TM, Yang C, Endres BT, Klotz J, Liang M, Cowley AW Jr. Maternal diet during gestation and lactation modifies the severity of salt-induced hypertension and renal injury in Dahl salt-sensitive rats. Hypertension 65: 447–455, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Geurts AM, Moreno C. Zinc-finger nucleases: new strategies to target the rat genome. Clin Sci (Lond) 119: 303–311, 2010. doi: 10.1042/CS20100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Gillen CM, Brill S, Payne JA, Forbush B III. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J Biol Chem 271: 16237–16244, 1996. doi: 10.1074/jbc.271.27.16237. [DOI] [PubMed] [Google Scholar]
- 201.Giménez I, Forbush B. Short-term stimulation of the renal Na-K-Cl cotransporter (NKCC2) by vasopressin involves phosphorylation and membrane translocation of the protein. J Biol Chem 278: 26946–26951, 2003. doi: 10.1074/jbc.M303435200. [DOI] [PubMed] [Google Scholar]
- 202.Giménez I, Isenring P, Forbush B. Spatially distributed alternative splice variants of the renal Na-K-Cl cotransporter exhibit dramatically different affinities for the transported ions. J Biol Chem 277: 8767–8770, 2002. doi: 10.1074/jbc.C200021200. [DOI] [PubMed] [Google Scholar]
- 203.Gingras V, Leroux C, Fortin A, Legault L, Rabasa-Lhoret R. Predictors of cardiovascular risk among patients with type 1 diabetes: a critical analysis of the metabolic syndrome and its components. Diabetes Metab 43: 217–222, 2017. doi: 10.1016/j.diabet.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 204.Gong G, Dobin A, Johnson ML, Pettinger WA. Sexual dimorphism of renal alpha2-adrenergic receptor regulation in Dahl rats. Hypertens Res 19: 83–89, 1996. doi: 10.1291/hypres.19.83. [DOI] [PubMed] [Google Scholar]
- 205.Gonzalez-Vicente A, Cabral PD, Garvin JL. Resveratrol increases nitric oxide production in the rat thick ascending limb via Ca2+/calmodulin. PLoS One 9: e110487, 2014. doi: 10.1371/journal.pone.0110487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Gonzalez-Vicente A, Cabral PD, Hong NJ, Asirwatham J, Yang N, Berthiaume JM, Dominici FP, Garvin JL. Dietary fructose enhances the ability of low concentrations of angiotensin II to stimulate proximal tubule Na+ reabsorption. Nutrients 9: 885, 2017. doi: 10.3390/nu9080885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Gonzalez-Vicente A, Garvin JL. Angiotensin II-induced hypertension increases plasma membrane Na pump activity by enhancing Na entry in rat thick ascending limbs. Am J Physiol Renal Physiol 305: F1306–F1314, 2013. doi: 10.1152/ajprenal.00064.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Gonzalez-Vicente A, Garvin JL. Effects of reactive oxygen species on tubular transport along the nephron. Antioxidants (Basel) 6: E23, 2017. doi: 10.3390/antiox6020023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gonzalez-Vicente A, Hopfer U, Garvin JL. Developing tools for analysis of renal genomic data: an invitation to participate. J Am Soc Nephrol 28: 3438–3440, 2017. doi: 10.1681/ASN.2017070811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gonzalez-Vicente A, Saikumar JH, Massey KJ, Hong NJ, Dominici FP, Carretero OA, Garvin JL. Angiotensin II stimulates superoxide production by nitric oxide synthase in thick ascending limbs. Physiol Rep 4: 4, 2016. doi: 10.14814/phy2.12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE, McDonough AA. The absence of intrarenal ACE protects against hypertension. J Clin Invest 123: 2011–2023, 2013. doi: 10.1172/JCI65460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Good DW. PGE2 reverses AVP inhibition of in rat MTAL by activation of protein kinase C. Am J Physiol Renal Physiol 270: F978–F985, 1996. doi: 10.1152/ajprenal.1996.270.6.F978. [DOI] [PubMed] [Google Scholar]
- 213.Good DW, Caflisch CR, George T. Prostaglandin E2 regulation of ion transport is absent in medullary thick ascending limbs from SHR. Am J Physiol Renal Physiol 269: F47–F54, 1995. doi: 10.1152/ajprenal.1995.269.1.F47. [DOI] [PubMed] [Google Scholar]
- 214.Good DW, George T. Regulation of absorption by prostaglandin E2 and G proteins in rat medullary thick ascending limb. Am J Physiol Renal Physiol 270: F711–F717, 1996. doi: 10.1152/ajprenal.1996.270.5.F711. [DOI] [PubMed] [Google Scholar]
- 215.Good DW, George T, Wang DH. Angiotensin II inhibits absorption via a cytochrome P-450-dependent pathway in MTAL. Am J Physiol Renal Physiol 276: F726–F736, 1999. doi: 10.1152/ajprenal.1999.276.5.F726. [DOI] [PubMed] [Google Scholar]
- 216.Good DW, George T, Watts BA III. Aldosterone inhibits absorption via a nongenomic pathway in medullary thick ascending limb. Am J Physiol Renal Physiol 283: F699–F706, 2002. doi: 10.1152/ajprenal.00133.2002. [DOI] [PubMed] [Google Scholar]
- 217.Good DW, George T, Watts BA III. Nongenomic regulation by aldosterone of the epithelial NHE3 Na+/H+ exchanger. Am J Physiol Cell Physiol 290: C757–C763, 2006. doi: 10.1152/ajpcell.00391.2005. [DOI] [PubMed] [Google Scholar]
- 218.Good DW, Knepper MA, Burg MB. Ammonia and bicarbonate transport by thick ascending limb of rat kidney. Am J Physiol Renal Physiol 247: F35–F44, 1984. doi: 10.1152/ajprenal.1984.247.1.F35. [DOI] [PubMed] [Google Scholar]
- 219.Gopalakrishnan K, Kumarasamy S, Abdul-Majeed S, Kalinoski AL, Morgan EE, Gohara AF, Nauli SM, Filipiak WE, Saunders TL, Joe B. Targeted disruption of Adamts16 gene in a rat genetic model of hypertension. Proc Natl Acad Sci USA 109: 20555–20559, 2012. doi: 10.1073/pnas.1211290109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Graham LA, Padmanabhan S, Fraser NJ, Kumar S, Bates JM, Raffi HS, Welsh P, Beattie W, Hao S, Leh S, Hultstrom M, Ferreri NR, Dominiczak AF, Graham D, McBride MW. Validation of uromodulin as a candidate gene for human essential hypertension. Hypertension 63: 551–558, 2014. doi: 10.1161/HYPERTENSIONAHA.113.01423. [DOI] [PubMed] [Google Scholar]
- 221.Granger J, Novak J, Schnackenberg C, Williams S, Reinhart GA. Role of renal nerves in mediating the hypertensive effects of nitric oxide synthesis inhibition. Hypertension 27: 613–618, 1996. doi: 10.1161/01.HYP.27.3.613. [DOI] [PubMed] [Google Scholar]
- 222.Granger JP, Alberola AM, Salazar FJ, Nakamura T. Control of renal hemodynamics during intrarenal and systemic blockade of nitric oxide synthesis in conscious dogs. J Cardiovasc Pharmacol 20, Suppl 12: S160–S162, 1992. doi: 10.1097/00005344-199204002-00045. [DOI] [PubMed] [Google Scholar]
- 223.Granger JP, Schnackenberg CG. Renal mechanisms of angiotensin II-induced hypertension. Semin Nephrol 20: 417–425, 2000. [PubMed] [Google Scholar]
- 224.Greene AS, Yu ZY, Roman RJ, Cowley AW Jr. Role of blood volume expansion in Dahl rat model of hypertension. Am J Physiol Heart Circ Physiol 258: H508–H514, 1990. doi: 10.1152/ajpheart.1990.258.2.H508. [DOI] [PubMed] [Google Scholar]
- 225.Greger R. Cation selectivity of the isolated perfused cortical thick ascending limb of Henle’s loop of rabbit kidney. Pflugers Arch 390: 30–37, 1981. doi: 10.1007/BF00582707. [DOI] [PubMed] [Google Scholar]
- 226.Greger R, Schlatter E. Properties of the basolateral membrane of the cortical thick ascending limb of Henle’s loop of rabbit kidney. A model for secondary active chloride transport. Pflugers Arch 396: 325–334, 1983. doi: 10.1007/BF01063938. [DOI] [PubMed] [Google Scholar]
- 227.Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, Harrison DG, Bhatnagar A; American Heart Association Council on Basic Cardiovascular Sciences . Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the American Heart Association. Circ Res 119: e39–e75, 2016. doi: 10.1161/RES.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Grim CE, Luft FC, Miller JZ, Meneely GR, Battarbee HD, Hames CG, Dahl LK. Racial differences in blood pressure in Evans County, Georgia: relationship to sodium and potassium intake and plasma renin activity. J Chronic Dis 33: 87–94, 1980. doi: 10.1016/0021-9681(80)90032-6. [DOI] [PubMed] [Google Scholar]
- 229.Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J 386: 401–416, 2005. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Gu R, Jin Y, Zhai Y, Yang L, Zhang C, Li W, Wang L, Kong S, Zhang Y, Yang B, Wang WH. PGE2 inhibits basolateral 50 pS potassium channels in the thick ascending limb of the rat kidney. Kidney Int 74: 478–485, 2008. doi: 10.1038/ki.2008.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Gu RM, Wang WH. Arachidonic acid inhibits K channels in basolateral membrane of the thick ascending limb. Am J Physiol Renal Physiol 283: F407–F414, 2002. doi: 10.1152/ajprenal.00002.2002. [DOI] [PubMed] [Google Scholar]
- 232.Gu RM, Yang L, Zhang Y, Wang L, Kong S, Zhang C, Zhai Y, Wang M, Wu P, Liu L, Gu F, Zhang J, Wang WH. CYP-omega-hydroxylation-dependent metabolites of arachidonic acid inhibit the basolateral 10 pS chloride channel in the rat thick ascending limb. Kidney Int 76: 849–856, 2009. doi: 10.1038/ki.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Guinamard R, Chraïbi A, Teulon J. A small-conductance Cl− channel in the mouse thick ascending limb that is activated by ATP and protein kinase A. J Physiol 485: 97–112, 1995. doi: 10.1113/jphysiol.1995.sp020715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Gunaratne R, Braucht DW, Rinschen MM, Chou CL, Hoffert JD, Pisitkun T, Knepper MA. Quantitative phosphoproteomic analysis reveals cAMP/vasopressin-dependent signaling pathways in native renal thick ascending limb cells. Proc Natl Acad Sci USA 107: 15653–15658, 2010. doi: 10.1073/pnas.1007424107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Guntupalli J, DuBose TD Jr. Effects of endothelin on rat renal proximal tubule Na+-Pi cotransport and Na+/H+ exchange. Am J Physiol Renal Physiol 266: F658–F666, 1994. doi: 10.1152/ajprenal.1994.266.4.F658. [DOI] [PubMed] [Google Scholar]
- 236.Günzel D, Stuiver M, Kausalya PJ, Haisch L, Krug SM, Rosenthal R, Meij IC, Hunziker W, Fromm M, Müller D. Claudin-10 exists in six alternatively spliced isoforms that exhibit distinct localization and function. J Cell Sci 122: 1507–1517, 2009. doi: 10.1242/jcs.040113. [DOI] [PubMed] [Google Scholar]
- 237.Günzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev 93: 525–569, 2013. doi: 10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Gurley SB, Riquier-Brison ADM, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab 13: 469–475, 2011. doi: 10.1016/j.cmet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Gutsche HU, Peterson LN, Levine DZ. In vivo evidence of impaired solute transport by the thick ascending limb in potassium-depleted rats. J Clin Invest 73: 908–916, 1984. doi: 10.1172/JCI111314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Guyton AC. Dominant role of the kidneys and accessory role of whole-body autoregulation in the pathogenesis of hypertension. Am J Hypertens 2: 575–585, 1989. doi: 10.1093/ajh/2.7.575. [DOI] [PubMed] [Google Scholar]
- 241.Guyton AC. Physiologic regulation of arterial pressure. Am J Cardiol 8: 401–407, 1961. doi: 10.1016/0002-9149(61)90159-X. [DOI] [PubMed] [Google Scholar]
- 242.Guyton AC, Hall JE. Regulation of potassium, calcium, phosphate, and magnesium: integration of renal mechanisms for control of blood volume and extracellular fluid volumes. : Textbook of Medical Physiology. Atlanta, GA: Elsevier, 2006, p. 365–382. [Google Scholar]
- 243.Hall DA, Varney DM. Effect of vasopressin on electrical potential difference and chloride transport in mouse medullary thick ascending limb of Henle’s loop. J Clin Invest 66: 792–802, 1980. doi: 10.1172/JCI109917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Hamaguchi A, Kim S, Yano M, Yamanaka S, Iwao H. Activation of glomerular mitogen-activated protein kinases in angiotensin II-mediated hypertension. J Am Soc Nephrol 9: 372–380, 1998. [DOI] [PubMed] [Google Scholar]
- 245.Hamilton KL, Devor DC. Basolateral membrane K+ channels in renal epithelial cells. Am J Physiol Renal Physiol 302: F1069–F1081, 2012. doi: 10.1152/ajprenal.00646.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Hancock ML II, Bichet DG, Eckert GJ, Bankir L, Wagner MA, Pratt JH. Race, sex, and the regulation of urine osmolality: observations made during water deprivation. Am J Physiol Regul Integr Comp Physiol 299: R977–R980, 2010. doi: 10.1152/ajpregu.00289.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Hannan FM, Babinsky VN, Thakker RV. Disorders of the calcium-sensing receptor and partner proteins: insights into the molecular basis of calcium homeostasis. J Mol Endocrinol 57: R127–R142, 2016. doi: 10.1530/JME-16-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Hao S, Bellner L, Ferreri NR. NKCC2A and NFAT5 regulate renal TNF production induced by hypertonic NaCl intake. Am J Physiol Renal Physiol 304: F533–F542, 2013. doi: 10.1152/ajprenal.00243.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Hao S, Zhao H, Darzynkiewicz Z, Battula S, Ferreri NR. Expression and function of NFAT5 in medullary thick ascending limb (mTAL) cells. Am J Physiol Renal Physiol 296: F1494–F1503, 2009. doi: 10.1152/ajprenal.90436.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Haque MZ, Ares GR, Caceres PS, Ortiz PA. High salt differentially regulates surface NKCC2 expression in thick ascending limbs of Dahl salt-sensitive and salt-resistant rats. Am J Physiol Renal Physiol 300: F1096–F1104, 2011. doi: 10.1152/ajprenal.00600.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Haque MZ, Ares GR, Ortiz PA. Enhanced phosphodiesterase 5 (PDE5) in thick ascending limbs of Dahl salt sensitive rats blunts NO-induced inhibition of transport (Abstract). Hypertension 60: A52, 2012. [DOI] [PubMed] [Google Scholar]
- 252.Haque MZ, Caceres PS, Ortiz PA. β-Adrenergic receptor stimulation increases surface NKCC2 expression in rat thick ascending limbs in a process inhibited by phosphodiesterase 4. Am J Physiol Renal Physiol 303: F1307–F1314, 2012. doi: 10.1152/ajprenal.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Harrap SB, Wang BZ, MacLellan DG. Renal transplantation between male and female spontaneously hypertensive rats. Hypertension 19: 431–434, 1992. doi: 10.1161/01.HYP.19.5.431. [DOI] [PubMed] [Google Scholar]
- 254.Harrison-Bernard LM, El-Dahr SS, O’Leary DF, Navar LG. Regulation of angiotensin II type 1 receptor mRNA and protein in angiotensin II-induced hypertension. Hypertension 33: 340–346, 1999. doi: 10.1161/01.HYP.33.1.340. [DOI] [PubMed] [Google Scholar]
- 255.Hart TC, Gorry MC, Hart PS, Woodard AS, Shihabi Z, Sandhu J, Shirts B, Xu L, Zhu H, Barmada MM, Bleyer AJ. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet 39: 882–892, 2002. doi: 10.1136/jmg.39.12.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Harvey AP, Montezano AC, Hood KY, Lopes RA, Rios F, Ceravolo G, Graham D, Touyz RM. Vascular dysfunction and fibrosis in stroke-prone spontaneously hypertensive rats: The aldosterone-mineralocorticoid receptor-Nox1 axis. Life Sci 179: 110–119, 2017. doi: 10.1016/j.lfs.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Hayashi H, Tamura A, Krishnan D, Tsukita S, Suzuki Y, Kocinsky HS, Aronson PS, Orlowski J, Grinstein S, Alexander RT. Ezrin is required for the functional regulation of the epithelial sodium proton exchanger, NHE3. PLoS One 8: e55623, 2013. doi: 10.1371/journal.pone.0055623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Hayyan M, Hashim MA, AlNashef IM. Superoxide ion: generation and chemical implications. Chem Rev 116: 3029–3085, 2016. doi: 10.1021/acs.chemrev.5b00407. [DOI] [PubMed] [Google Scholar]
- 259.He X, Liu Y, Usa K, Tian Z, Cowley AW Jr, Liang M. Ultrastructure of mitochondria and the endoplasmic reticulum in renal tubules of Dahl salt-sensitive rats. Am J Physiol Renal Physiol 306: F1190–F1197, 2014. doi: 10.1152/ajprenal.00073.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Healy V, Thompson C, Johns EJ. The adrenergic regulation of proximal tubular Na+/H+ exchanger 3 in the rat. Acta Physiol (Oxf) 210: 678–689, 2014. doi: 10.1111/apha.12181. [DOI] [PubMed] [Google Scholar]
- 261.Hebert SC. An ATP-regulated, inwardly rectifying potassium channel from rat kidney (ROMK). Kidney Int 48: 1010–1016, 1995. doi: 10.1038/ki.1995.383. [DOI] [PubMed] [Google Scholar]
- 262.Hebert SC. Bartter syndrome. Curr Opin Nephrol Hypertens 12: 527–532, 2003. doi: 10.1097/00041552-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 263.Hebert SC, Andreoli TE. Control of NaCl transport in the thick ascending limb. Am J Physiol Renal Physiol 246: F745–F756, 1984. doi: 10.1152/ajprenal.1984.246.6.F745. [DOI] [PubMed] [Google Scholar]
- 264.Hebert SC, Culpepper RM, Andreoli TE. NaCl transport in mouse medullary thick ascending limbs. I. Functional nephron heterogeneity and ADH-stimulated NaCl cotransport. Am J Physiol Renal Physiol 241: F412–F431, 1981. doi: 10.1152/ajprenal.1981.241.4.F412. [DOI] [PubMed] [Google Scholar]
- 265.Hebert SC, Culpepper RM, Andreoli TE. NaCl transport in mouse medullary thick ascending limbs. II. ADH enhancement of transcellular NaCl cotransport; origin of transepithelial voltage. Am J Physiol Renal Physiol 241: F432–F442, 1981. doi: 10.1152/ajprenal.1981.241.4.F432. [DOI] [PubMed] [Google Scholar]
- 266.Hebert SC, Desir G, Giebisch G, Wang W. Molecular diversity and regulation of renal potassium channels. Physiol Rev 85: 319–371, 2005. doi: 10.1152/physrev.00051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Hebert SC, Sun A. Hypotonic cell volume regulation in mouse medullary thick ascending limb: effects of ADH. Am J Physiol Renal Physiol 255: F962–F969, 1988. doi: 10.1152/ajprenal.1988.255.5.F962. [DOI] [PubMed] [Google Scholar]
- 268.Hebert SC, Wang WH. Structure and function of the low conductance KATP channel, ROMK. Wien Klin Wochenschr 109: 471–476, 1997. [PubMed] [Google Scholar]
- 269.Heinz F, Schlegel F, Krause PH. Enzymes of fructose metabolism in human kidney. Enzyme 19: 85–92, 1975. doi: 10.1159/000458977. [DOI] [PubMed] [Google Scholar]
- 270.Hennings JC, Andrini O, Picard N, Paulais M, Huebner AK, Cayuqueo IK, Bignon Y, Keck M, Cornière N, Böhm D, Jentsch TJ, Chambrey R, Teulon J, Hübner CA, Eladari D. The ClC-K2 chloride channel is critical for salt handling in the distal nephron. J Am Soc Nephrol 28: 209–217, 2017. doi: 10.1681/ASN.2016010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Herrera M, Garvin JL. Angiotensin II stimulates thick ascending limb NO production via AT(2) receptors and Akt1-dependent nitric-oxide synthase 3 (NOS3) activation. J Biol Chem 285: 14932–14940, 2010. doi: 10.1074/jbc.M110.109041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Herrera M, Garvin JL. Aquaporins as gas channels. Pflugers Arch 462: 623–630, 2011. doi: 10.1007/s00424-011-1002-x. [DOI] [PubMed] [Google Scholar]
- 273.Herrera M, Garvin JL. Endothelin stimulates endothelial nitric oxide synthase expression in the thick ascending limb. Am J Physiol Renal Physiol 287: F231–F235, 2004. doi: 10.1152/ajprenal.00413.2003. [DOI] [PubMed] [Google Scholar]
- 274.Herrera M, Garvin JL. A high-salt diet stimulates thick ascending limb eNOS expression by raising medullary osmolality and increasing release of endothelin-1. Am J Physiol Renal Physiol 288: F58–F64, 2005. doi: 10.1152/ajprenal.00209.2004. [DOI] [PubMed] [Google Scholar]
- 275.Herrera M, Garvin JL. Novel role of AQP-1 in NO-dependent vasorelaxation. Am J Physiol Renal Physiol 292: F1443–F1451, 2007. doi: 10.1152/ajprenal.00353.2006. [DOI] [PubMed] [Google Scholar]
- 276.Herrera M, Hong NJ, Garvin JL. Aquaporin-1 transports NO across cell membranes. Hypertension 48: 157–164, 2006. doi: 10.1161/01.HYP.0000223652.29338.77. [DOI] [PubMed] [Google Scholar]
- 277.Herrera M, Hong NJ, Ortiz PA, Garvin JL. Endothelin-1 inhibits thick ascending limb transport via Akt-stimulated nitric oxide production. J Biol Chem 284: 1454–1460, 2009. doi: 10.1074/jbc.M804322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Herrera M, Silva G, Garvin JL. A high-salt diet dissociates NO synthase-3 expression and NO production by the thick ascending limb. Hypertension 47: 95–101, 2006. doi: 10.1161/01.HYP.0000196274.78603.85. [DOI] [PubMed] [Google Scholar]
- 279.Herrera M, Silva GB, Garvin JL. Angiotensin II stimulates thick ascending limb superoxide production via protein kinase C(α)-dependent NADPH oxidase activation. J Biol Chem 285: 21323–21328, 2010. doi: 10.1074/jbc.M110.109157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Herrera VL, Lopez LV, Ruiz-Opazo N. Alpha1 Na,K-ATPase and Na,K,2Cl−cotransporte/D3mit3 loci interact to increase susceptibility to salt-sensitive hypertension in Dahl S(HSD) rats. Mol Med 7: 125–134, 2001. doi: 10.1007/BF03401946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90: 291–366, 2010. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 282.Higashi M, Shimokawa H, Hattori T, Hiroki J, Mukai Y, Morikawa K, Ichiki T, Takahashi S, Takeshita A. Long-term inhibition of Rho-kinase suppresses angiotensin II-induced cardiovascular hypertrophy in rats in vivo: effect on endothelial NAD(P)H oxidase system. Circ Res 93: 767–775, 2003. doi: 10.1161/01.RES.0000096650.91688.28. [DOI] [PubMed] [Google Scholar]
- 283.Hilliard LM, Chow CL, Mirabito KM, Steckelings UM, Unger T, Widdop RE, Denton KM. Angiotensin type 2 receptor stimulation increases renal function in female, but not male, spontaneously hypertensive rats. Hypertension 64: 378–383, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02809. [DOI] [PubMed] [Google Scholar]
- 284.Hingtgen SD, Tian X, Yang J, Dunlay SM, Peek AS, Wu Y, Sharma RV, Engelhardt JF, Davisson RL. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics 26: 180–191, 2006. doi: 10.1152/physiolgenomics.00029.2005. [DOI] [PubMed] [Google Scholar]
- 285.Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature 362: 31–38, 1993. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- 286.Hoagland KM, Flasch AK, Dahly-Vernon AJ, dos Santos EA, Knepper MA, Roman RJ. Elevated BSC-1 and ROMK expression in Dahl salt-sensitive rat kidneys. Hypertension 43: 860–865, 2004. doi: 10.1161/01.HYP.0000120123.44945.47. [DOI] [PubMed] [Google Scholar]
- 287.Hoagland KM, Flasch AK, Roman RJ. Inhibitors of 20-HETE formation promote salt-sensitive hypertension in rats. Hypertension 42: 669–673, 2003. doi: 10.1161/01.HYP.0000084634.97353.1A. [DOI] [PubMed] [Google Scholar]
- 288.Hoagland KM, Maier KG, Roman RJ. Contributions of 20-HETE to the antihypertensive effects of Tempol in Dahl salt-sensitive rats. Hypertension 41: 697–702, 2003. doi: 10.1161/01.HYP.0000047881.15426.DC. [DOI] [PubMed] [Google Scholar]
- 289.Holstein-Rathlou NH, Kanters JK, Leyssac PP. Exaggerated natriuresis and lithium clearance in spontaneously hypertensive rats. J Hypertens 6: 889–895, 1988. doi: 10.1097/00004872-198811000-00007. [DOI] [PubMed] [Google Scholar]
- 290.Holterman CE, Read NC, Kennedy CR. Nox and renal disease. Clin Sci (Lond) 128: 465–481, 2015. doi: 10.1042/CS20140361. [DOI] [PubMed] [Google Scholar]
- 291.Honegger KJ, Capuano P, Winter C, Bacic D, Stange G, Wagner CA, Biber J, Murer H, Hernando N. Regulation of sodium-proton exchanger isoform 3 (NHE3) by PKA and exchange protein directly activated by cAMP (EPAC). Proc Natl Acad Sci USA 103: 803–808, 2006. doi: 10.1073/pnas.0503562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Hong NJ, Garvin JL. Angiotensin II type 2 receptor-mediated inhibition of NaCl absorption is blunted in thick ascending limbs from Dahl salt-sensitive rats. Hypertension 60: 765–769, 2012. doi: 10.1161/HYPERTENSIONAHA.112.199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Hong NJ, Garvin JL. Endogenous flow-induced superoxide stimulates Na/H exchange activity via PKC in thick ascending limbs. Am J Physiol Renal Physiol 307: F800–F805, 2014. doi: 10.1152/ajprenal.00260.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Hong NJ, Garvin JL. Flow increases superoxide production by NADPH oxidase via activation of Na-K-2Cl cotransport and mechanical stress in thick ascending limbs. Am J Physiol Renal Physiol 292: F993–F998, 2007. doi: 10.1152/ajprenal.00383.2006. [DOI] [PubMed] [Google Scholar]
- 295.Hong NJ, Garvin JL. NADPH oxidase 4 mediates flow-induced superoxide production in thick ascending limbs. Am J Physiol Renal Physiol 303: F1151–F1156, 2012. doi: 10.1152/ajprenal.00181.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Hong NJ, Garvin JL. Nitric oxide reduces flow-induced superoxide production via cGMP-dependent protein kinase in thick ascending limbs. Am J Physiol Renal Physiol 296: F1061–F1066, 2009. doi: 10.1152/ajprenal.90707.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Hong NJ, Silva GB, Garvin JL. PKC-alpha mediates flow-stimulated superoxide production in thick ascending limbs. Am J Physiol Renal Physiol 298: F885–F891, 2010. doi: 10.1152/ajprenal.00543.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Hou J, Kato H, Cohen RA, Chobanian AV, Brecher P. Angiotensin II-induced cardiac fibrosis in the rat is increased by chronic inhibition of nitric oxide synthase. J Clin Invest 96: 2469–2477, 1995. doi: 10.1172/JCI118305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Hou J, Shan Q, Wang T, Gomes AS, Yan Q, Paul DL, Bleich M, Goodenough DA. Transgenic RNAi depletion of claudin-16 and the renal handling of magnesium. J Biol Chem 282: 17114–17122, 2007. doi: 10.1074/jbc.M700632200. [DOI] [PubMed] [Google Scholar]
- 300.Hou J, Renigunta A, Gomes AS, Hou M, Paul DL, Waldegger S, Goodenough DA. Claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc Natl Acad Sci USA 106: 15350–15355, 2009. doi: 10.1073/pnas.0907724106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Hu L, Manning RD Jr. Role of nitric oxide in regulation of long-term pressure-natriuresis relationship in Dahl rats. Am J Physiol Heart Circ Physiol 268: H2375–H2383, 1995. doi: 10.1152/ajpheart.1995.268.6.H2375. [DOI] [PubMed] [Google Scholar]
- 302.Huang DY, Boini KM, Friedrich B, Metzger M, Just L, Osswald H, Wulff P, Kuhl D, Vallon V, Lang F. Blunted hypertensive effect of combined fructose and high-salt diet in gene-targeted mice lacking functional serum- and glucocorticoid-inducible kinase SGK1. Am J Physiol Regul Integr Comp Physiol 290: R935–R944, 2006. doi: 10.1152/ajpregu.00382.2005. [DOI] [PubMed] [Google Scholar]
- 303.Hughes AK, Cline RC, Kohan DE. Alterations in renal endothelin-1 production in the spontaneously hypertensive rat. Hypertension 20: 666–673, 1992. doi: 10.1161/01.HYP.20.5.666. [DOI] [PubMed] [Google Scholar]
- 304.Hwang IS, Ho H, Hoffman BB, Reaven GM. Fructose-induced insulin resistance and hypertension in rats. Hypertension 10: 512–516, 1987. doi: 10.1161/01.HYP.10.5.512. [DOI] [PubMed] [Google Scholar]
- 305.Hyndman KA, Dugas C, Arguello AM, Goodchild TT, Buckley KM, Burch M, Yanagisawa M, Pollock JS. High salt induces autocrine actions of ET-1 on inner medullary collecting duct NO production via upregulated ETB receptor expression. Am J Physiol Regul Integr Comp Physiol 311: R263–R271, 2016. doi: 10.1152/ajpregu.00016.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Igarashi P, Vanden Heuvel GB, Payne JA, Forbush B III. Cloning, embryonic expression, and alternative splicing of a murine kidney-specific Na-K-Cl cotransporter. Am J Physiol Renal Physiol 269: F405–F418, 1995. doi: 10.1152/ajprenal.1995.269.3.F405. [DOI] [PubMed] [Google Scholar]
- 307.Imbert-Teboul M, Chabardès D, Montégut M, Clique A, Morel F. Vasopressin-dependent adenylate cyclase activities in the rat kidney medulla: evidence for two separate sites of action. Endocrinology 102: 1254–1261, 1978. doi: 10.1210/endo-102-4-1254. [DOI] [PubMed] [Google Scholar]
- 308.Imbert M, Chabardès D, Montegut M, Clique A, Morel F. Vasopressin dependent adenylate cyclase in single segments of rabbit kidney tubule. Pflugers Arch 357: 173–186, 1975. doi: 10.1007/BF00585973. [DOI] [PubMed] [Google Scholar]
- 309.Inoue A, Yanagisawa M, Kimura S, Kasuya Y, Miyauchi T, Goto K, Masaki T. The human endothelin family: three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc Natl Acad Sci USA 86: 2863–2867, 1989. doi: 10.1073/pnas.86.8.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Intengan HD, Smyth DD. Alpha-2a/d adrenoceptor subtype stimulation by guanfacine increases osmolar clearance. J Pharmacol Exp Ther 281: 48–53, 1997. [PubMed] [Google Scholar]
- 311.Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, Smith AV, Tobin MD, Verwoert GC, Hwang SJ, Pihur V, Vollenweider P, O’Reilly PF, Amin N, Bragg-Gresham JL, Teumer A, Glazer NL, Launer L, Zhao JH, Aulchenko Y, Heath S, Sõber S, Parsa A, Luan J, Arora P, , et al. ; International Consortium for Blood Pressure Genome-Wide Association Studies; CARDIoGRAM consortium; CKDGen Consortium; KidneyGen Consortium; EchoGen consortium; CHARGE-HF consortium . Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 478: 103–109, 2011. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Isacoff EY, Jan YN, Jan LY. Evidence for the formation of heteromultimeric potassium channels in Xenopus oocytes. Nature 345: 530–534, 1990. doi: 10.1038/345530a0. [DOI] [PubMed] [Google Scholar]
- 313.Ito O, Roman RJ. Role of 20-HETE in elevating chloride transport in the thick ascending limb of Dahl SS/Jr rats. Hypertension 33: 419–423, 1999. doi: 10.1161/01.HYP.33.1.419. [DOI] [PubMed] [Google Scholar]
- 314.Itoh Y, Yanagisawa M, Ohkubo S, Kimura C, Kosaka T, Inoue A, Ishida N, Mitsui Y, Onda H, Fujino M, Masaki T. Cloning and sequence analysis of cDNA encoding the precursor of a human endothelium-derived vasoconstrictor peptide, endothelin: identity of human and porcine endothelin. FEBS Lett 231: 440–444, 1988. doi: 10.1016/0014-5793(88)80867-6. [DOI] [PubMed] [Google Scholar]
- 315.Iwai J, Dahl LK, Knudsen KD. Genetic influence on the renin-angiotensin system: low renin activities in hypertension-prone rats. Circ Res 32: 678–684, 1973. doi: 10.1161/01.RES.32.6.678. [DOI] [PubMed] [Google Scholar]
- 316.Jakobsson B, Aperia A. High protein intake accelerates the maturation of Na,K-ATPase in rat renal tubules. Acta Physiol Scand 139: 1–7, 1990. doi: 10.1111/j.1748-1716.1990.tb08890.x. [DOI] [PubMed] [Google Scholar]
- 317.James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, Lackland DT, LeFevre ML, MacKenzie TD, Ogedegbe O, Smith SC Jr, Svetkey LP, Taler SJ, Townsend RR, Wright JT Jr, Narva AS, Ortiz E. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 311: 507–520, 2014. doi: 10.1001/jama.2013.284427. [DOI] [PubMed] [Google Scholar]
- 318.Jan LY, Jan YN. Potassium channels and their evolving gates. Nature 371: 119–122, 1994. doi: 10.1038/371119a0. [DOI] [PubMed] [Google Scholar]
- 319.Jang SW, Ihm SH, Choo EH, Kim OR, Chang K, Park CS, Kim HY, Seung KB. Imatinib mesylate attenuates myocardial remodeling through inhibition of platelet-derived growth factor and transforming growth factor activation in a rat model of hypertension. Hypertension 63: 1228–1234, 2014. doi: 10.1161/HYPERTENSIONAHA.113.01866. [DOI] [PubMed] [Google Scholar]
- 320.Jaykumar AB, Caceres PS, Sablaban I, Tannous BA, Ortiz PA. Real-time monitoring of NKCC2 endocytosis by total internal reflection fluorescence (TIRF) microscopy. Am J Physiol Renal Physiol 310: F183–F191, 2016. doi: 10.1152/ajprenal.00104.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Jeck N, Waldegger S, Lampert A, Boehmer C, Waldegger P, Lang PA, Wissinger B, Friedrich B, Risler T, Moehle R, Lang UE, Zill P, Bondy B, Schaeffeler E, Asante-Poku S, Seyberth H, Schwab M, Lang F. Activating mutation of the renal epithelial chloride channel ClC-Kb predisposing to hypertension. Hypertension 43: 1175–1181, 2004. doi: 10.1161/01.HYP.0000129824.12959.f0. [DOI] [PubMed] [Google Scholar]
- 322.Jensen ME, Odgaard E, Christensen MH, Praetorius HA, Leipziger J. Flow-induced [Ca2+]i increase depends on nucleotide release and subsequent purinergic signaling in the intact nephron. J Am Soc Nephrol 18: 2062–2070, 2007. doi: 10.1681/ASN.2006070700. [DOI] [PubMed] [Google Scholar]
- 323.Jiang J, Stec DE, Drummond H, Simon JS, Koike G, Jacob HJ, Roman RJ. Transfer of a salt-resistant renin allele raises blood pressure in Dahl salt-sensitive rats. Hypertension 29: 619–627, 1997. doi: 10.1161/01.HYP.29.2.619. [DOI] [PubMed] [Google Scholar]
- 324.Jin C, Sun J, Stilphen CA, Smith SM, Ocasio H, Bermingham B, Darji S, Guha A, Patel R, Geurts AM, Jacob HJ, Lambert NA, O’Connor PM. HV1 acts as a sodium sensor and promotes superoxide production in medullary thick ascending limb of Dahl salt-sensitive rats. Hypertension 64: 541–550, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, Lee YS, Jeong KS, Kim WB, Park JW, Song BJ, Huh TL. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J Biol Chem 276: 16168–16176, 2001. doi: 10.1074/jbc.M010120200. [DOI] [PubMed] [Google Scholar]
- 326.Johnson DK, Schillinger KJ, Kwait DM, Hughes CV, McNamara EJ, Ishmael F, O’Donnell RW, Chang MM, Hogg MG, Dordick JS, Santhanam L, Ziegler LM, Holland JA. Inhibition of NADPH oxidase activation in endothelial cells by ortho-methoxy-substituted catechols. Endothelium 9: 191–203, 2002. doi: 10.1080/10623320213638. [DOI] [PubMed] [Google Scholar]
- 327.Johnson ML, Ely DL, Turner ME. Genetic divergence between the Wistar-Kyoto rat and the spontaneously hypertensive rat. Hypertension 19: 425–427, 1992. doi: 10.1161/01.HYP.19.5.425. [DOI] [PubMed] [Google Scholar]
- 328.Judy WV, Farrell SK. Arterial baroreceptor reflex control of sympathetic nerve activity in the spontaneously hypertensive rat. Hypertension 1: 605–614, 1979. doi: 10.1161/01.HYP.1.6.605. [DOI] [PubMed] [Google Scholar]
- 329.Judy WV, Watanabe AM, Henry DP, Besch HR Jr, Murphy WR, Hockel GM. Sympathetic nerve activity: role in regulation of blood pressure in the spontaenously hypertensive rat. Circ Res 38, Suppl 2: 21–29, 1976. doi: 10.1161/01.RES.38.6.21. [DOI] [PubMed] [Google Scholar]
- 330.Juncos R, Garvin JL. Superoxide enhances Na-K-2Cl cotransporter activity in the thick ascending limb. Am J Physiol Renal Physiol 288: F982–F987, 2005. doi: 10.1152/ajprenal.00348.2004. [DOI] [PubMed] [Google Scholar]
- 331.Juncos R, Hong NJ, Garvin JL. Differential effects of superoxide on luminal and basolateral Na+/H+ exchange in the thick ascending limb. Am J Physiol Regul Integr Comp Physiol 290: R79–R83, 2006. doi: 10.1152/ajpregu.00447.2005. [DOI] [PubMed] [Google Scholar]
- 332.Jung J, Basile DP, Pratt JH. Sodium reabsorption in the thick ascending limb in relation to blood pressure: a clinical perspective. Hypertension 57: 873–879, 2011. doi: 10.1161/HYPERTENSIONAHA.108.120246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Jung J, Foroud TM, Eckert GJ, Flury-Wetherill L, Edenberg HJ, Xuei X, Zaidi SA, Pratt JH. Association of the calcium-sensing receptor gene with blood pressure and urinary calcium in African-Americans. J Clin Endocrinol Metab 94: 1042–1048, 2009. doi: 10.1210/jc.2008-1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Jung JS, Preston GM, Smith BL, Guggino WB, Agre P. Molecular structure of the water channel through aquaporin CHIP. The hourglass model. J Biol Chem 269: 14648–14654, 1994. [PubMed] [Google Scholar]
- 335.Jung JY, Song JH, Li C, Yang CW, Kang TC, Won MH, Jeong YG, Han KH, Choi KB, Lee SH, Kim J. Expression of epidermal growth factor in the developing rat kidney. Am J Physiol Renal Physiol 288: F227–F235, 2005. doi: 10.1152/ajprenal.00058.2004. [DOI] [PubMed] [Google Scholar]
- 336.Juraschek SP, Appel LJ, Anderson CA, Miller ER III. Effect of a high-protein diet on kidney function in healthy adults: results from the OmniHeart trial. Am J Kidney Dis 61: 547–554, 2013. doi: 10.1053/j.ajkd.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Kaji DM. Effect of membrane potential on K-Cl transport in human erythrocytes. Am J Physiol Cell Physiol 264: C376–C382, 1993. doi: 10.1152/ajpcell.1993.264.2.C376. [DOI] [PubMed] [Google Scholar]
- 338.Kaji DM, Chase HS Jr, Eng JP, Diaz J. Prostaglandin E2 inhibits Na-K-2Cl cotransport in medullary thick ascending limb cells. Am J Physiol Cell Physiol 271: C354–C361, 1996. doi: 10.1152/ajpcell.1996.271.1.C354. [DOI] [PubMed] [Google Scholar]
- 339.Kase H, Hashikabe Y, Uchida K, Nakanishi N, Hattori Y. Supplementation with tetrahydrobiopterin prevents the cardiovascular effects of angiotensin II-induced oxidative and nitrosative stress. J Hypertens 23: 1375–1382, 2005. doi: 10.1097/01.hjh.0000173520.13976.7d. [DOI] [PubMed] [Google Scholar]
- 340.Kassem KM, Ares G, Ortiz P. A role for sweet taste receptors (T1R2/T1R3) in fructose-induced increase in NKCC2 Phosphorylation in thick ascending limbs (TALs) (Abstract). FASEB J 31, Suppl 1: 853.852, 2017.28246297 [Google Scholar]
- 341.Katholi RE. Renal nerves in the pathogenesis of hypertension in experimental animals and humans. Am J Physiol Renal Physiol 245: F1–F14, 1983. doi: 10.1152/ajprenal.1983.245.1.F1. [DOI] [PubMed] [Google Scholar]
- 342.Kawakami K, Nagano K. The transmembrane segment of the human Na,K-ATPase beta-subunit acts as the membrane incorporation signal. J Biochem 103: 54–60, 1988. doi: 10.1093/oxfordjournals.jbchem.a122239. [DOI] [PubMed] [Google Scholar]
- 343.Kennedy-Lydon T, Crawford C, Wildman SS, Peppiatt-Wildman CM. Nonsteroidal anti-inflammatory drugs alter vasa recta diameter via pericytes. Am J Physiol Renal Physiol 309: F648–F657, 2015. doi: 10.1152/ajprenal.00199.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Kieferle S, Fong P, Bens M, Vandewalle A, Jentsch TJ. Two highly homologous members of the ClC chloride channel family in both rat and human kidney. Proc Natl Acad Sci USA 91: 6943–6947, 1994. doi: 10.1073/pnas.91.15.6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Kirchner KA. Greater loop chloride uptake contributes to blunted pressure natriuresis in Dahl salt sensitive rats. J Am Soc Nephrol 1: 180–186, 1990. [DOI] [PubMed] [Google Scholar]
- 346.Kirchner KA. Increased loop chloride uptake precedes hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 262: R263–R268, 1992. doi: 10.1152/ajpregu.1992.262.2.R263. [DOI] [PubMed] [Google Scholar]
- 347.Kirchner KA, Crosby BA, Patel AR, Granger JP. Segmental chloride transport in the Dahl-S rat kidney during L-arginine administration. J Am Soc Nephrol 5: 1567–1572, 1995. [DOI] [PubMed] [Google Scholar]
- 348.Kiroytcheva M, Cheval L, Carranza ML, Martin PY, Favre H, Doucet A, Féraille E. Effect of cAMP on the activity and the phosphorylation of Na+,K+-ATPase in rat thick ascending limb of Henle. Kidney Int 55: 1819–1831, 1999. doi: 10.1046/j.1523-1755.1999.00414.x. [DOI] [PubMed] [Google Scholar]
- 349.Kittikulsuth W, Pollock JS, Pollock DM. Sex differences in renal medullary endothelin receptor function in angiotensin II hypertensive rats. Hypertension 58: 212–218, 2011. doi: 10.1161/HYPERTENSIONAHA.111.172734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Kiuchi-Saishin Y, Gotoh S, Furuse M, Takasuga A, Tano Y, Tsukita S. Differential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segments. J Am Soc Nephrol 13: 875–886, 2002. [DOI] [PubMed] [Google Scholar]
- 351.Klein JD, Murrell BP, Tucker S, Kim YH, Sands JM. Urea transporter UT-A1 and aquaporin-2 proteins decrease in response to angiotensin II or norepinephrine-induced acute hypertension. Am J Physiol Renal Physiol 291: F952–F959, 2006. doi: 10.1152/ajprenal.00173.2006. [DOI] [PubMed] [Google Scholar]
- 352.Kobayashi K, Uchida S, Mizutani S, Sasaki S, Marumo F. Intrarenal and cellular localization of CLC-K2 protein in the mouse kidney. J Am Soc Nephrol 12: 1327–1334, 2001. [DOI] [PubMed] [Google Scholar]
- 353.Kobori H, Harrison-Bernard LM, Navar LG. Urinary excretion of angiotensinogen reflects intrarenal angiotensinogen production. Kidney Int 61: 579–585, 2002. doi: 10.1046/j.1523-1755.2002.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Kocinsky HS, Dynia DW, Wang T, Aronson PS. NHE3 phosphorylation at serines 552 and 605 does not directly affect NHE3 activity. Am J Physiol Renal Physiol 293: F212–F218, 2007. doi: 10.1152/ajprenal.00042.2007. [DOI] [PubMed] [Google Scholar]
- 355.Kohan DE. Endothelin synthesis by rabbit renal tubule cells. Am J Physiol Renal Physiol 261: F221–F226, 1991. doi: 10.1152/ajprenal.1991.261.2.F221. [DOI] [PubMed] [Google Scholar]
- 356.Kohan DE. The renal medullary endothelin system in control of sodium and water excretion and systemic blood pressure. Curr Opin Nephrol Hypertens 15: 34–40, 2006. doi: 10.1097/01.mnh.0000186852.15889.1a. [DOI] [PubMed] [Google Scholar]
- 357.Kohan DE, Padilla E. Endothelin-1 is an autocrine factor in rat inner medullary collecting ducts. Am J Physiol Renal Physiol 263: F607–F612, 1992. doi: 10.1152/ajprenal.1992.263.4.F607. [DOI] [PubMed] [Google Scholar]
- 358.Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev 91: 1–77, 2011. doi: 10.1152/physrev.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, Vitzthum H, Suzuki Y, Luk JM, Becker C, Schlingmann KP, Schmid M, Rodriguez-Soriano J, Ariceta G, Cano F, Enriquez R, Juppner H, Bakkaloglu SA, Hediger MA, Gallati S, Neuhauss SC, Nurnberg P, Weber S. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 79: 949–957, 2006. doi: 10.1086/508617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Kopkan L, Castillo A, Navar LG, Majid DS. Enhanced superoxide generation modulates renal function in ANG II-induced hypertensive rats. Am J Physiol Renal Physiol 290: F80–F86, 2006. doi: 10.1152/ajprenal.00090.2005. [DOI] [PubMed] [Google Scholar]
- 361.Kortenoeven ML, Pedersen NB, Rosenbaek LL, Fenton RA. Vasopressin regulation of sodium transport in the distal nephron and collecting duct. Am J Physiol Renal Physiol 309: F280–F299, 2015. doi: 10.1152/ajprenal.00093.2015. [DOI] [PubMed] [Google Scholar]
- 362.Krause G, Winkler L, Piehl C, Blasig I, Piontek J, Müller SL. Structure and function of extracellular claudin domains. Ann N Y Acad Sci 1165: 34–43, 2009. doi: 10.1111/j.1749-6632.2009.04057.x. [DOI] [PubMed] [Google Scholar]
- 363.Krishna GG, Miller E, Kapoor S. Increased blood pressure during potassium depletion in normotensive men N Engl J Med 320: 1177–1182, 1989. doi: 10.1056/NEJM198905043201804. [DOI] [PubMed] [Google Scholar]
- 364.Krum H, Schlaich M, Sobotka P. Renal sympathetic nerve ablation for treatment-resistant hypertension. Br J Clin Pharmacol 76: 495–503, 2013. doi: 10.1111/bcp.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Kumagai H, Averill DB, Khosla MC, Ferrario CM. Role of nitric oxide and angiotensin II in the regulation of sympathetic nerve activity in spontaneously hypertensive rats. Hypertension 21: 476–484, 1993. doi: 10.1161/01.HYP.21.4.476. [DOI] [PubMed] [Google Scholar]
- 366.Kunert MP, Drenjancevic-Peric I, Dwinell MR, Lombard JH, Cowley AW Jr, Greene AS, Kwitek AE, Jacob HJ. Consomic strategies to localize genomic regions related to vascular reactivity in the Dahl salt-sensitive rat. Physiol Genomics 26: 218–225, 2006. doi: 10.1152/physiolgenomics.00004.2006. [DOI] [PubMed] [Google Scholar]
- 367.Kuroda J, Ago T, Nishimura A, Nakamura K, Matsuo R, Wakisaka Y, Kamouchi M, Kitazono T. Nox4 is a major source of superoxide production in human brain pericytes. J Vasc Res 51: 429–438, 2014. doi: 10.1159/000369930. [DOI] [PubMed] [Google Scholar]
- 368.Kuroda J, Nakagawa K, Yamasaki T, Nakamura K, Takeya R, Kuribayashi F, Imajoh-Ohmi S, Igarashi K, Shibata Y, Sueishi K, Sumimoto H. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 10: 1139–1151, 2005. doi: 10.1111/j.1365-2443.2005.00907.x. [DOI] [PubMed] [Google Scholar]
- 369.Kuster B, Shainskaya A, Pu HX, Goldshleger R, Blostein R, Mann M, Karlish SJ. A new variant of the gamma subunit of renal Na,K-ATPase. Identification by mass spectrometry, antibody binding, and expression in cultured cells. J Biol Chem 275: 18441–18446, 2000. doi: 10.1074/jbc.M001411200. [DOI] [PubMed] [Google Scholar]
- 370.Kwon TH, Nielsen J, Kim YH, Knepper MA, Frøkiaer J, Nielsen S. Regulation of sodium transporters in the thick ascending limb of rat kidney: response to angiotensin II. Am J Physiol Renal Physiol 285: F152–F165, 2003. doi: 10.1152/ajprenal.00307.2002. [DOI] [PubMed] [Google Scholar]
- 371.Laakso J, Mervaala E, Himberg JJ, Teräväinen TL, Karppanen H, Vapaatalo H, Lapatto R. Increased kidney xanthine oxidoreductase activity in salt-induced experimental hypertension. Hypertension 32: 902–906, 1998. doi: 10.1161/01.HYP.32.5.902. [DOI] [PubMed] [Google Scholar]
- 372.Lahera V, Salom MG, Miranda-Guardiola F, Moncada S, Romero JC. Effects of NG-nitro-L-arginine methyl ester on renal function and blood pressure. Am J Physiol Renal Physiol 261: F1033–F1037, 1991. doi: 10.1152/ajprenal.1991.261.6.F1033. [DOI] [PubMed] [Google Scholar]
- 373.Lal-Nag M, Morin PJ. The claudins. Genome Biol 10: 235, 2009. doi: 10.1186/gb-2009-10-8-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, Tarpey M, Fukai T, Harrison DG. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 103: 1282–1288, 2001. doi: 10.1161/01.CIR.103.9.1282. [DOI] [PubMed] [Google Scholar]
- 375.Le Moellic C, Boulkroun S, González-Nunez D, Dublineau I, Cluzeaud F, Fay M, Blot-Chabaud M, Farman N. Aldosterone and tight junctions: modulation of claudin-4 phosphorylation in renal collecting duct cells. Am J Physiol Cell Physiol 289: C1513–C1521, 2005. doi: 10.1152/ajpcell.00314.2005. [DOI] [PubMed] [Google Scholar]
- 376.Le MT, Frye RF, Rivard CJ, Cheng J, McFann KK, Segal MS, Johnson RJ, Johnson JA. Effects of high-fructose corn syrup and sucrose on the pharmacokinetics of fructose and acute metabolic and hemodynamic responses in healthy subjects. Metabolism 61: 641–651, 2012. doi: 10.1016/j.metabol.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Lee JW, Chou CL, Knepper MA. Deep Sequencing in Microdissected Renal Tubules Identifies Nephron Segment-Specific Transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Leipziger J, MacGregor GG, Cooper GJ, Xu J, Hebert SC, Giebisch G. PKA site mutations of ROMK2 channels shift the pH dependence to more alkaline values. Am J Physiol Renal Physiol 279: F919–F926, 2000. doi: 10.1152/ajprenal.2000.279.5.F919. [DOI] [PubMed] [Google Scholar]
- 379.Lerolle N, Bourgeois S, Leviel F, Lebrun G, Paillard M, Houillier P. Angiotensin II inhibits NaCl absorption in the rat medullary thick ascending limb. Am J Physiol Renal Physiol 287: F404–F410, 2004. doi: 10.1152/ajprenal.00265.2003. [DOI] [PubMed] [Google Scholar]
- 380.Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, Barhanin J. TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J 15: 1004–1011, 1996. doi: 10.1002/j.1460-2075.1996.tb00437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Levine DZ, Burns KD, Jaffey J, Iacovitti M. Short-term modulation of distal tubule fluid nitric oxide in vivo by loop NaCl reabsorption. Kidney Int 65: 184–189, 2004. doi: 10.1111/j.1523-1755.2004.00361.x. [DOI] [PubMed] [Google Scholar]
- 382.Li J, Wang DH. Role of TRPV1 channels in renal haemodynamics and function in Dahl salt-sensitive hypertensive rats. Exp Physiol 93: 945–953, 2008. doi: 10.1113/expphysiol.2008.042036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Li N, Yi FX, Spurrier JL, Bobrowitz CA, Zou AP. Production of superoxide through NADH oxidase in thick ascending limb of Henle’s loop in rat kidney. Am J Physiol Renal Physiol 282: F1111–F1119, 2002. doi: 10.1152/ajprenal.00218.2001. [DOI] [PubMed] [Google Scholar]
- 385.Liang B, Leenen FH. Prevention of salt induced hypertension and fibrosis by angiotensin converting enzyme inhibitors in Dahl S rats. Br J Pharmacol 152: 903–914, 2007. doi: 10.1038/sj.bjp.0707472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Liang M, Yuan B, Rute E, Greene AS, Olivier M, Cowley AW Jr. Insights into Dahl salt-sensitive hypertension revealed by temporal patterns of renal medullary gene expression. Physiol Genomics 12: 229–237, 2003. doi: 10.1152/physiolgenomics.00089.2002. [DOI] [PubMed] [Google Scholar]
- 387.Liao TD, Yang XP, Liu YH, Shesely EG, Cavasin MA, Kuziel WA, Pagano PJ, Carretero OA. Role of inflammation in the development of renal damage and dysfunction in angiotensin II-induced hypertension. Hypertension 52: 256–263, 2008. doi: 10.1161/HYPERTENSIONAHA.108.112706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 9: 861–871, 1992. doi: 10.1016/0896-6273(92)90239-A. [DOI] [PubMed] [Google Scholar]
- 389.Lin Z, Jin S, Duan X, Wang T, Martini S, Hulamm P, Cha B, Hubbard A, Donowitz M, Guggino SE. Chloride channel (Clc)-5 is necessary for exocytic trafficking of Na+/H+ exchanger 3 (NHE3). J Biol Chem 286: 22833–22845, 2011. doi: 10.1074/jbc.M111.224998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Liška F, Landa V, Zídek V, Mlejnek P, Šilhavý J, Šimáková M, Strnad H, Trnovská J, Škop V, Kazdová L, Starker CG, Voytas DF, Izsvák Z, Mancini M, Šeda O, Křen V, Pravenec M. Downregulation of Plzf gene ameliorates metabolic and cardiac traits in the spontaneously hypertensive rat. Hypertension 69: 1084–1091, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08798. [DOI] [PubMed] [Google Scholar]
- 391.Liu HJ, Wei Y, Ferreri NR, Nasjletti A, Wang WH. Vasopressin and PGE(2) regulate activity of apical 70 pS K+ channel in thick ascending limb of rat kidney [Erratum in Am J Physiol Cell Physiol 279: Ca1, 2000.] Am J Physiol Cell Physiol 278: C905–C913, 2000. doi: 10.1152/ajpcell.2000.278.5.C905. [DOI] [PubMed] [Google Scholar]
- 392.Liu R, Carretero OA, Ren Y, Garvin JL. Increased intracellular pH at the macula densa activates nNOS during tubuloglomerular feedback. Kidney Int 67: 1837–1843, 2005. doi: 10.1111/j.1523-1755.2005.00282.x. [DOI] [PubMed] [Google Scholar]
- 393.Liu R, Carretero OA, Ren Y, Wang H, Garvin JL. Intracellular pH regulates superoxide production by the macula densa. Am J Physiol Renal Physiol 295: F851–F856, 2008. doi: 10.1152/ajprenal.90204.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Liu R, Ren Y, Garvin JL, Carretero OA. Superoxide enhances tubuloglomerular feedback by constricting the afferent arteriole. Kidney Int 66: 268–274, 2004. doi: 10.1111/j.1523-1755.2004.00727.x. [DOI] [PubMed] [Google Scholar]
- 395.Liu Y, Singh RJ, Usa K, Netzel BC, Liang M. Renal medullary 11 beta-hydroxysteroid dehydrogenase type 1 in Dahl salt-sensitive hypertension. Physiol Genomics 36: 52–58, 2008. doi: 10.1152/physiolgenomics.90283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Llinás MT, González JD, Nava E, Salazar FJ. Role of angiotensin II in the renal effects induced by nitric oxide and prostaglandin synthesis inhibition. J Am Soc Nephrol 8: 543–550, 1997. [PubMed] [Google Scholar]
- 397.Longenecker G, Kulkarni AB. Generation of gene knockout mice by ES cell microinjection. Curr Protoc Cell Biol 19: 1–36, 2009. doi: 10.1002/0471143030.cb1914s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Lorenz JN, Weihprecht H, Schnermann J, Skøtt O, Briggs JP. Renin release from isolated juxtaglomerular apparatus depends on macula densa chloride transport. Am J Physiol Renal Physiol 260: F486–F493, 1991. doi: 10.1152/ajprenal.1991.260.4.F486. [DOI] [PubMed] [Google Scholar]
- 399.Louis WJ, Howes LG. Genealogy of the spontaneously hypertensive rat and Wistar-Kyoto rat strains: implications for studies of inherited hypertension. J Cardiovasc Pharmacol 16, Suppl 7: S1–S5, 1990. doi: 10.1097/00005344-199006167-00002. [DOI] [PubMed] [Google Scholar]
- 400.Lu M, Wang T, Yan Q, Yang X, Dong K, Knepper MA, Wang W, Giebisch G, Shull GE, Hebert SC. Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Bartter’s) knockout mice. J Biol Chem 277: 37881–37887, 2002. doi: 10.1074/jbc.M206644200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Lu M, Wang T, Yan Q, Wang W, Giebisch G, Hebert SC. ROMK is required for expression of the 70-pS K channel in the thick ascending limb. Am J Physiol Renal Physiol 286: F490–F495, 2004. doi: 10.1152/ajprenal.00305.2003. [DOI] [PubMed] [Google Scholar]
- 402.Lu M, Wang X, Wang W. Nitric oxide increases the activity of the apical 70-pS K+ channel in TAL of rat kidney. Am J Physiol Renal Physiol 274: F946–F950, 1998. doi: 10.1152/ajprenal.1998.274.5.F946. [DOI] [PubMed] [Google Scholar]
- 403.Lu X, Wang F, Liu M, Yang KT, Nau A, Kohan DE, Reese V, Richardson RS, Yang T. Activation of ENaC in collecting duct cells by prorenin and its receptor PRR: involvement of Nox4-derived hydrogen peroxide. Am J Physiol Renal Physiol 310: F1243–F1250, 2016. doi: 10.1152/ajprenal.00492.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Lu Y, Wei J, Stec DE, Roman RJ, Ge Y, Cheng L, Liu EY, Zhang J, Hansen PB, Fan F, Juncos LA, Wang L, Pollock J, Huang PL, Fu Y, Wang S, Liu R. Macula densa nitric oxide synthase 1β protects against salt-sensitive hypertension. J Am Soc Nephrol 27: 2346–2356, 2016. doi: 10.1681/ASN.2015050515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Ludwig DS, Peterson KE, Gortmaker SL. Relation between consumption of sugar-sweetened drinks and childhood obesity: a prospective, observational analysis. Lancet 357: 505–508, 2001. doi: 10.1016/S0140-6736(00)04041-1. [DOI] [PubMed] [Google Scholar]
- 406.Luft FC, Miller JZ, Grim CE, Fineberg NS, Christian JC, Daugherty SA, Weinberger MH. Salt sensitivity and resistance of blood pressure. Age and race as factors in physiological responses. Hypertension 17, Suppl: I102–I108, 1991. doi: 10.1161/01.HYP.17.1_Suppl.I102. [DOI] [PubMed] [Google Scholar]
- 407.Luft FC, Rankin LI, Bloch R, Weyman AE, Willis LR, Murray RH, Grim CE, Weinberger MH. Cardiovascular and humoral responses to extremes of sodium intake in normal black and white men. Circulation 60: 697–706, 1979. doi: 10.1161/01.CIR.60.3.697. [DOI] [PubMed] [Google Scholar]
- 408.Lukaszewicz KM, Paudyal MP, Falck JR, Lombard JH. Role of vascular reactive oxygen species in regulating cytochrome P450-4A enzyme expression in Dahl salt-sensitive rats. Microcirculation 23: 540–548, 2016. doi: 10.1111/micc.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassègue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res 105: 249–259, 2009. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Lynch IJ, Welch AK, Kohan DE, Cain BD, Wingo CS. Endothelin-1 inhibits sodium reabsorption by ET(A) and ET(B) receptors in the mouse cortical collecting duct. Am J Physiol Renal Physiol 305: F568–F573, 2013. doi: 10.1152/ajprenal.00613.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Lyon-Roberts B, Strait KA, van Peursem E, Kittikulsuth W, Pollock JS, Pollock DM, Kohan DE. Flow regulation of collecting duct endothelin-1 production. Am J Physiol Renal Physiol 300: F650–F656, 2011. doi: 10.1152/ajprenal.00530.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Ma YH, Schwartzman ML, Roman RJ. Altered renal P-450 metabolism of arachidonic acid in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 267: R579–R589, 1994. doi: 10.1152/ajpregu.1994.267.2.R579. [DOI] [PubMed] [Google Scholar]
- 413.Macica CM, Escalante BA, Conners MS, Ferreri NR. TNF production by the medullary thick ascending limb of Henle’s loop. Kidney Int 46: 113–121, 1994. doi: 10.1038/ki.1994.250. [DOI] [PubMed] [Google Scholar]
- 414.MacKinnon R. Pore loops: an emerging theme in ion channel structure. Neuron 14: 889–892, 1995. doi: 10.1016/0896-6273(95)90327-5. [DOI] [PubMed] [Google Scholar]
- 415.Madrigal G, Saborio P, Mora F, Rincon G, Guay-Woodford LM. Bartter syndrome in Costa Rica: a description of 20 cases. Pediatr Nephrol 11: 296–301, 1997. doi: 10.1007/s004670050280. [DOI] [PubMed] [Google Scholar]
- 416.Magyar CE, Zhang Y, Holstein-Rathlou NH, McDonough AA. Downstream shift in sodium pump activity along the nephron during acute hypertension. J Am Soc Nephrol 12: 2231–2240, 2001. [DOI] [PubMed] [Google Scholar]
- 417.Magyar CE, Zhang Y, Holstein-Rathlou NH, McDonough AA. Proximal tubule Na transporter responses are the same during acute and chronic hypertension. Am J Physiol Renal Physiol 279: F358–F369, 2000. doi: 10.1152/ajprenal.2000.279.2.F358. [DOI] [PubMed] [Google Scholar]
- 418.Majid DS, Navar LG. Nitric oxide in the mediation of pressure natriuresis. Clin Exp Pharmacol Physiol 24: 595–599, 1997. doi: 10.1111/j.1440-1681.1997.tb02098.x. [DOI] [PubMed] [Google Scholar]
- 419.Majid DS, Nishiyama A. Nitric oxide blockade enhances renal responses to superoxide dismutase inhibition in dogs. Hypertension 39: 293–297, 2002. doi: 10.1161/hy0202.104137. [DOI] [PubMed] [Google Scholar]
- 420.Majid DS, Williams A, Navar LG. Inhibition of nitric oxide synthesis attenuates pressure-induced natriuretic responses in anesthetized dogs. Am J Physiol Renal Physiol 264: F79–F87, 1993. doi: 10.1152/ajprenal.1993.264.1.F79. [DOI] [PubMed] [Google Scholar]
- 421.Makino A, Skelton MM, Zou AP, Cowley AW Jr. Increased renal medullary H2O2 leads to hypertension. Hypertension 42: 25–30, 2003. doi: 10.1161/01.HYP.0000074903.96928.91. [DOI] [PubMed] [Google Scholar]
- 422.Makino A, Skelton MM, Zou AP, Roman RJ, Cowley AW Jr. Increased renal medullary oxidative stress produces hypertension. Hypertension 39: 667–672, 2002. doi: 10.1161/hy0202.103469. [DOI] [PubMed] [Google Scholar]
- 423.Mandon B, Siga E, Roinel N, de Rouffignac C. Ca2+, Mg2+ and K+ transport in the cortical and medullary thick ascending limb of the rat nephron: influence of transepithelial voltage. Pflugers Arch 424: 558–560, 1993. doi: 10.1007/BF00374924. [DOI] [PubMed] [Google Scholar]
- 424.Manning RD Jr, Hu L. Nitric oxide regulates renal hemodynamics and urinary sodium excretion in dogs. Hypertension 23: 619–625, 1994. doi: 10.1161/01.HYP.23.5.619. [DOI] [PubMed] [Google Scholar]
- 425.Maranon RO, Lima R, Mathbout M, do Carmo JM, Hall JE, Roman RJ, Reckelhoff JF. Postmenopausal hypertension: role of the sympathetic nervous system in an animal model. Am J Physiol Regul Integr Comp Physiol 306: R248–R256, 2014. doi: 10.1152/ajpregu.00490.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Maranon RO, Reckelhoff JF. Mechanisms responsible for postmenopausal hypertension in a rat model: Roles of the renal sympathetic nervous system and the renin-angiotensin system. Physiol Rep 4: e12669, 2016. doi: 10.14814/phy2.12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Markadieu N, Delpire E. Physiology and pathophysiology of SLC12A1/2 transporters. Pflugers Arch 466: 91–105, 2014. doi: 10.1007/s00424-013-1370-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Martus W, Kim D, Garvin JL, Beierwaltes WH. Commercial rodent diets contain more sodium than rats need. Am J Physiol Renal Physiol 288: F428–F431, 2005. doi: 10.1152/ajprenal.00310.2004. [DOI] [PubMed] [Google Scholar]
- 429.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 18: 69–82, 2006. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 430.Massey KJ, Hong NJ, Garvin JL. Angiotensin II stimulates superoxide production in the thick ascending limb by activating NOX4. Am J Physiol Cell Physiol 303: C781–C789, 2012. doi: 10.1152/ajpcell.00457.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H, Nishiyama A, Ichikawa I. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol 23: 1181–1189, 2012. doi: 10.1681/ASN.2011121159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Mattson DL, Dwinell MR, Greene AS, Kwitek AE, Roman RJ, Jacob HJ, Cowley AW Jr. Chromosome substitution reveals the genetic basis of Dahl salt-sensitive hypertension and renal disease. Am J Physiol Renal Physiol 295: F837–F842, 2008. doi: 10.1152/ajprenal.90341.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Mattson DL, Higgins DJ. Influence of dietary sodium intake on renal medullary nitric oxide synthase. Hypertension 27: 688–692, 1996. doi: 10.1161/01.HYP.27.3.688. [DOI] [PubMed] [Google Scholar]
- 434.Mattson DL, Kunert MP, Kaldunski ML, Greene AS, Roman RJ, Jacob HJ, Cowley AW Jr. Influence of diet and genetics on hypertension and renal disease in Dahl salt-sensitive rats. Physiol Genomics 16: 194–203, 2004. doi: 10.1152/physiolgenomics.00151.2003. [DOI] [PubMed] [Google Scholar]
- 435.Mattson DL, Lu S, Nakanishi K, Papanek PE, Cowley AW Jr. Effect of chronic renal medullary nitric oxide inhibition on blood pressure. Am J Physiol Heart Circ Physiol 266: H1918–H1926, 1994. doi: 10.1152/ajpheart.1994.266.5.H1918. [DOI] [PubMed] [Google Scholar]
- 436.Mattson DL, Roman RJ, Cowley AW Jr. Role of nitric oxide in renal papillary blood flow and sodium excretion. Hypertension 19: 766–769, 1992. doi: 10.1161/01.HYP.19.6.766. [DOI] [PubMed] [Google Scholar]
- 437.Mazancová K, Miksík I, Kunes J, Zicha J, Pácha J. Sexual dimorphism of 11beta-hydroxysteroid dehydrogenase in hypertensive and normotensive rats. Hypertens Res 26: 333–338, 2003. doi: 10.1291/hypres.26.333. [DOI] [PubMed] [Google Scholar]
- 438.McCubbin JW, DeMoura RS, Page IH, Olmsted F. Arterial hypertension elicited by subpressor amounts of angiotensin. Science 149: 1394–1395, 1965. doi: 10.1126/science.149.3690.1394. [DOI] [PubMed] [Google Scholar]
- 439.McDonough AA, Nguyen MT. Maintaining balance under pressure: integrated regulation of renal transporters during hypertension. Hypertension 66: 450–455, 2015. doi: 10.1161/HYPERTENSIONAHA.115.04593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.McNicholas CM, MacGregor GG, Islas LD, Yang Y, Hebert SC, Giebisch G. pH-dependent modulation of the cloned renal K+ channel, ROMK. Am J Physiol Renal Physiol 275: F972–F981, 1998. doi: 10.1152/ajprenal.1998.275.6.F972. [DOI] [PubMed] [Google Scholar]
- 441.Meinild AK, Klaerke DA, Zeuthen T. Bidirectional water fluxes and specificity for small hydrophilic molecules in aquaporins 0-5. J Biol Chem 273: 32446–32451, 1998. doi: 10.1074/jbc.273.49.32446. [DOI] [PubMed] [Google Scholar]
- 442.Meister B, Fryckstedt J, Schalling M, Cortés R, Hökfelt T, Aperia A, Hemmings HC Jr, Nairn AC, Ehrlich M, Greengard P. Dopamine- and cAMP-regulated phosphoprotein (DARPP-32) and dopamine DA1 agonist-sensitive Na+,K+-ATPase in renal tubule cells. Proc Natl Acad Sci USA 86: 8068–8072, 1989. doi: 10.1073/pnas.86.20.8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Melzi ML, Bertorello A, Fukuda Y, Muldin I, Sereni F, Aperia A. Na,K-ATPase activity in renal tubule cells from Milan hypertensive rats. Am J Hypertens 2: 563–566, 1989. doi: 10.1093/ajh/2.7.563. [DOI] [PubMed] [Google Scholar]
- 444.Melzi ML, Syrén ML, Assael BM, Sereni F, Aperia A. Increased renal tubular Na-K-ATPase activity in Milan hypertensive rats in the prehypertensive period. Pediatr Nephrol 5: 700–703, 1991. doi: 10.1007/BF00857877. [DOI] [PubMed] [Google Scholar]
- 445.Mendizabal VE, Feleder EC, Adler-Graschinsky E. Effects of the chronic in vivo administration of l-NAME on the contractile responses of the rat perfused mesenteric bed. J Auton Pharmacol 19: 241–248, 1999. doi: 10.1046/j.1365-2680.1999.00140.x. [DOI] [PubMed] [Google Scholar]
- 446.Meng S, Cason GW, Gannon AW, Racusen LC, Manning RD Jr. Oxidative stress in Dahl salt-sensitive hypertension. Hypertension 41: 1346–1352, 2003. doi: 10.1161/01.HYP.0000070028.99408.E8. [DOI] [PubMed] [Google Scholar]
- 447.Meng S, Roberts LJ II, Cason GW, Curry TS, Manning RD Jr. Superoxide dismutase and oxidative stress in Dahl salt-sensitive and -resistant rats. Am J Physiol Regul Integr Comp Physiol 283: R732–R738, 2002. doi: 10.1152/ajpregu.00346.2001. [DOI] [PubMed] [Google Scholar]
- 448.Milatz S, Himmerkus N, Wulfmeyer VC, Drewell H, Mutig K, Hou J, Breiderhoff T, Müller D, Fromm M, Bleich M, Günzel D. Mosaic expression of claudins in thick ascending limbs of Henle results in spatial separation of paracellular Na+ and Mg2+ transport. Proc Natl Acad Sci USA 114: E219–E227, 2017. doi: 10.1073/pnas.1611684114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Miller JA, Anacta LA, Cattran DC. Impact of gender on the renal response to angiotensin II. Kidney Int 55: 278–285, 1999. doi: 10.1046/j.1523-1755.1999.00260.x. [DOI] [PubMed] [Google Scholar]
- 450.Mineta K, Yamamoto Y, Yamazaki Y, Tanaka H, Tada Y, Saito K, Tamura A, Igarashi M, Endo T, Takeuchi K, Tsukita S. Predicted expansion of the claudin multigene family. FEBS Lett 585: 606–612, 2011. doi: 10.1016/j.febslet.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 451.Mitic LL, Van Itallie CM, Anderson JM. Molecular physiology and pathophysiology of tight junctions I. Tight junction structure and function: lessons from mutant animals and proteins. Am J Physiol Gastrointest Liver Physiol 279: G250–G254, 2000. doi: 10.1152/ajpgi.2000.279.2.G250. [DOI] [PubMed] [Google Scholar]
- 452.Miyata N, Zou AP, Mattson DL, Cowley AW Jr. Renal medullary interstitial infusion of l-arginine prevents hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 275: R1667–R1673, 1998. doi: 10.1152/ajpregu.1998.275.5.R1667. [DOI] [PubMed] [Google Scholar]
- 453.Modlinger P, Chabrashvili T, Gill PS, Mendonca M, Harrison DG, Griendling KK, Li M, Raggio J, Wellstein A, Chen Y, Welch WJ, Wilcox CS. RNA silencing in vivo reveals role of p22phox in rat angiotensin slow pressor response. Hypertension 47: 238–244, 2006. doi: 10.1161/01.HYP.0000200023.02195.73. [DOI] [PubMed] [Google Scholar]
- 454.Molony DA, Reeves WB, Hebert SC, Andreoli TE ADH increases apical Na+, K+, 2Cl− entry in mouse medullary thick ascending limbs of Henle. Am J Physiol Renal Physiol 252: F177–F187, 1987. doi: 10.1152/ajprenal.1987.252.1.F177. [DOI] [PubMed] [Google Scholar]
- 455.Monzon CM, Garvin JL. Nitric oxide decreases the permselectivity of the paracellular pathway in thick ascending limbs. Hypertension 65: 1245–1250, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Monzon CM, Occhipinti R, Pignataro OP, Garvin JL. Nitric oxide reduces paracellular resistance in rat thick ascending limbs by increasing Na+ and Cl− permeabilities. Am J Physiol Renal Physiol 312: F1035–F1043, 2017. doi: 10.1152/ajprenal.00671.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Moreno C, Hoffman M, Stodola TJ, Didier DN, Lazar J, Geurts AM, North PE, Jacob HJ, Greene AS. Creation and characterization of a renin knockout rat. Hypertension 57: 614–619, 2011. doi: 10.1161/HYPERTENSIONAHA.110.163840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Moreno C, Kaldunski ML, Wang T, Roman RJ, Greene AS, Lazar J, Jacob HJ, Cowley AW Jr. Multiple blood pressure loci on rat chromosome 13 attenuate development of hypertension in the Dahl S hypertensive rat. Physiol Genomics 31: 228–235, 2007. doi: 10.1152/physiolgenomics.00280.2006. [DOI] [PubMed] [Google Scholar]
- 459.Moreno C, Williams JM, Lu L, Liang M, Lazar J, Jacob HJ, Cowley AW Jr, Roman RJ. Narrowing a region on rat chromosome 13 that protects against hypertension in Dahl SS-13BN congenic strains. Am J Physiol Heart Circ Physiol 300: H1530–H1535, 2011. doi: 10.1152/ajpheart.01026.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Mori T, Cowley AW Jr. Angiotensin II-NAD(P)H oxidase-stimulated superoxide modifies tubulovascular nitric oxide cross-talk in renal outer medulla. Hypertension 42: 588–593, 2003. doi: 10.1161/01.HYP.0000091821.39824.09. [DOI] [PubMed] [Google Scholar]
- 461.Mori T, O’Connor PM, Abe M, Cowley AW Jr. Enhanced superoxide production in renal outer medulla of Dahl salt-sensitive rats reduces nitric oxide tubular-vascular cross-talk. Hypertension 49: 1336–1341, 2007. doi: 10.1161/HYPERTENSIONAHA.106.085811. [DOI] [PubMed] [Google Scholar]
- 462.Morton JJ, Beattie EC, Speirs A, Gulliver F. Persistent hypertension following inhibition of nitric oxide formation in the young Wistar rat: role of renin and vascular hypertrophy. J Hypertens 11: 1083–1088, 1993. doi: 10.1097/00004872-199310000-00012. [DOI] [PubMed] [Google Scholar]
- 463.Mount DB. Thick ascending limb of the loop of Henle. Clin J Am Soc Nephrol 9: 1974–1986, 2014. doi: 10.2215/CJN.04480413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Mount DB, Delpire E, Gamba G, Hall AE, Poch E, Hoover RS, Hebert SC. The electroneutral cation-chloride cotransporters. J Exp Biol 201: 2091–2102, 1998. [DOI] [PubMed] [Google Scholar]
- 465.Mount DB, Mercado A, Song L, Xu J, George AL Jr, Delpire E, Gamba G. Cloning and characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J Biol Chem 274: 16355–16362, 1999. doi: 10.1074/jbc.274.23.16355. [DOI] [PubMed] [Google Scholar]
- 466.Munoz-Garcia R, Maeso R, Rodrigo E, Navarro J, Ruilope LM, Casal MC, Cachofeiro V, Lahera V. Acute renal excretory actions of losartan in spontaneously hypertensive rats: role of AT2 receptors, prostaglandins, kinins and nitric oxide. J Hypertens 13: 1779–1784, 1995. [PubMed] [Google Scholar]
- 467.Mutig K. Trafficking and regulation of the NKCC2 cotransporter in the thick ascending limb. Curr Opin Nephrol Hypertens 26: 392–397, 2017. doi: 10.1097/MNH.0000000000000351. [DOI] [PubMed] [Google Scholar]
- 468.Muto S, Furuse M, Kusano E. Claudins and renal salt transport. Clin Exp Nephrol 16: 61–67, 2012. doi: 10.1007/s10157-011-0491-4. [DOI] [PubMed] [Google Scholar]
- 469.Nakanishi K, Mattson DL, Cowley AW Jr. Role of renal medullary blood flow in the development of l-NAME hypertension in rats. Am J Physiol Regul Integr Comp Physiol 268: R317–R323, 1995. doi: 10.1152/ajpregu.1995.268.2.R317. [DOI] [PubMed] [Google Scholar]
- 470.Nakao A, Allen ML, Sonnenburg WK, Smith WL. Regulation of cAMP metabolism by PGE2 in cortical and medullary thick ascending limb of Henle’s loop. Am J Physiol Cell Physiol 256: C652–C657, 1989. doi: 10.1152/ajpcell.1989.256.3.C652. [DOI] [PubMed] [Google Scholar]
- 471.Nakayama T, Kosugi T, Gersch M, Connor T, Sanchez-Lozada LG, Lanaspa MA, Roncal C, Perez-Pozo SE, Johnson RJ, Nakagawa T. Dietary fructose causes tubulointerstitial injury in the normal rat kidney. Am J Physiol Renal Physiol 298: F712–F720, 2010. doi: 10.1152/ajprenal.00433.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol 122: 277–291, 2004. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- 473.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension 57: 355–362, 2011. doi: 10.1161/HYPERTENSIONAHA.110.163519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Nguyen MT, Han J, Ralph DL, Veiras LC, McDonough AA. Short-term nonpressor angiotensin II infusion stimulates sodium transporters in proximal tubule and distal nephron. Physiol Rep 3: e12496, 2015. doi: 10.14814/phy2.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Nguyen MT, Lee DH, Delpire E, McDonough AA. Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol 305: F510–F519, 2013. doi: 10.1152/ajprenal.00183.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Nguyen PV, Parent A, Deng LY, Flückiger JP, Thibault G, Schiffrin EL. Endothelin vascular receptors and responses in deoxycorticosterone acetate-salt hypertensive rats. Hypertension 19, Suppl: II98–II104, 1992. doi: 10.1161/01.HYP.19.2_Suppl.II98. [DOI] [PubMed] [Google Scholar]
- 477.Nguyen S, Choi HK, Lustig RH, Hsu CY. Sugar-sweetened beverages, serum uric acid, and blood pressure in adolescents. J Pediatr 154: 807–813, 2009. doi: 10.1016/j.jpeds.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Ni Z, Oveisi F, Vaziri ND. Nitric oxide synthase isotype expression in salt-sensitive and salt-resistant Dahl rats. Hypertension 34: 552–557, 1999. doi: 10.1161/01.HYP.34.4.552. [DOI] [PubMed] [Google Scholar]
- 479.Nielsen S, DiGiovanni SR, Christensen EI, Knepper MA, Harris HW. Cellular and subcellular immunolocalization of vasopressin-regulated water channel in rat kidney. Proc Natl Acad Sci USA 90: 11663–11667, 1993. doi: 10.1073/pnas.90.24.11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Nielsen S, Maunsbach AB, Ecelbarger CA, Knepper MA. Ultrastructural localization of Na-K-2Cl cotransporter in thick ascending limb and macula densa of rat kidney. Am J Physiol Renal Physiol 275: F885–F893, 1998. doi: 10.1152/ajprenal.1998.275.6.F885. [DOI] [PubMed] [Google Scholar]
- 481.Nishi A, Bertorello AM, Aperia A. High salt diet down-regulates proximal tubule Na+, K+-ATPase activity in Dahl salt-resistant but not in Dahl salt-sensitive rats: evidence of defective dopamine regulation. Acta Physiol Scand 144: 263–267, 1992. doi: 10.1111/j.1748-1716.1992.tb09295.x. [DOI] [PubMed] [Google Scholar]
- 482.Nishi A, Eklöf AC, Bertorello AM, Aperia A. Dopamine regulation of renal Na+,K+-ATPase activity is lacking in Dahl salt-sensitive rats. Hypertension 21: 767–771, 1993. doi: 10.1161/01.HYP.21.6.767. [DOI] [PubMed] [Google Scholar]
- 483.Nishimoto Y, Tomida T, Matsui H, Ito T, Okumura K. Decrease in renal medullary endothelial nitric oxide synthase of fructose-fed, salt-sensitive hypertensive rats. Hypertension 40: 190–194, 2002. doi: 10.1161/01.HYP.0000024267.71656.0D. [DOI] [PubMed] [Google Scholar]
- 484.Nisimoto Y, Diebold BA, Cosentino-Gomes D, Lambeth JD. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry 53: 5111–5120, 2014. doi: 10.1021/bi500331y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Nizar JM, Bhalla V. Molecular mechanisms of sodium-sensitive hypertension in the metabolic syndrome. Curr Hypertens Rep 19: 60, 2017. doi: 10.1007/s11906-017-0759-5. [DOI] [PubMed] [Google Scholar]
- 486.O’Connor PM, Cowley AW Jr. Medullary thick ascending limb buffer vasoconstriction of renal outer-medullary vasa recta in salt-resistant but not salt-sensitive rats. Hypertension 60: 965–972, 2012. doi: 10.1161/HYPERTENSIONAHA.112.195214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.O’Connor PM, Lu L, Liang M, Cowley AW Jr. A novel amiloride-sensitive H+ transport pathway mediates enhanced superoxide production in thick ascending limb of salt-sensitive rats, not Na+/H+ exchange. Hypertension 54: 248–254, 2009. doi: 10.1161/HYPERTENSIONAHA.109.134692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.O’Connor PM, Lu L, Schreck C, Cowley AW Jr. Enhanced amiloride-sensitive superoxide production in renal medullary thick ascending limb of Dahl salt-sensitive rats. Am J Physiol Renal Physiol 295: F726–F733, 2008. doi: 10.1152/ajprenal.00137.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Odgaard E, Praetorius HA, Leipziger J. AVP-stimulated nucleotide secretion in perfused mouse medullary thick ascending limb and cortical collecting duct. Am J Physiol Renal Physiol 297: F341–F349, 2009. doi: 10.1152/ajprenal.00190.2009. [DOI] [PubMed] [Google Scholar]
- 490.Ogden CL, Kit BK, Carroll MD, Park S. Consumption of sugar drinks in the United States, 2005-2008. NCHS Data Brief 71 1–8, 2011. [PubMed] [Google Scholar]
- 491.Ohsaki Y, O’Connor P, Mori T, Ryan RP, Dickinson BC, Chang CJ, Lu Y, Ito S, Cowley AW Jr. Increase of sodium delivery stimulates the mitochondrial respiratory chain H2O2 production in rat renal medullary thick ascending limb. Am J Physiol Renal Physiol 302: F95–F102, 2012. doi: 10.1152/ajprenal.00469.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Okamoto K, Aoki K. Development of a strain of spontaneously hypertensive rats. Jpn Circ J 27: 282–293, 1963. doi: 10.1253/jcj.27.282. [DOI] [PubMed] [Google Scholar]
- 493.Orias M, Velázquez H, Tung F, Lee G, Desir GV. Cloning and localization of a double-pore K channel, KCNK1: exclusive expression in distal nephron segments. Am J Physiol Renal Physiol 273: F663–F666, 1997. doi: 10.1152/ajprenal.1997.273.4.F663. [DOI] [PubMed] [Google Scholar]
- 494.Ormsbee HS III, Ryan CF. Production of hypertension with desoxycorticorticosterone acetate-impregnated silicone rubber implants. J Pharm Sci 62: 255–257, 1973. doi: 10.1002/jps.2600620215. [DOI] [PubMed] [Google Scholar]
- 495.Ortiz-Capisano MC, Ortiz PA, Garvin JL, Harding P, Beierwaltes WH. Expression and function of the calcium-sensing receptor in juxtaglomerular cells. Hypertension 50: 737–743, 2007. doi: 10.1161/HYPERTENSIONAHA.107.095158. [DOI] [PubMed] [Google Scholar]
- 496.Ortiz MC, Manriquez MC, Romero JC, Juncos LA. Antioxidants block angiotensin II-induced increases in blood pressure and endothelin. Hypertension 38: 655–659, 2001. doi: 10.1161/01.HYP.38.3.655. [DOI] [PubMed] [Google Scholar]
- 497.Ortiz P, Stoos BA, Hong NJ, Boesch DM, Plato CF, Garvin JL. High-salt diet increases sensitivity to NO and eNOS expression but not NO production in THALs. Hypertension 41: 682–687, 2003. doi: 10.1161/01.HYP.0000047872.07864.20. [DOI] [PubMed] [Google Scholar]
- 498.Ortiz PA. cAMP increases surface expression of NKCC2 in rat thick ascending limbs: role of VAMP. Am J Physiol Renal Physiol 290: F608–F616, 2006. doi: 10.1152/ajprenal.00248.2005. [DOI] [PubMed] [Google Scholar]
- 499.Ortiz PA. A small fraction of total NKCC2 is located in the apical membrane of thick ascending limbs under basal conditions (Abstract). FASEB J 20: A1223, 2006. [Google Scholar]
- 500.Ortiz PA, Garvin JL. Autocrine effects of nitric oxide on HCO(3)(−) transport by rat thick ascending limb. Kidney Int 58: 2069–2074, 2000. doi: 10.1111/j.1523-1755.2000.00379.x. [DOI] [PubMed] [Google Scholar]
- 501.Ortiz PA, Garvin JL. Interaction of O(2)(−) and NO in the thick ascending limb. Hypertension 39: 591–596, 2002. doi: 10.1161/hy0202.103287. [DOI] [PubMed] [Google Scholar]
- 502.Ortiz PA, Garvin JL. NO Inhibits NaCl absorption by rat thick ascending limb through activation of cGMP-stimulated phosphodiesterase. Hypertension 37: 467–471, 2001. doi: 10.1161/01.HYP.37.2.467. [DOI] [PubMed] [Google Scholar]
- 503.Ortiz PA, Garvin JL. Superoxide stimulates NaCl absorption by the thick ascending limb. Am J Physiol Renal Physiol 283: F957–F962, 2002. doi: 10.1152/ajprenal.00102.2002. [DOI] [PubMed] [Google Scholar]
- 504.Ortiz PA, Hong NJ, Garvin JL. Luminal flow induces eNOS activation and translocation in the rat thick ascending limb. Am J Physiol Renal Physiol 287: F274–F280, 2004. doi: 10.1152/ajprenal.00382.2003. [DOI] [PubMed] [Google Scholar]
- 505.Ortiz PA, Hong NJ, Garvin JL. Luminal flow induces eNOS activation and translocation in the rat thick ascending limb. II. Role of PI3-kinase and Hsp90. Am J Physiol Renal Physiol 287: F281–F288, 2004. doi: 10.1152/ajprenal.00383.2003. [DOI] [PubMed] [Google Scholar]
- 506.Ortiz PA, Hong NJ, Garvin JL. NO decreases thick ascending limb chloride absorption by reducing Na+-K+-2Cl− cotransporter activity. Am J Physiol Renal Physiol 281: F819–F825, 2001. doi: 10.1152/ajprenal.0075.2001. [DOI] [PubMed] [Google Scholar]
- 507.Ortiz PA, Hong NJ, Wang D, Garvin JL. Gene transfer of eNOS to the thick ascending limb of eNOS-KO mice restores the effects of L-arginine on NaCl absorption. Hypertension 42: 674–679, 2003. doi: 10.1161/01.HYP.0000085561.00001.81. [DOI] [PubMed] [Google Scholar]
- 508.Osborn JL, Holdaas H, Thames MD, DiBona GF. Renal adrenoceptor mediation of antinatriuretic and renin secretion responses to low frequency renal nerve stimulation in the dog. Circ Res 53: 298–305, 1983. doi: 10.1161/01.RES.53.3.298. [DOI] [PubMed] [Google Scholar]
- 509.Padmanabhan S, Melander O, Johnson T, Di Blasio AM, Lee WK, Gentilini D, Hastie CE, Menni C, Monti MC, Delles C, Laing S, Corso B, Navis G, Kwakernaak AJ, van der Harst P, Bochud M, Maillard M, Burnier M, Hedner T, Kjeldsen S, Wahlstrand B, Sjögren M, Fava C, Montagnana M, Danese E, Torffvit O, Hedblad B, Snieder H, Connell JM, Brown M, Samani NJ, Farrall M, Cesana G, Mancia G, Signorini S, Grassi G, Eyheramendy S, Wichmann HE, Laan M, Strachan DP, Sever P, Shields DC, Stanton A, Vollenweider P, Teumer A, Völzke H, Rettig R, Newton-Cheh C, Arora P, Zhang F, Soranzo N, Spector TD, Lucas G, Kathiresan S, Siscovick DS, Luan J, Loos RJ, Wareham NJ, Penninx BW, Nolte IM, McBride M, Miller WH, Nicklin SA, Baker AH, Graham D, McDonald RA, Pell JP, Sattar N, Welsh P, Munroe P, Caulfield MJ, Zanchetti A, Dominiczak AF, Dominiczak AF; Global BPgen Consortium . Genome-wide association study of blood pressure extremes identifies variant near UMOD associated with hypertension. PLoS Genet 6: e1001177, 2010. doi: 10.1371/journal.pgen.1001177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Paillard M. H+ and transporters in the medullary thick ascending limb of the kidney: molecular mechanisms, function and regulation Kidney Int Suppl 65: S36–S41, 1998. [PubMed] [Google Scholar]
- 511.Palacios C, Wigertz K, Martin BR, Braun M, Pratt JH, Peacock M, Weaver CM. Racial differences in potassium homeostasis in response to differences in dietary sodium in girls. Am J Clin Nutr 91: 597–603, 2010. doi: 10.3945/ajcn.2009.28400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Palacios C, Wigertz K, Martin BR, Jackman L, Pratt JH, Peacock M, McCabe G, Weaver CM. Sodium retention in black and white female adolescents in response to salt intake. J Clin Endocrinol Metab 89: 1858–1863, 2004. doi: 10.1210/jc.2003-031446. [DOI] [PubMed] [Google Scholar]
- 513.Pallone TL, Zhang Z, Rhinehart K. Physiology of the renal medullary microcirculation. Am J Physiol Renal Physiol 284: F253–F266, 2003. doi: 10.1152/ajprenal.00304.2002. [DOI] [PubMed] [Google Scholar]
- 514.Pandit MM, Gao Y, van Hoek A, Kohan DE. Osmolar regulation of endothelin-1 production by the inner medullary collecting duct. Life Sci 159: 135–139, 2016. doi: 10.1016/j.lfs.2015.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Pandit MM, Inscho EW, Zhang S, Seki T, Rohatgi R, Gusella L, Kishore B, Kohan DE. Flow regulation of endothelin-1 production in the inner medullary collecting duct. Am J Physiol Renal Physiol 308: F541–F552, 2015. doi: 10.1152/ajprenal.00456.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Pandit MM, Strait KA, Matsuda T, Kohan DE. Na delivery and ENaC mediate flow regulation of collecting duct endothelin-1 production. Am J Physiol Renal Physiol 302: F1325–F1330, 2012. doi: 10.1152/ajprenal.00034.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Panel on Dietary Reference Intakes for Electrolytes and Water, Standing Committee on the Scientific Evaluation of Dietary Reference Intakes, and Academies Dietary Reference Intakes for Water, Potassium, Sodium, Chloride, and Sulfate. Washington, DC: National Academies, 2005. [Google Scholar]
- 518.Parekh N, Dobrowolski L, Zou AP, Steinhausen M. Nitric oxide modulates angiotensin II- and norepinephrine-dependent vasoconstriction in rat kidney. Am J Physiol Regul Integr Comp Physiol 270: R630–R635, 1996. doi: 10.1152/ajpregu.1996.270.3.R630. [DOI] [PubMed] [Google Scholar]
- 519.Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol 119: 229–240, 2006. doi: 10.1016/j.clim.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 520.Passmore JC, Jimenez AE. Effect of chloride on renal blood flow in angiotensin II induced hypertension. Can J Physiol Pharmacol 69: 507–511, 1991. doi: 10.1139/y91-076. [DOI] [PubMed] [Google Scholar]
- 521.Patel A, Layne S, Watts D, Kirchner KA. L-arginine administration normalizes pressure natriuresis in hypertensive Dahl rats. Hypertension 22: 863–869, 1993. doi: 10.1161/01.HYP.22.6.863. [DOI] [PubMed] [Google Scholar]
- 522.Patel RP, McAndrew J, Sellak H, White CR, Jo H, Freeman BA, Darley-Usmar VM. Biological aspects of reactive nitrogen species. Biochim Biophys Acta 1411: 385–400, 1999. doi: 10.1016/S0005-2728(99)00028-6. [DOI] [PubMed] [Google Scholar]
- 523.Paulais M, Lachheb S, Teulon J. A Na+- and Cl−-activated K+ channel in the thick ascending limb of mouse kidney. J Gen Physiol 127: 205–215, 2006. doi: 10.1085/jgp.200509360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Pavlov TS, Chahdi A, Ilatovskaya DV, Levchenko V, Vandewalle A, Pochynyuk O, Sorokin A, Staruschenko A. Endothelin-1 inhibits the epithelial Na+ channel through betaPix/14-3-3/Nedd4-2. J Am Soc Nephrol 21: 833–843, 2010. doi: 10.1681/ASN.2009080885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Pavlov TS, Ilatovskaya DV, Levchenko V, Mattson DL, Roman RJ, Staruschenko A. Effects of cytochrome P-450 metabolites of arachidonic acid on the epithelial sodium channel (ENaC). Am J Physiol Renal Physiol 301: F672–F681, 2011. doi: 10.1152/ajprenal.00597.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Pavlov TS, Levchenko V, Ilatovskaya DV, Moreno C, Staruschenko A. Renal sodium transport in renin-deficient Dahl salt-sensitive rats. J Renin Angiotensin Aldosterone Syst 17 1470320316653858, 2016. doi: 10.1177/1470320316653858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Payne JA, Forbush B III. Alternatively spliced isoforms of the putative renal Na-K-Cl cotransporter are differentially distributed within the rabbit kidney. Proc Natl Acad Sci USA 91: 4544–4548, 1994. doi: 10.1073/pnas.91.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Payne JA, Forbush B III. Molecular characterization of the epithelial Na-K-Cl cotransporter isoforms. Curr Opin Cell Biol 7: 493–503, 1995. doi: 10.1016/0955-0674(95)80005-0. [DOI] [PubMed] [Google Scholar]
- 529.Peng JJ, Liu B, Xu JY, Peng J, Luo XJ. NADPH oxidase: its potential role in promotion of pulmonary arterial hypertension. Naunyn Schmiedebergs Arch Pharmacol 390: 331–338, 2017. doi: 10.1007/s00210-017-1359-2. [DOI] [PubMed] [Google Scholar]
- 530.Perez-Rojas JM, Kassem KM, Beierwaltes WH, Garvin JL, Herrera M. Nitric oxide produced by endothelial nitric oxide synthase promotes diuresis. Am J Physiol Regul Integr Comp Physiol 298: R1050–R1055, 2010. doi: 10.1152/ajpregu.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Perico N, Cornejo RP, Benigni A, Malanchini B, Ladny JR, Remuzzi G. Endothelin induces diuresis and natriuresis in the rat by acting on proximal tubular cells through a mechanism mediated by lipoxygenase products. J Am Soc Nephrol 2: 57–69, 1991. [DOI] [PubMed] [Google Scholar]
- 532.Peterson LN, De Rouffignac C, Sonnenberg H, Levine DZ. Thick ascending limb response to dDAVP and atrial natriuretic factor in vivo. Am J Physiol Renal Physiol 252: F374–F381, 1987. doi: 10.1152/ajprenal.1987.252.3.F374. [DOI] [PubMed] [Google Scholar]
- 533.Peti-Peterdi J, Harris RC. Macula densa sensing and signaling mechanisms of renin release. J Am Soc Nephrol 21: 1093–1096, 2010. doi: 10.1681/ASN.2009070759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Peti-Peterdi J, Warnock DG, Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT(1) receptors. J Am Soc Nephrol 13: 1131–1135, 2002. doi: 10.1097/01.ASN.0000013292.78621.FD. [DOI] [PubMed] [Google Scholar]
- 535.Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem 277: 50812–50819, 2002. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- 536.Piehl C, Piontek J, Cording J, Wolburg H, Blasig IE. Participation of the second extracellular loop of claudin-5 in paracellular tightening against ions, small and large molecules. Cell Mol Life Sci 67: 2131–2140, 2010. doi: 10.1007/s00018-010-0332-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Piontek J, Winkler L, Wolburg H, Müller SL, Zuleger N, Piehl C, Wiesner B, Krause G, Blasig IE. Formation of tight junction: determinants of homophilic interaction between classic claudins. FASEB J 22: 146–158, 2008. doi: 10.1096/fj.07-8319com. [DOI] [PubMed] [Google Scholar]
- 538.Plato CF. Alpha-2 and beta-adrenergic receptors mediate NE’s biphasic effects on rat thick ascending limb chloride flux. Am J Physiol Regul Integr Comp Physiol 281: R979–R986, 2001. doi: 10.1152/ajpregu.2001.281.3.R979. [DOI] [PubMed] [Google Scholar]
- 539.Plato CF, Garvin JL. Alpha(2)-adrenergic-mediated tubular NO production inhibits thick ascending limb chloride absorption. Am J Physiol Renal Physiol 281: F679–F686, 2001. doi: 10.1152/ajprenal.2001.281.4.F679. [DOI] [PubMed] [Google Scholar]
- 540.Plato CF, Osborn JL. Chronic renal neuroadrenergic hypertension is associated with increased renal norepinephrine sensitivity and volume contraction. Hypertension 28: 1034–1040, 1996. doi: 10.1161/01.HYP.28.6.1034. [DOI] [PubMed] [Google Scholar]
- 541.Plato CF, Pollock DM, Garvin JL. Endothelin inhibits thick ascending limb chloride flux via ET(B) receptor-mediated NO release. Am J Physiol Renal Physiol 279: F326–F333, 2000. doi: 10.1152/ajprenal.2000.279.2.F326. [DOI] [PubMed] [Google Scholar]
- 542.Plato CF, Shesely EG, Garvin JL. eNOS mediates L-arginine-induced inhibition of thick ascending limb chloride flux. Hypertension 35: 319–323, 2000. doi: 10.1161/01.HYP.35.1.319. [DOI] [PubMed] [Google Scholar]
- 543.Plato CF, Stoos BA, Wang D, Garvin JL. Endogenous nitric oxide inhibits chloride transport in the thick ascending limb. Am J Physiol Renal Physiol 276: F159–F163, 1999. doi: 10.1152/ajprenal.1999.276.1.F159. [DOI] [PubMed] [Google Scholar]
- 544.Pollock DM. Endothelin Receptor Subtypes and Tissue Distribution. In: Endothelin, edited by Highsmith RF. Totowa, NJ: Humana, 1998, p. 1–29. doi: 10.1007/978-1-4757-2783-8_1 [DOI] [Google Scholar]
- 545.Pollock DM. Renal endothelin in hypertension Curr Opin Nephrol Hypertens 9: 157–164, 2000. doi: 10.1097/00041552-200003000-00010. [DOI] [PubMed] [Google Scholar]
- 546.Pollock DM, Allcock GH, Krishnan A, Dayton BD, Pollock JS. Upregulation of endothelin B receptors in kidneys of DOCA-salt hypertensive rats. Am J Physiol Renal Physiol 278: F279–F286, 2000. doi: 10.1152/ajprenal.2000.278.2.F279. [DOI] [PubMed] [Google Scholar]
- 547.Pollock DM, Pollock JS. Evidence for endothelin involvement in the response to high salt. Am J Physiol Renal Physiol 281: F144–F150, 2001. doi: 10.1152/ajprenal.2001.281.1.F144. [DOI] [PubMed] [Google Scholar]
- 548.Ponce-Coria J, San-Cristobal P, Kahle KT, Vazquez N, Pacheco-Alvarez D, de Los Heros P, Juárez P, Muñoz E, Michel G, Bobadilla NA, Gimenez I, Lifton RP, Hebert SC, Gamba G. Regulation of NKCC2 by a chloride-sensing mechanism involving the WNK3 and SPAK kinases. Proc Natl Acad Sci USA 105: 8458–8463, 2008. doi: 10.1073/pnas.0802966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Powell DW. Barrier function of epithelia. Am J Physiol Gastro Intestinal Physiol 241: G275–G288, 1981. doi: 10.1152/ajpgi.1981.241.4.G275. [DOI] [PubMed] [Google Scholar]
- 550.Prabhakar P, Thatte HS, Goetz RM, Cho MR, Golan DE, Michel T. Receptor-regulated translocation of endothelial nitric-oxide synthase. J Biol Chem 273: 27383–27388, 1998. doi: 10.1074/jbc.273.42.27383. [DOI] [PubMed] [Google Scholar]
- 551.Praticó D, FitzGerald GA. Generation of 8-epiprostaglandin F2alpha by human monocytes. Discriminate production by reactive oxygen species and prostaglandin endoperoxide synthase-2. J Biol Chem 271: 8919–8924, 1996. doi: 10.1074/jbc.271.15.8919. [DOI] [PubMed] [Google Scholar]
- 552.Pratt JH, Ambrosius WT, Agarwal R, Eckert GJ, Newman S. Racial difference in the activity of the amiloride-sensitive epithelial sodium channel. Hypertension 40: 903–908, 2002. doi: 10.1161/01.HYP.0000039749.75068.F4. [DOI] [PubMed] [Google Scholar]
- 553.Pratt JH, Jones JJ, Miller JZ, Wagner MA, Fineberg NS. Racial differences in aldosterone excretion and plasma aldosterone concentrations in children. N Engl J Med 321: 1152–1157, 1989. doi: 10.1056/NEJM198910263211703. [DOI] [PubMed] [Google Scholar]
- 554.Pratt JH, Manatunga AK, Hanna MP, Ambrosius WT. Effect of administered potassium on the renin-aldosterone axis in young blacks compared with whites. J Hypertens 15: 877–883, 1997. doi: 10.1097/00004872-199715080-00012. [DOI] [PubMed] [Google Scholar]
- 555.Pratt JH, Manatunga AK, Peacock M. A comparison of the urinary excretion of bone resorptive products in white and black children. J Lab Clin Med 127: 67–70, 1996. doi: 10.1016/S0022-2143(96)90167-5. [DOI] [PubMed] [Google Scholar]
- 556.Preston GM, Agre P. Isolation of the cDNA for erythrocyte integral membrane protein of 28 kilodaltons: member of an ancient channel family. Proc Natl Acad Sci USA 88: 11110–11114, 1991. doi: 10.1073/pnas.88.24.11110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Procino G, Carmosino M, Tamma G, Gouraud S, Laera A, Riccardi D, Svelto M, Valenti G. Extracellular calcium antagonizes forskolin-induced aquaporin 2 trafficking in collecting duct cells. Kidney Int 66: 2245–2255, 2004. doi: 10.1111/j.1523-1755.2004.66036.x. [DOI] [PubMed] [Google Scholar]
- 558.Pu HX, Cluzeaud F, Goldshleger R, Karlish SJ, Farman N, Blostein R. Functional role and immunocytochemical localization of the gamma a and gamma b forms of the Na,K-ATPase gamma subunit. J Biol Chem 276: 20370–20378, 2001. doi: 10.1074/jbc.M010836200. [DOI] [PubMed] [Google Scholar]
- 559.Quamme GA, de Rouffignac C. Epithelial magnesium transport and regulation by the kidney. Front Biosci 5: D694–D711, 2000. doi: 10.2741/A544. [DOI] [PubMed] [Google Scholar]
- 560.Ramseyer VD, Cabral PD, Garvin JL. Role of endothelin in thick ascending limb sodium chloride transport. Contrib Nephrol 172: 76–83, 2011. doi: 10.1159/000328686. [DOI] [PubMed] [Google Scholar]
- 561.Ramseyer VD, Garvin JL. Angiotensin II decreases nitric oxide synthase 3 expression via nitric oxide and superoxide in the thick ascending limb. Hypertension 53: 313–318, 2009. doi: 10.1161/HYPERTENSIONAHA.108.124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Ramseyer VD, Gonzalez-Vicente A, Carretero OA, Garvin JL. Angiotensin II-induced hypertension blunts thick ascending limb NO production by reducing NO synthase 3 expression and enhancing threonine 495 phosphorylation. Am J Physiol Renal Physiol 308: F149–F156, 2015. doi: 10.1152/ajprenal.00279.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Ramseyer VD, Ortiz PA, Carretero OA, Garvin JL. Angiotensin II-mediated hypertension impairs nitric oxide-induced NKCC2 inhibition in thick ascending limbs. Am J Physiol Renal Physiol 310: F748–F754, 2016. doi: 10.1152/ajprenal.00473.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Ranieri M, Tamma G, Di Mise A, Russo A, Centrone M, Svelto M, Calamita G, Valenti G. Negative feedback from CaSR signaling to aquaporin-2 sensitizes vasopressin to extracellular Ca2. J Cell Sci 128: 2350–2360, 2015. doi: 10.1242/jcs.168096. [DOI] [PubMed] [Google Scholar]
- 565.Rapp JP, Dene H. Development and characteristics of inbred strains of Dahl salt-sensitive and salt-resistant rats. Hypertension 7: 340–349, 1985. [PubMed] [Google Scholar]
- 566.Rapp JP, Wang SM, Dene H. A genetic polymorphism in the renin gene of Dahl rats cosegregates with blood pressure. Science 243: 542–544, 1989. doi: 10.1126/science.2563177. [DOI] [PubMed] [Google Scholar]
- 567.Rastogi R, Geng X, Li F, Ding Y. NOX activation by subunit interaction and underlying mechanisms in disease. Front Cell Neurosci 10: 301, 2017. doi: 10.3389/fncel.2016.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Reckelhoff JF, Zhang H, Srivastava K. Gender differences in development of hypertension in spontaneously hypertensive rats: role of the renin-angiotensin system. Hypertension 35: 480–483, 2000. doi: 10.1161/01.HYP.35.1.480. [DOI] [PubMed] [Google Scholar]
- 569.Reed MJ, Ho H, Donnelly R, Reaven GM. Salt-sensitive and carbohydrate-sensitive rodent hypertension: evidence of strain differences. Blood Press 3: 197–201, 1994. doi: 10.3109/08037059409102253. [DOI] [PubMed] [Google Scholar]
- 570.Reeves AS, Collins JH, Schwartz A. Isolation and characterization of (Na,K)-ATPase proteolipid. Biochem Biophys Res Commun 95: 1591–1598, 1980. doi: 10.1016/S0006-291X(80)80080-5. [DOI] [PubMed] [Google Scholar]
- 571.Reeves WB, McDonald GA, Mehta P, Andreoli TE. Activation of K+ channels in renal medullary vesicles by cAMP-dependent protein kinase. J Membr Biol 109: 65–72, 1989. doi: 10.1007/BF01870791. [DOI] [PubMed] [Google Scholar]
- 572.Reeves WB, Winters CJ, Andreoli TE. Chloride channels in the loop of Henle. Annu Rev Physiol 63: 631–645, 2001. doi: 10.1146/annurev.physiol.63.1.631. [DOI] [PubMed] [Google Scholar]
- 573.Reid JJ, Rand MJ. Renal vasoconstriction is modulated by nitric oxide. Clin Exp Pharmacol Physiol 19: 376–379, 1992. doi: 10.1111/j.1440-1681.1992.tb00476.x. [DOI] [PubMed] [Google Scholar]
- 574.Ren H, Gu L, Andreasen A, Thomsen JS, Cao L, Christensen EI, Zhai XY. Spatial organization of the vascular bundle and the interbundle region: three-dimensional reconstruction at the inner stripe of the outer medulla in the mouse kidney. Am J Physiol Renal Physiol 306: F321–F326, 2014. doi: 10.1152/ajprenal.00429.2013. [DOI] [PubMed] [Google Scholar]
- 575.Ren Y, Garvin JL, Carretero OA. Vasodilator action of angiotensin-(1-7) on isolated rabbit afferent arterioles. Hypertension 39: 799–802, 2002. doi: 10.1161/hy0302.104673. [DOI] [PubMed] [Google Scholar]
- 576.Ren Y, Carretero OA, Garvin JL. Mechanism by which superoxide potentiates tubuloglomerular feedback. Hypertension 39: 624–628, 2002. doi: 10.1161/hy0202.103299. [DOI] [PubMed] [Google Scholar]
- 577.Ren YL, Garvin JL, Carretero OA. Role of macula densa nitric oxide and cGMP in the regulation of tubuloglomerular feedback. Kidney Int 58: 2053–2060, 2000. doi: 10.1111/j.1523-1755.2000.00377.x. [DOI] [PubMed] [Google Scholar]
- 578.Rettig R, Folberth C, Stauss H, Kopf D, Waldherr R, Unger T. Role of the kidney in primary hypertension: a renal transplantation study in rats. Am J Physiol Renal Physiol 258: F606–F611, 1990. doi: 10.1152/ajprenal.1990.258.3.F606. [DOI] [PubMed] [Google Scholar]
- 579.Rettig R, Folberth CG, Stauss H, Kopf D, Waldherr R, Baldauf G, Unger T. Hypertension in rats induced by renal grafts from renovascular hypertensive donors. Hypertension 15: 429–435, 1990. doi: 10.1161/01.HYP.15.4.429. [DOI] [PubMed] [Google Scholar]
- 580.Riazi S, Tiwari S, Sharma N, Rash A, Ecelbarger CM. Abundance of the Na-K-2Cl cotransporter NKCC2 is increased by high-fat feeding in Fischer 344 X Brown Norway (F1) rats. Am J Physiol Renal Physiol 296: F762–F770, 2009. doi: 10.1152/ajprenal.90484.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Ribeiro MO, Antunes E, de Nucci G, Lovisolo SM, Zatz R. Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension. Hypertension 20: 298–303, 1992. doi: 10.1161/01.HYP.20.3.298. [DOI] [PubMed] [Google Scholar]
- 582.Ribou AC. Synthetic sensors for reactive oxygen species detection and quantification: a critical review of current methods. Antioxid Redox Signal 25: 520–533, 2016. doi: 10.1089/ars.2016.6741. [DOI] [PubMed] [Google Scholar]
- 583.Riccardi D, Lee WS, Lee K, Segre GV, Brown EM, Hebert SC. Localization of the extracellular Ca2+-sensing receptor and PTH/PTHrP receptor in rat kidney. Am J Physiol Renal Physiol 271: F951–F956, 1996. doi: 10.1152/ajprenal.1996.271.4.F951. [DOI] [PubMed] [Google Scholar]
- 584.Richardson C, Sakamoto K, de los Heros P, Deak M, Campbell DG, Prescott AR, Alessi DR. Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J Cell Sci 124: 789–800, 2011. doi: 10.1242/jcs.077230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Roger F, Martin PY, Rousselot M, Favre H, Féraille E. Cell shrinkage triggers the activation of mitogen-activated protein kinases by hypertonicity in the rat kidney medullary thick ascending limb of the Henle’s loop. Requirement of p38 kinase for the regulatory volume increase response. J Biol Chem 274: 34103–34110, 1999. doi: 10.1074/jbc.274.48.34103. [DOI] [PubMed] [Google Scholar]
- 586.Roman RJ. Abnormal renal hemodynamics and pressure-natriuresis relationship in Dahl salt-sensitive rats. Am J Physiol Renal Physiol 251: F57–F65, 1986. doi: 10.1152/ajprenal.1986.251.1.F57. [DOI] [PubMed] [Google Scholar]
- 587.Roman RJ. Altered pressure-natriuresis relationship in young spontaneously hypertensive rats. Hypertension 9: III130–III136, 1987. doi: 10.1161/01.HYP.9.6_Pt_2.III130. [DOI] [PubMed] [Google Scholar]
- 588.Roman RJ, Cowley AW Jr. Abnormal pressure-diuresis-natriuresis response in spontaneously hypertensive rats. Am J Physiol Renal Physiol 248: F199–F205, 1985. doi: 10.1152/ajprenal.1985.248.2.F199. [DOI] [PubMed] [Google Scholar]
- 589.Roman RJ, Kaldunski ML. Enhanced chloride reabsorption in the loop of Henle in Dahl salt-sensitive rats. Hypertension 17: 1018–1024, 1991. doi: 10.1161/01.HYP.17.6.1018. [DOI] [PubMed] [Google Scholar]
- 590.Roman RJ, Kaldunski ML. Renal cortical and papillary blood flow in spontaneously hypertensive rats. Hypertension 11: 657–663, 1988. doi: 10.1161/01.HYP.11.6.657. [DOI] [PubMed] [Google Scholar]
- 591.Roman RJ, Osborn JL. Renal function and sodium balance in conscious Dahl S and R rats. Am J Physiol Regul Integr Comp Physiol 252: R833–R841, 1987. doi: 10.1152/ajpregu.1987.252.5.R833. [DOI] [PubMed] [Google Scholar]
- 592.Romano G, Giagu P, Favret G, Bartoli E. Effect of endothelin 1 on proximal reabsorption and tubuloglomerular feedback. Kidney Blood Press Res 23: 360–365, 2000. doi: 10.1159/000025984. [DOI] [PubMed] [Google Scholar]
- 593.Roos JC, Kirchner KA, Abernethy JD, Langford HG. Differential effect of salt loading on sodium and lithium excretion in Dahl salt-resistant and -sensitive rats. Hypertension 6: 420–424, 1984. doi: 10.1161/01.HYP.6.3.420. [DOI] [PubMed] [Google Scholar]
- 594.Rosa RM, De Jesus E, Sperling K, Suh A, Gmurczyk A, Myrie KA, Rosner K, Lerma E, Yu W, Breuer R, Young JB. Gastrointestinal and renal excretion of potassium in African-Americans and White Americans. J Hypertens 30: 2373–2377, 2012. doi: 10.1097/HJH.0b013e32835a27b3. [DOI] [PubMed] [Google Scholar]
- 595.Rossoni G, Manfredi B, De Gennaro Colonna V, Berti M, Guazzi M, Berti F. Sildenafil reduces l-NAME-induced severe hypertension and worsening of myocardial ischaemia-reperfusion damage in the rat. Br J Pharmacol 150: 567–576, 2007. doi: 10.1038/sj.bjp.0707131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Rouse D, Williams S, Suki WN. Clonidine inhibits fluid absorption in the rabbit proximal convoluted renal tubule. Kidney Int 38: 80–85, 1990. doi: 10.1038/ki.1990.170. [DOI] [PubMed] [Google Scholar]
- 597.Ruan X, Purdy KE, Oliverio MI, Coffman TM, Arendshorst WJ. Effects of candesartan on angiotensin II-induced renal vasoconstriction in rats and mice. J Am Soc Nephrol 10, Suppl 11: S202–S207, 1999. [PubMed] [Google Scholar]
- 598.Rugale C, Delbosc S, Cristol JP, Mimran A, Jover B. Sodium restriction prevents cardiac hypertrophy and oxidative stress in angiotensin II hypertension. Am J Physiol Heart Circ Physiol 284: H1744–H1750, 2003. doi: 10.1152/ajpheart.00864.2002. [DOI] [PubMed] [Google Scholar]
- 599.Ruppersberg JP, Schröter KH, Sakmann B, Stocker M, Sewing S, Pongs O. Heteromultimeric channels formed by rat brain potassium-channel proteins. Nature 345: 535–537, 1990. doi: 10.1038/345535a0. [DOI] [PubMed] [Google Scholar]
- 600.Saez F, Hong NJ, Garvin JL. Luminal flow induces NADPH oxidase 4 translocation to the nuclei of thick ascending limbs. Physiol Rep 4: e12724, 2016. doi: 10.14814/phy2.12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Salardi S, Modica R, Ferrandi M, Ferrari P, Torielli L, Bianchi G. Characterization of erythrocyte adducin from the Milan hypertensive strain of rats. J Hypertens Suppl 6: S196–S198, 1988. doi: 10.1097/00004872-198812040-00058. [DOI] [PubMed] [Google Scholar]
- 602.Salazar FJ, Alberola A, Pinilla JM, Romero JC, Quesada T. Salt-induced increase in arterial pressure during nitric oxide synthesis inhibition. Hypertension 22: 49–55, 1993. doi: 10.1161/01.HYP.22.1.49. [DOI] [PubMed] [Google Scholar]
- 603.Salazar FJ, Pinilla JM, López F, Romero JC, Quesada T. Renal effects of prolonged synthesis inhibition of endothelium-derived nitric oxide. Hypertension 20: 113–117, 1992. doi: 10.1161/01.HYP.20.1.113. [DOI] [PubMed] [Google Scholar]
- 604.Salom MG, Lahera V, Miranda-Guardiola F, Romero JC. Blockade of pressure natriuresis induced by inhibition of renal synthesis of nitric oxide in dogs. Am J Physiol Renal Physiol 262: F718–F722, 1992. doi: 10.1152/ajprenal.1992.262.5.F718. [DOI] [PubMed] [Google Scholar]
- 605.Sánchez-Lozada LG, Tapia E, Jiménez A, Bautista P, Cristóbal M, Nepomuceno T, Soto V, Avila-Casado C, Nakagawa T, Johnson RJ, Herrera-Acosta J, Franco M. Fructose-induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. Am J Physiol Renal Physiol 292: F423–F429, 2007. doi: 10.1152/ajprenal.00124.2006. [DOI] [PubMed] [Google Scholar]
- 606.Sander M, Hansen J, Victor RG. The sympathetic nervous system is involved in the maintenance but not initiation of the hypertension induced by N(omega)-nitro-L-arginine methyl ester Hypertension 30: 64–70, 1997. doi: 10.1161/01.HYP.30.1.64. [DOI] [PubMed] [Google Scholar]
- 607.Sasaki S, Imai M. Effects of vasopressin on water and NaCl transport across the in vitro perfused medullary thick ascending limb of Henle’s loop of mouse, rat, and rabbit kidneys. Pflugers Arch 383: 215–221, 1980. doi: 10.1007/BF00587521. [DOI] [PubMed] [Google Scholar]
- 608.Sasser JM, Sullivan JC, Elmarakby AA, Kemp BE, Pollock DM, Pollock JS. Reduced NOS3 phosphorylation mediates reduced NO/cGMP signaling in mesenteric arteries of deoxycorticosterone acetate-salt hypertensive rats. Hypertension 43: 1080–1085, 2004. doi: 10.1161/01.HYP.0000122804.32680.c9. [DOI] [PubMed] [Google Scholar]
- 609.Sawyer DT. The chemistry of dioxygen species (O2, ·, HOO·, and HOOH) and their activation by transition metals. Int Rev Exp Pathol 31: 109–131, 1990. doi: 10.1016/B978-0-12-364931-7.50009-4. [DOI] [PubMed] [Google Scholar]
- 610.Schießl IM, Castrop H. Regulation of NKCC2 splicing and phosphorylation. Curr Opin Nephrol Hypertens 24: 457–462, 2015. doi: 10.1097/MNH.0000000000000150. [DOI] [PubMed] [Google Scholar]
- 611.Schiessl IM, Rosenauer A, Kattler V, Minuth WW, Oppermann M, Castrop H. Dietary salt intake modulates differential splicing of the Na-K-2Cl cotransporter NKCC2. Am J Physiol Renal Physiol 305: F1139–F1148, 2013. doi: 10.1152/ajprenal.00259.2013. [DOI] [PubMed] [Google Scholar]
- 612.Schnackenberg CG, Wilcox CS. Two-week administration of tempol attenuates both hypertension and renal excretion of 8-Iso prostaglandin f2α. Hypertension 33: 424–428, 1999. doi: 10.1161/01.HYP.33.1.424. [DOI] [PubMed] [Google Scholar]
- 613.Schneider MP, Ge Y, Pollock DM, Pollock JS, Kohan DE. Collecting duct-derived endothelin regulates arterial pressure and Na excretion via nitric oxide. Hypertension 51: 1605–1610, 2008. doi: 10.1161/HYPERTENSIONAHA.107.108126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614.Schnermann J, Briggs JP. Tubular control of renin synthesis and secretion. Pflugers Arch 465: 39–51, 2013. doi: 10.1007/s00424-012-1115-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Schröder K, Weissmann N, Brandes RP. Organizers and activators: Cytosolic Nox proteins impacting on vascular function. Free Radic Biol Med 109: 22–32, 2017. doi: 10.1016/j.freeradbiomed.2017.03.017. [DOI] [PubMed] [Google Scholar]
- 616.Schulze MB, Manson JE, Ludwig DS, Colditz GA, Stampfer MJ, Willett WC, Hu FB. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. JAMA 292: 927–934, 2004. doi: 10.1001/jama.292.8.927. [DOI] [PubMed] [Google Scholar]
- 617.Schuster VL. Molecular mechanisms of prostaglandin transport. Annu Rev Physiol 60: 221–242, 1998. doi: 10.1146/annurev.physiol.60.1.221. [DOI] [PubMed] [Google Scholar]
- 618.Sechi LA. Mechanisms of insulin resistance in rat models of hypertension and their relationships with salt sensitivity. J Hypertens 17: 1229–1237, 1999. doi: 10.1097/00004872-199917090-00001. [DOI] [PubMed] [Google Scholar]
- 619.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Fórró L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 406: 105–114, 2007. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Shi LB, Skach WR, Verkman AS. Functional independence of monomeric CHIP28 water channels revealed by expression of wild-type mutant heterodimers. J Biol Chem 269: 10417–10422, 1994. [PubMed] [Google Scholar]
- 621.Shindo A, Maki T, Mandeville ET, Liang AC, Egawa N, Itoh K, Itoh N, Borlongan M, Holder JC, Chuang TT, McNeish JD, Tomimoto H, Lok J, Lo EH, Arai K. Astrocyte-derived pentraxin 3 supports blood-brain barrier integrity under acute phase of stroke. Stroke 47: 1094–1100, 2016. doi: 10.1161/STROKEAHA.115.012133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S, Hattori M, Sakaki Y, Sumimoto H. A novel superoxide-producing NAD(P)H oxidase in kidney. J Biol Chem 276: 1417–1423, 2001. doi: 10.1074/jbc.M007597200. [DOI] [PubMed] [Google Scholar]
- 623.Shirley DG, Walter SJ, Unwin RJ, Giebisch G. Contribution of Na+-H+ exchange to sodium reabsorption in the loop of henle: a microperfusion study in rats. J Physiol 513: 243–249, 1998. doi: 10.1111/j.1469-7793.1998.243by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Shull GE, Schwartz A, Lingrel JB. Amino-acid sequence of the catalytic subunit of the (Na+ + K+)ATPase deduced from a complementary DNA. Nature 316: 691–695, 1985. doi: 10.1038/316691a0. [DOI] [PubMed] [Google Scholar]
- 625.Sile S, Gillani NB, Velez DR, Vanoye CG, Yu C, Byrne LM, Gainer JV, Brown NJ, Williams SM, George AL Jr. Functional BSND variants in essential hypertension. Am J Hypertens 20: 1176–1182, 2007. doi: 10.1016/j.amjhyper.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 626.Sile S, Velez DR, Gillani NB, Narsia T, Moore JH, George AL Jr, Vanoye CG, Williams SM. CLCNKB-T481S and essential hypertension in a Ghanaian population. J Hypertens 27: 298–304, 2009. doi: 10.1097/HJH.0b013e3283140c9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Silldorff EP, Yang S, Pallone TL. Prostaglandin E2 abrogates endothelin-induced vasoconstriction in renal outer medullary descending vasa recta of the rat. J Clin Invest 95: 2734–2740, 1995. doi: 10.1172/JCI117976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Silva GB, Garvin JL. Akt1 mediates purinergic-dependent NOS3 activation in thick ascending limbs. Am J Physiol Renal Physiol 297: F646–F652, 2009. doi: 10.1152/ajprenal.00270.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Silva GB, Garvin JL. Angiotensin II-dependent hypertension increases Na transport-related oxygen consumption by the thick ascending limb. Hypertension 52: 1091–1098, 2008. doi: 10.1161/HYPERTENSIONAHA.108.120212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Silva GB, Garvin JL. Extracellular ATP inhibits transport in medullary thick ascending limbs: role of P2X receptors. Am J Physiol Renal Physiol 297: F1168–F1173, 2009. doi: 10.1152/ajprenal.00325.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Silva GB, Garvin JL. Rac1 mediates NaCl-induced superoxide generation in the thick ascending limb. Am J Physiol Renal Physiol 298: F421–F425, 2010. doi: 10.1152/ajprenal.00472.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Silva GB, Juncos LI, Baigorria ST, Garcia NH. Angiotensin II inhibits ADH-stimulated cAMP: role on and transport-related oxygen consumption in the loop of Henle. J Biol Regul Homeost Agents 27: 569–578, 2013. [PubMed] [Google Scholar]
- 633.Silva GB, Juncos LI, Garcia NH. Angiotensin II and anti diuretic hormone exert synergistic effects on thick ascending limb transport in spontaneously hypertensive rats. Nephron 132: 153–160, 2016. doi: 10.1159/000444025. [DOI] [PubMed] [Google Scholar]
- 634.Silva GB, Ortiz PA, Hong NJ, Garvin JL. Superoxide stimulates NaCl absorption in the thick ascending limb via activation of protein kinase C. Hypertension 48: 467–472, 2006. doi: 10.1161/01.HYP.0000236646.83354.51. [DOI] [PubMed] [Google Scholar]
- 635.Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, Lifton RP. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet 17: 171–178, 1997. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- 636.Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13: 183–188, 1996. doi: 10.1038/ng0696-183. [DOI] [PubMed] [Google Scholar]
- 637.Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, Sanjad SA, Lifton RP. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet 14: 152–156, 1996. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 638.Sonalker PA, Jackson EK. Norepinephrine, via beta-adrenoceptors, regulates bumetanide-sensitive cotransporter type 1 expression in thick ascending limb cells. Hypertension 49: 1351–1357, 2007. doi: 10.1161/HYPERTENSIONAHA.107.088732. [DOI] [PubMed] [Google Scholar]
- 639.Sonalker PA, Tofovic SP, Jackson EK. Cellular distribution of the renal bumetanide-sensitive Na-K-2Cl cotransporter BSC-1 in the inner stripe of the outer medulla during the development of hypertension in the spontaneously hypertensive rat. Clin Exp Pharmacol Physiol 34: 1307–1312, 2007. doi: 10.1111/j.1440-1681.2007.04747.x. [DOI] [PubMed] [Google Scholar]
- 640.Sonalker PA, Tofovic SP, Jackson EK. Increased expression of the sodium transporter BSC-1 in spontaneously hypertensive rats. J Pharmacol Exp Ther 311: 1052–1061, 2004. doi: 10.1124/jpet.104.071209. [DOI] [PubMed] [Google Scholar]
- 641.Song J, Lu Y, Lai EY, Wei J, Wang L, Chandrashekar K, Wang S, Shen C, Juncos LA, Liu R. Oxidative status in the macula densa modulates tubuloglomerular feedback responsiveness in angiotensin II-induced hypertension. Acta Physiol (Oxf) 213: 249–258, 2015. doi: 10.1111/apha.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Speed JS, LaMarca B, Berry H, Cockrell K, George EM, Granger JP. Renal medullary endothelin-1 is decreased in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 301: R519–R523, 2011. doi: 10.1152/ajpregu.00207.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.St Lezin EM, Pravenec M, Wong A, Wang JM, Merriouns T, Newton S, Stec DE, Roman RJ, Lau D, Morris RC Jr. Genetic contamination of Dahl SS/Jr rats. Impact on studies of salt-sensitive hypertension Hypertension 23: 786–790, 1994. doi: 10.1161/01.HYP.23.6.786. [DOI] [PubMed] [Google Scholar]
- 644.St Lezin EM, Pravenec M, Wong AL, Liu W, Wang N, Lu S, Jacob HJ, Roman RJ, Stec DE, Wang JM, Reid IA, Kurtz TW. Effects of renin gene transfer on blood pressure and renin gene expression in a congenic strain of Dahl salt-resistant rats. J Clin Invest 97: 522–527, 1996. doi: 10.1172/JCI118444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645.Starremans PG, Kersten FF, Knoers NV, van den Heuvel LP, Bindels RJ. Mutations in the human Na-K-2Cl cotransporter (NKCC2) identified in Bartter syndrome type I consistently result in nonfunctional transporters. J Am Soc Nephrol 14: 1419–1426, 2003. doi: 10.1097/01.ASN.0000064948.39199.A0. [DOI] [PubMed] [Google Scholar]
- 646.Stec DE, Deng AY, Rapp JP, Roman RJ. Cytochrome P4504A genotype cosegregates with hypertension in Dahl S rats. Hypertension 27: 564–568, 1996. doi: 10.1161/01.HYP.27.3.564. [DOI] [PubMed] [Google Scholar]
- 647.Sterling H, Lin DH, Gu RM, Dong K, Hebert SC, Wang WH. Inhibition of protein-tyrosine phosphatase stimulates the dynamin-dependent endocytosis of ROMK1. J Biol Chem 277: 4317–4323, 2002. doi: 10.1074/jbc.M109739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Sterling H, Lin DH, Wei Y, Wang WH. Tetanus toxin abolishes exocytosis of ROMK1 induced by inhibition of protein tyrosine kinase. Am J Physiol Renal Physiol 284: F510–F517, 2003. doi: 10.1152/ajprenal.00309.2002. [DOI] [PubMed] [Google Scholar]
- 649.Stern MD, Bowen PD, Parma R, Osgood RW, Bowman RL, Stein JH. Measurement of renal cortical and medullary blood flow by laser-Doppler spectroscopy in the rat. Am J Physiol Renal Physiol 236: F80–F87, 1979. doi: 10.1152/ajprenal.1979.236.1.F80. [DOI] [PubMed] [Google Scholar]
- 650.Stewart AP, Smith GD, Sandford RN, Edwardson JM. Atomic force microscopy reveals the alternating subunit arrangement of the TRPP2-TRPV4 heterotetramer. Biophys J 99: 790–797, 2010. doi: 10.1016/j.bpj.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 11: 95–102, 1994. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- 652.Stoos BA, Carretero OA, Farhy RD, Scicli G, Garvin JL. Endothelium-derived relaxing factor inhibits transport and increases cGMP content in cultured mouse cortical collecting duct cells. J Clin Invest 89: 761–765, 1992. doi: 10.1172/JCI115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653.Stoos BA, Garcia NH, Garvin JL. Nitric oxide inhibits sodium reabsorption in the isolated perfused cortical collecting duct. J Am Soc Nephrol 6: 89–94, 1995. [DOI] [PubMed] [Google Scholar]
- 654.Sugawara N, Morimoto T, Farajov EI, Kumagai N, Aslanova UF, Rai T, Uchida S, Sasaki S, Tsuchiya S, Kondo Y. Calcium and calcimimetics regulate paracellular Na+ transport in the thin ascending limb of Henle’s loop in mouse kidney. Pflugers Arch 460: 197–205, 2010. doi: 10.1007/s00424-010-0836-y. [DOI] [PubMed] [Google Scholar]
- 655.Suki WN, Rouse D, Ng RC, Kokko JP. Calcium transport in the thick ascending limb of Henle. Heterogeneity of function in the medullary and cortical segments. J Clin Invest 66: 1004–1009, 1980. doi: 10.1172/JCI109928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Sullivan JC, Sasser JM, Pollock DM, Pollock JS. Sexual dimorphism in renal production of prostanoids in spontaneously hypertensive rats. Hypertension 45: 406–411, 2005. doi: 10.1161/01.HYP.0000156879.83448.93. [DOI] [PubMed] [Google Scholar]
- 657.Sullivan JC, Sasser JM, Pollock JS. Sexual dimorphism in oxidant status in spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol 292: R764–R768, 2007. doi: 10.1152/ajpregu.00322.2006. [DOI] [PubMed] [Google Scholar]
- 658.Sun A, Grossman EB, Lombardi M, Hebert SC. Vasopressin alters the mechanism of apical Cl− entry from Na+: Cl− to Na+:K+:2Cl− cotransport in mouse medullary thick ascending limb. J Membr Biol 120: 83–94, 1991. doi: 10.1007/BF01868594. [DOI] [PubMed] [Google Scholar]
- 659.Sun AM, Liu Y, Dworkin LD, Tse CM, Donowitz M, Yip KP. Na+/H+ exchanger isoform 2 (NHE2) is expressed in the apical membrane of the medullary thick ascending limb. J Membr Biol 160: 85–90, 1997. doi: 10.1007/s002329900297. [DOI] [PubMed] [Google Scholar]
- 660.Sun P, Yue P, Wang WH. Angiotensin II stimulates epithelial sodium channels in the cortical collecting duct of the rat kidney. Am J Physiol Renal Physiol 302: F679–F687, 2012. doi: 10.1152/ajprenal.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661.Sventek P, Li JS, Grove K, Deschepper CF, Schiffrin EL. Vascular structure and expression of endothelin-1 gene in l-NAME-treated spontaneously hypertensive rats. Hypertension 27: 49–55, 1996. doi: 10.1161/01.HYP.27.1.49. [DOI] [PubMed] [Google Scholar]
- 662.Szentiványi M Jr, Maeda CY, Cowley AW Jr. Local renal medullary l-NAME infusion enhances the effect of long-term angiotensin II treatment. Hypertension 33: 440–445, 1999. doi: 10.1161/01.HYP.33.1.440. [DOI] [PubMed] [Google Scholar]
- 663.Szentiványi M Jr, Zou AP, Maeda CY, Mattson DL, Cowley AW Jr. Increase in renal medullary nitric oxide synthase activity protects from norepinephrine-induced hypertension. Hypertension 35: 418–423, 2000. doi: 10.1161/01.HYP.35.1.418. [DOI] [PubMed] [Google Scholar]
- 664.Szentiványi M Jr, Zou AP, Mattson DL, Soares P, Moreno C, Roman RJ, Cowley AW Jr. Renal medullary nitric oxide deficit of Dahl S rats enhances hypertensive actions of angiotensin II. Am J Physiol Regul Integr Comp Physiol 283: R266–R272, 2002. doi: 10.1152/ajpregu.00461.2001. [DOI] [PubMed] [Google Scholar]
- 665.Takabatake T, Ise T, Ohta K, Kobayashi K. Endothelin effects on renal function and tubuloglomerular feedback. Kidney Int Suppl 32: S122–S124, 1991. [PubMed] [Google Scholar]
- 666.Takac I, Schröder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, Brandes RP. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem 286: 13304–13313, 2011. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667.Takenaka T, Nobe K, Okayama M, Kojima E, Nodaira Y, Sueyoshi K, Hoshi H, Watanabe Y, Takane H, Suzuki H. Aliskiren reduces morning blood pressure in hypertensive patients with diabetic nephropathy. Clin Exp Hypertens 34: 243–248, 2012. doi: 10.3109/10641963.2012.681080. [DOI] [PubMed] [Google Scholar]
- 668.Tanaka M, Kamata R, Sakai R. EphA2 phosphorylates the cytoplasmic tail of Claudin-4 and mediates paracellular permeability. J Biol Chem 280: 42375–42382, 2005. doi: 10.1074/jbc.M503786200. [DOI] [PubMed] [Google Scholar]
- 669.Tatum R, Zhang Y, Lu Q, Kim K, Jeansonne BG, Chen YH. WNK4 phosphorylates ser(206) of claudin-7 and promotes paracellular Cl(−) permeability. FEBS Lett 581: 3887–3891, 2007. doi: 10.1016/j.febslet.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 670.Taylor NE, Cowley AW Jr. Effect of renal medullary H2O2 on salt-induced hypertension and renal injury. Am J Physiol Regul Integr Comp Physiol 289: R1573–R1579, 2005. doi: 10.1152/ajpregu.00525.2005. [DOI] [PubMed] [Google Scholar]
- 671.Taylor NE, Glocka P, Liang M, Cowley AW Jr. NADPH oxidase in the renal medulla causes oxidative stress and contributes to salt-sensitive hypertension in Dahl S rats. Hypertension 47: 692–698, 2006. doi: 10.1161/01.HYP.0000203161.02046.8d. [DOI] [PubMed] [Google Scholar]
- 672.Taylor NE, Maier KG, Roman RJ, Cowley AW Jr. NO synthase uncoupling in the kidney of Dahl S rats: role of dihydrobiopterin. Hypertension 48: 1066–1071, 2006. doi: 10.1161/01.HYP.0000248751.11383.7c. [DOI] [PubMed] [Google Scholar]
- 673.Thai TL, Yu L, Eaton DC, Duke BJ, Al-Khalili O, Lam HY, Ma H, Bao HF. Basolateral P2X4 channels stimulate ENaC activity in Xenopus cortical collecting duct A6 cells. Am J Physiol Renal Physiol 307: F806–F813, 2014. doi: 10.1152/ajprenal.00350.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674.Thallas-Bonke V, Jandeleit-Dahm KA, Cooper ME. Nox-4 and progressive kidney disease. Curr Opin Nephrol Hypertens 24: 74–80, 2015. doi: 10.1097/MNH.0000000000000082. [DOI] [PubMed] [Google Scholar]
- 675.Tharaux PL, Dussaule JC, Pauti MD, Vassitch Y, Ardaillou R, Chatziantoniou C. Activation of renin synthesis is dependent on intact nitric oxide production. Kidney Int 51: 1780–1787, 1997. doi: 10.1038/ki.1997.245. [DOI] [PubMed] [Google Scholar]
- 676.Theilig F, Goranova I, Hirsch JR, Wieske M, Unsal S, Bachmann S, Veh RW, Derst C. Cellular localization of THIK-1 (K2P13.1) and THIK-2 (K2P12.1) K+ channels in the mammalian kidney. Cell Physiol Biochem 21: 63–74, 2008. doi: 10.1159/000113748. [DOI] [PubMed] [Google Scholar]
- 677.Therien AG, Blostein R. Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol 279: C541–C566, 2000. doi: 10.1152/ajpcell.2000.279.3.C541. [DOI] [PubMed] [Google Scholar]
- 678.Thomas DD. Breathing new life into nitric oxide signaling: a brief overview of the interplay between oxygen and nitric oxide. Redox Biol 5: 225–233, 2015. doi: 10.1016/j.redox.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 679.Tian N, Thrasher KD, Gundy PD, Hughson MD, Manning RD Jr. Antioxidant treatment prevents renal damage and dysfunction and reduces arterial pressure in salt-sensitive hypertension. Hypertension 45: 934–939, 2005. doi: 10.1161/01.HYP.0000160404.08866.5a. [DOI] [PubMed] [Google Scholar]
- 680.Tian W, Salanova M, Xu H, Lindsley JN, Oyama TT, Anderson S, Bachmann S, Cohen DM. Renal expression of osmotically responsive cation channel TRPV4 is restricted to water-impermeant nephron segments. Am J Physiol Renal Physiol 287: F17–F24, 2004. doi: 10.1152/ajprenal.00397.2003. [DOI] [PubMed] [Google Scholar]
- 681.Tian Z, Liu Y, Usa K, Mladinov D, Fang Y, Ding X, Greene AS, Cowley AW Jr, Liang M. Novel role of fumarate metabolism in dahl-salt sensitive hypertension. Hypertension 54: 255–260, 2009. doi: 10.1161/HYPERTENSIONAHA.109.129528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682.Tiwari S, Li L, Riazi S, Halagappa VK, Ecelbarger CM. Sex differences in adaptive downregulation of pre-macula densa sodium transporters with ANG II infusion in mice. Am J Physiol Renal Physiol 298: F187–F195, 2010. doi: 10.1152/ajprenal.00088.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 683.Tolins JP, Shultz PJ. Endogenous nitric oxide synthesis determines sensitivity to the pressor effect of salt. Kidney Int 46: 230–236, 1994. doi: 10.1038/ki.1994.264. [DOI] [PubMed] [Google Scholar]
- 684.Tomita K, Nonoguchi H, Terada Y, Marumo F. Effects of ET-1 on water and chloride transport in cortical collecting ducts of the rat. Am J Physiol Renal Physiol 264: F690–F696, 1993. doi: 10.1152/ajprenal.1993.264.4.F690. [DOI] [PubMed] [Google Scholar]
- 685.Treeck B, Aukland K. Effect of l-NAME on glomerular filtration rate in deep and superficial layers of rat kidneys. Am J Physiol Renal Physiol 272: F312–F318, 1997. doi: 10.1152/ajprenal.1997.272.3.F312. [DOI] [PubMed] [Google Scholar]
- 686.Treeck B, Roald AB, Tenstad O, Aukland K. Effect of exogenous and endogenous angiotensin II on intrarenal distribution of glomerular filtration rate in rats. J Physiol 541: 1049–1057, 2002. doi: 10.1113/jphysiol.2002.018390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687.Trepiccione F, Zacchia M, Capasso G. The role of the kidney in salt-sensitive hypertension. Clin Exp Nephrol 16: 68–72, 2012. doi: 10.1007/s10157-011-0489-y. [DOI] [PubMed] [Google Scholar]
- 688.Tripodi G, Modica R, Reina C, Bianchi G. Tissue-specific modulation of beta-adducin transcripts in Milan hypertensive rats. Biochem Biophys Res Commun 303: 230–237, 2003. doi: 10.1016/S0006-291X(03)00330-9. [DOI] [PubMed] [Google Scholar]
- 689.Tripodi G, Valtorta F, Torielli L, Chieregatti E, Salardi S, Trusolino L, Menegon A, Ferrari P, Marchisio PC, Bianchi G. Hypertension-associated point mutations in the adducin alpha and beta subunits affect actin cytoskeleton and ion transport. J Clin Invest 97: 2815–2822, 1996. doi: 10.1172/JCI118737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690.Trudu M, Janas S, Lanzani C, Debaix H, Schaeffer C, Ikehata M, Citterio L, Demaretz S, Trevisani F, Ristagno G, Glaudemans B, Laghmani K, Dell’Antonio G, Loffing J, Rastaldi MP, Manunta P, Devuyst O, Rampoldi L; SKIPOGH team . Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat Med 19: 1655–1660, 2013. doi: 10.1038/nm.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 691.Tsuruoka S, Koseki C, Muto S, Tabei K, Imai M. Axial heterogeneity of potassium transport across hamster thick ascending limb of Henle’s loop. Am J Physiol Renal Physiol 267: F121–F129, 1994. doi: 10.1152/ajprenal.1994.267.1.F121. [DOI] [PubMed] [Google Scholar]
- 692.Tu W, Eckert GJ, Decker BS, Howard Pratt J. Varying Influences of aldosterone on the plasma potassium concentration in blacks and whites. Am J Hypertens 30: 490–494, 2017. doi: 10.1093/ajh/hpx006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 693.Tucker SJ, Imbrici P, Salvatore L, D’Adamo MC, Pessia M. pH dependence of the inwardly rectifying potassium channel, Kir5.1, and localization in renal tubular epithelia. J Biol Chem 275: 16404–16407, 2000. doi: 10.1074/jbc.C000127200. [DOI] [PubMed] [Google Scholar]
- 694.Tumlin JA, Hoban CA, Medford RM, Sands JM. Expression of Na-K-ATPase alpha- and beta-subunit mRNA and protein isoforms in the rat nephron. Am J Physiol Renal Physiol 266: F240–F245, 1994. doi: 10.1152/ajprenal.1994.266.2.F240. [DOI] [PubMed] [Google Scholar]
- 695.Turban S, Miller ER III, Ange B, Appel LJ. Racial differences in urinary potassium excretion. J Am Soc Nephrol 19: 1396–1402, 2008. doi: 10.1681/ASN.2007101142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696.Turban S, Thompson CB, Parekh RS, Appel LJ. Effects of sodium intake and diet on racial differences in urinary potassium excretion: results from the Dietary Approaches to Stop Hypertension (DASH)-Sodium trial. Am J Kidney Dis 61: 88–95, 2013. doi: 10.1053/j.ajkd.2012.08.036. [DOI] [PubMed] [Google Scholar]
- 697.Turban S, Wang XY, Knepper MA. Regulation of NHE3, NKCC2, and NCC abundance in kidney during aldosterone escape phenomenon: role of NO. Am J Physiol Renal Physiol 285: F843–F851, 2003. doi: 10.1152/ajprenal.00110.2003. [DOI] [PubMed] [Google Scholar]
- 698.Uchida S, Sasaki S, Furukawa T, Hiraoka M, Imai T, Hirata Y, Marumo F. Molecular cloning of a chloride channel that is regulated by dehydration and expressed predominantly in kidney medulla. J Biol Chem 268: 3821–3824, 1993. [PubMed] [Google Scholar]
- 699.Vallon V, Peterson OW, Gabbai FB, Blantz RC, Thomson SC. Interactive control of renal function by alpha 2-adrenergic system and nitric oxide: role of angiotensin II. J Cardiovasc Pharmacol 26: 916–922, 1995. doi: 10.1097/00005344-199512000-00010. [DOI] [PubMed] [Google Scholar]
- 700.Van den Berghe G. Fructose: metabolism and short-term effects on carbohydrate and purine metabolic pathways. Prog Biochem Pharmacol 21: 1–32, 1986. [PubMed] [Google Scholar]
- 701.Van Itallie CM, Rogan S, Yu A, Vidal LS, Holmes J, Anderson JM. Two splice variants of claudin-10 in the kidney create paracellular pores with different ion selectivities. Am J Physiol Renal Physiol 291: F1288–F1299, 2006. doi: 10.1152/ajprenal.00138.2006. [DOI] [PubMed] [Google Scholar]
- 702.van Vark LC, Bertrand M, Akkerhuis KM, Brugts JJ, Fox K, Mourad JJ, Boersma E. Angiotensin-converting enzyme inhibitors reduce mortality in hypertension: a meta-analysis of randomized clinical trials of renin-angiotensin-aldosterone system inhibitors involving 158,998 patients. Eur Heart J 33: 2088–2097, 2012. doi: 10.1093/eurheartj/ehs075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703.Van Vliet BN, Chafe LL, Halfyard SJ, Leonard AM. Distinct rapid and slow phases of salt-induced hypertension in Dahl salt-sensitive rats. J Hypertens 24: 1599–1606, 2006. doi: 10.1097/01.hjh.0000239296.25260.e0. [DOI] [PubMed] [Google Scholar]
- 704.Van Vliet BN, Montani JP. The time course of salt-induced hypertension, and why it matters. Int J Obes 32, Suppl 6: S35–S47, 2008. doi: 10.1038/ijo.2008.205. [DOI] [PubMed] [Google Scholar]
- 705.Vásquez-Vivar J, Kalyanaraman B, Martásek P. The role of tetrahydrobiopterin in superoxide generation from eNOS: enzymology and physiological implications. Free Radic Res 37: 121–127, 2003. doi: 10.1080/1071576021000040655. [DOI] [PubMed] [Google Scholar]
- 706.Velázquez H, Silva T. Cloning and localization of KCC4 in rabbit kidney: expression in distal convoluted tubule. Am J Physiol Renal Physiol 285: F49–F58, 2003. doi: 10.1152/ajprenal.00389.2002. [DOI] [PubMed] [Google Scholar]
- 707.Vos MB, Kimmons JE, Gillespie C, Welsh J, Blanck HM. Dietary fructose consumption among US children and adults: the Third National Health and Nutrition Examination Survey. Medscape J Med 10: 160, 2008. [PMC free article] [PubMed] [Google Scholar]
- 708.Wakui H, Tamura K, Masuda S, Tsurumi-Ikeya Y, Fujita M, Maeda A, Ohsawa M, Azushima K, Uneda K, Matsuda M, Kitamura K, Uchida S, Toya Y, Kobori H, Nagahama K, Yamashita A, Umemura S. Enhanced angiotensin receptor-associated protein in renal tubule suppresses angiotensin-dependent hypertension. Hypertension 61: 1203–1210, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 709.Wald H, Scherzer P, Rubinger D, Popovtzer MM. Effect of indomethacin in vivo and PGE2 in vitro on MTAL Na-K-ATPase of the rat kidney. Pflugers Arch 415: 648–650, 1990. doi: 10.1007/BF02583521. [DOI] [PubMed] [Google Scholar]
- 710.Waldegger S, Jeck N, Barth P, Peters M, Vitzthum H, Wolf K, Kurtz A, Konrad M, Seyberth HW. Barttin increases surface expression and changes current properties of ClC-K channels. Pflugers Arch 444: 411–418, 2002. doi: 10.1007/s00424-002-0819-8. [DOI] [PubMed] [Google Scholar]
- 711.Walder RY, Morgan DA, Haynes WG, Sigmund RD, McClain AM, Stokes JB, Mark AL. Genetic characterization of the “new” Harlan Sprague Dawley Dahl salt-sensitive rats. Hypertension 27: 546–551, 1996. doi: 10.1161/01.HYP.27.3.546. [DOI] [PubMed] [Google Scholar]
- 712.Wang D, An SJ, Wang WH, McGiff JC, Ferreri NR. CaR-mediated COX-2 expression in primary cultured mTAL cells. Am J Physiol Renal Physiol 281: F658–F664, 2001. doi: 10.1152/ajprenal.2001.281.4.F658. [DOI] [PubMed] [Google Scholar]
- 713.Wang D, Pedraza PL, Abdullah HI, McGiff JC, Ferreri NR. Calcium-sensing receptor-mediated TNF production in medullary thick ascending limb cells. Am J Physiol Renal Physiol 283: F963–F970, 2002. doi: 10.1152/ajprenal.00108.2002. [DOI] [PubMed] [Google Scholar]
- 714.Wang H, Carretero OA, Garvin JL. Nitric oxide produced by THAL nitric oxide synthase inhibits TGF. Hypertension 39: 662–666, 2002. doi: 10.1161/hy0202.103470. [DOI] [PubMed] [Google Scholar]
- 715.Wang L, Shen C, Liu H, Wang S, Chen X, Roman RJ, Juncos LA, Lu Y, Wei J, Zhang J, Yip KP, Liu R. Shear stress blunts tubuloglomerular feedback partially mediated by primary cilia and nitric oxide at the macula densa. Am J Physiol Regul Integr Comp Physiol 309: R757–R766, 2015. doi: 10.1152/ajpregu.00173.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 716.Wang M, Luan H, Wu P, Fan L, Wang L, Duan X, Zhang D, Wang WH, Gu R. Angiotensin II stimulates basolateral 50-pS K channels in the thick ascending limb. Am J Physiol Renal Physiol 306: F509–F516, 2014. doi: 10.1152/ajprenal.00476.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717.Wang T. The effects of the potassium channel opener minoxidil on renal electrolytes transport in the loop of henle. J Pharmacol Exp Ther 304: 833–840, 2003. doi: 10.1124/jpet.102.043380. [DOI] [PubMed] [Google Scholar]
- 718.Wang T, Wang WH, Klein-Robbenhaar G, Giebisch G. Effects of a novel KATP channel blocker on renal tubule function and K channel activity. J Pharmacol Exp Ther 273: 1382–1389, 1995. [PubMed] [Google Scholar]
- 719.Wang W, Lu M. Effect of arachidonic acid on activity of the apical K+ channel in the thick ascending limb of the rat kidney. J Gen Physiol 106: 727–743, 1995. doi: 10.1085/jgp.106.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720.Wang W, Lu M, Balazy M, Hebert SC. Phospholipase A2 is involved in mediating the effect of extracellular Ca2+ on apical K+ channels in rat TAL. Am J Physiol Renal Physiol 273: F421–F429, 1997. doi: 10.1152/ajprenal.1997.273.3.F421. [DOI] [PubMed] [Google Scholar]
- 721.Wang W, Mitra A, Poole B, Falk S, Lucia MS, Tayal S, Schrier R. Endothelial nitric oxide synthase-deficient mice exhibit increased susceptibility to endotoxin-induced acute renal failure. Am J Physiol Renal Physiol 287: F1044–F1048, 2004. doi: 10.1152/ajprenal.00136.2004. [DOI] [PubMed] [Google Scholar]
- 722.Wang WH. Regulation of ROMK (Kir1.1) channels: new mechanisms and aspects. Am J Physiol Renal Physiol 290: F14–F19, 2006. doi: 10.1152/ajprenal.00093.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723.Wang WH. Two types of K+ channel in thick ascending limb of rat kidney. Am J Physiol Renal Physiol 267: F599–F605, 1994. doi: 10.1152/ajprenal.1994.267.4.F599. [DOI] [PubMed] [Google Scholar]
- 724.Wang WH, Lu M, Hebert SC. Cytochrome P-450 metabolites mediate extracellular Ca2+-induced inhibition of apical K+ channels in the TAL. Am J Physiol Cell Physiol 271: C103–C111, 1996. doi: 10.1152/ajpcell.1996.271.1.C103. [DOI] [PubMed] [Google Scholar]
- 725.Wang Y, Wang DH. A novel mechanism contributing to development of Dahl salt-sensitive hypertension: role of the transient receptor potential vanilloid type 1. Hypertension 47: 609–614, 2006. doi: 10.1161/01.HYP.0000197390.10412.c4. [DOI] [PubMed] [Google Scholar]
- 726.Wangensteen R, Rodríguez-Gomez I, Moreno JM, Vargas F, Alvarez-Guerra M. Chronic nitric oxide blockade modulates renal Na-K-2Cl cotransporters. J Hypertens 24: 2451–2458, 2006. doi: 10.1097/01.hjh.0000251907.93298.44. [DOI] [PubMed] [Google Scholar]
- 727.Watanabe S, Fukumoto S, Chang H, Takeuchi Y, Hasegawa Y, Okazaki R, Chikatsu N, Fujita T. Association between activating mutations of calcium-sensing receptor and Bartter’s syndrome. Lancet 360: 692–694, 2002. doi: 10.1016/S0140-6736(02)09842-2. [DOI] [PubMed] [Google Scholar]
- 728.Watt PA, Thurston H. Endothelium-dependent relaxation in resistance vessels from the spontaneously hypertensive rats. J Hypertens 7: 661–666, 1989. doi: 10.1097/00004872-198908000-00010. [DOI] [PubMed] [Google Scholar]
- 729.Watts BA III, George T, Good DW. Aldosterone inhibits apical NHE3 and absorption via a nongenomic ERK-dependent pathway in medullary thick ascending limb. Am J Physiol Renal Physiol 291: F1005–F1013, 2006. doi: 10.1152/ajprenal.00507.2005. [DOI] [PubMed] [Google Scholar]
- 730.Weder AB, Gleiberman L, Sachdeva A. Whites excrete a water load more rapidly than blacks. Hypertension 53: 715–718, 2009. doi: 10.1161/HYPERTENSIONAHA.108.121665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 731.Wei Y, Babilonia E, Pedraza PL, Ferreri NR, Wang WH. Acute application of TNF stimulates apical 70-pS K+ channels in the thick ascending limb of rat kidney. Am J Physiol Renal Physiol 285: F491–F497, 2003. doi: 10.1152/ajprenal.00104.2003. [DOI] [PubMed] [Google Scholar]
- 732.Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension 27: 481–490, 1996. doi: 10.1161/01.HYP.27.3.481. [DOI] [PubMed] [Google Scholar]
- 733.Welch WJ, Blau J, Xie H, Chabrashvili T, Wilcox CS. Angiotensin-induced defects in renal oxygenation: role of oxidative stress. Am J Physiol Heart Circ Physiol 288: H22–H28, 2005. doi: 10.1152/ajpheart.00626.2004. [DOI] [PubMed] [Google Scholar]
- 734.Welch WJ, Tojo A, Wilcox CS. Roles of NO and oxygen radicals in tubuloglomerular feedback in SHR. Am J Physiol Renal Physiol 278: F769–F776, 2000. doi: 10.1152/ajprenal.2000.278.5.F769. [DOI] [PubMed] [Google Scholar]
- 735.Welling PA, Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol 297: F849–F863, 2009. doi: 10.1152/ajprenal.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736.Wengert M, Ribeiro MC, Abreu TP, Coutinho-Silva R, Leão-Ferreira LR, Pinheiro AA, Caruso-Neves C. Protein kinase C-mediated ATP stimulation of Na+-ATPase activity in LLC-PK1 cells involves a P2Y2 and/or P2Y4 receptor. Arch Biochem Biophys 535: 136–142, 2013. doi: 10.1016/j.abb.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 737.Wigertz K, Palacios C, Jackman LA, Martin BR, McCabe LD, McCabe GP, Peacock M, Pratt JH, Weaver CM. Racial differences in calcium retention in response to dietary salt in adolescent girls. Am J Clin Nutr 81: 845–850, 2005. doi: 10.1093/ajcn/81.4.845. [DOI] [PubMed] [Google Scholar]
- 738.Williams JM, Fan F, Murphy S, Schreck C, Lazar J, Jacob HJ, Roman RJ. Role of 20-HETE in the antihypertensive effect of transfer of chromosome 5 from Brown Norway to Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 302: R1209–R1218, 2012. doi: 10.1152/ajpregu.00604.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 739.Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med 27: 322–328, 1999. doi: 10.1016/S0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 740.Wronski T, Seeliger E, Persson PB, Forner C, Fichtner C, Scheller J, Flemming B. The step response: a method to characterize mechanisms of renal blood flow autoregulation. Am J Physiol Renal Physiol 285: F758–F764, 2003. doi: 10.1152/ajprenal.00420.2002. [DOI] [PubMed] [Google Scholar]
- 741.Wu P, Gao Z, Ye S, Qi Z. Nitric oxide inhibits basolateral 10-pS Cl− channels through the cGMP/PKG signaling pathway in the thick ascending limb of C57BL/6 mice. Am J Physiol Renal Physiol 310: F755–F762, 2016. doi: 10.1152/ajprenal.00270.2015. [DOI] [PubMed] [Google Scholar]
- 742.Wu P, Wang M, Luan H, Li L, Wang L, Wang WH, Gu R. Angiotensin II stimulates basolateral 10-pS Cl channels in the thick ascending limb. Hypertension 61: 1211–1217, 2013. doi: 10.1161/HYPERTENSIONAHA.111.01069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 743.Wu R, Lamontagne D, de Champlain J. Antioxidative properties of acetylsalicylic Acid on vascular tissues from normotensive and spontaneously hypertensive rats. Circulation 105: 387–392, 2002. doi: 10.1161/hc0302.102609. [DOI] [PubMed] [Google Scholar]
- 744.Wu R, Millette E, Wu L, de Champlain J. Enhanced superoxide anion formation in vascular tissues from spontaneously hypertensive and desoxycorticosterone acetate-salt hypertensive rats. J Hypertens 19: 741–748, 2001. doi: 10.1097/00004872-200104000-00011. [DOI] [PubMed] [Google Scholar]
- 745.Xu JZ, Hall AE, Peterson LN, Bienkowski MJ, Eessalu TE, Hebert SC. Localization of the ROMK protein on apical membranes of rat kidney nephron segments. Am J Physiol Renal Physiol 273: F739–F748, 1997. doi: 10.1152/ajprenal.1997.273.5.F739. [DOI] [PubMed] [Google Scholar]
- 746.Yamada SS, Sassaki AL, Fujihara CK, Malheiros DM, De Nucci G, Zatz R. Effect of salt intake and inhibitor dose on arterial hypertension and renal injury induced by chronic nitric oxide blockade. Hypertension 27: 1165–1172, 1996. doi: 10.1161/01.HYP.27.5.1165. [DOI] [PubMed] [Google Scholar]
- 747.Yamada Y, Tsuboi K, Hattori T, Murase T, Ohtake M, Furukawa M, Ueyama J, Nishiyama A, Murohara T, Nagata K. Mechanism underlying the efficacy of combination therapy with losartan and hydrochlorothiazide in rats with salt-sensitive hypertension. Hypertens Res 34: 809–816, 2011. doi: 10.1038/hr.2011.34. [DOI] [PubMed] [Google Scholar]
- 748.Yamauchi K, Rai T, Kobayashi K, Sohara E, Suzuki T, Itoh T, Suda S, Hayama A, Sasaki S, Uchida S. Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc Natl Acad Sci USA 101: 4690–4694, 2004. doi: 10.1073/pnas.0306924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 749.Yan Y, Haller S, Shapiro A, Malhotra N, Tian J, Xie Z, Malhotra D, Shapiro JI, Liu J. Ouabain-stimulated trafficking regulation of the Na/K-ATPase and NHE3 in renal proximal tubule cells. Mol Cell Biochem 367: 175–183, 2012. doi: 10.1007/s11010-012-1331-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 750.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells [see comments]. Nature 332: 411–415, 1988. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 751.Yanes LL, Reckelhoff JF. Postmenopausal hypertension. Am J Hypertens 24: 740–749, 2011. doi: 10.1038/ajh.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Yanes LL, Romero DG, Iles JW, Iliescu R, Gomez-Sanchez C, Reckelhoff JF. Sexual dimorphism in the renin-angiotensin system in aging spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol 291: R383–R390, 2006. doi: 10.1152/ajpregu.00510.2005. [DOI] [PubMed] [Google Scholar]
- 753.Yanes LL, Romero DG, Iliescu R, Zhang H, Davis D, Reckelhoff JF. Postmenopausal hypertension: role of the Renin-Angiotensin system. Hypertension 56: 359–363, 2010. doi: 10.1161/HYPERTENSIONAHA.110.152975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 754.Yanes LL, Sartori-Valinotti JC, Iliescu R, Romero DG, Racusen LC, Zhang H, Reckelhoff JF. Testosterone-dependent hypertension and upregulation of intrarenal angiotensinogen in Dahl salt-sensitive rats. Am J Physiol Renal Physiol 296: F771–F779, 2009. doi: 10.1152/ajprenal.90389.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 755.Yang C, Stingo FC, Ahn KW, Liu P, Vannucci M, Laud PW, Skelton M, O’Connor P, Kurth T, Ryan RP, Moreno C, Tsaih SW, Patone G, Hummel O, Jacob HJ, Liang M, Cowley AW Jr. Increased proliferative cells in the medullary thick ascending limb of the loop of Henle in the Dahl salt-sensitive rat. Hypertension 61: 208–215, 2013. doi: 10.1161/HYPERTENSIONAHA.112.199380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 756.Yang S, Silldorff EP, Pallone TL. Effect of norepinephrine and acetylcholine on outer medullary descending vasa recta. Am J Physiol Heart Circ Physiol 269: H710–H716, 1995. doi: 10.1152/ajpheart.1995.269.2.H710. [DOI] [PubMed] [Google Scholar]
- 757.Yang T, Hassan S, Huang YG, Smart AM, Briggs JP, Schnermann JB. Expression of PTHrP, PTH/PTHrP receptor, and Ca2+-sensing receptor mRNAs along the rat nephron. Am J Physiol Renal Physiol 272: F751–F758, 1997. doi: 10.1152/ajprenal.1997.272.6.F751. [DOI] [PubMed] [Google Scholar]
- 758.Yang T, Huang YG, Singh I, Schnermann J, Briggs JP. Localization of bumetanide- and thiazide-sensitive Na-K-Cl cotransporters along the rat nephron. Am J Physiol Renal Physiol 271: F931–F939, 1996. doi: 10.1152/ajprenal.1996.271.4.F931. [DOI] [PubMed] [Google Scholar]
- 759.Yavuzer H, Yavuzer S, Cengiz M, Erman H, Doventas A, Balci H, Erdincler DS, Uzun H. Biomarkers of lipid peroxidation related to hypertension in aging. Hypertens Res 39: 342–348, 2016. doi: 10.1038/hr.2015.156. [DOI] [PubMed] [Google Scholar]
- 760.Yip KP, Tse CM, Donowitz M, Marsh DJ. Immunolocalization of NAVH exchanger isoform NHE-2 in rat kidney with confocal fluorescence microscopy. FASEB J 10: A370, 1996. [Google Scholar]
- 761.Yoshikawa M, Uchida S, Yamauchi A, Miyai A, Tanaka Y, Sasaki S, Marumo F. Localization of rat CLC-K2 chloride channel mRNA in the kidney. Am J Physiol Renal Physiol 276: F552–F558, 1999. doi: 10.1152/ajprenal.1999.276.4.F552. [DOI] [PubMed] [Google Scholar]
- 762.Yu L, Bao HF, Self JL, Eaton DC, Helms MN. Aldosterone-induced increases in superoxide production counters nitric oxide inhibition of epithelial Na channel activity in A6 distal nephron cells. Am J Physiol Renal Physiol 293: F1666–F1677, 2007. doi: 10.1152/ajprenal.00444.2006. [DOI] [PubMed] [Google Scholar]
- 763.Yu M, Lopez B, Dos Santos EA, Falck JR, Roman RJ. Effects of 20-HETE on Na+ transport and Na+-K+-ATPase activity in the thick ascending loop of Henle. Am J Physiol Regul Integr Comp Physiol 292: R2400–R2405, 2007. doi: 10.1152/ajpregu.00791.2006. [DOI] [PubMed] [Google Scholar]
- 764.Yuan B, Cowley AW Jr. Evidence that reduced renal medullary nitric oxide synthase activity of dahl s rats enables small elevations of arginine vasopressin to produce sustained hypertension. Hypertension 37: 524–528, 2001. doi: 10.1161/01.HYP.37.2.524. [DOI] [PubMed] [Google Scholar]
- 765.Yun CH, Tse CM, Nath SK, Levine SA, Brant SR, Donowitz M. Mammalian Na+/H+ exchanger gene family: structure and function studies. Am J Physiol Gastrointestinal Physiol 269: G1–G11, 1995. doi: 10.1113/jphysiol.1995.sp020558. [DOI] [PubMed] [Google Scholar]
- 766.Zacchia M, Capolongo G, Rinaldi L, Capasso G. The importance of the thick ascending limb of Henle’s loop in renal physiology and pathophysiology. Int J Nephrol Renovasc Dis 11: 81–92, 2018. doi: 10.2147/IJNRD.S154000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 767.Zamofing D, Rossier BC, Geering K. Inhibition of N-glycosylation affects transepithelial Na+ but not Na+-K+-ATPase transport. Am J Physiol Cell Physiol 256: C958–C966, 1989. doi: 10.1152/ajpcell.1989.256.5.C958. [DOI] [PubMed] [Google Scholar]
- 768.Zamofing D, Rossier BC, Geering K. Role of the Na,K-ATPase beta-subunit in the cellular accumulation and maturation of the enzyme as assessed by glycosylation inhibitors. J Membr Biol 104: 69–79, 1988. doi: 10.1007/BF01871903. [DOI] [PubMed] [Google Scholar]
- 769.Zeidel ML, Brady HR, Kone BC, Gullans SR, Brenner BM. Endothelin, a peptide inhibitor of Na+-K+-ATPase in intact renaltubular epithelial cells. Am J Physiol Cell Physiol 257: C1101–C1107, 1989. doi: 10.1152/ajpcell.1989.257.6.C1101. [DOI] [PubMed] [Google Scholar]
- 770.Zhang C, Wang L, Su XT, Lin DH, Wang WH. KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the cortical thick ascending limb. Am J Physiol Renal Physiol 308: F1288–F1296, 2015. doi: 10.1152/ajprenal.00687.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 771.Zhang Q, Lin L, Lu Y, Liu H, Duan Y, Zhu X, Zou C, Manning RD Jr, Liu R. Interaction between nitric oxide and superoxide in the macula densa in aldosterone-induced alterations of tubuloglomerular feedback. Am J Physiol Renal Physiol 304: F326–F332, 2013. doi: 10.1152/ajprenal.00501.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 772.Zhang ZR, Chu WF, Song B, Gooz M, Zhang JN, Yu CJ, Jiang S, Baldys A, Gooz P, Steele S, Owsianik G, Nilius B, Komlosi P, Bell PD. TRPP2 and TRPV4 form an EGF-activated calcium permeable channel at the apical membrane of renal collecting duct cells. PLoS One 8: e73424, 2013. doi: 10.1371/journal.pone.0073424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 773.Zheleznova NN, Yang C, Cowley AW Jr. Role of Nox4 and p67phox subunit of Nox2 in ROS production in response to increased tubular flow in the mTAL of Dahl salt-sensitive rats. Am J Physiol Renal Physiol 311: F450–F458, 2016. doi: 10.1152/ajprenal.00187.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 774.Zheleznova NN, Yang C, Ryan RP, Halligan BD, Liang M, Greene AS, Cowley AW Jr. Mitochondrial proteomic analysis reveals deficiencies in oxygen utilization in medullary thick ascending limb of Henle in the Dahl salt-sensitive rat. Physiol Genomics 44: 829–842, 2012. doi: 10.1152/physiolgenomics.00060.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 775.Zheng W, Verlander JW, Lynch IJ, Cash M, Shao J, Stow LR, Cain BD, Weiner ID, Wall SM, Wingo CS. Cellular distribution of the potassium channel KCNQ1 in normal mouse kidney. Am J Physiol Renal Physiol 292: F456–F466, 2007. doi: 10.1152/ajprenal.00087.2006. [DOI] [PubMed] [Google Scholar]
- 776.Zhou X, Forrest MJ, Sharif-Rodriguez W, Forrest G, Szeto D, Urosevic-Price O, Zhu Y, Stevenson AS, Zhou Y, Stribling S, Dajee M, Walsh SP, Pasternak A, Sullivan KA. Chronic inhibition of renal outer medullary potassium channel not only prevented but also reversed development of hypertension and end-organ damage in Dahl salt-sensitive rats. Hypertension 69: 332–338, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08358. [DOI] [PubMed] [Google Scholar]
- 777.Zhou X, Zhang Z, Shin MK, Horwitz SB, Levorse JM, Zhu L, Sharif-Rodriguez W, Streltsov DY, Dajee M, Hernandez M, Pan Y, Urosevic-Price O, Wang L, Forrest G, Szeto D, Zhu Y, Cui Y, Michael B, Balogh LA, Welling PA, Wade JB, Roy S, Sullivan KA. Heterozygous disruption of renal outer medullary potassium channel in rats is associated with reduced blood pressure. Hypertension 62: 288–294, 2013. doi: 10.1161/HYPERTENSIONAHA.111.01051. [DOI] [PubMed] [Google Scholar]
- 778.Zou AP, Drummond HA, Roman RJ. Role of 20-HETE in elevating loop chloride reabsorption in Dahl SS/Jr rats. Hypertension 27: 631–635, 1996. doi: 10.1161/01.HYP.27.3.631. [DOI] [PubMed] [Google Scholar]
- 779.Zou AP, Li N, Cowley AW Jr. Production and actions of superoxide in the renal medulla. Hypertension 37: 547–553, 2001. doi: 10.1161/01.HYP.37.2.547. [DOI] [PubMed] [Google Scholar]
- 780.Zou LX, Imig JD, von Thun AM, Hymel A, Ono H, Navar LG. Receptor-mediated intrarenal angiotensin II augmentation in angiotensin II-infused rats. Hypertension 28: 669–677, 1996. doi: 10.1161/01.HYP.28.4.669. [DOI] [PubMed] [Google Scholar]
- 781.Zuo E, Cai YJ, Li K, Wei Y, Wang BA, Sun Y, Liu Z, Liu J, Hu X, Wei W, Huo X, Shi L, Tang C, Liang D, Wang Y, Nie YH, Zhang CC, Yao X, Wang X, Zhou C, Ying W, Wang Q, Chen RC, Shen Q, Xu GL, Li J, Sun Q, Xiong ZQ, Yang H. One-step generation of complete gene knockout mice and monkeys by CRISPR/Cas9-mediated gene editing with multiple sgRNAs. Cell Res 27: 933–945, 2017. doi: 10.1038/cr.2017.81. [DOI] [PMC free article] [PubMed] [Google Scholar]