

Abstract
The renin-angiotensin-aldosterone system plays crucial roles in cardiovascular physiology and pathophysiology. However, many of the signaling mechanisms have been unclear. The angiotensin II (ANG II) type 1 receptor (AT1R) is believed to mediate most functions of ANG II in the system. AT1R utilizes various signal transduction cascades causing hypertension, cardiovascular remodeling, and end organ damage. Moreover, functional cross-talk between AT1R signaling pathways and other signaling pathways have been recognized. Accumulating evidence reveals the complexity of ANG II signal transduction in pathophysiology of the vasculature, heart, kidney, and brain, as well as several pathophysiological features, including inflammation, metabolic dysfunction, and aging. In this review, we provide a comprehensive update of the ANG II receptor signaling events and their functional significances for potential translation into therapeutic strategies. AT1R remains central to the system in mediating physiological and pathophysiological functions of ANG II, and participation of specific signaling pathways becomes much clearer. There are still certain limitations and many controversies, and several noteworthy new concepts require further support. However, it is expected that rigorous translational research of the ANG II signaling pathways including those in large animals and humans will contribute to establishing effective new therapies against various diseases.
I. INTRODUCTION
The renin-angiotensin-aldosterone system (RAS or RAAS, hereafter RAS) is involved in numerous physiological functions that regulate vasoconstriction, fluid volume regulation, cardiac output, cell growth, and vascular wall integrity, to name a few. Production of angiotensin peptides is first initiated by the synthesis and processing of preprorenin in juxtaglomerular cells neighboring the renal glomerulus with subsequent proteolytic cleavage of the signal peptide, intracellular sorting of prorenin to dense-core secretory vesicles, and cleavage of the prosegment, producing catalytically active renin that is secreted in the systemic circulation. Classically, renin cleaves liver-derived angiotensinogen (AGT) into angiotensin I (ANG I), a decapeptide [ANG (1–10)] which is then further processed by angiotensin-converting enzyme (ACE) into the octapeptide ANG II [Asp-Arg-Val-Tyr-Ile-His-Pro-Phe, ANG (1–8)] (462).
In addition to the systemic circulating RAS (also known as the endocrine RAS), the importance of local generation and action of the RAS has been well documented (177, 547, 919). Although ANG II is a vasoactive peptide, it regulates many aspects of cellular function unrelated to vasoconstriction in different tissues. Under pathological circumstances, the RAS also contributes to various diseases. At its most basic function, ANG II is known to physiologically regulate blood pressure and is a key player in hypertension. The continued clinical effectiveness of blockers of RAS as antihypertensives together with their benefits for target organ protection evidences the continued clinical importance of RAS research (189, 1042). The complexity of the system is further illustrated by recent accumulating findings supporting the production and action of alternative angiotensin peptides such as angiotensin-1–7 [ANG (1–7)], which is believed to exert its action through a G protein-coupled receptor (GPCR), Mas. The effects of ANG (1–7)/Mas activation are generally considered to be counter to those of ANG II (46).
Historically, the first non-peptide ANG II receptor antagonist, losartan-sensitive receptor was termed the ANG II type 1/AT1 receptor (AT1R), and the losartan-insensitive receptor was termed AT2 receptor (AT2R) (102). In 1991, two research groups isolated cDNA (termed AGTR1) encoding the mammalian AT1R (728, 916). AT2R cDNA (AGTR2) was then cloned in 1993 (452, 720). Both receptor subtypes belong to the seven-transmembrane GPCR superfamily. In the early 1990s, induction of protein tyrosine phosphorylation and activation of mitogen-activated protein kinase (p42/p44 MAPK)/extracellular signal regulated kinase (ERK1/2) through AT1R were reported and the concept that ANG II has direct growth promoting effects on cardiac myocytes, fibroblasts, and vascular smooth muscle cells (VSMCs) causing cardiovascular remodeling was established (317, 897). These findings led to identification of several AT1R signaling mechanisms shared with growth factor receptors (252, 337, 926, 1060). Another historical discovery is identification of reactive oxygen species (ROS) produced via NAD(P)H oxidase activation as a critical second messenger of AT1R (336). This finding became a critical foundation for the concept that ANG II acts like an inflammatory cytokine (99). The basic understanding that the AT1R contributes to hypertension and various complications via second messengers, activation of various kinases, and induction of remodeling and inflammation remains solid (470, 1098).
Over the past two decades, several seminal review articles have highlighted the unique signal transduction mechanisms of ANG II in the cardiovascular system (379, 682, 1060). Since then, research has further uncovered many key molecular and cellular mechanisms regarding ANG II and its signaling pathways beyond their roles in blood pressure regulation (189, 1042, 1098). This review aims to build upon and expand previous reports and highlights the recently explored novel mechanisms by which ANG II influences physiology and pathophysiology. The review will place an emphasis on signal transduction and how it relates to resultant physiological and pathophysiological phenotypes in response to ANG II in distinct cell types and tissues, including those in the vasculature, heart, kidney, adipose tissue, brain, and skeletal muscle (TABLE 1). We acknowledge that there are many other important areas and topics in RAS signaling research that are not covered in this review. Please refer to recent review articles for ANG II mechanisms involved in regulation of these topic areas (TABLE 2).
Table 1.
ANG II physiology and pathophysiology covered in this article
| System | Physiology/Pathophysiology |
|---|---|
| Physiology | |
| Vascular | Increase in vascular resistance |
| Heart | Increase in cardiac output with positive inotropic and chronotropic effects |
| Kidney | Increase in salt/water intake and suppression of renin secretion |
| Brain | Stimulate sympathetic nervous system, increase in fluid intake and metabolic rate |
| Pathophysiology | |
| Vascular | Hypertension, hypertrophy, fibrosis, atherosclerosis, aneurysms, and endothelial dysfunction |
| Heart | Hypertrophy, fibrosis, ventricular arrhythmias, and atrial fibrillation |
| Kidney | Hypertension, fibrosis, and inflammation |
| Brain | Hypertension, cognitive dysfunction, Alzheimer disease, and stroke |
| Adipose tissue | Inflammation and insulin resistance |
| Skeletal muscle | Wasting/atrophy and insulin resistance |
| Immune cells | Inflammation and cytokine production |
Table 2.
ANG II signal topics not covered in this article
| Topics | Reference Nos. |
|---|---|
| Adrenal hormone secretion | 755 |
| Angiogenesis | 1150 |
| AT1R agonistic autoantibodies | 1169 |
| Autoimmune diseases | 138 |
| Cancer/malignancies | 877 |
| Chronic obstructive pulmonary disease | 959 |
| Diabetic retinopathy | 1148 |
| Erectile dysfunction | 291 |
| Gastrointestinal system | 270 |
| Gender differences | 1008 |
| Hematopoiesis | 364 |
| Insulin secretion | 617 |
| Lipid metabolism | 841 |
| Nonalcoholic liver disease | 335, 717a |
| Preeclampsia | 1091 |
| Pulmonary arterial hypertension | 97 |
| Reproduction system | 376 |
| Stem/progenitor cells | 248 |
| Thrombosis | 290 |
A. New and Old Angiotensin Family Ligands
ANG II was originally thought to be the main product of the RAS and primary ligand of the ANG II receptor. An update regarding the molecular understanding of classical ANG II regulation via AGT, renin, and ACE has been described recently (985). However, the discovery of additional RAS components has changed the paradigm of ANG II signaling. The family of RAS has expanded to include peptides consisting of angiotensin-1–7 [ANG (1–7)], angiotensin-2–8 (ANG III), angiotensin-3–8 (ANG IV), and angiotensin-1–12 [ANG (1–12)]. ANG (1–7) is formed by the catalytic action of ACE2 on ANG II and is thought to balance the RAS system by promoting an antagonistic effect on the responses elicited by ANG II-AT1R such as vasodilation (278, 280). ANG III is produced from ANG II by enzymatic cleavage by an aminopeptidase which fuels similar physiological responses as ANG II (1228). ANG IV is cleaved from ANG III by another aminopeptidase and contributes to blood flow regulation, learning and memory, and neuronal development (363). ANG (1–12) has been reported to serve as an upstream renin-independent precursor peptide for ANG I and ANG II (15, 734). ANG (1–9) is generated from ANG I by several carboxypeptidase-type enzymes, including carboxypeptidase A, cathepsin A, and ACE2 (767). In addition to being converted to ANG (1–7) by ACE or ANG (2–9) by aminopeptidase A, evidence suggests that ANG (1–9) exerts direct actions that counterregulate ANG II. ANG (1–9) may also be a competitive inhibitor of ACE (767). Alamandine [Ala1-ANG (1–7)] is endogenously synthesized from ANG (1–7) and possesses many of the functional properties of ANG (1–7) (121). Angiotensin A has been identified in human plasma (428), and it differs from ANG II by substitution of Asp for Ala in the first amino acid position. Angiotensin A has similar physiological functions as ANG II in the cardiovascular system (187). ANG II-like peptide angioprotectin has also been identified in human plasma. It differs from ANG II by substitution of Pro1 and Glu2 in place of Asp1 and Arg2. This peptide antagonizes contractile actions of ANG II via binding to Mas receptor. A more in-depth review of the basic, as well as new, components of the RAS system can be found elsewhere (49, 140, 938).
B. Receptors for the Angiotensin Peptides
While ANG II-dependent signaling is the focus of this review, significant advancement has also been made for signaling mechanisms utilized by new members of the angiotensin family (FIGURE 1). ANG (1–7) has been reported to elicit its effects through the GPCR Mas receptor whereby it mediates antagonistic effects of ANG II, including vasodilation and antihypertensive and antifibrosis effects (800). Similar to Mas activation by ANG (1–7), alamandine also promotes antihypertensive effects through the Mas-related GPCR member D, MrgD, to produce nitric oxide (NO) (542). Interestingly, MrgD appears to be a second receptor for ANG (1–7). Both Mas and MrgD produce cAMP upon ANG (1–7) binding (1044). ANG III binds and activates the classical angiotensin receptors AT1R and AT2R (682). ANG (1–9) exerts its direct effects via AT2R (767). While still controversial, AT2R is recognized as a counter balance to AT1R-dependent signaling. General functions of the AT2R include vasodilation through NO and cGMP stimulation, natriuresis, antiangiogenesis, antiproliferation, and decreased fibrosis, and these effects are observed in various tissues including endothelium, vascular smooth muscle, heart, brain, and kidney (660, 790, 1011) . ANG IV binds to AT4R, which is also now known as insulin-regulated aminopeptidase (IRAP). ANG IV activation of AT4R causes vasorelaxation in renal and cerebral vascular beds, stimulates endothelial nitric oxide synthase (eNOS), enhances protein synthesis in cardiac fibroblasts, and can increase renal cortical blood flow and urinary sodium excretion (132).
FIGURE 1.

Identified angiotensin (ANG) peptides and receptors in RAS signaling. Prorenin and renin have both been identified to bind to the (pro)renin receptor. ANG I is cleaved by angiotensin converting enzyme (ACE) to ANG II which can stimulate the AT1R and AT2R. ANG I can also be cleaved into ANG (1–9) which can bind to the AT2R. ANG II can further diverge into either ANG (1–7) through ACE2 or ANG III through an aminopeptidase. ANG (1–7) has been identified to bind to the AT2R, MAS receptor, and the MrgD receptor. ANG (1–7) can also form alamandine which binds to the MrgD receptor. ANG III binds to the AT1R and AT2R. ANG III can be further cleaved into ANG IV which binds to the AT4R.
C. Limitations and Pitfalls in ANG II Signaling Research
It is important to acknowledge several limitations and pitfalls in ANG II signal transduction research. Pharmacological doses of ANG II are most often used in research, which may or may not mimic physiological conditions. Most of the in vitro studies use single high-dose stimulation by ANG II (~100 nM) whose half-life is quite short in culture medium (note that a constitutive ANG II producing vector is available) (1032). Alternatively, to maintain chronic ANG II stimulation, one can repeatedly add ANG II to the cell culture. However, rapid conversion of ANG II to ANG III and ANG (1–7) in culture medium (61) will further complicate the interpretation.
Although systemic ANG II infusion via osmotic mini-pump may increase the circulating levels of ANG II, it may or may not reconstitute increasing tissue/local RAS activity under pathophysiology in humans. In humans, plasma ANG II concentrations in healthy volunteers were reported to be 5~35 fmol/ml (249). Recent human studies reported that ANG II and ANG (1–7) concentrations in healthy volunteers were ~8.7 fmol/ml and 3.4–5.6 fmol/ml, respectively (385, 761). Physiological plasma and kidney concentrations of ANG II were reported to be ~50 fmol/ml and ~160 fmol/g in rats, which increased to ~130 fmol/ml and ~350 fmol/g, respectively, upon 2 wk ANG II infusion (40 ng/min per 180–200 g) (1280). In C57BL/6 mice, physiological plasma and kidney concentrations of ANG II were reported to be ~130 fmol/ml and ~520 fmol/g, which increased to ~370 fmol/ml and ~1000 fmol/g, respectively, upon 12 days of ANG II infusion (400 ng·min−1·kg−1) (333). ANG II infusion is also expected to increase ANG II metabolites in plasma and tissue as shown in elevation of ANG (1–7) in kidney cortex (1009). ANG (1–7) concentration in human, mouse, and rat plasma were quite low and averaged 4–80 fmol/ml (140, 1165). However, it is important to remember that ANG II stimulation or infusion has indirect effects on cells and animals via its metabolites including ANG (1–7), ANG III, and ANG IV, which are regulated cell/tissue specifically (140, 509).
Biochemical, molecular, and pharmacological tools have been used without rigorous validations, as have AT2R agonist/antagonists. Moreover, the specificity and selectivity of the antibodies targeting the receptors remain hotly debated (69, 259, 351, 377).
Rag1−/− mice (lacking both T and B lymphocytes) were reported to demonstrate significant reduction in blood pressure in response to chronic ANG II infusion (490 ng·min−1·kg−1) for 2 wk compared with control C57BL/6 mice (348). Inhibition of ANG II-induced hypertension in Rag1−/− mice have been confirmed by several distinct institutions (432, 829, 1074, 1095). One laboratory, however, could not confirm their finding (432) when the mice were repurchased recently (431), suggesting that spontaneous mutations might be occurring in the inbred strain. Distinct expression levels of glomerular ANG II receptors were suggested to explain the difference according to binding studies. The readers should be aware of the limitations and pitfalls that are applicable to many of the publications referenced in this review and are advised to be careful in reconciling the maze that is ANG II receptor signal transduction.
Regardless of receptor composition and activation, signal transduction is at the crux of physiological and pathological functions carried out by ANG II. The following section will discuss the basics of angiotensin receptor signaling by focusing on the AT1R, AT2R, Mas, MrgD, and pro(renin) receptor.
II. BASICS OF ANGIOTENSIN RECEPTORS AND SIGNALING
A. Angiotensin Type 1 Receptor
1. Structure, internalization, and expression
The majority of investigation into ANG II signal transduction has focused on ANG II stimulation of the ANG II type 1 and type 2 receptors (AT1R, AT2R). The AT1R is present in various tissues, including vascular smooth muscle, endothelium, heart, brain, kidney, adrenal gland, and adipose tissue and facilitates most of the physiological functions induced by ANG II (220). The AT1R is a seven-transmembrane (TM) protein belonging to the GPCR superfamily. The 359 amino acid (~41 kDa) AT1R contains several contact sites for ANG II binding. Hormone docking is facilitated by salt bridges connecting the Arg2 side chain of ANG II and Asp281 of AT1R along with the ANG II COOH terminus and Lys199 of AT1R. Receptor activation by ANG II is mediated through Phe8(ANG II)/His256(AT1R) and Tyr4(ANG II)/Asn111(AT1R) (462). There are updated review articles available describing structural determinants for binding, activation, and functional selectivity of AT1R (50, 970). Moreover, crystal structure of the AT1R has been recently demonstrated with an antagonist, candesartan precursor ZD7155. Three new AT1R residues were found to form critical interactions, Tyr35, Trp84 and Arg167, in which Arg167 may be essential in determining ligand binding and selectivity. The study further supports the notion that AT1R has a sodium binding pocket, and Asn111 and Asn295 in transmembrane domain III and VII, respectively, facilitate receptor activation (1245, 1246). In addition, AT1R activation by ANG II promotes a conformational change in the TM3-TM6 region causing an interaction between TM2 and TM7 (703). Aside from components for ANG II binding, AT1R contains an extracellular domain, with two predicted disulfide bonds, consisting of an NH2 terminus and extracellular loops which contain three N-glycosylation sites. Intracellular loops form the domain responsible for G protein activation, and the COOH-terminal tail consists of several sites for serine/threonine kinase phosphorylation.
ANG II-dependent activation of AT1R is transient and results in receptor internalization upon COOH-terminal phosphorylation by PKC and GPCR kinases (GRK2 and GRK5). The components of AT1R internalization include GTP hydrolysis-mediated dynamin fission of clathrin-coated vesicles and β-arrestin-dependent mechanisms, but can vary depending on AT1R dimer composition (see sect. IIA6) (5, 300, 462). Recently, peroxidase-catalyzed proximity labeling combined with mass spectrometry has enabled unbiased quantitation of intracellular protein interactions of AT1R. Upon ANG II stimulation, AT1R rapidly and transiently interacts with known regulators of receptor internalization including β-arrestin 2, adaptor protein complex 2 β1 subunit, clathrin, FCH domain only 2, and intersectin 2, wheras it exhibits sustained interaction with endosomal markers, Rab5 and Rab7. Lysosomal entry is also confirmed at later time points (10 min) with LAMP1. In addition, AT1R rapidly (10 s) dissociates from various Gα proteins (792).
Expression of the AT1R can be regulated by numerous factors and conditions with complex transcriptional and posttranscriptional mechanisms (411). Recent data provide additional information about these mechanisms. Specifically, purinergic GPCR P2Y2 receptor-mediated inducible NOS induction in cardiac fibroblasts reduces AT1R expression via suppression of nuclear factor κB (NF-κB) by p65 S-nitrosylation (751), insulin-induced upregulation via HuR-dependent 3′-UTR stabilization (805), and ER stress causing 3′-UTR stabilization via T cell-restricted intracellular antigen-1 (45). AT1R activity and protein levels are affected by posttranslational modification as well. For instance, evidence suggests that tissue transglutaminase (TG2)-mediated modification of AT1R contributes to AT1R autoantibody production and hypertension associated with preeclampsia. Posttranslational modification of Q187 in the second extracellular loop creates a neo-epitope that encourages production of an autoantibody by the adaptive immune system that can activate the receptor (596). In addition, isopeptide modification of Q315 in the cytoplasmic tail by TG2 prevents ubiquitination-dependent receptor degradation (597). The subsequent increased abundance of AT1R on the cell surface likely contributes to hypertension as well, due to enhanced ANG II sensitivity and the increased likelihood of autoantibody production.
The rodent AT1Rs are products of two separate genes and share substantial sequence homology. AT1AR is dominantly expressed in most organs. AT1AR mediates vasoconstrictor responses and is essential in regulation of blood pressure. Therefore, the AT1AR is believed to be the closest homologue to the human AT1R. AT1BR has a unique role in mediating thirst responses and is dominantly expressed in the adrenal gland and certain regions of the central nerves system (reviewed in Ref. 985).
2. G protein coupling, second messengers, and activation of protein kinases
ANG II activation of AT1R promotes signaling that is diverse, convergent, and convoluted. However, scientific evidence has established key signaling components that are necessary for ANG II-dependent mechanisms of physiology and pathophysiology. Briefly, ANG II binding to AT1R causes AT1R interaction with heterotrimeric G proteins, including Gq/11, G12/13, and Gi. Subsequent second messenger signaling includes inositol trisphosphate, diacylglycerol, arachidonic acid, and reactive oxygen species (ROS), leading to activation of downstream effectors, phospholipases C, A, and D. Depending on the tissue in which these signals are activated, the response may differ, such as in vascular smooth muscle cells (VSMCs) where these components typically regulate contraction through Gq/11 Ca2+-sensitive myosin light chain kinase (MLCK) activation and G12/13 Rho/Rho kinase-mediated inhibition of myosin light chain phosphatase (MLCP). Importantly, AT1R activates various intracellular protein kinases including receptor and non-receptor tyrosine kinases, and serine/threonine kinases including mitogen-activated protein kinase (MAPK) family kinases, Akt, and PKC. FIGURE 2 lists classically known AT1R-activated tyrosine and serine/threonine kinases described in past ANG II signal transduction reviews published in 2007 (379, 682). This review article has updated this list with additional tyrosine and serine/threonine kinases that appear to be activated by ANG II via AT1R or presumably through AT1R. TABLE 3 summarizes the novel ANG II-activated kinases and downstream substrates together with the functional consequences on signal transduction. The regulatory mechanism and pathophysiological significance of several of these key new kinases will also be described in the following sections.
FIGURE 2.

List of tyrosine and serine threonine kinases activated by the AT1R. RTK, receptor tyrosine kinase; nRTK, non-receptor tyrosine kinase.
Table 3.
Newly identified protein kinases in ANG II signaling
| Kinase | Substrates | Function | Reference Nos. |
|---|---|---|---|
| Ser/Thr kinase | |||
| ALK1 | Unstudied | Central regulation of hypertensiona | 331 |
| ALK2 | SMAD1/5 | Cardiac hypertrophy, cardiac fibrosis | 942 |
| ALK4 | SMAD2/3 | Cardiac fibroblast proliferation, collagen synthesis | 561 |
| AMPK (α1) | ACC | Endothelial protection, limit Nox2 induction and ROS | 931 |
| AMPK (α2) | AP2α, ACC | AAA protection, limit VSMC MMP2 induction | 1117 |
| BCR | PPARγ | VSMC PPARγ inhibition and NF-κB activation | 20 |
| DAPK | Unstudied | Vascular constrictiona | 1080 |
| ILK | Unstudied | Cardiac myocyte hypertrophya | 76 |
| Unstudied | Cardiac fibroblast NF-κB activation and collagen synthesisa | 1045 | |
| Unstudied | Renal NF-κB activation and inflammationa | 23 | |
| Unstudied | VSMC ROS production, migration, and proliferationa | 714 | |
| MKK4 | JNK | Atrial fibrosisa | 210 |
| MNK1 | Sprouty2c | Suppression of cardiac hypertrophy and fibrosis | 1225 |
| PAK1 | Unstudied | VSMC migration | 384 |
| Unstudied | Suppression of cardiac hypertrophy and fibrosis | 602 | |
| PERK | Unstudied | SFO-mediated hypertensiond | 1216 |
| PKD1 | Unstudied | Cardiac hypertrophy and perivascular fibrosisa | 281 |
| HDAC4/5/7 | Intestinal epithelial cell proliferation | 974 | |
| HDAC5c | Skeletal muscle atrophya | 245 | |
| SGK1 | FOXO3A | Fibroblast survival | 56 |
| Cardiac remodeling and inflammation via macrophage | 1193 | ||
| SPAK | NCC | Sodium reabsorption | 127 |
| NKCC1 | Vascular contraction and hypertension | 1238 | |
| WNK3 | SPAK | Vascular contraction and hypertensiona | 1237, 1238 |
| WNK4 | SPAK | Sodium reabsorptiona | 127 |
| Tyr kinase | |||
| BMX | Unstudied | Mediates EGFR transactivationa | 313 |
| Unstudied | Endothelial activation to mediate cardiac hypertrophy | 389 | |
| CSKb | FYN | Podocyte apoptosis | 1253 |
Activation of the kinase is predicted. bActivation of CSK has been predicted in VSMCs (600, 1061). cPredicted substrate. dFunction is predicted. ALK, activin receptor-like kinase; SMAD, small mothers against decapentaplegic; AMPK, AMP-activated protein kinase; ACC, acetyl-CoA carboxylase; AP2α, activator protein 2α; BCR, breakpoint cluster region protein; CSK, COOH-terminal Src kinase; DAPK, death-associated protein kinase; MKK4, mitogen-activated protein kinase kinase 4; MNK1, mitogen-activated protein kinase-interacting kinase 1; PAK1, p21-activated kinase 1; PERK, protein kinase R-like ER kinase; SFO, subfornical organ; PKD1, protein kinase D1; HDAC, histone deacetylase; SGK1, serum-glucocorticoid regulated kinase 1; FOXO3a, Forkhead box group O 3a; SPAK, STE20/SPS1-related proline/alanine-rich kinase; NCC, NaCl cotransporter; NKCC1, Na/K/Cl cotransporter isoform 1; WNK, with no lysine kinase.
3. Growth factor receptor transactivation
Recent research has uncovered the dynamic phenomena of AT1R-dependent growth factor receptor transactivation. Intermediary signals, including Ca2+, ROS, and downstream activation of A Disintegrin and Metalloproteinase (ADAM) trigger epidermal growth factor (EGF) receptor (EGFR) transactivation (289). In cultured VSMCs, ADAM17 cleaves heparin-binding EGF-like growth factor (HB-EGF) thereby producing ligands necessary for EGFR transactivation. EGFR transactivation by ANG II promotes Ras-GTP formation and activation of the MARK/ERK and Akt/mechanistic target of rapamycin (mTOR)/p70 S6 kinase (p70S6K) cascades resulting in increased protein synthesis that contributes to hypertrophy and cell migration (251, 253, 254, 258, 771). Although some reports show Gq-independent EGFR transactivation (275, 704), ADAM17-mediated HB-EGF shedding by ANG II requires Gq activation (697, 772). A yet unidentified tyrosine kinase likely phosphorylates ADAM17 at Tyr702 upon ANG II stimulation leading to ADAM17 activation in VSMCs (258). In addition, BMX (bone marrow kinase), CHKA (choline kinase alpha), and TRIO [triple functional domain (PTPRF interacting)] have been identified as upstream signaling molecules required for ANG II-induced EGFR transactivation by siRNA library screening (313).
ANG II is also known to transactivate the platelet-derived growth factor (PDGF) receptor (PDGFR) (370) and the insulin-like growth factor I (IGF-I) receptor (IGF-IR) (246) in VSMCs. PDGFR transactivation is reported to mediate ERK activation (708), vascular hypertrophy, and fibrosis (925). The ANG II-induced IGF-IR transactivation appears to be required for phosphatidylinositol 3-kinase (PI3K) and p70S6K activation, but not for ERK activation (1230). IGF-IR transactivation is also important for Src kinase-mediated cortactin phosphorylation and cytoskeletal reorganization in response to ANG II (1231). IGF-IR partially mediates ROS production by ANG II leading to p38MAPK and ERK5 activation in VSMCs (1058). However, unlike the EGFR, information is still limited regarding the role of ANG II-induced transactivation of PDGFR and IGF-IR in cardiovascular pathophysiology.
4. ROS generation by Nox
ANG II is known to stimulate nicotinamide adenine dinucleotide phosphate (NADPH) oxidases to produce superoxide and H2O2 (682). The catalytic subunit of the NADPH oxidases, the Nox family consists of seven members, Nox1-Nox5, and dual oxidases, Duox1 and Duox2. The Nox family interacts with shared as well as specific regulatory subunits which affect their catalytic activity. Nox1, Nox2, and Nox3 interact with p22phox regulatory subunit and Nox1 and Nox2 interacts with Rac1. Nox activity is also significantly altered by interaction with specific regulatory proteins, Noxa1 for Nox1, p67phox for Nox2, and calmodulin for Nox5. Nox4 is considered a constitutively active enzyme. Nox1, Nox2, and Nox5 mainly produce superoxide, whereas Nox4 mainly produces H2O2 (29).
In general, ANG II is believed to increase ROS production via the Nox family proteins by increasing their expression as well as the catalytic activity (308). For example, in rat aortic VSMCs, ANG II appears to increase superoxide via Nox1 leading to both superoxide and H2O2 production, whereas Nox4 maintains basal H2O2 production (231). Ezrin-radixin-moesin (ERM) binding phosphoprotein 50 has been shown to be a positive regulator of Nox1 activation by ANG II in VSMC (18). ANG II has also increased several Nox proteins and subunits. AP-1 and c-Jun activation are critical for induction of Nox1, Nox4, p67phox, p47phox, and p22phox in human aortic VSMC (644). While ANG II-induced ROS production was completely blunted in VSMC cultured from Nox1-deficient mice, Nox1 silencing may cause global inhibition of AT1R signaling in VSMCs including attenuation of acute intracellular Ca2+ response. The mechanism likely involves reduced H2O2-dependent phosphorylation of caveolin 1 (Cav1) at Tyr14, altering AT1R membrane expression (57). Therefore, Nox1 abundance in VSMCs will determine physiological function of ANG II. It has been demonstrated that the G protein-coupled estrogen receptor activity specifically maintains Nox1 abundance in VSMCs. ANG II-induced intracellular Ca2+ elevation, superoxide production, and hypertension were attenuated in mice deficient in G protein-coupled estrogen receptor (693).
In contrast, Nox2 appears to mediate ANG II-induced superoxide production in cytosol and mitochondrial fractions in cultured aortic endothelial cells (233). In mesangial cells, Nox4-derived ROS contribute to fibrotic responses, which require downstream eNOS uncoupling (552). ANG II also activates Nox5 via Ca2+ and calmodulin leading to growth and inflammatory responses in human microvascular endothelial cells (710). Thus distinct Nox isoform are involved in ANG II-induced ROS production and downstream function in given cell types. However, the relation between ANG II signaling and Nox3, Duox1, and Duox2 remains unclear. Functional significance of Nox isoforms in ANG II pathophysiology including hypertension and cardiovascular remodeling will be described in tissue-specific context in the subsequent sections, where the complexity and controversy surrounding Nox/ROS signaling via AT1R are also included. The mechanism and significance of ROS production in ANG II signal transduction have also been reviewed recently (746).
5. G protein-independent signal via β-arrestin
While the above signal transduction events are mostly known to be G protein dependent, it is increasingly recognized that AT1R also elicits several G protein-independent signal transduction cascades. One of the well-documented cascades is through GRKs and β-arrestin, which are classically known to terminate GPCR signaling by uncoupling GPCR from a G protein and targeting the receptor for internalization (960). Recruitment of β-arrestin 2 to AT1R is required for full activation of ERK and Akt in parallel with G protein-dependent mechanisms leading to protein synthesis in VSMCs (482, 1052). A study in human embryonic kidney (HEK)-293 cells utilizing the mutant AT1R-DRY/AAY or a biased agonist Sar1, Ire4, Ile8-ANG II (SII) suggest distinct active confirmations of the AT1R. The mutant AT1R or SII via wild-type AT1R induces G protein-independent, but β-arrestin 2-dependent ERK activation (1132). However, a more sensitive G protein assay via bioluminescence resonance energy transfer (BRET) demonstrated Gq and Gi activation by SII, whereas the mode of the activation appears distinct from those by ANG II (920).
In the field of GPCR signaling, it is increasingly recognized that specific ligands selectively bind and favor the activation of some signaling pathways over others, a concept termed ligand bias or “functional selectivity.” In the SII study (1132), detailed relationships between functional selectivity and positions 1, 4, and 8 of ANG II were studied in HEK293 cells. While position 1 does not confer functional selectivity, position 8 appears critical for Gq activation. Position 8 mutations also reduce G12 activation. Position 8 is also important to partially mediate EGFR-dependent as well as PKC-dependent ERK activation (239). As mentioned above, SII also activates Akt leading to mTOR-dependent protein synthesis in HEK293 cells (482). A recent BRET assay demonstrated that canonical (ANG II) and biased agonists including SII cause distinct conformational changes of AT1R, involving intracellular loop 3 and 2, respectively. The canonical change further requires Gαq/11 proteins (230). Phosphoproteomic studies to look for distinct signaling molecules regulated by ANG II or SII have also been reported (see sect. IIIF). Biased agonism also differently affects for the receptor fate. Biased AT1R agonists accelerated receptor internalization compared with ANG II stimulation. This is due to the differences in plasma membrane phosphatidylinositol 4,5-bisphosphate depletion (1019).
It is interesting to note that such biased agonistic mechanism is utilized with mechanical stretch-induced ANG II-independent AT1R activation and EGFR transactivation contributing to cardiac myocyte survival (853). This novel information has been recently translated to create a β-arrestin-biased AT1R agonist to potentially treat heart failure and hypertension (83, 84, 707) (see also the cardiac section of this review regarding the mechanism and function of this cascade). TRV120027, which is a β-arrestin biased AT1R ligand, promotes β-arrestin 2 recruitment to AT1R but fails to elicit G protein coupling and prevents ANG II-induced elevation in mean arterial pressure in rats (1094). Thus an importance for β-arrestin signaling has emerged in ANG II-induced pathophysiology. However, studies suggest both benefit and harm of β-arrestin signals. Stimulation of β-arrestin 1 or inhibition of β-arrestin 2 seems to be a potential treatment for VSMC hypertrophy and hyperplasia. However, β-arrestin 2 stimulation appears to be a desirable therapeutic strategy for cardiac hypertrophy and heart failure. Importantly, adrenal AT1R stimulation mediates ERK1/2-dependent aldosterone production via β-arrestin 1 (619). This mechanism may also explain aldosterone escape, which occurs in certain patients with AT1R blocker treatment (620). Thus selective β-arrestin 1 inhibition in adrenal gland may be beneficial. It is important to note that several AT1R blockers have been classified as G protein-selective inhibitors or dual G protein/β-arrestin inhibitors (198). The mechanosensing role of AT1R has also been reported in VSMC, mediating myogenic tone independently of ANG II secretion (681). Both AT1AR and AT1BR appear mechanosensitive (81, 928). RGS5 appears to inhibit mechano-activated AT1R in VSMCs (390). For a comprehensive layout of basic ANG II/AT1R signaling mechanisms, please see FIGURE 3.
FIGURE 3.

Classical ANG II signal transduction. Readers are encouraged to review previous review articles for a more in-depth understanding of classical ANG II signal transduction. Basic ANG II signaling involves Gq-mediated Nox, phospholipase C (PLC), and protein kinase C (PKC)-δ signaling. PKC-δ and G12/13-induced RhoA elicits downstream MLC-mediated contraction. PLC stimulates IP3-mediated Ca2+ release and subsequent MLCK activation. ANG II and Nox-dependent ROS are involved in HB-EGF shedding through ADAMs leading to EGFR transactivation. EGFR transactivation is responsible for ANG II-induced ERK1/2 and Akt signaling. ANG II promotes ER stress and downstream inflammatory activation through NF-κB, apoptosis, and fibrosis through transforming growth factor (TGF)-β signaling. ANG II also stimulates organelle stress and activation of clearance pathways including mitochondria respiratory dysfunction and autophagy. ROCK induction through Nox-dependent ROS has also been implicated in microparticle formation in response to ANG II.
6. AT1R-GPCR heterodimer
GPCRs, including AT1R, are hypothesized to activate G protein-dependent signaling in monomeric and homodimeric forms (1020, 1146). AT1R has been reported to also exist in a heterodimeric form with other GPCRs, including the AT2R, α1D adrenergic receptor, β1 adrenergic receptor, β2 adrenergic receptor, bradykinin receptor B2, chemokine (C-C motif) receptor 2 (CCR2), dopamine receptor D1, endothelin B receptor, Mas, prostaglandin F receptor, and P2Y purinergic receptor 6 (4, 5, 42, 55, 330, 334, 512, 752, 1234, 1235). For most receptor dimerization pairs, potential alteration to AT1R signaling has been described (462). For example, dimerization with AT2R, endothelin B receptor, and Mas is considered inhibitory, whereas dimerization with β1 adrenergic receptor, β2 adrenergic receptor, bradykinin receptor B2, CCR2, prostaglandin F receptor, and P2Y purinergic receptor 6 are considered stimulatory. The mechanisms by which the heterodimers function are diverse. The AT1R-AT2R inhibitory interaction requires PKC-dependent AT2R phosphorylation (416). The AT1R-apelin receptor interaction inhibits AT1R ANG II binding (966). AT1R-α2c adrenergic receptor interaction appears to stimulate Gs-cAMP signal (65). Thus ANG II-induced signal transduction may vary depending on receptor dimer composition. However, most of the studies were limited to artificial cell lines with overexpression of the GPCRs. Peroxidase-catalyzed proximity labeling further identified AT1R interacting with two orphan GPCRs, GPRC5A and GPRC5C (792). Functional relevance and molecular mechanisms of the receptor dimers remain largely unclear.
7. Other AT1R interacting proteins
In addition to the GPCRs, AT1R also interacts directly with other proteins that are not part of the G protein family. These previously identified AT1R interacting proteins include Ca2+/calmodulin, JAK2, phospholipase C (PLC)-γ1, AT1 receptor associated protein (ATRAP), type 1 angiotensin II receptor-associated protein (ARAP1), and GEF-like protein (GLP) (379, 706, 1049). The COOH-terminal cytoplasmic domain of AT1R is known to interact with Ca2+/calmodulin, JAK2, and PLC-γ1 (21, 1049, 1090). Ca2+/calmodulin also interacts with third intracellular loop of AT1R and competes with Gβγ binding (1255). ATRAP binds to the COOH-terminal cytoplasmic domain of the AT1R, mediates AT1R internalization (195, 1040), and negatively regulates AT1R signal transduction (610, 1071). ATRAP attenuates AT1R-mediated vascular senescence via calcineurin/nuclear factor of activated T cells (NFAT) pathway (700). ATRAP transgenic mice do not reveal a significant phenotype; however, neointima formation induced by vascular injury is inhibited together with ERK, STAT1, and STAT3 activity (782). ATRAP also mediates ventricular relaxation through SERCA2a (680) and cardiac-specific ATRAP transgenic mice are protected from ANG II-induced cardiac hypertrophy (1101). Unlike ATRAP which is generally inhibitory, ARAP1 binds to AT1R and mediates AT1R recycling to the plasma membrane and thus is generally stimulatory (344). Proximal tubule-specific ARAP1 transgenic mice exhibit hypertension and kidney hypertrophy through enhancement of AT1R signaling (343). Overexpression of GLP, a cytosolic protein, causes hypertrophy in VSMCs and renal proximal tubular cells via activation of Akt and inhibition of p28kip1 protein expression (345).
The list of AT1R interacting proteins has now been expanded to include filamin A, a cross-linking signal transducer (1055), tubulin (1258), Coatomer subunit β (β-COP) (1275), and GABA receptor-associated protein (GABARAP) (182). Filamin A and tubulin are associated with the cellular cytoskeleton, and the tubulin/AT1R interaction contributes to AT1R trafficking to the cell surface (1055, 1258). β-COP is a component of Coat Protein I (COPI) transport vesicles involved in the transport between different Golgi stacks and transport from the Golgi to the ER. β-COP and AT1R interaction is dependent on Lys308 in the cytoplasmic domain of AT1R and functions to repress AT1R forward trafficking as a Lys308 mutation enhances AT1R cell surface expression (1275). GABARAP is involved in GABAA receptor trafficking through microtubule networks. GABARAP binds the COOH-terminal domain of the AT1R and promotes AT1R trafficking to the plasma membrane (182).
In addition, AT1R associates with the EGFR (774) and lectin-like oxidized low-density lipoprotein (oxLDL) receptor (LOX1) (1185). oxLDL activates AT1R and ERK through the interaction in cultured ECs, potentially causing endothelial dysfunction (1185). Peroxidase-catalyzed proximity labeling also identified LMBR1 domain containing 2, a seven-transmembrane protein, as a novel AT1R interacting protein (792). Cumulatively, newly identified interacting proteins for the AT1R have further highlighted the complexity and diverse signaling behind the AT1R cell surface expression and activation. An altered interaction between AT1R and receptor binding proteins likely affects physiological and pathophysiological conditions induced by ANG II.
B. Angiotensin Type 2 Receptor
While AT1R is the most studied, it has been increasingly attractive to study AT2R due to its role in opposing the effects of AT1R (168, 660, 790, 1011). However, signaling mechanisms of AT2R are still speculative compared with those of AT1R. Similar to AT1R, AT2R belongs to the family of GPCRs with diverse downstream signaling mechanisms depending on the cell type. AT2R stimulates MAPK phophatase1 (MKP-1) and SH2 domain-containing phosphatase 1 (SHP-1) resulting in decreased tyrosine phosphorylation throughout the cell with SHP-1 requiring Gαs for activation (75). AT2R mediates programmed cell death as inhibition of AT2R-dependent MKP-1 prevents cell death in PC12W and R3T3 cells (462), which supports a potential involvement of AT2R in cancer protection (469). In addition, inhibition of ERK MAPK by AT2R appears to induce myoblast differentiation and potentiate skeletal muscle regeneration providing a new therapeutic target in muscle wasting disorders (1211). Within the cardiovascular system, AT2R promotes vasorelaxation through PKA-dependent eNOS activation and paracrine signaling through bradykinin/cGMP/NO production (790). The physiological consequences of this is evidenced in AT2R null mice, as ANG II stimulation promotes an exacerbated pressor response (1073). Similarly, overexpression of AT2R promotes vasodilation. Furthermore, pharmacological stimulation of AT2R by compound 21 (C21) causes natriuresis, lowers blood pressure, and protects against ANG II-induced hypertension in rats (479, 480). In addition, AT2R-interacting protein family (759) (ATIP1–ATIP4) has been observed to promote macrophage polarization and an anti-inflammatory environment in adipose tissue in mice fed a high-cholesterol diet (443), and to contribute to normal brain function (872). ATIP can also prevent vascular remodeling and ANG II-dependent vascular senescence, as well as suppress the growth of various forms of tumors (699, 873). Interestingly, ATIP1 has also been identified as mitochondrial tumor suppressor gene 1 (MTSG1 coded by Mtus1A) whose overexpression suppresses mitochondrial ROS production, ERK activation, and hypertrophy in response to phenylephrine in cardiac myocytes. However, the functional relation of the phenotype with AT2R remains unclear (424).
Inquiry has also indicated conflicting mechanisms of AT2R signaling. Spontaneously hypertensive rats show vasoconstriction that is mediated by AT2R, which questions the typical view that AT2R stimulation is vasodilatory (1214). Cardiac overexpression of AT2R prevents ANG II-induced perivascular fibrosis, but does not alter cardiac hypertrophy (526). However, the mice also developed dilated cardiomyopathy and heart failure without any intervention (1188). The mechanism explaining the discrepancy has been proposed recently with a mouse model of myocardial infarction. The beneficial or detrimental effect of AT2R in the heart is largely dependent on AT2R expression levels and possibly via regulation of Nox2 and transforming growth factor β1 (TGF-β1) signaling pathways (1178). AT2Rs on inguinal adipocytes have also been implicated in opposing the induction of uncoupling protein-1 (UCP1) production by norepinephrine and aspects of cellular respiration, all of which has implications for the regulation of resting metabolic rate by the sympathetic nervous system (595).
Despite the studies referenced above, a growing number of studies suggest that AT2R signals primarily via noncanonical, G protein- and β-arrestin-independent pathways (834). The crystal structure of human AT2R with ligands has recently been reported. The data suggest an active-like conformation, but with the helix VIII preventing recruitment of G proteins or β-arrestins, which is in agreement with lack of signaling responses in standard assays (1244). While decades of research have established the distinct signaling mechanisms of AT1R and AT2R, future experiments should establish precise signaling mechanisms by which ANG II modulates physiological and pathophysiological functions. Please also refer to recent review articles for accumulating literature regarding the protections and potential mechanisms utilized by AT2R against hypertension, vascular remodeling, and end organ damage in heart, kidney, and brain (168, 660, 1011).
C. Mas Receptor
While knowledge of signal transduction is limited, evidence exists for a defined role of the ANG (1–7)/Mas signaling axis. ANG (1–7) generation results from ANG II conversion by ACE2 into ANG (1–7). ANG (1–7) has been reported to exert its effects through the Mas receptor in various tissues, including the kidneys, heart, vasculature, and brain. Mas receptor activation by ANG (1–7) binding has been implicated in physiological and pathophysiological processes, including leukocyte recruitment and inflammation, cardiac remodeling, renal function, and vascular alteration (141, 969). However, evidence suggests ANG (1–7) may promote signaling via other receptor(s) or Mas in a G protein-independent manner. Mas appears to have strong constitutive activities with Gq and G12. ANG (1–7) does not stimulate these G proteins via Mas, while neuropeptide FF activates Gq and G12 in Mas-expressing cells (1054). Recent research also points to protective functions of ANG (1–7) mediated through AT2R. It is shown that ANG (1–7) mediates vasodilation via AT2R in the presence of AT1R blocker (1102). This link has been expanded in the apolipoprotein E apoE−/− mouse model of atherosclerosis and isolated aorta with pharmacological interventions (820). Moreover, in a model of intracranial aneurysm rupture, ANG (1–7) protected against rupture that was attenuated in AT2R-deficient mice and in mice treated with AT2R antagonist, but not with Mas antagonist (950). Clearly, more research is needed to determine how ANG (1–7)/Mas signaling and ANG (1–7)/AT2R signaling affects various physiological and pathophysiological processes. Clinical studies are also needed to assess the relative importance of ANG (1–7). Please note that a detailed review article covering controversies with ANG (1–7)/Mas signaling and function has been published (461).
D. MrgD
A member of Mas-related GPCR family, MrgD, has been shown to react with ANG (1–7) to produce arachidonic acid (461). In addition, the MrgD receptor is responsive to Alamandine. Alamandine stimulates NO production in cells transfected with MrgD and not Mas. Moreover, Alamandine administration results in an antihypertensive response in SHRs (542). In addition, ANG (1–7) stimulates cAMP production resulting in increased PKA-dependent cAMP response element-binding protein (CREB) phosphorylation contributing to vasorelaxation. Likewise, MrgD KO mice show no reduction in mean arterial blood pressure in response to a bolus injection of ANG (1–7) (1044). Taken together, the MrgD receptor has emerged as a counterbalance to the signal transduction elicited by ANG II and AT1R.
E. (Pro)renin Receptor
Previously identified as a protein associated with the vacuolar H+-ATPase, the (pro)renin receptor (PRR) was discovered to bind both renin and its inactive proenzyme form prorenin. Renin binding induces an increase in the catalytic efficiency of angiotensin conversion to ANG I, further increasing ANG II generation as part of a tissue-specific RAS system. Binding of prorenin to the PRR induces a conformational change in prorenin rendering it enzymatically active (748). PRR colocalizes with vacuolar H+-ATPase at the apex of acid-secreting cells in the collecting duct where it participates in distal nephron H+ transport (12). In addition, a novel intracellular activity of PRR has been studied in the kidneys where its activation by prorenin binding (as a ligand) initiates ERK1/2 signaling leading to downstream activation of genes associated with fibrosis including TGF-β1, fibronectin, collagen, and plasminogen activator inhibitor-1 (PAI1) (748). This intracellular PRR activity also induces p38MAPK-HSP27 and PI3K-p85 signaling. The PI3K-p85 pathway leads to downstream nuclear translocation and activity of the zinc finger transcription factor, which represses PRR transcription (748). ANG II induces PRR expression in the collecting duct thereby increasing renin activity and contributing to local ANG II generation. This ANG II-induced PRR is elicited through a cascade involving COX2 and PGE2 (1104). Furthermore, collecting duct PRR mediates the ANG II-induced hypertensive response, most likely through increased α-ENaC-dependent Na+ transport (818). Transgenic rats overexpressing human PRR ubiquitously are normotensive, but show progressive development of nephropathy (454), whereas transgenic rats with smooth muscle specific PRR overexpression are hypertensive at 6 mo of age, which could be due to intra-adrenal ANG II-induced aldosterone generation (103). Likewise, brain PRR is hypothesized to regulate ANG II-dependent hypertensive responses (576, 1179). In addition, PRR-dependent, but AT1R-indepednent effects have been reported in neuronal cells, including ROS production (814) and iNOS mRNA induction (406). Despite the growth of this area of investigation, the relative importance of the ANG II-dependent and ANG II-independent actions of PRR in many tissues remains unclear. For a comprehensive review on the PRR, please see the following references (781, 855).
III. NOVEL AND EXPANDING ANG II SIGNAL TRANSDUCTION
Although the other receptor members have received attention recently, decades of research have accumulated regarding the signal transduction components of the AT1R. Historically, past investigation of AT1R has explored the critical contribution of several tyrosine and serine threonine kinases, phosphorylation of their substrates, and ROS in various ANG II target cells and tissues. While these investigations are still expanding, including the identification of new pathways and their in vivo significance, there has been remarkable paradigm shift in ANG II signal transduction research. The key expanding areas of novel investigations include intracellular as well as extracellular/spatial and temporal organelle signal communications, cellular and tissue metabolic modulation, signaling through posttranslational protein modification, microRNAs, and long noncoding RNAs. In addition, tissue specific targeting of the receptor and downstream signal transduction in genetically modified rodents has enabled us to obtain fundamental new knowledge in signaling mechanisms, physiology, and pathophysiology of the ANG II receptors.
A. New ANG II Signaling Pathways
1. Wnt/β-catenin pathway
Signaling by the Wnt family of secreted glycolipoproteins is one of the fundamental mechanisms that direct cell responses during embryonic development and tissue homeostasis. Wnt/β-catenin pathway is activated when a Wnt ligand binds to the seven-transmembrane Frizzled (Fz or Fzd) receptor and its coreceptor, low-density lipoprotein receptor-related protein 6 (LRP6), or its close relative LRP5. The formation of Wnt-Fz-LRP6 complex, together with recruitment of the scaffolding protein Dishevelled, results in LRP6 phosphorylation, activation, and the recruitment of the Axin complex to the receptors. These events lead to inhibition of axin-mediated β-catenin phosphorylation and thereby to the stabilization of β-catenin, which translocates to the nucleus to form complexes with T cell factor/lymphoid enhancer factor (TCF/LEF) and stimulate Wnt target gene expression (627). With the use of cardiac myocyte specific β-catenin depletion in mice and β-catenin stabilization in mice, it has been shown that β-catenin stabilization promotes cardiac dysfunction and decreased cardiac hypertrophy with ANG II infusion, whereas β-catenin depletion elicits cardiac hypertrophy, suggesting the requirement of β-catenin for adaptive cardiac remodeling (62). Wnt-induced secreted protein-1 (WISP1) has been identified as a TCF/LEF-target to promote cardiac hypertrophy. ANG II induces WISP1 via CREB and NOX2/Akt/GSK-3β/β-catenin-dependent TCF/LEF transcriptional activation through AT1R in cardiac myocytes (944).
The Wnt/β-catenin pathway has also been implicated in ANG II-related renal injury and renal fibrosis (1270). In immortalized mouse podocytes, ANG II induces Wnt1 expression, β-catenin nuclear translocation, and TCF reporter transcription. Inhibition of the Wnt/β-catenin pathway by β-catenin siRNA or overexpression of endogenous Wnt inhibitor Dickkopf1 attenuates ANG II-induced podocyte injury as assessed by declines in podocin and nephrin expression. Ca2+/calmodulin kinase II (CaMKII) and CREB appear to mediate induction of Wnt1 mRNA. Induction of Wnt1/3 and nuclear accumulation of β-catenin are confirmed in mice infused with ANG II, suggesting involvement of the Wnt/β-catenin pathway in renal injury by ANG II (439). In addition, in the mouse collecting duct cell line M1, ANG II induces β-catenin protein, fibronectin mRNA, and collagen I mRNA, which are attenuated with β-catenin destabilization reagent pyrvinium pamoate (192). Moreover, pyrvinium pamoate treatment ameliorates kidney induction of β-catenin, collagen I, fibronectin, and osteopontin in a rat model of renovascular hypertension, suggesting the role of the Wnt/β-catenin pathway in ANG II-dependent renal fibrosis (193). It is also interesting to note that multiple RAS genes are targets of the Wnt/β-catenin pathway. Bioinformatics suggests the presence of a conserved TCF/LEF binding site in the promoter of AGT, renin, ACE, AT1R, and AT2R. Subsequent in vitro and in vivo experiments confirmed upregulation of the RAS components in proximal tubular cells and kidney upon specific activation of the Wnt/β-catenin pathway (1269). Thus activation of the Wnt/β-catenin pathway by ANG II could be further amplified with multiple feed-forward mechanisms via the RAS component in vivo, contributing to chronic kidney disease.
A few manuscripts implicate the Wnt/β-catenin pathway in vascular pathophysiology. In VSMCs, ANG II rapidly stimulates β-catenin nuclear accumulation. Vascular protective nuclear orphan receptor Nur77 induces proteosomal degradation of β-catenin and inhibits the Wnt/β-catenin pathway. Loss of β-catenin downregulation could be involved in enhanced vascular remodeling in Nur77−/− mice infused with ANG II (194). The Wnt pathway has also been reported to contribute to atherosclerosis and abdominal aortic aneurysm (AAA) progression. Overexpression of sclerostin, a secreted binding inhibitor for the Wnt coreceptor LRP4/5/6, attenuates atherosclerosis and AAA development in apoE−/− mice infused with ANG II, which is associated with reduced glycogen synthase kinase 3β (GSK3β) Ser9 phosphorylation. In addition, sclerostin expression is reduced in human aortic aneurysm samples (516). β-Catenin-dependent, but Wnt-independent ANG II function is also noted in mice with deletion of VSMC β-catenin. While ANG II cannot induce proliferation of human aortic VSMCs in vitro, ANG II infusion in mice causes macrophage-dependent complement c1q production, which in turn stimulates β-catenin pathway and proliferation of VSMCs in vivo. However, hypertension induced by ANG II is unaltered in mice with VSMC β-catenin deletion (1010).
2. Notch pathway
The Notch pathway is crucial in several developmental processes. However, it has been recently implicated in cardiovascular pathophysiology (226). In the Notch pathway, the Jagged and Delta family of ligands bind to a family of four Notch receptors. In response to ligand binding at the cell surface, the Notch1 receptor is activated resulting in canonical γ-secretase-dependent cleavage and translocation of a transcriptional activator, Notch1 intracellular domain (NICD), to the nucleus. While suppression of the Notch3 cascade by ANG II in VSMCs was reported more than a decade ago (114), there has been significant progress regarding the Notch pathway playing a role in ANG II signaling and function recently.
In contrast to the earlier report (114), activation of the Notch pathway via the AT1R has been confirmed in HEK293 cells expressing AT1R and in human VSMCs. In these cells, enhanced nuclear translocation of NICD is observed with ANG II stimulation, which is sensitive to a γ-secretase inhibitor. The γ-secretase inhibitor further attenuates NICD-dependent transcriptional activation, VSMC migration, and vascular remodeling in response to ANG II (785). In Notch3−/− mice, renal vascular constriction induced by ANG II is significantly reduced. The vessel wall thickness was reduced in Notch3−/− mice, and development of ANG II-induced hypertension was also attenuated. However, ANG II-induced cardiac hypertrophy was enhanced in Notch3−/− mice, which correlated with greater mortality due to heart failure (851). The kidney also shows enhanced tubular dilation and fibrosis suggesting that Notch3 is necessary for end organ adaptation in hypertension (92). However, the phenotype could involve developmental and/or constitutive suppression of Notch3. In inducible γ-secretase complex silencing mice, ANG II-induced blood pressure elevation and left ventricular hypertrophy are both attenuated (871). In addition, it is well recognized that NICD partners with the transcriptional activator Mastermind and a DNA-binding protein, CBF1/Su(H)/Lag2 protein (CSL), targeting the complex to gene promoters containing the consensus motif GTGGGAA, for transcriptional activation. In smooth muscle-specific dominant-negative Mastermind-like protein transgenic mice (inhibits CSL-dependent transcription as a pan Notch inhibitor), ANG II-induced blood pressure elevation is significantly attenuated. This is because of a transcriptional suppression of the expression of MLCK, a master regulator of VSMC contraction (60).
Notch has also been implicated in other ANG II pathologies. In human and an ANG II-dependent mouse model of AAA, expression of NICD appears enhanced. In Notch1+/− apoE−/− mice, ANG II-induced AAA formation is attenuated. The mechanism involves macrophage Notch1 regulating macrophage infiltration and inflammatory activation (353). ANG II-induced AAA formation and vascular inflammatory responses are also reduced with γ-secretase inhibitors (158, 1267). A growing body of evidence suggests kidney proximal tubule epithelial cells (PTECs) undergo epithelial cell-mesenchymal cell transition (EMT) to contribute to interstitial fibrosis in progressive renal disease. Notch has been shown to promote EMT in the development of cardiac valves via repression of E-cadherin through upregulated Snail expression. Notch1 appears to contribute to ANG II plus TGF-β-dependent EMT via Snail induction in human tubular epithelial cells (892). However, negative data regarding Notch participating in EMT and kidney fibrosis by ANG II have also been reported (548). Notch signaling has also been implicated in development and expression of renin progenitor cells in both zebrafish (868) and mouse (66, 128) and was reported to be an important regulator of renin transcription (793).
3. NLRP3 inflammasome
Nucleotide-binding domain and leucine-rich repeat containing PYD-3 (NLRP3) is a pattern recognition receptor that is implicated in the pathogenesis of inflammation and chronic diseases. Upon activation by danger/damage associated molecular patterns, NLRP3 induces the formation of the inflammasome, a multiprotein complex that consists of the adaptor molecule apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and the effector enzyme caspase 1. Activated caspase 1 mediates the maturation of the proinflammatory cytokines IL-1β and IL-18 for secretion by innate immune cells (859). However, the role of NLRP3 in nonimmune cells is unclear. In NLRP3−/− cardiac fibroblasts, ANG II-induced myofibroblast differentiation is impaired. Moreover, ANG II-induced cardiac fibrosis, but not hypertension or cardiac hypertrophy, is attenuated in NLRP3−/− mice. An additional TGF-β study suggests involvement of mitochondrial NLRP3 which augments ROS production independently from the inflammasome (96). In contrast, in a mouse model of preeclampsia with ANG II infusion, hypertension is inhibited with restored IL-6 upregulation in NLRP3−/− but not ASC−/− mice (957). Moreover, in a mouse model of AAA with apoE−/− mice with ANG II infusion, AAA development is attenuated in apoE−/−NLRP3−/− mice, apoE−/−Asc−/− mice, and apoE−/−Casp1−/− mice, suggesting a critical role for the NLRP3 inflammasome in ANG II-dependent AAA formation. Further investigation suggests additional involvements of mitochondrial ROS generation by macrophages in the NLRP3 pathway (1079).
4. Hippo pathway
The Hippo signaling pathway controls organ size by mediating the balance between proliferation, differentiation, and apoptosis. The core components of the Hippo pathway are the cotranscription factor Yes-associated protein (YAP) in mammals. The activity of YAP is controlled by the large tumor suppressor kinase (LATS), which phosphorylates and thereby inactivates YAP. For years, factors controlling the Hippo pathway upstream of LATS kinase were elusive. However, recently several groups independently discovered that GPCRs are able to act as modulators of Hippo signaling (689). In HEK293 cells expressing AT1R, ANG II induces dephosphorylation of LATS and YAP, leading to nuclear translocation of YAP. In contrast, ANG II does not affect Hippo pathway activity in podocytes (1140). The functional significance of the Hippo pathway activation by ANG II remains elusive.
B. Novel and Expanding Organelle Communications Induced by ANG II
1. Mitochondria
Under cellular stress both “forward grade” signaling to and “retrograde” signaling from the mitochondria are generated to respond to cellular stress. ANG II has been shown to induce mitochondrial dysfunction leading to mitochondrial ROS generation, which modulates various ANG II responses, including experimental hypertension (232). The ANG II-induced mitochondrial alteration has also been implicated in metabolic disease and aging (218). Mitochondrial ROS production induced by ANG II appears to require NADH/NADPH oxidase such as Nox2 in ECs (233, 242). It has been demonstrated that mitochondrial cyclophilin D, which acts as Ca2+ sensitizer for mitochondrial permeability transition pore opening, mediates ANG II-induced mitochondrial superoxide production in ECs as well as in mouse aorta. Pharmacological and genetic inhibition of mitochondrial ROS (233, 1147) or cyclophilin D (421) are effective in reducing ANG II-induced hypertension and vascular dysfunction in rodents. ANG II-induced hypertension and vascular dysfunction also involve mitochondrial ROS-dependent activation of the L-type Ca2+ channel in VSMCs (139). In contrast, mitochondrial targeted antioxidant peptide or genetic mitochondrial catalase transgene attenuates mitochondrial oxidative damage and cardiac hypertrophy, but not hypertension induced by ANG II in mice (200, 201). Such tissue-specific protection via general mitochondrial protection has also been shown in ANG II-enhanced atherosclerosis. A mitochondria-targeted polyphenol mito-esculetin activates AMPK and eNOS and attenuates ANG II-induced plaque formation in apoE−/− mice, whereas ANG II-induced aortic aneurysm development is unaffected (460).
In mice infused with ANG II, cardiac hypertrophy and diastolic dysfunction are associated with reductions in cardiac glucose oxidation and ATP production, confirming that ANG II can alter mitochondrial function in vivo. This is explained by pyruvate dehydrogenase kinase 4 upregulation via activation of the cyclin/cyclin-dependent kinase-retinoblastoma protein-E2F pathway in response to ANG II (716). Fatty acid oxidation (FAO) is a major energy source for mammalian heart. Decreased FAO contributes to the reappearance of the fetal metabolic pattern in failing hearts that leads to increased reliance on glycolysis and anaplerosis to maintain the TCA cycle. The rate-limiting step of FAO is mitochondrial import of fatty acids through carnitine palmitoyl transferase I. This carnitine palmitoyl transferase I function is inhibited by malonyl CoA, which is formed via acetyl CoA carboxylase (ACC). Thus deletion of ACC2, the primary ACC isoform in oxidative tissues, leads to increases in FAO (506). It has been reported that ANG II infusion reduces cardiac FAO associated with enhanced glycolysis. These effects are reversed by inducible cardiac-specific deletion of ACC2. Moreover, associated cardiac hypertrophy, fibrosis, and diastolic dysfunctions are improved (167). Therefore, the mitochondrial metabolic shift induced by ANG II appears to be a cause of cardiac pathophysiology.
It has been demonstrated that mitochondrial quality control is regulated through a fission and fusion cycle to replace dysfunctional mitochondria. A GTPase dynamin-related protein 1 (Drp1) promotes mitochondrial fission. ANG II induces mitochondrial fission/fragmentation via Drp1 phosphorylation in VSMCs and neuroblastoma cells (590, 844). siRNA silencing of Drp1 attenuates ANG II-induced ERK activation and matrix metalloproteinase (MMP)-2/9 induction in VSMCs. Drp1 inhibitor mdivi1 also attenuates ANG II-induced VSMC migration and proliferation (590). PKC-δ is proposed to phosphorylate Drp1 Ser616 for activation (590, 844). Mitofusion 2 (MFN2), another GTPase, controls mitochondrial fusion, and MFN2 overexpression attenuates ANG II-induced cardiac myocyte hypertrophy in vitro and in vivo (1220). In addition, several RAS components have also been detected in mitochondria and these mitochondrial RAS may mediate ANG II actions (see sect. IIIB6). These data suggest a presence of alternative forward and retrograde signals to and from mitochondria, respectively, upon ANG II stimulation in regulating cardiovascular pathophysiology, which may involve the mitochondrial RAS.
2. Endoplasmic reticulum
The endoplasmic reticulum (ER) is a complex membranous network that is vital for protein synthesis, folding, and the secretory pathway. The discovery of the unfolded protein response (UPR) has highlighted the influence of ER in the development of cardiovascular and metabolic pathology (702, 788). ER stress promotes induction of a three-branched UPR initiating from three ER transmembrane sensors, protein kinase-like ER kinase (PERK), transcription factor-activating transcription factor 6 (ATF6), and inositol requiring kinase 1 (IRE1). All three sensors have luminal domains that bind to the ER chaperone glucose-regulated protein 78 kDa (GRP78), which is released from the sensors upon ER stress, allowing them to initiate the three UPR pathways. 1) PERK phosphorylates eukaryotic translation initiation factor (eIF)2α upon ER stress resulting in the inhibition of most cap-dependent translation, but paradoxically increasing mRNA translation of ATF4. ATF4 induces UPR-target genes including chaperones, antioxidants, and proapoptotic basic-leucine zipper transcription factor, CCAAT/enhancer-binding protein homologous protein (CHOP). 2) ATF6 translocates to the Golgi apparatus where it is cleaved. Cleaved ATF6 binds to and activates the ER stress response element in promoters of UPR target genes, including chaperones, CHOP, and a transcriptional factor, X box-binding protein-1 (XBP1). 3) IRE1 promotes splicing of XBP1 mRNA. Active (spliced) XBP1 binds to the ER stress response element to induce chaperones and the ER-associated protein degradation pathway. In addition, IRE1 interacts with TNF receptor-associated factor 2 (TRAF2), thereby activating NF-κB, JNK, and subsequent inflammatory responses (702, 788).
ANG II infusion increases eIF2α phosphorylation, ATF4 and CHOP mRNA (465) and protein (989) in aorta, as well as CHOP protein in coronary arteries (1035). ANG II infusion increases phosphorylated PERK in brain circumventricular subfornical organ (SFO). Moreover, ER stress and UPR have been shown to play causative roles in various ANG II-induced pathologies, including brain (1216) and vascular (465, 587) regulation of hypertension, endothelial dysfunction (465), cardiac hypertrophy and fibrosis (465), and vascular hypertrophy and fibrosis (465, 989, 1035). Specifically, CHOP−/− mice are protected from ANG II-induced NADPH oxidase activation, hypertension, and cardiovascular pathology (464). ANG II-induced ER stress exists downstream of the ADAM17/EGFR transactivation cascade (1034, 1035) and stromal interaction molecule 1 (STIM1) induction (464), and upstream of cardiac (465) and renal TGF-β induction (1118), vascular apoptotic pathway (989), and brain SFO NF-κB activation (1218). While these studies suggest that ER stress is vital for ANG II-induced signal transduction and pathologies, it remains elusive whether AT1R stimulation increases unfolded proteins in ER, or if so, how it happens.
3. Membrane rafts, caveolae, and caveolins
The lipid bilayer of the plasma membrane is organized into liquid-ordered and disordered domains by virtue of clustering of particular lipids. Liquid-ordered domains are enriched in sphingolipids and cholesterol which provide a preferential environment for targeting of receptors and acetylated signaling molecules. Thus these membrane regions form “rafts,” which compartmentalize sets of signaling molecules that float on the plasma membrane. This molecular arrangement confers specificity to signaling responses elicited by the myriad factors that interact with cells and tissue (123, 939). The incorporation of the caveolin and cavin family of proteins into these lipid domains form caveolae organelles. Caveolae are 50–70 nm sigma-shaped invaginations of the plasma membrane and, similar to membrane rafts, house signaling messengers. Molecular homing of signaling molecules to caveolae is not only achieved via interaction with caveolar membrane lipids but also direct association with caveolin proteins, an interaction which can regulate signaling molecule function. Both membrane rafts and caveolae can fuse, cluster, and internalize, thus forming mobile compartments which can regulate surface expression of signaling domains (161, 799).
The concept that caveolae can serve as both transport vesicles and signaling platforms initiated a wave of exploration into the relationship between specific receptors and these plasma membrane microdomains in the late 1990s. Given the observation that G proteins as well as many other second messengers that are activated in response to ANG II are localized within caveolae and/or interact with caveolins prompted investigation into the relationship between ANG II receptors and these organelles. Early studies showed that signaling and coupling proteins utilized by AT1R are present in membrane rafts and coprecipitated with caveolin-1 (Cav1) following ANG II stimulation in cultured VSMCs (419). Moreover, Cav1 serves as a chaperone for AT1R shuttling to the plasma membrane. The loss of Cav1 (1164) or mutation in the caveolin consensus binding site of AT1R (550) results in improper targeting of AT1R and defective ANG II signaling. Rat aortas treated with methyl-β-cyclodextrin, a compound which ablates raft/caveolae structure, show a perturbation in AT1R endocytosis and prolonged receptor expression on the plasma membrane following stimulation with ANG II. This results in loss of the normal tachyphylactic or desensitization response in vessels repeatedly exposed to ANG II (594). AT1R-mediated intracellular ANG II uptake is also partially dependent on the presence of Cav1/caveolae in proximal convoluted tubule cells (577). Cav1-dependent internalization of ANG II appears necessary for activation of ERK1/2 and the Na+/H+ exchanger in these cells.
It is highly likely from the above studies that caveolae and caveolins govern key aspects of ANG II/AT1R signaling. The relationship between ANG II receptor and caveolae-based signaling adds an additional layer of cellular regulation. Introduction of the Cav1 scaffolding domain peptide (residues 82–101), a sequence which recognizes and interacts with consensus binding motifs found within many proteins, including AT1R, attenuates ANG II signaling responses involving Ca2+ flux and the PI3K/Akt pathway (687). The localization of AT1R, Gq/11, NADPH subunit Nox1, and cSrc to caveolae is also important for vascular tone regulation. The loss of caveolae integrity with Cav1 siRNA treatment disturbs the compartmentalization of these elements on the plasma membrane and attenuates ANG II-induced inhibition of BK channel activity in VSMCs (614). A similar pathway has been described for ANG II-induced vasoconstriction, where AT1R, Gq/11, PKC-ε, and KATP channels are clustered in caveolae (906). In VSMCs (979) and ECs (608), ANG II induces ROS production through the translocation of p47phox to caveolae, where it associates with other NADPH oxidase subunits. In VSMCs, these responses are positively regulated through cyclophilin A (979). In ECs, reduction of Cav1 protein attenuates p47phox translocation and localized ROS production. The reduction in oxidative stress reversed eNOS uncoupling and preserved endothelial cell function (608). Cav1-mediated ROS production may also be important to explain endothelial dysfunction associated with hypercholesterolemia. Free cholesterol promotes enrichment of the lipid raft fraction with Tyr14 phosphorylated Cav1 and Rac1, a positive regulatory component of the NADPH oxidase complex, and enhances ANG II-induced ROS production and eNOS uncoupling (30). EGFR and ADAM17, a metalloprotease that generates mature ligands for EGFR, are also localized in Cav1-containing membrane fractions in VSMCs. Overexpression of Cav1 inhibits ANG II-induced transactivation of EGFR and VSMC hypertrophic responses (1027). In addition, AT1R translocates to the caveolae of podocytes following ANG II stimulation. Depletion of Cav1 via siRNA blocks signaling pathways involving modification of nephrin, an important structural element of slit diaphragms, and prevents podocyte apoptosis (865).
The Cav1 knockout mouse has proven to be a useful tool for deciphering the physiological function of these membrane organelles. Global Cav1 knockout mice are viable and reproduce but show a wide variety of phenotypic defects, including cardiomyopathy, enhanced vascular permeability, airway hypersensitivity, and maladaptive angiogenesis (852). In response to ANG II plus the NOS inhibitor, N-omega-nitro-l-arginine methyl ester (l-NAME), Cav1-deficient mice on the 129/Sv background show a reduction in biventricular damage and transcript levels of the cardiac pro-inflammatory marker plasminogen activator inhibitor type I (PAI-1), and no increase in cardiac mineralocorticoid receptor levels, despite basal cardiac hypertrophy and higher blood pressure response (826). A reduced blood pressure response is reported with ANG II infusion in Cav1+/− mice on the C57BL/6 background at 16 wk of age (608). In contrast, no reduction in ANG II-increased blood pressure, nor in ANG II-induced cardiac hypertrophy (although basal hypertrophy was present), was observed in Cav1−/− mice on the C57Bl6 background at 8 wk of age. Yet, global Cav1 silencing prevents ANG II-induced vascular hypertrophy, perivascular fibrosis, and vascular cell adhesion molecule 1 (VCAM-1) induction (288). ANG II-dependent formation of AAA is prevented in these Cav1 knockout mice, which is correlated with reduced vascular inflammation, ER stress, and oxidative stress (1033). ANG II-induced brain microvascular hypertrophy is also prevented in global Cav1-deficient mice (1077). These data suggest the importance of mouse genetic background and age in influencing Cav1 null mouse phenotypes when they are stressed. The data also highlight that Cav1 knockout mice are specifically protected from vascular inflammation and remodeling induced by ANG II. Targeting this pathway in the vasculature may hold some therapeutic promise.
4. Microparticle and exosome
The research field of microparticles and exosomes is rapidly expanding due to their possible involvement in various diseased states, as well as their potential as biomarkers and therapies. Vesicles larger than 100 nm in diameter originating from plasma membranes are usually called microparticles, while smaller vesicles originating from the multivesicular bodies are described as exosomes. These cell-derived vesicles carry microRNAs, hormonal factors, and cell surface receptors, which can transmit information and signal transduction from originating cells to receiving cells (105, 180, 514). While the exosome field is relatively new, several recent reports have suggested interesting roles for microparticles in ANG II signal transduction and functions.
Hypertensive patients display an increase in circulating microparticles, along with increased chemokine and soluble adhesion marker expression. Strikingly, treatment with angiotensin II receptor blockers (ARBs) in these patients showed significant reductions in microparticle number, as well as other measured markers, suggesting a causative role for ANG II in microparticle release in humans (530, 757). Furthermore, hypertensive and hypercholesterolemic hamsters treated with irbesartan exhibit a decline in microparticle infiltration into the vascular wall. In addition, isolated microparticles show a decrease in expression of inflammatory markers (315). Conversely, there may be some benefit to microparticles. Microparticles derived from human T lymphocytes show protective effects against ANG II-induced hypertension and vascular dysfunction. ANG II-infused mice show aortic endothelial dysfunction associated with increased superoxide, and microparticle injection reverses this increase as well as promotes increased NO production. The Sonic hedgehog receptor antagonist attenuates these benefits suggesting that the protection involves Sonic hedgehog-positive microparticles (653).
At the cellular level, ANG II appears to stimulate Nox-dependent ROS generation with downstream Rho kinase activation, resulting in enhanced microparticle release at caveolae/lipid rafts in cultured mouse aortic ECs. In addition, microparticles derived from untreated ECs carry HB-EGF and stimulate VCAM-1 and PECAM induction and macrophage adhesion to ECs. These effects involve EGFR activation and subsequent ROS production. It has been confirmed that ANG II infusion in apoE−/− mice enhances production of EC-derived microparticles, which is markedly reduced with apocynin, an NADPH/NADH oxidase inhibitor (104). Senescent ECs and ECs from patients with acute coronary syndrome produce procoagulant microparticles that also stimulate endothelial senescence in vitro. This effect is due to microparticles carrying ACE, which activate EC AT1R, Nox, and downstream p38MAPK and ERK1/2 (3). Likewise, ANG II stimulates procoagulant microparticle release from human mononuclear cells that is dependent upon AT2R-mediated mobilization of intracellular Ca2+ (185). In addition, high glucose plus ANG II combination enhances production of extracellular vesicles (mixture of microparticles and exosomes), which in turn stimulate ERK1/2 and reduce eNOS expression and NO production in human umbilical endothelial cells (HUVECs) (1026).
Regarding exosomes and ANG II, a recent study showed that AT1R-expressing HEK 293T cells release exosomes upon osmotic stretch or ANG II stimulation containing functional AT1Rs that stimulate ERK. Moreover, injection of AT1R KO mice with exosomes from the serum of mice that underwent transverse aortic constriction (TAC) causes increases in systolic blood pressure and ERK activation within the heart via the exosome transfer of the AT1Rs. β-Arrestin is necessary for the packaging of the receptor to exosomes and cardiac myocytes are the major source of AT1R containing serum exosomes (824). In addition, ANG II stimulation enhances exosome release from cardiac fibroblasts. These exosomes exhibit hypertrophic responses in cardiac myocytes ostensibly via upregulation of a local RAS by activation of MAPKs and Akt. Moreover, pharmacological treatment to block exosome secretion attenuates ANG II-induced cardiac hypertrophy and fibrosis (621). Clearly, further study is needed in both the field of microparticle and exosome research. In ANG II-related diseases, microparticle/exosome formation and release may contribute to organ dysfunction. However, microparticles/exosomes from healthy or even artificially modified vesicles may be valuable as potential therapeutic options.
5. Autophagy and mitophagy
Autophagy is a critical mechanism needed to maintain cell and organ homeostasis through the stimulation of catabolism. It is initiated by formation of a double-membrane vesicle termed the autophagosome, which is responsible for degrading organelles and large protein aggregates (963). Inquiries into the fundamental mechanisms of autophagy for adaptation under stress have shed light into how this pathway may be regulated through various cellular conditions including starvation, ER stress, and oxidative stress (1, 468). In addition to its importance in protein catabolism during starvation, basal autophagy is now recognized as a critical housekeeping pathway even in nutrient-rich conditions. This protein quality control mechanism is particularly crucial, where autophagy is important for the removal of aggregated proteins. For example, loss of autophagy in cardiac muscle can result in the accumulation of ubiquitinated proteins and inclusion bodies, leading to cardiac hypertrophy. For general reviews on autophagy in cardiovascular and metabolic diseases, please refer to the following reviews (328, 543, 762).
ANG II has been shown to enhance autophagy via AT1R in several cell types. In neonatal rat cardiac myocytes infected with AT1R-encoding adenovirus, ANG II-induced myocardial hypertrophy is associated with increased autophagy. However, while addition of AT2R-encoding adenovirus in conjunction with AT1R augments ANG II-induced hypertrophy, enhancement in autophagy is attenuated by AT2R (833). In the HL-1 cardiomyocyte cell line, ANG II at concentrations between 10 and 100 nM stimulates autophagy, whereas higher concentrations cause apoptosis and decrease autophagy, suggesting a protective role for autophagy (1122). ANG II-enhanced autophagy is associated with induction of beclin-1, microtubule-associated protein light chain 3 (LC3), and autophagy protein 9A (Atg9A), proteins essential for autophagosome formation (401, 1122). Inhibition of beclin-1 or Atg9A with siRNA attenuates the ANG II-induced hypertrophic response in rat neonatal cardiac myocytes (401, 794). The ANG II-enhanced autophagy and hypertrophic responses are associated with downregulation of miR-30 and miR-34a, where miR-30 appears to regulate beclin-1 expression and miR-34a regulates Atg9A expression (401, 794). Furthermore, AT1R activation promotes Nox4 activation at the mitochondria leading to mitochondrial dysfunction, mitochondrial ROS production, and downstream induction of autophagy in cardiac myocytes (201). ANG II-induced in vivo cardiac hypertrophy is also highlighted by increased autophagy as measured by LC3b-II and p62, which can be inhibited by the ANG (1–7)/Mas receptor signaling axis by reducing ROS (592). In line with these reports, deletion of the cardiac anti-inflammatory cytokine IL-10 enhances cardiac autophagy and hypertrophy induced by ANG II (500). Likewise, in a swine model of renovascular hypertension, ARBs alone or in combination with non-RAS antihypertensive agents similarly lower blood pressure; however, ARBs but not the combination alleviates left ventricular hypertrophy, myocardial autophagy and mitophagy, and increased mitochondrial biogenesis (1256).
The role of ANG II-enhanced autophagy in cardiovascular pathophysiology is unsettled. Autophagy protein 5 (Atg5) is an E3 ubiquitin ligase that is necessary for formation of autophagosomes and autophagy. ANG II infusion increases cardiac Atg5 expression and autophagy and mitophagy in infiltrated macrophages. In Atg5+/− mice, reduction in macrophage autophagy and mitophagy is associated with enhancement of cardiac hypertrophy, fibrosis, ROS production, and NF-κB activation (1265). Likewise, adiponectin induces macrophage autophagy, thereby inhibiting ANG II-induced inflammatory responses and cardiac fibrosis in mice (843). These data suggest a protective role of autophagy against ANG II-induced proinflammatory responses.
In cultured VSMCs, ANG II increases the LC3-II to LC3-I ratio and beclin-1 expression and decreases p62, suggesting stimulation of autophagy. These responses are attenuated with a NADPH oxidase inhibitor and mitochondrial KATP channel inhibitor (1221). Kelch-like protein 3 (KLHL3)-Cullin3 complex was identified as an E3 ubiquitin ligase for with-no-lysine kinases (WNK) (see detail of this system in the kidney section). In mouse VSMCs, KLHL3 is not expressed, but its close homolog KLHL2 is expressed. Acute (30 min) ANG II infusion reduces KLHL2 in the aorta with a concomitant increase in WNK3 expression, an effect confirmed in mouse VSMCs. This WNK3 accumulation leads to Ste20/SPS1-related proline/alanine-rich kinase (SPAK) activation and Na-K-Cl cotransporter isoform 1 (NKCC1) phosphorylation and vascular contraction. Mechanistically, KLHL2 is degraded by p62 via enhanced autophagy with ANG II stimulation, suggesting a role for autophagy in ANG II-induced hypertension (1237).
6. Intracellular/organelle RAS
In addition to the role of ANG II in systemic and tissue RAS, the presence and functional significance of intracellular/intracrine ANG II has been described. The importance of the intracellular RAS was highlighted recently in key review articles (181, 260, 520). Intracellular ANG II binds to the AT1R expressed in the nuclear membrane causing growth promoting signal transduction. However, there is controversy over whether intracellular ANG II and its receptors are internalized or originate within the cells they affect. Both AT1R and AT2R are also reported to be present within mitochondria (2). However, cautious reading is recommended as many ANG II receptor antibodies used in these studies have been reported to be nonspecific. No evidence has been obtained regarding RAS component expression in liver mitochondria with proteomic approaches (38).
It is important to confirm that genetically expressed intracellular ANG II can cause hypertension in AT1AR knockout mice, which would suggest a novel intracellular receptor mechanism for ANG II (260). By using UV light activating intracellular caged ANG II, intracrine ANG II was shown to activate ERK in AT1R-expressing HEK cells (1023). Intracellular injection of ANG II also confirms ANG II-dependent intracellular Ca2+ signaling in hypothalamic neurons (228). ANG II intracrine activation of nuclear AT1R is also shown to mediate cardiac fibroblast proliferation and collagen synthesis (1024).
C. Novel Posttranslational Modification in ANG II Signal Transduction
1. Acetylation/deacetylation
Histone acetylation/deacetylation is a fundamental mechanism for posttranslational regulation of gene expression. Histone deacetylases (HDACs) deacetylate histones, condense chromatin, and thereby repress transcription. In contrast, histone acetyltransferases acetylate histones, resulting in relaxation of nucleosomes and stimulation of transcription. Importantly, class II HDACs interact with myocyte enhancer factor 2 (MEF2) and appear to repress cardiac hypertrophy. In VSMCs, ANG II induces PKC/PKD1-dependent class IIa HDAC5 phosphorylation and nuclear export, leading to MEF2 activation and protein synthesis (1181). Class IIa HDAC4 and HDAC5 are phosphorylated by CaMKIIδ2 to activate MEF2-dependent induction of Nur77 and monocyte chemoattractant protein-1 (MCP1) in VSMCs (320). A scaffold protein GRK2 interacting protein 1 mediates ANG II-induced HDAC phosphorylation in VSMC (795). siRNA studies further confirm the roles of HDAC4 and GATA6 in VSMC hypertrophy induced by ANG II (492). Global deletion of HDAC6 in class IIb HDAC6-deficient mice protected mice from ANG II-induced cardiac systolic dysfunction and skeletal muscle wasting. HDAC6-deficient hearts infused with ANG II are associated with reduced autophagy (229). In addition, ANG II-induced cardiac hypertrophy is blocked with a class I HDAC inhibitor (477). Subsequent study shows that ANG II infusion increases class I HDAC2 activity in mouse hearts (476). Class I HDAC inhibitors also attenuate ANG II-induced cardiac fibrosis (1149).
Histone acetyltransferases, such as CREB-binding protein (CBP)/p300, steroid receptor coactivator-1 (SRC-1), and p/CAF mediate nuclear histone lysine acetylation and activate transcription. Chromatin immunoprecipitation assays show that ANG II increases histone H3 Lys9/14 acetylation on the IL-6 promoter. IL-6 transcriptional activity is attenuated by ERK-phosphorylation site deficient mutants of CBP/p300 and SRC-1, suggesting activation of these histone acetyltransferases in rat VSMCs (899). In addition, ANG II-induced acetylation of cyclophilin A stimulates its secretion in VSMCs enhancing ERK activation and ROS production (978).
Sirtuin-1 (SIRT1; mammalian homolog of silent information regulator in yeast) is a nicotinamide adenine dinucleotide+ deacetylase and molecular target of polyphenols and calorie restriction. SIRT1 is generally associated with beneficial metabolic effects by virtue of antioxidant and anti-inflammatory effects. Peroxisome proliferator-activated receptor gamma (PPARγ) coactivator-1α (PGC-1α) is a master regulator of mitochondrial biogenesis and function, oxidative stress, and insulin resistance. ANG II stimulates Akt-dependent phosphorylation of PGC-1α on Ser570, which is required for binding of the histone acetyltransferase GCN5 (general control nonderepressible 5) to PGC-1α and for its lysine acetylation. These sequential posttranslational modifications suppress PGC-1α activity and prevent its binding to the catalase promoter through the forkhead box O1 (FoxO1) transcription factor, thus decreasing catalase expression and enhancing VSMC ROS generation and hypertrophy (1174). ANG II also induces prolonged lysine acetylation of PGC-1α and releases the PGC-1α·FoxO1 complex from the SIRT1 promoter, thus reducing SIRT1 expression in VSMCs (1173). Vascular protective effects of SIRT1 against ANG II will be described in detail in the vascular section of this review. Mitochondrial SIRT3 and SIRT4 play a role in the longevity network that maintains mitochondrial vitality. In AT1AR−/− mice, SIRT3 is upregulated in the kidney, which may contribute to mouse longevity. ANG II stimulation also downregulates SIRT3 mRNA in cultured tubular epithelial cells (70). A subsequent study demonstrates that acetyl-l-carnitine supplementation improved ANG II-induced insulin resistance in L6 muscle cells, which is associated with SIRT3 reexpression, mitochondrial protein deacetylation, and reduced mitochondrial ROS production (626). A recent study further demonstrated that ANG II causes SIRT3 S-glutathionylation, with resultant increased SOD2 acetylation and inactivation contributing to ANG II-induced hypertension due to vascular oxidative stress (235).
2. S-Nitrosylation
NO is an important signaling molecule in the cardiovascular system and a regulator of vascular tone by binding and activating soluble guanylate cyclase (sGC). NO also reacts indirectly with biological molecules, and these chemical reactions include S-nitrosylation. ANG II has been shown to enhance S-nitrosylation of various proteins in rat aorta (164). ANG II induced S-nitrosylation of sGC is associated with reduced cGMP production in VSMCs (188). It is interesting to note that AT1R is also regulated by S-nitrosylation of Cys289, which reduces ANG II binding affinity (551).
3. O-GlcNAcylation
Cytoplasmic and nuclear protein O-GlcNAcylation on serine and threonine residues by the O-linked attachment of the monosaccharide N-acetylglucosamine (O-GlcNAc) is rapidly emerging as a critical signaling event in cell biology. Moreover, enhanced protein O-GlcNAcylation has been implicated in glucose toxicity and insulin resistance (531). In cardiac myocytes, the ANG II-induced increase in intracellular Ca2+ is inhibited by glucosamine and PUGNAc [N-acetylglucosaminono-1,5-lactone O-(phenylcarbamoyl)oxime], both of which are associated with increased protein O-GlcNAc levels (736). Moreover, activation of the hexosamine biosynthesis pathway and increased protein O-GlcNAcylation in nondiabetic cardiomyocytes suppresses the hypertrophic signaling response to ANG II (654).
4. SUMOylation
SUMOylation is a posttranslational modification in which a member of the small ubiquitin-like modifier (SUMO) family of proteins conjugates to lysine residues in target proteins. SUMOylation alters the activity of target proteins and cross talks with ubiquitination, acetylation, and phosphorylation. Protein SUMOylation is a dynamic process that can be readily reversed by a family of sentrin/SUMO-specific proteases (SENPs). ATF3 is an adaptive-response protein induced by various environmental stresses. ANG II upregulates ATF3 and SUMO1 in endothelial cells in vitro and in vivo. ANG II induces lysine SUMOylation of ATF3 which is required for endothelial inflammatory responses (1260).
D. MicroRNA
MicroRNAs (miRNA) regulate diverse cellular processes with important roles in physiology and pathophysiology, including tissue remodeling (1085, 1092). miRNAs interfere with mRNA translation via inhibition of translation elongation and/or targeting of mRNAs for degradation. Under baseline conditions, miRNAs appear to function as a fine tuner of gene expression. However, under stress or disease conditions, mRNAs exert pronounced silencing functions (1085). Note that the RAS components including AT1R are targets of several miRNAs, which can be regulated with ANG II stimulation (481, 764).
Recent investigation into ANG II-linked signal transduction has shed light on a functional role for miRNAs in pathophysiology. For example, numerous miRNAs have been identified as playing a role in regulating ANG II-mediated cardiac hypertrophy including miR-98 (target: cyclin D2) (1197), miR-21 (target unknown) (159), miR-21–3p (target: HDAC8) (1187), miR-26a (target: GATA4) (605), miR-410 and miR-495 (targets: unknown) (175), miR-101 (target: Rab1a)(1133), miR-30a (target: beclin-1) (794), miR-34a (target: Atg9A) (401), and miR-99a (target: mTOR) (574). And the list may keep increasing. How all of these miRNAs fit together, and how they are regulated in ANG II-induced cardiac hypertrophy has yet to be determined, but remains a continued area of investigation.
miRNAs have also been implicated in cardiac fibrosis. MiR-125b negatively regulates p53 thereby promoting fibroblast proliferation. In cardiac fibroblasts, miR-125b directly binds to the 3′UTR of the Apelin gene, which is a negative repressor of the fibrogenic pathway. Cumulatively, miR-125b expression stimulates fibroblast proliferation as well as cardiac fibroblast-to-myofibroblast differentiation, and in vivo abolishment of miR-125b prevents ANG II-induced cardiac and perivascular fibrosis (735). In addition, ANG II decreases miR-let-7i expression in the mouse heart, and delivery of miR-let-7i in ANG II-infused mice reduces cardiac inflammation and fibrosis by targeting IL-6 and collagens (1124). In ANG II-treated cardiac fibroblasts, osteopontin is essential for AP-1-mediated transcription of miR-21. MiR-21 inhibits SMAD7, the endogenous inhibitor of TGF-β signaling, and promotes downstream ERK phosphorylation, contributing to fibroblast survival and extracellular matrix (ECM) production (611). MiR-29 also targets TGF-β1 and ANG II reduces miR-29 expression in cardiac fibroblasts. Therefore, a decrease in miR-29b enhances ANG II-induced cardiac fibrosis (1259). Central to the inflammatory and fibrotic response, ANG II-induced NF-κB activation suppresses miR-26a-mediated negative regulation of collagen I and connective tissue growth factor (CTGF), thereby promoting cardiac fibrosis. Reexpression of miR-26a prevents this response and establishes a negative feedback loop, which suppresses NF-κB (1131). Thus ANG II appears to stimulate a dynamic cardiac remodeling program whereby ANG II induces a distinct miRNA expression profile where profibrotic gene expression is favored.
Separate from cardiac-based disease, many miRNAs have been implicated in modulating vascular pathologies including endothelial dysfunction, hypertension, and atherosclerosis (274, 914). Thus how miRNAs regulate vascular homeostasis or its disruption under RAS activation remains an area of much interest. In cultured rat VSMCs, small RNA deep sequencing was performed to identify ANG II-responsive miRNAs. An miR-132 and miR-212 cluster was identified as the most highly induced miRNAs, in which miR-132 targets phosphatase and tensin homolog (PTEN) and p120 Ras GAP to enhance MCP1 induction and CREB activation, respectively (442). In vivo approaches utilizing miRNA-deficient mice have further advanced this field. MiR-143/145-deficient VSMCs exhibit a synthetic phenotype with reduced contractile proteins. MiR-143/145-deficient mice exhibit reduced vascular media thickness. As such, these mice have a reduced contractile response to ANG II. Unbiased quantitative proteomics combined with microarray-based transcriptional profiling revealed the vascular targets of miR143/145, including tropomyosin 4 and ACE (85). Furthermore, coculture with EC induces VSMC miR-145 expression, which suppresses TGF-β-mediated ECM accumulation by targeting the TGF-β receptor II. Thus miR-145-deficient mice have enhanced cardiac and perivascular fibrosis with ANG II infusion (1262). ANG II also decreases miR-145-mediated suppression of Kruppel-like factor 4 (KLF4) in VSMCs. Increased KLF4 reduces myocardin expression thereby increasing VSMC proliferation and migration (962).
Regarding endothelial biology, EC miR-155 targets AT1R expression, decreases HUVEC migration and leukocyte adhesion with ANG II stimulation (1273), and promotes anti-angiogenic and pro-arteriogenic functions during neovascularization (797). ANG II-induced oxidative stress is also known to promote SREBP2 transactivation of miR-92a in HUVECs. EC miR-92a suppresses expression of SIRT1, KLF2, KLF4, and eNOS leading to inflammasome activation, stimulation of endothelial innate immunity, and reduction of NO production. Thus ANG II-enhanced atherosclerosis in apoE−/− mice is attenuated with miR-92a inhibition. Levels of circulating miR-92a are inversely correlated with endothelial function in humans (157).
In regards to aortic dilation and aneurysm formation, biopsies of human thoracic aneurysms show increased miR-29, whereas miR-29 inhibition induces ECM expression and inhibits ANG II-induced aortic dilation and aneurysm development/progression in mouse models of thoracic and abdominal aortic aneurysms, although conflicting data are reported regarding miR-29 regulation by ANG II (86, 632). Murine-specific miR-712 and the human/murine homolog miR-205 are induced by ANG II and stimulate MMP activity through repression of tissue inhibitor of metalloproteinase 3 (TIMP3) and reversion-inducing cysteine-rich protein with kazal motifs (RECK). MiR-205 is expressed in human AAA biopsies, and silencing of miR-712 and miR-205 prevents AAA development in ANG II-infused apoE(−/−) mice (491). Mir-21 is also responsive to ANG II, and overexpression of miR-21 in an elastase AAA infusion model induces cell proliferation within the aortic wall, but prevents apoptosis and AAA expansion (631). In addition, miR-195 contributes to ANG II-dependent AAA in mice by targeting ECM proteins (1233). TABLE 4 summarizes ANG II-regulated miRNAs, their targets, and the cell/tissue types involved, together with the functional consequences of miRNA regulation.
Table 4.
ANG II-regulated microRNA and pathology
| MicroRNA | Target | MicroRNA Function | Reference Nos. |
|---|---|---|---|
| Upregulated | |||
| miR-21 | Unstudied | Promote cardiac myocyte hypertrophy | 159 |
| PTEN/SMAD7 | Enhance Akt/ERK and cardiac fibrosis | 611 | |
| PTEN | Enhance Akt and promote AAA | 631 | |
| miR-29b | ECM proteins | Promote TAA and AAA | 86 |
| miR-92a | SIRT1/KLF2/KLF4 | Induce endothelial inflammasome and atherosclerosis | 157 |
| miR-98 | Cyclin D2 | Negative feedback mechanism to suppress cardiac hypertrophy | 1197 |
| miR-99a | mTOR | Negative feedback mechanism to suppress cardiac hypertrophy | 574 |
| miR-125b | Apelin/p53 | Promote cardiac fibrosis | 735 |
| miR-132 | PTEN/RASA1 | MCP1 induction and CREB activation in VSMCs | 442 |
| miR-195 | ECM proteins | Promote AAA | 1233 |
| miR-410 | Unstudied | Promote cardiac myocyte hypertrophy | 175 |
| miR-495 | Unstudied | Promote cardiac myocyte hypertrophy | 175 |
| miR-712/-205* | TIMP3/RECK | Activate MMP and induce inflammation and AAA | 491 |
| Downregulated | |||
| miR-21–3p | HDAC8 | Inhibit cardiac myocyte hypertrophy | 1187 |
| miR-26a | GATA4 | Inhibit cardiac myocyte hypertrophy | 605 |
| CTGF/collagen I | Inhibit cardiac fibrosis | 1131 | |
| miR-29 | TGF-β | Inhibit cardiac fibrosis | 1259 |
| miR-29b | ECM proteins | Promote TAA and AAA | 632 |
| miR-30a | Becline-1 | Inhibit autophagy and cardiac myocyte hypertrophy | 794 |
| miR-34a | ATG9A | Inhibit autophagy and cardiac myocyte hypertrophy | 401 |
| miR-101 | Rab1a | Inhibit cardiac myocyte hypertrophy | 1133 |
| miR-145 | KLF4 | Induce myocardin and inhibit VSMC proliferation | 962 |
| miR-let-7i | IL-6/collagens | Inhibit cardiac fibrosis and inflammation | 1124 |
miR-205 is the human homologue of murine miR-712.
E. Long Noncoding RNA
Molecular functions of long noncoding RNA (lncRNA; >200 nucleotides) include cis- or trans-acting transcriptional regulation, competing with miRNAs, and stabilizing mRNAs (558). lncRNAs can interfere with gene expressions and signaling pathways at several stages and are increasingly recognized for their roles in cardiovascular diseases (293, 1075). While lncRNA research is a new field in ANG II signal transduction, a few interesting findings have been made in regards to how lncRNAs are potentially involved in ANG II pathophysiology. With the use of RNA-seq, it has been uncovered that ANG II can promote dynamic and transient changes in lncRNA expression. The responsive lncRNAs appear to be proximal to various genes. In particular, lncRNA-Ang362 sits proximal to both miR-221 and miR-222, both of which are involved in vascular proliferation and inflammation. lncRNA-Ang362 is needed for proper expression of mature forms of both miR-221 and miR-222, and silencing of lncRNA-Ang362 reduces VSMC proliferation suggesting its regulatory role in vascular remodeling (559). In addition, a bioinformatics approach used to predict lncRNA-disease associations predicted four specific lncRNAs associated with hypertension. Of these four, lncRNAs SLC17A9, SPATA9, and C16orf95 were downregulated in human VSMCs treated with ANG II (1108).
lncRNA CHRF in cardiac myocytes acts as an antagomir for miR-489 during ANG II stimulation and increases expression of the miR-489 target, Myd88, which appears to drive cardiac hypertrophy (1109). In addition, expression of lncRNA PVT1 is increased in hypertrophied mouse hearts with TAC. In cultured cardiac myocytes, ANG II increases PVT1, and siRNA silencing of this lncRNA prevents ANG II-induced cardiomyocyte hypertrophy (1224). Expression profiling in cardiac fibroblasts in response to ANG II shows a robust expression in lncRNAs such as NR024118 and Cdkn1c via AT1R (440, 441), whereas their functional significance remain unstudied.
F. System Biology and Bioinformatics
The advent of unbiased system biology and bioinformatic approaches have paved the way for exploration into transcriptomics and proteomics, which link distinctly regulated genes and proteins to a pathological phenotype. These fields have already greatly enhanced our knowledge in all fields, and ANG II is no exception. Expanding information and big data sets have been vital in elucidating our understanding of ANG II signaling and its effect on global gene and protein expression in a wide array of tissues in various diseased states. An earlier study with DNA microarray analysis with subsequent bioinformatics identified ANG II responsive genes and their respective clusters. Furthermore, in vivo analysis indicated calpactin I and osteopontin are both involved in the vascular injury response of ANG II (115). Microarrays have also identified novel transcripts associated with the RAS system in human atheroma development (744). Likewise, serial analysis of gene expression (SAGE) on mouse kidneys following ANG II infusion identified cathepsin D as a new gene regulated by ANG II that was functionally characterized to interact and affect blood pressure regulation (932). Furthermore, RNA-seq has led to identification of novel protein-coding and noncoding transcripts associated with ANG II stimulation in VSMCs (559). As mentioned, bioinformatic analysis has been used to identify lncRNAs associated with hypertension and ANG II stimulation of VSMCs (1108). Genome-wide Chip-seq analysis identified over 2,000 enhancers regulated by ANG II in VSMCs (206). In addition, unbiased gene screening approaches have been employed to explore ANG II signal transduction. With the use of a siRNA kinome library targeting 720 kinase genes, siRNA screening was performed to identify a kinase required for EGFR transactivation by ANG II. Results yielded Trio, Chka, and Bmx as kinases upstream of the EGFR, supporting the usefulness of this approach for signal transduction research (313).
Recently, genome-wide transcriptome analyses have been performed in vivo with ANG II infusion. Two days after the infusion, differently expressed genes are identified in the kidney. Genes in metabolism and ion transport pathways are upregulated, while genes protective against oxidative stress, including glutathione synthetase and mitochondrial SOD2, are downregulated (637). Gene categories specifically regulated in ANG II-induced AAA in apoE−/− mice have also been reported, including cytokine-cytokine receptor interaction, leukocyte transendothelial migration, natural killer cell-mediated cytotoxicity, and hematopoietic cell lineage (888). Another study on the same subject also demonstrates widespread upregulation of inflammatory, immune, and matrix remodeling genes with ANG II treatment (988). In addition, vascular transcriptome analysis 2 wk after ANG II infusion in mice demonstrates sphingosine kinase 1 in the highest number of pathways affected. Subsequent analysis confirms contribution of sphingosine kinase 1 to ANG II-induced hypertension as well as endothelial dysfunction (967).
Mass spectrometry has shown robust changes in the VSMC proteome in response to ANG II. Coupled with an algorithm used to study protein coordination allowing for quantitative proteomic analysis, ANG II-stimulated VSMCs show significant upregulation of proteins related to protein synthesis, folding, and turnover. Alongside this, ANG II elicits protein coordination associated with contraction and migration. Some of these proteins include those involved in mitochondria biogenesis, including PGC1-α. As contraction is a calcium-dependent and ATP consuming process, increased mitochondria would allow for continual ATP production and act as an energy source (307). ANG II also mediates suppression of the homocysteine biosynthesis pathway, a pathway largely involved in vascular smooth muscle proliferation (1281), and increased gluconeogenesis pathway activation with a concomitant decrease in the glycolytic pathway (307). Investigation into the ANG II-induced secretome in VSMCs has also shed light into how ANG II signaling may affect neighboring cells as ANG II is known to affect ECM homeostasis and participate in vascular inflammation and fibrosis. Sixty-four ECM and cell adhesion-related proteins were identified as VSMC-secreted proteins with ANG II eliciting significant upregulation of specific proteins including collagen α-1(VI), osteopontin, TGF-β, and lysl oxidase-like 2 (304). In human proximal tubular cells, proteome analysis using stable isotope labeling with amino acids reveals heme oxygenase-1 as the most significantly upregulated protein in response to ANG II (508).
Recent phospho-proteome analysis on an AT1R expressing cell line has significantly contributed to our understanding of global signaling mechanisms utilized by ANG II, as well as G protein-independent biased agonists. One study identified more than 1,000 phosphorylation sites regulated by ANG II or the biased ligand SII-ANG II. PKD is considered as a key kinase regulating both G protein-dependent and -independent phosphorylation events of the AT1R (170). Another study identified more than 1,500 phosphorylated proteins with SII-ANG II stimulation, which include cytoskeletal reorganization network proteins, cofilin, and slingshot (1170). A gel-based phosphoproteomic analysis has identified much smaller pools of SII or ANG II-induced protein phosphorylation in HEK cells demonstrating a link between protein phosphorylation and inhibition of protein phosphatase 2A associated with β-arrestin 2 (483). In addition, relevant to ANG II signal transduction and functions in kidney, phospho-proteomic analysis performed in proximal tubules of rats infused with ANG II, identified several signaling proteins phosphorylated upon ANG II infusion (581).
Under sustained metabolic and mechanical stress, such as observed in the heart with ANG II infusion or aging, enhanced misfolding of proteins leads to protein aggregation. Proteome analysis of hearts from aged or young mice, compared with ANG II-treated mice, shows commonly recognized aggregated proteins linked to the ubiquitin-proteasome system and autophagy machinery, suggesting the clearance process for proteins is impaired in ANG II or age-related cardiac pathologies. Further analysis using a gene ontology meta-analysis pointed to increases in genes related to ECM remodeling, the proteasome, and Alzheimer's disease-related genes, and a decrease in genes related to mitochondrial and cell respiration. Together these data suggest the presence of common mechanisms underlying disease pathology in aging and ANG II-induced hypertension (43).
A report also used proteomic screening and gene ontology to observe and delineate specific protein groups that are affected by ANG II treatment, such as proteins including HO-1 involved in the ER-mediated apoptotic cascade in proximal epithelial tubule cells (508). ANG II infusion in rats stimulates protein degradation without de novo protein synthesis in the platelet proteome, suggesting that ANG II may cause accelerated platelet aging (312). As introduced in the exosome section, proteomic analysis was performed on exosomes derived from cardiac fibroblasts treated with ANG II, which contain several factors (osteopontin, EGFR, etc.) linked to hypertrophic PI3K/Akt and MAPK pathways (621). Furthermore, proteomic studies have shed light on the critical control that ANG II-mediated AT1R signaling has in other disease states, including diabetic retinopathy. Proteomic analysis uncovered robust changes in proteins involved in translation, electron transport, and protein transport, of which 72% were downregulated when mice were treated with AT1R antagonist candesartan (303). Proteome peptide pattern profiling in diabetic patient urine has been used as a clinical indicator of diabetic nephropathy, and patients receiving ARBs show normalization of these peptide patterns (879).
ANG II interferes with mitochondrial function and alters target cell and tissue metabolism (219), which can influence ANG II-linked pathophysiology. Metabolomic study of hearts from double transgenic rats harboring both the human renin and angiotensinogen genes showed changes in 112 metabolites associated with decreased fatty acid synthesis; increased hypoxanthine, which is suggestive of enhanced purine degradation; alterations in ketogenic amino acids indicating impaired glucose use; and decreased cytochrome c oxidase activity. Strikingly, mice treated with ARB valsartan display a reduction in hypoxanthine and a recovery of cytochrome c oxidase activity (692). In addition, translational research was reported to use serum metabolomics data to predict albuminuria response to ARBs in type II diabetic patients (812).
In summary, the field of systems biology has started to greatly enhance our knowledge of the diverse signaling that is carried out by ANG II. Continually pursuing and exploring this area of research will further identify clusters of gene or proteins that may regulate system function in terms of metabolism, inflammatory signaling, and apoptotic signaling to name a few. Using these approaches, we may be able to identify novel and candidate genes/proteins that may be suitable for therapeutic intervention in a wide-array of ANG II-related diseases.
G. Tissue/Cell Type Specific ANG II Signaling
The emergence of genetic engineering has paid dividends in understanding genetic and molecular links to disease pathophysiology including ANG II-based diseases. Tissue/cell type-specific knockouts of the AT1R have been used to investigate the tissue-specific role of ANG II signaling. Recently, the use of several distinct tissue-specific knockout mice for the AT1R identified adventitial fibroblast-specific AT1AR signaling but not VSMC or EC as the driving mechanism in ANG II-induced medial thickening and hyperplasia in the ascending aorta (825). Likewise, AT1AR depletion in ECs or VSMCs in LDLr −/− mice does not affect AAA or atherosclerosis (858), but EC AT1AR depletion in LDLr −/− mice attenuates TAA development (857). In these studies with Cre-loxP technology, Sm22α Cre, Tie2 Cre, or S100A4 Cre driver was used to knockout smooth muscle, endothelial, or fibroblast AT1AR. Vascular smooth muscle AT1AR was found to be dispensable for ANG II increasing blood pressure (986). However, in a recent study, using Cre transgenes with robust expression in both conductance and resistance arteries (Sm22α Cre knock in), it appears that vascular smooth AT1AR deficiency is preventive against ANG II-induced elevation in blood pressure, which is associated with increased urinary sodium excretion and decreased vasoconstrictor responses in the renal vasculature (987). Tie2 Cre is known to be expressed in hematopoietic cells, and S100A4 expression in vascular smooth muscle cells was also reported (163). Therefore, cell type specificity should be more carefully interpreted, and negative study with Sm22α Cre mice should be confirmed with other types of smooth muscle Cre driver mouse. In addition, there is a report that mice with kidney principal cell-specific AT1AR deletion show a slight reduction in BP elevation in the initial phase of ANG II-dependent hypertension that is correlated with decreased α-epithelial sodium channel activation and increased natriuresis (147).
Hypoxia-inducible factor 1 alpha (HIF-1α) and PPARγ are also considered to regulate ANG II functions with tissue/cell type specifically. ANG II upregulates HIF-1α expression and induces ADAM17 promoter activation and protein expression in VSMCs (766). Moreover, in smooth muscle cell-specific HIF-1α knockout mice, ANG II-induced vascular medial hypertrophy and hypertension are reduced (414). Increased vascular AT1R expression and elevated blood pressure with downregulation of PPARγ in VSMC are also reported in smooth muscle cell-specific HIF-1α knockout mice (404). It has also been reported that smooth muscle-specific overexpression of a dominant negative PPARγ mutant (P467L) leads to enhanced vasoconstriction (485) and ERK activation (124) in response to ANG II. ANG II-induced PKC-ζ activation, ERK activation, KLF5 expression, and VSMC proliferation are also inhibited by PPARγ agonist (305). ANG II-induced vascular remodeling, contractility, inflammation, and endothelial dysfunction are enhanced in inducible smooth muscle-specific PPARγ-deficient mice, which is associated with decreased expression of SOD3 (649). Endothelial dysfunction and augmented pressor response to ANG II were observed in endothelial-specific expression of dominant negative PPARγ (V290M) (77). The mouse phenotype is associated with enhanced oxidative stress and reduced expression of catalase and SOD3 (399). Moreover, the antioxidant effects of PPARγ against ANG II-mediated ROS in endothelial cells requires the PPARγ target gene PPARγ cofactor retinol binding protein 7 (398). The above findings illustrate the complex relations among the AT1R, HIF-1α, and PPARγ in mediating hypertension, vascular remodeling, and endothelial dysfunction.
These studies have helped to specify the potential tissue specific role of ANG II in mediating not only vasoconstriction and blood pressure elevation, but also cardiovascular remodeling. The following sections covering ANG II-mediated pathophysiology will cover in depth how ANG II signaling leads to downstream alterations in organ and cell homeostasis and resultant dysfunction.
IV. UPDATE OF VASCULAR SIGNALING AND PATHOPHYSIOLOGY OF ANG II
A. Vasoconstriction
One of the most understood signaling components of how ANG II mediates vascular physiology and pathophysiology is through the control of vascular tone. Readers are referred to previous reviews on this subject to explain basic signaling mechanisms (747, 1163).
1. Gq/11- and G12/13-mediated pathways converging on MLC phosphorylation
Briefly, ANG II promotes vasoconstriction through AT1R-dependent heterotrimeric G protein activation of Gq/11 and G12/13, and subsequent activation of the G protein effectors, PLC-β and a Rho guanine nucleotide exchange factor (GEF), LARG, respectively. This in turn leads to IP3 receptor-dependent intracellular Ca2+ elevation and Rho kinase/Rho-associated protein kinase (ROCK) activation, resulting in increased MLC phosphorylation through increased MLCK and decreased MLCP/MYPT1 activity, respectively, culminating in smooth muscle contraction. The essential roles of these G proteins and downstream effectors in ANG II-induced VSMC signal transduction and vascular contraction have been well established with in vitro (772, 1018) and in vivo (593, 846, 1152) support. It is also important to note that while ANG II-induced hypertension is attenuated in mice with smooth muscle-specific IP3 receptor triple knockout (IP3 has 3 genes and subtypes), ANG II-induced vascular remodeling is unaltered, suggesting that ANG II-induced hypertension is dispensable for vascular remodeling in certain experimental conditions (593). Noteworthy new regulators of these converging cascades include PKC-potentiated myosin phosphatase inhibitor of 17 kDa (CPI-17) and a RhoGAP, GRAF3. CPI-17, a myosin phosphatase inhibitory protein, predominantly expressed in VSMCs, enhances ANG II-induced vasoconstriction along with increases in ROCK2 and PKC-α/δ (1003). GRAF3 deletion in mice enhances Rho-dependent vascular contractile response to ANG II (48). However, whether ANG II modulates GRAF3 activity or expression remains unknown.
2. Tyrosine and serine/threonine kinases involved in vasoconstriction
While the above cascades represent major signaling pathways for VSMC contraction in response to ANG II stimulation of AT1R, there have been additional key modulators reported. The PDZ-RhoGEF/RhoA/Rho kinase signaling cascade has been shown to promote ANG II-induced contractile responses in mesenteric arteries through MLCP inhibition (380). PYK2-dependent tyrosine phosphorylation of PDZ-RhoGEF via intracellular Ca2+ elevation appears to mediate ANG II-induced RhoA activation in cultured rat VSMCs (1206). Similarly, Ras-related protein 1 (Rap1b) knockdown promotes vascular contraction induced by ANG II through inhibition of MLCP (533). In addition, ANG II stimulates JAK2-dependent phosphorylation (Tyr738) of smooth muscle expressed Arhgef1, a Rho exchange factor, which in turn activates RhoA and mediates ANG II-induced blood pressure elevation and vascular tone regulation in mice (342). Further research has uncovered the activation of Arhgef1 by ANG II in human VSMCs and peripheral mononuclear cells (119). Importance of this pathway is also confirmed with VSMC JAK2-deficient mice infused with ANG II. VSMC silencing of JAK2 attenuates ANG II-induced vascular contraction, which is associated with decreases in Rho kinase activity, intracellular Ca2+, and ROS (499). In addition, Src family tyrosine kinases have a role in ANG II-induced vascular contractility. A Src family kinase inhibitor prevents ANG II-induced MLC phosphorylation and vascular contraction (847). Moreover, ANG II-dependent hypertension, but not vascular remodeling, is attenuated in c-Src+/− mice (111). ANG II is known to activate IKK2/IKK-β leading to NF-κB activation in VSMCs (1252), and inhibition of IKK2 attenuates the contractile response to ANG II. Furthermore, mice harboring a smooth muscle-specific deletion of IKK2 exhibit a blunted hypertensive response to ANG II, which is explained by direct phosphorylation of MLC by IKK2. The negative effect with NF-κB RelA/p65 translocation inhibitor ex vivo suggests that the role of VSMC IKK2 in ANG II-induced hypertension is independent from NF-κB activation (1205). A new role for CaMKII has also emerged as smooth muscle specific inhibition of CaMKII prevents L-type Ca2+ channel activity and intracellular Ca2+ elevation, but is unable to prevent ANG II-induced contraction of mesenteric arteries. CaMKII inhibition increases MLCK activity pointing to an alternative route of vasoconstriction separate from CaMKII-dependent mechanisms of Ca2+ homeostasis (837). In addition, a mitotic kinase PLK1 expressed in VSMCs has recently been shown to mediate RhoA activity, ANG II-induced hypertension, and vascular integrity (216).
3. Voltage-gated Ca2+ channel and Cl− regulation by WNK
In regards to Ca2+ regulation of vascular tone, the importance of L-type Ca2+ channel in blood pressure regulation has been well-established pharmacologically and clinically and, more recently, has been confirmed genetically in mice. Mice deficient in the regulatory subunit of L-type Ca2+ channel, Cavβ3, show a reduced hypertensive response upon ANG II infusion. ANG II also increases vascular expression of the Ca2+ channel subunits in wild-type mice (489). How does ANG II cause membrane depolarization to activate the voltage-dependent L-type Ca2+ channel? This long unanswered question seems to have been solved recently. Na-K-Cl cotransporter 1 (NKCC1) mediates intracellular Cl− accumulation, leading to membrane depolarization and activation of voltage-dependent Ca2+ channels (779). WNKs phosphorylate and activate oxidative stress-responsive kinase 1 (OSR1) and SPAK, leading to downstream phosphorylation and activation of NKCC and Na-Cl cotransporter (NCC) (215). ANG II has been shown to stimulate phosphorylation of SPAK and NKCC1 in mouse aorta via AT1R, which is attenuated in WNK3 knockout mouse. Moreover, ANG II-induced vasoconstriction and hypertension are attenuated in WNK3 knockout mice (1238). Bumetanide, an NKCC1 inhibitor, also attenuates ANG II-induced mesenteric artery tension in hypertensive rats (162). ANG II infusion increases NKCC1 expression in arteries, which involves transcriptional activation of the promoter via histone modification.
Cullin3 and Kelch-like proteins (KLHL) are part of the ubiquitin-proteasome system that regulates WNK activity. In mouse aortic VSMCs and aorta, ANG II decreases KLHL2 by selective autophagy via sequestosome-1 (aka ubiquitin-binding protein p62), leading to enhanced WNK3 expression and NKCC1 phosphorylation in VSMCs. In addition, cullin3 silencing enhances WNK3 expression induced by ANG II in VSMCs (1237). In humans, a cullin3 mutation (CUL3Δ9) causes pseudohypoaldosteronism type II, a dominant monogenetic form of hypertension (95). In smooth muscle-specific transgenic mice expressing CUL3Δ9, ANG II-induced vasoconstriction is enhanced, which is associated with increased RhoA, a substrate of cullin3, and aortic stiffness (13, 409). These data suggest that cullin3 mediates ANG II-induced hypertension via 2 pathways (WNK3/NKCC1 and RhoA/ROCK).
Opening of Cl− channels within plasma membrane of VSMCs causes Cl− efflux and membrane depolarization. Ca2+-activated chloride currents have been described in VSMCs and can activate voltage-gated Ca2+ channels. The Ca2+-activated chloride channel ANO1 (anoctamin-1)/TMEM16A has been identified to mediate Ca2+-activated chloride currents. ANG II-induced development of hypertension is significantly suppressed in smooth muscle-specific ANO1-deficient mice (371). In cultured VSMCs, ANG II increases expression of ANO1, which in part mediates intracellular Ca2+ elevation (1103).
4. Transient receptor potential canonical channels and stromal interaction molecule 1
In addition, the canonical subfamily transient receptor potential canonical channels (TRPC) are thought of as putative candidates for receptor-operated cation channels (ROCCs). Among the seven members of vertebrate TRPCs (TRPC1 to TRPC7), TRPC2, TRPC3, TRPC6, and TRPC7 are activated by DAG. Sustained cation influx induced by activation of DAG-activated TRPC channels leads to membrane depolarization, resulting in activation of voltage-dependent L-type Ca2+ channels. The Ca2+ influx-mediated Ca2+ response induced by ANG II is suppressed by knockdown of TRPC6 in VSMCs, but not TRPC3 or TRPC7 (753). TRPC channel blocker SKF96365 blocks increased myogenic tone in response to ANG II infusion, and is associated with cytochrome P-450 omega-hydroxylase and 20-hydroxyeicosatetraenoic acid (20-HETE) production (1057). STIM1 is an ER Ca2+ sensor, which plays a critical role in the activation of Orai1 channels at the plasma membrane that mediate store-operated Ca2+ entry. Smooth muscle-specific STIM1-deficient mice do not develop hypertension upon ANG II infusion; the vasculature of STIM1-deficient mice exhibits impaired ER stress responses and is protected from ANG II-induced endothelial dysfunction (464).
5. Contribution of other receptors
As mentioned, AT1R interacts and forms a heterodimer with other GPCRs including the prostaglandin F2α receptor and purinergic P2Y6 receptor. Treatment with a prostaglandin F2α receptor antagonist reduces ANG II-induced contraction. Contractile responses elicited through ANG II or prostaglandin F2α likely involve heterodimer-dependent activation of the Gαq-PLC-Ca2+ cascade as antagonism of either receptor reduces IP1 production (334). Similar relation is found between AT1R and inflammation-inducible P2Y6 receptor. The receptor antagonist or genetic knockdown attenuates ANG II-induced hypertensive responses, including ROS production and endothelial dysfunction. Mechanistically, P2Y6 receptor converts VSMC AT1R signal from β-arrestin-dependent proliferation to G protein-dependent hypertrophy (752).
Likewise, downstream of ANG II and AT1R signaling, VSMC mineralocorticoid receptor (MR) stimulation promotes increased blood pressure, oxidative stress, vascular contraction, and expression of L-type Ca2+ channels. Potential crosstalks between AT1R signaling and MR signaling independently from aldosterone actions have been reported (677). Also, VSMC MR-deficient mice show decreased elevation in systolic blood pressure and oxidative stress during ANG II infusion, suggesting a regulatory role of MR on ANG II-induced BP elevation (678).
6. ROS, Nox, and SOD
As mentioned in an earlier section, ROS production via NADPH oxidase, Nox, induced by ANG II has been strongly implicated in ANG II pathophysiology, including enhanced vasoconstriction, endothelial dysfunction, vascular remodeling, and hypertension. There are excellent reviews focusing on the signaling and cellular mechanisms of this topic (308, 537, 711, 746, 911). The readers is suggested to refer to these articles for molecular understanding of ROS regulation in the vasculature.
The roles of ROS in mediating endothelial dysfunction are well established [at least experimentally (378, 628)]. Recent accumulating findings further support the roles of EC- and VSMC-derived ROS in mediating hypertension induced by ANG II in vivo, although there are many controversial findings. It has been shown that smooth muscle specific Nox1 transgenic mice demonstrate enhanced ROS production, hypertension, and vascular remodeling in response to ANG II infusion, which are attenuated with an antioxidant Tempol (234). As such, ANG II-induced hypertension is attenuated in Nox1-deficient mice, whereas conflicting data are observed regarding the prevention of vascular hypertrophy (310, 665). Smooth muscle CYP1B1-mediated ROS production has also been implicated in ANG II-induced hypertension (430, 823). In p47phox-deficient mice, ANG II-induced hypertension (535) and afferent arteriolar contraction response are attenuated together with renal cortical NADPH oxidase activation (532). Nox2 deletion in ECs, but not in myeloid cells, attenuates ANG II-induced hypertension in mice (898). Thus, in EC-specific Nox2 transgenic mice, ANG II-induced endothelial dysfunction, hypertension, and cardiac remodeling are augmented (68, 725, 726).
In contrast, in a transgenic mouse that produces human active renin in the liver, deletion of Nox1 (1208) or Nox2 (1059) does not alter hypertension, while oxidative stress is attenuated. In endothelial cell-targeted Nox4 transgenic mice, enhancement of basal H2O2 production was observed. However, ANG II-induced elevation in blood pressure was reduced in the mice (860). In smooth muscle-specific SOD3 knockout mice, ANG II-induced vascular superoxide production is enhanced, and NO levels are reduced. However, vascular inflammation and hypertension are not augmented. In contrast, silencing of SOD3 in circumventricular organs, augments hypertension induced by ANG II infusion emphasizing the role of brain ROS in ANG II-induced hypertension (607).
ROS are also produced by immune cells in response to ANG II stimulation. Lysozyme M‐positive monocytes mediate ANG II‐induced arterial hypertension, vascular dysfunction, and ROS production (1141). Specifically, T cells appear to contribute to ANG II-induced hypertension in mice (54, 1093). Highly reactive isoketals-dependent dendritic cell activation is proposed for ANG II-induced T cell proliferation and activation (498). Mice lacking a transcription factor, T‐box, expressed in T cells, which mediates IFN-γ formation in immune cells, are protected from ANG II‐induced vascular injury (511). Additionally, T cell-derived IL‐17A mediates ANG II‐induced hypertension and inflammation‐associated vascular dysfunction (630). In close relation to the role of immune response in ANG II-induced hypertension, the absence of microbiota in germ‐free mice protects from ANG II-induced vascular ROS production and hypertension (459).
The role of copper transport protein antioxidant 1 in ANG II-induced hypertension has also been demonstrated. SOD3/extracellular Cu/ZnSOD is known to protect against ANG II-induced endothelial dysfunction and hypertension and is induced by ANG II in VSMC (326). VSMC antioxidant 1 is increased by ANG II and appears to upregulate SOD3 transcription by binding the promoter. Antioxidant 1 also participates in copper transport for SOD3 via binding with ATP7A leading to SOD3 activation. Thus, in antioxidant 1-deficient mice, enhanced hypertension and worsened endothelial function are observed with ANG II infusion (789).
Cumulatively, significant advances have been made in determining the direct signaling ANG II mediates to regulate vasoconstriction in vivo. Evidence appears to indicate tight regulation of Ca2+ and Cl− through multiple avenues (FIGURE 4) as well as provide clarity into G protein and Rho kinase signaling (FIGURE 5). However, additional investigations are desired regarding the potential roles of putative kinases, other receptors, and ROS/Nox in ANG II-induced hypertension.
FIGURE 4.

Ion regulation and ANG II-induced vasoconstriction. AT1R stimulation leads to NCC and NKCC activation via a WNK/SPAK/OSR1 signaling cascade. Furthermore, AT1R-stimulated Ca2+ release can occur through an ORAI1/STIM1 mechanism leading to ER Ca2+ release from the IP3 receptor. Likewise, AT1R-induced PLC/DAG induces TPRC and LTCC leading to increased Ca2+ entry. Elevated Ca2+ can also induce ANO1 and VDCC leading to further Ca2+ entry. AT1R-dependent activation of Nox and the MR induces ROS leading to vasoconstriction. Importance of AT1R/GPCR heterodimer formation in enhancing vasoconstriction has also been recognized.
FIGURE 5.

AT1R-mediated vasoconstriction cascades. ANG II-induced vasoconstriction signaling involves both G protein and protein kinase signaling. Myosin light-chain kinase (MLCK) activity can result from both Gq/PLC-β/IP3-dependent intracellular Ca2+ elevation as well as JAK2-induced ROS production and downstream Ca2+ release. Ca2+/calmodulin protein kinase II (CaMKII) has also been observed to activate MLCK. MLC can be activated via classical G12/13/RhoA signaling and phosphorylation of MYPT. RhoA is also inhibited by GRAF3 and activated by JAK2-dependent Arghef1 and the Ca2+/PYK2/PDZ-RhoGEF cascade. Recent evidence also points towards c-Src and IKK2 as MLC activators.
B. Vascular Hypertrophy and Hyperplasia
1. EGFR, JAK2, and hypertension-independent mechanisms
One of the most implicated mechanisms in which ANG II promotes vascular hypertrophy and hyperplasia is through downstream ERK1/2 signaling. Specifically, ANG II phosphorylates and activates a transmembrane metalloproteinase ADAM17, promoting HB-EGF shedding and transactivation of the EGFR. EGFR stimulation promotes downstream ERK signaling and ER stress, as well as working in a feedback loop to promote ADAM17 induction (289). Blockade of EGFR or ER stress attenuates ANG II-induced vascular wall thickening in mice, which highlights the importance of this signaling cascade in hypertensive vascular remodeling (1035). The VSMC specific mechanism of EGFR transactivation is further supported by data with smooth muscle specific ADAM17-deficient mice where ANG II-induced EGFR transactivation and vascular remodeling, but not hypertension, are attenuated (1034). Note that ADAM17 colocalizes with Cav1 at lipid rafts in VSMCs (1027), and ANG II infusion increases Cav1 expression in mouse aorta (288). The link between EGFR activation and Cav1 induction is further supported by the fact that ANG II-dependent Cav1 expression, EGFR activation, and cerebral artery hypertrophy are attenuated in mice carrying the waved-2 mutation in EGFR (136). Thus Cav1−/− mice are protected from ANG II-dependent vascular remodeling (288, 1077). Commensurate with a role in vasoconstriction, JAK2 and downstream Rho kinase/ROCK activation can promote increased vascular wall thickening in response to ANG II as supported by findings with smooth muscle JAK2-deficient mice infused with ANG II (499). These mice also show reduced ROS production. Therefore, EGFR and JAK2 appear to be key upstream tyrosine kinases in mediating vascular remodeling induced by ANG II.
One may consider that a decrease in vascular remodeling should reduce the high blood pressure response to ANG II; however, in a 2 wk ANG II (1 µg·kg−1·min−1) infusion model with both genetic [Cav1 or ADAM17 deletion (288, 1034)] and pharmacological [EGFR inhibitor erlotinib or ER stress inhibitor PBA, 4-phenylbutyrate (1035)] interventions, suppression of vascular remodeling does not alter hypertension. The concentration of ANG II used in these protocols (1 µg·kg−1·min−1) may maintain hypertension even with a reduction in vascular remodeling. Many published articles using a similar dose of ANG II demonstrate that near-maximal hypertensive responses occur within a few days (190, 986). Likewise, time course analysis of vascular remodeling in response to ANG II in mice shows gradual arterial hypertrophy initiating at day 3, and developing for 4 wk (1263), suggesting that vascular remodeling may not be required for the establishment of hypertension in response to a high dose of continuous ANG II infusion. In contrast, at a 60% lower dose of ANG II, 4-phenylbutyrate reduces both cardiac fibrosis and hypertension (465). These data can be interpreted to mean that the mechanism of vascular remodeling induced by ANG II is at least in part hypertension independent, which is also consistent with other studies showing that ANG II-induced arterial medial thickening is independent of blood pressure in mice (784).
2. Serine threonine kinases
In vivo pharmacological investigation in mice together with in vitro VSMC data suggests that CaMKII contributes to hypertension and the hypertrophic response elicited by ANG II. CaMKII phosphorylates HDAC4, promoting its nuclear export and activation of hypertrophic transcriptional activator, MEF2 (565). The role of CaMKII in ANG II-induced arterial remodeling is further supported by transgenic mice constitutively expressing a CaMKII inhibitory peptide specifically in smooth muscle cells. In addition to suppression of ANG II-induced vascular remodeling, enhanced arterial stiffness, ECM gene expression, baroreceptor activity, and hypertension are reduced (836). Other serine threonine kinases implicated in ANG II-induced vascular hypertrophic responses include mitogen-activated protein kinase-activated protein kinase 2 (MK2) and PKC-δ. MK2 is a downstream target of p38 MAPK and is phosphorylated in response to ANG II. MK2−/− mice exhibit a delay in ANG II-induced hypertension associated with reduced NADPH oxidase activation, VSMC proliferation, and inflammatory markers (250). PKC-δ may also play a role in the hypertrophic response (740). ANG II/AT1R signaling induces phosphorylation (Ser181) and degradation of SM22α through PKC-δ, which in turn promotes PKC-δ binding to p47phox, leading to ROS production, and hypertrophy and hyperplasia of VSMCs (618). However, there is concern over in vivo findings regarding the role of SM22α, since the adenoviral vector used to target vascular SM22α may not be effectively transduced.
3. ROS and the NADPH oxidases
Among the components of NADPH oxidase implicated in ANG II pathology, p47phox deficiency attenuates medial expansion of the aorta, and a redox-sensitive gene, ID3, deficiency reduces hyperplasia within the ascending aorta (784). As mentioned, it has been shown that smooth muscle specific Nox1 transgenic mice demonstrate enhanced ROS production, hypertension, and vascular remodeling in response to ANG II infusion, which are attenuated with the antioxidant Tempol (234). In Nox1 null mice, ANG II-induced development of hypertension and vascular medial hypertrophy is blunted (310). The role of ANG II in the production of superoxide in hypertension and vascular remodeling has been further confirmed in SOD1 knockout and transgenic mice. ANG II-induced hypertension and inward vascular remodeling are exaggerated in SOD1 knockout mice, whereas these responses are reduced in SOD1 transgenic mice (122). In contrast, ANG II-induced hypertension is unaltered in Nox2-deficient mice (1059), while inward vascular remodeling is attenuated in cerebral arterioles (135). In mice with inducible Nox4 deletion, ANG II-induced vascular hypertrophy is exaggerated whereas hypertension is unaltered. The mechanism seems to involve reduced Nrf2-dependent heme oxygenase-1 expression in endothelial cells (930). Overall, while these findings suggest a potential contribution of ROS in hypertension and/or vascular remodeling, the exact roles of VSMC ROS and Nox components in hypertensive vascular remodeling remain uncertain.
4. Transcription factors
A role for transcriptional factors has emerged in regulation of vascular hypertrophy and hyperplasia induced by ANG II. HIF-1α is upregulated in response to ANG II in mouse aorta and induces ADAM17 through a transcriptional mechanism (765). Smooth muscle knockout of HIF-1α attenuates medial wall thickening in response to ANG II (414). While suppression of ANG II-dependent hypertension development is reported in the same study (414), other studies report that smooth muscle HIF-1α-deficient mice develop hypertension and hyperresponsiveness to ANG II, which involves PPARγ-mediated upregulation of vascular AT1R (404). Therefore, further research is needed to explore the role and mechanism of VSMC HIF-1α in modulating hypertensive vascular remodeling. It has also been reported that ANG II infusion leads to vascular Ets-1 expression, and ANG II-induced vascular remodeling, but not hypertension, which is completely blocked in Ets-1-deficient mice (1239). MCP1, VCAM-1, PAI1, CREG, p47phox, and ROS production are proposed to be the downstream targets of Ets-1, mediating ANG II-dependent vascular remodeling (583, 750, 1239).
A few negative regulatory transcriptional factors against ANG II-induced vascular remodeling have also been reported. ESE-1 belongs to the Ets family of transcription factors and is induced by ANG II; however, ESE-1 appears to be a counterregulatory factor of vascular remodeling. In ESE-1-deficient mice, ANG II-induced vascular remodeling and hypertension are enhanced compared with control mice (1240). Nur77 is a nuclear orphan receptor, and ANG II induces Nur77 in VSMCs via MAPK/CREB activation, which contributes to VSMC phenotype switching. Likewise, Nur77−/− mice exhibit increased aortic collagen deposition, medial thinking, and disruption of elastin. The current rationale for Nur77 involvement in ANG II-induced vascular remodeling is through its suppression of β-catenin (194). SIRT1, the closest homologue to yeast silent information regulator 2 (Sir2) protein in the humans, functions as a nicotinamide adenine dinucleotide (NAD+)-dependent protein and histone deacetylase, which has been implicated in the processes of aging, metabolism, and tolerance to oxidative stress. SIRT1 expression and activity declines in ANG II-infused aorta. Smooth muscle specific SIRT1 transgenic mice are protected against ANG II-induced vascular remodeling, which is associated with reduced MCP1 induction (306). In addition, VSMC SIRT1 overexpression in apoE−/− mice protects against ANG II-dependent AAA, which is associated with inhibition of NF-κB activation, MCP1 induction, and senescence (149).
5. Phospholipase A2 and vascular remodeling
ANG II is a known activator of phospholipase A2 (PLA2) and arachidonic acid production, resulting in downstream eicosanoid production that can exert pro- or antihypertensive effects. Intriguingly, cPLA2α-deficient mice are protected from ANG II-induced hypertension and show significant prevention of vascular remodeling (aortic hypertrophy, fibrosis) commensurate with reductions in ERK1/2 and cSrc expression (486). In addition, ANG II infusion increases vascular expression of Ca2+-independent PLA2, iPLA2β. In smooth muscle specific iPLA2β transgenic mice, ANG II-induced hypertension and vascular remodeling are enhanced, which likely involves c-Jun activation via the 12/15-lipoxygenase pathway (110).
Overall, significant advancements have been made in understanding how ANG II can instigate vascular hypertrophy and hyperplasia, and a key hallmark to this cascade appears to be ADAM17 activation and downstream ERK1/2 activation (FIGURE 6). However, there is still controversy in this field highlighted by the evidence that EC or VSMC AT1R depletion does not prevent ANG II-induced medial thickening. This mechanism can be largely dependent on neural- and fibroblast-specific AT1R in the ascending aorta based on the data with cell type specific AT1AR-deficient mice (825), although the promoter used for fibroblast-specific targeting appears to be expressed in other cell types (163). TABLE 5 summarizes cell type specific signal mediators thus far reported in ANG II-induced vascular remodeling. The EGFR pathway appears central in mediating ANG II-induced vascular hypertrophy, whereas additional evidence is needed for other kinases and Nox/ROS in mediating ANG II-dependent vascular remodeling. Furthermore, investigation is needed into the precise role of tissue specific ANG II signal transduction to elucidate the key roles these tissues play in pathogenesis of vascular hypertrophy and hyperplasia.
FIGURE 6.

ANG II signaling in vascular hypertrophy/hyperplasia. Recent studies have uncovered numerous pathways to ANG II-induced vascular growth. ANG II induces ADAM17 activity and resultant EGFR activation leading to downstream ERK/Akt/p70S6K activation, HIF-1α induction, and ER stress resulting in hypertrophy. AT1R activates p38, MK2, and Nox leading to ROS production. ROS can also be induced through JAK2-dependent mechanism, whereby ROS induces ROCK activity resulting in hypertrophy. Furthermore, ANG II induces CaMKII activation leading to HDAC2 phosphorylation, nuclear export, and resultant MEF2 activity leading to hypertrophic gene transcription. ANG II has also been found to regulate hypertrophic/proliferative signaling via cPLA2α/c-Src/ERK and iPLA2B/lipoxygenase/c-Jun signaling.
Table 5.
Cell type-specific signal mediators in ANG II-induced vascular structural remodeling
| Cell Type | Mediator | Model | Remodeling | BP | Reference Nos. |
|---|---|---|---|---|---|
| VSMC | ADAM17 | f/f-sm22α Cre | ↓ | ↔ | 1034 |
| VSMC | CaMKII | Inhi* sm22α-Tg | ↓ | ↓ | 836 |
| VSMC | HIF1α | f/f-sm22α Cre | ↓ | ↓ | 414 |
| VSMC | iPLA2β | smmhc-Tg | ↑ | ↑ | 110 |
| VSMC | JAK2 | f/f-sm22α Cre | ↓ | ↓ | 499 |
| VSMC | Nox1 | smmhc-Tg | ↑ | ↑ | 234 |
| VSMC | Sirt1 | sm22α Tg | ↓ | ↓ | 306 |
| Fibroblast | AT1AR | S100A4 Tg | ↓ | ↔ | 825 |
| Macrophage | C1q | C1qa−/− | ↓ | ↔ | 1010 |
| B cell | IgG | BAFF-R−/− | ↓ | ↓ | 134 |
Ca2+/calmodulin protein kinase II (CaMKII) peptide inhibitor.
C. Vascular/Perivascular Fibrosis and Arterial Stiffness
Arterial stiffness precedes the development of hypertension and is associated with aging. It is caused by excessive fibrosis and reduced elasticity, which is associated with increased collagen deposition, increased elastin fiber degeneration, calcification, and cross-linking of collagen molecules. There is an excellent review article regarding the molecular mechanism of vascular fibrosis, where the critical roles and balance of MMPs and TIMPs, and contribution of TGF-β, PAI1, CTGF, and galectin-3 are discussed. Moreover, ANG II appears to be central among other factors (i.e., aldosterone and ET-1) in inducing vascular (pro)fibrotic responses and is capable of activating and/or inducing all of the above factors (360).
1. Relation between vascular fibrosis and hypertrophy
While the progression of ANG II-stimulated vascular fibrosis may result from stimulation of various cascades, almost all studies demonstrating attenuation of vascular hypertrophic responses by ANG II are associated with suppression of perivascular fibrosis, suggesting a close causal relationship between these responses. However, one study reports that N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP), an alternative target of ACE, prevents vascular fibrosis without altering hypertension or arterial medial thickening in rats infused with ANG II. This is associated with inhibition of vascular ROS and TGF-β, suggesting central roles for these factors in vascular fibrosis induced by ANG II (591). Genetic replacement of IκBα with IκBβ also attenuates ANG II-induced NF-κB activation, inflammation, and perivascular fibrosis but not vascular hypertrophy (1180). However, as mentioned in the previous section, tissue-specific genetic engineering has highlighted the role of fibroblast AT1R in promoting ANG II-dependent vascular hypertrophy in the ascending aorta (825). This highlights the significance and novelty of adventitial fibroblast signaling in promoting paracrine signaling whereby activated fibroblasts mediate changes in endothelial and vascular smooth muscle cell function through an “outside-in” mechanism in addition to promoting perivascular fibrosis. Recent reviews have shed light on this topic (634, 683).
2. Cytokines and EGFR
Of importance in perivascular fibrosis is the induction of cytokine and growth factor responses by ANG II. TGF-β and downstream Smad3 are required for ANG II-induced vascular fibrosis (1119). ANG II also induces TNF-α expression, contributing to increased blood pressure and cardiac hypertrophy (991). ANG II-induced TNF-α facilitates cardiac interstitial and perivascular fibrosis through increased collagen, CTGF, and TGF-β production. In cardiac tissue, this response is dependent on TNF-α-induced ROS production through upregulation of Nox2, p22phox, p47phox, p67phox, and downstream activation of NF-κB, p38MAPK, and JNK (990). Furthermore, TNF-α exacerbates the ANG II response through feedback regulation of AT1R (817). AT1R signaling appears to be at the crux of fibrotic signaling as inhibition of prolyl hydroxylase domain-containing protein (PHD, HIF-1α target) through cobalt chloride administration reduces AT1R expression, ERK1/2 activation, and perivascular fibrosis in response to ANG II (670). Direct inhibition of AT1R is also protective against perivascular fibrosis (217). In addition, ANG II-dependent activation of ADAM17 mediates EGFR transactivation, ER stress, oxidative stress, and perivascular fibrosis. Activation of ADAM17 promotes both HB-EGF and TNF-α production, and the ADAM17 promoter contains an ER stress response element, promoting feedback regulation within this cascade (766, 1034). Showing causality, suppression of ER stress attenuates ANG II-induced collagen accumulation in VSMCs (1034).
ANG II infusion in aged rats causes vascular calpain 1 induction leading to MMP2 activation (438). Based on the findings using calpastatin (an endogenous calpain inhibitor) transgenic mice infused with ANG II as well as cultured VSMC, calpains have been proposed as downstream of EGFR transactivation, which mediates ANG II-induced vascular hypertrophy, fibrosis, and NF-κB activation/inflammation as well as cardiac hypertrophy (557). In addition, ANG II-induced cardiac perivascular fibrosis is attenuated in heterozygous Rho-kinase 1 (ROCK1+/−) mice without altering cardiac hypertrophy (869). This cardiac phenotype is associated with suppression of mRNAs for CTGF, TGF-β, and collagen type III. A role for vascular Notch signaling through γ-secretase in perivascular fibrosis and vascular remodeling has also been reported (785). A recent study has pointed to a role for EC-derived ET-1 in ANG II-induced perivascular fibrosis as well (11).
3. Mineralocorticoid receptor
The transgenic mRen2 (27) rat is a unique model of enhanced RAS activity in which tissue renin, ANG II, and circulating aldosterone are increased leading to hypertensive cardiovascular remodeling and inflammation. Investigation of this model has contributed to our understanding of the relationship between the RAS, oxidative stress, and perivascular fibrosis. In young mRen2 (27) rats, an antioxidant, Tempol, prevents cardiac oxidative stress and perivascular fibrosis without altering hypertension (1144). A subsequent study also showed that low-dose MR blocker, spironolactone, attenuates perivascular fibrosis and cardiac NADPH oxidase activity, suggesting the contribution of MR activation in this model (350). In addition, vascular MR-deficient mice develop less vascular fibrosis from aging and are protected from ANG II-induced vascular oxidative stress and hypertension (678).
The mechanism by which aldosterone stimulates perivascular fibrosis via MR appears to require apoptosis-signal regulating kinase 1 (ASK1), which mediates vascular ROS production (739). Requirements of smooth muscle MR and galectin-3 have also been described (112, 838). Taken together, these reports suggest an important contribution of VSMC MR activation and subsequent ROS generation in ANG II- or aldosterone-induced perivascular fibrosis.
4. ANG II mechanisms of arterial stiffness
The clinical understanding of this topic has been reviewed before and includes the efficacies of RAS inhibitors (633). Recent in vivo measurements of arterial stiffness in animal models of hypertension have advanced our knowledge by which ANG II contributes to the development of vascular stiffness, including mechanisms involving tissue-specific RAS (34).
A selective AT2R agonist, C21, reduces vascular stiffness in two distinct hypertension models in rats [stroke-prone SHR (864) and Wister rats treated with l-NAME (806)] without altering blood pressure. In the study with stroke-prone SHR, reduced vascular collagen accumulation by C21 is observed along with a reduction in oxidative stress and inflammatory cell infiltration. These findings suggest that the mechanism causing arterial stiffness in hypertension could be separate from blood pressure. However, the signaling mechanism by which AT2R stimulation reduces arterial stiffness remains obscure. In contrast, enhanced arterial stiffness induced by ANG II infusion in mice is attenuated with normalization of blood pressure by hydralazine and hydrochlorothiazide, suggesting a blood pressure-dependent mechanism. The mechanism is attributed to p38 activation in arterial fibroblasts, as mechanical stretch-induced collagen expression is prevented with p38 inhibition. Moreover, p38 inhibition attenuates hypertension, adventitial collagen contents, and arterial stiffness in ANG II-infused mice (1161). Involvement of inflammatory T cells and cytokine IL-17 in the development of hypertension and arterial stiffness are also noted (630, 1161). ANG II reduces the number of immune inhibitory regulatory T cells (Treg), and adoptive transfer of Tregs reduces blood pressure, oxidative stress/inflammation, endothelial dysfunction, and arterial stiffness in mice infused with ANG II (54, 263). Furthermore, stimulation of Treg proliferation in ANG II-infused mice via an IL-2/mAbCD25 immune complex rescues mice from collagen accumulation and arterial stiffening, as well as reduces IL-17 gene expression and immune cell infiltration (636). However, hypertension is not inhibited in this study, suggesting a complex relationship among adventitial mechanical stress, inflammation, and hypertension. In addition, Sca-1+ progenitor cells and bone marrow-derived infiltrating fibrocytes are also required for arterial fibrosis in ANG II-induced hypertension (1160).
Among the regulators to vascular stiffness, the roles of MMPs and TIMPs in ECM homeostasis appear essential. Specifically, enhanced degradation of ECM enables vascular smooth muscle cells to migrate and proliferate, and inflammatory cells to infiltrate the arterial wall during vascular remodeling (1112). ANG II infusion enhances vascular MMP2 activity through a mechanism involving ADAM17 and MMP7, and MMP2 siRNA infusion attenuates ANG II-induced hypertension in mice (768). Alternatively, if TIMPs override MMPs, a depressed degradation of collagen type I and elastin may facilitate aortic stiffness and organ fibrosis (850). In TIMP3 knockout mice, ANG II-induced hypertension is attenuated, which is explained by adverse vascular remodeling associated with reduced vascular ECM contents due to enhanced MMP2 activity. However, no phenotypic alteration is observed with TIMP1, TIMP2, or TIMP4 knockout mice infused with ANG II (59).
As introduced in the fibrosis section, ER stress also plays a role in enhancing arterial stiffness. An ER stress inducer, tunicamycin, causes hypertension and arterial stiffness, and inhibition of ER stress using 4-phenylbutyric acid (PBA) prevents arterial stiffness and hypertension. VSMC apoptosis is proposed as a potential mechanism regulated by ER stress (989). Other potential VSMC mechanisms contributing to arterial stiffness include arginase-1, lysyl oxidase (Lox), cullin-3, and COX-2 (see below). Arginase-1 inhibits NO production by reducing l-arginine availability. It also elevates ornithine, a substrate for ornithine decarboxylase and ornithine aminotransferase to potentiate cell proliferation and collagen formation, respectively. In heterozygous Arg1+/− mice, ANG II-induced increases in arterial stiffness, vascular stiffness, perivascular fibrosis, and endothelial dysfunction are prevented. In addition, ANG II increases expression and activity of arginase-1 in VSMCs (78). Lox cross-links ECM to stabilize fibrous ECM proteins and produces H2O2. Lox inhibitor, β aminopropionitrile (BAPN), reduces ANG II-induced arterial stiffness, and cross-linked collagen in mice. This is associated with reduction of adventitial thickness, but not medial thickness. The contribution of Lox to arterial stiffness is confirmed with smooth muscle specific Lox overexpressed mice, which show higher vascular stiffness, but normal blood pressure (656). ANG II further enhances vascular Lox expression, and BAPN reduces ANG II-induced arterial stiffness, oxidative stress, and p38 MAPK activation in mice (656). As mentioned, loss of cullin-3 mediates hypertension as well as arterial stiffness (13). Although enhanced RhoA/ROCK signaling mediated vascular dysfunction, it remains unclear if it also mediates vascular stiffness. ANG II is known to induce COX2 and COX2-derived prostanoids such as PGE2. COX2−/− mice, COX2 inhibitor celecoxibm, and EP1R antagonist SC19220, all of which have been shown to reduce ANG II-induced vascular stiffness along with collagen deposition (40).
Another mechanism by which ANG II contributes to arterial stiffness is through vascular calcification. When rabbits fed an atherogenic diet to induce atherosclerosis and arterial calcification are administrated with an ARB, calcification together with expression of BMP2 and osteocalcin are attenuated (33). Rats treated with warfarin and vitamin K with or without AT1R blocker losartan also demonstrate that the ARB reduces aortic calcification with suppression of BMP2 and Runx2 (570). ANG II also upregulates the receptor activator of NF-κB ligand (RANKL) system in VSMCs, which is known to increase BMP2 expression, and bone-related genes cbfa1 and msx2, which cumulatively promote vascular calcification, and enhanced ACE expression and AT1R mRNA in vivo. Blockade of AT1R prevents RANKL expression and vascular calcification in ovariectomized apoE−/− mice fed with high-fat diet (780).
Overall, ANG II-enhanced vascular fibrosis and arterial stiffness require mechanisms involving adventitial fibroblasts, VSMCs, immune cells, and ECM modulation interacting with inflammatory mediators and related signal transduction (FIGURE 7). Among them, emphasis should be placed on the roles of ADAM17, TNF-α, TGF-β, EGFR, MR, and ER stress.
FIGURE 7.

ANG II signaling and vascular fibrosis. Transforming growth factor (TGF)-β1 plays a central role in ANG II-induced vascular fibrosis. AT1R can induce TGF-β1 through an ADAM17/TNF-α pathway as well as through EGFR transactivation leading to ER stress and ROS generation. Likewise, AT1R can induce p38 activation and Arg-1 expression, both of which lead to collagen production. ANG II-induced MR activation also induces ASK1 and downstream Nox induction leading to ROS and TGF-β1 signaling.
D. Atherosclerosis
1. Clinical evidence and research limitation
Inhibition of the RAS is one of the key pharmacological approaches in both primary and secondary prevention of atherosclerotic cardiovascular diseases in humans. Emerging evidence indicates that the beneficial effects of RAS inhibitors are not only due to their effects on blood pressure, but due to a direct inhibition of other ANG II actions, including its inflammatory activity (709). Clinical evidence suggests that ANG II contributes to initiation and progression of atherosclerosis by inducing endothelial dysfunction, inflammation, oxidative stress, thrombosis, and plaque destabilization in humans (1081). Less is clear regarding the molecular and cellular mechanisms by which ANG II promotes atherosclerosis. It has been proposed that AT1R activation on inflammatory cells can be a major mechanism of ANG II contribution to atherosclerosis. However, while there is a consensus that systemic AT1R deletion prevents atherosclerosis in mice (1128), no consistent data have been accumulated for bone marrow deletion of the receptor. In addition, inconsistent data have been reported regarding the role of AT2 receptor in atherosclerosis (208). There is also a limitation in studying downstream signal transduction of the AT1R in promoting atherosclerosis. Available atherosclerosis models require hypercholesteremia to gain ANG II-dependent enhancement. Thus modulation of the atherosclerotic phenotype with signal manipulation may not be directly attributed to a potential AT1R mechanism. For the most part, recent core review articles have few descriptions of RAS signaling and molecular and cellular mechanisms of atherosclerosis (72, 196, 484, 982, 1021) except a few [ANG II promotes hypertension and ROS to cause endothelial dysfunction (319) and contribution of leukocyte ACE (see below) (612)]. Taking these limitations in mind, we will describe recent progresses toward understanding the molecular mechanism of atherosclerosis involving RAS.
2. Controversies in the cell type specific ANG II mechanisms
It has been shown that in apoE−/− mice, even with low-fat diet, DOCA/salt treatment accelerates atherosclerosis that is protected with ARB treatment. Adventitial macrophages are proposed to express local/tissue ANG II in this model (1137). However, ACE in bone marrow-derived cells has been shown to contribute to ANG I-enhanced atherosclerosis, but not necessarily ANG II (154). In addition, smooth muscle ACE activity appears to be critical for hypocholesteremia-induced atherosclerosis, as smooth muscle specific ACE knockout mice on an LDR −/− background are resistant against high-fat diet-induced atherosclerosis, a finding that is not recapitulated in endothelial specific ACE knockout mice (153). While there may be a beneficial effect due to smooth muscle specific deletion of ACE, other components of the RAS may be dispensable in the development of atherosclerosis, as neither smooth muscle nor endothelial AT1AR deletion prevents atherosclerosis (858). These studies suggest that VSMC-derived ANG II acts on other cell types (possibly fibroblasts) or an ANG II-independent action of VSMC ACE may play a role.
3. Inflammatory ANG II signals
To support the central role of IFN-γ in vascular inflammatory activation, suppression of ANG II-enhanced atherosclerosis is observed in IFN-γ-deficient apoE−/− mice, which is associated with reduced CXCL10 expression. However, AAA phenotype is enhanced in the IFN-γ deficient mice, suggesting a distinct molecular mechanism for ANG II-induced AAA (497). Similar phenotypic modulation is observed with an ET-1 receptor antagonist in apoE−/− mice (1006). The role of TNF-α has also been tested in ANG II-enhanced atherosclerosis. In p55 TNFR-deficient LDLr−/− mice, ANG II-enhanced atherosclerosis is significantly reduced, which is associated with downregulation of adhesion molecules, inflammatory cytokines, and chemokines; however, the AAA phenotype is unaltered (1166). CCR2 is a cognate receptor for MCP1, and ANG II-enhanced atherosclerosis and AAA (as well as TAA) are attenuated in CCR2 apoE double-knockout mice (209). CD31-derived T cell inhibitory peptide has also been used to assess the role of T cell activation in atherosclerosis and AAA induced by ANG II. Peptide treatment reduces both pathologies and inhibition of T cell activation and macrophage recruitment (287). Calpain is a Ca2+-sensitive protease with several unique substrates. Transgenic mice on LDR−/− background overexpressing the endogenous calpain inhibitor calpastatin in bone marrow-derived cells were resistant to ANG II-induced atherosclerosis but not AAA. Myeloid cell-specific calpain 1 or 2 deletion also attenuates ANG II-enhanced atherosclerosis, but not AAA. This is attributed to reduced inflammatory responses, including NF-κB activation and macrophage accumulation (395). However, while bone marrow-derived calpain activity may be dispensable for AAA development, pharmacological inhibition of calpain with BDA-410 prevents atherosclerosis and AAA development in LDR−/− mice fed a high-fat diet with ANG II infusion (1005). In LDLr−/− mice, calpain 2 appears to compensate for loss of calpain 1 for ANG II-induced AAA formation (1004). In line with the role of NF-κB activation in atherosclerosis, ANG II has been shown to activate the CARM1-Bcl10-MALT1 (CBM) signaling complex leading to NF-κB activation in vascular smooth muscle and endothelial cells (see also sect. VC). In apoE−/− Bcl10−/− mice infused with ANG II, there is significant reduction of development of atherosclerosis and AAA compared with control apoE−/− mice infused with ANG II (673). Mitochondrial ROS are also required for ANG II enhanced atherosclerosis (460)
While the above findings strongly support the role of leukocytes and inflammatory responses in the development of ANG II-enhanced atherosclerosis, there are a few conflicting reports in addition to the inconsistent findings of bone marrow AT1R deletion. As mentioned in the hypertension section, T cell-derived IL-17A appears to be critical for ANG II-induced hypertension (630). While IL-17A/apoE double-knockout mice fed a high-fat diet show reduced IFN-γ production and oxidative stress, they develop similar levels of atherosclerosis and AAA in response to ANG II infusion (629). Endothelial Nox2 transgenic mice bred onto the apoE−/− genetic background show enhanced vascular ROS production, VCAM-1 induction, and macrophage accumulation. However, ANG II-enhanced atherosclerosis or hypertension is unaltered (243).
4. Athero-protective signaling mechanisms
Several factors have been shown to protect against ANG II-enhanced atherosclerosis. Ligands to the PPAR family of nuclear receptors have potent anti-inflammatory capacity. Expression of dominant negative PPARγ specifically in either smooth muscle or endothelium worsens atherosclerotic lesions in apoE−/− mice fed a western diet, although the cell type specific mechanism may differ (808). Increased expression of inflammatory markers was a consistent observation, and a recent study showed that wild-type PPARγ in smooth muscle cells limits NF-κB activation by facilitating nuclear export of p65 (719). Expression of PPARδ is increased in the vasculature of LDLr−/− mice infused with ANG II, and treatment with a PPARδ agonist attenuates vascular inflammation and atherosclerosis possibly through a suppression of ERK and p38 MAPK activation (1030). Apelin and its cognate GPCR APJ have opposing physiological roles to the RAS. Apelin treatment prevents ANG II-enhanced atherosclerosis, which seems to involve enhanced NO bioavailability. Apelin also attenuates ANG II-induced ERK activation and promoter activation of NF-κB, AP1, NFAT, and SRE in cultured VSMCs, and dimerization between APJ and AT1R has been proposed as an inhibitory mechanism (172). Kinin B1 receptor is expressed in human atherosclerosis tissue and has been implicated in chronic inflammation. Both atherosclerosis and AAA, as well as inflammatory responses are enhanced in apoE kinin B1 receptor double-knockout mice infused with ANG II, suggesting this receptor elicits protective signaling against ANG II pathology (691). In addition, a recent study demonstrated suppression of ANG II-enhanced atherosclerosis as well as AAA by inhibitors of factor Xa, which catalyze the conversion of FII to thrombin (715).
E. Abdominal Aortic Aneurysm
Aneurysm development and rupture is a significant cause of mortality, whereas no absolute pharmacological treatment is currently available. Abdominal aortic aneurysms (AAA) are more common than thoracic aortic aneurysms (TAA). Both AAA and TAA have strong genetic components. AAA represent multifactorial genetic variations. Family history is the second strongest risk factor (next to smoking) for AAA (915). ANG II and the RAS have been strongly implicated in development of AAA (613). While generally supportive, there are some contradictory data regarding the clinical use of ARBs or ACE inhibitors for prevention of AAA development or rupture (639), suggesting the need for additional research to fill the gap between bench and bedside. Historically, the development of an AAA model in hypercholesteremic mouse (apoE−/− or LDLr−/− background) with ANG II infusion (207) has contributed to the research field, dissecting the molecular mechanisms leading to AAA. Several additional ANG II-dependent and ANG II-independent models of AAA have been described with their pathophysiological features (937). Elastase-induced AAA in rats was prevented by ACE inhibitors but not by losartan (588). In the mouse model, however, AAA was prevented by AT1AR deletion or an ARB, telmisartan (1182). Whether calcium chloride-induced AAA is also prevented by ARB remains unclear. ANG II infusion also causes TAA mainly at ascending aorta of hypercholesteremic mouse (209). Distinct phenotypes in ANG II-dependent AAA and TAA have been recognized. As these topics have been reviewed recently (598, 613), we will focus on recent key advancements in the signaling aspects of the ANG II-dependent AAA and TAA. In addition, ANG II has been implicated in cerebral/intracranial aneurysm development and rupture, which is supported with clinical as well as experimental studies. There are many similarities among the mechanistic findings in cerebral aneurysms and AAA/TAA. We will then discuss recent progress regarding the signaling mechanism of ANG II in cerebral aneurysm development and rupture.
1. Cell type specific signaling mechanisms leading to AAA
Recent years have shed light on the role of ANG II signaling in AAA development. While the exact molecular mechanism(s) by which ANG II promotes AAA development remains unclear, it seems to involve multiple cell types (leukocytes, VSMCs, and endothelial cells) and signaling responses, such as oxidative stress, induction of inflammatory cytokines, and activation of MMPs (212, 261). However, limited information is available regarding the cell/tissue type specific signaling mechanism critical for AAA development in response to ANG II. LDLr−/− mice with a systemic AT1AR deletion are protected from ANG II-induced AAA development. However, AT1AR depletion in either ECs or VSMCs does not protect against ANG II-induced AAA development in LDLr−/− mice, providing speculation of different cell-type specific mechanisms (858). In contrast, apoE−/− mice expressing an EC-specific dominant negative IκBα mutant are protected against ANG II-induced AAA development and blood pressure elevation. In addition, inflammatory responses, ROS production, and MMP induction are attenuated, suggesting that ANG II signaling via NF-κB within the endothelium is required for ANG II-dependent AAA (900). As well, selectively silencing of NF-κB/RelA in mesenchymal cell by Col1a2 promoter-driven Cre recombinase expression leads to RelA deletion in VSMCs and fibroblasts and protects mice from ANG II-induced AAA, but not hypertension (412). ApoE−/− mice overexpressing SMC-specific catalase are also protected from early aortic structural remodeling that underlies AAA development, further highlighting the role of SMC-related ROS production in AAA pathophysiology (635). As described below, VSMC ADAM17-deficient mice are also protected from ANG II-induced AAA, and this was associated with reduced oxidative and ER stress in abdominal aorta (471). VSMC KLF4-deficient mice are also protected from ANG II-dependent AAA development. The protection was attributed to KLF4 function promoting inflammation and phenotype switching of VSMCs (905).
Recent studies provided evidence of immune cell mechanisms contributing to AAA. Spleen removal or lymphocyte deficiency in apoE−/−Rag2−/− mice similarly impairs early monocyte-mediated increases in blood pressure in response to ANG II, and protects against AAA development, independently of blood pressure (685). In line with the role of immune cells in AAA, adoptive transfer of regulatory T cells reduces ANG II-induced AAA development in apoE−/− mice via inhibiting macrophage infiltration and induction of pro-inflammatory cytokines and MMPs (688). The AAA protective role of endogenous regulatory T cells has also been confirmed (1207). A terminator of heterotrimeric Gαi activation, regulator of G protein signal 1 (Rgs1), appears to be upregulated in activated monocytes in apoE−/− mice. Systemic Rgs1 deletion protected mice from atherosclerosis. Moreover, leukocyte Rgs1 deficiency protected mice from ANG II-induced AAA development and rupture. The proposed mechanism was that Rgs1 terminates chemokine signaling, thereby reducing chemotaxis to maintain inflammatory cell (macrophage) retention in the subintimal space in AAA (801). In addition, leukocyte KLF5 deficiency protected ANG II-induced AAA formation, podosome formation, and associated inflammation linked to the KLF5-Myo9b-RhoA pathway (623).
In addition, the role of perivascular visceral adipose tissue AT1AR contributing to ANG II-induced AAA has been reported. Adipocyte-derived osteopontin via AT1AR stimulation appears to contribute to AAA through inflammatory actions involving macrophage migration and polarization (902). Taken together, these studies highlight the roles of distinct cell type-specific signaling mechanisms by which ANG II may contribute to AAA development and the need for further investigation.
2. EGFR, Toll-like receptor 4, and Notch contributing to AAA induced by ANG II
Separate from tissue-specific signaling, several known components of ANG II signaling appear to regulate AAA development that include membrane receptors, cytosolic kinases, and intermediators, as well as nuclear transcriptional regulators. The membrane receptors contributing to ANG II-dependent AAA include EGFR, Toll-like receptor 4 (TLR4), and Notch1 (353) (for Notch1, see sect. IIIA2 for its role in AAA). Induction of ADAM17 and EGFR Tyr1068 phosphorylation are enhanced in human AAA and AAA in C57BL/6 mice with ANG II plus BAPN treatment. EGFR inhibition by erlotinib attenuates AAA development and downstream mediators of AAA including IL-6, MMP2, ER stress, and oxidative stress (766). Building upon this, deletion of Cav1, which is required for ANG II-induced ADAM17 activation, prevents AAA development in mice (1033). In line with these observations, deletion of VSMC ADAM17 attenuated AAA formation in mice, which is associated with reduction in aortic EGFR activation, oxidative stress, ER stress, and immune cell accumulation (471). An ER stress inhibitor is also effective in preventing ANG II-induced AAA (848).
TLRs, critically involved in innate immune responses, have been implicated in inflammatory response induced by ANG II (971). TLR4, but not TLR2, deletion protects LDLr−/− mice from developing AAA and atherosclerosis. TLR4 deficiency in bone marrow-derived cells had no effect on either pathology. Myeloid differentiation factor 88 (MyD88) is the major mediator of TLR4 signaling and is indispensable for both AAA and enhancement of atherosclerosis by ANG II. Deficiency in bone marrow-derived cells also attenuates both pathologies. It is therefore unlikely that TLR4 mediates its effect on AAA via MyD88, but TLR4 and MyD88 independently contribute to the AAA promoting mechanisms of ANG II (783). IAXO-102 is a novel TLR4 antagonist. ANG II-infused mice treated with IAXO-102 show reduced aortic expansion, incidence, and rupture of AAAs. IAXO-102 also prevented AAA-associated signal transduction in aorta, which include phosphorylation of p65, ERK, p38, and JNK (407). Toll-interleukin receptor domain-containing adaptor protein-inducing interferon-β (TRIF) is a cytoplasmic adaptor protein for TLR4, and signals NF-κB and interferon regulation factor-3 activation. It has been demonstrated using apoE−/− TRIF−/− mice that TRIF mediates ANG II-dependent AAA (1097). STAT3 activation in AAA is also inhibited in apoE−/− TLR4−/− mice, as well as in Eritoran (a TLR4 inhibitor)-treated mice infused with ANG II. Moreover, STAT3 inhibitor attenuates AAA development, associated enhancement of M1/M2 macrophage ratio, and expression of MMP2/9. Human aneurysm tissue also shows TLR4 expression and STAT3 phosphorylation (849). However, the signaling link between TLR4 and STAT3 remains obscure. In vitro experiments using THP1 monocytes suggest that AMPK negatively regulates STAT3. An insulin sensitizing AMPK activator, metformin, attenuates ANG II-induced AAA, atherosclerosis, and associated inflammatory activation in apoE−/− mice infused with ANG II (1087). CD14 is a GPI-linked surface protein pattern recognition receptor known to participate in signal transduction via TLR4. CD14-deficient apoE−/− mice develop less AAA compared with the control apoE−/− mice infused with ANG II (82).
3. Other signal intermediates contributing to AAA
Additional noteworthy signal intermediates involved in ANG II-dependent AAA include Wnt/LRPs (see sect. IIIA1 and Ref. 516), NLRP3 inflammasome (see sect. IIIA3 and Ref. 1079), β arrestin-2, focal adhesion protein hydrogen peroxide-inducible clone 5 (Hic-5), L-type Ca2+ channel, AMPKα2, and Nox. β Arrestin-2−/− apoE−/− mice treated with ANG II show attenuation of AAA formation with decreased cyclooxygenase-2 (COX2), MCP1, monocyte infiltration, and phosphorylated ERK1/2 in the aorta along with reduced MMP production (1065). ANG II-induced ROS in VSMCs promotes expression of Hic-5, which serves as a scaffold for MAPKK4/MKK4 and p54 JNK. The Hic-5/MKK4/JNK pathway appears to regulate MMP production, and disruption of this signaling scaffold using Hic5−/− mice prevents AAA development in an ANG II apoE−/− mouse model (556). L-type Ca2+ channel also plays a role in ANG II-induced AAA development. Diltiazem prevents AAA and aortic inflammation in apoE−/− mice with ANG II infusion, and this effect appears independent of blood pressure (696). L-type Ca2+ channel inhibitor, nifedipine, reduces AAA incidence, attenuates superoxide production, and recouples eNOS, which points to a ROS-dependent mechanism of AAA formation in this mouse model (695). Hyperphenylalaninemia (hph)-1 mice with enhanced uncoupling of eNOS develop AAA with ANG II infusion. It has been reported that deletion of p47phox, which activates Nox1 and Nox2, prevents ANG II-induced AAA formation in apoE−/− mice (1048). Deletion of Nox1, Nox2, or Nox4 further diminished ANG II-dependent AAA and eNOS uncoupling in these mice (975). Systemic AMPKα2 deficiency, but not bone marrow deficiency, in apoE−/− mice prevents ANG II-induced AAA development. The mechanism involves ROS-dependent VSMC AMPKα2 activation, leading to activator protein 2α-mediated MMP2 induction (1117). However, there is a conflicting report demonstrating that ANG II-induced AAA is exacerbated in apoE−/− Nox2−/− mice compared with control apoE−/− mice, whereas ROS generation in AAA is attenuated. The mechanism was reported to involve enhanced macrophage-derived IL-1β production (490). Cumulatively, signal transduction in AAA pathology is complex, but studies point towards key mechanisms whereby ANG II induces AAA via EGFR transactivation, the TLR4 pathway, and eNOS uncoupling. However, further research is required for defining the roles of ROS and Nox isoforms in modulating AAA.
4. Signal intermediates and ECM associated proteins protecting AAA
A few endogenous signaling intermediates are known to protect against formation of AAA. As mentioned, SMAD3 is involved in canonical TGF-β signaling. SMAD3 mutations have been reported in patients with aortic dissection. SMAD3−/− mice develop TAA, AAA, and dissection associated with augmented systemic and vascular inflammation upon ANG II infusion. Enhancement of NF-κB, C/EBPβ, and iNOS induction is observed, suggesting the protective mechanism utilized by activated SMAD3 (1037). CCN (Cyr61, Ctgf, Nov) proteins are a group of secreted ECM-associated, partially identical cysteine-rich proteins that mediate diverse biologic functions. CCN3 expression is reduced in ApoE−/− mouse AAA with ANG II infusion and in human AAA samples. CCN3-deficient mice develop AAA in response to ANG II infusion, whereas CCN3 overexpression attenuates ANG II-dependent AAA. The mechanism appears to involve suppression of ERK activation and ROS production by CCN3 in VSMC, which is shown with CCN3−/− ERK1−/− mice (1243). TIMP3 is the only ECM-bound TIMP and is increased in abdominal aorta upon ANG II infusion in mice. ANG II infusion for 4 wk causes AAA in TIMP3−/− mice, but not in wild-type control mice. The AAA is associated with reduced ECM contents and enhanced MMP2 activity (58). Interestingly, AAA protective role of TGF-β has been demonstrated, as cotreatment of C57BL/6 mice with ANG II plus TGF-β neutralizing antibody develop AAA (1125). Subsequent studies show that both AAA and TAA as well as their rupture are induced with the combination (155). Systemic TGF-β receptor II protects from AAA, whereas the VSMC TGF-β receptor II protects from TAA in mice infused with ANG II (32).
5. Abdominal aortic dissection
Abdominal aortic dissection (AAD) occurs when AAA rupture. A few studies describe signaling pathways leading to abdominal aortic dissection, one of which involves the vascular inflammatory response. IL-6 deficiency in mice is known to protect against ANG II-induced AAD in mice. ANG II also promotes accumulation of CD4+-T-helper (Th) 17 cells in aorta, which are characterized by IL-17A secretion. The IL-6/STAT3 signaling pathway mediates ANG II-induced Th17 formation; and IL-17A neutralizing antibody or systemic IL-17A deletion protected mice from aortic dissection (447). Furthermore, endothelial Nox2 transgenic mice were used to demonstrate that ANG II-induced endothelial ROS release promotes vascular inflammation and cyclophilin A secretion, which mediates VSMC ERK1/2 activation leading to AAD (269).
F. Thoracic Aortic Aneurysm, Aortic Dissection, and Limitations
Being localized to the ascending aorta, it is speculated that the most common area for TAA development is the aortic root, and is a consequence of medial layer degeneration and degradation of elastic fiber. Smooth muscle and elastic fiber degeneration creates an environment favoring vessel expansion and dilation, leading to aneurysm development (420). TAA are known to be associated with certain inherited connective tissue disorders (syndromic TAA) including Marfan syndrome and Loeys-Dietz syndrome (915). However, nonsyndromic TAA are also associated with significant genetic alterations. While ANG II appears to play a critical role in both syndromic and nonsyndromic TAA, insight into signaling mechanisms of ANG II in TAA is less clear than AAA (613).
Associated with TAA, aortic dissection (AD or TAD) occurs when a tear in the intimal and medial area of the aorta allows blood to flow between the layers. Surgical intervention is usually needed, and untreated aortic dissections often result in death (1050). Type A AD involves ascending aorta and type B AA involves descending aorta. However, AD can occur without aneurysms (324). As ANG II-dependent models of AAA and TAA also develop all types of AD as well as rupture of the aneurysms, it is difficult to dissect responsible signal transduction independent for AD. In addition, there is a report demonstrating that most commonly used ANG II-infusion model in ApoE−/− mice may primarily represent a model for AD (1063). Nevertheless, we will describe recent noteworthy findings regarding the potential role of ANG II signaling potentially involved in TAA and AD.
1. Cell type specific signaling mechanisms of ANG II leading to TAA
Unlike the AAA model, EC-specific deletion of AT1AR in LDLr−/− mice infused with ANG II show reductions in medial thickness, elastin breaks, aortic arch area, and length compared with controls. In contrast, VSMC-specific AT1AR-specific deletion in LDLr−/− mice has no protective effect (857). However, these tissue-specific findings are controversial and need to be verified in other mouse models of TAA. VSMC-specific deletion of low-density lipoprotein receptor-related protein 1 (LRP1) in mice results in elastin fragmentation and aortic dilation in the ascending aorta in response to ANG II infusion (211). In another model of TAA, VSMCs do appear to play a role, as VSMC-specific fibulin-4 (Fbln4)-deficient mice show TAA development, which can be prevented using AT1R blocker losartan or ACE inhibitor captopril. Fbln4 deficiency in VSMCs elicits downstream activation of classical ANG II signaling components, including ERK1/2, Smad2/3, and Smad1/5/8, which are blocked with losartan (402). Subsequent study further shows Egr1-dependent ACE induction, ANG II production, and PI3K-dependent activation of a phosphatase, slingshot-1, causing activation of actin depolymerizing factor cofilin as a VSMC mechanism of TAA (1186). Mice deficient in the smooth muscle specific isoform of α-actin (SM α-actin), Acta2, have also been used as a model of ARB-preventive TAA and AD. Loss of Acta2 in VSMCs is accompanied by ROS and NF-κB-dependent induction of AT1R (152), suggesting the importance of VSMC mechanism leading to ANG II-dependent TAA and AD. Cumulatively, these studies highlight the need for further exploration into tissue-specific mechanisms by which ANG II may contribute to TAA progression.
2. Controversy of TGF-β contribution to TAA
Other than tissue-specific studies, abnormal action of TGF-β has been implicated in ANG II/AT1R-dependent development of TAA and AD. AT1R blockade using losartan or TGF-β neutralizing antibody protects against aortic expansion and rupture through inhibition of TGF-β-dependent ERK1/2 signaling, which is dependent on AT2R in a Marfan syndrome mouse model of fibrillin1 heterozygous mutation Fbn1C1039G/+ (349). Loeys-Dietz syndrome is a connective tissue disorder with significant phenotypic overlap with Marfan syndrome, including a high risk to develop TAA and AD. TGF-β acts via a heteromeric complex of TGF-β type 1 and 2 receptors (TGFBR1 or 2). Mouse models of Loeys-Dietz syndrome with a knock-in mutation of TGFBR1 or 2 leads to compensatory enhancement of TGF-β signaling with upregulation of TGF-β, which is dependent on AT1R (301). TGF-β is secreted from cells complexed with latent TGF-β binding protein (LTBP), a protein that targets TGF-β to the ECM through interaction with fibrillin-1. LBPT3 deletion in mouse with hypomorphic fibrillin-1 homozygous mutation prevents TAA (1276). While these studies strongly support the contribution of TGF-β signaling, the role of TGF-β in TAA and AD pathology remains controversial (155, 640). The mouse with hypomorphic fibrillin-1 homozygous mutation develops AT1R-dependent TAA and AD, when treated with TGF-β neutralizing antibody (183). Inducible smooth muscle specific TGFBR2 deletion in mice causes TAA-like phenotype with decreased SMAD signaling and enhanced ERK signaling, while the role of ANG II in this model remains unstudied (575). Moreover, inducible smooth muscle selective TGFBR1 deletion leads to severe TAA phenotype with enhanced ERK activation, which can be rescued with AT1R antagonist (1194).
3. Signal intermediates protecting ANG II-induced TAA and/or TAD
Several signaling intermediates appear to protect from development of TAA and/or TAD in response to ANG II infusion, which include Akt2, MMP2, and SIRT1. Impaired Akt2 signaling appears to increase susceptibility to TAA and AD associated with ANG II signaling. Expression of Akt2, but not Akt1, is reduced in human samples of TAA and TAD. Akt2-deficient mice develop both AAA and TAA with their dissections that are associated with enhanced MMP9 expression and reduced TIMP1 expression through FOXO1-dependent reciprocal transcriptional regulation (946). In human TAA samples and ANG II-infused mouse aorta, significant reduction of a Rho GAP, Arhgap18, was observed. Upon ANG II infusion, Arhgap18−/− mice develops TAA with a higher frequency, whereas the incidence of AAA was eliminated while ANG II-induced hypertension was unaltered. Arhgap18−/− deficient VSMCs demonstrate synthetic and inflammatory phenotype associated with reduction of Akt2 expression (601). Induction and activation of MMP2 and MMP9 has been described to contribute to AAA. However, mice lacking MMP2 develop TAA in ascending aorta upon ANG II infusion due to impairment in ANG II-induced TGF-β/Smad2/3 signaling and LTBT1 cleavage (945). In addition, a missense mutation of MMP17 is found in patients with TAA. MMP17-deficient mice have enhanced susceptibility to TAA and AD upon ANG II infusion. Mechanistically, reduced osteopontin cleavage by MMP17 reduces JNK activity and alters VSMC maturation (655). A role for SIRT1 has also appeared in AD pathophysiology. SIRT1 suppresses AT1R expression in VSMCs, and VSMC SIRT1 knockout mice show enhanced ROS production, MMP2/9 gene expression, and reduced TIMP1 expression, which was associated with increased AD in these mice. However, VSMC SIRT1 knockout mice fail to show an elevation in blood pressure (296). Nicotinamide phosphoribosyltransferase (Nampt) is the rate-limiting enzyme in the nicotinamide adenine dinucleotide (NAD+) salvage pathway that converts nicotinamide to nicotinamide mononucleotide in mammals to enable NAD+ biosynthesis. VSMC Nampt deletion cause TAD and AAD upon ANG II infusion, which involves oxidative DNA damage and premature senescence of VSMCs (1129).
4. Signal intermediates contributing to TAA and/or TAD
While signal transduction research into TAA development is in its infancy, there already exists substantial evidence of a role for ANG II signaling in mediating TAA pathophysiology. Chemotaxis likely contributes to ascending aorta dilation and aneurysm development, as deletion of the receptor for MCP1, CC chemokine receptor 2 (CCR2), prevents ANG II-induced ascending aorta expansion and elastin fiber destruction in ApoE−/− mice, in addition to prevention of AAA and atherosclerosis (209). In patients with TAD, enhanced smooth muscle contractile protein degradation is observed with the evidence of activation of NLRP3/caspase-1 inflammasome. Caspase-1 is responsible for the protein degradation. Thus AAA, TAA, and AD are attenuated in NLRP3−/− or caspase-1−/− mice treated with ANG II plus high-fat diet (1158). In addition, in a novel model of TAD induced by ANG II infusion with BAPN pretreatment, neutrophil-derived MMP9 is proposed to contribute to TAD (525). Nox1-deficient mice are also protected from ANG II-induced AD in which Nox1 suppresses vascular TIMP1 expression (311).
Overall, there have been significant advances in recent years in our understanding of atherosclerosis, AAA, TAA, and AD pathophysiology. ANG II has emerged as a crucial mediator of signal transduction resulting in vascular pathological complication that appears to be distinct based on the aortic area and the pathology being studied. TABLE 6 summarizes genetic as well as pharmacological interventions distinctly altering these vascular pathologies induced by ANG II infusion.
Table 6.
ANG II infusion studies to look for key mediators in atherosclerosis, AAA, and TAA
| Protein | Model/Intervention | Athero | AAA | TAA | Rupture | BP | Reference Nos. |
|---|---|---|---|---|---|---|---|
| ADAM17 | ADAM17ff sm22α Cre/BAPN | ? | ↓ | ? | ↓ | ↔ | 471 |
| AMPKα2 | apoE−/− AMPKα2−/− | ? | ↓ | ? | ? | ↔ | 1117 |
| Arhgap18 | Arhgap18−/− | ? | ↓ | ↑ | ? | ↔ | 601 |
| AT1AR | LDLr−/− AT1ARff Tie2-Crea | ↔ | ↔ | ? | ? | ↔ | 858 |
| LDLr−/− AT1ARff SM22-Crea | ↔ | ↔ | ? | ? | ↔ | 858 | |
| βarr2 | apoE−/− βarr2−/− | ? | ↓ | ? | ? | ↔ | 1065 |
| Bcl10 | apoE−/− Bcl10−/− | ↓ | ↓ | ? | ? | ↔ | 673 |
| Calpain-1/2 | LDLr−/− BMT CAST-Tgb | ↓ | ↔ | ? | ↔ | ↔ | 395 |
| LDLr−/− calpainff LysM Crec | ↓ | ↔ | ? | ↔ | ↔ | 395 | |
| LDLr−/− calpain inhibitor | ↓ | ↓ | ? | ? | ↔ | 1005 | |
| CCR2 | apoE−/− CCR2−/− | ↓ | ↓ | ↓ | ? | ↔ | 209 |
| CD14 | apoE−/− CD14−/− | ? | ↓ | ↓ | ? | ↔ | 82 |
| CD31 | apoE−/− CD31551–574 | ↓ | ↓ | ? | ? | ? | 287 |
| CXCL10 | apoE−/− CXCL10−/− | ↓ | ↑ | ↑ | ↑ | ? | 497 |
| ETA/BR | apoE−/− ETA/BR antagonist | ↓ | ↑ | ? | ↑ | ↔ | 1006 |
| Factor Xa | apoE−/− FXa inhibitors | ↓ | ↓ | ? | ↓ | ? | 715 |
| Hic-5 | apoE−/− Hic-5ff SMMHC Cre | ? | ↓ | ? | ↓ | ↔ | 556 |
| IκBα | apoE−/− Tie2-dnIκBα-Tg | ? | ↓ | ? | ? | ↓ | 900 |
| IFN-γ | apoE−/− IFN-γ−/− | ↓ | ↑ | ? | ↑ | ↔ | 497 |
| IL-17A | apoE−/− IL-17A−/− | ↔ | ↔ | ? | ? | ? | 629 |
| KLF4 | apoE−/− KLF4ff SMMHC Cre | ? | ↓ | ? | ↓ | ? | 905 |
| MyD88 | apoE−/− / LDLr−/− MyD88−/−d | ↓ | ↓ | ? | ↓ | ↔ | 783 |
| NLRP3 | apoE−/− Nlrp3−/− | ? | ↓ | ? | ? | ↔ | 1079 |
| Notch1 | apoE−/− Notch1+/− BMT | ? | ↓ | ? | ↓ | ↔ | 353 |
| Nox1 | hph-1 Nox1−/− | ? | ↓ | ? | ? | ? | 975 |
| Nox2 | hph-1 Nox2−/− | ? | ↓ | ? | ? | ? | 975 |
| Nox2 | LDLr−/− Nox2-/y | ↔ | ↑ | ↔ | ↔ | ↔ | 490 |
| Nox2 | apoE−/− Tie2 Nox2-Tg | ↔ | ? | ? | ? | ↔ | 243 |
| Nox4 | hph-1 Nox4−/− | ? | ↓ | ? | ? | ? | 975 |
| p47phox | apoE−/− p47phox−/− | ? | ↓ | ↔ | ? | ↓ | 1048 |
| RGS1 | apoE−/− Rgs1−/− | ↓ | ↓ | ? | ↓ | ↔ | 801 |
| RelA | RelAf/f Col1a2-Cre | ? | ↓ | ? | ? | ↔ | 412 |
| SIRT1 | Sirt1f/f sm22α-Cre | ? | ↑ | ? | ? | ? | 149 |
| TGF-β receptor II | Tgfbr2f/f Acta2-Cre | ? | ↔ | ↑ | ↓ | ↔ | 32 |
| TLR2 | LDLr−/− TLR2−/− | ↓ | ↔ | ? | ↔ | ↔ | 783 |
| TLR4 | LDLr−/− TLR4−/−e | ↓ | ↓ | ? | ↔ | ↓f | 783 |
| TNFR1 p55 | LDLr−/− TNFR1−/− | ↓ | ↔ | ? | ↔ | ? | 1166 |
| TRIF | apoE−/− Trif−/− | ? | ↓ | ? | ? | ? | 1097 |
| Wnt | apoE−/− SOST-Tgg | ↓ | ↓ | ↓ | ↓ | ↔ | 516 |
In attempt to test the role of EC or VSMC AT1R, Tie2 or SM22 promoter was used to delete EC or VSMC AT1R, respectively. bBone marrow transplant of calpastatin transgenic mice. cBoth calpain-1 flox/flox LysM Cre+/− or calpain-2 flox/flox LysM Cre+/− mice are compared with the control Cre−/− littermates. dThe phenotype was confirmed in both apoE−/− MyD88−/− mice and LDLr−/− MyD88−/− mice. In addition, the phenotype was confirmed with bone marrow MyD88 deficiency. eThe phenotype was not confirmed with bone marrow TLR4 deficiency. fA slight reduction in blood pressure was observed. gSOST is an endogenous Wnt inhibitor.
G. Intracranial Aneurysm
Literature in basic, clinical, and population studies suggests a strong relationship between the RAS and intracranial aneurysm; however, they are largely circumstantial (958). While several animal models are available for intracranial aneurysm (1126), there is a model of this disease in mice with injection of elastase into the cerebrospinal fluid at the right basal cistern combined with systemic ANG II infusion (760). This model and others have been employed in the effort to elucidate potential signaling mechanisms by which ANG II may contribute to intracranial aneurysm development and rupture. As observed in AAA and TAA, inflammation plays an essential role. Incidence of intracranial aneurysm is reduced in MCP1-deficient mice and in mice treated with clodronate liposomes, which induce macrophage depletion (453). A general MMP inhibitor, doxycycline, but not a specific MMP2/9 inhibitor, prevents aneurysm rupture while showing no effect on aneurysm development (638). In addition, TNF-α deficiency strongly protects mice from intracranial aneurysm development and rupture (994). Myeloperoxidase is a major oxidative enzyme produced by activated neutrophils, monocytes, and macrophages. In myeloperoxidase-deficient mice, elastase/ANG II-induced intracranial aneurysm development and rupture are significantly reduced. The protection is associated with reduced inflammatory responses including induction of VCAM-1, intercellular adhesion molecule 1 (ICAM-1), and TNF-α (171). Tissue-type plasminogen activator (tPA) is a serine protease expressed and released by endothelial cells, where it displays its fibrinolytic role through activation of plasminogen into plasmin. The tPA-plasmin axis is known to promote inflammatory processes and to enhance extracellular matrix degradation. tPA-deficient mice are also protected from ANG II-dependent intracranial aneurysm (529). Hepatocyte growth factor and its receptor cMet are also identified as being protective against intracranial aneurysm rupture, but are not preventative against intracranial aneurysm development (813). In addition, smooth muscle, but not endothelial, PPARγ activity has been identified to protect mice from intracranial aneurysm development and rupture due to a similar signaling mechanism as observed in atherosclerosis (361). A transcriptional factor, Sox17, is essential for vascular development and is highly expressed in intracranial ECs. Sox17 deficiency in addition to ANG II infusion induces cranial aneurysm development and rupture in mice (553). These data suggest the presence of a partially overlapping mechanism by which ANG II promotes intracranial aneurysm development and rupture similar to that observed in AAA and TAA.
H. ANG II/AT1R Mechanism of Endothelial Dysfunction
The endothelium constitutes the inner lining of all blood vessels. As such, the endothelial cells are exposed to blood constituents and the hemodynamic forces generated by blood flow. By virtue of this unique anatomical positioning, the endothelium forms a critical interface between humoral factors and the organ parenchyma. Thus endothelial cells are viewed as a gateway for both local and systemic regulation of tissue and organ function. In the cardiovascular system, loss of normal endothelial cell structure or function is an early and prime indicator of future cardiovascular pathology. In fact, endothelial dysfunction is an independent risk factor for development of cardiovascular diseases (CVDs) such as hypertension and coronary artery diseases. The endothelial dysfunction in humans is characterized by impaired endothelium-dependent vasodilation due to decreased NO bioavailability. Endothelial inflammation and oxidative stress are well established mechanisms leading to endothelial dysfunction (203).
1. Classical understanding and controversy
The relationship between the RAS and the endothelium is apparent on several levels. An initial key step in the production of ANG II is the metabolism of ANG I to ANG II by ACE. While this enzyme is present in several cell types, it is prominently expressed on the surface of lung endothelia. Thus the functional status of lung endothelium would be expected to impact both normal and pathological production of ANG II. In addition, the local production of ANG II may have paracrine effects on neighboring endothelium. Given the associated pathology with AT1R activation, it is important to understand the general mechanisms that lead to endothelial phenotype shift observed following ANG II exposure. As is the case in VSMCs, ANG II rapidly and potently induces overproduction of ROS in the endothelium. While there are multiple sources of ROS in these cells, ANG II induces early activation of the superoxide generating enzyme NADPH oxidase. This process involves ANG II-dependent activation of PKC and Src which, in turn, phosphorylates the p47phox and activate Rac-1, respectively. Each is a necessary subunit for NADPH oxidase complex formation and ROS production in endothelial cells (746). ANG II has also been shown to increase production of mitochondrial ROS. Interestingly, both pharmacological (apocynin) and genetic (p22phox siRNA) inhibition of NADPH oxidase decrease mitochondrial ROS production, suggesting a feedforward mechanism through which ANG II induces oxidative stress in the endothelium (232). An important pathophysiological consequence of enhanced ROS production in endothelial cells is the loss of NO bioavailability. NO exerts a host of beneficial effects on the endothelium and across the vessel wall including regulation of cell survival and apoptosis, mediation of vascular tone and factor into antithrombogenic and anti-inflammatory pathways. NO can be rapidly sequestered by superoxide and converted into a long-lived, toxic reactive compound, peroxynitrite. These reactive oxygen and nitrogen species can interact with lipids and proteins, modify their structure and function, and induce endothelial cell dysfunction. An example is the oxidation of tetrahydrobiopterin, an eNOS cofactor that is necessary for NO production. This alteration in biopterin results in the uncoupling of eNOS to a state where the enzyme no longer produces NO but instead adds to the ROS pool by producing superoxide (100). Thus there are several mechanisms through which ANG II can reduce beneficial NO levels and induce endothelial dysfunction via ROS production (reviewed in Refs. 379, 746). In addition, ANG II has also been strongly implicated in endothelial inflammation (648). In endothelial cells in vitro and endothelium in vivo, ANG II activates the NF-κB cascade, induces VCAM-1 and ICAM-1, and enhances leukocyte endothelial interaction (648, 922). The roles ROS, inflammatory cytokines and immune cells play in ANG II-induced endothelial dysfunction are also described in the previous sections and section X.
While targeting these mechanisms have been shown to protect endothelial function in several distinct animal models, there are still some controversies regarding the pathophysiological significance of endothelial AT1R function. AT1AR depletion in endothelial cells did not protect mice from ANG II-induced hypertension, medial thickening, AAA formation, and atherosclerosis (825, 858). However, depletion of AT1AR in endothelial cells attenuates TAA formation (857). ANG II has been shown to produce NO via eNOS activation in cultured endothelial cells (1017). Transgenic expression of constitutively active AT1R mutant (N111G) in endothelial cells with Tie1 promoter in mice exhibited reduced systolic blood pressure. Note that this mutant AT1R can be further activated upon ANG II binding. Acute pressor response of ANG II was inhibited in the mice associated with enhanced NO production, thus confirming eNOS activation by the mutant AT1R in vivo (854).
2. Novel mechanisms of ANG II causing endothelial dysfunction
While most of the accumulating literature supports the mechanism of ANG II-induced endothelial dysfunction as described above, there are some noteworthy new mechanisms by which ANG II may modulate endothelial function. These new mechanisms include AT1R/P2Y6 receptor heterodimer (752), AT1R/LOX1 receptor heterodimer (1185), ER stress/UPR (465), miR-92A (157), sphingosine kinase 1(Sphk1) (967), Sphk2 (1171), STIM1 (464), SIRT3 S-glutathionylation/SOD2 acetylation (235), and c-Src (111) as described in the above sections. Here, we will describe additional noteworthy signaling mechanisms by which ANG II promotes endothelial dysfunction.
In addition to the well-established eNOS regulatory phosphorylation sites such as Ser1177 (stimulatory) and Thr495 (inhibitory) (845), ANG II has been shown to induce eNOS phosphorylation at Tyr657 via PYK2, which is activated through Nox2-derived ROS in endothelium. This phosphorylation inactivates eNOS, leading to endothelial dysfunction (609). In line with the importance of sphingosine kinases in endothelial dysfunction induced by ANG II, Nogo-B, which inhibits endothelial S1P production, appears crucial for endothelial dysfunction. Mice lacking endothelial Nogo-B are protected from ANG II-induced endothelial dysfunction and hypertension and have preserved NO release (117). The protective role of endothelial S1P receptor (S1PR1) against ANG II-induced hypertension has also been demonstrated (116).
V. ANG II SIGNALING IN CARDIAC PATHOPHYSIOLOGY
Similar to vascular dysfunction, ANG II is a potent modulator of cardiac function and activates signaling mechanisms that alter cardiac function in regards to contractility, hypertrophy, fibrosis, heart failure, cardiac protection, and arrhythmias. A local RAS has been demonstrated for the heart and, along with the circulating system, implicated in the adverse remodeling associated with various pathological conditions, including myocardial infarction, hypertensive heart disease, and heart failure (227). The major RAS components are expressed in the heart; however, the source and functional relevance of renin are debatable (279, 521). In addition, chymase from either mast cells or cardiac myocytes may be more important than ACE in generating ANG II in the human heart from either ANG I or the dodecapeptide ANG (1–12) (279, 1076). Local ANG II levels are increased in the heart in pathological conditions, particularly in diabetes, driven in part by increased expression of AGT. In diabetes, cardiac myocyte chymase may be responsible for the intracellular generation and intracrine actions of ANG II (521). This section will focus on recent in vivo and in vitro analysis of ANG II signal transduction in the heart. ANG II signal transduction in cardiac tissue is well established, and readers are directed towards a previous review on the subject (494). However, there seems to be very limited review of this topic recently except for one article covering the role of signaling by cardiac GPCRs, including AT1R, in modulating cardiac contractility (118). As with vascular ANG II signal transduction, general mediators include the ERK1/2, JNK, and p38MAPK signaling networks. Recent advances in the past decade will be discussed to broaden these and other new cascades.
A. Cardiac Hypertrophy
1. ANG II in cardiac hypertrophy
Cardiac hypertrophy is one of the most extensively studied mechanisms of ANG II signaling and generally reflects the increase in size of individual cardiac myocytes. ANG II has been implicated in left ventricular hypertrophy associated with hypertension (277), the remote myocardium after myocardial infraction (MI) (1096), and diabetic cardiomyopathy (408). Increased blood pressure causes a concentric pattern of LV hypertrophy, which may progress to ventricular dilation and heart failure with reduced ejection fraction (HFrEF) by poorly understood means. Concentric hypertrophy is also a feature of heart failure with preserved ejection fraction (HFpEF), which generally is associated with hypertension but does not progress to dilation. While debatable, unlike with HFrEF, ANG II does not seem to play a central role in the etiology of HFpEF (941). At the cellular level, concentric hypertrophy reflects the addition of new sarcomeres in parallel to existing sarcomeres. In contrast, volume overload due to fluid retention or impaired kidney function may cause a dilated pattern of eccentric LV hypertrophy with new sarcomeres added in series to existing sarcomeres. The mouse model of ANG II infusion at a high pressor dose exhibits concentric left ventricular hypertrophy due to hypertension with a variable contribution of volume overload caused by aortic valve insufficiency (375). Unlike the cardiac hypertrophy observed with exercise or pregnancy, cardiac hypertrophy observed with hypertension, diabetes, or MI is pathological due to inappropriate alteration in the gene expression profile, including reexpression of fetal genes, and induction of death-related signaling mechanisms (87, 142, 276). Pathological cardiac hypertrophy is also associated with endothelial dysfunction, perivascular and interstitial fibrosis, and capillary rarefaction or a decrease in the capillary to myocyte number (951). Key signaling pathways required for ANG II-induced pathological cardiac hypertrophy will be discussed in the following sections.
a) blood pressure and cardiac hypertrophy.
Although a plethora of in vitro studies have proven that ANG II has direct growth-promoting action on cardiac myocytes, the question of whether ANG II has any direct role in vivo in cardiac hypertrophy, independent of increased blood pressure, has been controversial (866). Two lines of evidence from KO mice suggest that cardiac AT1Rs are not obligatory for cardiac hypertrophy in response to increased blood pressure: extrarenal AT1ARs were found to be dispensable for hypertension or cardiac hypertrophy in response to ANG II infusion (190), and pressure overload by either abdominal or transverse aorta constriction still induced cardiac hypertrophy in AT1AR KO mice (355, 356). Of course, issues such as genetic compensation with germline gene KO, potential experimental immunosuppression or preconditioning with renal transplants, and the sheer magnitude of the stress on the heart with pressure overload, could be raised to question the broad applicability of these findings. Several recent studies support the conclusion that ANG II can act independently of, or in synergy with, increased blood pressure to induce cardiac hypertrophy (27, 523). Three examples are cited here. First, cardiomyocyte-specific expression of AT1R-associated protein (ATRAP), which promotes constitutive internalization of the AT1R, suppresses development of cardiac hypertrophy, activation of p38MAPK, and induction of hypertrophy-related genes in the mouse ANG II infusion model without affecting increases in blood pressure (1101). However, the possibility cannot be discounted that AT1Rs in cardiac myocytes function in an agonist-independent manner, as the AT1R contains mechanosensing sites that can lead to receptor activation (437). Second and as mentioned in the vascular section, targeting MMP2 in the ANG II-infused mouse was reported to block hypertension, but not cardiac hypertrophy and fibrosis (768). Third, deletion of ADAM17 specifically in VSMCs was shown to prevent cardiac hypertrophy, perivascular fibrosis, and vascular medial hypertrophy, in mice infused with ANG II, without affecting the induced hypertension (1034). In addition, while focus has largely been on the role of AT1R in cardiac hypertrophy, evidence was reported that AT2R overexpression promotes ligand-independent, constitutive cardiomyocyte hypertrophy in vitro (197) and, based on knockout studies, AT2R couples to pathological cardiac hypertrophy in the mouse in response to ANG II infusion or pressure overload by aortic banding (see sect. VA2 and Refs. 410, 934).
b) nf-κb and nox/ros.
ANG II has been linked to cardiac hypertrophy both in vitro and in vivo via the activation of a number of canonical signaling cascades, including ERK1/2, calcineurin/nuclear factor of activated T cells (NFAT), PKC-α, and CaMKII/MEF2 (951, 1046). Although AT1R may couple directly to these cascades based on studies on cultured cells, there is ample evidence that the growth-promoting actions of ANG II on cardiac myocytes in vivo are amplified and sustained by the upregulation of an innate immune response and inflammation (295, 652, 971). This involves induction or activation of a number of growth-promoting/proinflammatory factors, including IFN-γ (652), IL-18 (1082), NF-κB (294, 473), and increased ROS formation due to the engagement of Nox2 (1254) and Nox4 (1264), as well as downregulation of the TLR4 inhibitor signal regulatory protein-α (SIRPA) (436). An adaptor molecule, connection to IKK and SAPK/JNK (CIKS), which plays a prominent role in autoimmune and inflammatory diseases, may couple the AT1R to cardiac hypertrophy and fibrosis, downstream of Nox2 activation. ANG II enhances expression and activation of CIKS in cardiac myocytes leading to IKK/NF-κB and JNK/AP1 activation and subsequent induction of MMP9 and IL-18, a potent growth factor for cardiac myocytes. Notably, genetic ablation of CIKS blocked ANG II-induced cardiac remodeling and dysfunction, but had no effect on the increase in blood pressure (1082). Cardiac hypertrophy in the mouse from both ANG II infusion and pressure overload is also promoted by the ubiquitin E3 ligase, tumor necrosis factor receptor-associated factor 6 (TRAF6), which mediates signal transduction from members of the TNF receptor superfamily and Toll/IL-1 receptor family. TRAF6 expression is increased in human and murine hypertrophied hearts and is induced by ROS, which also stimulates its auto-ubiquitination. This is turn facilitates recruitment of the adaptor protein TAB2 and subsequent binding of TRAF6 to TGF-β-activated kinase 1 (TAK1), which undergoes ubiquitination and activation thereby initiating JNK1/2/p38 signaling that ostensibly results in cardiac hypertrophy (433). Secretion of Nampt from cardiac myocytes, which functions extracellularly as a proinflammatory cytokine, may also contribute to ANG II-induced cardiac hypertrophy and fibrosis (822).
ROS, through NADPH oxidase, stimulates CCL2 (MCP1) expression through TLR4, and TLR4 KO mice treated with ANG II are protected from cardiac hypertrophy, whereas the increase in blood pressure is not affected (662). SIRPA is an upstream signaling molecule in this response, as cardiac overexpression of SIRPA protects against cardiac hypertrophy through inhibition of ANG II-induced TLR4/NF-κB signaling (436). Surprisingly, TLR4 antagonism in the brain also reduces ANG II infusion-induced cardiac remodeling and elevations of TNF-α, IL-1β, NF-κB, and iNOS in the heart, although central blockade of TLR4 delays progression of hypertension (205).
The significant role of inflammation to ANG II-induced cardiac remodeling is illustrated by the protective counter effects of pentoxifylline, a phosphodiesterase inhibitor with anti-inflammatory actions. Pentoxifylline attenuates cardiac fibrosis, hypertrophy, and diastolic dysfunction in ANG II-infused rats, as well as myocardial macrophage infiltration, NF-κB activation, and proinflammatory cytokine expression, while not affecting ANG II-induced hypertension (1257). However, some of the inflammatory actions of ANG II on the heart may be beneficial for dealing with the increased wall stress associated with hypertension. ANG II-induced upregulation of osteoprotegerin by cardiac cells, including infiltrating macrophages, was found to protect against the development of eccentric hypertrophy and dilated cardiomyopathy by enhancing collagen synthesis and inhibiting cardiac myocyte apoptosis. Osteoprotegerin is a secretory glycoprotein belonging to the TNF receptor superfamily that generally acts as a decoy receptor for RANKL, but that was not so in this case (1070). Although not well studied, the engagement of adaptive immunity by ANG II may also impact on cardiac hypertrophy and overall structural remodeling of the heart. For instance, the pathogenesis of ANG II-induced dilated cardiomyopathy was reported to depend on the T cell Th2 phenotype (816) and to involve the Th2 polarizing cytokine IL-4 (815). Others recently showed that B cells have a contributory role in an ANG II-induced model of heart failure (186).
Oxidative stress from enhanced activity of Nox2 (1254) and Nox4 (1264) may contribute to ANG II-induced cardiac hypertrophy and fibrosis. ANG II activates Nox2 by canonical means, and Nox4 by enhancing its expression (14), likely via ROS (108, 201). Cardiac Nox2 also contributes to cardiac contractility in response to ANG II. This is explained by ROS-mediated inhibition of protein phosphatase 1 and phospholamban phosphorylation leading to increased SR Ca2+ uptake (1254). In addition, AT1R associates with both Nox2 (1082) and Nox4 (980), although the consequences of this interaction are not established. ANG II infusion enhances cardiac Nox4 expression. Cardiac specific Nox4 transgene enhances ROS production and NF-κB and Akt/mTOR signaling, which exaggerate ANG II-induced cardiac hypertrophy and fibrosis (1264). Recent evidence has also highlighted a beneficial role for PPARγ in blocking ANG II-induced upregulation of Nox2/Nox4 and nuclear translocation of NF-κB p65 (368). Mitochondrial oxidative stress with enhanced ROS formation contributes to ANG II-induced cardiac remodeling as well, likely reflecting in part the ROS-induced ROS phenomenon downstream of Nox activation (496). A mitochondrial targeted antioxidant peptide attenuates ANG II-induced cardiac hypertrophy, fibrosis, and diastolic dysfunction. Interestingly, this peptide also attenuates ANG II-induced Nox4 expression (200). Others report that caloric restriction, which is expected to tamp down ROS generation, protects against ANG II-induced mitochondrial remodeling, oxidative stress, and cardiac hypertrophy (282). Recently, ANG II-induced mitochondrial ROS that was linked to cardiac hypertrophy and fibrosis was attributed to reduced SOD2 activity resulting from interference by SIRT4 in SIRT3-mediated SOD2 deacetylation (616).
While accumulated evidence referenced above supports the direct contribution of Nox and mitochondria-derived ROS in ANG II-induced cardiac hypertrophy there are controversial reports as well. Cardiac Nox4 transgene increases mitochondria-derived superoxide and subsequent myocyte apoptosis but not hypertrophy, thus enhancing compensatory cardiac remodeling (14). Slight reductions in systolic pressure and pulse pressure were observed in Nox4 knockout mice infused with ANG II, whereas no protection of cardiac remodeling was detected (90). In Nox2 knockout mice, ANG II-dependent cardiac hypertrophy was unaffected, while cardiac ROS levels were decreased (1059).
c) rhoa, g proteins, and ca2+.
The small GTPase RhoA is a key effector in promoting prohypertrophic gene expression. Of note, Gα13 regulates RhoA activation in the heart in response to ANG II (1036), which is linked to activation of myocardin-related transcription factors, cardiac hypertrophy, and eventual heart failure. This Gα13-initiated cascade is independent of ANG II-activated Gαq/11 signaling that was earlier linked to pathological cardiac hypertrophy (1143). RhoA is also a downstream effector of Nox-induced ROS in response to ANG II treatment (49, 746), as are various redox-sensitive kinases and transcription factors linked to cardiac hypertrophy (148, 567, 1107).
A-kinase anchoring protein (AKAP)-Lbc serves as a GEF for activating RhoA and is a scaffold for various proteins involved in classical signal transduction including PKC, PKD1, and PKA, and also tethers the tyrosine phosphatase Shp2. Mice harboring a gene-trap expressing AKAP-Lbc with a truncated COOH terminus preventing PKD1 binding show reduced HDAC5 phosphorylation in response to ANG II and are protected against cardiac hypertrophy, but show increased cardiac fibrosis (1025). As an effector of RhoA, ROCK2 has been reported to mediate ANG II-induced cardiac hypertrophy via its substrate, formin homology 2 domain containing 3 (FHOD3), which is a cardiac-restricted member of diaphanous-related formins crucial in regulating myofibrillogenesis in cardiomyocytes (1272). However, cardiac fibroblast ROCK2 may also mediate cardiac hypertrophy and fibrosis induced by ANG II through a paracrine mechanism involving CTGF and FGF2 (952). G protein-dependent MAPK/ERK kinase (MEK)-ERK1/2 signaling is also under negative regulation by regulator of G protein signaling 3 (RGS3) and RGS5, which attenuate cardiac hypertrophy in vivo from aortic banding or in vitro from ANG II (564, 603).
Isoprenylation is important for the signaling function of members of the Rho GTPase family, such as Rac1 (requisite component of the NADPH oxidases) and RhoA, and is dependent on the formation of isoprenoids from mevalonate, which is synthesized by HMG-CoA reductase (HMGCR). Evidence indicates that MMP2 may act as an initial brake on ANG II-induced cardiac hypertrophy by attenuating sterol regulatory element-binding protein 2 (SREBP-2)-mediated transcription of HMGCR (1121). Cardiac MMP2 is upregulated by hypertrophic stimuli, and compared with wild-type mice, MMP2 knockout mice develop cardiac hypertrophy at an earlier time and to a greater extent with ANG II infusion, as well as greater expression of inflammatory genes and perivascular fibrosis. This response was associated with increased expression of SREBP-2 and HMGC, although the mechanism by which MMP2 attenuates SREBP-2 expression was not defined. Nor was it established whether MMP2 ablation increases isoprenylation of small GTPases with ANG II treatment. In any case, these findings highlight the protective role of MMP2 in the heart in response to ANG II infusion, in contrast to its prohypertrophic and profibrotic actions in the heart under pressure overload (666) or its proinflammatory and ECM disruptive effects in the infarcted myocardium (664).
ANG II induces cardiac hypertrophy in part via IP3-mediated Ca2+ release downstream of GPCR signaling (742). Ca2+ contributes to cardiac hypertrophy by several means including activation of NFAT and CaMKII (524, 951). Recently, a background Ca2+ entry pathway in cardiac myocytes formed by TRPC1 and TRPC4 channels was implicated in ANG II-induced hypertrophy as well (113). In contrast, the Ca2+-activated cation channel TRPM4 was identified as a negative regulator of ANG II-induced cardiac hypertrophy, ostensibly by inducing a depolarizing current that attenuates Ca2+ sarcolemmal entry, thus limiting filling of the IP3-sensitive Ca2+ stores (475).
d) β-arrestin.
AT1R may also couple mechanical forces to cardiac hypertrophy independent of G proteins via β-arrestin2-biased signaling and Src-mediated activation of ERK1/2 (1116). Indeed, recent evidence reveals that the Frank-Starling mechanism of the heart by which increased cardiac filling leads to enhanced cardiac contractility is a manifestation of β-arrestin-biased signaling of AT1R (6). TRV120027 (Sar-Arg-Val-Tyr-Ile-His-Pro-D-Ala-OH) is a selective β-arrestin-biased ligand of AT1R that competitively blocks ANG II-stimulated G protein signaling, but activates several kinase pathways via β-arrestin coupling, including Src, ERK1/2, and eNOS phosphorylation (1094). TRV120027 enhances cardiac contractility without increasing myocardial oxygen demand, as this ligand lowers blood pressure and afterload, and thus preserves cardiac stroke volume. The inotropic actions of β-arrestin-biased AT1R ligands may be due to phosphorylation of components of the contractile machinery, including ventricular myosin regulatory light chain 2 (MLC2v) (890, 1041). AT1R β-arrestin-biased signaling leads to MLC2v phosphorylation via the ERK1/2-RSK3 cascade, directly or through inhibition of myosin phosphatase targeting protein 1/2 (MYPT1/2) (890). RSK3 may also couple to activation of SRF and induction of SERCA2 protein expression. Its unique pharmacological profile suggests that TRV120027 may be a useful therapeutic agent for treating acute heart failure, a condition of poor heart contractile performance with neurohormonal activation, including activation of the RAS, and associated increased blood pressure (388, 413). In normal and heart failure dogs, TRV120027 was shown to have cardiac unloading actions, while renal function is preserved (83). However, in the recently completed BLAST-AHF trial on patients with acute heart failure, TRV120027 did not exhibit any benefit over placebo in the primary composite end point or the individual components, but notably plasma renin activity was unexpectedly low in the patients enrolled in this study (796).
e) egfr and adam17.
ANG II-induced transactivation of the EGFR is instrumental in facilitating cardiac hypertrophy (289, 819). EGFR transactivation is dependent on Src phosphorylation and activation of ADAM17, which has sheddase activity for cleaving the ectodomain of several precursor molecules, including heparin-binding EGF-like growth factor. EGFR signaling enhances ERK1/2 and Akt activation which upregulate cardiac hypertrophy markers including MyHC, ANP, BNP, SKA, and β/α-MyHC (819). EGFR transactivation also elicits downstream ER stress leading to cardiac hypertrophy and fibrosis (465, 1035). In vitro analysis indicates ANG II-induced EGFR transactivation is dependent on Nox2/Nox4-mediated upregulation of ADAM17 (1236). Knockdown of ADAM17 with siRNA attenuated cardiac hypertrophy in the spontaneously hypertensive rat without diminishing blood pressure, and cardiac hypertrophy and fibrosis in the mouse with ANG II infusion, although the increase in blood pressure was moderately attenuated (1123). ADAM17 also plays additional roles separate from EGFR transactivation in myocardial hypertrophy. ROS derived from Nox2 mediates ANG II-dependent activation of p38MAPK and subsequent phosphorylation and activation of ADAM17. ADAM17 activation causes cleavage of ACE2 potentially preventing ANG (1–7) generation and antihypertrophic signaling of the Mas receptor (803). On the other hand, cardiomyocyte-specific ADAM17 silencing enhances pressure overload-induced myocardial hypertrophy, cardiac fibrosis, and contractile dysfunction by reducing the proteolytic processing of integrin β1, thereby increasing activation of the focal adhesion kinase (FAK) pathway (268). However, cardiac hypertrophy induced by a subpressor dose of ANG II is not affected by ADAM17 knockdown.
f) paracrine and autocrine mechanisms.
ANG II acts in a paracrine manner to induce hypertrophy of cardiac myocytes by the release of growth factors from endothelial cells and fibroblasts. Endothelial cell-produced endothelin-1 (ET-1) contributes to ANG II-induced cardiac hypertrophy and fibrosis, independently from blood pressure (11). ANG II induces ET-1 expression by activation of c-Jun/c-Fos (AP1) and formation of chromatin remodeling complex that includes AP1-mediated recruitment of a histone H3K4 trimethyltransferase, either SET1 or MRTF-A (1138, 1139, 1222). Others report that endothelial tyrosine kinase Bmx and subsequent activation of STAT3 is necessary for ANG II-induced cardiac hypertrophy and fibrosis due to the induction of proinflammatory cytokines (389). ANG II stimulates cardiac fibroblasts to produce FGF-2, which is important for concentric hypertrophy and preventing dilated cardiomyopathy (810). Fibroblasts also produce TGF-β, which contributes to cardiac hypertrophy at subpressor doses of ANG II. Fibulin-2 is a likely matricellular protein that is upregulated by ANG II and is required in turn for upregulation of TGF-β. Knockout of fibulin-2 prevents ANG II-induced cardiac fibrosis at subpressor and pressor doses, but cardiac hypertrophy only at subpressor doses of ANG II (487, 1247). Recently, evidence was presented that ANG II induces cardiac hypertrophy in vivo through the release of exosomes from cardiac fibroblasts that are enriched in passenger strand miR-21*, causing the downregulation of sorbin and SH3 domain-containing protein 2 (SORBS2) and PDZ and LIM domain 5 (PDLIM5) in cardiac myocytes (52). Release of exosomes from cardiac fibroblasts in response to ANG II stimulation may be mediated through both AT1R and AT2R and may lead to the upregulation of the local renin angiotensin system in cardiac myocytes that would further drive cardiac remodeling (621).
Bone morphogenetic protein 4 (BMP4) signaling contributes to the development of ANG II- and pressure overload-induced cardiac hypertrophy via Smad1/5 phosphorylation and DNA-binding protein inhibitor (Id1) induction (942). Cardiac hypertrophy and fibrosis are attenuated in ANG II-infused mice treated with a BMP inhibitor or in cardiac-specific BMP type 1 receptor ALK2-deficient mice. In addition to Smad1/5 activation, BMP-induced hypertrophy also involves calcineurin-NFAT signaling and Nox4-induced ROS in cardiac myocytes (942, 1012). These findings indicate that ANG II elicits an upregulation of BMP4 expression, ALK2 receptor binding, and activation of Smad1/5 and Id1 signaling, thereby constituting an autocrine loop for cardiac hypertrophy (942).
As mentioned in regards to perivascular fibrosis, a role for TNF-α is emerging in the ANG II response pathway. TNF-α knockout mice treated with ANG II show reduced cardiac hypertrophy (991), although an impact on blood pressure elevation must be taken into consideration. Likewise, the TNF-α inhibitor etanercept blocks ANG II-dependent activation of p38MAPK and JNK (990). TRAF2 is a required component for TNF-α induced downstream signaling, and in vitro analysis indicates that TRAF2 enhances ANG II-dependent upregulation in cardiomyocyte area and ANP/BNP expression through Akt activation and subsequent inhibition of GSK3β activation (405).
g) immunoproteasome.
Generation and activation of the immunoproteasome is implicated in ANG II-induced cardiac hypertrophy. ANG II increases association of TR3 (also termed Nur77), a member of the steroid/thyroid/retinoid receptor family, with the tuberous sclerosis (TSC) TSC1/TSC2 complex, which promotes TSC2 degradation through the proteasome/ubiquitination pathway. This in turn activates mTORC1, which increases protein synthesis (1115). Proteasome activation is also implicated in the degradation of ATRAP in the heart in response to ANG II infusion, which seemingly is a contributing factor to AT1R-mediated cardiac hypertrophy and fibrosis (571). However, this conclusion is based on the use of the proteasome inhibitor bortezomib, which significantly attenuates ANG II-induced increases in blood pressure. Finally, ANG II-induced expression of IGF receptor II (IGF-IIR), which has been implicated in hypertensive ANG II-induced cardiac myocyte apoptosis, is attributed to proteasome-mediated degradation of SIRT1 downstream of JNK activation via AT1R. Loss of SIRT1 in turn prevents deacetylation of heat-shock transcription factor 1 (HSF1), which impairs its ability to act as a repressor at the IGF-IIR promoter. Consequently, the subsequent expression of IGF-IIR contributes to ANG II-induced cardiac hypertrophy and apoptosis (400).
Key signaling pathways required for ANG II-induced pathological cardiac hypertrophy are illustrated in FIGURE 8, which are classified to include four major myocyte mechanisms: 1) canonical G protein and downstream signaling, 2) the transactivation pathway, 3) inflammatory signaling, and 4) the noncanoncal/β-arrestin mechanism.
FIGURE 8.

ANG II induces cardiac hypertrophy by multiple means involving direct actions on cardiac myocytes (cm) or the release of paracrine factors from other cell types, such as endothelial cells and fibroblasts/myofibroblasts. Developments within the last 5 yr are noted in darker text and are highlighted here. Three processes are dominant in cardiac myocytes: canonical signaling, transactivation of EGFR, and inflammatory signaling. The later may involve other cell types as the source of ROS. For canonical signaling, Gα13 was recently shown to regulate RhoA activation that was linked to activation of myocardin-related transcription factors. A background Ca2+ entry pathway formed by TRPC1 and TRPC4 channels was also implicated in ANG II-induced cardiac hypertrophy via canonical means. AT1R may couple to cardiac hypertrophy as a mechanosensor for increased blood pressure (BP) via β-arrestin 2 biased signaling that involves activation of Src and ERK1/2. As a mechanosensor, AT1R may conceivably activate canonical signaling as well. Inflammation is driven by Nox4- and Nox2-derived ROS that leads to the engagement of a number of processes that sustain hypertrophic signaling, such as the downregulation of the TLR4 inhibitor, SIRPA, as well as the activation of RhoA and Akt/mTOR signaling. Mitochondria are an additional source of ROS for cardiac hypertrophy (MitoROS). The adaptor molecule CIKS may couple AT1R to cardiac hypertrophy and fibrosis, downstream of Nox2 activation. ANG II enhances expression and activation of CIKS in cardiac myocytes leading to IKK/NF-κB and JNK/AP-1 activation and subsequent induction of MMP-9 and IL-18, a growth factor for cardiac myocytes. Secreted Nampt from cardiac myocytes, as well as the generation and activation of the immunoproteasome, may also contribute to ANG II-induced cardiac hypertrophy. In response to ANG II, endothelial cells release ET-1 and inflammatory cytokines that induce cardiac myocyte hypertrophy. Activated fibroblasts stimulate hypertrophy by releasing FGF-2, fibulin-2, TGF-β, and exosomes that are enriched in passenger strand miR-21*.
h) counterregulatory mechanisms.
Besides ATRAP, a number of additional endogenous counterregulatory mechanisms that attenuate ANG II-induced cardiac remodeling and could be exploited therapeutically have recently been identified (TABLE 7):
Table 7.
Newly identified antihypertrophic molecules
| Molecule | Overview | Mode of Action | Reference Nos. |
|---|---|---|---|
| Dusp14 | Dual-specificity phosphatase 14; in vivo loss enhanced, while overexpression attenuated, pressure overload-induced cardiac remodeling/hypertrophy; knockdown in vitro enhanced ANG II-induced hypertrophic gene expression | Targets TAK1/mitogen-activated protein kinase kinase kinase 7 in JNK1/2 and p38 cascades | 562 |
| miR-133a | Downregulated by ANG II-induced ERK1/2 activation; thyroid hormone-mediated cardiac hypertrophy partially attributed to AT1R-mediated miR-133a reduction; adiponectin opposes ANG II’s actions on miR-133a | Implicated in silencing expression of prohypertrophic/fibrotic genes such as CTGF, inositol 1,4,5-trisphosphate receptor II (IP3RII) calcium channel, collagen 1a1 (Col1A1), and nuclear factor of activated T cells, cytoplasmic (4NFATc4) | 129, 236, 244, 572, 582 |
| TIEG1 | Transforming growth factor (TGF)-β-inducible early gene; member of the Sp1/Krüppel-like family of transcription factors | Inhibits expression and transcriptional activity of transcription factor GATA4 in cardiac myocytes | 573 |
| IL-10 | Interleukin-10; anti-inflammatory cytokine | IL-10 deficiency enhances ANG II-induced gene expression of TNF-α, IL-6, and MMP-2/9, as well as Akt phosphorylation; IL-10 deficiency increases p38 phosphorylation; IL-10 activates Akt/mTORC1 signaling and attenuates ANG II-induced pathological autophagy | 500, 528 |
| IRF7 | Interferon regulatory factor 7; heart-specific IRF7 overexpression attenuated pressure overload-induced cardiac hypertrophy, fibrosis, and dysfunction, while loss had opposite effects; protected against ANG II-induced cardiac myocyte hypertrophy in vitro | Binds IKKβ with subsequent NF-κB inactivation | 435 |
| IL-33 | Biomechanically induced by cardiac fibroblasts; antagonizes ANG II- and phenylephrine-induced cardiac myocyte hypertrophy | Inhibits phosphorylation of inhibitor of NF-κB alpha (IκBα) and NF-κB nuclear binding activity | 908 |
| Elabela (ELA) | Endogenous peptide ligand for APJ; antagonizes pressure overload-induced cardiac hypertrophy, fibrosis, and dysfunction | Transcriptionally downregulates ACE expression by downregulating expression of the FoxM1 transcription factor | 918 |
-
STAT3 signaling, which is activated by ANG II in the heart either directly or indirectly via AT1R, is suggested to be a protective mechanism in hypertrophy. Mice harboring an S727A mutation, which decreases STAT3 activity, show less of an increase in heart weight in response to ANG II, and this was associated with reduced myofibril number and density, as well as foci of necrotic cardiomyocytes and reparative fibrosis (1283). These observations suggest that STAT3 may serve an adaptive and protective role in ANG II-associated cardiac hypertrophy. A contribution of noncardiomyocytes seems likely in this regard, as cardiomyocyte-targeted STAT3 deletion does not attenuate ANG II-induced cardiac hypertrophy, although contractile function was impaired and metabolic homeostasis was disturbed with ANG II treatment (28). These findings are somewhat reminiscent of a previous report that global deficiency of either AT1R subtype in the mouse does not affect normal cardiac morphology, but lack of both receptors leads to atrophic changes in the heart, with reduced baseline coronary flow and left ventricular systolic pressure in the presence of normal morphology of medium-sized vessels and no overt fibrosis (1084). Together, these findings indicate that AT1R may couple to protective prosurvival signaling in the heart, besides the better characterized pathways that are linked to adverse remodeling.
However, noncanonical STAT3 signaling has been implicated in ANG II-induced pathological cardiac hypertrophy and inflammation (1282). In canonical STAT3 signaling, Tyr705 phosphorylation favors formation of STAT3 parallel dimers that tightly bind GAS-like elements in promoters, while Ser727 phosphorylation may further enhance transcription by recruiting p300. Noncanonical STAT3 signaling involves unphosphorylated STAT3 (U-STAT3), which arises from canonical STAT3-induced STAT3 expression, forms antiparallel dimers, and perhaps has a distinct acetylation profile. Nuclear U-STAT3 levels were shown to increase in hearts overexpressing human AT1R and in neonatal rat ventricular myocytes and fibroblasts treated with ANG II (1226). In heart-restricted AT1R transgenic mice, U-STAT3 levels correlated with cardiac hypertrophy and dysfunction, as well as abnormal expression of osteopontin and regulator of G protein signaling 2 (RGS2).
Xin actin-binding repeat-containing protein 2 (Xirp2) is a protein that is highly expressed in cardiac muscle and plays a role in the formation of intercalated disks (ICDs) of the heart, which subsequently influence the surface expression or distribution of ion channels (1114). The Xirp2 gene of cardiomyocytes is an early direct target of ANG II stimulation via activation of the MEF2A transcription factor. Surprisingly, hearts of Xirp2 hypomorphic mice infused with ANG II exhibit less hypertrophy, as well as markedly reduced fibrosis and apoptosis, although Xirp2 deficiency is associated with basal cardiac hypertrophy in unstressed mice ostensibly through increased calcineurin activity (1114). These findings indicate that a reduction in Xirp2 is beneficial in countering ANG II-induced maladaptive cardiac remodeling. Xirp2 deficiency does not affect ANG II-induced activation of ERK1/2, JNK, p38, or PKD1; however, cardiac GSK3β S9 phosphorylation is significantly less in ANG II-infused Xirp2 hypomorphic mice (1114). GSK3β kinase activity opposes cardiac hypertrophy, and GSK3β is normally inhibited with ANG II treatment by S9 phosphorylation, although in this case no differences were noted in the activity of the kinase Akt generally responsible for GSK3β phosphorylation. In any case, increased GSK3β activity was postulated to attenuate the hypertrophic actions of ANG II due to reduced levels of β-catenin, which is targeted for ubiquitination and degradation following phosphorylation by GSK3β. However, this explanation is at odds with the previously mentioned finding (62; see sect. IIIA1) that cardiomyocyte-specific deletion of β-catenin in the mouse enhances the hypertrophic response to ANG II infusion. On the other hand, as discussed elsewhere (1043), several in vitro and in vivo studies positively associate β-catenin expression to cardiac hypertrophy after TAC or agonist stimulation, and overall the role of β-catenin in cardiac hypertrophy is unsettled. Notably, Xirp2 appears to be able to bind β-catenin (165), which in cardiac muscle localizes to adherens junctions in ICD structures, and thus Xirp2 might normally influence expression levels of β-catenin by thwarting its degradation. This might explain why cardiac β-catenin is not increased in ANG II-treated Xirp2-deficient mice. Besides that consideration, active GSK3β also prevents hypertrophic growth by additional means, such as phosphorylating and inhibiting transcriptional regulators, NFAT and GATA4, and translation initiation factor EIF2B5 (1043).
ANG II-dependent JNK1/2 and p38MAPK activation is dependent on upstream mitogen-activated protein kinase kinase (MKK) activation (MKK4/7 and MKK3/6, respectively). Activation of these MKKs depends on activation of TAK1, which can be repressed by dual-specificity phosphatase 14 (Dusp14). In vivo genetic loss of Dusp14 aggravates, while cardiac-specific Dusp14 overexpression attenuates, aortic banding-induced cardiac dysfunction and remodeling, including hypertrophy. Interestingly, Dusp14 knockdown in vitro promotes elevated cardiac hypertrophic gene regulation in response to ANG II, suggesting that Dusp14 is a negative regulator of cardiac remodeling in response to ANG II (562). Note, however, that the contributions of p38 and JNK to cardiac hypertrophy are uncertain with JNK signaling perhaps being antihypertrophic (951).
ANG II-induced ERK1/2 activation in cardiac myocytes downregulates miR-133a levels (582), an event that results in cardiac hypertrophy (120, 244). Recent evidence reveals that thyroid hormone-mediated cardiac hypertrophy, which is known to be partly mediated by AT1R, proceeds via an AT1R-mediated reduction in miR-133a levels (236). However, ERK1/2 signaling, rather than being essential for cardiac hypertrophy, may coordinate concentric and eccentric growth of the heart (951). Cardiac myocytes from hearts not expressing ERK1/2 demonstrate preferential eccentric growth with TAC or ANG II infusion, while cardiac myocytes from MEK1 transgenic hearts exhibit concentric growth (478).
Interferon regulatory factor 7 (IRF7) is downregulated in the heart with aortic banding-induced hypertrophy and in cardiac myocytes treated with ANG II. Conversely, overexpression of IRF7 protects against banding-induced cardiac hypertrophy and fibrosis in vivo and ANG II-induced cardiac myocyte hypertrophy in vitro. IRF7-mediated attenuation of hypertrophy occurs through binding of IRF7 to inhibitor of κB kinase-β (IKKβ) and subsequent inactivation of NF-κB (435). In contrast, IRF3 is activated by ANG II in cardiac fibroblasts via canonical ERK signaling and contributes to cardiac fibrosis, but plays no role in cardiac hypertrophy (1072). In addition, ANG II increases IRF4 leading to upregulation of CREB and hypertrophy-related genes in cardiac myocytes (434).
Elabela (ELA) is a novel endogenous peptide ligand for the apelin receptor (APJ). In the TAC mouse model, continuous infusion of ELA improved cardiac performance and attenuated both cardiac hypertrophy and fibrosis, and their associated gene expression. ELA may exert beneficial effects on the heart by opposing stress-induced ACE expression via negative regulation of the FoxM1 transcription factor. ELA also attenuates ANG II-induced cardiac remodeling in the heart, although blood pressure is normalized as well (918).
TGF-β-inducible early gene (TIEG1), a member of the Sp1/Krüppel-like family of transcription factors, is another potential negative regulator of ANG II-induced cardiac myocyte hypertrophy. In cultured cardiac myocytes, ANG II downregulates TIEG1 expression in association with the induction of hypertrophy; moreover, by inhibiting the expression and transcriptional activity of transcription factor GATA4, TIEG1 was shown to block hypertrophic gene expression in cardiomyocytes (573).
The counteraction of adiponectin on ANG II-induced cardiac hypertrophy involves antagonistic actions on miR-133a levels. Overexpression of adiponectin prevents cardiac hypertrophy in rats infused with ANG II by increasing AMPK activity, which in turn attenuates ERK1/2 activation and subsequent miR-133a downregulation. However, ANG II also suppresses adiponectin receptor 1 expression (582).
Induction of the anti-inflammatory cytokine IL-10 in the heart by ANG II may serve to temper its adverse effects on cardiac structure and function (528). Loss of IL-10 worsens ANG II-induced cardiac fibrosis, hypertrophy, pathological autophagy, and contractile dysfunction (500, 528).
IL-33 is a biomechanically induced cytokine that is produced by endothelial cells and cardiac fibroblasts and attenuates ANG II-induced cardiac hypertrophy and fibrosis (908).
Adrenomedullin, which is secreted by cardiac myocytes, fibroblasts, and endothelial cells, inhibits ANG II-induced cardiac hypertrophy and fibrosis by suppressing ERK1/2 activation through PKC inhibition and PKA activation (754).
2. AT2R in cardiac hypertrophy
The heart has a complete local RAS that generates angiotensin peptides from hepatic or endogenously produced angiotensinogen (861, 1042). However, the issue of renin expression in the heart is controversial, and evidence for non-renin-dependent mechanisms in the local formation of cardiac angiotensin peptides is reported (16). In addition to the canonical ACE/chymase-ANG II/AT1R metabolic pathway that is implicated in adverse cardiac remodeling, the heart possesses both the ACE2/ANG (1–7)/Mas axis and the ACE2/ANG (1–9)/AT2R (alternatively ANG II/ANG III-AT2R) axis that have for the most part protective and anti-inflammatory actions (798, 1042). Like AT1R, AT2R is primarily expressed in the heart by endothelial cells, fibroblasts, and VSMCs, with lower levels in cardiac myocytes (863, 1153). Levels of AT2R in the heart increase with hypertension, MI, and in the failing heart, most likely due to expression by cardiac fibroblasts, with a variable contribution from cardiac myocytes. Uncertainty still surrounds the role of AT2R in the heart, which signals through activation of three main signaling pathways: 1) serine/threonine and tyrosine phosphatases, 2) phospholipase A2, and 3) bradykinin/NO/cGMP pathway (272). Downstream consequences include (for pathway 1) decreased NAD(P)H oxidase activity and ROS formation, reduced inflammatory cytokine activity and synthesis, as well as increased apoptosis; (for pathway 2) cell hyperpolarization and reduced sympathetic activity; and (for pathway 3) vasodilation. Most studies have shown increased mortality and impaired cardiac function post-MI with AT2R deletion with variable effects in attenuating cardiac hypertrophy or fibrosis (463). Conversely, cardiac overexpression of AT2R is associated with improved contractile function after MI, which may be due to increased levels of NO. Little if any effect is observed on cardiac myocyte size or fibrosis (93, 94, 1198). In contrast, in animal models of ischemic heart disease, an AT2R agonist C21 improves systolic and diastolic heart functions, lessens myocardial fibrosis and oxidative stress, and attenuates inflammatory immune cell infiltration (540, 864). Interestingly, C21 reinforces post-MI compensatory hypertrophy of cardiac myocytes (540); however, off-target effects of C21 may be a concern (675).
Conflicting findings have also been reported on the effect of overexpressing or deleting AT2R in hypertension-induced cardiac remodeling (41). Overexpression of AT2R in cardiac myocytes improves contractile function and attenuates cardiac hypertrophy and fibrosis in response to chronic pressure overload (1189). However, others report no effect on ANG II-induced cardiac remodeling with high levels of AT2R expression, and a beneficial effect on cardiac hypertrophy and fibrosis in response to aortic stenosis only at lower levels of overexpression. Although controversial, AT2R has been implicated as a contributing factor in cardiac hypertrophy (998). Unexpectedly, AT2R knockout in mice was reported to prevent cardiac hypertrophy and reduce fibrosis in response to chronic ANG II infusion or pressure overload from either TAC or aortic stenosis, although a strain effect may influence outcome at least with aortic stenosis (41). AT2R is reported to couple to cardiac hypertrophy via the transcription factor promyelocytic zinc finger protein (PLZF), which is highly expressed in the heart and shown to bind to the COOH terminus of AT2R (935, 1113). With stimulation-induced internalization of AT2R, PLZF localizes in the nucleus and upregulates both GATA4, a master transcription factor of cardiac hypertrophy, and the p85α regulatory subunit of PI3K. With the latter, the subsequent activation of PI3K/Akt signaling leads to cellular hypertrophy partially from enhanced protein synthesis by p70S6K, as well as by inhibition of GSK3β, which normally phosphorylates GATA4 to promote its nuclear export. Knockout of PLZF in the mouse was found to prevent both cardiac hypertrophy and fibrosis from ANG II infusion. The hypertrophic actions of AT2R on the heart may also require increased blood pressure, suggesting that AT2R is a mechanosensor (1113). Finally, the role of AT2R in modulating the immune system is an area of ongoing interest. Recently, a novel regulatory T cell subset that expresses AT2R was found to have a beneficial impact on infarct size and heart function after MI in mice (976).
3. ACE2, ANG (1–7), and Mas in cardiac hypertrophy
In the heart, ACE2 is primarily expressed by vascular endothelium and smooth muscle, and Mas mostly by cardiac myocytes (46). Loss of ACE2 is associated with cardiac hypertrophy and fibrosis (1261); loss of Mas, with impaired cardiac function and increased fibrosis (913). Overexpression of ACE2 attenuates cardiac hypertrophy, fibrosis, and dysfunction induced by diabetes (241), myocardial infarction (1266), and ANG II infusion (1268). Conversely, ACE2 deficiency exacerbates adverse cardiac remodeling caused by diabetes (802), MI (466), and ANG II (1268), as well as other pathologies or treatments (46). Notably, ANG (1–7) infusion was found to prevent cardiac hypertrophy and reduce cardiac fibrosis in pressure-overloaded ACE2 KO mice by suppressing NAD(P)H oxidase activity. ANG (1–7) attenuates cardiac hypertrophy and fibrosis and improves diastolic dysfunction in a model of diabetic cardiomyopathy (802). ANG (1–7) attenuates cardiac hypertrophy and fibrosis caused by ANG II infusion, and this is associated with upregulation of DUSP1 and decreased ERK1/2 activation (676). The intermediate-conductance Ca2+-activated K+ (KCa3.1) channel, which is essential for ANG II-induced fibrosis, is a downstream target of ERK1/2 activation in cardiac myofibroblasts and is inhibited by the ACE2/ANG (1–7)/Mas axis (1111).
Under stress conditions, ACE is upregulated in the heart, whereas ACE2 is downregulated. Recent evidence has shown that the “ACE2-to ACE pathological switch” is under transcriptional control in endothelial cells via a complex formed by the Brahma-related gene-1 (Brg1) chromatin remodeler and forkhead box M1 (FoxM1) transcription factor. Endothelial cell deletion of Brg1 or FoxM1, or chemical inhibition of FoxM1, markedly attenuate ANG II-induced cardiac remodeling, including hypertrophy and fibrosis. Of note, endothelial cell expression of Brg1 and FoxM1 is increased in human hypertrophic hearts and correlates with the ACE/ACE2 expression ratio (1191).
B. Cardiac Fibrosis
1. Roles of ECM and MR
As discussed with cardiac hypertrophy, pathological cardiac remodeling is a severe alteration in the structure of the heart due to changes at the molecular and cellular level that not only impair contractility and heart function, but enhance the vulnerability of the heart to lethal injury (951). Cardiac fibroblasts are integral contributors to adverse cardiac remodeling in their capacity to increase ECM production and turnover, and to alter its composition and mechanical properties. Humoral and mechanical stimulation of cardiac fibroblasts may cause fibrosis that leads to myocardial stiffness (eventually manifesting as diastolic dysfunction) and arrhythmias, or contribute to tissue inflammation by the formation of proteolytic fragments of the ECM (36, 267, 811, 1064). A principal hormone contributing to ECM deposition is ANG II, which has been implicated in both reparative fibrosis of scar tissue that is seen post-MI (835, 1130) and reactive fibrosis that occurs for example with hypertension or aortic valve stenosis (256, 723). Some of the pro-fibrotic effects of ANG II on the left ventricle may be mediated by aldosterone, particularly in the presence of a high-salt diet (131). In fact, high salt may activate the mineralocorticoid receptor, due to increased Rac1 activity, independent of aldosterone (101). Recent evidence has shown that deoxycorticosterone/salt-induced cardiac fibrosis results from activation of the mineralocorticoid receptors of macrophages (867).
In response to various external stimuli, including ANG II, cardiac fibroblasts are activated and commonly adapt a myofibroblast phenotype characterized by the presence of alpha-smooth muscle actin (α-SMA) microfilaments (213, 549, 1130). Historically, expression of α-SMA was used to identify activated cardiac fibroblasts both in vitro and in vivo, but recent evidence shows that periostin expression is a more comprehensive marker for these cells (456, 467). Periostin is a secreted ECM protein that functions in cellular adhesion and migration as a ligand for αV/β3 and αV/β5 integrins via its FAS1–2 domains (778). ANG II increases periostin expression in adult rat cardiac fibroblasts directly via a Ras/p38MAPK/CREB signaling pathway and indirectly via induction of TGF-β1 expression and Smad2/3 activation (568). ANG II-induced periostin expression, like α-SMA expression, involves oxidative stress (1159). ANG II also increases αV/β3 and αV/β5 integrin expression and signaling in rat cardiac fibroblasts (472).
2. Fibroblast lineage
Until recently, the identity of the source of activated fibroblasts within the heart was unresolved. Extensive lineage tracing studies have now shown that with either pressure overload or ANG II infusion, interstitial and perivascular myofibroblasts in the mouse heart arise from the expansion of resident cells of epicardial embryonic origins (456, 712, 713). This is also the case for the myocardial infarction scar (456, 886). A second population of endocardial embryonic origins contributes to fibrosis in the interventricular septum and left ventricular free wall (712). Previously studied sources of cardiac myofibroblasts, namely, circulating hematopoietic progenitors, endothelial to mesenchymal transition (EndoMT), and epithelial to mesenchymal transdifferentiation (EMT), were found to have a negligible contribution. This conclusion is seemingly at odds with previous findings of several groups showing that the infiltration of bone marrow-derived progenitor cells (“fibrocytes”) into the myocardium with ANG II plays a critical role in ECM deposition (362, 981, 1149, 1176).
3. ANG II concentration
Although multiple in vitro studies have shown that ANG II has direct stimulatory actions on cardiac fibroblasts that are profibrotic, the situation in vivo has been controversial due in part to the wide range in dosage levels for ANG II used in different mouse studies (523, 866). A few reports (using high levels of 1,000–1,500 ng·kg−1·min−1) have indicated the requirement for increased blood pressure to mediate or be permissive for the profibrotic and hypertrophic actions of ANG II in the heart (190, 842); for intermediate levels of ANG II infusion (400–1,000 ng·kg−1·min−1) increased blood pressure is dependent on induction of IL-6 and IL-6-mediated upregulation of ANG II within the kidney (645), which conceivably could have a role in activation of the local cardiac RAS. Higher levels of ANG II appear to downregulate this positive feedback loop in the kidney. When blood pressure was elevated by an IL-6-independent means with high-salt diet and high ANG II (>3,000 ng·kg−1·min−1), cardiac dysfunction, myocardial inflammation, and fibrosis were prevented by genetic deletion of IL-6, although hypertension and cardiac hypertrophy were not affected (329). This result indicates that ANG II has blood pressure-independent effects on the fibroblast compartment of the heart that are mediated by IL-6. Supraphysiological levels of ANG II (≥400 ng·kg−1·min−1) are associated with endothelial dysfunction and vascular injury in the heart concomitant with inflammation and the recruitment of macrophages that drive fibrosis (325, 995). Surprisingly, the influence of a subpressor dose of ANG II (~200 ng·kg−1·min−1) on cardiac structure has been less well studied, but reported to induce both cardiac myocyte hypertrophy and increased interstitial fibrosis or collagen type I (Col I) deposition (67, 862, 1247). It was recently proposed that chronic infusion of the mouse with low-dose ANG II may serve as a model for studying the pathogenesis of heart failure with preserved ejection fraction (HFpEF), which is characterized by concentric cardiac hypertrophy and diastolic dysfunction due in part to increased interstitial fibrosis (862).
4. EGFR, Nox, and ROS
As with vascular and cardiac hypertrophy, EGFR plays a role in cardiac fibrosis at the level of the whole heart. Transactivation of EGFR by ANG II is dependent on c-Src phosphorylation and EGFR inhibition prevents cardiac fibrosis through inhibition of Akt/ERK1/2 signaling (819) and ER stress (465). In addition, ANG II increases physical association of Nox4 and the AT1R in adult mouse cardiac fibroblasts (980); however, the functional consequences of this association on ROS production by Nox4, which is thought to be constitutively active (541), awaits investigation. In mouse cardiac fibroblasts, activation of redox-sensitive NF-κB and AP1 by Nox4-induced ROS is implicated in ANG II-induced expression of IL-18 and collagen, as well as activation of MMP9 and LOX (980). Proinflammatory cytokine IL-18 has been found to be responsible for ANG II-induced cardiac fibroblast proliferation, migration, and collagen production; MMP9 induction for migration. Recently, evidence shows ANG II-induced Nox4 activity contributes to cardiac hypertrophy and fibrosis in vivo via Akt/mTOR and NF-κB signaling (1264). However, no protection of cardiac fibrosis was detected in Nox4-deficient mice infused with ANG II (90).
As discussed elsewhere (732), a number of in vivo studies have implicated enhanced Nox2 activity to ANG II-induced cardiac remodeling. For example, genetic deletion of Nox2−/− in mice inhibits cardiac interstitial fibrosis observed with a pressor dose of ANG II, although cardiac hypertrophy is unaffected (445). A similar study with Nox2−/− mice reporting attenuation of hypertrophy might be explained by differences in the levels of hypertension that were achieved (108). Nox2 deletion inhibits ANG II-induced cardiac NF-κB activation, NADPH oxidase and MMP2 activities, and expression of fibronectin, procollagen I, and CTGF. Similar findings have been reported in another study using a subpressor dose of ANG II, although in this case cardiac hypertrophy was inhibited (67). Nox2 is expressed by coronary endothelial cells and cardiomyocytes, as well as by cardiac fibroblasts in mice though seemingly not in humans (44, 732), but the cell-specific role of Nox2 in ANG II-induced ventricular fibrosis and cardiac dysfunction is unclear. New evidence obtained with transgenic mice indicates a prominent contribution of endothelial cell Nox2 to ANG II-induced cardiac fibrosis. Mice overexpressing Nox2 in endothelial cells are phenotypically normal and develop similar cardiac hypertrophy and systolic hypertension with ANG II infusion as wild-type mice; however, with endothelial Nox2 overexpression, ANG II induced greater cardiac fibrosis, which is associated with LV diastolic stiffness and isolated LV dysfunction as seen in HFpEF. The increased fibrosis is attributed to increased infiltration of inflammatory cells into the heart as a result of enhanced endothelial expression of the VCAM-1 adhesion molecule (726).
Other mechanisms besides activation of redox-sensitive transcription factors may explain the relationship between ANG II-induced Nox2/4 activity and cardiac fibrosis. For example, ANG II-induced collagen production is linked to ROS-induced activation of the RNA binding protein HuR and stabilization of TGF-β mRNA in mouse cardiac fibroblasts (47). Also, mitochondria may constitute an additional source of ROS for driving fibrosis. Mouse cardiac fibroblasts express NLRP3, a pattern recognition receptor. Levels of NLRP3 are increased by TGF-β and ANG II and NLRP3 is important for myofibroblast differentiation independent of its function in the inflammasome. Evidence indicates that NLRP3 localizes to mitochondria and regulates mitochondrial ROS production, which in turn is important for canonical TGF-β signaling via Smad2 phosphorylation and induction of profibrotic proteins, such as CTGF and MMP9. Furthermore, NLRP3-deficient mice are protected from ANG II-induced fibrosis and exhibit reduced Col1 deposition (96).
5. Src, transcriptional factors, and MAPK family
Although c-Src is implicated in both ANG II-induced NADPH oxidase activity (1062) and EGFR transactivation (819), its exact role in ANG II-induced cardiac fibrosis is unclear. c-Src deficiency or pharmacological inhibition was found to attenuate ANG II-induced hypertension, cardiac hypertrophy, and oxidative stress, as well as improve cardiac and endothelial function; however, both vascular and cardiac fibrosis are enhanced (111). While not fully explained, these findings do provide further support to the conclusion that ANG II causes cardiac fibrosis independent of increased blood pressure.
AT1R-dependent activation of Gq/11 activates RhoA through p63RhoGEF. RhoA activation through this pathway increases serum response factor (SRF) activity, leading to increased CTGF production and eventual secretion (777). ANG II-dependent ERK1/2 activation through Nox-generated ROS activates both AP1 and STAT3. AP1/STAT3 upregulation increases miR-21 which in turn suppresses MMP regulator reversion-inducing cysteine-rich protein with Kazal motifs (RECK), Sprouty homologue 1 (SPRY1), and PTEN. Reduction of these proteins increases MMP2 production or activity and encourages cardiac fibroblast migration (965). AP1 and NF-κB also contribute to fibroblast migration through collagen synthesis and MMP production, respectively (964).
Interestingly, p38MAPK has opposing roles as witnessed in cardiac hypertrophy (951). In primary cardiac fibroblasts, ANG II suppresses p38MAPK activation through PKC-βII and -δ stimulating proliferation and collagen accumulation (160). However, the role of p38MAPK is complex and controversial as ANG II-dependent periostin expression within areas of cardiac fibrosis is dependent on RAS-induced p38MAPK activation and downstream CREB (568). Nox-generated ROS and activation of CREB via p38MAPK and ERK1/2 is implicated in ANG II-induced IL-6 expression by cardiac fibroblasts (910). ANG II-induced p38MAPK activity may be linked as well to gene expression in myofibroblasts via SRF (213). Recently, ANG II-induced expression of the collagen-specific tyrosine kinase receptor discoidin domain receptor 2 (DDR2) in cardiac fibroblasts was linked to activation of NF-κB by p38MAPK, downstream of PLC and PKC activation, which in turn are linked to NADPH oxidase-dependent ROS generation. Intriguingly, DDR2 is critical for ANG II-induced ERK1/2 activation and subsequent collagen expression (314).
ANG II activates ERK1/2 in cardiac fibroblasts by increased intracellular Ca2+ or PKC-δ (88, 775) and stimulates downstream TGF-β1/Smad signaling leading to increased periostin expression (568). Interestingly, EGFR transactivation in cardiac myocytes and not fibroblasts may contribute to fibrosis, since cardiac fibroblast EGFR transactivation is dispensable for ANG II-induced ERK1/2 activation (775) while EGFR inhibitors attenuate ANG II-induced cardiac remodeling in vivo (819). ANG II increases intracellular Ca2+ in cardiac fibroblasts by enhancing store-operated Ca2+ entry (SOCE), which is critical for the induction of Col I, fibronectin, α-SMA, and CTGF expression (1241). SOCE activity is associated with increased Smad2/3 phosphorylation, nuclear translocation of NFATc4, and TGF-β1 protein expression. Likewise, ANG II signals activation of an NF-κB/TGF-β1/TRIF/TRAF6 pathway in atrial fibroblasts. Attenuation of this pathway using pioglitazone, a PPARγ agonist, protects mice against ANG II-induced atrial fibrosis (156). NF-κB is also part of the SP1/TGFβ/Smad3 pathway involved in cardiac fibrosis (403, 1134). In addition, the ANG II-induced MAPK/TGFβ1/TRAF6 pathway is necessary for CTGF production and proliferation in atrial fibroblasts (341). The relationship between TGF-β1 and ANG II in mediating myofibroblast transformation and expression of ECM target genes is highly complex and integrated with many of the profibrotic actions of ANG II mediated by the upregulation of TGF-β1 (213, 1064). ANG II can enhance both canonical and noncanonical TGF-β1 signaling in cardiac fibroblasts by inducing Smad2/3 phosphorylation via ERK1/2 activation or by increasing p38MAPK activity via Ras or other means (213, 776). Recently, ventricular myocytes were identified as an important source of TGF-β that acts in an early autocrine manner to induce hypertrophy and in a late paracrine manner to induce fibrosis in response to ANG II infusion of mice (284). Expression of TGF-β by cardiac myocytes, which can be induced by either TGF-β or ANG II, was found to be normally dampened by TGF-β-induced expression of the serine protease inhibitor PAI-1 via a negative feedback loop. Notably, while ANG II-induced hypertension was attenuated in mice with PAI-1 deficiency, cardiac hypertrophy was enhanced. In addition, with PAI-1 deficiency, BMP-7 was found to stimulate activation of inhibitory Smad6 and reduce TGF-β generation and signaling by cardiac myocytes, thereby inhibiting ANG II-induced fibrosis and cardiomyopathy. A recent study showed that IL-11 is required for the profibrotic effects of TGF-β1 in the heart, acting posttranscriptionally to induce fibrogenic protein synthesis via noncanonical, ERK-dependent autocrine signaling (923). Like IL-6, IL-11 signals by causing dimerization of the transmembrane glycoprotein 130 (gp130), and upregulation of IL-11 was found to be the dominant transcriptional response of cardiac fibroblasts to TGF-β1, which also express its receptor IL-11RA.
Past and recently identified signaling pathways required for ANG II-dependent cardiac fibrosis are illustrated in FIGURE 9, in which ROS, TGF-β, EGFR, and MAPKs remain central mediators. However, recent studies have also pointed out the importance of inflammatory responses in cardiac fibrosis induced by ANG II (see below).
FIGURE 9.

ROS has a critical role in ANG II-induced fibrosis of the heart. In the context of fibrosis, AT1R induces ROS formation primarily via engagement of Nox2 and Nox4, but also via mitochondria in a process involving the pattern recognition receptor NLRP3. ROS leads to the induction of various signaling pathways, including NF-κB, ERK1/2, and Akt/mTOR that are linked to myofibroblast target genes. NF-κB and AP1 are implicated in cardiac fibroblast proliferation, migration, and collagen production through the induction of IL-18. ERK1/2 activation is linked to both AP1 and STAT3 activation and the upregulation of miR-21, which in turn suppresses MMP regulator reversion-inducing cysteine-rich protein with Kazal motifs (RECK), Sprouty homologue 1 (SPRY1), and PTEN. Reduction of these proteins increases MMP-2 production or activity and encourages cardiac fibroblast migration. ANG II-induced ROS is implicated as well in both canonical and noncanonical TGF-β signaling. The former involves association of phosphorylated Smad2 and 3 with Smad4; the latter activation of SRF or CREB via p38MAPK signaling, which is engaged directly by an AT1R-linked cascade or indirectly through ROS formation. Increased TGF-β levels may contribute to ANG II-induced fibrosis because of the stabilization of TGF-β mRNA resulting from ROS-induced activation of RNA binding protein HuR. Lastly, EGFR transactivation may contribute to ANG II-induced fibrosis, although cardiac myocytes and not fibroblasts may be involved in this mechanism. *May involve additional cell types besides cardiac fibroblasts/myofibroblasts.
6. Inflammation and cardiac fibrosis
ANG II-induced fibrosis of the heart (perivascular and interstitial) was shown to be dependent on the recruitment of bone marrow-derived monocytic fibroblast precursors with conflicting evidence reported on the importance of endothelial cell produced MCP1 (CCL2) as a chemotactic factor (265, 362). These so-called fibrocytes express both hematopoietic (CD45, CD34, and CD133) and mesenchymal markers (Col-I, αSMA, vimentin). Based on the expression of CD (cluster of differentiation) cell surface markers, recruited fibrocytes were shown to have initially a “classical” pro-inflammatory M1 phenotype, while at a later time cells with an “alternatively activated” M2 phenotype and also positive for procollagen type I were predominant. Prevalence of M1 and M2 was associated with a proinflammatory and profibrotic cytokine milieu, respectively, and evidence was reported that formation of M2 cells was dependent on production of TNF-α by M1 cells (247). However, the exact identity and function of the myeloid fibrocytes in ANG II-induced cardiac fibrosis is unclear given the previously discussed results of lineage tracing and the relative transient presence of the myeloid fibrocytes in the heart (672).
ANG II-induced cardiac fibrosis is dependent on the infiltration of monocyte-derived macrophages originating from the bone marrow, which comprise the largest proportion of leukocytes infiltrating the heart (264). Macrophage levels in the heart with ANG II infusion are somewhat dose-dependent, but blood pressure-independent (264, 325). Accumulation is MCP1/CCL2-dependent or at least modulated by MCP1 (624). Activation of the complement system is involved as well in the recruitment and activation of monocytes/macrophages (1242). From an early time point, most infiltrating macrophages exhibit an alternatively activated M2 phenotype generally considered profibrotic due to associated TGF-β production; however, reducing M2 macrophage infiltration by knocking out the fractalkine receptor Cx3cr1 increases M1 macrophage infiltration, favors a pro-inflammatory myocardial environment, and worsens cardiac fibrosis even though myocardial TGF-β transcript levels are reduced as expected (264). Thus M2 macrophages may exert a protective role in ANG II-induced fibrosis by tamping down the proinflammatory actions of infiltrating M1 macrophages.
Others previously showed that ANG II-induced cardiac fibrosis is dependent on macrophages and IL-6, with activated macrophages stimulating cardiac fibroblasts to produce IL-6, leading to an autocrine loop for TGF-β1 production and fibrosis (625). ANG II-induced expression of IL-6 and collagens in cardiac fibroblasts is dependent on the downregulation of microRNA Let-7i (1124). ANG II infusion may activate macrophages to cause cardiac fibrosis as a result of the production of cellular debris by injured tissue cells and the engagement of TLR2, a pattern recognition receptor of innate immunity (1110). Evidence suggests that other routes for macrophage activation by ANG II occur in the heart as well. The upregulation of the ECM glycoprotein tenascin C by cardiac fibroblasts with ANG II stimulation is implicated in fibrosis ostensibly by enhancing the migration and activation of macrophages, as well as their IL-6 production, downstream of integrin αVβ3 activation (953). With ANG II infusion, monocyte-secreted IL-1β stimulates IL-17A production by γδT cells that in turn induces IL-6 production by cardiac fibroblasts leading to their differentiation into myofibroblasts and fibrosis (584). Others report that ANG II causes the infiltration of IFN-γ producing T cells into the heart that on direct interaction with macrophages produce IFN-γ, which in turn activates CCL2 production by macrophages that is postulated to fuel inflammation by recruiting more macrophages and T cells (352). IFN-γ also polarizes macrophages to the M1 phenotype. The differentiation of naive T cells into IFN-γ-producing proinflammatory T-helper 1 (Th1) cells is dependent on IL-12, a heterodimeric cytokine of two covalently linked p35 and p40 subunits. ANG II infusion markedly increases the expression of IL12p35 in cardiac macrophages, and deletion of IL12p35 leads to polarization of CD4+ T cells to the Th2 phenotype and Th2-dependent accumulation of TGF-β expressing M2 macrophages in the heart in response to ANG II. Moreover, IL12p35 knockout M2 macrophages can stimulate differentiation of cardiac fibroblasts into collagen producing myofibroblasts, and hearts of IL12p35-deficient mice exhibited greater fibrosis with ANG II infusion (585). On the other hand, activation of serum and glucocorticoid-regulated kinase 1 (SGK1) by ANG II is implicated in macrophage recruitment to the heart and polarization of macrophages, upstream of STAT3 activation, to the M2 phenotype. Loss of SGK1 attenuates the increase in profibrotic cytokines in the heart in response to ANG II treatment and blocks cardiac hypertrophy and fibrosis. ANG II-induced cardiac fibroblast to myofibroblast transformation is observed in cocultures with wild-type bone marrow-derived macrophages, but not with SGK1-deficient macrophages (1193).
Myocardial recruitment and activation of CD8+ T cells (cytotoxic T cells), due to IFN-γ production by parenchymal cells (including vascular endothelial cells and fibroblasts), is required for ANG II-induced cardiac recruitment and activation of macrophages towards a phenotype characterized by the production of pro-inflammatory and profibrotic cytokines, including CCL2. Knocking out either IFN-γ or CD8+ T cells markedly attenuates ANG II-induced cardiac fibrosis independent of increased blood pressure or cardiac hypertrophy. Recruitment and activation of macrophages by CD8+ T cells does not require IFN-γ, but does require their physical contact and is TCR-independent, consistent with an innate immune cell-like function of CD8+ cells (624). Finally, others report that production of extracellular heterodimeric proteins S100a8/a9 by CD11b+Gr1+ neutrophils may be an early initiation factor in ANG II-induced cardiac fibrosis. S100a8/a9 activates NF-κB signaling in cardiac fibroblasts via the receptor for advanced glycation end products (RAGE) and induces expression of multiple chemokines and cytokines. Blocking S100a8/a9 prevents ANG II infusion-induced cardiac cytokine production, inflammatory cell infiltration, perivascular and interstitial fibrosis, and hypertrophy without affecting increases in blood pressure (1162). Contributions of the immune cell types implicated in ANG II-induced cardiac fibrosis are summarized in TABLE 8.
Table 8.
Role of myeloid or immune cells in ANG II-induced cardiac fibrosis
| Cell Type | Notes (In Response to ANG II Infusion) | Reference Nos. |
|---|---|---|
| Fibrocytes | CD45+CD34+CD133+ and “mesenchymal” markers | 247, 265, 362 |
| Temporal transition from proinflammatory M1 to collagen producing M2 phenotype | ||
| Macrophages (Mφ) | Activated by cell debris via TLR2 engagement | 264, 585, 625, 953, 1110, 1193 |
| Also activated and induced to produce IL-6 by fibroblast-derived tenascin-C | ||
| Activated Mφ stimulate cardiac fibroblasts to produce IL-6, leading to autocrine loop of TGF-β1 and fibrosis | ||
| Polarize T cells to inflammatory Th1 phenotype by ANG II-induced IL-12p35 production | ||
| Relative contribution of M1 and M2 phenotypes to cardiac fibrosis somewhat unsettled | ||
| Th1 cells | Produce IFN-γ that induces Mφ CCL2 production to sustain inflammation | 352 |
| Polarize Mφ to M1 phenotype | ||
| Th2 cells | Possibly contribute to polarization of Mφ to TGF-β producing M2 phenotype | 585 |
| γδ T cells | Produce IL-17A in response to monocyte IL-1β | 584 |
| IL-17A-induced IL-6 production by cardiac fibroblasts leads to myofibroblast phenotype | ||
| Cytotoxic T cell (CTL) | Recruited/activated by parenchymal cell IFN-γ | 624 |
| Polarize macrophages to M1 phenotype via direct interaction/TCR independent | ||
| Neutrophils | Activate RAGE signaling and cytokine/chemokine signaling in fibroblasts via S100a8/a9 production/release | 1162 |
| Early event |
7. Role of TIMPs
Recent evidence reveals that synthesis of TIMP1 from cardiac fibroblasts in response to ANG II or pressure overload contributes to collagen synthesis and cardiac fibrosis in a MMP-independent manner (1031). Extracellular TIMP1 fosters an interaction in cardiac fibroblasts between the cell surface TIMP1 receptor CD63 and integrin β1, leading to the phosphorylation and release of β-catenin from N-cadherin, which in association with activated SMAD2/3 from TGF-β signaling, translocates to the nucleus and induces collagen gene transcription. The roles of TIMP2 and TIMP3 are similarly complicated in ANG II-induced cardiac remodeling, with loss of either leading to diastolic dysfunction in the context of a preserved ejection fraction with ANG II infusion, but for different reasons (266). Knockout of TIMP2 impairs active relaxation by enhancing ANG II-induced cardiac hypertrophy (there is no effect on ANG II-induced hypertension), while cardiac fibrosis is eliminated due to suppressed collagen deposition attributed to a reduction in cross-linking enzymes LOX and PLOD1. TIMP3 knockout increases LV passive stiffness due to excess fibrosis resulting from posttranslational stabilization and deposition of collagen by matricellular proteins SPARC and osteopontin. In addition, loss of TIMP3 eliminates ANG II-cardiac hypertrophy, not simply because ANG II-induced hypertension is reduced by TIMP3 knockout, but elimination of TIMP3 attenuates the prohypertrophic actions of cardiac fibroblasts on cardiomyocytes (266).
C. Ventricular Arrhythmias
Activation of inflammation and an immune response through engagement of NF-κB contributes to ANG II-induced cardiac fibrosis and electrical remodeling of the left ventricle. AT1R couples to NF-κB activation in nonimmune cells via the CBM signalosome, consisting of three principal proteins: caspase recruitment domain (CARD) 10 (CARMA3, caspase recruitment domain membrane-associated guanylate kinase), B cell lymphoma/leukemia 10 (Bcl10), and mucosa-associated lymphoid tissue lymphoma translocation protein (MALT1) (674). The CBN signalosome transmits the signal from AT1R to the IκB kinase (IKK) complex, which is activated by MALT1-mediated ubiquitination of the regulatory subunit, IKKγ. The activated IKK complex phosphorylates IκB to mark it for proteosomal degradation, thereby releasing NF-κB. A similar system for AT1R involving CARMA1 may occur in immune cells (674). Compared with wild-type mice, ANG II-infused Bcl10 knockout mice exhibited reduced cardiac fibrosis and cellular infiltration, as well as improved arrhythmogenic electrical remodeling, despite similar degrees of hypertension and cardiac hypertrophy (651). NF-κB activation in both cardiac and immune cells contributed to adverse cardiac remodeling by ANG II as evidenced by attenuated inflammatory cytokine/chemokine production by the heart, reduced expression of adhesion molecules on endothelial cells, impaired chemotaxis of monocytes/macrophages, and lessened infiltration of bone marrow-derived myofibroblasts in Bcl10 knockout mice. QRS interval prolongation with ANG II treatment can lead to ventricular arrhythmias, and besides hypertrophy and fibrosis, delocalization of connexin-43 (Cx43) contributes to slowing of ventricular conductance. In Bcl10 knockout mice, Cx43 remained localized at ICD regions after ANG II infusion, whereas in wild-type mice Cx43 showed partial redistribution toward the lateral borders of cardiac myocytes. Reduced Cx43 levels may also contribute to ANG II-induced ventricular arrhythmias and sudden cardiac death. Mice with cardiac-restricted ACE expression show an increased incidence of arrhythmic deaths resulting from ventricular tachycardia degenerating to ventricular fibrillation. This is attributed to reduced ventricular levels of Cx43 due to the activation of c-Src tyrosine kinase, as phosphorylated c-Src competes with Cx43 for a binding site on the ICD scaffolding protein, zonula occludens-1 (984). Displacement of Cx43 from gap junctions and its diffusion away from the ICD results in its enzymatic degradation. Preclinical evidence implicates AT1R in the increased susceptibility to ventricular tachycardia in cardiac hypertrophy independent of structural remodeling, highlighting the importance of Cx43 alterations in this regard (1199). ANG II-induced oxidative activation of CaMKII and PKA via Nox2 likely contributes as well to ventricular arrhythmias due to increased diastolic SR Ca2+ leakage and disturbed Na+ and Ca2+ currents, respectively (1099).
D. Atrial Fibrillation
Cardiac remodeling can lead to profound changes in cardiac function including the development of arrhythmias. Atrial fibrillation (AF) is considered to be the most common sustained type of cardiac arrhythmia and is regulated by focal ectopic firing and/or reentry manifesting in irregular atrial electrical activity (425). Cardiac remodeling is a major player in the development of AF, and hypertension is a major risk factor for AF, which can be attributed to increased cardiac fibrosis and left ventricular hypertrophy that can occur with untreated hypertension (646). Thus investigation into the mechanisms and etiology of AF has pointed to a role for ANG II. A meta-analysis conducted on AF prevention showed ACE inhibitors and ARBs were moderately protective against AF after cardioversion, although the beneficial effect of these drugs was limited to patients with LVH or systolic left ventricular dysfunction. Likewise, hypertensive patients receiving losartan in the LIFE study showed significant risk reduction in new-onset AF and associated stroke compared with hypertensive patients treated with atenolol, suggesting a role for ANG II signaling in the development of AF (369). These findings were further confirmed in the VALUE trial (929). Furthermore, AT1R blockade reduces the atrial effective refractory period in a dog model of rapid atrial pacing (741) and delays promotion of AF most likely through reductions in interstitial fibrosis (519). Human atrial tissue samples of patients with AF also show increased expression of ACE and ERK1/2 along with increased atrial fibrosis (323). While specifics of how ANG II may promote AF are still unclear, it appears there is a correlation between ANG II-dependent fibrosis and AF.
Whether the fibrotic response is reactive in which fibrotic tissue separates muscle bundles, or reparative, where the fibrotic response replaces dead cardiomyocytes, fibrosis can slow conduction by interfering with electric continuity (425). As such, elucidating ANG II-dependent mechanisms of atrial fibrosis and how they affect the development of AF is critical. For general mechanisms of AF, please see a recent review (107). Human AF samples show marked elevation in STAT3 phosphorylation, and in rat atrial myocytes ANG II induces interaction between Rac1 and STAT3 causing STAT3 phosphorylation (1067). Mice with cardiac-specific overexpression of Rac1 at 16 mo of age display AF alongside of increased atrial fibrosis (9). Patients with AF show decreased NO production and eNOS−/− mice infused with ANG II develop cardiac remodeling and AF through Rac1-induced TGF-β1 signaling (1184). As discussed in earlier sections, TGF-β1 is a key player in mediating fibrosis, and recent evidence has highlighted MKK4 as a negative regulator of TGF-β1. MKK4-deficient mice show susceptibility to atrial remodeling and arrythmogenesis and primary cultured cardiomyocytes stimulated with ANG II show increased TGF-β1 induction (210). Furthermore, rapid atrial pacing suppresses Smad7, a negative regulator of TGF-β1, through an ANG II/AT1R-dependent TGF-β1 and ERK/Smad2/3 signaling mechanism that activates the Arkadia ubiquitin proteasome pathway. AT2R seems not involved (367). CTGF is upregulated by ANG II through Rac1 in atrial myocytes and promotes AF and cardiac remodeling through downstream upregulation of N-cadheirn and Cx43 (10).
With regard to ROS and AF, ANG II-dependent Nox2/4 induction (1215) and Nox-derived ROS activates CaMKII through methionine oxidation (sites 281/282). Oxidized CaMKII increases Ca2+ leak from RyR2 through serine phosphorylation (site 2814), thus promoting increased susceptibility to AF (839). Likewise, in atrial myocytes, ANG II induces spontaneous release of Ca2+ from the sarcoplasmic reticulum coupled with electrophysiological changes which could lead to AF susceptibility (309). Nox2-dependent superoxide production is considered to originate from both atrial fibroblasts and macrophages, as macrophages are recruited to the atria as a result of AF and release superoxide through p38-mediated activation of Nox2 (884). Inflammatory cell infiltration during AF may also be a result of ANG II-induced inflammatory activation as rapid arterial pacing increases endocardial VCAM-1 expression through the AT1R (322). In addition, myeloperoxidase-deficient mice were used to demonstrate a crucial role of neutrophil derived myeloperoxidase in ANG II induced activation of MMPs, atrial fibrosis, and AF (885). Ca2+ influx is also dependent on TRPC3 as blockade using pyrazole-3 prevents ANG II-induced α-SMA expression and ERK1/2-dependent proliferation of cardiac fibroblasts. Pyrazole-3 administration in vivo prevents AF development (357). ANG II also enhances promoter methylation leading to decreased protein expression of Pitx2c, a gene involved in AF, through AT1R (457). Recent evidence implicates intracrine ANG II signaling as well in the pathogenesis of AF. Nuclear AT1R and AT2R that are linked to IP3 receptor- and NO-dependent pathways, respectively, were identified in canine atrial fibroblasts, and intracellular ANG II was found to induce proliferation and collagen gene transcription. Moreover, levels of intracellular ANG II and nuclear AT1R in canine atrial fibroblasts are increased with pacing-induced heart failure (1024). Taken together, significant progress is made regarding the signal transduction mechanisms and cell communications by which ANG II contribute to cardiac pathologies including cardiac hypertrophy, fibrosis, and AF. However, further research in large animals and humans to translate the latest information is desired.
VI. ANG II SIGNALING IN RENAL PHYSIOLOGY AND PATHOPHYSIOLOGY
A. Basic Understanding of ANG II Effects on Kidney
A major physiological role of the RAS is to regulate salt and water balance, particularly in circumstances of dehydration and volume depletion (985). Renal control of sodium and water handling by ANG II is critical to these actions. Furthermore, dysregulation of the RAS can cause hypertension, and its activity in the kidney is believed to be a central player in development and maintenance of essential hypertension (179). In this regard, kidney cross-transplantation studies have shown that AT1R in the kidney are necessary and sufficient for development of ANG II-dependent hypertension (190). Angiotensin receptors are expressed throughout the kidney in vasculature, a range of tubular epithelial populations and the juxtaglomerular apparatus (JGA), as well as podocytes and mesangial cells in the glomerulus (985). This pattern of angiotensin receptor expression portends the broad impact of ANG II on renal function. ANG II has a direct effect on renal VSMCs, causing vasoconstriction of afferent and efferent arterioles, resulting in reduced renal blood flow, favoring sodium reabsorption. The role of renal vascular AT1R in reducing RBF and contributing to ANG II-induced hypertension has been recently confirmed with VSMC AT1AR-deficient mice (987). A few studies suggest a contribution of oxidative stress in ANG II-enhanced vasoconstriction of afferent arterioles (569). In p47phox knockout mice, ANG II-mediated afferent arteriole contraction is blunted, suggesting that Nox-derived ROS are important to maintain enhanced renal vascular resistance induced by ANG II (532). ANG II also directly stimulates reabsorption of salt and water in multiple nephron segments. Moreover, ANG II stimulates aldosterone secretion to further augment salt and water retention in mineralocorticoid-sensitive segments of the nephron (690).
The local events at the JGA regulating glomerular filtration rate (GFR) by tubuloglomerular feedback (TGF) cause a major realignment of glomerular arteriolar vasomotor tone. TGF is an intrarenal regulatory system in which NaCl concentration at a distal nephron site is sensed as a signal of alterations in glomerular filtration, initiating a response that acutely modifies GFR. Increased intrarenal ANG II levels (see below) amplify the sensitivity of TGF to reduce GFR. As in other target organs, ANG II also has critical nonhemodynamic functions to alter hypertensive renal pathophysiology, including enhancement of immune and inflammatory responses, oxidative stress, endothelial dysfunction, mesangial cell growth, and interstitial fibrosis in the kidney. In addition to inflammation and oxidative stress, ANG II-induced generations of ET-1 and TGF-β are considered to be other major mechanisms in ANG II-induced renal pathophysiology (690).
B. Intrarenal RAS and Hypertension
Kidneys express all necessary components [AGT, renin, (pro)renin receptor, and ACE] for ANG II generation. Studies have suggested that this intrarenal RAS is subject to local regulation (502), and intrarenal ANG II production, which is paradoxically stimulated under several pathophysiological conditions, including ANG II infusion, has been implicated in renal ANG II physiology and pathophysiology. Regulation and impact of intrarenal RAS in renal physiology, pathophysiology including hypertension, fibrosis, and inflammation have been reviewed extensively (316, 502, 855, 919). The significance of this intrarenal system has been demonstrated in studies showing that two distinct genetic deletions of renal ACE attenuate hypertension induced with ANG II infusion or l-NAME treatment. These modified phenotypes are associated with inhibition of activation of renal sodium transporters and reduced sodium reabsorption (316, 332). Note that there are some recent controversies in this field, such as the possible extrarenal origins of AGT and other RAS components (667, 668, 875).
C. Salt and Water Balance
As mentioned above, AT1R are expressed in tubular epithelia across the nephron, where they mediate ANG II-dependent regulation of sodium and water handling. However, the effects and mechanisms of tubular actions of ANG II may vary in different nephron segments. For example, actions of ANG II to affect proximal tubule functions have been long-recognized, where it targets key sodium transporters. There is evidence of a role for altered transport preceding blood pressure elevation in ANG II infusion models. Within 20 min, ANG II treatment stimulates trafficking of Na+/H+ exchanger (NHE3), Na+/Pi cotransporter 2 (NaPi2) and NHE regulatory factor (NHERF)1 into the proximal tubule brush-border microvilli, where they likely contribute to rapid increase in salt and water reabsorption (870). A 3-day nonpressor dose of ANG II also increases proximal tubule NHE3 cortical NKCC2 and ENaC (749). NHE3 plays a pivotal role in salt and fluid reabsorption in the proximal tubule, and is required for ANG II-induced hypertension in mice (580). It should also be noted that renal sympathetic nerve stimulation leads to NHE3-dependent proximal tubule sodium reabsorption via enhanced generation of intrarenal ANG II (831, 832). ANG II activates NHE3 via Ca2+, calmodulin (CaM), and CaMKII. This pathway regulates NHE3 interaction with ROS-mediated JAK2-CaM complex (51). Upon ANG II stimulation, CaMKII also phosphorylates a NHE3 interacting protein, inositol 1,4,5-trisphosphate receptor-binding protein released with inositol 1,4,5-trisphosphate (IRBIT) to mediate trafficking and activation of NHE3 in cultured opossum kidney proximal tubule epithelial cells (365). It was recently shown that ANG II induces NHERF1 interaction with NHE3 and IRBIT, and NHERF1 silencing attenuates NHE3 activation in these cells (366).
Consistent with these signaling and mechanistic studies, cell-specific deletion of AT1AR from proximal tubule epithelia lowers baseline blood pressure and attenuates ANG II-dependent hypertension (347). These effects on blood pressure are associated with impaired renal sodium reabsorption and reduced abundance of key sodium transporters, including NHE3 and the sodium-phosphate cotransporter. These findings are in line with the reports demonstrating attenuation of ANG II-induced hypertension in NHE3-deficient mice (579, 580). Finally, the role of proximal tubule AT1AR in blood pressure regulation is also demonstrated in transgenic mice expressing a constitutively active AT1AR specifically in proximal tubule, which have increased blood pressure (566).
ANG II also modulates NaCl handling in the thick ascending limb of Henle (THAL), which avidly reabsorbs NaCl and contributes to the generation of renal osmotic gradient. These effects of ANG II on THAL function are mediated by both direct and indirect mechanisms. In contrast, NO, another key regulator of THAL function, decreases NaCl reabsorption via cGMP-dependent inhibition of the Na/K/2Cl cotransporter (NKCC2) (718). WNK kinases make up crucial molecular pathway connecting ANG II and aldosterone control of renal sodium and potassium transport (391, 881). WNK kinases have broad actions to regulate trafficking and activation of NKCC2, the sodium-chloride cotransporter (NCC) in the distal convoluted tubule, and the renal outer medullary potassium channel (ROMK). In general, the WNK kinase-dependent modulation of the transporters involves phosphorylation of the transporters via SPAK and OSR1, two highly homologous kinases that are phosphorylated and activated by WNK kinases (391). In patients with pseudohypoaldosteronism type II, mutations in WNK4 or WNK1, or in either Cullin 3 or Kelch-like 3 (KLHL3) components of an E3 ubiquitin ligase complex that targets WNKs for degradation, cause constitutively increased renal salt reabsorption and impaired K+ secretion, resulting in hypertension and hyperkalemia. It has been shown that ANG II induces PKC-dependent KLHL3 phosphorylation, preventing WNK4 degradation and phenocopying the KLHL3 mutation (949). In addition, aldosterone stabilizes PY motif-containing WNK1 isoform in the kidney via SGK1-dependent inhibition of E3 ubiquitin ligase NEDD4–2 (880). ANG II infusion increases renal phosphorylation of NKCC2 and SPAK (451, 749), suggesting a direct mechanism of NKCC2 via WNK kinases. The direct mechanism may also involve Src and the ERK cascade (717). It was recently shown that ANG II also promotes NaCl reabsorption through enhanced phosphodiesterase 5 (PDE5)-driven degradation of cGMP (856). In addition, in ANG II-induced hypertension, IL-1 receptor stimulation augments hypertension and NKCC2 activity via prevention of the maturation of intrarenal myeloid cells into Ly6C+Ly6G− macrophages, thereby inhibiting macrophage-dependent NO secretion (1249).
NCC activity in the distal convoluted tubule plays an essential role in Na+ handling, reflected by its importance as a target for thiazide diuretics, one of the most effective treatments for essential hypertension. ANG II promotes NCC-dependent sodium reabsorption through WNK4 and SPAK/OSR1-dependent signaling (127, 907, 1083). Interestingly, lack of IFN-γ in mice prevents ANG II-induced elevations in blood pressure, and this is associated with reduced phosphorylation of NKCC2, NCC, and SPAK in the distal convoluted tubule. Similarly, deficiency of IL-17A reduces proximal transporter activity in response to ANG II (451), suggesting a contribution of inflammatory responses to enhance sodium reabsorption in hypertension.
The collecting duct is a key target of aldosterone, where it has potent effects to enhance epithelial sodium channel (ENaC) activity. ANG II also has direct actions to influence handling of salt and water by collecting duct epithelia. ENaC is activated by ANG II in collecting duct, potentially contributing to sodium reabsorption (643). ANG II-dependent increases in ENaC activity require NADPH oxidase activation and generation of ROS (642), Ca2+-independent PKC activation (1014), and (pro)renin receptor activity (818). ATRAP promotes AT1R internalization and is protective against ANG II-induced pathological signaling. ATRAP deficiency enhances AT1R signaling, which stimulates ENaC and enhances sodium retention in distal tubules (769). It is also important to note that unlike aldosterone, which induces and stimulates ENaC as well as renal outer medullary channel (ROMK) to accelerate sodium potassium exchange (1232), ANG II uncouples ENaC-dependent sodium reabsorption by inhibiting ROMK (1136). The mechanism of ROMK inhibition by ANG II appears to involve WNK4, SGK1, and c-Src-dependent ROMK phosphorylation (1227). Despite this evidence of direct effects of ANG II on ENaC to promote sodium reabsorption, cell-specific deletion of AT1AR from principal cells of the collecting duct does not affect baseline blood pressure and only modestly diminishes ANG II-dependent hypertension (147). In contrast, deletion of AT1AR from both principal and intercalated cells of the collecting duct is associated with an exaggerated blood pressure increase with chronic ANG II infusion, due to elimination of AT1AR-dependent generation of vasodilator prostanoids that normally resist the development of hypertension (999).
Systemic AT1AR knockout mice have an impaired capacity to concentrate the urine, which is associated with reduced aquaporin-2 (AQP2) expression in the inner medulla (578). The AQP2 water channel is the major protein regulating the permeability of the collecting duct to water. Specific elimination of AT1AR from both principal and intercalated cells in the collecting duct impairs urinary concentrating capacity. This is associated with a reduction in levels of AQP2 protein in the medulla after water deprivation, suggesting that AT1AR in epithelial cells of the collecting duct regulate the concentration of urine via AQP2 (1000). Several studies suggest that ANG II and AT1R are important regulators of AQPs including AQP2. ANG II infusion in rats reduces AQP2 expression in outer medulla, which is associated with diuresis (501). In inner medullary collecting duct cells from rats, ANG II rapidly stimulates membrane trafficking of AQP2 via increases in cAMP through AT1R (554). ANG II also increased AQP2 protein expression through AT1R in the immortalized mouse renal collecting duct principal cells via PKA and PKC (560). ANG II via AT1R also increases AQP1 expression in renal proximal tubules in vitro and in vivo (91).
Cumulatively, ANG II signal transduction has a significant role in regulation of renal ion and water handling. Critical components of these signal transduction pathways include AT1R, WNKs, SPAK, NKCC2, NCC, NHE3, ENaC, and AQP2 (FIGURE 10). Future research will hopefully further elucidate the specific molecular mechanisms leading to the development of altered renal function and hypertension elicited by ANG II.
FIGURE 10.

Renal ion regulation in response to ANG II. In the thick ascending limb (TAL), ANG II-induced Ca2+ elevation via a c-Src or PLC/PKC/ERK pathway leads to NKCC2 activity which can be inhibited through NO-dependent cGMP elevation. ANG II-dependent IL-1R induction in intra-myeloid cells inhbitis NO secretion. Furthermore, ANG II-induced PDE5 converts cGMP to GMP. In the PCT, AT1R induced PLC and resultant Ca2+ elevation. Ca2+ can induce NHE3 through a JAK2/CaM pathway or a CaMKII/NHERF1/IRBIT pathway leading to NHE3 trafficking to the cell membrane. DCT ANG II signaling leads to WNK-4/SPAK-mediate NCC activation. Furthermore, AT1R stimulated PKC induces Nox and resultant ROS leading to ENaC activation.
D. Renal Fibrosis and Inflammation
ANG II-induced renal inflammation and fibrosis are significant contributors to chronic kidney injury and the development of end-stage kidney disease. The importance of these pathways in chronic kidney disease is reflected in the clinical efficacy of ACE inhibitors and angiotensin receptor blockers to slow the progression of renal damage. As with cardiac and vascular fibrosis, TGF-β plays a central role for this ANG II-dependent pathology. TGF-β contributes to epithelial cell-mesenchymal cell transition to promote ANG II-induced fibrosis in kidney. TGF-β1 induction is regulated by p38MAPK activation and JNK/thrombospondin-1 signaling (889). Moreover, EGFR transactivation by ANG II in renal proximal tubule appears critical for ANG II-induced renal TGF-β induction, epithelial cell-mesenchymal cell transition, and fibrosis (150, 151). In addition to EGFR, a recent study demonstrated that ER stress-induced activation of a transcription factor sterol regulatory element-binding protein-1 (SREBP-1) contributes to TGF-β induction and renal fibrosis in ANG II-infused mice (1118).
Along with TGF-β signaling, ANG II-induced fibrogenesis is also mediated through CTGF, ET-1, COX-2, PAI-1, osteopontin, and various inflammatory effectors including VCAM-1, IL-6, TNF-α, and Lox1, which affect ECM production and inflammatory responses, resulting in renal fibrosis (889). CTGF and downstream collagen I production by ANG II is mediated through an ERK/p38MAPK/Smad3 axis (889) and Rho kinase (887). While CTGF/CCN2 has also been implicated in ANG II-induced renal fibrosis, its role in general fibrotic responses has been challenged recently (286). HIF-1α has been implicated in ANG II-induced renal fibrosis (1274). EC-selective HIF-1α knockout mice are protected against ANG II-induced hypertension and renal fibrosis, which involves NF-κB-dependent HIF-1α induction (615). In addition to the well-established transcriptional induction of the inflammatory effectors (including those by Wnt/β-catenin; sect. IIIA1), ANG II also enhances stabilization of PAI-1 and COX2 mRNA via PKC-δ-mediated HuR binding to 3′UTR AU-rich destabilization elements in the kidney (238).
In an ANG II-dependent model of chronic kidney disease (5/6 nephrectomy with ANG II infusion), TLR4 mutant mice are protected from markers of renal dysfunction including albuminuria, increased serum BUN and creatinine, glomerulosclerosis, and interstitial fibrosis compared with control mice. Systemic low-grade inflammation was also reduced in these mice, in part due to attenuation of TLR4-dependent inflammasome signaling (983). In line with the role of inflammation in ANG II-induced renal injury, NF-κB inhibition also protects against ANG II-induced renal injury, and reduces TGF-β induction (786). It is noteworthy that AT1R on T lymphocytes appears protective for ANG II-induced renal injury, but not hypertension (1251), whereas blocking T cell accumulation in the kidney limits renal damage and the hypertensive response to ANG II (348). The proinflammatory factor produced from Th1 T cells in the kidney contributing to hypertensive responses seems to be TNF-α, according to kidney transplant experiments with TNF-α-deficient mice (1248), and TNF receptor 2 appears to mediate renal inflammation induced by ANG II (972). In addition, CCR2 knockout mice show attenuated macrophage accumulation in the kidney, resulting in reduced local oxidative stress and renal fibrosis upon ANG II infusion (589). IL-6-dependent activation of the renal JAK2 and STAT3 pathway also contributes to ANG II-induced hypertension (98).
In contrast, it has been demonstrated that ANG II-induced hypertension and renal inflammatory cell infiltration are enhanced in bone marrow AT1AR-deficient mice (191). In line with this finding, macrophage-specific AT1AR deficiency enhances production of proinflammatory M1 cytokines and exacerbates kidney fibrosis induced by unilateral ureteral obstruction, in part through enhanced IL-1 receptor stimulation (1250). In addition, deficiency of AT1ARs on T cells also potentiated kidney injury during hypertension with exaggerated renal expression of chemokines and enhanced accumulation of T cells in the kidney (1251). TNF receptor 1-deficient mice also show higher blood pressure and aggravated renal damage after ANG II infusion (146). Taken together, these studies suggest that, depending on the circumstance, ANG II can enhance or inhibit inflammatory responses in the kidney.
Overall, ANG II is a well-known instigator of renal fibrosis and inflammation (FIGURE 11). Key players in this cascade include TGF-β1 and various mediators of inflammation, involving several cell types in the kidney, as well as leukocytes and other immune cells. It is also interesting to note that an NHE3 inhibitor, S3236, reduces ANG II-stimulated induction of several cytokines in cultured proximal tubular cells (1172), suggesting a close relationship between renal inflammation and enhanced sodium reabsorption.
FIGURE 11.
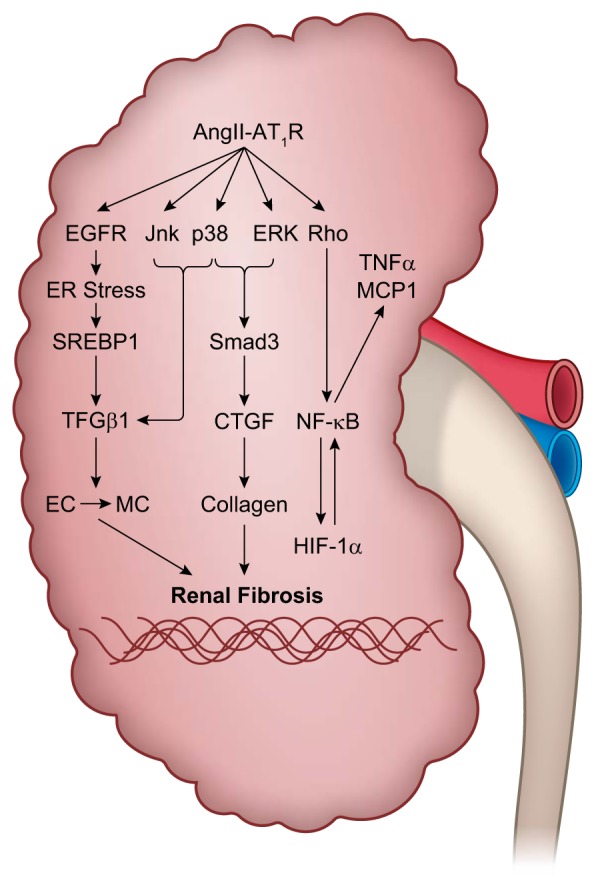
ANG II signaling in renal fibrosis. EGFR transactivation leads to ER stress, SREBP1 and TGF-β1 signaling. Likewise, ANG II signals Jnk and p38 leading to TGF-β1 induction. TGF-β1 then stimulates epithelial cell-mesenchymal cell transition. ERK and p38 both induce Smad3 signaling leading to CTGF-dependent collagen expression. ANG II-induced Rho activity also induces NF-κB leading to CTGF and HIF-1α expression.
E. AT2R in Renal Physiology
In the kidney, as in other organ and cell systems, AT2R seems to have effects that oppose actions of ANG II-mediated through the dominant AT1R. In ANG II-dependent hypertension, renal AT2R activation prevents Na+ retention and reduces blood pressure via natriuresis in a bradykinin-NO-cGMP-dependent manner in rats. The mechanism appears to involve translocation of AT2R to the apical membrane and internalization of NHE3 and Na+-K+-ATPase (479, 480). This mechanism is extinguished in spontaneously hypertensive rats, where AT2R does not translocate to the apical plasma membrane of proximal tubules upon intrarenal ANG II infusion (791). Stimulation of natriuresis by AT2R also involves inhibition of NADPH oxidase and ROS (896). In addition, in proximal tubule cells, the heterodimer of AT2R and dopamine D1-like receptor on the plasma membrane increases cAMP and cGMP production, protein phosphatase 2A activation, and Na+-K+-ATPase internalization, while inhibiting sodium transport (318). In addition, suppression of renal fibrosis in response to unilateral ureteric obstruction by relaxin appears to require a heterodimer of AT2R and relaxin family peptide receptor 1 (169). AT2R also has anti-inflammatory and antioxidative functions in the kidney in obese Zucker rats but proinflammatory and pro-oxidative functions in lean Zucker rats (895). An AT2R agonist may also prevent diabetic nephropathy through a similar mechanism (130, 513). However, there are certain limitations in these studies such as specificity issues of pharmacological AT2R agonists/antagonists and the AT2R antibody utilized.
In agreement with these studies, a few studies genetically demonstrate that AT2R can be beneficial for renal pathophysiology. After renal ablation, AT2R-deficient mice show greater impairment of renal function, glomerular injury, albuminuria, and mortality, along with increased fibronectin expression and inflammation (71). With a high-fat diet, AT2R-deficient mice also show enhanced kidney injury with reduced expression of renal ACE2, ANG (1–7) and Mas (22). Finally, AT2R deficiency accelerates the development of nephropathy in type I diabetes via oxidative stress and increased ACE/ACE2 ratio (137).
F. ANG II, Aldosterone, MR, and Renal Pathology
ANG II is a major regulator of aldosterone generated by the adrenal gland, and ANG II-dependent aldosterone release plays a significant role in renal physiology and pathophysiology. The overall actions of aldosterone to enhance renal sodium reabsorption are congruent with those of ANG II, and there is significant communication and convergence of ANG II and aldosterone signaling pathways in the kidney. Similar to ANG II regulation of Na+ reabsorption, aldosterone promotes mineralocorticoid receptor activation in principal cells and enhances Na+ reabsorption in the distal nephron through upregulation of ENaC activity via SGK1 (391, 1232). The actions of aldosterone and ANG II to influence sodium reabsorption across the nephron are complementary, where ANG II predominantly affects the proximal tubule (347), whereas the major impact of aldosterone is in the collecting duct. For example, the combined presence of ANG II and aldosterone simultaneously, as would occur in hypovolemia, promotes proximal Na+ reabsorption by ANG II and distal Na+ reabsorption by aldosterone through SPAK/WNK signaling (391, 1232). In the setting of low Na+ intake, upregulation of renin and ANG II and downstream AT1R activation increases aldosterone secretion and subsequent SGK1 activation and WNK4 phosphorylation, thereby inhibiting the renal outer medullary K+ channel (1120).
The MR-Rac1 pathway also promotes renal injury, exemplified in Tsukuba hypertensive mice that have elevated serum ANG II levels. In this setting, MR and Rac1 inhibition both prevent ANG II-induced renal injury, and it has been suggested that ROS mediates induction of Rac1 (474). In addition to Rac1, EGFR transactivation has been implicated in several nongenomic effects of MR (101). Pharmacological inhibition of EGFR by erlotinib in mice attenuates renal profibrotic responses induced by aldosterone (947).
VII. ANG II SIGNALING IN THE BRAIN
The brain is the central regulator of autonomic function and plays a major role in blood pressure regulation through its effects on fluid homeostasis and sympathetic nerve activity. Like other major organs and tissues already discussed, the brain is responsive to the effects of ANG II. The brain contains both AT1R and AT2R and is responsive to ANG II, which gains entry to the brain through the circumventricular organs, regions of the brain lacking a blood-brain barrier. In addition, the brain contains its own intrinsic RAS, defined by the capacity for both de novo synthesis and action of ANG II inside the blood-brain barrier (340, 893). Indeed, a preponderance of evidence suggests the brain has the capacity for both. However, recent studies and editorial commentary highlight the surprisingly controversial nature of this conclusion (968, 1086). Due to the complexity and vastness of this subject, we direct readers to other reviews that also cover the brain RAS (177, 222, 1156, 1217).
A. Localization of RAS Components in Brain
AGT, the substrate for renin, is expressed widely in glial cells in nearly all regions of the brain (1002). The importance of glial AGT is illustrated by the dramatic physiological effects of its deficiency in transgenic rats (338, 903, 927) and mice (948). AGT is also expressed in neurons, at least in those regions of the brain controlling the cardiovascular system (1190). Similarly, ACE is expressed in the subfornical organ (SFO), organum vasculosum of the lamina terminalis (OVLT), paraventricular nucleus (PVN), and supraoptic nucleus (SON) (133). Renin is expressed at the lowest level of all RAS genes, and its localization remains controversial (737). Several studies using easily detectable reporters driven by the renin promoter have identified potential sites of synthesis (24, 545, 1175). Importantly, such studies provided evidence that renin and AGT were either coexpressed or expressed in neighboring cells in several regions of the brain (544). Recent studies also suggest that an alternative form of renin is specifically expressed in the brain. This form of renin, termed renin-b, lacks both the signal peptide and should remain intracellular, and the first third of the prosegment and should therefore be enzymatically active. In vitro and in vivo experiments suggest this form of renin is indeed active (546, 555, 973). Nevertheless, the physiological significance of endogenous intracellular renin remains unclear because, surprisingly, deletion of renin-b caused hypertension and increased activity of the brain RAS (954, 955).
Numerous studies have identified AT1R expressing cells in many of the key nuclei regulating cardiovascular function, and some of those are sites of renin and AGT synthesis (25). However, as mentioned earlier in this review, the lack of specificity of AT1R antisera (377) remains a limitation in identifying AT1R containing cells in the brain. To facilitate identification of AT1R containing cells, the GENSAT project developed a mouse (NZ44) carrying a BAC transgene expressing enhanced green fluorescence protein (eGFP) inserted into the AT1AR locus (327). According to its website, the GENSAT project aims to map the expression of genes in the central nervous system of the mouse, using both in situ hybridization and transgenic mouse techniques (http://www.gensat.org/about.jsp). In this model, eGFP serves as a surrogate marker for activity of the AT1AR promoter. Studies of the NZ44 mouse revealed that AT1AR is highly expressed in neurons of many nuclei controlling drinking and blood pressure, including the SFO, OVLT, SON, PVN, amygdala (CeA), rostral ventrolateral medulla (RVLM), and nucleus tractus solitaries (NTS). Recall rodents contain two genes encoding AT1R, AT1A and AT1B which may serve separate functions (214). Unfortunately, there is no model in which expression of AT1BR is easily detected. However, a similar BAC transgenic mouse driving eGFP from the AT2R promoter has facilitated the identification of neurons expressing this receptor (224).
B. Brain AT1R Signaling in Blood Pressure, Fluid Intake, and Metabolism
1. Sympathoexcitatory action
Brain renin-angiotensin is well established to regulate arterial pressure through its effects on the sympathetic nervous system (SNS) and fluid homeostasis. Increased activity of the SNS has also been implicated in playing an important role in heart failure (1284). It is now widely recognized that AT1Rs in the brain play a critical role in this process. For example, whereas specific deletion of AT1R from catecholaminergic cells does not alter baseline blood pressure, it delayed the full pressor response to ANG II and blunted the ANG II-induced SNS response (427).
Since the demonstration that AT1Rs are important mediators of SNS activation by ANG II, numerous studies have examined pathways downstream of ANG II/AT1R activation involved in modulating its activity. As examples, ANG II has been shown to upregulate brain-derived neurotrophic factor (BDNF) and its receptor TrkB in catecholaminergic cells, leading to p38MAPK-dependent reduction in voltage-gated K+ current (64). TrkB blockade decreases the pressor and sympathoexcitatory effects of centrally administered ANG II, suggesting that the BDNF/TrkB pathway is required to mediate ANG II-dependent responses (63). Interestingly, central infusion of an AT1R blocker, losartan, or an ACE inhibitor, lisinopril, as well as ganglionic blockade attenuates central BDNF-induced blood pressure elevation (924), further suggesting a feed-forward relationship in brain between the BDNF/TrKB pathway and the RAS.
PPARβ activation attenuates the pressor and sympathoexcitatory actions of ANG II in the brain by inhibiting NADPH oxidase activity through a mechanism involving regulator of G protein signaling 5 (RGS5) (876). ANG II infusion decreases PPARγ DNA-binding activity in the brain, while activation of PPARγ in the brain blunts pressor response to systemically administered ANG II (1223). Intracerebroventricular (ICV) injection of ANG II activates Rho kinase in brain of conscious rabbits, and the Rho kinase inhibitor Fasudil attenuates blood pressure elevation and sympathetic activation in response to the ANG II injection, suggesting the role of Rho kinase in brain ANG II function (809). Estradiol, through the estrogen receptor, is reported to reduce ANG II-induced sodium intake and water intake within the lamina terminalis through inhibition of ERK1/2 and JNK, respectively. In addition, estradiol prevents arginine vasopressin (AVP) secretion in the hypothalamus through MKP1-mediated ERK1/2 dephosphorylation (26). With the use of double transgenic mice to cause ANG II-dependent hypertension, it has been demonstrated that AT1R mediates brain ACE2 inhibition (1167). ACE2 is a known substrate of ADAM17 (534). The importance of neuronal AT1Rs in mediating upregulation of ADAM17 and downregulation of ACE2 in humans and in DOCA-salt hypertension highlights the importance of neuronal AT1R clinically and in experimental hypertension (1168, 1177).
2. SFO regulation of fluid and sodium intake and blood pressure
The SFO, a component of the lamina terminalis, has long been known to be critical for induction of polydipsia, sodium intake, and hypertension. The SFO-dependent mechanisms affecting fluid and sodium intake have been reviewed previously (177, 383, 386). Recent optogenetic studies confirm the exquisite sensitivity of SFO neurons to mediate a drinking response (773), anticipate homeostatic imbalances leading to drinking (1278), and differentially control thirst and salt appetite (663). Deletion of ANG II production in the SFO in a model of brain RAS overexpression blunts drinking (901).
Deletion of AT1R in the SFO also impaired the blood pressure response induced by DOCA-salt (381). Blockade of AT1R in the SFO of rats prevented pressor response to infusion of sodium-rich cerebrospinal fluid (1053). AT1R in the SFO is also required to mediate a sustained elevation in blood pressure caused by intermittent hypoxia, a model of sleep apnea (921). Expression of AT1R in the SFO (and PVN) may be mediated by GRP4, which encodes a proton-sensing receptor (1015). Because of the importance of ANG II in the SFO, the responses mediated by its signaling and the downstream pathways induced by ANG II has been the frequent topic of investigation.
The mTOR pathway, which responds to nutritional and environmental cues, has been implicated in ANG II signaling in the SFO. A single ICV injection or chronic subcutaneous infusion of ANG II activates mTORC1 signaling in SFO, as evidenced by p70S6K phosphorylation and nuclear c-Fos accumulation (730). Interestingly, whereas treatment with the mTOR inhibitor rapamycin does not alter the pressor response to ICV ANG II, it attenuates ANG II-induced water intake. This suggests some selectivity in the ANG II-dependent response mediated by mTOR, which is conceptually interesting as other ANG II-dependent pathways exhibit a similar selectivity. For example, selective activation of ANG II production in the SFO is sufficient to stimulate increased fluid consumption, but is insufficient to increase arterial blood pressure (176). The absence of a pressor response in this model may be due to the low expression level of AGT in the SFO. Indeed, higher level expression of ANG II in the SFO of transgenic mice leads to both increased fluid intake and hypertension (339, 901). Mechanistically, the dipsogenic response to either increased ANG II production in the brain or DOCA-salt hypertension requires PKC-α (178). ANG II-induced drinking via SFO may also be regulated by α1- and β-adrenergic receptors (1028).
ANG II-induced ROS production via Rac1-dependent Nox activation in SFO has been implicated in the pressor response and hypertension (1217). Both Nox2 and Nox4 are involved in the acute pressor response to ICV ANG II, whereas the water intake response is dependent on Nox2 (821). SFO silencing of a Nox subunit, p22phox, also attenuated hypertension and vascular inflammation in response to systemic ANG II infusion for 2 wk (606). Interestingly, SFO-specific delivery of an siRNA targeting the mineralocorticoid receptor prevented brain ROS production and the hypertension caused by systemic ANG II infusion in Wistar rats (1106).
ER stress is also induced in the SFO in response to systemic ANG II infusion (1216). ICV injection of the chemical chaperone TUDCA or SFO-targeted adenoviral delivery of the molecular chaperone GRP78 attenuates ANG II-induced ROS production and hypertension. A subsequent study suggested NF-κB as a downstream mediator of ROS and ER stress in the SFO (1218). Surprisingly, however, attenuation of ER stress through ICV delivery of TUDCA suppresses increased water intake, but did not affect blood pressure in the DOCA-salt model of hypertension (444). The effects of ER stress in the SFO may extend beyond blood pressure, as recent evidence suggests that hepatic steatosis induced by high-fat diet is mediated by ER stress in the SFO (393).
3. Metabolic regulation via SFO AT1R
Recent studies also implicate AT1R in the SFO as a regulator of systemic metabolism. For instance, selective deletion of AT1R in the SFO blunted the weight loss induced by leptin by impairing sympathetic activation of brown adipose tissue and brown adipose tissue thermogenesis (1219). A physiological link between leptin and AT1R signaling in the brain was previously proposed, and recent studies suggest that AT1R on leptin-receptor containing neurons may regulate metabolism (174, 382, 977). This is consistent with studies showing that increased activity of the brain RAS, particularly in the SFO, results in increased resting metabolic rate (339).
4. Regulation via PVN AT1R
The PVN is a major integrating center for cardiovascular and metabolic signaling. Elimination of AT1R expression in the PVN using viruses expressing siRNAs targeting AT1R mRNA impairs the blood pressure increase caused by subcutaneous ANG II (143). The pressor actions of ANG II are mediated in part by N-methyl-d-aspartic acid (NMDA) receptors in the PVN, as PVN-specific deletion of NMDA receptor GluN1 subunit blunts pressor actions of systemically infused ANG II (321). This is consistent with the activation of NMDA-evoked inward currents in PVN neurons induced by ANG II and oxidative stress (1105).
According to one study, AT1Rs are predominantly expressed in parvocellular neurosecretory neurons in the PVN, and optogenetic stimulation of these neurons causes elevated blood pressure and activation of the hypothalamic-pituitary-adrenal and hypothalamic-pituitary-thyroid axes (225). Conceptually, the study implicates this specific AT1R-containing neuronal population in the response to stress. In another study by the same authors, loss of AT1R in the PVN decreased inflammation in the hypothalamus, augmented weight gain by increased food intake and reduced energy expenditure, and simultaneously decreased blood pressure, suggesting a complex interplay between the myriad of physiological effects regulated by ANG II/AT1R (223).
More recently, neuroglial signaling through AT1R-containing astrocytes in the PVN has been identified as a key mediator of ANG II-mediated sympathoexcitation (1001). This is conceptually important, as the study implicates AT1R on astrocytes in the PVN rather than direct action on PVN neurons themselves (686). Similarly, a link between AT1R and TLR4 in medicating microglial activation in the PVN has been reported (79). Interestingly, AT1R is upregulated in glial astrocytes after myocardial infarction, and glial-cell specific deletion of AT1R blunted sympathoexcitation in response to myocardial infarction (417). These studies implicate both neuroglial signaling and AT1R expression and action in cells other than neurons in the brain as mediating some of the pathological action of ANG II.
C. ANG II Signaling in Cognitive Dysfunction, Dementia, and Alzheimer’s Disease
A growing interest in brain research is the role and mechanism that RAS plays in the development of cognitive dysfunction, dementia, and Alzheimer’s disease (705, 1155). Much of what is known is based on hypertension as an independent risk factor for cognitive decline and dementia. Although robust mechanistic human data are scant, it has been generally assumed that elevated ANG II is detrimental because RAS blockade, particularly by the ACE inhibitors, reduces cognitive decline in the elderly. ACE inhibitors which cross the blood-brain barrier are thought to reduce cognitive decline through anti-inflammatory actions that are independent of blood pressure lowering effects, whereas those that act peripherally (cannot cross the blood-brain barrier) act through lowering blood pressure (891). Moreover, ARBs are effective in preventing many risk factors for and are potential therapeutic agents for Alzheimer's disease (894).
Chronic ANG II activation decreases cognitive function in animals (415) and synaptic plasticity in mice through activation of p38MAPK (202). Hippocampal ANG II is elevated in animal models of postoperative cognitive dysfunction and promotes NF-κB signaling and alterations in MMP/TIMP ratio, which contribute to impaired blood-brain barrier integrity (586). Central neuroinflammation often involves activation of microglial cells. AT1R participates in microglial activation and migration primarily through NADPH oxidase activation, NF-κB activation, and Rho kinase/ROCK induction. Rho kinase/ROCK induction promotes a feed-forward mechanism for NADPH oxidase activation through p38MAPK, and NF-κB activation promotes a feedback mechanism of AT1R upregulation (874). AT1R blockade elicits deactivation of the hypothalamic pituitary adrenal axis and increased BDNF expression in a mouse model of chronic psychological stress (1151). However, the role of BDNF is controversial as increased expression of BDNF is noted in PVN in hypertensive settings, and BDNF administration into the PVN increases mean arterial pressure (262).
ANG II signaling through AT1R promotes neuroinflammation in a mouse model of Alzheimer’s disease termed 5X familial AD (5XFAD). ICV infusion of ANG II impaired cognitive function in 5XFAD mice, which was associated with hippocampal inflammation, oxidative stress, and increased amyloid-β deposition (1029). Telmisartan prevents neuroinflammation by suppression of lipopolysaccharide-induced NO production from inducible NO synthase, and TNF-α and IL-1β secretion by BV2 microglia. Telmisartan is also protective against amyloid burden and glial activation in vivo (1056). Whereas ANG II increased amyloid-β protein in a transgenic mouse model of Alzheimer’s disease (mice expressing human amyloid precursor protein bearing specific mutations), deletion of AT1R inhibited amyloid-β protein generation and amyloid plaque formation (599). Transcriptomic analysis of primary neuronal cultures suggests AT1R inhibition may also provide neuroprotection independent of effects on blood pressure (257).
D. ANG II Signaling in Brain Ischemia and Stroke
While direct mechanistic findings regarding a concrete role for ANG II in stroke and ischemia-related events remain elusive, there have been a few reports indicating a direct correlation between ANG II signaling, stroke susceptibility, and patient outcomes. For a detailed review on ANG II and AT1R signaling in ischemic brain damage, please see Reference 392. In addition, the ANG (1–7)-Mas axis is an active area of investigation for the treatment of stroke (73, 74).
ANG II vaccination induces the production of anti-ANG II antibody, reduces Nox2 mRNA, and promotes an antioxidant environment, reducing infarct volume in a model of permanent middle cerebral artery in rats (1100). AT2R agonism with CGP42112 reduced infarct volume, and induction of the proinflammatory cytokines, TNF-α and IL-1β, in a rat model of middle cerebral artery occlusion, whereas the opposite occurred with AT2R blockade (622). Activation of AT2R with C21 accelerated recovery from middle cerebral artery occlusion through a mechanism that may require VEGF upregulation and mTORC1 signaling (661). In another study, C21 was similarly effective in wild-type mice but had no effect in AT2R-deficient mice (933).
AT1R antagonism with olmesartan in 5XFAD mice, a model of Alzheimer's disease, protects from ischemia-induced cognitive decline after transient bilateral common carotid artery occlusion. This was associated with a protection of neurons and the blood-brain barrier, and a reduction of oxidative stress within the hippocampus (738). This finding is consistent with studies in transgenic mice overexpressing AT1R showing increased susceptibility to strokelike injury caused by injection of ET-1 into the striatum (426). Moreover, in the two transgenic models studied, there was a worsening of the phenotype in the model with high levels of AT1R expression. On the contrary, AT1R deletion was protective against ischemia-induced pathophysiology (733). Furthermore, pretreatment of stroke-resistant spontaneously hypertensive rats with telmisartan before transient middle cerebral artery occlusion is protective against stroke-accelerated neuroinflammation (507). Overall, clinical and animal studies have pointed towards a benefit in using AT1R antagonists in the treatment of ischemic stroke (35), although this is not without controversy (909) and thus the need for additional mechanistic insight.
VIII. ANG II SIGNALING IN ADIPOCYTES AND METABOLIC DISORDERS
A link between RAS signaling and metabolic disorders was first identified by randomized clinical studies that demonstrated an increased incidence of new-onset diabetes in patients treated with β-blockers compared with those treated with ACE inhibitors or ARBs (199, 354). Treatment of rats with the ACE inhibitor captopril further demonstrated that ACE inhibition protects against the development of diet-induced obesity and glucose intolerance (221). Moreover, mice deficient in ACE appear to have increased energy expenditure with reduced fat mass and improved glucose clearance (429). Importantly, rodents as well as human adipocyte express AGT, renin, ACE, AT1R, and AT2R constituting the local adipocyte RAS. The adipocyte RAS has been implicated in hypertension, obesity, and metabolic disorders (126). While white adipose tissue is the most abundant source of AGT next to the liver, both white and brown adipose tissue express the local RAS components as in white and brown perivascular adipose tissue (302). A mechanistic confirmation of the role that the RAS plays in adipocyte dysfunction in obesity was obtained through genetic manipulation of AGT in the mouse. Transgenic mice expressing AGT in adipose tissue have increased fat mass (658), whereas mice deficient in AGT show reduced weight gain in response to high-fat diet (659). In vitro studies also indicate stimulation of lipogenesis in 3T3-L1 and human adipocyte by ANG II (446). Mice overexpressing AGT in white adipose tissue (via aP2 promoter) exhibited systemic insulin resistance and adipose tissue inflammation (450). Adipocyte deficiency of AGT does not alter body weight or fat mass in mice fed with normal diet but reduce systolic blood pressure (1203) and prevents high-fat diet-induced hypertension (1202). However, hepatocytes appear to be the major source of systemic AGT regulating blood pressure in both lean and obese mice (996, 997, 1204). With regard to the ANG II receptor, one study reported that mice lacking the AT2R have reduced adipose cell size and are protected from diet-induced obesity and insulin resistance (1229). However, a recent study with detailed metabolic analyses in AT2R-deficient mice found metabolic dysfunction in white adipose tissue (756). As indicated above, inguinal adipocyte AT2Rs oppose the induction of uncoupling protein-1 (UCP1) production by norepinephrine, suggesting a brain-adipose axis regulated by AT2R (595). These clinical and experimental observations have sparked intensive research into the mechanisms by which the RAS regulates whole body metabolism and insulin sensitivity.
Whole body energy homeostasis is a complex phenomenon involving many organ systems cooperating to orchestrate food intake and nutrient distribution, storage, and metabolism. Alterations of the RAS with ANG II signaling have been reported in each of the major tissues involved in energy homeostasis, i.e., the brain, digestive tract, liver, pancreas, adipose tissues, muscle, and cells of the immune system. The reader is encouraged to visit recent reviews on the role that the RAS of the brain (173), skeletal muscle (373), heart (89, 1145), and microcirculation (721) play in the control of energy balance and overall metabolism. Additionally, others have recently reviewed the role of RAS in glucose homeostasis and insulin resistance (273, 539, 647, 650, 1078).
A. Molecular Mechanisms Mediating RAS-Induced Insulin Resistance
Although the role that the RAS plays in cardiovascular insulin resistance has been extensively reviewed, the molecular mechanisms behind insulin resistance remain incompletely understood. It is clear though that cross-talk between insulin and ANG II (285, 1089) can modulate the final outcome of the action of insulin. Studies demonstrating a close connection between insulin resistance and cardiovascular disease support the notion that alterations within the RAS could be related to dysregulation of the action of insulin (240, 538, 1271). Mechanistically, ANG II negatively regulates several steps of the insulin-signaling cascade via AT1R (FIGURE 12), including insulin-induced phosphorylation of the insulin receptor (IR), IR substrate-1 (IRS-1), and activation of Akt by phosphoinositide 3-kinase (PI3K) (31, 493, 729). ANG II also directly interferes with the AMPK signaling cascade causing insulin resistance (956), as well as indirectly via inhibition of adiponectin secretion (298). The inhibition of AMPK signaling and subsequent insulin resistance caused by ANG II likely involves SIRT3 dysfunction in skeletal muscle (626).
FIGURE 12.

ANG II signaling in insulin resistance. ANG II negatively regulates the insulin signaling cascade at multiple steps, including IRS-1, PI3K, and Akt, causing insulin resistance. ANG II also inhibits the AMPK signaling cascade, which together with the associated reduction in Sirt3 expression and resultant mitochondrial oxidative stress, contributes to insulin resistance. Furthermore, ANG II indirectly causes insulin resistance via induction of pro-inflammatory cytokines, as well as by suppression of adiponectin secretion.
Interestingly, though, cytokines have been implicated in mediating the effects of ANG II on insulin resistance (770, 807), and in states of obesity with insulin resistance cytokines are largely produced by dysfunctional adipocytes (394). Accordingly, here we review the most recent studies, which advance our understanding of the signaling mechanism(s) through which the RAS primarily affects adipocyte function.
B. Roles of ANG II Receptor Signaling in Adipose Tissue
Data discussed in the review articles referenced above demonstrate that ANG II has a powerful metabolic effect on adipose tissue in experimental animal models and human obesity. During chronic positive energy balance, adipose tissue expansion occurs by adipocyte hypertrophy (enlargement of adipocytes) and hyperplasia (adipogenesis). A reduced adipogenic capacity leads to adipocyte hypertrophy associated with dysfunctions of adipose tissue endocrine function, which can lead to metabolic derangements. Formation of new adipocytes (adipogenesis) is, in fact, a physiological adaptive response to overnutrition aimed at maintaining an overall healthier adipose tissue because newly differentiated adipocytes have better energy storage capacity, thus remaining more insulin-sensitive (204). Hypertrophic adipocytes, instead, are less insulin-sensitive (518). Recent data have emphasized a role for ANG II in opposing adipogenesis. Studies into the intracellular mechanisms involved in the anti-adipogenic response of human preadipose cells from omental fat exposed to ANG II revealed a role for the ERK1/2 pathway (297). AGT silencing via shRNA results in reduced preadipocyte differentiation and reduced lipid accumulation (1201). Data in the literature support the existence of counteracting interplay between ACE2/ANG (1–7)/Mas and ANG II/AT1R/ERK signaling upon adipogenesis (1047). Recent studies have revealed that the AT2R inhibits adipogenic differentiation in murine mesenchymal stem cells (MSC) (669), a major source of adipocyte generation. This inhibitory effect of the AT2R was associated with activation of Wnt10b/β-catenin signaling (669), an important regulator of mesenchymal stem cell fate (917) necessary for the adipogenic differentiation of preadipocytes (878). In particular, Wnt10b is considered to act as a brake on adipogenic differentiation by activating the canonical Wnt signaling pathway that leads to stabilization of cytosolic β-catenin (878). In line with these data, adipocyte size in obese Zucker rat is reduced following long-term treatment with an ARB (722).
Signaling through the AT1Rs/AT2Rs, ANG II activates different calcium signaling pathways in adipocytes, an area of investigation that still remains poorly characterized (237). This study first demonstrated that ANG II initiates periodic Ca2+ oscillations and transient responses by activating AT1Rs/AT2Rs and involving branched signaling cascades. Since the Са2+ signaling system participates in lipogenesis, lipolysis, adipokine secretion, proliferation and differentiation of the white fat adipocytes, studies of Са2+ signaling may uncover novel therapeutics in hypertension, obesity, and type 2 diabetes. Related to Са2+ signaling, it has been shown that ANG II/AT1R signaling activates the calcium-dependent protease calpain in the visceral fat microcirculation of the mesentery of rats and mice (922). Activation of calpain causes inflammatory leukocyte trafficking in the splanchnic microcirculation via upregulation of endothelial expressed cell adhesion molecules (993). Interestingly, there is evidence of increased calpain activity in the Zucker diabetic fatty rat (992), a rat model of obesity and type 2 diabetes in which inhibition of ANG II signaling improves insulin resistance and glucose hemostasis (743).
Another important aspect of ANG II signaling in adipocytes is related to the adipocyte production of inflammatory mediators and chemoattractant that cause low-grade adipose tissue inflammation, and obesity and insulin resistance are closely associated with a state of low-grade inflammation in adipose tissues (394). Data demonstrate that ANG II enhances TNF-α-induced MCP1 expression in 3T3-L1 preadipocytes via a ERK1/2- and p38MAPK-dependent pathways (37). An ARB with PPARγ receptor agonist activity, telmisartan, attenuated the release of the proinflammatory factors secreted from 3T3-L1 adipocytes and improved lipid metabolism possibly via activation of the MAPK pathway and upregulation of eNOS/NOS3 and carnitine palmitoyltransferase 1α. Telmisartan also reduced lipid storage and increased glucose uptake in 3T3-L1 adipocytes (455). Silencing of AGT also reduced markers of lipid accumulation and inflammation in cultured adipocytes (125). Specifically, AGT gene silencing in 3T3-L1 adipocytes significantly reduced the intracellular level of pro-inflammatory adipokines, including MCP1, IL-6, and TNF-α. In the same study, microarray analyses identified that AGT gene silencing decreased expression of several genes involved in adipose inflammation including Saa3, Nod1, Stat1, and CXCL12. Saa3, which encodes for the acute phase protein-serum amyloid A3, was recently identified as a critical proinflammatory adipokine involved in obesity-associated metabolic disorders (1195). Others have found that several other chemokines were altered by AGT inactivation. These include CXCL12 and Ptx3. CXCL12 is mainly expressed in stromal cells, but is also detected in 3T3-L1 adipocytes (166). In line with these studies, palmitic acid, a common saturated fatty acid, can stimulate upregulation of local RAS (ANG II secretion and AT1R) in 3T3-L1 adipocytes, via TLR4 and NF-κB signaling (1013).
In vivo data have confirmed the impact of ANG II signaling on adipose tissue lipid metabolism. Activation of the AT2Rs has been shown to promote adipocyte differentiation and restore adipocyte size in high-fat/high-fructose diet-induced insulin resistance in rats (961). In particular, stimulation of AT2R was found to restore the adipocyte cell size and to reduce the insulin resistance induced by high-fat/high-fructose diet. Therefore, AT2R agonists may be an attractive therapeutic tool for patients with metabolic complications of obesity. Putnam et al. (840) have instead emphasized a role for adipocyte differentiation in the absence of changes in lipid uptake and storage. They, in fact, demonstrate that mice with adipocyte AT1AR deficiency develop striking adipocyte hypertrophy even though total fat mass is not different from wild-type mice.
Recent literature has emphasized a role for the RAS in the “browning” of white adipose tissue, and browning of white adipose tissue has been highlighted as a new possible therapeutic target for obesity, diabetes, and lipid metabolic disorders, because white adipose tissue browning could increase energy expenditure and reduce adiposity. Interestingly, deletion of ANG II receptor subtypes, especially the AT1AR induces white-to-beige fat conversion in vivo (1069). Therefore, it is possible that blockade of AT1R may be useful for the treatment of obesity and metabolic syndrome by enhancing adipocyte browning.
C. Roles of (Pro)Renin Receptor Signaling in Adipose Tissue
Human adipose tissue synthesizes the (pro)renin receptor, with increased expression in visceral adipose tissue. Binding of this receptor by prorenin or renin induces an increase in the catalytic efficiency for AGT conversion to AngI and an intracellular signal with phosphorylation of serine and tyrosine residues associated with an activation of MAPKs, ERK1 and ERK2. Specifically, the binding of renin or prorenin to (pro)renin receptor in 3T3-L1 preadipocytes initiates an intracellular signaling cascade associated with the activation of the ERK1/2 pathway (7). Data also show that association of postnatal overfeeding and high-fat diet increased plasma renin activity and adipose (pro)renin receptor expression (8). Although the mechanism through which overfeeding increases (pro)renin receptor expression remains unclear, such phenomenon could explain, at least in part, the associated disproportionate adipocyte hypertrophy and its accompanying increased glucose intolerance. In line with this concept, Tan et al. (1038) have demonstrated that administration of the handle region peptide (HRP), a (pro)renin receptor blocker, decreases body weight gain and visceral adipose tissue (VAT) in high-fat/high-carbohydrate diet-fed mice. Data from this study also suggest that the effects of (pro)renin signaling are different in VAT versus subcutaneous adipose tissue (SAT) since blockade of the (pro)renin receptor normalized adipocyte size in VAT while increasing adipocyte size in SAT. These differences were linked to the upregulation of both lipogenesis and lipolysis in subcutaneous fat promoting a triglyceride (TG)/free fatty acid (FFA) futile cycling, similar to what was reported in rats treated with ACE inhibitor or ARB (1279). The activation of TG/FFA cycling by (pro)renin receptor signaling in SAT could also affect “beiging”. Interestingly, blocking of the (pro)renin receptor increased PRDM16 and PGC-1α mRNA levels (1038), which suggests that activation of “beiging” of SAT could also contribute to the reduced body weight gain observed in mice receiving (pro)renin receptor blockers (1039).
The role of adipose tissue (pro)renin receptor gene deletion on energy balance has been studied in adipose tissue specific (pro)renin receptor knockout mice, which have decreased body weight and fat mass (943). In addition to a reduction of adipose tissue mass, deletion of adipocyte-(pro)renin receptor led to an increase in lipid deposition in liver, suggesting lipodystrophy accompanied by liver steatosis (1157). In vitro data further revealed that (pro)renin receptor silencing significantly decreases PPARγ and Fabp4 expression, thus suggesting that (pro)renin receptor is a master regulator of adipocytes differentiation (1157). Because fatty acid-binding proteins are important carriers for fatty acid uptake and fatty acid transport to sites of esterification into triglycerides (1277), the lipodystrophy phenotype seen in mice deficient in (pro)renin receptor also suggests an important role of (pro)renin receptor in fatty acid trafficking and storage in adipocytes. Overall, emerging evidence suggests that adipose tissue (pro)renin receptor is involved in the development of obesity and its associated complications. This indicates that adipose tissue (pro)renin receptor may be an important target for the development of new drugs for the treatment of complicated obesity.
D. Roles of ACE Signaling in Adipose Tissue
A paucity of data exists in the literature to support a primary role for ACE in the regulation of adipose tissue function in health and disease. It has been demonstrated that an ACE inhibiton has beneficial effects on adipose tissue function that are associated with improved insulin sensitivity (299). ACE gene insertion/deletion polymorphism has also been associated with obesity in the Turkish population (17). Data in the literature have demonstrated that ACE expression levels are comparable among different fat depots (302). Interestingly, it has been demonstrated that ACE has unique signaling mechanism independent from its enzymatic activity. ACE inhibitor binding to ACE leads to phosphorylation of ACE at Ser1270 via CK2 and c-Jun-dependent gene regulation in endothelial cells (283). Accordingly, it remains difficult to dissect the effects of ACE signaling from those obtained by blockade of its downstream mediator ANG II or stabilization of bradykinin. Nevertheless, ACE inhibition has been shown to enhance circulating adiponectin levels in vitro and in vivo and potentiate the adiponectin-enhancing effect of the PPARγ agonist rosiglitazone in ob/ob mice (504). In this study, the signaling cascade underlying this effect was linked to the expression of CRBP1, which was identified as a gene regulated by the ACE signaling cascade, as it was attenuated in cells expressing the ACE S1270A mutant. Increases in the CRBP1 protein led to the upregulation of adiponectin most probably via activation of retinol-dependent nuclear receptor proteins (RAR/RXR) and cross-activation of PPARs that are activated via heterodimerization with RAR/RXR. Similar results were confirmed in humans, where plasma adiponectin was increased dramatically as a result of ACE inhibition and ARB treatment (1051). It has been also shown that adipocyte-derived lipids increase ACE expression via AMPK signaling in macrophages (505). Further studies are necessary to understand how alterations in lipid composition of adipocytes affects the RAS expression and the signaling activities so as to contribute to metabolic disorders including obesity and type 2 diabetes.
IX. ANG II SIGNALING IN AGING
A definitive link between RAS and longevity was first established by Benigni et al. (70) by demonstrating that disruption of the AT1AR promotes longevity in mice, possibly through the attenuation of oxidative stress and induction of pro-survival genes. On the basis of data obtained in Caenorhabditis elegans and humans, three major mechanisms of aging and senescence have been postulated: the free radical theory (358), programmed cellular senescence (39, 745), and the inflammaging (184). ANG II has been shown to induce premature senescence in VSMCs (522). A study demonstrated that ANG II causes DNA damage and telomere-independent acute stress-induced premature senescence as well as telomere attrition-dependent chronic replicative senescence, and both mechanisms require AT1R-mediated ROS production in VSMCs (374). Nox1-derived ROS appears to mediate ANG II-induced VSMC senescence (1068). Importance of intracellular zinc in ANG II-induced senescence via Nox1 induction has also been demonstrated in VSMCs (804, 904). The role that activation of the RAS plays in free radical production and cardiovascular inflammation has been discussed elsewhere in this review. Accordingly, this section will focus on the role that RAS plays in cellular senescence.
Alteration in certain genes as well as treatments with compounds targeting genes have been shown to extend the lifespan of Caenorhabditis elegans, Drosophila, and mice, suggesting that signaling cascades actually regulate cell senescence and overall aging (359, 397, 1154). In the past decades, three major signaling cascades have been extensively studied in cellular senescence (745): 1) the insulin/IGF-I pathway, 2) TSC/mTOR pathway, and 3) sirtuins. As the insulin/IGF-I pathway is described elsewhere in this review, here we will discuss the effect that RAS signaling has on the TSC/mTOR and sirtuin pathways. It should be noted that the TSC/mTOR pathway is closely related to macromolecular degradation and inflammation-induced aging, termed as inflammaging (292). The signaling pathways required for ANG II enhancement of aging are illustrated in FIGURE 13. In addition, notable antiaging mechanisms and the ANG II signaling mechanism of aging-related muscle wasting will be described.
FIGURE 13.
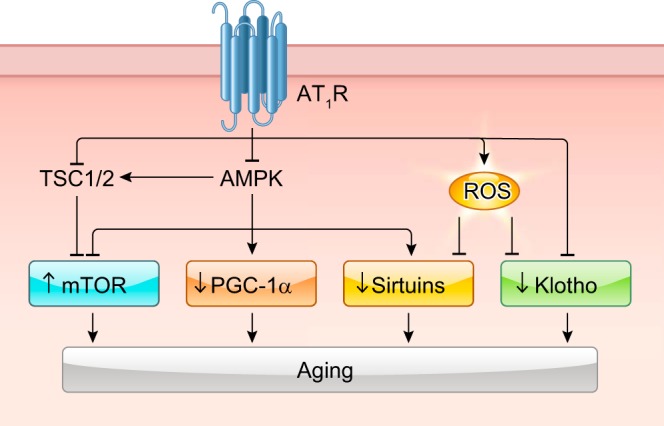
ANG II signaling in aging. ANG II stimulates mTOR pathway by inhibiting TSC1/2. ANG II also inhibits the PGC-1α, sirtuin, and Klotho pathways via inhibition of AMPK or stimulation of ROS production.
A. mTOR Pathway, ANG II, and Aging
The serine/threonine kinase mTOR is highly conserved from yeast to humans. In general, mTOR regulates nutrient and growth factor signaling (536). However, mTOR signaling appears to have a negative impact on longevity. In Caenorhabditis elegans, the loss of mTOR signaling extends lifespan by 2.5-fold, which is independent of the insulin/IGF-I signaling pathway (1088). Moreover, functional inhibition of multiple mTOR substrates leads to an extended lifespan in worms (144). Several studies have confirmed that mTOR signaling pathways are involved in longevity in mice (458).
Evidence indicating a role for the RAS in mTOR signaling in aging is mostly circumstantial and derived from aging-associated disease conditions (219). Recent data show that inhibition of ANG II signaling with the ACE inhibitor enalapril started late in life mitigates several adverse age-related changes in skeletal muscle, including increased oxidative stress and proinflammatory signaling via a NO-dependent mechanism. However, mTOR protein levels are increased by enalapril (657).
B. Sirtuin Interference by ANG II in Aging
The sirtuin pathway was originally identified in yeast as a regulator of lifespan (449). Since then, a large body of work has demonstrated that sirtuins play an important role in mediating aging and age-related diseases like cancer, inflammation, cardiac function, and cognition through the regulation of diverse molecular/cellular process, including genomic stability, senescence, DNA repair, mitochondrial function, metabolic homeostasis, and stem cell exhaustion. In general, it is accepted that activation of sirtuins by agonists like resveratrol promotes longevity by inducing physiological adaptations and changes in gene expression that are similar to dietary restriction (372). Aging-associated diseases, such as atherosclerosis, obesity, type II diabetes, and neurodegeneration (Alzheimer’s and Parkinson’s disease), also correlate with SIRT2 polymorphisms (827, 828). Thus understanding how systemic regulators of multiple organ functions such as the RAS impact on function of sirtuins is of value, and indeed, recent studies have identified a link between sirtuins and RAS signaling in aging. In the first study to definitively define the role of AT1R in aging, deletion of AT1AR was associated with upregulation of nicotinamide phosphoribosyltransferase (Nampt) and SIRT3 in the kidney. In SIRT3-deficient mice, ANG II-induced cardiovascular pathology, but not hypertension, was markedly enhanced. The phenotype was associated with cardiac mitochondrial dysfunction. This is because SIRT3 inhibition suppresses Pink/Parkin-mediated mitophagy and impairs angiogenic capacity in endothelial cells (1135). Regarding SIRT1, a recent study demonstrates that in the mouse, caloric restriction protects against aortic aneurysm formation induced by ANG II via a SIRT1-dependent mechanism in vascular smooth muscle (604). Although ANG II infusion did not significantly affect SIRT1 expression, ANG II treatment decreased SIRT1 deacetylase activity and NAD+ availability in the aortas. As mentioned in the vascular section, ANG II-induced vascular remodeling and hypertension in mice were attenuated by overexpression of SIRT1 (306). It has also been shown that raising (NAD+)-dependent SIRT1 activity attenuates ANG II-Induced senescence in VSMCs (563).
C. Anti-Aging Effects by Klotho, PGC-1α, ATRAP, ATIP, AT2R, and Mdm2
Klotho gene mutations have been associated with aging. Transgenic mice that overexpress Klotho live longer than wild-type mice (527). Klotho overexpression also suppresses ANG II-induced Nox2 protein expression through a cAMP-PKA pathway and prevents VSMC oxidative damage and cellular senescence (1127). The transcriptional factor nuclear factor erythroid 2-related factor 2 (Nrf2) mediates antioxidant cell defense. Soluble klotho protected against ANG II-mediated human aortic VSMC apoptosis and senescence via activation of Nrf2 (641).
PGC-1α, a target of AMPK, regulates mitochondrial homeostasis, and mice deficient in PGC-1α have enhanced ANG II-induced vascular inflammation and cell senescence that is largely dependent on mitochondrial ROS production (517). ANG II promotes PGC-1α acetylation thereby inhibiting its cotranscriptional activity and Fox-O1-SIRT1 signaling. Suppression of SIRT1 activity in turn decreases catalase expression resulting in increased intracellular ROS (1173).
The regulation of the AT1R through ATRAP is also a critical component to the VSMC senescent phenotype. ATRAP promotes AT1R internalization, along with reductions in CAML, calcineurin and NFAT induced-p53 expression, thus preventing VSMC senescence (700). Stimulation of AT2R upregulates of methyl methanesulfonate-sensitive 2, a DNA repair factor, in VSMCs and prevents ANG II/AT1R-induced senescence (701). In line with this finding, AT2R-interacting ATIP overexpression attenuates VSMC senescence with upregulation of methyl methanesulfonate-sensitive 2 and Src homology 2 domain-containing protein-tyrosine phosphatase 1 (699).
Mdm2 (mouse double minute 2 homolog) is an E3 ubiquitin-protein ligase, which protects against senescence by promoting p53 degradation. ANG II increases SM22α in VSMC in vitro and in vivo, which attenuates Akt-dependent Mdm2 phosphorylation and p53 degradation leading to VSMC senescence (694).
D. ANG II Signaling in Promoting Skeletal Muscle Atrophy
Skeletal muscle shows functional and physiological declines with aging resulting in skeletal muscle wasting and atrophy. Muscle atrophy (cachexia) also frequently associates with several pathological conditions including heart failure, diabetes, cancer, and renal failure. In these conditions, enhanced RAS appears to be critical in mediating muscle wasting. Specifically, ANG II causes skeletal muscle proteolysis through the ubiquitin-proteasome system via elevation of ROS derived by NADPH oxidase as well as mitochondria (1007, 1213). Additionally, losartan was found to combat disuse atrophy in muscle sarcopenia (106). The protective mechanism appears to involve the activation of the IGF-I/Akt/mTOR pathway. Furthermore, AT1AR-deficient mice show a lack of an age-related decline in skeletal muscle function and enhanced regenerative capacity after injury through suppression of the AT1R/C1q-Wnt/β-catenin pathway (1183). However, there is a conflicting report regarding the regenerative capacity of AT1AR KO mice. A previous study showed enhanced muscle function in AT1AR KO mice despite the presence of reduced muscle mass, but these mice showed a decline in muscle reparative capacity following injury (727). The explanation for the discrepancy between these two studies regarding the regenerative capacity of AT1AR KO mice skeletal muscle is currently unknown.
In skeletal muscle satellite cells, proliferation is also inhibited by ANG II through AT1R-dependent Notch and MyoD suppression (1210). In contrast, AT2R stimulation leads to myoblast differentiation and skeletal muscle regeneration (1211). The AT2R-mediated muscle regeneration capacity seems blocked in congestive heart failure via suppression of AT2R gene transcription (1209). Muscle RING-finger-1 (MuRF1) is a muscle specific E3 ubiquitin ligase and mediator of muscle atrophy, and ANG II induces its expression. This signaling cascade involves ANG II/AT1R-induced PKD1 activation, which relieves HDAC5-mediated inhibition of TFEB resulting in TFEB-dependent MuRF1 promoter activation. PKD1 KO mice are unable to induce MuRF1 expression in response to ANG II and are spared from ANG II-dependent skeletal muscle atrophy (245). Atrogin-1 is another E3 ubiquitin-ligase that can be increased in skeletal muscle upon stimulation of ANG II. ANG II promotes Akt dephosphorylation and activation of Foxo which enables transcriptional upregulation of Atrogin-1. This signaling cascade can be blocked with IGF-I administration (1212).
Similar to cardiovascular aging mechanisms, AMPK plays a role in mitochondrial maintenance and skeletal muscle homeostasis. ANG II induces PP2Cα, a negative regulator of AMPK, with downstream phosphorylation inhibition of AMPK and reduced PGC-1α expression. Suppression of AMPK/PGC-1α signaling decreases NRF1, TFAM, mitochondrial complex IV activity, and ATP production, resulting in mitochondrial dysfunction associated with increased mitochondrial ROS. Knockdown of PP2Cα normalized AMPK activity, PGC-1α, NRF1, and TFAM levels and blocked ANG II inhibition of autophagy-regulating kinase ULK1, leading to improved mitochondrial biogenesis/recycling/function, energy production, and inhibition of ANG II-induced wasting (1022).
Overall, ANG II/AT1R promotes every aspect of the cardiovascular aging and is a major player in the signaling cascades leading to skeletal muscle atrophy. The cardiovascular system and skeletal muscle share similar signaling mechanisms activated by ANG II that promote their aging phenotype, including inflammatory activation, Nox induction, mitochondrial dysfunction, and decline in SIRTs activity.
X. ANG II SIGNALING IN IMMUNE CELLS AND INFLAMMATION
As described elsewhere, ANG II has been recognized to promote proinflammatory responses in target organs via AT1R expressed in both immune and nonimmune cell types. Induction of inflammatory signaling enhances pathophysiology of various CVDs as mentioned in previous sections. For example, monocytes and macrophages play an important role in ANG II-induced hypertension and end organ damage. MCP1 mediates monocyte recruitment to the site of inflammation. Bone marrow deletion of MCP1 receptor CCR2 diminished ANG II-induced vascular inflammatory responses, vascular hypertrophy, and vascular fibrosis, but not cardiac hypertrophy (418). ANG II-induced hypertension was also attenuated in CCR2 knockout mice (589). Bone marrow deletion of MMP2 also prevented ANG II-induced hypertension in mice (53). Depletion of lysosome M (LysM), which is expressed by activated monocytes and macrophages, attenuates blood pressure elevation, endothelial dysfunction, and vascular ROS production induced by chronic ANG II infusion (1141). A subsequent study demonstrated that ANG II infusion increases LysM+ monocytes, enhances iNOS expression, and uncouples eNOS, resulting in aggravated vascular nitro-oxidative stress (510).
In this section, we describe signal transduction pathways elicited by ANG II in distinct leukocyte populations together with their functional significances including those in gut inflammation and microbiota. The readers are advised to refer to previous sections for the ANG II signaling mechanisms of immune cells in vasoconstriction and ROS (see sect. IVA6), arterial stiffness (see sect. IVC4), atherosclerosis (sect. IVD3), AAA (see sect. IVE1), cardiac fibrosis (see sect. VB6 and TABLE 8), and renal fibrosis/inflammation (see sect. VID). Please also refer to recent review articles for ANG II mechanisms involved in regulation of immune cells such as those in vascular inflammation (648), hypertension (679, 882, 1142), and cardiac hypertrophy (295). In addition, it has been demonstrated that ANG II infusion stimulates proliferation, differentiation, and activation of hematopoietic stem cells (495). The roles of immune cell subtypes implicated in ANG II pathophysiology described in the previous sections and this section are summarized in TABLE 9 (For cardiac fibrosis, see TABLE 8).
Table 9.
Roles of immune cell subtypes in ANG II pathophysiology
| Cell Type | Pathophysiology | Model | Mediators | Reference Nos. |
|---|---|---|---|---|
| Macrophages | Hypertension ↓ | IL-1R1−/− | Renal NKCC2 | 1249 |
| VSMC proliferation ↓ | Clodronate | C1q/β-catenin | 1010 | |
| AAA ↓ | apoE−/− Notch+/− BMT | Notch1 | 353 | |
| AAA ↑ | LDLr−/− Nox2−/− BMT | ROS/IL-1β | 490 | |
| Intracranial aneurysm ↓ | Clodronate | MCP1 | 453 | |
| Cardiac inflammation ↑ | Atg5+/− | Autophagy/ROS | 1265 | |
| Renal fibrosis (UUO) ↑ | AT1ARf/f RysM-Cre | M1 cytokines/IL-1R | 1250 | |
| Cognitive dysfunction ↓ | Clodronate or AT1AR−/− BMT | Nox2/ROS | 271 | |
| T cells | Hypertension ↓ | AT1AR−/− T cell transfer | Nox/ROS/TNF-α | 348 |
| Hypertension ↓ | CTLA4-Ig, B7(CD80/86)−/− | Inflammation | 1093 | |
| Hypertension ↓ | IL-17−/− | ROS/cytokines | 630 | |
| Hypertension ↓ | AT1ARff CD4-Cre | Th1 cytokines | 1251 | |
| Vascular dysfunction ↓ | Tbx21−/− | IFN-γ/ROS/NK cell | 511 | |
| T cells (CD8+) | Hypertension ↓ | CD8−/−, CD8+ T cell transfer | Kidney T cell R | 1066 |
| γδ T cells | Hypertension ↓ | TCRδ−/− | EC dysfunction | 109 |
| Tregs | Hypertension ↓ | Treg adoptive transfer | ROS/Inflammation | 54 |
| Hypertension/fibrosis ↓ | Treg adoptive transfer | CD39/neutrophils | 263 | |
| B cells | Hypertension ↓ | BAFF-R−/− | IgG/macrophage | 134 |
| Atherosclerosis | apoE−/− BAFF-R−/− | IL-10 Bregs | 830 | |
| NK cells | Vascular dysfunction ↓ | NK1.1 depletion antibody | Inflammation | 511 |
| Neutrophils | Aortic dissection ↓ | Anti-Gr-1 antibody | MMP9 | 525 |
| MDSCs | Hypertension ↑ | MDSC depletion | H2O2 | 940 |
However, caution is needed when interpreting these findings. Many of the strategies utilized remove a specific subset of immune cells. It may be difficult to conclude if the outcomes are due to ANG II signal transduction in the immune cell, if the cell function lay downstream of signal transduction elicited in nonimmune cells, or removing the specific cell type is affecting the phenotype indirectly. AT1R deletion in bone marrow cells augmented hypertension and renal inflammation in mice (191) and did not protect against atherosclerosis in apoE−/− mice infused with ANG II (503). A few studies are available utilizing immune cell targeted conditional AT1R knockout mice demonstrating protective roles of immune cell AT1R (see below).
A. ANG II Signaling in Macrophages
Macrophage infiltration caused by ANG II infusion has been implicated in target organ damage such as seen in kidney (787). Stimulation of mouse macrophage cell line RAW264.7 cells by ANG II causes both NF-κB and AP1 activation, ROS production, and secretion of inflammatory cytokines including TNF-α, IL-1β, IL-6, and IL-10 through AT1R (346). AT1R-dependent IL-6 production has been confirmed in rat lung macrophage-derived cell line, NR8383 (763). As mentioned in section IIIA1, macrophage depletion and C1qa gene deletion attenuated ANG II-induced β-catenin signaling and arterial remodeling (1010). In addition, a recent study demonstrated that AT1R-mediated Nox2-dependent ROS production in perivascular macrophages contributed to cognitive dysfunction associated with hypertension in mice (271). However, macrophage AT1R may be reno-protective (1250) as described in the kidney section.
B. ANG II Signaling in Dendritic Cells
ANG II has been shown to activate dendritic cells via NF-κB activation (145). ANG II induces IL-23 by in dendritic cell /natural killer cell coculture system, which is dependent on JNK stimulation (1192). As mentioned in section IVA6, ANG II-induced dendritic cell activation has been demonstrated to contribute to hypertension via ANG II-induced T cell proliferation and activation (498).
C. ANG II Signaling in T Lymphocytes
T cells express RAS components including AT1R. T cells also proliferate in response to ANG II (448). As described in the vascular section (see sect. IVA6), T cell AT1R activation by ANG II appears crucial for ANG II causing vascular inflammation, ROS production, and hypertension (348). T cell activation by B7 ligands is also required for ANG II-induced hypertension (1093). In addition, primary cultured T cells produce ANG II, which stimulates production of superoxide and TNF-α via both AT1R and AT2R (387).
T lymphocyte activation by ANG II increases CC chemokine receptor (CCR) expression including CCR1, CCR3, and CCR5, which are receptors for the RANTES chemokine. RANTES knockout mice have blunted vascular leukocyte infiltration in response to ANG II (698). Furthermore, knockout of T cells as well as Nox2 inactivation is protective from ANG II-induced thrombus formation (936). Involvement of T cell SGK1 in ANG II-induced hypertension and end organ damage has also been demonstrated (758). γδ T cells appears to mediate ANG II-induced hypertension, vascular injury, and T cell activation in mice (109). Repeated infusion of ANG II induces memory T cells in mice. CD70 mediates formation of memory T cells, and CD70−/− mice are protected from effector memory T cell accumulation, hypertension, and renal damage upon ANG II infusion (423).
C-C motif chemokine 5 (CCL5), which binds to CCR3, CCR4, and CCR5, drives recruitment of macrophages and T lymphocytes into injured tissues. Unexpectedly, ANG II-induced albuminuria and glomerular injury, but not hypertension or cardiac hypertrophy, was worsened in CCL5-deficient mice. The phenotype was associated with renal macrophage infiltration via enhanced CCL2 generation (883). In addition, T cell AT1R activation in kidney may be reno-protective in hypertension by attenuating polarization of T lymphocytes towards the pro-inflammatory Th1 phenotype (1251). ANG II-induced T cell mobilization also requires generation of sphingosine-1-phosphate (S1P) via S1P-generating enzyme type 2 (SphK2) and S1P receptor S1P1. This is because S1P1 mediates lymphocyte egress and homing. ANG II-induced vascular inflammation, endothelial dysfunction, and hypertension were attenuated in Sphk2−/− mice (684). In addition, renal denervation experiments suggest the importance of renal sympathetic nerve stimulation in ANG II-induced T cell infiltration and end organ damage (1171).
CD4+ T helper cells are subset of T cells important for adaptive immune responses. T helper cells are subclassified to three types based on their cytokine secretion profiles, Th1, Th2, and Th17, which secrete IFN-γ, IL-4, and IL-17, respectively. As described elsewhere, IFN-γ and IL-17 appear to participate in ANG II-induced hypertension (451, 630). In contrast, IL-4 contributes to cardiac fibrosis induced by ANG II, but has no effect on hypertension induced by ANG II (815). These data suggest the distinct involvement of T helper cell subsets in ANG II pathophysiology. However, T-helper 17 cells may also be necessary for protective effects in response to DOCA plus ANG II as IL-17 knockout mice show increased albuminuria, glomerular injury, and renal infiltration compared with wild-type mice, suggesting that IL-17 also serves a protective role (515). Importance of T cell and T cell-derived IL-17A in ANG II-induced hypertension was also confirmed in humanized mice with replaced human immune system (422). Th22 cells are a recently identified subpopulation of Th cells secreting proinflammatory IL-22. ANG II infusion increases Th22 cells and serum IL-22. IL-22 neutralizing antibody decreased blood pressure inflammation and endothelial dysfunction induced by ANG II in mice (1200). CD8+ (cytotoxic) T cells have also been shown to play a critical role in ANG II-induced hypertension. CD8−/− mice are protected from ANG II-induced hypertension, endothelial dysfunction, and vascular remodeling (1066). In CD4-Cre mediated Tcell MR knockout mice, ANG II-induced hypertension and CD8+ cell accumulation in the kidney were blunted. In CD8+ T cells, MR seems to regulate IFN-γ production via NFAT1 and AP1 (1016).
D. ANG II and T Regulatory Lymphocytes (Tregs)
Tregs suppresses innate and adaptive immune responses. Adoptive transfer of Tregs prevented ANG II-induced hypertension, endothelial dysfunction, vascular inflammation, and ROS production, but not cardiac hypertrophy in mice. The vascular effects were associated with reduced T cell infiltration to adventitia and periadventitial fat (54). However, immunosuppression-independent mechanism of tissue protection by Tregs has also been reported. This mechanism involves the direct apoptosis of tissue-resident neutrophils by the ecto-ATP diphosphohydrolase activity of CD39, which protects mice from cardiac and renal fibrosis and hypertension induced by ANG II (263).
E. ANG II Signaling in B Lymphocytes
B lymphocytes play an important role in ANG II-induced blood pressure elevation and aortic macrophage accumulation and TGF-β expression. These effects are attributed to IgG production by B lymphocytes (134). However, infusion of ANG II in B cell replenished ApoE−/− Baffr−/− mice unexpectedly prevented the progression of atherosclerosis (830).
F. ANG II and Natural Killer Cells
Natural killer cells (NK-cells) express RAS components including AT1R. NK-cells also proliferate in response to ANG II (448). As mentioned in the vascular section, mice lacking T‐box, which mediates IFN-γ formation in immune cells, are protected from ANG II‐induced vascular injury (511). In these mice as well as in IFN-γ−/− mice, ANG II-induced NK-cell recruitment in the aortic wall was eliminated. Depression of NK-cells by NK1.1 antibody injection protected mice from vascular dysfunction in response to ANG II infusion (511).
G. ANG II Signaling in Neutrophils
Neutrophil accumulation is one of the earliest inflammatory responses to ANG II stimulation (731). ANG II stimulation of isolated human neutrophils leads to AT1R-dependent ROS production via ERK and p38MAPK activation, and activation of calcineurin and NF-κB (255). ANG II also inhibits Nrf2 nuclear translocation and heme oxygenase 1 expression in neutrophils, whereas these responses are independent of ROS or MAPKs (19). As mentioned in the vascular and cardiac sections, neutrophil activation is crucial for ANG II-induced aortic dissection (525), cardiac fibrosis (1162), and atrial fibrillation (885). Interestingly, ACE knockout mice and mice treated with ACE inhibitor are more susceptible to bacterial infection. Neutrophil ACE is required for ROS production and this effect is AT1R independent (488).
H. Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells and have the ability to suppress T cell activation and limit inflammation. Circulating MDSCs are increased in animal models of hypertension including ANG II infusion in mice. MDSCs appear to act as negative regulators of ANG II-induced hypertension and renal inflammation via H2O2 production (940).
I. ANG II, Gut Inflammation, and Microbiota
Gut microbiota has been implicated in several pathophysiological processes including CVDs via alteration in intestinal as well as systemic immune systems (396). ANG II infusion significantly increased rat microbiota Fimicutes/Bacteroides ratio, which was diminished with minocycline treatment. Interestingly minocycline significantly lowers ANG II-elevated blood pressure in rats (1196). Subsequent study demonstrated ANG II infusion increases bone marrow-derived T cells and macrophage in small intestine and colon, increases sympathetic drive to the gut, and reduces mesenteric blood flow in rats (912). As mentioned, the absence of microbiota in germ‐free mice protects from ANG II-induced vascular ROS production and hypertension, which is associated with reduced MCP1-dependent chemotaxis of myelomonocytic cells (459).
XI. CONCLUDING REMARKS
It has been reported that optimally treated hypertension patients still have 50% greater cardiovascular risk than untreated normotensive subjects (80). The 2017 American College of Cardiology/American Heart Association guideline has provided new recommendations for the definition of hypertension, systolic and diastolic blood pressure thresholds for initiation of antihypertensive medication, and blood pressure target goals. Compared with the past JNC7 guideline prevalence of hypertension among United States adults will increase to 45.6% from 31.9%, and antihypertensive medication will be recommended for 36.2% of adults in the United States (724). While more patients will be treated with ARB and other antihypertensives, the statistics indicate that currently available antihypertensive therapies have certain limitations to sufficiently lower the risk of cardiovascular mortality. Therefore, further efforts are desired to look for novel molecular mechanisms of ANG II causing cardiovascular complications independently from blood pressure regulation, which will provide an alternative therapeutic target. ANG II stands at the center of systemic, various local or tissue-confined, and intracellular systems, which integrate multiple cellular signaling events that broadly have opposing or counterregulatory actions. Perturbation in the balance of these processes can lead to organ dysfunctions and numerous diverse pathologies. Understanding how these processes are regulated and integrated with each other and other physiological systems in real time still remains a formidable challenge. While many studies have taken a “hammer” approach with regard to defining the actions of ANG II, there is ample evidence that its actions are often subtle and both context- and time-dependent, for instance, ANG II-induced modulations of various immune cells. Further studies with cell-specific knockouts and transgenics are needed to unravel the role of innate and adaptive immunity and their interplay in ANG II-induced tissue remodeling. Many questions still remain as well regarding the workings of the local RAS, particularly those of the heart, kidney, and brain, with regard to the interactions among different cell types. The role of ANG II in fetal development and genetic/epigenetic programing impacting on adult-onset cardiovascular disease is an emerging area of study. Another difficult challenge is providing rigorous evidence for the intracellular/intracrine, autocrine, and paracrine actions of ANG II and the receptors affecting organelle communication such as those between mitochondria and nucleus, leading to cardiovascular pathophysiology. Questions remain as to whether the reported intracrine actions of ANG II occur at reasonable levels of expression, and if so, do they occur by activating nuclear, mitochondrial, or other pools of AT1R or AT2R, or by some as yet unidentified means. Finally, a systems biology and bioinformatics approach that integrates elements of genomics, epigenomics, proteomics, and metabolomics is likely to reveal novel, as yet unimagined, aspects of ANG II signal transduction.
GRANTS
S. Eguchi is funded by National Institutes of Health (NIH) Grants RO1 HL128324, R01 HL133248, and R01 DK111042 and American Heart Association Grant 16GRNT30410007. V. Rizzo is funded by NIH Grant RO1 HL128324 and American Heart Association Grant 16GRNT30130013. C. Sigmund is funded by NIH Grants PO1 HL084207, R01 HL125603, and R01 HL131689; American Heart Association Grant 15SFRN23480000; and Roy J. Carver Trust. R. Scalia is funded by NIH Grants RO1 DK096521 and R01 DK111042. S. J. Forrester is supported by a predoctoral fellowship F31HL127971 from NIH. T. Kawai is supported by a postdoctoral fellowship 16POST3051004 from the Great Rivers Affiliate of American Heart Association.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
The creative assistance of Dr. Raffaele Altara with FIGURES 8 and 9 is gratefully acknowledged.
Present address of S. J. Forrester: Department of Medicine, Emory University, Atlanta, GA.
Address for reprint requests and other correspondence: S. Eguchi, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, 3500 N. Broad St., Philadelphia, PA 19140 (e-mail: seguchi@temple.edu).
REFERENCES
- 1.Abada A, Elazar Z. Getting ready for building: signaling and autophagosome biogenesis. EMBO Rep 15: 839–852, 2014. doi: 10.15252/embr.201439076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, Smith BJ, Burks TN, Cohn RD, Fedarko NS, Carey RM, O’Rourke B, Walston JD. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci USA 108: 14849–14854, 2011. doi: 10.1073/pnas.1101507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abbas M, Jesel L, Auger C, Amoura L, Messas N, Manin G, Rumig C, León-González AJ, Ribeiro TP, Silva GC, Abou-Merhi R, Hamade E, Hecker M, Georg Y, Chakfe N, Ohlmann P, Schini-Kerth VB, Toti F, Morel O. Endothelial Microparticles From Acute Coronary Syndrome Patients Induce Premature Coronary Artery Endothelial Cell Aging and Thrombogenicity: Role of the Ang II/AT1 Receptor/NADPH Oxidase-Mediated Activation of MAPKs and PI3-Kinase Pathways. Circulation 135: 280–296, 2017. doi: 10.1161/CIRCULATIONAHA.116.017513. [DOI] [PubMed] [Google Scholar]
- 4.AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J Biol Chem 276: 39721–39726, 2001. doi: 10.1074/jbc.M105253200. [DOI] [PubMed] [Google Scholar]
- 5.AbdAlla S, Lother H, Quitterer U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 407: 94–98, 2000. doi: 10.1038/35024095. [DOI] [PubMed] [Google Scholar]
- 6.Abraham DM, Davis RT III, Warren CM, Mao L, Wolska BM, Solaro RJ, Rockman HA. β-Arrestin mediates the Frank-Starling mechanism of cardiac contractility. Proc Natl Acad Sci USA 113: 14426–14431, 2016. doi: 10.1073/pnas.1609308113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Achard V, Boullu-Ciocca S, Desbriere R, Nguyen G, Grino M. Renin receptor expression in human adipose tissue. Am J Physiol Regul Integr Comp Physiol 292: R274–R282, 2007. doi: 10.1152/ajpregu.00439.2005. [DOI] [PubMed] [Google Scholar]
- 8.Achard V, Tassistro V, Boullu-Ciocca S, Grino M. Expression and nutritional regulation of the (pro)renin receptor in rat visceral adipose tissue. J Endocrinol Invest 34: 840–846, 2011. doi: 10.3275/7627. [DOI] [PubMed] [Google Scholar]
- 9.Adam O, Frost G, Custodis F, Sussman MA, Schäfers HJ, Böhm M, Laufs U. Role of Rac1 GTPase activation in atrial fibrillation. J Am Coll Cardiol 50: 359–367, 2007. doi: 10.1016/j.jacc.2007.03.041. [DOI] [PubMed] [Google Scholar]
- 10.Adam O, Lavall D, Theobald K, Hohl M, Grube M, Ameling S, Sussman MA, Rosenkranz S, Kroemer HK, Schäfers H-J, Böhm M, Laufs U. Rac1-induced connective tissue growth factor regulates connexin 43 and N-cadherin expression in atrial fibrillation. J Am Coll Cardiol 55: 469–480, 2010. doi: 10.1016/j.jacc.2009.08.064. [DOI] [PubMed] [Google Scholar]
- 11.Adiarto S, Heiden S, Vignon-Zellweger N, Nakayama K, Yagi K, Yanagisawa M, Emoto N. ET-1 from endothelial cells is required for complete angiotensin II-induced cardiac fibrosis and hypertrophy. Life Sci 91: 651–657, 2012. doi: 10.1016/j.lfs.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Advani A, Kelly DJ, Cox AJ, White KE, Advani SL, Thai K, Connelly KA, Yuen D, Trogadis J, Herzenberg AM, Kuliszewski MA, Leong-Poi H, Gilbert RE. The (Pro)renin receptor: site-specific and functional linkage to the vacuolar H+-ATPase in the kidney. Hypertension 54: 261–269, 2009. doi: 10.1161/HYPERTENSIONAHA.109.128645. [DOI] [PubMed] [Google Scholar]
- 13.Agbor LN, Ibeawuchi SC, Hu C, Wu J, Davis DR, Keen HL, Quelle FW, Sigmund CD. Cullin-3 mutation causes arterial stiffness and hypertension through a vascular smooth muscle mechanism. JCI Insight 1: e91015, 2016. doi: 10.1172/jci.insight.91015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res 106: 1253–1264, 2010. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmad S, Varagic J, Groban L, Dell’Italia LJ, Nagata S, Kon ND, Ferrario CM. Angiotensin-(1-12): a chymase-mediated cellular angiotensin II substrate. Curr Hypertens Rep 16: 429, 2014. doi: 10.1007/s11906-014-0429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahmad S, Wei CC, Tallaj J, Dell’Italia LJ, Moniwa N, Varagic J, Ferrario CM. Chymase mediates angiotensin-(1-12) metabolism in normal human hearts. J Am Soc Hypertens 7: 128–136, 2013. doi: 10.1016/j.jash.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akin F, Turgut S, Bastemir M, Turgut G, Kursunluoglu R, Karasu U, Guclu A. Angiotensin-converting enzyme gene polymorphism in overweight and obese Turkish patients with insulin resistance. DNA Cell Biol 29: 207–212, 2010. doi: 10.1089/dna.2009.0934. [DOI] [PubMed] [Google Scholar]
- 18.Al Ghouleh I, Meijles DN, Mutchler S, Zhang Q, Sahoo S, Gorelova A, Henrich Amaral J, Rodríguez AI, Mamonova T, Song GJ, Bisello A, Friedman PA, Cifuentes-Pagano ME, Pagano PJ. Binding of EBP50 to Nox organizing subunit p47phox is pivotal to cellular reactive species generation and altered vascular phenotype. Proc Natl Acad Sci USA 113: E5308–E5317, 2016. doi: 10.1073/pnas.1514161113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alba G, El Bekay R, Chacón P, Reyes ME, Ramos E, Oliván J, Jiménez J, López JM, Martín-Nieto J, Pintado E, Sobrino F. Heme oxygenase-1 expression is down-regulated by angiotensin II and under hypertension in human neutrophils. J Leukoc Biol 84: 397–405, 2008. doi: 10.1189/jlb.0108035. [DOI] [PubMed] [Google Scholar]
- 20.Alexis JD, Wang N, Che W, Lerner-Marmarosh N, Sahni A, Korshunov VA, Zou Y, Ding B, Yan C, Berk BC, Abe J-i. Bcr kinase activation by angiotensin II inhibits PPARγ transcriptional activity in vascular smooth muscle cells. Circ Res 104: 69–78, 2009. doi: 10.1161/CIRCRESAHA.108.188409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ali MS, Sayeski PP, Dirksen LB, Hayzer DJ, Marrero MB, Bernstein KE. Dependence on the motif YIPP for the physical association of Jak2 kinase with the intracellular carboxyl tail of the angiotensin II AT1 receptor. J Biol Chem 272: 23382–23388, 1997. doi: 10.1074/jbc.272.37.23382. [DOI] [PubMed] [Google Scholar]
- 22.Ali Q, Dhande I, Samuel P, Hussain T. Angiotensin type 2 receptor null mice express reduced levels of renal angiotensin II type 2 receptor/angiotensin (1-7)/Mas receptor and exhibit greater high-fat diet-induced kidney injury. J Renin Angiotensin Aldosterone Syst 17: 1470320316661871, 2016. doi: 10.1177/1470320316661871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alique M, Civantos E, Sanchez-Lopez E, Lavoz C, Rayego-Mateos S, Rodrigues-Díez R, García-Redondo AB, Egido J, Ortiz A, Rodríguez-Puyol D, Rodríguez-Puyol M, Ruiz-Ortega M. Integrin-linked kinase plays a key role in the regulation of angiotensin II-induced renal inflammation. Clin Sci (Lond) 127: 19–31, 2014. doi: 10.1042/CS20130412. [DOI] [PubMed] [Google Scholar]
- 24.Allen AM, O’Callaghan EL, Hazelwood L, Germain S, Castrop H, Schnermann J, Bassi JK. Distribution of cells expressing human renin-promoter activity in the brain of a transgenic mouse. Brain Res 1243: 78–85, 2008. doi: 10.1016/j.brainres.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 25.Allen AM, Zhuo J, Mendelsohn FA. Localization and function of angiotensin AT1 receptors. Am J Hypertens 13: 31S–38S, 2000. doi: 10.1016/S0895-7061(99)00249-6. [DOI] [PubMed] [Google Scholar]
- 26.Almeida-Pereira G, Coletti R, Mecawi AS, Reis LC, Elias LLK, Antunes-Rodrigues J. Estradiol and angiotensin II crosstalk in hydromineral balance: Role of the ERK1/2 and JNK signaling pathways. Neuroscience 322: 525–538, 2016. doi: 10.1016/j.neuroscience.2016.02.067. [DOI] [PubMed] [Google Scholar]
- 27.Altara R, Booz GW. Deleting Vascular ADAM17 Sheds New Light on Hypertensive Cardiac Hypertrophy. Hypertension 68: 849–850, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07715. [DOI] [PubMed] [Google Scholar]
- 28.Altara R, Harmancey R, Didion SP, Booz GW, Zouein FA. Cardiac STAT3 Deficiency Impairs Contractility and Metabolic Homeostasis in Hypertension. Front Pharmacol 7: 436, 2016. doi: 10.3389/fphar.2016.00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Altenhöfer S, Kleikers PW, Radermacher KA, Scheurer P, Rob Hermans JJ, Schiffers P, Ho H, Wingler K, Schmidt HH. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci 69: 2327–2343, 2012. doi: 10.1007/s00018-012-1010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amiya E, Watanabe M, Takeda N, Saito T, Shiga T, Hosoya Y, Nakao T, Imai Y, Manabe I, Nagai R, Komuro I, Maemura K. Angiotensin II impairs endothelial nitric-oxide synthase bioavailability under free cholesterol-enriched conditions via intracellular free cholesterol-rich membrane microdomains. J Biol Chem 288: 14497–14509, 2013. doi: 10.1074/jbc.M112.448522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andreozzi F, Laratta E, Sciacqua A, Perticone F, Sesti G. Angiotensin II impairs the insulin signaling pathway promoting production of nitric oxide by inducing phosphorylation of insulin receptor substrate-1 on Ser312 and Ser616 in human umbilical vein endothelial cells. Circ Res 94: 1211–1218, 2004. doi: 10.1161/01.RES.0000126501.34994.96. [DOI] [PubMed] [Google Scholar]
- 32.Angelov SN, Hu JH, Wei H, Airhart N, Shi M, Dichek DA. TGF-β (Transforming Growth Factor-β) Signaling Protects the Thoracic and Abdominal Aorta From Angiotensin II-Induced Pathology by Distinct Mechanisms. Arterioscler Thromb Vasc Biol 37: 2102–2113, 2017. doi: 10.1161/ATVBAHA.117.309401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armstrong ZB, Boughner DR, Drangova M, Rogers KA. Angiotensin II type 1 receptor blocker inhibits arterial calcification in a pre-clinical model. Cardiovasc Res 90: 165–170, 2011. doi: 10.1093/cvr/cvq391. [DOI] [PubMed] [Google Scholar]
- 34.Aroor AR, Demarco VG, Jia G, Sun Z, Nistala R, Meininger GA, Sowers JR. The role of tissue Renin-Angiotensin-aldosterone system in the development of endothelial dysfunction and arterial stiffness. Front Endocrinol (Lausanne) 4: 161, 2013. doi: 10.3389/fendo.2013.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arroja MM, Reid E, McCabe C. Therapeutic potential of the renin angiotensin system in ischaemic stroke. Exp Transl Stroke Med 8: 8, 2016. doi: 10.1186/s13231-016-0022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arroyo AG, Iruela-Arispe ML. Extracellular matrix, inflammation, and the angiogenic response. Cardiovasc Res 86: 226–235, 2010. doi: 10.1093/cvr/cvq049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asamizu S, Urakaze M, Kobashi C, Ishiki M, Norel Din AK, Fujisaka S, Kanatani Y, Bukahari A, Senda S, Suzuki H, Yamazaki Y, Iwata M, Usui I, Yamazaki K, Ogawa H, Kobayashi M, Tobe K. Angiotensin II enhances the increase in monocyte chemoattractant protein-1 production induced by tumor necrosis factor-alpha from 3T3-L1 preadipocytes. J Endocrinol 202: 199–205, 2009. doi: 10.1677/JOE-08-0363. [DOI] [PubMed] [Google Scholar]
- 38.Astin R, Bentham R, Djafarzadeh S, Horscroft JA, Kuc RE, Leung PS, Skipworth JR, Vicencio JM, Davenport AP, Murray AJ, Takala J, Jakob SM, Montgomery H, Szabadkai G. No evidence for a local renin-angiotensin system in liver mitochondria. Sci Rep 3: 2467, 2013. doi: 10.1038/srep02467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aubert G, Lansdorp PM. Telomeres and aging. Physiol Rev 88: 557–579, 2008. doi: 10.1152/physrev.00026.2007. [DOI] [PubMed] [Google Scholar]
- 40.Avendaño MS, Martínez-Revelles S, Aguado A, Simões MR, González-Amor M, Palacios R, Guillem-Llobat P, Vassallo DV, Vila L, García-Puig J, Beltrán LM, Alonso MJ, Cachofeiro MV, Salaices M, Briones AM. Role of COX-2-derived PGE2 on vascular stiffness and function in hypertension. Br J Pharmacol 173: 1541–1555, 2016. doi: 10.1111/bph.13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avila MD, Morgan JP, Yan X. Genetically modified mouse models used for studying the role of the AT2 receptor in cardiac hypertrophy and heart failure. J Biomed Biotechnol 2011: 141039, 2011. doi: 10.1155/2011/141039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ayoub MA, Zhang Y, Kelly RS, See HB, Johnstone EK, McCall EA, Williams JH, Kelly DJ, Pfleger KD. Functional interaction between angiotensin II receptor type 1 and chemokine (C-C motif) receptor 2 with implications for chronic kidney disease. PLoS One 10: e0119803, 2015. doi: 10.1371/journal.pone.0119803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ayyadevara S, Mercanti F, Wang X, Mackintosh SG, Tackett AJ, Prayaga SV, Romeo F, Shmookler Reis RJ, Mehta JL. Age- and Hypertension-Associated Protein Aggregates in Mouse Heart Have Similar Proteomic Profiles. Hypertension 67: 1006–1013, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bache RJ, Chen Y. NOX2-induced myocardial fibrosis and diastolic dysfunction: role of the endothelium. J Am Coll Cardiol 63: 2742–2744, 2014. doi: 10.1016/j.jacc.2014.01.070. [DOI] [PubMed] [Google Scholar]
- 45.Backlund M, Paukku K, Kontula KK, Lehtonen JY. Endoplasmic reticulum stress increases AT1R mRNA expression via TIA-1-dependent mechanism. Nucleic Acids Res 44: 3095–3104, 2016. doi: 10.1093/nar/gkv1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bader M. ACE2, angiotensin-(1–7), and Mas: the other side of the coin. Pflugers Arch 465: 79–85, 2013. doi: 10.1007/s00424-012-1120-0. [DOI] [PubMed] [Google Scholar]
- 47.Bai D, Ge L, Gao Y, Lu X, Wang H, Yang G. Cytoplasmic translocation of HuR contributes to angiotensin II induced cardiac fibrosis. Biochem Biophys Res Commun 463: 1273–1277, 2015. doi: 10.1016/j.bbrc.2015.06.101. [DOI] [PubMed] [Google Scholar]
- 48.Bai X, Lenhart KC, Bird KE, Suen AA, Rojas M, Kakoki M, Li F, Smithies O, Mack CP, Taylor JM. The smooth muscle-selective RhoGAP GRAF3 is a critical regulator of vascular tone and hypertension. Nat Commun 4: 2910, 2013. doi: 10.1038/ncomms3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Balakumar P, Jagadeesh G. A century old renin-angiotensin system still grows with endless possibilities: AT1 receptor signaling cascades in cardiovascular physiopathology. Cell Signal 26: 2147–2160, 2014. doi: 10.1016/j.cellsig.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 50.Balakumar P, Jagadeesh G. Structural determinants for binding, activation, and functional selectivity of the angiotensin AT1 receptor. J Mol Endocrinol 53: R71–R92, 2014. doi: 10.1530/JME-14-0125. [DOI] [PubMed] [Google Scholar]
- 51.Banday AA, Lokhandwala MF. Oxidative stress causes renal angiotensin II type 1 receptor upregulation, Na+/H+ exchanger 3 overstimulation, and hypertension. Hypertension 57: 452–459, 2011. doi: 10.1161/HYPERTENSIONAHA.110.162339. [DOI] [PubMed] [Google Scholar]
- 52.Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A, Just A, Remke J, Zimmer K, Zeug A, Ponimaskin E, Schmiedl A, Yin X, Mayr M, Halder R, Fischer A, Engelhardt S, Wei Y, Schober A, Fiedler J, Thum T. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest 124: 2136–2146, 2014. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barhoumi T, Fraulob-Aquino JC, Mian MOR, Ouerd S, Idris-Khodja N, Huo KG, Rehman A, Caillon A, Dancose-Giambattisto B, Ebrahimian T, Lehoux S, Paradis P, Schiffrin EL. Matrix metalloproteinase-2 knockout prevents angiotensin II-induced vascular injury. Cardiovasc Res 113: 1753–1762, 2017. doi: 10.1093/cvr/cvx115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension 57: 469–476, 2011. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 55.Barki-Harrington L, Luttrell LM, Rockman HA. Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor-receptor interaction in vivo. Circulation 108: 1611–1618, 2003. doi: 10.1161/01.CIR.0000092166.30360.78. [DOI] [PubMed] [Google Scholar]
- 56.Baskin R, Sayeski PP. Angiotensin II mediates cell survival through upregulation and activation of the serum and glucocorticoid inducible kinase 1. Cell Signal 24: 435–442, 2012. doi: 10.1016/j.cellsig.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Basset O, Deffert C, Foti M, Bedard K, Jaquet V, Ogier-Denis E, Krause KH. NADPH oxidase 1 deficiency alters caveolin phosphorylation and angiotensin II-receptor localization in vascular smooth muscle. Antioxid Redox Signal 11: 2371–2384, 2009. doi: 10.1089/ars.2009.2584. [DOI] [PubMed] [Google Scholar]
- 58.Basu R, Fan D, Kandalam V, Lee J, Das SK, Wang X, Baldwin TA, Oudit GY, Kassiri Z. Loss of Timp3 gene leads to abdominal aortic aneurysm formation in response to angiotensin II. J Biol Chem 287: 44083–44096, 2012. doi: 10.1074/jbc.M112.425652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Basu R, Lee J, Morton JS, Takawale A, Fan D, Kandalam V, Wang X, Davidge ST, Kassiri Z. TIMP3 is the primary TIMP to regulate agonist-induced vascular remodelling and hypertension. Cardiovasc Res 98: 360–371, 2013. doi: 10.1093/cvr/cvt067. [DOI] [PubMed] [Google Scholar]
- 60.Basu S, Srinivasan DK, Yang K, Raina H, Banerjee S, Zhang R, Fisher SA, Proweller A. Notch transcriptional control of vascular smooth muscle regulatory gene expression and function. J Biol Chem 288: 11191–11202, 2013. doi: 10.1074/jbc.M112.442996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Basu U, Seravalli J, Madayiputhiya N, Adamec J, Case AJ, Zimmerman MC. Rapid metabolism of exogenous angiotensin II by catecholaminergic neuronal cells in culture media. Physiol Rep 3: e12287, 2015. doi: 10.14814/phy2.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baurand A, Zelarayan L, Betney R, Gehrke C, Dunger S, Noack C, Busjahn A, Huelsken J, Taketo MM, Birchmeier W, Dietz R, Bergmann MW. Beta-catenin downregulation is required for adaptive cardiac remodeling. Circ Res 100: 1353–1362, 2007. doi: 10.1161/01.RES.0000266605.63681.5a. [DOI] [PubMed] [Google Scholar]
- 63.Becker BK, Wang H, Zucker IH. Central TrkB blockade attenuates ICV angiotensin II-hypertension and sympathetic nerve activity in male Sprague-Dawley rats. Auton Neurosci 205: 77–86, 2017. doi: 10.1016/j.autneu.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Becker BK, Wang HJ, Tian C, Zucker IH. BDNF contributes to angiotensin II-mediated reductions in peak voltage-gated K+ current in cultured CATH.a cells. Physiol Rep 3: e12598, 2015. doi: 10.14814/phy2.12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bellot M, Galandrin S, Boularan C, Matthies HJ, Despas F, Denis C, Javitch J, Mazères S, Sanni SJ, Pons V, Seguelas MH, Hansen JL, Pathak A, Galli A, Sénard JM, Galés C. Dual agonist occupancy of AT1-R-α2C-AR heterodimers results in atypical Gs-PKA signaling. Nat Chem Biol 11: 271–279, 2015. doi: 10.1038/nchembio.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Belyea BC, Xu F, Sequeira-Lopez ML, Ariel Gomez R. Loss of Jagged1 in renin progenitors leads to focal kidney fibrosis. Physiol Rep 3: e12544, 2015. doi: 10.14814/phy2.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 105: 293–296, 2002. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 68.Bendall JK, Rinze R, Adlam D, Tatham AL, de Bono J, Channon KM. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: studies in endothelial-targeted Nox2 transgenic mice. Circ Res 100: 1016–1025, 2007. doi: 10.1161/01.RES.0000263381.83835.7b. [DOI] [PubMed] [Google Scholar]
- 69.Benicky J, Hafko R, Sanchez-Lemus E, Aguilera G, Saavedra JM. Six commercially available angiotensin II AT1 receptor antibodies are non-specific. Cell Mol Neurobiol 32: 1353–1365, 2012. doi: 10.1007/s10571-012-9862-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benigni A, Corna D, Zoja C, Sonzogni A, Latini R, Salio M, Conti S, Rottoli D, Longaretti L, Cassis P, Morigi M, Coffman TM, Remuzzi G. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest 119: 524–530, 2009. doi: 10.1172/JCI36703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Benndorf RA, Krebs C, Hirsch-Hoffmann B, Schwedhelm E, Cieslar G, Schmidt-Haupt R, Steinmetz OM, Meyer-Schwesinger C, Thaiss F, Haddad M, Fehr S, Heilmann A, Helmchen U, Hein L, Ehmke H, Stahl RA, Böger RH, Wenzel UO. Angiotensin II type 2 receptor deficiency aggravates renal injury and reduces survival in chronic kidney disease in mice. Kidney Int 75: 1039–1049, 2009. doi: 10.1038/ki.2009.2. [DOI] [PubMed] [Google Scholar]
- 72.Bennett MR, Sinha S, Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res 118: 692–702, 2016. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bennion DM, Haltigan E, Regenhardt RW, Steckelings UM, Sumners C. Neuroprotective mechanisms of the ACE2-angiotensin-(1-7)-Mas axis in stroke. Curr Hypertens Rep 17: 3, 2015. doi: 10.1007/s11906-014-0512-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bennion DM, Haltigan EA, Irwin AJ, Donnangelo LL, Regenhardt RW, Pioquinto DJ, Purich DL, Sumners C. Activation of the Neuroprotective Angiotensin-Converting Enzyme 2 in Rat Ischemic Stroke. Hypertension 66: 141–148, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Berk BC. Angiotensin type 2 receptor (AT2R): a challenging twin. Sci STKE 2003: PE16, 2003. doi: 10.1126/stke.2003.181.pe16. [DOI] [PubMed] [Google Scholar]
- 76.Bettink SI, Werner C, Chen CH, Müller P, Schirmer SH, Walenta KL, Böhm M, Laufs U, Friedrich EB. Integrin-linked kinase is a central mediator in angiotensin II type 1- and chemokine receptor CXCR4 signaling in myocardial hypertrophy. Biochem Biophys Res Commun 397: 208–213, 2010. doi: 10.1016/j.bbrc.2010.05.086. [DOI] [PubMed] [Google Scholar]
- 77.Beyer AM, de Lange WJ, Halabi CM, Modrick ML, Keen HL, Faraci FM, Sigmund CD. Endothelium-specific interference with peroxisome proliferator activated receptor gamma causes cerebral vascular dysfunction in response to a high-fat diet. Circ Res 103: 654–661, 2008. doi: 10.1161/CIRCRESAHA.108.176339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bhatta A, Yao L, Toque HA, Shatanawi A, Xu Z, Caldwell RB, Caldwell RW. Angiotensin II-induced arterial thickening, fibrosis and stiffening involves elevated arginase function. PLoS One 10: e0121727, 2015. doi: 10.1371/journal.pone.0121727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Biancardi VC, Stranahan AM, Krause EG, de Kloet AD, Stern JE. Cross talk between AT1 receptors and Toll-like receptor 4 in microglia contributes to angiotensin II-derived ROS production in the hypothalamic paraventricular nucleus. Am J Physiol Heart Circ Physiol 310: H404–H415, 2016. doi: 10.1152/ajpheart.00247.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blacher J, Evans A, Arveiler D, Amouyel P, Ferrières J, Bingham A, Yarnell J, Haas B, Montaye M, Ruidavets JB, Ducimetière P; PRIME Study Group . Residual cardiovascular risk in treated hypertension and hyperlipidaemia: the PRIME Study. J Hum Hypertens 24: 19–26, 2010. doi: 10.1038/jhh.2009.34. [DOI] [PubMed] [Google Scholar]
- 81.Blodow S, Schneider H, Storch U, Wizemann R, Forst AL, Gudermann T, Mederos y Schnitzler M. Novel role of mechanosensitive AT1B receptors in myogenic vasoconstriction. Pflugers Arch 466: 1343–1353, 2014. doi: 10.1007/s00424-013-1372-3. [DOI] [PubMed] [Google Scholar]
- 82.Blomkalns AL, Gavrila D, Thomas M, Neltner BS, Blanco VM, Benjamin SB, McCormick ML, Stoll LL, Denning GM, Collins SP, Qin Z, Daugherty A, Cassis LA, Thompson RW, Weiss RM, Lindower PD, Pinney SM, Chatterjee T, Weintraub NL. CD14 directs adventitial macrophage precursor recruitment: role in early abdominal aortic aneurysm formation. J Am Heart Assoc 2: e000065, 2013. doi: 10.1161/JAHA.112.000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boerrigter G, Lark MW, Whalen EJ, Soergel DG, Violin JD, Burnett JC Jr. Cardiorenal actions of TRV120027, a novel ß-arrestin-biased ligand at the angiotensin II type I receptor, in healthy and heart failure canines: a novel therapeutic strategy for acute heart failure. Circ Heart Fail 4: 770–778, 2011. doi: 10.1161/CIRCHEARTFAILURE.111.962571. [DOI] [PubMed] [Google Scholar]
- 84.Boerrigter G, Soergel DG, Violin JD, Lark MW, Burnett JC Jr. TRV120027, a novel β-arrestin biased ligand at the angiotensin II type I receptor, unloads the heart and maintains renal function when added to furosemide in experimental heart failure. Circ Heart Fail 5: 627–634, 2012. doi: 10.1161/CIRCHEARTFAILURE.112.969220. [DOI] [PubMed] [Google Scholar]
- 85.Boettger T, Beetz N, Kostin S, Schneider J, Krüger M, Hein L, Braun T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest 119: 2634–2647, 2009. doi: 10.1172/JCI38864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E, Horrevoets AJ, Vinciguerra M, Rosenthal N, Sciacca S, Pilato M, van Heijningen P, Essers J, Brandes RP, Zeiher AM, Dimmeler S. MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res 109: 1115–1119, 2011. doi: 10.1161/CIRCRESAHA.111.255737. [DOI] [PubMed] [Google Scholar]
- 87.Booz GW. Putting the brakes on cardiac hypertrophy: exploiting the NO-cGMP counter-regulatory system. Hypertension 45: 341–346, 2005. doi: 10.1161/01.HYP.0000156878.17006.02. [DOI] [PubMed] [Google Scholar]
- 88.Booz GW, Dostal DE, Singer HA, Baker KM. Involvement of protein kianse C and Ca2+ in angiotensin II-induced mitogenesis of cardiac fibroblasts. Am J Physiol Cell Physiol 267: C1308–C1318, 1994. doi: 10.1152/ajpcell.1994.267.5.C1308. [DOI] [PubMed] [Google Scholar]
- 89.Borghi F, Sevá-Pessôa B, Grassi-Kassisse DM. The adipose tissue and the involvement of the renin-angiotensin-aldosterone system in cardiometabolic syndrome. Cell Tissue Res 366: 543–548, 2016. doi: 10.1007/s00441-016-2515-6. [DOI] [PubMed] [Google Scholar]
- 90.Bouabout G, Ayme-Dietrich E, Jacob H, Champy MF, Birling MC, Pavlovic G, Madeira L, Fertak LE, Petit-Demoulière B, Sorg T, Herault Y, Mudgett J, Monassier L. Nox4 genetic inhibition in experimental hypertension and metabolic syndrome. Arch Cardiovasc Dis 111: 41–52, 2018. doi: 10.1016/j.acvd.2017.03.011. [DOI] [PubMed] [Google Scholar]
- 91.Bouley R, Palomino Z, Tang SS, Nunes P, Kobori H, Lu HA, Shum WW, Sabolic I, Brown D, Ingelfinger JR, Jung FF. Angiotensin II and hypertonicity modulate proximal tubular aquaporin 1 expression. Am J Physiol Renal Physiol 297: F1575–F1586, 2009. doi: 10.1152/ajprenal.90762.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boulos N, Helle F, Dussaule JC, Placier S, Milliez P, Djudjaj S, Guerrot D, Joutel A, Ronco P, Boffa JJ, Chatziantoniou C. Notch3 is essential for regulation of the renal vascular tone. Hypertension 57: 1176–1182, 2011. doi: 10.1161/HYPERTENSIONAHA.111.170746. [DOI] [PubMed] [Google Scholar]
- 93.Bove CM, Gilson WD, Scott CD, Epstein FH, Yang Z, Dimaria JM, Berr SS, French BA, Bishop SP, Kramer CM. The angiotensin II type 2 receptor and improved adjacent region function post-MI. J Cardiovasc Magn Reson 7: 459–464, 2005. doi: 10.1081/JCMR-200053461. [DOI] [PubMed] [Google Scholar]
- 94.Bove CM, Yang Z, Gilson WD, Epstein FH, French BA, Berr SS, Bishop SP, Matsubara H, Carey RM, Kramer CM. Nitric oxide mediates benefits of angiotensin II type 2 receptor overexpression during post-infarct remodeling. Hypertension 43: 680–685, 2004. doi: 10.1161/01.HYP.0000115924.94236.91. [DOI] [PubMed] [Google Scholar]
- 95.Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, Tikhonova IR, Bjornson R, Mane SM, Colussi G, Lebel M, Gordon RD, Semmekrot BA, Poujol A, Välimäki MJ, De Ferrari ME, Sanjad SA, Gutkin M, Karet FE, Tucci JR, Stockigt JR, Keppler-Noreuil KM, Porter CC, Anand SK, Whiteford ML, Davis ID, Dewar SB, Bettinelli A, Fadrowski JJ, Belsha CW, Hunley TE, Nelson RD, Trachtman H, Cole TR, Pinsk M, Bockenhauer D, Shenoy M, Vaidyanathan P, Foreman JW, Rasoulpour M, Thameem F, Al-Shahrouri HZ, Radhakrishnan J, Gharavi AG, Goilav B, Lifton RP. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 482: 98–102, 2012. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bracey NA, Gershkovich B, Chun J, Vilaysane A, Meijndert HC, Wright JR Jr, Fedak PW, Beck PL, Muruve DA, Duff HJ. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J Biol Chem 289: 19571–19584, 2014. doi: 10.1074/jbc.M114.550624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bradford CN, Ely DR, Raizada MK. Targeting the vasoprotective axis of the renin-angiotensin system: a novel strategic approach to pulmonary hypertensive therapy. Curr Hypertens Rep 12: 212–219, 2010. doi: 10.1007/s11906-010-0122-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension 56: 879–884, 2010. doi: 10.1161/HYPERTENSIONAHA.110.158071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brasier AR, Recinos A III, Eledrisi MS. Vascular inflammation and the renin-angiotensin system. Arterioscler Thromb Vasc Biol 22: 1257–1266, 2002. doi: 10.1161/01.ATV.0000021412.56621.A2. [DOI] [PubMed] [Google Scholar]
- 100.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res 116: 531–549, 2015. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol 9: 459–469, 2013. doi: 10.1038/nrneph.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bumpus FM, Catt KJ, Chiu AT, DeGasparo M, Goodfriend T, Husain A, Peach MJ, Taylor DG Jr, Timmermans PB. Nomenclature for angiotensin receptors. A report of the Nomenclature Committee of the Council for High Blood Pressure Research. Hypertension 17: 720–721, 1991. doi: 10.1161/01.HYP.17.5.720. [DOI] [PubMed] [Google Scholar]
- 103.Burcklé CA, Jan Danser AH, Müller DN, Garrelds IM, Gasc JM, Popova E, Plehm R, Peters J, Bader M, Nguyen G. Elevated blood pressure and heart rate in human renin receptor transgenic rats. Hypertension 47: 552–556, 2006. doi: 10.1161/01.HYP.0000199912.47657.04. [DOI] [PubMed] [Google Scholar]
- 104.Burger D, Montezano AC, Nishigaki N, He Y, Carter A, Touyz RM. Endothelial microparticle formation by angiotensin II is mediated via Ang II receptor type I/NADPH oxidase/ Rho kinase pathways targeted to lipid rafts. Arterioscler Thromb Vasc Biol 31: 1898–1907, 2011. doi: 10.1161/ATVBAHA.110.222703. [DOI] [PubMed] [Google Scholar]
- 105.Burger D, Schock S, Thompson CS, Montezano AC, Hakim AM, Touyz RM. Microparticles: biomarkers and beyond. Clin Sci (Lond) 124: 423–441, 2013. doi: 10.1042/CS20120309. [DOI] [PubMed] [Google Scholar]
- 106.Burks TN, Andres-Mateos E, Marx R, Mejias R, Van Erp C, Simmers JL, Walston JD, Ward CW, Cohn RD. Losartan restores skeletal muscle remodeling and protects against disuse atrophy in sarcopenia. Sci Transl Med 3: 82ra37, 2011. doi: 10.1126/scitranslmed.3002227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol 51: 802–809, 2008. doi: 10.1016/j.jacc.2007.09.064. [DOI] [PubMed] [Google Scholar]
- 108.Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, Cave AC, Shah AM. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res 93: 802–805, 2003. doi: 10.1161/01.RES.0000099504.30207.F5. [DOI] [PubMed] [Google Scholar]
- 109.Caillon A, Mian MOR, Fraulob-Aquino JC, Huo KG, Barhoumi T, Ouerd S, Sinnaeve PR, Paradis P, Schiffrin EL. γδ T Cells Mediate Angiotensin II-Induced Hypertension and Vascular Injury. Circulation 135: 2155–2162, 2017. doi: 10.1161/CIRCULATIONAHA.116.027058. [DOI] [PubMed] [Google Scholar]
- 110.Calderon LE, Liu S, Su W, Xie Z, Guo Z, Eberhard W, Gong MC. iPLA2β overexpression in smooth muscle exacerbates angiotensin II-induced hypertension and vascular remodeling. PLoS One 7: e31850, 2012. doi: 10.1371/journal.pone.0031850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Callera GE, Antunes TT, He Y, Montezano AC, Yogi A, Savoia C, Touyz RM. c-Src Inhibition Improves Cardiovascular Function but not Remodeling or Fibrosis in Angiotensin II-Induced Hypertension. Hypertension 68: 1179–1190, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07699. [DOI] [PubMed] [Google Scholar]
- 112.Calvier L, Miana M, Reboul P, Cachofeiro V, Martinez-Martinez E, de Boer RA, Poirier F, Lacolley P, Zannad F, Rossignol P, López-Andrés N. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler Thromb Vasc Biol 33: 67–75, 2013. doi: 10.1161/ATVBAHA.112.300569. [DOI] [PubMed] [Google Scholar]
- 113.Camacho Londoño JE, Tian Q, Hammer K, Schröder L, Camacho Londoño J, Reil JC, He T, Oberhofer M, Mannebach S, Mathar I, Philipp SE, Tabellion W, Schweda F, Dietrich A, Kaestner L, Laufs U, Birnbaumer L, Flockerzi V, Freichel M, Lipp P. A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur Heart J 36: 2257–2266, 2015. doi: 10.1093/eurheartj/ehv250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Campos AH, Wang W, Pollman MJ, Gibbons GH. Determinants of Notch-3 receptor expression and signaling in vascular smooth muscle cells: implications in cell-cycle regulation. Circ Res 91: 999–1006, 2002. doi: 10.1161/01.RES.0000044944.99984.25. [DOI] [PubMed] [Google Scholar]
- 115.Campos AH, Zhao Y, Pollman MJ, Gibbons GH. DNA microarray profiling to identify angiotensin-responsive genes in vascular smooth muscle cells: potential mediators of vascular disease. Circ Res 92: 111–118, 2003. doi: 10.1161/01.RES.0000049100.22673.F6. [DOI] [PubMed] [Google Scholar]
- 116.Cantalupo A, Gargiulo A, Dautaj E, Liu C, Zhang Y, Hla T, Di Lorenzo A. S1PR1 (Sphingosine-1-Phosphate Receptor 1) Signaling Regulates Blood Flow and Pressure. Hypertension 70: 426–434, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cantalupo A, Zhang Y, Kothiya M, Galvani S, Obinata H, Bucci M, Giordano FJ, Jiang XC, Hla T, Di Lorenzo A. Nogo-B regulates endothelial sphingolipid homeostasis to control vascular function and blood pressure. Nat Med 21: 1028–1037, 2015. doi: 10.1038/nm.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Capote LA, Mendez Perez R, Lymperopoulos A. GPCR signaling and cardiac function. Eur J Pharmacol 763, Pt B: 143–148, 2015. doi: 10.1016/j.ejphar.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 119.Carbone ML, Brégeon J, Devos N, Chadeuf G, Blanchard A, Azizi M, Pacaud P, Jeunemaître X, Loirand G. Angiotensin II activates the RhoA exchange factor Arhgef1 in humans. Hypertension 65: 1273–1278, 2015. doi: 10.1161/HYPERTENSIONAHA.114.05065. [DOI] [PubMed] [Google Scholar]
- 120.Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Høydal M, Autore C, Russo MA, Dorn GW II, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med 13: 613–618, 2007. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 121.Carey RM. Newly discovered components and actions of the renin-angiotensin system. Hypertension 62: 818–822, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01111. [DOI] [PubMed] [Google Scholar]
- 122.Carlström M, Lai EY, Ma Z, Steege A, Patzak A, Eriksson UJ, Lundberg JO, Wilcox CS, Persson AE. Superoxide dismutase 1 limits renal microvascular remodeling and attenuates arteriole and blood pressure responses to angiotensin II via modulation of nitric oxide bioavailability. Hypertension 56: 907–913, 2010. doi: 10.1161/HYPERTENSIONAHA.110.159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Carquin M, D’Auria L, Pollet H, Bongarzone ER, Tyteca D. Recent progress on lipid lateral heterogeneity in plasma membranes: From rafts to submicrometric domains. Prog Lipid Res 62: 1–24, 2016. doi: 10.1016/j.plipres.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Carrillo-Sepulveda MA, Keen HL, Davis DR, Grobe JL, Sigmund CD. Role of vascular smooth muscle PPARγ in regulating AT1 receptor signaling and angiotensin II-dependent hypertension. PLoS One 9: e103786, 2014. doi: 10.1371/journal.pone.0103786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Carroll WX, Kalupahana NS, Booker SL, Siriwardhana N, Lemieux M, Saxton AM, Moustaid-Moussa N. Angiotensinogen gene silencing reduces markers of lipid accumulation and inflammation in cultured adipocytes. Front Endocrinol (Lausanne) 4: 10, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cassis LA, Police SB, Yiannikouris F, Thatcher SE. Local adipose tissue renin-angiotensin system. Curr Hypertens Rep 10: 93–98, 2008. doi: 10.1007/s11906-008-0019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Castañeda-Bueno M, Cervantes-Pérez LG, Vázquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA 109: 7929–7934, 2012. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Castellanos Rivera RM, Monteagudo MC, Pentz ES, Glenn ST, Gross KW, Carretero O, Sequeira-Lopez ML, Gomez RA. Transcriptional regulator RBP-J regulates the number and plasticity of renin cells. Physiol Genomics 43: 1021–1028, 2011. doi: 10.1152/physiolgenomics.00061.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Castoldi G, Di Gioia CR, Bombardi C, Catalucci D, Corradi B, Gualazzi MG, Leopizzi M, Mancini M, Zerbini G, Condorelli G, Stella A. MiR-133a regulates collagen 1A1: potential role of miR-133a in myocardial fibrosis in angiotensin II-dependent hypertension. J Cell Physiol 227: 850–856, 2012. doi: 10.1002/jcp.22939. [DOI] [PubMed] [Google Scholar]
- 130.Castoldi G, di Gioia CR, Bombardi C, Maestroni S, Carletti R, Steckelings UM, Dahlöf B, Unger T, Zerbini G, Stella A. Prevention of diabetic nephropathy by compound 21, selective agonist of angiotensin type 2 receptors, in Zucker diabetic fatty rats. Am J Physiol Renal Physiol 307: F1123–F1131, 2014. doi: 10.1152/ajprenal.00247.2014. [DOI] [PubMed] [Google Scholar]
- 131.Catena C, Colussi G, Brosolo G, Novello M, Sechi LA. Aldosterone and Left Ventricular Remodeling. Horm Metab Res 47: 981–986, 2015. doi: 10.1055/s-0035-1565055. [DOI] [PubMed] [Google Scholar]
- 132.Chai SY, Fernando R, Peck G, Ye SY, Mendelsohn FA, Jenkins TA, Albiston AL. The angiotensin IV/AT4 receptor. Cell Mol Life Sci 61: 2728–2737, 2004. doi: 10.1007/s00018-004-4246-1. [DOI] [PubMed] [Google Scholar]
- 133.Chai SY, Mendelsohn FA, Paxinos G. Angiotensin converting enzyme in rat brain visualized by quantitative in vitro autoradiography. Neuroscience 20: 615–627, 1987. doi: 10.1016/0306-4522(87)90114-X. [DOI] [PubMed] [Google Scholar]
- 134.Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, Kim HA, Krishnan SM, Lewis CV, Salimova E, Tipping P, Vinh A, Samuel CS, Peter K, Guzik TJ, Kyaw TS, Toh BH, Bobik A, Drummond GR. Obligatory Role for B Cells in the Development of Angiotensin II-Dependent Hypertension. Hypertension 66: 1023–1033, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05779. [DOI] [PubMed] [Google Scholar]
- 135.Chan SL, Baumbach GL. Deficiency of Nox2 prevents angiotensin II-induced inward remodeling in cerebral arterioles. Front Physiol 4: 133, 2013. doi: 10.3389/fphys.2013.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chan SL, Umesalma S, Baumbach GL. Epidermal growth factor receptor is critical for angiotensin II-mediated hypertrophy in cerebral arterioles. Hypertension 65: 806–812, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04794. [DOI] [PubMed] [Google Scholar]
- 137.Chang SY, Chen YW, Chenier I, Tran SM, Zhang SL. Angiotensin II type II receptor deficiency accelerates the development of nephropathy in type I diabetes via oxidative stress and ACE2. Exp Diabetes Res 2011: 521076, 2011. doi: 10.1155/2011/521076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chang Y, Wei W. Angiotensin II in inflammation, immunity and rheumatoid arthritis. Clin Exp Immunol 179: 137–145, 2015. doi: 10.1111/cei.12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chaplin NL, Nieves-Cintrón M, Fresquez AM, Navedo MF, Amberg GC. Arterial Smooth Muscle Mitochondria Amplify Hydrogen Peroxide Microdomains Functionally Coupled to L-Type Calcium Channels. Circ Res 117: 1013–1023, 2015. doi: 10.1161/CIRCRESAHA.115.306996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? Am J Physiol Heart Circ Physiol 310: H137–H152, 2016. doi: 10.1152/ajpheart.00618.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chappell MC. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS receptor axis: more than regulation of blood pressure? Hypertension 50: 596–599, 2007. doi: 10.1161/HYPERTENSIONAHA.106.076216. [DOI] [PubMed] [Google Scholar]
- 142.Chatterjee A, Mir SA, Dutta D, Mitra A, Pathak K, Sarkar S. Analysis of p53 and NF-κB signaling in modulating the cardiomyocyte fate during hypertrophy. J Cell Physiol 226: 2543–2554, 2011. doi: 10.1002/jcp.22599. [DOI] [PubMed] [Google Scholar]
- 143.Chen A, Huang BS, Wang HW, Ahmad M, Leenen FH. Knockdown of mineralocorticoid or angiotensin II type 1 receptor gene expression in the paraventricular nucleus prevents angiotensin II hypertension in rats. J Physiol 592: 3523–3536, 2014. doi: 10.1113/jphysiol.2014.275560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chen AT, Guo C, Dumas KJ, Ashrafi K, Hu PJ. Effects of Caenorhabditis elegans sgk-1 mutations on lifespan, stress resistance, and DAF-16/FoxO regulation. Aging Cell 12: 932–940, 2013. doi: 10.1111/acel.12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chen C, Meng Y, Wang L, Wang HX, Tian C, Pang GD, Li HH, Du J. Ubiquitin-activating enzyme E1 inhibitor PYR41 attenuates angiotensin II-induced activation of dendritic cells via the IκBa/NF-κB and MKP1/ERK/STAT1 pathways. Immunology 142: 307–319, 2014. doi: 10.1111/imm.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen CC, Pedraza PL, Hao S, Stier CT, Ferreri NR. TNFR1-deficient mice display altered blood pressure and renal responses to ANG II infusion. Am J Physiol Renal Physiol 299: F1141–F1150, 2010. doi: 10.1152/ajprenal.00344.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen D, Stegbauer J, Sparks MA, Kohan D, Griffiths R, Herrera M, Gurley SB, Coffman TM. Impact of Angiotensin Type 1A Receptors in Principal Cells of the Collecting Duct on Blood Pressure and Hypertension. Hypertension 67: 1291–1297, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen G, Pan SQ, Shen C, Pan SF, Zhang XM, He QY. Puerarin inhibits angiotensin II-induced cardiac hypertrophy via the redox-sensitive ERK1/2, p38 and NF-κB pathways. Acta Pharmacol Sin 35: 463–475, 2014. doi: 10.1038/aps.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chen HZ, Wang F, Gao P, Pei JF, Liu Y, Xu TT, Tang X, Fu WY, Lu J, Yan YF, Wang XM, Han L, Zhang ZQ, Zhang R, Zou MH, Liu DP. Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ Res 119: 1076–1088, 2016. doi: 10.1161/CIRCRESAHA.116.308895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chen J, Chen JK, Harris RC. Angiotensin II induces epithelial-to-mesenchymal transition in renal epithelial cells through reactive oxygen species/Src/caveolin-mediated activation of an epidermal growth factor receptor-extracellular signal-regulated kinase signaling pathway. Mol Cell Biol 32: 981–991, 2012. doi: 10.1128/MCB.06410-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chen J, Chen JK, Nagai K, Plieth D, Tan M, Lee TC, Threadgill DW, Neilson EG, Harris RC. EGFR signaling promotes TGFβ-dependent renal fibrosis. J Am Soc Nephrol 23: 215–224, 2012. doi: 10.1681/ASN.2011070645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chen J, Peters A, Papke CL, Villamizar C, Ringuette LJ, Cao J, Wang S, Ma S, Gong L, Byanova KL, Xiong J, Zhu MX, Madonna R, Kee P, Geng YJ, Brasier AR, Davis EC, Prakash S, Kwartler CS, Milewicz DM. Loss of Smooth Muscle α-Actin Leads to NF-κB-Dependent Increased Sensitivity to Angiotensin II in Smooth Muscle Cells and Aortic Enlargement. Circ Res 120: 1903–1915, 2017. doi: 10.1161/CIRCRESAHA.117.310563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chen X, Howatt DA, Balakrishnan A, Moorleghen JJ, Wu C, Cassis LA, Daugherty A, Lu H. Angiotensin-Converting Enzyme in Smooth Muscle Cells Promotes Atherosclerosis-Brief Report. Arterioscler Thromb Vasc Biol 36: 1085–1089, 2016. doi: 10.1161/ATVBAHA.115.307038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chen X, Lu H, Zhao M, Tashiro K, Cassis LA, Daugherty A. Contributions of leukocyte angiotensin-converting enzyme to development of atherosclerosis. Arterioscler Thromb Vasc Biol 33: 2075–2080, 2013. doi: 10.1161/ATVBAHA.113.301777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chen X, Rateri DL, Howatt DA, Balakrishnan A, Moorleghen JJ, Cassis LA, Daugherty A. TGF-β Neutralization Enhances AngII-Induced Aortic Rupture and Aneurysm in Both Thoracic and Abdominal Regions. PLoS One 11: e0153811, 2016. doi: 10.1371/journal.pone.0153811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chen XQ, Liu X, Wang QX, Zhang MJ, Guo M, Liu F, Jiang WF, Zhou L. Pioglitazone inhibits angiotensin II-induced atrial fibroblasts proliferation via NF-κB/TGF-β1/TRIF/TRAF6 pathway. Exp Cell Res 330: 43–55, 2015. doi: 10.1016/j.yexcr.2014.08.021. [DOI] [PubMed] [Google Scholar]
- 157.Chen Z, Wen L, Martin M, Hsu CY, Fang L, Lin FM, Lin TY, Geary MJ, Geary GG, Zhao Y, Johnson DA, Chen JW, Lin SJ, Chien S, Huang HD, Miller YI, Huang PH, Shyy JY. Oxidative stress activates endothelial innate immunity via sterol regulatory element binding protein 2 (SREBP2) transactivation of microRNA-92a. Circulation 131: 805–814, 2015. doi: 10.1161/CIRCULATIONAHA.114.013675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cheng J, Koenig SN, Kuivaniemi HS, Garg V, Hans CP. Pharmacological inhibitor of notch signaling stabilizes the progression of small abdominal aortic aneurysm in a mouse model. J Am Heart Assoc 3: e001064, 2014. doi: 10.1161/JAHA.114.001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol 170: 1831–1840, 2007. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chichger H, Vang A, O’Connell KA, Zhang P, Mende U, Harrington EO, Choudhary G. PKC δ and βII regulate angiotensin II-mediated fibrosis through p38: a mechanism of RV fibrosis in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L827–L836, 2015. doi: 10.1152/ajplung.00184.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chidlow JH Jr, Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovasc Res 86: 219–225, 2010. doi: 10.1093/cvr/cvq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cho HM, Lee DY, Kim HY, Lee HA, Seok YM, Kim IK. Upregulation of the Na(+)-K(+)-2Cl(-) cotransporter 1 via histone modification in the aortas of angiotensin II-induced hypertensive rats. Hypertens Res 35: 819–824, 2012. doi: 10.1038/hr.2012.37. [DOI] [PubMed] [Google Scholar]
- 163.Choe N, Kwon DH, Shin S, Kim YS, Kim YK, Kim J, Ahn Y, Eom GH, Kook H. The microRNA miR-124 inhibits vascular smooth muscle cell proliferation by targeting S100 calcium-binding protein A4 (S100A4). FEBS Lett 591: 1041–1052, 2017. doi: 10.1002/1873-3468.12606. [DOI] [PubMed] [Google Scholar]
- 164.Choi H, Allahdadi KJ, Tostes RC, Webb RC. Augmented S-nitrosylation contributes to impaired relaxation in angiotensin II hypertensive mouse aorta: role of thioredoxin reductase. J Hypertens 29: 2359–2368, 2011. doi: 10.1097/HJH.0b013e32834d2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Choi S, Gustafson-Wagner EA, Wang Q, Harlan SM, Sinn HW, Lin JL, Lin JJ. The intercalated disk protein, mXinalpha, is capable of interacting with beta-catenin and bundling actin filaments [corrected]. J Biol Chem 282: 36024–36036, 2007. doi: 10.1074/jbc.M707639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Choi S, Kim J, Yea K, Suh PG, Kim J, Ryu SH. Targeted label-free quantitative analysis of secretory proteins from adipocytes in response to oxidative stress. Anal Biochem 401: 196–202, 2010. doi: 10.1016/j.ab.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 167.Choi YS, de Mattos AB, Shao D, Li T, Nabben M, Kim M, Wang W, Tian R, Kolwicz SC Jr. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J Mol Cell Cardiol 100: 64–71, 2016. doi: 10.1016/j.yjmcc.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chow BS, Allen TJ. Angiotensin II type 2 receptor (AT2R) in renal and cardiovascular disease. Clin Sci (Lond) 130: 1307–1326, 2016. doi: 10.1042/CS20160243. [DOI] [PubMed] [Google Scholar]
- 169.Chow BS, Kocan M, Bosnyak S, Sarwar M, Wigg B, Jones ES, Widdop RE, Summers RJ, Bathgate RA, Hewitson TD, Samuel CS. Relaxin requires the angiotensin II type 2 receptor to abrogate renal interstitial fibrosis. Kidney Int 86: 75–85, 2014. doi: 10.1038/ki.2013.518. [DOI] [PubMed] [Google Scholar]
- 170.Christensen GL, Kelstrup CD, Lyngsø C, Sarwar U, Bøgebo R, Sheikh SP, Gammeltoft S, Olsen JV, Hansen JL. Quantitative phosphoproteomics dissection of seven-transmembrane receptor signaling using full and biased agonists. Mol Cell Proteomics 9: 1540–1553, 2010. doi: 10.1074/mcp.M900550-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chu Y, Wilson K, Gu H, Wegman-Points L, Dooley SA, Pierce GL, Cheng G, Pena Silva RA, Heistad DD, Hasan D. Myeloperoxidase is increased in human cerebral aneurysms and increases formation and rupture of cerebral aneurysms in mice. Stroke 46: 1651–1656, 2015. doi: 10.1161/STROKEAHA.114.008589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chun HJ, Ali ZA, Kojima Y, Kundu RK, Sheikh AY, Agrawal R, Zheng L, Leeper NJ, Pearl NE, Patterson AJ, Anderson JP, Tsao PS, Lenardo MJ, Ashley EA, Quertermous T. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest 118: 3343–3354, 2008. doi: 10.1172/JCI34871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Claflin KE, Grobe JL. Control of energy balance by the brain renin-angiotensin system. Curr Hypertens Rep 17: 38, 2015. doi: 10.1007/s11906-015-0549-x. [DOI] [PubMed] [Google Scholar]
- 174.Claflin KE, Sandgren JA, Lambertz AM, Weidemann BJ, Littlejohn NK, Burnett CM, Pearson NA, Morgan DA, Gibson-Corley KN, Rahmouni K, Grobe JL. Angiotensin AT1A receptors on leptin receptor-expressing cells control resting metabolism. J Clin Invest 127: 1414–1424, 2017. doi: 10.1172/JCI88641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Clark AL, Maruyama S, Sano S, Accorsi A, Girgenrath M, Walsh K, Naya FJ. miR-410 and miR-495 Are Dynamically Regulated in Diverse Cardiomyopathies and Their Inhibition Attenuates Pathological Hypertrophy. PLoS One 11: e0151515, 2016. doi: 10.1371/journal.pone.0151515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Coble JP, Cassell MD, Davis DR, Grobe JL, Sigmund CD. Activation of the renin-angiotensin system, specifically in the subfornical organ is sufficient to induce fluid intake. Am J Physiol Regul Integr Comp Physiol 307: R376–R386, 2014. doi: 10.1152/ajpregu.00216.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Coble JP, Grobe JL, Johnson AK, Sigmund CD. Mechanisms of brain renin angiotensin system-induced drinking and blood pressure: importance of the subfornical organ. Am J Physiol Regul Integr Comp Physiol 308: R238–R249, 2015. doi: 10.1152/ajpregu.00486.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Coble JP, Johnson RF, Cassell MD, Johnson AK, Grobe JL, Sigmund CD. Activity of protein kinase C-α within the subfornical organ is necessary for fluid intake in response to brain angiotensin. Hypertension 64: 141–148, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Coffman TM. The inextricable role of the kidney in hypertension. J Clin Invest 124: 2341–2347, 2014. doi: 10.1172/JCI72274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30: 255–289, 2014. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- 181.Cook JL, Re RN. Lessons from in vitro studies and a related intracellular angiotensin II transgenic mouse model. Am J Physiol Regul Integr Comp Physiol 302: R482–R493, 2012. doi: 10.1152/ajpregu.00493.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Cook JL, Re RN, deHaro DL, Abadie JM, Peters M, Alam J. The trafficking protein GABARAP binds to and enhances plasma membrane expression and function of the angiotensin II type 1 receptor. Circ Res 102: 1539–1547, 2008. doi: 10.1161/CIRCRESAHA.108.176594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, Nelson CA, Cheng SH, Wentworth BM, Ramirez F. Dimorphic effects of transforming growth factor-β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol 35: 911–917, 2015. doi: 10.1161/ATVBAHA.114.305150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118, 2010. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cordazzo C, Neri T, Petrini S, Lombardi S, Balìa C, Cianchetti S, Carmazzi Y, Paggiaro P, Pedrinelli R, Celi A. Angiotensin II induces the generation of procoagulant microparticles by human mononuclear cells via an angiotensin type 2 receptor-mediated pathway. Thromb Res 131: e168–e174, 2013. doi: 10.1016/j.thromres.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 186.Cordero-Reyes AM, Youker KA, Trevino AR, Celis R, Hamilton DJ, Flores-Arredondo JH, Orrego CM, Bhimaraj A, Estep JD, Torre-Amione G. Full Expression of Cardiomyopathy Is Partly Dependent on B-Cells: A Pathway That Involves Cytokine Activation, Immunoglobulin Deposition, and Activation of Apoptosis. J Am Heart Assoc 5: e002484, 2016. doi: 10.1161/JAHA.115.002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Coutinho DC, Foureaux G, Rodrigues KD, Salles RL, Moraes PL, Murça TM, De Maria ML, Gomes ER, Santos RA, Guatimosim S, Ferreira AJ. Cardiovascular effects of angiotensin A: a novel peptide of the renin-angiotensin system. J Renin Angiotensin Aldosterone Syst 15: 480–486, 2014. doi: 10.1177/1470320312474856. [DOI] [PubMed] [Google Scholar]
- 188.Crassous PA, Couloubaly S, Huang C, Zhou Z, Baskaran P, Kim DD, Papapetropoulos A, Fioramonti X, Durán WN, Beuve A. Soluble guanylyl cyclase is a target of angiotensin II-induced nitrosative stress in a hypertensive rat model. Am J Physiol Heart Circ Physiol 303: H597–H604, 2012. doi: 10.1152/ajpheart.00138.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Crowley SD, Coffman TM. Recent advances involving the renin-angiotensin system. Exp Cell Res 318: 1049–1056, 2012. doi: 10.1016/j.yexcr.2012.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA 103: 17985–17990, 2006. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Crowley SD, Song YS, Sprung G, Griffiths R, Sparks M, Yan M, Burchette JL, Howell DN, Lin EE, Okeiyi B, Stegbauer J, Yang Y, Tharaux PL, Ruiz P. A role for angiotensin II type 1 receptors on bone marrow-derived cells in the pathogenesis of angiotensin II-dependent hypertension. Hypertension 55: 99–108, 2010. doi: 10.1161/HYPERTENSIONAHA.109.144964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cuevas CA, Gonzalez AA, Inestrosa NC, Vio CP, Prieto MC. Angiotensin II increases fibronectin and collagen I through the β-catenin-dependent signaling in mouse collecting duct cells. Am J Physiol Renal Physiol 308: F358–F365, 2015. doi: 10.1152/ajprenal.00429.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Cuevas CA, Tapia-Rojas C, Cespedes C, Inestrosa NC, Vio CP. β-Catenin-Dependent Signaling Pathway Contributes to Renal Fibrosis in Hypertensive Rats. BioMed Res Int 2015: 726012, 2015. doi: 10.1155/2015/726012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cui M, Cai Z, Chu S, Sun Z, Wang X, Hu L, Yi J, Shen L, He B. Orphan Nuclear Receptor Nur77 Inhibits Angiotensin II-Induced Vascular Remodeling via Downregulation of β-Catenin. Hypertension 67: 153–162, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06114. [DOI] [PubMed] [Google Scholar]
- 195.Cui T, Nakagami H, Iwai M, Takeda Y, Shiuchi T, Tamura K, Daviet L, Horiuchi M. ATRAP, novel AT1 receptor associated protein, enhances internalization of AT1 receptor and inhibits vascular smooth muscle cell growth. Biochem Biophys Res Commun 279: 938–941, 2000. doi: 10.1006/bbrc.2000.4055. [DOI] [PubMed] [Google Scholar]
- 196.Cybulsky MI, Cheong C, Robbins CS. Macrophages and Dendritic Cells: Partners in Atherogenesis. Circ Res 118: 637–652, 2016. doi: 10.1161/CIRCRESAHA.115.306542. [DOI] [PubMed] [Google Scholar]
- 197.D’Amore A, Black MJ, Thomas WG. The angiotensin II type 2 receptor causes constitutive growth of cardiomyocytes and does not antagonize angiotensin II type 1 receptor-mediated hypertrophy. Hypertension 46: 1347–1354, 2005. doi: 10.1161/01.HYP.0000193504.51489.cf. [DOI] [PubMed] [Google Scholar]
- 198.Dabul S, Bathgate-Siryk A, Valero TR, Jafferjee M, Sturchler E, McDonald P, Koch WJ, Lymperopoulos A. Suppression of adrenal βarrestin1-dependent aldosterone production by ARBs: head-to-head comparison. Sci Rep 5: 8116, 2015. doi: 10.1038/srep08116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Dahlöf B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H; LIFE Study Group . Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 359: 995–1003, 2002. doi: 10.1016/S0140-6736(02)08089-3. [DOI] [PubMed] [Google Scholar]
- 200.Dai DF, Chen T, Szeto H, Nieves-Cintrón M, Kutyavin V, Santana LF, Rabinovitch PS. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol 58: 73–82, 2011. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintrón M, Chen T, Marcinek DJ, Dorn GW II, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 108: 837–846, 2011. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Dai HL, Hu WY, Jiang LH, Li L, Gaung XF, Xiao ZC. p38 MAPK Inhibition Improves Synaptic Plasticity and Memory in Angiotensin II-dependent Hypertensive Mice. Sci Rep 6: 27600, 2016. doi: 10.1038/srep27600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Daiber A, Steven S, Weber A, Shuvaev VV, Muzykantov VR, Laher I, Li H, Lamas S, Münzel T. Targeting vascular (endothelial) dysfunction. Br J Pharmacol 174: 1591–1619, 2017. doi: 10.1111/bph.13517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Danforth E., Jr Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet 26: 13, 2000. doi: 10.1038/79111. [DOI] [PubMed] [Google Scholar]
- 205.Dange RB, Agarwal D, Masson GS, Vila J, Wilson B, Nair A, Francis J. Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II-induced hypertension. Cardiovasc Res 103: 17–27, 2014. doi: 10.1093/cvr/cvu067. [DOI] [PubMed] [Google Scholar]
- 206.Das S, Senapati P, Chen Z, Reddy MA, Ganguly R, Lanting L, Mandi V, Bansal A, Leung A, Zhang S, Jia Y, Wu X, Schones DE, Natarajan R. Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells. Nat Commun 8: 1467, 2017. doi: 10.1038/s41467-017-01629-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest 105: 1605–1612, 2000. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Daugherty A, Poduri A, Chen X, Lu H, Cassis LA. Genetic variants of the Renin Angiotensin system: effects on atherosclerosis in experimental models and humans. Curr Atheroscler Rep 12: 167–173, 2010. doi: 10.1007/s11883-010-0109-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Daugherty A, Rateri DL, Charo IF, Owens AP III, Howatt DA, Cassis LA. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE-/- mice. Clin Sci (Lond) 118: 681–689, 2010. doi: 10.1042/CS20090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Davies L, Jin J, Shen W, Tsui H, Shi Y, Wang Y, Zhang Y, Hao G, Wu J, Chen S, Fraser JA, Dong N, Christoffels V, Ravens U, Huang CLH, Zhang H, Cartwright EJ, Wang X, Lei M. Mkk4 is a negative regulator of the transforming growth factor beta 1 signaling associated with atrial remodeling and arrhythmogenesis with age. J Am Heart Assoc 3: e000340, 2014. doi: 10.1161/JAHA.113.000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Davis FM, Rateri DL, Balakrishnan A, Howatt DA, Strickland DK, Muratoglu SC, Haggerty CM, Fornwalt BK, Cassis LA, Daugherty A. Smooth muscle cell deletion of low-density lipoprotein receptor-related protein 1 augments angiotensin II-induced superior mesenteric arterial and ascending aortic aneurysms. Arterioscler Thromb Vasc Biol 35: 155–162, 2015. doi: 10.1161/ATVBAHA.114.304683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Davis FM, Rateri DL, Daugherty A. Abdominal aortic aneurysm: novel mechanisms and therapies. Curr Opin Cardiol 30: 566–573, 2015. doi: 10.1097/HCO.0000000000000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Davis J, Molkentin JD. Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol 70: 9–18, 2014. doi: 10.1016/j.yjmcc.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Davisson RL, Oliverio MI, Coffman TM, Sigmund CD. Divergent functions of angiotensin II receptor isoforms in the brain. J Clin Invest 106: 103–106, 2000. doi: 10.1172/JCI10022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Dbouk HA, Huang CL, Cobb MH. Hypertension: the missing WNKs. Am J Physiol Renal Physiol 311: F16–F27, 2016. doi: 10.1152/ajprenal.00358.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.De Cárcer G, Wachowicz P, Martínez-Martínez S, Oller J, Méndez-Barbero N, Escobar B, González-Loyola A, Takaki T, El Bakkali A, Cámara JA, Jiménez-Borreguero LJ, Bustelo XR, Cañamero M, Mulero F, de Los Ángeles Sevilla M, Montero MJ, Redondo JM, Malumbres M. Plk1 regulates contraction of postmitotic smooth muscle cells and is required for vascular homeostasis. Nat Med 23: 964–974, 2017. doi: 10.1038/nm.4364. [DOI] [PubMed] [Google Scholar]
- 217.De Cavanagh EM, Ferder LF, Ferder MD, Stella IY, Toblli JE, Inserra F. Vascular structure and oxidative stress in salt-loaded spontaneously hypertensive rats: effects of losartan and atenolol. Am J Hypertens 23: 1318–1325, 2010. doi: 10.1038/ajh.2010.167. [DOI] [PubMed] [Google Scholar]
- 218.De Cavanagh EM, Inserra F, Ferder L. Angiotensin II blockade: a strategy to slow ageing by protecting mitochondria? Cardiovasc Res 89: 31–40, 2011. doi: 10.1093/cvr/cvq285. [DOI] [PubMed] [Google Scholar]
- 219.De Cavanagh EM, Inserra F, Ferder L. Angiotensin II blockade: how its molecular targets may signal to mitochondria and slow aging. Coincidences with calorie restriction and mTOR inhibition. Am J Physiol Heart Circ Physiol 309: H15–H44, 2015. doi: 10.1152/ajpheart.00459.2014. [DOI] [PubMed] [Google Scholar]
- 220.De Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 52: 415–472, 2000. [PubMed] [Google Scholar]
- 221.De Kloet AD, Krause EG, Kim DH, Sakai RR, Seeley RJ, Woods SC. The effect of angiotensin-converting enzyme inhibition using captopril on energy balance and glucose homeostasis. Endocrinology 150: 4114–4123, 2009. doi: 10.1210/en.2009-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.De Kloet AD, Liu M, Rodríguez V, Krause EG, Sumners C. Role of neurons and glia in the CNS actions of the renin-angiotensin system in cardiovascular control. Am J Physiol Regul Integr Comp Physiol 309: R444–R458, 2015. doi: 10.1152/ajpregu.00078.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.De Kloet AD, Pati D, Wang L, Hiller H, Sumners C, Frazier CJ, Seeley RJ, Herman JP, Woods SC, Krause EG. Angiotensin type 1a receptors in the paraventricular nucleus of the hypothalamus protect against diet-induced obesity. J Neurosci 33: 4825–4833, 2013. doi: 10.1523/JNEUROSCI.3806-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.De Kloet AD, Wang L, Ludin JA, Smith JA, Pioquinto DJ, Hiller H, Steckelings UM, Scheuer DA, Sumners C, Krause EG. Reporter mouse strain provides a novel look at angiotensin type-2 receptor distribution in the central nervous system. Brain Struct Funct 221: 891–912, 2016. doi: 10.1007/s00429-014-0943-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.De Kloet AD, Wang L, Pitra S, Hiller H, Smith JA, Tan Y, Nguyen D, Cahill KM, Sumners C, Stern JE, Krause EG. A Unique “Angiotensin-Sensitive” Neuronal Population Coordinates Neuroendocrine, Cardiovascular, and Behavioral Responses to Stress. J Neurosci 37: 3478–3490, 2017. doi: 10.1523/JNEUROSCI.3674-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.De la Pompa JL, Epstein JA. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev Cell 22: 244–254, 2012. doi: 10.1016/j.devcel.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.De Mello WC, Frohlich ED. Clinical perspectives and fundamental aspects of local cardiovascular and renal Renin-Angiotensin systems. Front Endocrinol (Lausanne) 5: 16, 2014. doi: 10.3389/fendo.2014.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Deliu E, Brailoiu GC, Eguchi S, Hoffman NE, Rabinowitz JE, Tilley DG, Madesh M, Koch WJ, Brailoiu E. Direct evidence of intracrine angiotensin II signaling in neurons. Am J Physiol Cell Physiol 306: C736–C744, 2014. doi: 10.1152/ajpcell.00131.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Demos-Davies KM, Ferguson BS, Cavasin MA, Mahaffey JH, Williams SM, Spiltoir JI, Schuetze KB, Horn TR, Chen B, Ferrara C, Scellini B, Piroddi N, Tesi C, Poggesi C, Jeong MY, McKinsey TA. HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. Am J Physiol Heart Circ Physiol 307: H252–H258, 2014. doi: 10.1152/ajpheart.00149.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Devost D, Sleno R, Pétrin D, Zhang A, Shinjo Y, Okde R, Aoki J, Inoue A, Hébert TE. Conformational Profiling of the AT1 Angiotensin II Receptor Reflects Biased Agonism, G Protein Coupling, and Cellular Context. J Biol Chem 292: 5443–5456, 2017. doi: 10.1074/jbc.M116.763854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med 45: 1340–1351, 2008. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Dikalov SI, Nazarewicz RR. Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal 19: 1085–1094, 2013. doi: 10.1089/ars.2012.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Dikalov SI, Nazarewicz RR, Bikineyeva A, Hilenski L, Lassègue B, Griendling KK, Harrison DG, Dikalova AE. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid Redox Signal 20: 281–294, 2014. doi: 10.1089/ars.2012.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Dikalova A, Clempus R, Lassègue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HH, Owens GK, Lambeth JD, Griendling KK. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 112: 2668–2676, 2005. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 235.Dikalova AE, Itani HA, Nazarewicz RR, McMaster WG, Flynn CR, Uzhachenko R, Fessel JP, Gamboa JL, Harrison DG, Dikalov SI. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ Res 121: 564–574, 2017. doi: 10.1161/CIRCRESAHA.117.310933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Diniz GP, Lino CA, Guedes EC, Moreira LN, Barreto-Chaves ML. Cardiac microRNA-133 is down-regulated in thyroid hormone-mediated cardiac hypertrophy partially via Type 1 Angiotensin II receptor. Basic Res Cardiol 110: 49, 2015. doi: 10.1007/s00395-015-0504-7. [DOI] [PubMed] [Google Scholar]
- 237.Dolgacheva LP, Turovskaya MV, Dynnik VV, Zinchenko VP, Goncharov NV, Davletov B, Turovsky EA. Angiotensin II activates different calcium signaling pathways in adipocytes. Arch Biochem Biophys 593: 38–49, 2016. doi: 10.1016/j.abb.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 238.Doller A, Gauer S, Sobkowiak E, Geiger H, Pfeilschifter J, Eberhardt W. Angiotensin II induces renal plasminogen activator inhibitor-1 and cyclooxygenase-2 expression post-transcriptionally via activation of the mRNA-stabilizing factor human-antigen R. Am J Pathol 174: 1252–1263, 2009. doi: 10.2353/ajpath.2009.080652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Domazet I, Holleran BJ, Richard A, Vandenberghe C, Lavigne P, Escher E, Leduc R, Guillemette G. Characterization of Angiotensin II Molecular Determinants Involved in AT1 Receptor Functional Selectivity. Mol Pharmacol 87: 982–995, 2015. doi: 10.1124/mol.114.097337. [DOI] [PubMed] [Google Scholar]
- 240.Dominici FP, Burghi V, Muñoz MC, Giani JF. Modulation of the action of insulin by angiotensin-(1-7). Clin Sci (Lond) 126: 613–630, 2014. doi: 10.1042/CS20130333. [DOI] [PubMed] [Google Scholar]
- 241.Dong B, Yu QT, Dai HY, Gao YY, Zhou ZL, Zhang L, Jiang H, Gao F, Li SY, Zhang YH, Bian HJ, Liu CX, Wang N, Xu H, Pan CM, Song HD, Zhang C, Zhang Y. Angiotensin-converting enzyme-2 overexpression improves left ventricular remodeling and function in a rat model of diabetic cardiomyopathy. J Am Coll Cardiol 59: 739–747, 2012. doi: 10.1016/j.jacc.2011.09.071. [DOI] [PubMed] [Google Scholar]
- 242.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 102: 488–496, 2008. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 243.Douglas G, Bendall JK, Crabtree MJ, Tatham AL, Carter EE, Hale AB, Channon KM. Endothelial-specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE−/− mice. Cardiovasc Res 94: 20–29, 2012. doi: 10.1093/cvr/cvs026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Drawnel FM, Wachten D, Molkentin JD, Maillet M, Aronsen JM, Swift F, Sjaastad I, Liu N, Catalucci D, Mikoshiba K, Hisatsune C, Okkenhaug H, Andrews SR, Bootman MD, Roderick HL. Mutual antagonism between IP(3)RII and miRNA-133a regulates calcium signals and cardiac hypertrophy. J Cell Biol 199: 783–798, 2012. doi: 10.1083/jcb.201111095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Du Bois P, Pablo Tortola C, Lodka D, Kny M, Schmidt F, Song K, Schmidt S, Bassel-Duby R, Olson EN, Fielitz J. Angiotensin II Induces Skeletal Muscle Atrophy by Activating TFEB-Mediated MuRF1 Expression. Circ Res 117: 424–436, 2015. doi: 10.1161/CIRCRESAHA.114.305393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Du J, Sperling LS, Marrero MB, Phillips L, Delafontaine P. G-protein and tyrosine kinase receptor cross-talk in rat aortic smooth muscle cells: thrombin- and angiotensin II-induced tyrosine phosphorylation of insulin receptor substrate-1 and insulin-like growth factor 1 receptor. Biochem Biophys Res Commun 218: 934–939, 1996. doi: 10.1006/bbrc.1996.0165. [DOI] [PubMed] [Google Scholar]
- 247.Duerrschmid C, Trial J, Wang Y, Entman ML, Haudek SB. Tumor necrosis factor: a mechanistic link between angiotensin-II-induced cardiac inflammation and fibrosis. Circ Heart Fail 8: 352–361, 2015. doi: 10.1161/CIRCHEARTFAILURE.114.001893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Durik M, Sevá Pessôa B, Roks AJ. The renin-angiotensin system, bone marrow and progenitor cells. Clin Sci (Lond) 123: 205–223, 2012. doi: 10.1042/CS20110660. [DOI] [PubMed] [Google Scholar]
- 249.Düsterdieck G, McElwee G. Estimation of angiotensin II concentration in human plasma by radioimmunoassay. Some applications to physiological and clinical states. Eur J Clin Invest 2: 32–38, 1971. doi: 10.1111/j.1365-2362.1971.tb00565.x. [DOI] [PubMed] [Google Scholar]
- 250.Ebrahimian T, Li MW, Lemarié CA, Simeone SM, Pagano PJ, Gaestel M, Paradis P, Wassmann S, Schiffrin EL. Mitogen-activated protein kinase-activated protein kinase 2 in angiotensin II-induced inflammation and hypertension: regulation of oxidative stress. Hypertension 57: 245–254, 2011. doi: 10.1161/HYPERTENSIONAHA.110.159889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Eguchi S, Dempsey PJ, Frank GD, Motley ED, Inagami T. Activation of MAPKs by angiotensin II in vascular smooth muscle cells. Metalloprotease-dependent EGF receptor activation is required for activation of ERK and p38 MAPK but not for JNK. J Biol Chem 276: 7957–7962, 2001. doi: 10.1074/jbc.M008570200. [DOI] [PubMed] [Google Scholar]
- 252.Eguchi S, Frank GD, Mifune M, Inagami T. Metalloprotease-dependent ErbB ligand shedding in mediating EGFR transactivation and vascular remodelling. Biochem Soc Trans 31: 1198–1202, 2003. doi: 10.1042/bst0311198. [DOI] [PubMed] [Google Scholar]
- 253.Eguchi S, Iwasaki H, Ueno H, Frank GD, Motley ED, Eguchi K, Marumo F, Hirata Y, Inagami T. Intracellular signaling of angiotensin II-induced p70 S6 kinase phosphorylation at Ser(411) in vascular smooth muscle cells. Possible requirement of epidermal growth factor receptor, Ras, extracellular signal-regulated kinase, and Akt. J Biol Chem 274: 36843–36851, 1999. doi: 10.1074/jbc.274.52.36843. [DOI] [PubMed] [Google Scholar]
- 254.Eguchi S, Numaguchi K, Iwasaki H, Matsumoto T, Yamakawa T, Utsunomiya H, Motley ED, Kawakatsu H, Owada KM, Hirata Y, Marumo F, Inagami T. Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. J Biol Chem 273: 8890–8896, 1998. doi: 10.1074/jbc.273.15.8890. [DOI] [PubMed] [Google Scholar]
- 255.El Bekay R, Alvarez M, Monteseirín J, Alba G, Chacón P, Vega A, Martin-Nieto J, Jiménez J, Pintado E, Bedoya FJ, Sobrino F. Oxidative stress is a critical mediator of the angiotensin II signal in human neutrophils: involvement of mitogen-activated protein kinase, calcineurin, and the transcription factor NF-kappaB. Blood 102: 662–671, 2003. doi: 10.1182/blood-2002-09-2785. [DOI] [PubMed] [Google Scholar]
- 256.Elder DH, McAlpine-Scott V, Choy AM, Struthers AD, Lang CC. Aortic valvular heart disease: Is there a place for angiotensin-converting-enzyme inhibitors? Expert Rev Cardiovasc Ther 11: 107–114, 2013. doi: 10.1586/erc.12.143. [DOI] [PubMed] [Google Scholar]
- 257.Elkahloun AG, Hafko R, Saavedra JM. An integrative genome-wide transcriptome reveals that candesartan is neuroprotective and a candidate therapeutic for Alzheimer’s disease. Alzheimers Res Ther 8: 5, 2016. doi: 10.1186/s13195-015-0167-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Elliott KJ, Bourne AM, Takayanagi T, Takaguri A, Kobayashi T, Eguchi K, Eguchi S. ADAM17 silencing by adenovirus encoding miRNA-embedded siRNA revealed essential signal transduction by angiotensin II in vascular smooth muscle cells. J Mol Cell Cardiol 62: 1–7, 2013. doi: 10.1016/j.yjmcc.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Elliott KJ, Kimura K, Eguchi S. Lack of specificity of commercial antibodies leads to misidentification of angiotensin type-1 receptor protein. Hypertension 61: e31, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00943. [DOI] [PubMed] [Google Scholar]
- 260.Ellis B, Li XC, Miguel-Qin E, Gu V, Zhuo JL. Evidence for a functional intracellular angiotensin system in the proximal tubule of the kidney. Am J Physiol Regul Integr Comp Physiol 302: R494–R509, 2012. doi: 10.1152/ajpregu.00487.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Emeto TI, Moxon JV, Au M, Golledge J. Oxidative stress and abdominal aortic aneurysm: potential treatment targets. Clin Sci (Lond) 130: 301–315, 2016. doi: 10.1042/CS20150547. [DOI] [PubMed] [Google Scholar]
- 262.Erdos B, Backes I, McCowan ML, Hayward LF, Scheuer DA. Brain-derived neurotrophic factor modulates angiotensin signaling in the hypothalamus to increase blood pressure in rats. Am J Physiol Heart Circ Physiol 308: H612–H622, 2015. doi: 10.1152/ajpheart.00776.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Fabbiano S, Menacho-Márquez M, Robles-Valero J, Pericacho M, Matesanz-Marín A, García-Macías C, Sevilla MA, Montero MJ, Alarcón B, López-Novoa JM, Martín P, Bustelo XR. Immunosuppression-Independent Role of Regulatory T Cells against Hypertension-Driven Renal Dysfunctions. Mol Cell Biol 35: 3528–3546, 2015. doi: 10.1128/MCB.00518-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Falkenham A, de Antueno R, Rosin N, Betsch D, Lee TD, Duncan R, Légaré JF. Nonclassical resident macrophages are important determinants in the development of myocardial fibrosis. Am J Pathol 185: 927–942, 2015. doi: 10.1016/j.ajpath.2014.11.027. [DOI] [PubMed] [Google Scholar]
- 265.Falkenham A, Sopel M, Rosin N, Lee TD, Issekutz T, Légaré JF. Early fibroblast progenitor cell migration to the AngII-exposed myocardium is not CXCL12 or CCL2 dependent as previously thought. Am J Pathol 183: 459–469, 2013. doi: 10.1016/j.ajpath.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 266.Fan D, Takawale A, Basu R, Patel V, Lee J, Kandalam V, Wang X, Oudit GY, Kassiri Z. Differential role of TIMP2 and TIMP3 in cardiac hypertrophy, fibrosis, and diastolic dysfunction. Cardiovasc Res 103: 268–280, 2014. doi: 10.1093/cvr/cvu072. [DOI] [PubMed] [Google Scholar]
- 267.Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 5: 15, 2012. doi: 10.1186/1755-1536-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Fan D, Takawale A, Shen M, Samokhvalov V, Basu R, Patel V, Wang X, Fernandez-Patron C, Seubert JM, Oudit GY, Kassiri Z. A Disintegrin and Metalloprotease-17 Regulates Pressure Overload-Induced Myocardial Hypertrophy and Dysfunction Through Proteolytic Processing of Integrin β1. Hypertension 68: 937–948, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07566. [DOI] [PubMed] [Google Scholar]
- 269.Fan LM, Douglas G, Bendall JK, McNeill E, Crabtree MJ, Hale AB, Mai A, Li JM, McAteer MA, Schneider JE, Choudhury RP, Channon KM. Endothelial cell-specific reactive oxygen species production increases susceptibility to aortic dissection. Circulation 129: 2661–2672, 2014. doi: 10.1161/CIRCULATIONAHA.113.005062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Fändriks L. The renin-angiotensin system and the gastrointestinal mucosa. Acta Physiol (Oxf) 201: 157–167, 2011. doi: 10.1111/j.1748-1716.2010.02165.x. [DOI] [PubMed] [Google Scholar]
- 271.Faraco G, Sugiyama Y, Lane D, Garcia-Bonilla L, Chang H, Santisteban MM, Racchumi G, Murphy M, Van Rooijen N, Anrather J, Iadecola C. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J Clin Invest 126: 4674–4689, 2016. doi: 10.1172/JCI86950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Faria-Costa G, Leite-Moreira A, Henriques-Coelho T. Cardiovascular effects of the angiotensin type 2 receptor. Rev Port Cardiol 33: 439–449, 2014. doi: 10.1016/j.repc.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 273.Favre GA, Esnault VL, Van Obberghen E. Modulation of glucose metabolism by the renin-angiotensin-aldosterone system. Am J Physiol Endocrinol Metab 308: E435–E449, 2015. doi: 10.1152/ajpendo.00391.2014. [DOI] [PubMed] [Google Scholar]
- 274.Feinberg MW, Moore KJ. MicroRNA Regulation of Atherosclerosis. Circ Res 118: 703–720, 2016. doi: 10.1161/CIRCRESAHA.115.306300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Feng YH, Ding Y, Ren S, Zhou L, Xu C, Karnik SS. Unconventional homologous internalization of the angiotensin II type-1 receptor induced by G-protein-independent signals. Hypertension 46: 419–425, 2005. doi: 10.1161/01.HYP.0000172621.68061.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Ferrari R, Ceconi C, Campo G, Cangiano E, Cavazza C, Secchiero P, Tavazzi L. Mechanisms of remodelling: a question of life (stem cell production) and death (myocyte apoptosis). Circ J 73: 1973–1982, 2009. doi: 10.1253/circj.CJ-09-0573. [DOI] [PubMed] [Google Scholar]
- 277.Ferrario CM. Cardiac remodelling and RAS inhibition. Ther Adv Cardiovasc Dis 10: 162–171, 2016. doi: 10.1177/1753944716642677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ferrario CM. New physiological concepts of the renin-angiotensin system from the investigation of precursors and products of angiotensin I metabolism. Hypertension 55: 445–452, 2010. doi: 10.1161/HYPERTENSIONAHA.109.145839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Ferrario CM, Ahmad S, Varagic J, Cheng CP, Groban L, Wang H, Collawn JF, Dell'Italia LJ. Intracrine angiotensin II functions originate from noncanonical pathways in the human heart. Am J Physiol Heart Circ Physiol 311: H404–H414, 2016. doi: 10.1152/ajpheart.00219.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Ferreira AJ, Santos RA, Bradford CN, Mecca AP, Sumners C, Katovich MJ, Raizada MK. Therapeutic implications of the vasoprotective axis of the renin-angiotensin system in cardiovascular diseases. Hypertension 55: 207–213, 2010. doi: 10.1161/HYPERTENSIONAHA.109.140145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Fielitz J, Kim M-S, Shelton JM, Qi X, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc Natl Acad Sci USA 105: 3059–3063, 2008. doi: 10.1073/pnas.0712265105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Finckenberg P, Eriksson O, Baumann M, Merasto S, Lalowski MM, Levijoki J, Haasio K, Kytö V, Muller DN, Luft FC, Oresic M, Mervaala E. Caloric restriction ameliorates angiotensin II-induced mitochondrial remodeling and cardiac hypertrophy. Hypertension 59: 76–84, 2012. doi: 10.1161/HYPERTENSIONAHA.111.179457. [DOI] [PubMed] [Google Scholar]
- 283.Fleming I. Signaling by the angiotensin-converting enzyme. Circ Res 98: 887–896, 2006. doi: 10.1161/01.RES.0000217340.40936.53. [DOI] [PubMed] [Google Scholar]
- 284.Flevaris P, Khan SS, Eren M, Schuldt AJT, Shah SJ, Lee DC, Gupta S, Shapiro AD, Burridge PW, Ghosh AK, Vaughan DE. Plasminogen Activator Inhibitor Type I Controls Cardiomyocyte Transforming Growth Factor-β and Cardiac Fibrosis. Circulation 136: 664–679, 2017. doi: 10.1161/CIRCULATIONAHA.117.028145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Folli F, Kahn CR, Hansen H, Bouchie JL, Feener EP. Angiotensin II inhibits insulin signaling in aortic smooth muscle cells at multiple levels. A potential role for serine phosphorylation in insulin/angiotensin II crosstalk. J Clin Invest 100: 2158–2169, 1997. doi: 10.1172/JCI119752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Fontes MS, Kessler EL, van Stuijvenberg L, Brans MA, Falke LL, Kok B, Leask A, van Rijen HV, Vos MA, Goldschmeding R, van Veen TA. CTGF knockout does not affect cardiac hypertrophy and fibrosis formation upon chronic pressure overload. J Mol Cell Cardiol 88: 82–90, 2015. doi: 10.1016/j.yjmcc.2015.09.015. [DOI] [PubMed] [Google Scholar]
- 287.Fornasa G, Clement M, Groyer E, Gaston AT, Khallou-Laschet J, Morvan M, Guedj K, Kaveri SV, Tedgui A, Michel JB, Nicoletti A, Caligiuri G. A CD31-derived peptide prevents angiotensin II-induced atherosclerosis progression and aneurysm formation. Cardiovasc Res 94: 30–37, 2012. doi: 10.1093/cvr/cvs076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Forrester SJ, Elliott KJ, Kawai T, Obama T, Boyer MJ, Preston KJ, Yan Z, Eguchi S, Rizzo V. Caveolin-1 Deletion Prevents Hypertensive Vascular Remodeling Induced by Angiotensin II. Hypertension 69: 79–86, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Forrester SJ, Kawai T, O’Brien S, Thomas W, Harris RC, Eguchi S. Epidermal Growth Factor Receptor Transactivation: Mechanisms, Pathophysiology, and Potential Therapies in the Cardiovascular System. Annu Rev Pharmacol Toxicol 56: 627–653, 2016. doi: 10.1146/annurev-pharmtox-070115-095427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Fraga-Silva RA, Da Silva DG, Montecucco F, Mach F, Stergiopulos N, da Silva RF, Santos RA. The angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas receptor axis: a potential target for treating thrombotic diseases. Thromb Haemost 108: 1089–1096, 2012. doi: 10.1160/TH12-06-0396. [DOI] [PubMed] [Google Scholar]
- 291.Fraga-Silva RA, Montecucco F, Mach F, Santos RA, Stergiopulos N. Pathophysiological role of the renin-angiotensin system on erectile dysfunction. Eur J Clin Invest 43: 978–985, 2013. doi: 10.1111/eci.12117. [DOI] [PubMed] [Google Scholar]
- 292.Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 908: 244–254, 2000. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 293.Freedman JE, Miano JM; National Heart, Lung, and Blood Institute Workshop Participants . Challenges and Opportunities in Linking Long Noncoding RNAs to Cardiovascular, Lung, and Blood Diseases. Arterioscler Thromb Vasc Biol 37: 21–25, 2017. doi: 10.1161/ATVBAHA.116.308513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Freund C, Schmidt-Ullrich R, Baurand A, Dunger S, Schneider W, Loser P, El-Jamali A, Dietz R, Scheidereit C, Bergmann MW. Requirement of nuclear factor-kappaB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation 111: 2319–2325, 2005. doi: 10.1161/01.CIR.0000164237.58200.5A. [DOI] [PubMed] [Google Scholar]
- 295.Frieler RA, Mortensen RM. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation 131: 1019–1030, 2015. doi: 10.1161/CIRCULATIONAHA.114.008788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Fry JL, Shiraishi Y, Turcotte R, Yu X, Gao YZ, Akiki R, Bachschmid M, Zhang Y, Morgan KG, Cohen RA, Seta F. Vascular Smooth Muscle Sirtuin-1 Protects Against Aortic Dissection During Angiotensin II-Induced Hypertension. J Am Heart Assoc 4: e002384, 2015. doi: 10.1161/JAHA.115.002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Fuentes P, Acuña MJ, Cifuentes M, Rojas CV. The anti-adipogenic effect of angiotensin II on human preadipose cells involves ERK1,2 activation and PPARG phosphorylation. J Endocrinol 206: 75–83, 2010. doi: 10.1677/JOE-10-0049. [DOI] [PubMed] [Google Scholar]
- 298.Furuhashi M, Ura N, Higashiura K, Murakami H, Tanaka M, Moniwa N, Yoshida D, Shimamoto K. Blockade of the renin-angiotensin system increases adiponectin concentrations in patients with essential hypertension. Hypertension 42: 76–81, 2003. doi: 10.1161/01.HYP.0000078490.59735.6E. [DOI] [PubMed] [Google Scholar]
- 299.Furuhashi M, Ura N, Takizawa H, Yoshida D, Moniwa N, Murakami H, Higashiura K, Shimamoto K. Blockade of the renin-angiotensin system decreases adipocyte size with improvement in insulin sensitivity. J Hypertens 22: 1977–1982, 2004. doi: 10.1097/00004872-200410000-00021. [DOI] [PubMed] [Google Scholar]
- 300.Gáborik Z, Szaszák M, Szidonya L, Balla B, Paku S, Catt KJ, Clark AJ, Hunyady L. Beta-arrestin- and dynamin-dependent endocytosis of the AT1 angiotensin receptor. Mol Pharmacol 59: 239–247, 2001. doi: 10.1124/mol.59.2.239. [DOI] [PubMed] [Google Scholar]
- 301.Gallo EM, Loch DC, Habashi JP, Calderon JF, Chen Y, Bedja D, van Erp C, Gerber EE, Parker SJ, Sauls K, Judge DP, Cooke SK, Lindsay ME, Rouf R, Myers L, ap Rhys CM, Kent KC, Norris RA, Huso DL, Dietz HC. Angiotensin II-dependent TGF-β signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest 124: 448–460, 2014. doi: 10.1172/JCI69666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Gálvez-Prieto B, Bolbrinker J, Stucchi P, de Las Heras AI, Merino B, Arribas S, Ruiz-Gayo M, Huber M, Wehland M, Kreutz R, Fernandez-Alfonso MS. Comparative expression analysis of the renin-angiotensin system components between white and brown perivascular adipose tissue. J Endocrinol 197: 55–64, 2008. doi: 10.1677/JOE-07-0284. [DOI] [PubMed] [Google Scholar]
- 303.Gao B-B, Phipps JA, Bursell D, Clermont AC, Feener EP. Angiotensin AT1 receptor antagonism ameliorates murine retinal proteome changes induced by diabetes. J Proteome Res 8: 5541–5549, 2009. doi: 10.1021/pr9006415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Gao BB, Stuart L, Feener EP. Label-free quantitative analysis of one-dimensional PAGE LC/MS/MS proteome: application on angiotensin II-stimulated smooth muscle cells secretome. Mol Cell Proteomics 7: 2399–2409, 2008. doi: 10.1074/mcp.M800104-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Gao D, Hao G, Meng Z, Ning N, Yang G, Liu Z, Dong X, Niu X. Rosiglitzone suppresses angiotensin II-induced production of KLF5 and cell proliferation in rat vascular smooth muscle cells. PLoS One 10: e0123724, 2015. doi: 10.1371/journal.pone.0123724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Gao P, Xu TT, Lu J, Li L, Xu J, Hao DL, Chen HZ, Liu DP. Overexpression of SIRT1 in vascular smooth muscle cells attenuates angiotensin II-induced vascular remodeling and hypertension in mice. J Mol Med (Berl) 92: 347–357, 2014. doi: 10.1007/s00109-013-1111-4. [DOI] [PubMed] [Google Scholar]
- 307.García-Marqués F, Trevisan-Herraz M, Martínez-Martínez S, Camafeita E, Jorge I, Lopez JA, Méndez-Barbero N, Méndez-Ferrer S, Del Pozo MA, Ibáñez B, Andrés V, Sánchez-Madrid F, Redondo JM, Bonzon-Kulichenko E, Vázquez J. A Novel Systems-Biology Algorithm for the Analysis of Coordinated Protein Responses Using Quantitative Proteomics. Mol Cell Proteomics 15: 1740–1760, 2016. doi: 10.1074/mcp.M115.055905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Garrido AM, Griendling KK. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol 302: 148–158, 2009. doi: 10.1016/j.mce.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Gassanov N, Brandt MC, Michels G, Lindner M, Er F, Hoppe UC. Angiotensin II-induced changes of calcium sparks and ionic currents in human atrial myocytes: potential role for early remodeling in atrial fibrillation. Cell Calcium 39: 175–186, 2006. doi: 10.1016/j.ceca.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 310.Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, Krause KH. Decreased blood pressure in NOX1-deficient mice. FEBS Lett 580: 497–504, 2006. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- 311.Gavazzi G, Deffert C, Trocme C, Schäppi M, Herrmann FR, Krause KH. NOX1 deficiency protects from aortic dissection in response to angiotensin II. Hypertension 50: 189–196, 2007. doi: 10.1161/HYPERTENSIONAHA.107.089706. [DOI] [PubMed] [Google Scholar]
- 312.Gebhard S, Steil L, Peters B, Gesell-Salazar M, Hammer E, Kuttler B, Clemetson KJ, Scharf C, Peters J, Völker U, Rettig R, Greinacher A. Angiotensin II-dependent hypertension causes reversible changes in the platelet proteome. J Hypertens 29: 2126–2137, 2011. doi: 10.1097/HJH.0b013e32834b1991. [DOI] [PubMed] [Google Scholar]
- 313.George AJ, Purdue BW, Gould CM, Thomas DW, Handoko Y, Qian H, Quaife-Ryan GA, Morgan KA, Simpson KJ, Thomas WG, Hannan RD. A functional siRNA screen identifies genes modulating angiotensin II-mediated EGFR transactivation. J Cell Sci 126: 5377–5390, 2013. doi: 10.1242/jcs.128280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.George M, Vijayakumar A, Dhanesh SB, James J, Shivakumar K. Molecular basis and functional significance of Angiotensin II-induced increase in Discoidin Domain Receptor 2 gene expression in cardiac fibroblasts. J Mol Cell Cardiol 90: 59–69, 2016. doi: 10.1016/j.yjmcc.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 315.Georgescu A, Alexandru N, Nemecz M, Titorencu I, Popov D. Irbesartan administration therapeutically influences circulating endothelial progenitor cell and microparticle mobilization by involvement of pro-inflammatory cytokines. Eur J Pharmacol 711: 27–35, 2013. doi: 10.1016/j.ejphar.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 316.Giani JF, Shah KH, Khan Z, Bernstein EA, Shen XZ, McDonough AA, Gonzalez-Villalobos RA, Bernstein KE. The intrarenal generation of angiotensin II is required for experimental hypertension. Curr Opin Pharmacol 21: 73–81, 2015. doi: 10.1016/j.coph.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Gibbons GH, Pratt RE, Dzau VJ. Vascular smooth muscle cell hypertrophy vs. hyperplasia. Autocrine transforming growth factor-beta 1 expression determines growth response to angiotensin II. J Clin Invest 90: 456–461, 1992. doi: 10.1172/JCI115881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Gildea JJ, Wang X, Shah N, Tran H, Spinosa M, Van Sciver R, Sasaki M, Yatabe J, Carey RM, Jose PA, Felder RA. Dopamine and angiotensin type 2 receptors cooperatively inhibit sodium transport in human renal proximal tubule cells. Hypertension 60: 396–403, 2012. doi: 10.1161/HYPERTENSIONAHA.112.194175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Gimbrone MA Jr, García-Cardeña G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res 118: 620–636, 2016. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Ginnan R, Sun LY, Schwarz JJ, Singer HA. MEF2 is regulated by CaMKIIδ2 and a HDAC4-HDAC5 heterodimer in vascular smooth muscle cells. Biochem J 444: 105–114, 2012. doi: 10.1042/BJ20120152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Glass MJ, Wang G, Coleman CG, Chan J, Ogorodnik E, Van Kempen TA, Milner TA, Butler SD, Young CN, Davisson RL, Iadecola C, Pickel VM. NMDA Receptor Plasticity in the Hypothalamic Paraventricular Nucleus Contributes to the Elevated Blood Pressure Produced by Angiotensin II. J Neurosci 35: 9558–9567, 2015. doi: 10.1523/JNEUROSCI.2301-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Goette A, Bukowska A, Lendeckel U, Erxleben M, Hammwöhner M, Strugala D, Pfeiffenberger J, Röhl FW, Huth C, Ebert MP, Klein HU, Röcken C. Angiotensin II receptor blockade reduces tachycardia-induced atrial adhesion molecule expression. Circulation 117: 732–742, 2008. doi: 10.1161/CIRCULATIONAHA.107.730101. [DOI] [PubMed] [Google Scholar]
- 323.Goette A, Staack T, Röcken C, Arndt M, Geller JC, Huth C, Ansorge S, Klein HU, Lendeckel U. Increased expression of extracellular signal-regulated kinase and angiotensin-converting enzyme in human atria during atrial fibrillation. J Am Coll Cardiol 35: 1669–1677, 2000. doi: 10.1016/S0735-1097(00)00611-2. [DOI] [PubMed] [Google Scholar]
- 324.Goldfinger JZ, Halperin JL, Marin ML, Stewart AS, Eagle KA, Fuster V. Thoracic aortic aneurysm and dissection. J Am Coll Cardiol 64: 1725–1739, 2014. doi: 10.1016/j.jacc.2014.08.025. [DOI] [PubMed] [Google Scholar]
- 325.Gomolak JR, Didion SP. Angiotensin II-induced endothelial dysfunction is temporally linked with increases in interleukin-6 and vascular macrophage accumulation. Front Physiol 5: 396, 2014. doi: 10.3389/fphys.2014.00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Gongora MC, Qin Z, Laude K, Kim HW, McCann L, Folz JR, Dikalov S, Fukai T, Harrison DG. Role of extracellular superoxide dismutase in hypertension. Hypertension 48: 473–481, 2006. doi: 10.1161/01.HYP.0000235682.47673.ab. [DOI] [PubMed] [Google Scholar]
- 327.Gonzalez AD, Wang G, Waters EM, Gonzales KL, Speth RC, Van Kempen TA, Marques-Lopes J, Young CN, Butler SD, Davisson RL, Iadecola C, Pickel VM, Pierce JP, Milner TA. Distribution of angiotensin type 1a receptor-containing cells in the brains of bacterial artificial chromosome transgenic mice. Neuroscience 226: 489–509, 2012. doi: 10.1016/j.neuroscience.2012.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Gonzalez CD, Lee MS, Marchetti P, Pietropaolo M, Towns R, Vaccaro MI, Watada H, Wiley JW. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy 7: 2–11, 2011. doi: 10.4161/auto.7.1.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.González GE, Rhaleb NE, D’Ambrosio MA, Nakagawa P, Liu Y, Leung P, Dai X, Yang XP, Peterson EL, Carretero OA. Deletion of interleukin-6 prevents cardiac inflammation, fibrosis and dysfunction without affecting blood pressure in angiotensin II-high salt-induced hypertension. J Hypertens 33: 144–152, 2015. doi: 10.1097/HJH.0000000000000358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.González-Hernández ML, Godínez-Hernández D, Bobadilla-Lugo RA, López-Sánchez P. Angiotensin-II type 1 receptor (AT1R) and alpha-1D adrenoceptor form a heterodimer during pregnancy-induced hypertension. Auton Autacoid Pharmacol 30: 167–172, 2010. doi: 10.1111/j.1474-8673.2009.00446.x. [DOI] [PubMed] [Google Scholar]
- 331.González-Núñez M, Riolobos AS, Castellano O, Fuentes-Calvo I, de los Ángeles Sevilla M, Oujo B, Pericacho M, Cruz-Gonzalez I, Pérez-Barriocanal F, ten Dijke P, López-Novoa JM. Heterozygous disruption of activin receptor-like kinase 1 is associated with increased arterial pressure in mice. Dis Model Mech 8: 1427–1439, 2015. doi: 10.1242/dmm.019695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE, McDonough AA. The absence of intrarenal ACE protects against hypertension. J Clin Invest 123: 2011–2023, 2013. doi: 10.1172/JCI65460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Gonzalez-Villalobos RA, Satou R, Seth DM, Semprun-Prieto LC, Katsurada A, Kobori H, Navar LG. Angiotensin-converting enzyme-derived angiotensin II formation during angiotensin II-induced hypertension. Hypertension 53: 351–355, 2009. doi: 10.1161/HYPERTENSIONAHA.108.124511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Goupil E, Fillion D, Clément S, Luo X, Devost D, Sleno R, Pétrin D, Saragovi HU, Thorin É, Laporte SA, Hébert TE. Angiotensin II type I and prostaglandin F2α receptors cooperatively modulate signaling in vascular smooth muscle cells. J Biol Chem 290: 3137–3148, 2015. doi: 10.1074/jbc.M114.631119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Grace JA, Herath CB, Mak KY, Burrell LM, Angus PW. Update on new aspects of the renin-angiotensin system in liver disease: clinical implications and new therapeutic options. Clin Sci (Lond) 123: 225–239, 2012. doi: 10.1042/CS20120030. [DOI] [PubMed] [Google Scholar]
- 336.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 74: 1141–1148, 1994. doi: 10.1161/01.RES.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 337.Griendling KK, Ushio-Fukai M, Lassègue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension 29: 366–370, 1997. doi: 10.1161/01.HYP.29.1.366. [DOI] [PubMed] [Google Scholar]
- 338.Groban L, Wang H, Machado FS, Trask AJ, Kritchevsky SB, Ferrario CM, Diz DI. Low glial angiotensinogen improves body habitus, diastolic function, and exercise tolerance in aging male rats. Cardiovasc Endocrinol 1: 49–58, 2012. doi: 10.1097/XCE.0b013e32835a2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Grobe JL, Grobe CL, Beltz TG, Westphal SG, Morgan DA, Xu D, de Lange WJ, Li H, Sakai K, Thedens DR, Cassis LA, Rahmouni K, Mark AL, Johnson AK, Sigmund CD. The brain Renin-angiotensin system controls divergent efferent mechanisms to regulate fluid and energy balance. Cell Metab 12: 431–442, 2010. doi: 10.1016/j.cmet.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Grobe JL, Xu D, Sigmund CD. An intracellular renin-angiotensin system in neurons: fact, hypothesis, or fantasy. Physiology (Bethesda) 23: 187–193, 2008. doi: 10.1152/physiol.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Gu J, Liu X, Wang QX, Tan HW, Guo M, Jiang WF, Zhou L. Angiotensin II increases CTGF expression via MAPKs/TGF-β1/TRAF6 pathway in atrial fibroblasts. Exp Cell Res 318: 2105–2115, 2012. doi: 10.1016/j.yexcr.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 342.Guilluy C, Brégeon J, Toumaniantz G, Rolli-Derkinderen M, Retailleau K, Loufrani L, Henrion D, Scalbert E, Bril A, Torres RM, Offermanns S, Pacaud P, Loirand G. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med 16: 183–190, 2010. doi: 10.1038/nm.2079. [DOI] [PubMed] [Google Scholar]
- 343.Guo DF, Chenier I, Lavoie JL, Chan JS, Hamet P, Tremblay J, Chen XM, Wang DH, Inagami T. Development of hypertension and kidney hypertrophy in transgenic mice overexpressing ARAP1 gene in the kidney. Hypertension 48: 453–459, 2006. doi: 10.1161/01.HYP.0000230664.32874.52. [DOI] [PubMed] [Google Scholar]
- 344.Guo DF, Chenier I, Tardif V, Orlov SN, Inagami T. Type 1 angiotensin II receptor-associated protein ARAP1 binds and recycles the receptor to the plasma membrane. Biochem Biophys Res Commun 310: 1254–1265, 2003. doi: 10.1016/j.bbrc.2003.09.154. [DOI] [PubMed] [Google Scholar]
- 345.Guo DF, Tardif V, Ghelima K, Chan JS, Ingelfinger JR, Chen X, Chenier I. A novel angiotensin II type 1 receptor-associated protein induces cellular hypertrophy in rat vascular smooth muscle and renal proximal tubular cells. J Biol Chem 279: 21109–21120, 2004. doi: 10.1074/jbc.M401544200. [DOI] [PubMed] [Google Scholar]
- 346.Guo F, Chen XL, Wang F, Liang X, Sun YX, Wang YJ. Role of angiotensin II type 1 receptor in angiotensin II-induced cytokine production in macrophages. J Interferon Cytokine Res 31: 351–361, 2011. doi: 10.1089/jir.2010.0073. [DOI] [PubMed] [Google Scholar]
- 347.Gurley SB, Riquier-Brison ADM, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab 13: 469–475, 2011. doi: 10.1016/j.cmet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science 332: 361–365, 2011. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Habibi J, DeMarco VG, Ma L, Pulakat L, Rainey WE, Whaley-Connell AT, Sowers JR. Mineralocorticoid receptor blockade improves diastolic function independent of blood pressure reduction in a transgenic model of RAAS overexpression. Am J Physiol Heart Circ Physiol 300: H1484–H1491, 2011. doi: 10.1152/ajpheart.01000.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Hafko R, Villapol S, Nostramo R, Symes A, Sabban EL, Inagami T, Saavedra JM. Commercially available angiotensin II At2 receptor antibodies are nonspecific. PLoS One 8: e69234, 2013. doi: 10.1371/journal.pone.0069234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Han YL, Li YL, Jia LX, Cheng JZ, Qi YF, Zhang HJ, Du J. Reciprocal interaction between macrophages and T cells stimulates IFN-γ and MCP-1 production in Ang II-induced cardiac inflammation and fibrosis. PLoS One 7: e35506, 2012. doi: 10.1371/journal.pone.0035506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Hans CP, Koenig SN, Huang N, Cheng J, Beceiro S, Guggilam A, Kuivaniemi H, Partida-Sánchez S, Garg V. Inhibition of Notch1 signaling reduces abdominal aortic aneurysm in mice by attenuating macrophage-mediated inflammation. Arterioscler Thromb Vasc Biol 32: 3012–3023, 2012. doi: 10.1161/ATVBAHA.112.254219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Hansson L, Lindholm LH, Niskanen L, Lanke J, Hedner T, Niklason A, Luomanmäki K, Dahlöf B, de Faire U, Mörlin C, Karlberg BE, Wester PO, Björck JE. Effect of angiotensin-converting-enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPPP) randomised trial. Lancet 353: 611–616, 1999. doi: 10.1016/S0140-6736(98)05012-0. [DOI] [PubMed] [Google Scholar]
- 355.Harada K, Komuro I, Shiojima I, Hayashi D, Kudoh S, Mizuno T, Kijima K, Matsubara H, Sugaya T, Murakami K, Yazaki Y. Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation 97: 1952–1959, 1998. doi: 10.1161/01.CIR.97.19.1952. [DOI] [PubMed] [Google Scholar]
- 356.Harada K, Komuro I, Zou Y, Kudoh S, Kijima K, Matsubara H, Sugaya T, Murakami K, Yazaki Y. Acute pressure overload could induce hypertrophic responses in the heart of angiotensin II type 1a knockout mice. Circ Res 82: 779–785, 1998. doi: 10.1161/01.RES.82.7.779. [DOI] [PubMed] [Google Scholar]
- 357.Harada M, Luo X, Qi XY, Tadevosyan A, Maguy A, Ordog B, Ledoux J, Kato T, Naud P, Voigt N, Shi Y, Kamiya K, Murohara T, Kodama I, Tardif J-C, Schotten U, Van Wagoner DR, Dobrev D, Nattel S. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 126: 2051–2064, 2012. doi: 10.1161/CIRCULATIONAHA.112.121830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Harman D. Free radical theory of aging: an update: increasing the functional life span. Ann N Y Acad Sci 1067: 10–21, 2006. doi: 10.1196/annals.1354.003. [DOI] [PubMed] [Google Scholar]
- 359.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460: 392–395, 2009. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Harvey A, Montezano AC, Lopes RA, Rios F, Touyz RM. Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Can J Cardiol 32: 659–668, 2016. doi: 10.1016/j.cjca.2016.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Hasan DM, Starke RM, Gu H, Wilson K, Chu Y, Chalouhi N, Heistad DD, Faraci FM, Sigmund CD. Smooth Muscle Peroxisome Proliferator-Activated Receptor γ Plays a Critical Role in Formation and Rupture of Cerebral Aneurysms in Mice In Vivo. Hypertension 66: 211–220, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial J, Taffet GE, Entman ML. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol 49: 499–507, 2010. doi: 10.1016/j.yjmcc.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Haulica I, Bild W, Serban DN. Angiotensin peptides and their pleiotropic actions. J Renin Angiotensin Aldosterone Syst 6: 121–131, 2005. doi: 10.3317/jraas.2005.018. [DOI] [PubMed] [Google Scholar]
- 364.Haznedaroglu IC, Beyazit Y. Local bone marrow renin-angiotensin system in primitive, definitive and neoplastic haematopoiesis. Clin Sci (Lond) 124: 307–323, 2013. doi: 10.1042/CS20120300. [DOI] [PubMed] [Google Scholar]
- 365.He P, Klein J, Yun CC. Activation of Na+/H+ exchanger NHE3 by angiotensin II is mediated by inositol 1,4,5-triphosphate (IP3) receptor-binding protein released with IP3 (IRBIT) and Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 285: 27869–27878, 2010. doi: 10.1074/jbc.M110.133066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.He P, Zhao L, No YR, Karvar S, Yun CC. The NHERF1 PDZ1 domain and IRBIT interact and mediate the activation of Na+/H+ exchanger 3 by ANG II. Am J Physiol Renal Physiol 311: F343–F351, 2016. doi: 10.1152/ajprenal.00247.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.He X, Gao X, Peng L, Wang S, Zhu Y, Ma H, Lin J, Duan DD. Atrial fibrillation induces myocardial fibrosis through angiotensin II type 1 receptor-specific Arkadia-mediated downregulation of Smad7. Circ Res 108: 164–175, 2011. doi: 10.1161/CIRCRESAHA.110.234369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.He Z, Zhang X, Chen C, Wen Z, Hoopes SL, Zeldin DC, Wang DW. Cardiomyocyte-specific expression of CYP2J2 prevents development of cardiac remodelling induced by angiotensin II. Cardiovasc Res 105: 304–317, 2015. doi: 10.1093/cvr/cvv018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Healey JS, Baranchuk A, Crystal E, Morillo CA, Garfinkle M, Yusuf S, Connolly SJ. Prevention of atrial fibrillation with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: a meta-analysis. J Am Coll Cardiol 45: 1832–1839, 2005. doi: 10.1016/j.jacc.2004.11.070. [DOI] [PubMed] [Google Scholar]
- 370.Heeneman S, Haendeler J, Saito Y, Ishida M, Berk BC. Angiotensin II induces transactivation of two different populations of the platelet-derived growth factor beta receptor. Key role for the p66 adaptor protein Shc. J Biol Chem 275: 15926–15932, 2000. doi: 10.1074/jbc.M909616199. [DOI] [PubMed] [Google Scholar]
- 371.Heinze C, Seniuk A, Sokolov MV, Huebner AK, Klementowicz AE, Szijártó IA, Schleifenbaum J, Vitzthum H, Gollasch M, Ehmke H, Schroeder BC, Hübner CA. Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. J Clin Invest 124: 675–686, 2014. doi: 10.1172/JCI70025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Heiss C, Spyridopoulos I, Haendeler J. Interventions to slow cardiovascular aging: Dietary restriction, drugs and novel molecules [In press]. Exp Gerontol S0531-5565(17)30293-0, 2017. doi: 10.1016/j.exger.2017.06.015. [DOI] [PubMed] [Google Scholar]
- 373.Henriksen EJ, Prasannarong M. The role of the renin-angiotensin system in the development of insulin resistance in skeletal muscle. Mol Cell Endocrinol 378: 15–22, 2013. doi: 10.1016/j.mce.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 374.Herbert KE, Mistry Y, Hastings R, Poolman T, Niklason L, Williams B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ Res 102: 201–208, 2008. doi: 10.1161/CIRCRESAHA.107.158626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Hermans H, Swinnen M, Pokreisz P, Caluwé E, Dymarkowski S, Herregods MC, Janssens S, Herijgers P. Murine pressure overload models: a 30-MHz look brings a whole new “sound” into data interpretation. J Appl Physiol (1985) 117: 563–571, 2014. doi: 10.1152/japplphysiol.00363.2014. [DOI] [PubMed] [Google Scholar]
- 376.Herr D, Bekes I, Wulff C. Local Renin-Angiotensin system in the reproductive system. Front Endocrinol (Lausanne) 4: 150, 2013. doi: 10.3389/fendo.2013.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Herrera M, Sparks MA, Alfonso-Pecchio AR, Harrison-Bernard LM, Coffman TM. Lack of specificity of commercial antibodies leads to misidentification of angiotensin type 1 receptor protein. Hypertension 61: 253–258, 2013. doi: 10.1161/HYPERTENSIONAHA.112.203679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Higashi Y, Maruhashi T, Noma K, Kihara Y. Oxidative stress and endothelial dysfunction: clinical evidence and therapeutic implications. Trends Cardiovasc Med 24: 165–169, 2014. doi: 10.1016/j.tcm.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 379.Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci (Lond) 112: 417–428, 2007. doi: 10.1042/CS20060342. [DOI] [PubMed] [Google Scholar]
- 380.Hilgers RH, Todd J Jr, Webb RC. Increased PDZ-RhoGEF/RhoA/Rho kinase signaling in small mesenteric arteries of angiotensin II-induced hypertensive rats. J Hypertens 25: 1687–1697, 2007. doi: 10.1097/HJH.0b013e32816f778d. [DOI] [PubMed] [Google Scholar]
- 381.Hilzendeger AM, Cassell MD, Davis DR, Stauss HM, Mark AL, Grobe JL, Sigmund CD. Angiotensin type 1a receptors in the subfornical organ are required for deoxycorticosterone acetate-salt hypertension. Hypertension 61: 716–722, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Hilzendeger AM, Morgan DA, Brooks L, Dellsperger D, Liu X, Grobe JL, Rahmouni K, Sigmund CD, Mark AL. A brain leptin-renin angiotensin system interaction in the regulation of sympathetic nerve activity. Am J Physiol Heart Circ Physiol 303: H197–H206, 2012. doi: 10.1152/ajpheart.00974.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Hindmarch CC, Ferguson AV. Physiological roles for the subfornical organ: a dynamic transcriptome shaped by autonomic state. J Physiol 594: 1581–1589, 2016. doi: 10.1113/JP270726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Hinoki A, Kimura K, Higuchi S, Eguchi K, Takaguri A, Ishimaru K, Frank GD, Gerthoffer WT, Sommerville LJ, Autieri MV, Eguchi S. p21-activated kinase 1 participates in vascular remodeling in vitro and in vivo. Hypertension 55: 161–165, 2010. doi: 10.1161/HYPERTENSIONAHA.109.143057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Hisatake S, Kiuchi S, Kabuki T, Oka T, Dobashi S, Ikeda T. Serum angiotensin-converting enzyme 2 concentration and angiotensin-(1-7) concentration in patients with acute heart failure patients requiring emergency hospitalization. Heart Vessels 32: 303–308, 2017. doi: 10.1007/s00380-016-0877-z. [DOI] [PubMed] [Google Scholar]
- 386.Hiyama TY, Noda M. Sodium sensing in the subfornical organ and body-fluid homeostasis. Neurosci Res 113: 1–11, 2016. doi: 10.1016/j.neures.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 387.Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol 296: R208–R216, 2009. doi: 10.1152/ajpregu.90521.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Hodavance SY, Gareri C, Torok RD, Rockman HA. G Protein-coupled Receptor Biased Agonism. J Cardiovasc Pharmacol 67: 193–202, 2016. doi: 10.1097/FJC.0000000000000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Holopainen T, Räsänen M, Anisimov A, Tuomainen T, Zheng W, Tvorogov D, Hulmi JJ, Andersson LC, Cenni B, Tavi P, Mervaala E, Kivelä R, Alitalo K. Endothelial Bmx tyrosine kinase activity is essential for myocardial hypertrophy and remodeling. Proc Natl Acad Sci USA 112: 13063–13068, 2015. doi: 10.1073/pnas.1517810112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Hong K, Li M, Nourian Z, Meininger GA, Hill MA. Angiotensin II Type 1 Receptor Mechanoactivation Involves RGS5 (Regulator of G Protein Signaling 5) in Skeletal Muscle Arteries: Impaired Trafficking of RGS5 in Hypertension. Hypertension 70: 1264–1272, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Hoorn EJ, Nelson JH, McCormick JA, Ellison DH. The WNK kinase network regulating sodium, potassium, and blood pressure. J Am Soc Nephrol 22: 605–614, 2011. doi: 10.1681/ASN.2010080827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Horiuchi M, Mogi M. Role of angiotensin II receptor subtype activation in cognitive function and ischaemic brain damage. Br J Pharmacol 163: 1122–1130, 2011. doi: 10.1111/j.1476-5381.2010.01167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Horwath JA, Hurr C, Butler SD, Guruju M, Cassell MD, Mark AL, Davisson RL, Young CN. Obesity-induced hepatic steatosis is mediated by endoplasmic reticulum stress in the subfornical organ of the brain. JCI Insight 2: e90170, 2017. doi: 10.1172/jci.insight.90170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Hotamisligil GS. Inflammation and metabolic disorders. Nature 444: 860–867, 2006. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 395.Howatt DA, Balakrishnan A, Moorleghen JJ, Muniappan L, Rateri DL, Uchida HA, Takano J, Saido TC, Chishti AH, Baud L, Subramanian V. Leukocyte Calpain Deficiency Reduces Angiotensin II-Induced Inflammation and Atherosclerosis But Not Abdominal Aortic Aneurysms in Mice. Arterioscler Thromb Vasc Biol 36: 835–845, 2016. doi: 10.1161/ATVBAHA.116.307285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Howitt MR, Garrett WS. A complex microworld in the gut: gut microbiota and cardiovascular disease connectivity. Nat Med 18: 1188–1189, 2012. doi: 10.1038/nm.2895. [DOI] [PubMed] [Google Scholar]
- 397.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425: 191–196, 2003. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 398.Hu C, Keen HL, Lu KT, Liu X, Wu J, Davis DR, Ibeawuchi SC, Vogel S, Quelle FW, Sigmund CD. Retinol-binding protein 7 is an endothelium-specific PPARγ cofactor mediating an antioxidant response through adiponectin. JCI Insight 2: e91738, 2017. doi: 10.1172/jci.insight.91738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Hu C, Lu KT, Mukohda M, Davis DR, Faraci FM, Sigmund CD. Interference with PPARγ in endothelium accelerates angiotensin II-induced endothelial dysfunction. Physiol Genomics 48: 124–134, 2016. doi: 10.1152/physiolgenomics.00087.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Huang CY, Kuo WW, Yeh YL, Ho TJ, Lin JY, Lin DY, Chu CH, Tsai FJ, Tsai CH, Huang CY. ANG II promotes IGF-IIR expression and cardiomyocyte apoptosis by inhibiting HSF1 via JNK activation and SIRT1 degradation. Cell Death Differ 21: 1262–1274, 2014. doi: 10.1038/cdd.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Huang J, Sun W, Huang H, Ye J, Pan W, Zhong Y, Cheng C, You X, Liu B, Xiong L, Liu S. miR-34a modulates angiotensin II-induced myocardial hypertrophy by direct inhibition of ATG9A expression and autophagic activity. PLoS One 9: e94382, 2014. doi: 10.1371/journal.pone.0094382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Huang J, Yamashiro Y, Papke CL, Ikeda Y, Lin Y, Patel M, Inagami T, Le VP, Wagenseil JE, Yanagisawa H. Angiotensin-converting enzyme-induced activation of local angiotensin signaling is required for ascending aortic aneurysms in fibulin-4-deficient mice. Sci Transl Med 5: 183ra58, 2013. doi: 10.1126/scitranslmed.3005025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Huang XR, Chung AC, Yang F, Yue W, Deng C, Lau CP, Tse HF, Lan HY. Smad3 mediates cardiac inflammation and fibrosis in angiotensin II-induced hypertensive cardiac remodeling. Hypertension 55: 1165–1171, 2010. doi: 10.1161/HYPERTENSIONAHA.109.147611. [DOI] [PubMed] [Google Scholar]
- 404.Huang Y, Di Lorenzo A, Jiang W, Cantalupo A, Sessa WC, Giordano FJ. Hypoxia-inducible factor-1α in vascular smooth muscle regulates blood pressure homeostasis through a peroxisome proliferator-activated receptor-γ-angiotensin II receptor type 1 axis. Hypertension 62: 634–640, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Huang Y, Wu D, Zhang X, Jiang M, Hu C, Lin J, Tang J, Wu L. Cardiac-specific Traf2 overexpression enhances cardiac hypertrophy through activating AKT/GSK3β signaling. Gene 536: 225–231, 2014. doi: 10.1016/j.gene.2013.12.052. [DOI] [PubMed] [Google Scholar]
- 406.Huber MJ, Basu R, Cecchettini C, Cuadra AE, Chen QH, Shan Z. Activation of the (pro)renin receptor in the paraventricular nucleus increases sympathetic outflow in anesthetized rats. Am J Physiol Heart Circ Physiol 309: H880–H887, 2015. doi: 10.1152/ajpheart.00095.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Huggins C, Pearce S, Peri F, Neumann F, Cockerill G, Pirianov G. A novel small molecule TLR4 antagonist (IAXO-102) negatively regulates non-hematopoietic toll like receptor 4 signalling and inhibits aortic aneurysms development. Atherosclerosis 242: 563–570, 2015. doi: 10.1016/j.atherosclerosis.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 408.Huynh K, Bernardo BC, McMullen JR, Ritchie RH. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther 142: 375–415, 2014. doi: 10.1016/j.pharmthera.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 409.Ibeawuchi SR, Agbor LN, Quelle FW, Sigmund CD. Hypertension-causing Mutations in Cullin3 Protein Impair RhoA Protein Ubiquitination and Augment the Association with Substrate Adaptors. J Biol Chem 290: 19208–19217, 2015. doi: 10.1074/jbc.M115.645358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Ichihara S, Senbonmatsu T, Price E Jr, Ichiki T, Gaffney FA, Inagami T. Angiotensin II type 2 receptor is essential for left ventricular hypertrophy and cardiac fibrosis in chronic angiotensin II-induced hypertension. Circulation 104: 346–351, 2001. doi: 10.1161/01.CIR.104.3.346. [DOI] [PubMed] [Google Scholar]
- 411.Ichiki T. Regulation of angiotensin II receptor expression. Curr Pharm Des 19: 3013–3021, 2013. doi: 10.2174/1381612811319170007. [DOI] [PubMed] [Google Scholar]
- 412.Ijaz T, Sun H, Pinchuk IV, Milewicz DM, Tilton RG, Brasier AR. Deletion of NF-κB/RelA in Angiotensin II-Sensitive Mesenchymal Cells Blocks Aortic Vascular Inflammation and Abdominal Aortic Aneurysm Formation. Arterioscler Thromb Vasc Biol 37: 1881–1890, 2017. doi: 10.1161/ATVBAHA.117.309863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Ikeda Y, Kumagai H, Motozawa Y, Suzuki J, Komuro I. Biased Agonism of the Angiotensin II Type I Receptor. Int Heart J 56: 485–488, 2015. doi: 10.1536/ihj.15-256. [DOI] [PubMed] [Google Scholar]
- 414.Imanishi M, Tomita S, Ishizawa K, Kihira Y, Ueno M, Izawa-Ishizawa Y, Ikeda Y, Yamano N, Tsuchiya K, Tamaki T. Smooth muscle cell-specific Hif-1α deficiency suppresses angiotensin II-induced vascular remodelling in mice. Cardiovasc Res 102: 460–468, 2014. doi: 10.1093/cvr/cvu061. [DOI] [PubMed] [Google Scholar]
- 415.Inaba S, Iwai M, Furuno M, Tomono Y, Kanno H, Senba I, Okayama H, Mogi M, Higaki J, Horiuchi M. Continuous activation of renin-angiotensin system impairs cognitive function in renin/angiotensinogen transgenic mice. Hypertension 53: 356–362, 2009. doi: 10.1161/HYPERTENSIONAHA.108.123612. [DOI] [PubMed] [Google Scholar]
- 416.Inuzuka T, Fujioka Y, Tsuda M, Fujioka M, Satoh AO, Horiuchi K, Nishide S, Nanbo A, Tanaka S, Ohba Y. Attenuation of ligand-induced activation of angiotensin II type 1 receptor signaling by the type 2 receptor via protein kinase C. Sci Rep 6: 21613, 2016. doi: 10.1038/srep21613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Isegawa K, Hirooka Y, Katsuki M, Kishi T, Sunagawa K. Angiotensin II type 1 receptor expression in astrocytes is upregulated leading to increased mortality in mice with myocardial infarction-induced heart failure. Am J Physiol Heart Circ Physiol 307: H1448–H1455, 2014. doi: 10.1152/ajpheart.00462.2014. [DOI] [PubMed] [Google Scholar]
- 418.Ishibashi M, Hiasa K, Zhao Q, Inoue S, Ohtani K, Kitamoto S, Tsuchihashi M, Sugaya T, Charo IF, Kura S, Tsuzuki T, Ishibashi T, Takeshita A, Egashira K. Critical role of monocyte chemoattractant protein-1 receptor CCR2 on monocytes in hypertension-induced vascular inflammation and remodeling. Circ Res 94: 1203–1210, 2004. doi: 10.1161/01.RES.0000126924.23467.A3. [DOI] [PubMed] [Google Scholar]
- 419.Ishizaka N, Griendling KK, Lassègue B, Alexander RW. Angiotensin II type 1 receptor: relationship with caveolae and caveolin after initial agonist stimulation. Hypertension 32: 459–466, 1998. doi: 10.1161/01.HYP.32.3.459. [DOI] [PubMed] [Google Scholar]
- 420.Isselbacher EM. Thoracic and abdominal aortic aneurysms. Circulation 111: 816–828, 2005. doi: 10.1161/01.CIR.0000154569.08857.7A. [DOI] [PubMed] [Google Scholar]
- 421.Itani HA, Dikalova AE, McMaster WG, Nazarewicz RR, Bikineyeva AT, Harrison DG, Dikalov SI. Mitochondrial Cyclophilin D in Vascular Oxidative Stress and Hypertension. Hypertension 67: 1218–1227, 2016. doi: 10.1161/HYPERTENSIONAHA.115.07085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Itani HA, McMaster WG Jr, Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM, Konior A, Prejbisz A, Januszewicz A, Norlander AE, Chen W, Bonami RH, Marshall AF, Poffenberger G, Weyand CM, Madhur MS, Moore DJ, Harrison DG, Guzik TJ. Activation of Human T Cells in Hypertension: Studies of Humanized Mice and Hypertensive Humans. Hypertension 68: 123–132, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, Barbaro NR, Foss JD, Kirabo A, Montaniel KR, Norlander AE, Chen W, Sato R, Navar LG, Mallal SA, Madhur MS, Bernstein KE, Harrison DG. CD70 Exacerbates Blood Pressure Elevation and Renal Damage in Response to Repeated Hypertensive Stimuli. Circ Res 118: 1233–1243, 2016. doi: 10.1161/CIRCRESAHA.115.308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Ito S, Asakura M, Liao Y, Min KD, Takahashi A, Shindo K, Yamazaki S, Tsukamoto O, Asanuma H, Mogi M, Horiuchi M, Asano Y, Sanada S, Minamino T, Takashima S, Mochizuki N, Kitakaze M. Identification of the Mtus1 Splice Variant as a Novel Inhibitory Factor Against Cardiac Hypertrophy. J Am Heart Assoc 5: e003521, 2016. doi: 10.1161/JAHA.116.003521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Iwasaki YK, Nishida K, Kato T, Nattel S. Atrial fibrillation pathophysiology: implications for management. Circulation 124: 2264–2274, 2011. doi: 10.1161/CIRCULATIONAHA.111.019893. [DOI] [PubMed] [Google Scholar]
- 426.Jain S, Tulsulkar J, Rana A, Kumar A, Shah ZA. Transgenic Mice Overexpressing Human Angiotensin I Receptor Gene Are Susceptible to Stroke Injury. Mol Neurobiol 53: 1533–1539, 2016. doi: 10.1007/s12035-015-9109-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Jancovski N, Bassi JK, Carter DA, Choong YT, Connelly A, Nguyen TP, Chen D, Lukoshkova EV, Menuet C, Head GA, Allen AM. Stimulation of angiotensin type 1A receptors on catecholaminergic cells contributes to angiotensin-dependent hypertension. Hypertension 62: 866–871, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01474. [DOI] [PubMed] [Google Scholar]
- 428.Jankowski V, Vanholder R, van der Giet M, Tölle M, Karadogan S, Gobom J, Furkert J, Oksche A, Krause E, Tran TN, Tepel M, Schuchardt M, Schlüter H, Wiedon A, Beyermann M, Bader M, Todiras M, Zidek W, Jankowski J. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler Thromb Vasc Biol 27: 297–302, 2007. doi: 10.1161/01.ATV.0000253889.09765.5f. [DOI] [PubMed] [Google Scholar]
- 429.Jayasooriya AP, Mathai ML, Walker LL, Begg DP, Denton DA, Cameron-Smith D, Egan GF, McKinley MJ, Rodger PD, Sinclair AJ, Wark JD, Weisinger HS, Jois M, Weisinger RS. Mice lacking angiotensin-converting enzyme have increased energy expenditure, with reduced fat mass and improved glucose clearance. Proc Natl Acad Sci USA 105: 6531–6536, 2008. doi: 10.1073/pnas.0802690105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Jennings BL, Sahan-Firat S, Estes AM, Das K, Farjana N, Fang XR, Gonzalez FJ, Malik KU. Cytochrome P450 1B1 contributes to angiotensin II-induced hypertension and associated pathophysiology. Hypertension 56: 667–674, 2010. doi: 10.1161/HYPERTENSIONAHA.110.154518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Ji H, Pai AV, West CA, Wu X, Speth RC, Sandberg K. Loss of Resistance to Angiotensin II-Induced Hypertension in the Jackson Laboratory Recombination-Activating Gene Null Mouse on the C57BL/6J Background. Hypertension 69: 1121–1127, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Ji H, Zheng W, Li X, Liu J, Wu X, Zhang MA, Umans JG, Hay M, Speth RC, Dunn SE, Sandberg K. Sex-specific T-cell regulation of angiotensin II-dependent hypertension. Hypertension 64: 573–582, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Ji YX, Zhang P, Zhang XJ, Zhao YC, Deng KQ, Jiang X, Wang PX, Huang Z, Li H. The ubiquitin E3 ligase TRAF6 exacerbates pathological cardiac hypertrophy via TAK1-dependent signalling. Nat Commun 7: 11267, 2016. doi: 10.1038/ncomms11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Jiang DS, Bian ZY, Zhang Y, Zhang SM, Liu Y, Zhang R, Chen Y, Yang Q, Zhang XD, Fan GC, Li H. Role of interferon regulatory factor 4 in the regulation of pathological cardiac hypertrophy. Hypertension 61: 1193–1202, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Jiang DS, Liu Y, Zhou H, Zhang Y, Zhang XD, Zhang XF, Chen K, Gao L, Peng J, Gong H, Chen Y, Yang Q, Liu PP, Fan GC, Zou Y, Li H. Interferon regulatory factor 7 functions as a novel negative regulator of pathological cardiac hypertrophy. Hypertension 63: 713–722, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Jiang DS, Zhang XF, Gao L, Zong J, Zhou H, Liu Y, Zhang Y, Bian ZY, Zhu LH, Fan GC, Zhang XD, Li H. Signal regulatory protein-α protects against cardiac hypertrophy via the disruption of toll-like receptor 4 signaling. Hypertension 63: 96–104, 2014. doi: 10.1161/HYPERTENSIONAHA.113.01506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Jiang G, Gong H, Niu Y, Yang C, Wang S, Chen Z, Ye Y, Zhou N, Zhang G, Ge J, Zou Y. Identification of Amino Acid Residues in Angiotensin II Type 1 Receptor Sensing Mechanical Stretch and Function in Cardiomyocyte Hypertrophy. Cell Physiol Biochem 37: 105–116, 2015. doi: 10.1159/000430337. [DOI] [PubMed] [Google Scholar]
- 438.Jiang L, Wang M, Zhang J, Monticone RE, Telljohann R, Spinetti G, Pintus G, Lakatta EG. Increased aortic calpain-1 activity mediates age-associated angiotensin II signaling of vascular smooth muscle cells. PLoS One 3: e2231, 2008. doi: 10.1371/journal.pone.0002231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Jiang L, Xu L, Song Y, Li J, Mao J, Zhao AZ, He W, Yang J, Dai C. Calmodulin-dependent protein kinase II/cAMP response element-binding protein/Wnt/β-catenin signaling cascade regulates angiotensin II-induced podocyte injury and albuminuria. J Biol Chem 288: 23368–23379, 2013. doi: 10.1074/jbc.M113.460394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Jiang X, Zhang F, Ning Q. Losartan reverses the down-expression of long noncoding RNA-NR024118 and Cdkn1c induced by angiotensin II in adult rat cardiac fibroblasts. Pathol Biol (Paris) 63: 122–125, 2015. doi: 10.1016/j.patbio.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 441.Jiang XY, Ning QL. Expression profiling of long noncoding RNAs and the dynamic changes of lncRNA-NR024118 and Cdkn1c in angiotensin II-treated cardiac fibroblasts. Int J Clin Exp Pathol 7: 1325–1336, 2014. [PMC free article] [PubMed] [Google Scholar]
- 442.Jin W, Reddy MA, Chen Z, Putta S, Lanting L, Kato M, Park JT, Chandra M, Wang C, Tangirala RK, Natarajan R. Small RNA sequencing reveals microRNAs that modulate angiotensin II effects in vascular smooth muscle cells. J Biol Chem 287: 15672–15683, 2012. doi: 10.1074/jbc.M111.322669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Jing F, Mogi M, Min L-J, Ohshima K, Nakaoka H, Tsukuda K, Wang X, Iwanami J, Horiuchi M. Effect of angiotensin II type 2 receptor-interacting protein on adipose tissue function via modulation of macrophage polarization. PLoS One 8: e60067, 2013. doi: 10.1371/journal.pone.0060067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Jo F, Jo H, Hilzendeger AM, Thompson AP, Cassell MD, Rutkowski DT, Davisson RL, Grobe JL, Sigmund CD. Brain endoplasmic reticulum stress mechanistically distinguishes the saline-intake and hypertensive response to deoxycorticosterone acetate-salt. Hypertension 65: 1341–1348, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J 20: 1546–1548, 2006. doi: 10.1096/fj.05-4642fje. [DOI] [PubMed] [Google Scholar]
- 446.Jones BH, Standridge MK, Moustaid N. Angiotensin II increases lipogenesis in 3T3-L1 and human adipose cells. Endocrinology 138: 1512–1519, 1997. doi: 10.1210/endo.138.4.5038. [DOI] [PubMed] [Google Scholar]
- 447.Ju X, Ijaz T, Sun H, Ray S, Lejeune W, Lee C, Recinos A III, Guo DC, Milewicz DM, Tilton RG, Brasier AR. Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin II via the T-helper lymphocyte 17-interleukin 17 axis in C57BL/6 mice. Arterioscler Thromb Vasc Biol 33: 1612–1621, 2013. doi: 10.1161/ATVBAHA.112.301049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Jurewicz M, McDermott DH, Sechler JM, Tinckam K, Takakura A, Carpenter CB, Milford E, Abdi R. Human T and natural killer cells possess a functional renin-angiotensin system: further mechanisms of angiotensin II-induced inflammation. J Am Soc Nephrol 18: 1093–1102, 2007. doi: 10.1681/ASN.2006070707. [DOI] [PubMed] [Google Scholar]
- 449.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13: 2570–2580, 1999. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Kalupahana NS, Massiera F, Quignard-Boulange A, Ailhaud G, Voy BH, Wasserman DH, Moustaid-Moussa N. Overproduction of angiotensinogen from adipose tissue induces adipose inflammation, glucose intolerance, and insulin resistance. Obesity (Silver Spring) 20: 48–56, 2012. doi: 10.1038/oby.2011.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG, McDonough AA. Renal transporter activation during angiotensin II hypertension is blunted in IFN-γ(−/−) and IL-17A (−/−) mice. Hypertension 65: 569–576, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Kambayashi Y, Bardhan S, Takahashi K, Tsuzuki S, Inui H, Hamakubo T, Inagami T. Molecular cloning of a novel angiotensin II receptor isoform involved in phosphotyrosine phosphatase inhibition. J Biol Chem 268: 24543–24546, 1993. [PubMed] [Google Scholar]
- 453.Kanematsu Y, Kanematsu M, Kurihara C, Tada Y, Tsou TL, van Rooijen N, Lawton MT, Young WL, Liang EI, Nuki Y, Hashimoto T. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke 42: 173–178, 2011. doi: 10.1161/STROKEAHA.110.590976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Kaneshiro Y, Ichihara A, Sakoda M, Takemitsu T, Nabi AH, Uddin MN, Nakagawa T, Nishiyama A, Suzuki F, Inagami T, Itoh H. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J Am Soc Nephrol 18: 1789–1795, 2007. doi: 10.1681/ASN.2006091062. [DOI] [PubMed] [Google Scholar]
- 455.Kang C, Yijun L, Jingtao D, Changyu P, Wenhua Y, Baoan W, Fangling M, Xianling W, Guoqing Y, Yiming M, Juming L. Effects of telmisartan on lipid metabolisms and proinflammatory factors secretion of differentiated 3T3-L1 adipocytes. J Renin Angiotensin Aldosterone Syst 16: 1061–1068, 2015. doi: 10.1177/1470320313518252. [DOI] [PubMed] [Google Scholar]
- 456.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, J Lin SC, Aronow BJ, Tallquist MD, Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun 7: 12260, 2016. doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Kao YH, Chen YC, Chung CC, Lien GS, Chen SA, Kuo CC, Chen YJ. Heart failure and angiotensin II modulate atrial Pitx2c promotor methylation. Clin Exp Pharmacol Physiol 40: 379–384, 2013. doi: 10.1111/1440-1681.12089. [DOI] [PubMed] [Google Scholar]
- 458.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol 14: 885–890, 2004. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Karbach SH, Schönfelder T, Brandão I, Wilms E, Hörmann N, Jäckel S, Schüler R, Finger S, Knorr M, Lagrange J, Brandt M, Waisman A, Kossmann S, Schäfer K, Münzel T, Reinhardt C, Wenzel P. Gut Microbiota Promote Angiotensin II-Induced Arterial Hypertension and Vascular Dysfunction. J Am Heart Assoc 5: e003698, 2016. doi: 10.1161/JAHA.116.003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Karnewar S, Vasamsetti SB, Gopoju R, Kanugula AK, Ganji SK, Prabhakar S, Rangaraj N, Tupperwar N, Kumar JM, Kotamraju S. Mitochondria-targeted esculetin alleviates mitochondrial dysfunction by AMPK-mediated nitric oxide and SIRT3 regulation in endothelial cells: potential implications in atherosclerosis. Sci Rep 6: 24108, 2016. doi: 10.1038/srep24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Karnik SS, Singh KD, Tirupula K, Unal H. Significance of angiotensin 1-7 coupling with MAS1 receptor and other GPCRs to the renin-angiotensin system: IUPHAR Review 22. Br J Pharmacol 174: 737–753, 2017. doi: 10.1111/bph.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Karnik SS, Unal H, Kemp JR, Tirupula KC, Eguchi S, Vanderheyden PM, Thomas WG. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli. Pharmacol Rev 67: 754–819, 2015. doi: 10.1124/pr.114.010454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Kaschina E, Lauer D, Schmerler P, Unger T, Steckelings UM. AT2 receptors targeting cardiac protection post-myocardial infarction. Curr Hypertens Rep 16: 441, 2014. doi: 10.1007/s11906-014-0441-0. [DOI] [PubMed] [Google Scholar]
- 464.Kassan M, Ait-Aissa K, Radwan E, Mali V, Haddox S, Gabani M, Zhang W, Belmadani S, Irani K, Trebak M, Matrougui K. Essential Role of Smooth Muscle STIM1 in Hypertension and Cardiovascular Dysfunction. Arterioscler Thromb Vasc Biol 36: 1900–1909, 2016. doi: 10.1161/ATVBAHA.116.307869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Kassan M, Galán M, Partyka M, Saifudeen Z, Henrion D, Trebak M, Matrougui K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol 32: 1652–1661, 2012. doi: 10.1161/ATVBAHA.112.249318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Kassiri Z, Zhong J, Guo D, Basu R, Wang X, Liu PP, Scholey JW, Penninger JM, Oudit GY. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail 2: 446–455, 2009. doi: 10.1161/CIRCHEARTFAILURE.108.840124. [DOI] [PubMed] [Google Scholar]
- 467.Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Möllmann H, Looso M, Troidl C, Offermanns S, Wettschureck N. Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ Res 118: 1906–1917, 2016. doi: 10.1161/CIRCRESAHA.116.308643. [DOI] [PubMed] [Google Scholar]
- 468.Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 16: 461–472, 2015. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- 469.Kawabata A, Baoum A, Ohta N, Jacquez S, Seo GM, Berkland C, Tamura M. Intratracheal administration of a nanoparticle-based therapy with the angiotensin II type 2 receptor gene attenuates lung cancer growth. Cancer Res 72: 2057–2067, 2012. doi: 10.1158/0008-5472.CAN-11-3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Kawai T, Forrester SJ, O’Brien S, Baggett A, Rizzo V, Eguchi S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol Res 125, Pt A: 4–13, 2017. doi: 10.1016/j.phrs.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Kawai T, Takayanagi T, Forrester SJ, Preston KJ, Obama T, Tsuji T, Kobayashi T, Boyer MJ, Cooper HA, Kwok HF, Hashimoto T, Scalia R, Rizzo V, Eguchi S. Vascular ADAM17 (a Disintegrin and Metalloproteinase Domain 17) Is Required for Angiotensin II/β-Aminopropionitrile-Induced Abdominal Aortic Aneurysm. Hypertension 70: 959–963, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Kawano H, Cody RJ, Graf K, Goetze S, Kawano Y, Schnee J, Law RE, Hsueh WA. Angiotensin II enhances integrin and alpha-actinin expression in adult rat cardiac fibroblasts. Hypertension 35: 273–279, 2000. doi: 10.1161/01.HYP.35.1.273. [DOI] [PubMed] [Google Scholar]
- 473.Kawano S, Kubota T, Monden Y, Kawamura N, Tsutsui H, Takeshita A, Sunagawa K. Blockade of NF-kappaB ameliorates myocardial hypertrophy in response to chronic infusion of angiotensin II. Cardiovasc Res 67: 689–698, 2005. doi: 10.1016/j.cardiores.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 474.Kawarazaki W, Nagase M, Yoshida S, Takeuchi M, Ishizawa K, Ayuzawa N, Ueda K, Fujita T. Angiotensin II- and salt-induced kidney injury through Rac1-mediated mineralocorticoid receptor activation. J Am Soc Nephrol 23: 997–1007, 2012. doi: 10.1681/ASN.2011070734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Kecskés M, Jacobs G, Kerselaers S, Syam N, Menigoz A, Vangheluwe P, Freichel M, Flockerzi V, Voets T, Vennekens R. The Ca(2+)-activated cation channel TRPM4 is a negative regulator of angiotensin II-induced cardiac hypertrophy. Basic Res Cardiol 110: 43, 2015. doi: 10.1007/s00395-015-0501-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Kee HJ, Eom GH, Joung H, Shin S, Kim JR, Cho YK, Choe N, Sim BW, Jo D, Jeong MH, Kim KK, Seo JS, Kook H. Activation of histone deacetylase 2 by inducible heat shock protein 70 in cardiac hypertrophy. Circ Res 103: 1259–1269, 2008. doi: 10.1161/01.RES.0000338570.27156.84. [DOI] [PubMed] [Google Scholar]
- 477.Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, Kim JK, Kim KK, Epstein JA, Kook H. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 113: 51–59, 2006. doi: 10.1161/CIRCULATIONAHA.105.559724. [DOI] [PubMed] [Google Scholar]
- 478.Kehat I, Davis J, Tiburcy M, Accornero F, Saba-El-Leil MK, Maillet M, York AJ, Lorenz JN, Zimmermann WH, Meloche S, Molkentin JD. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ Res 108: 176–183, 2011. doi: 10.1161/CIRCRESAHA.110.231514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Kemp BA, Howell NL, Gildea JJ, Keller SR, Padia SH, Carey RM. AT2 receptor activation induces natriuresis and lowers blood pressure. Circ Res 115: 388–399, 2014. doi: 10.1161/CIRCRESAHA.115.304110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Kemp BA, Howell NL, Keller SR, Gildea JJ, Padia SH, Carey RM. AT2 Receptor Activation Prevents Sodium Retention and Reduces Blood Pressure in Angiotensin II-Dependent Hypertension. Circ Res 119: 532–543, 2016. doi: 10.1161/CIRCRESAHA.116.308384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Kemp JR, Unal H, Desnoyer R, Yue H, Bhatnagar A, Karnik SS. Angiotensin II-regulated microRNA 483-3p directly targets multiple components of the renin-angiotensin system. J Mol Cell Cardiol 75: 25–39, 2014. doi: 10.1016/j.yjmcc.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Kendall RT, Lee MH, Pleasant DL, Robinson K, Kuppuswamy D, McDermott PJ, Luttrell LM. Arrestin-dependent angiotensin AT1 receptor signaling regulates Akt and mTor-mediated protein synthesis. J Biol Chem 289: 26155–26166, 2014. doi: 10.1074/jbc.M114.595728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Kendall RT, Strungs EG, Rachidi SM, Lee MH, El-Shewy HM, Luttrell DK, Janech MG, Luttrell LM. The beta-arrestin pathway-selective type 1A angiotensin receptor (AT1A) agonist [Sar1,Ile4,Ile8]angiotensin II regulates a robust G protein-independent signaling network. J Biol Chem 286: 19880–19891, 2011. doi: 10.1074/jbc.M111.233080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Ketelhuth DF, Hansson GK. Adaptive Response of T and B Cells in Atherosclerosis. Circ Res 118: 668–678, 2016. doi: 10.1161/CIRCRESAHA.115.306427. [DOI] [PubMed] [Google Scholar]
- 485.Ketsawatsomkron P, Lorca RA, Keen HL, Weatherford ET, Liu X, Pelham CJ, Grobe JL, Faraci FM, England SK, Sigmund CD. PPARγ regulates resistance vessel tone through a mechanism involving RGS5-mediated control of protein kinase C and BKCa channel activity. Circ Res 111: 1446–1458, 2012. doi: 10.1161/CIRCRESAHA.112.271577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Khan NS, Song CY, Jennings BL, Estes AM, Fang XR, Bonventre JV, Malik KU. Cytosolic phospholipase A2α is critical for angiotensin II-induced hypertension and associated cardiovascular pathophysiology. Hypertension 65: 784–792, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Khan SA, Dong H, Joyce J, Sasaki T, Chu ML, Tsuda T. Fibulin-2 is essential for angiotensin II-induced myocardial fibrosis mediated by transforming growth factor (TGF)-β. Lab Invest 96: 773–783, 2016. doi: 10.1038/labinvest.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Khan Z, Shen XZ, Bernstein EA, Giani JF, Eriguchi M, Zhao TV, Gonzalez-Villalobos RA, Fuchs S, Liu GY, Bernstein KE. Angiotensin-converting enzyme enhances the oxidative response and bactericidal activity of neutrophils. Blood 130: 328–339, 2017. doi: 10.1182/blood-2016-11-752006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Kharade SV, Sonkusare SK, Srivastava AK, Thakali KM, Fletcher TW, Rhee SW, Rusch NJ. The β3 subunit contributes to vascular calcium channel upregulation and hypertension in angiotensin II-infused C57BL/6 mice. Hypertension 61: 137–142, 2013. doi: 10.1161/HYPERTENSIONAHA.112.197863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Kigawa Y, Miyazaki T, Lei XF, Nakamachi T, Oguchi T, Kim-Kaneyama JR, Taniyama M, Tsunawaki S, Shioda S, Miyazaki A. NADPH oxidase deficiency exacerbates angiotensin II-induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol 34: 2413–2420, 2014. doi: 10.1161/ATVBAHA.114.303086. [DOI] [PubMed] [Google Scholar]
- 491.Kim CW, Kumar S, Son DJ, Jang IH, Griendling KK, Jo H. Prevention of abdominal aortic aneurysm by anti-microRNA-712 or anti-microRNA-205 in angiotensin II-infused mice. Arterioscler Thromb Vasc Biol 34: 1412–1421, 2014. doi: 10.1161/ATVBAHA.113.303134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Kim GR, Cho SN, Kim HS, Yu SY, Choi SY, Ryu Y, Lin MQ, Jin L, Kee HJ, Jeong MH. Histone deacetylase and GATA-binding factor 6 regulate arterial remodeling in angiotensin II-induced hypertension. J Hypertens 34: 2206–2219, 2016. doi: 10.1097/HJH.0000000000001081. [DOI] [PubMed] [Google Scholar]
- 493.Kim JA, Jang HJ, Martinez-Lemus LA, Sowers JR. Activation of mTOR/p70S6 kinase by ANG II inhibits insulin-stimulated endothelial nitric oxide synthase and vasodilation. Am J Physiol Endocrinol Metab 302: E201–E208, 2012. doi: 10.1152/ajpendo.00497.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev 52: 11–34, 2000. [PubMed] [Google Scholar]
- 495.Kim S, Zingler M, Harrison JK, Scott EW, Cogle CR, Luo D, Raizada MK. Angiotensin II Regulation of Proliferation, Differentiation, and Engraftment of Hematopoietic Stem Cells. Hypertension 67: 574–584, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Suzuki T, Maeta H, Abe Y. Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension 45: 860–866, 2005. doi: 10.1161/01.HYP.0000163462.98381.7f. [DOI] [PubMed] [Google Scholar]
- 497.King VL, Lin AY, Kristo F, Anderson TJ, Ahluwalia N, Hardy GJ, Owens AP III, Howatt DA, Shen D, Tager AM, Luster AD, Daugherty A, Gerszten RE. Interferon-gamma and the interferon-inducible chemokine CXCL10 protect against aneurysm formation and rupture. Circulation 119: 426–435, 2009. doi: 10.1161/CIRCULATIONAHA.108.785949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J II, Harrison DG. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest 124: 4642–4656, 2014. doi: 10.1172/JCI74084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Kirabo A, Kearns PN, Jarajapu YP, Sasser JM, Oh SP, Grant MB, Kasahara H, Cardounel AJ, Baylis C, Wagner K-U, Sayeski PP. Vascular smooth muscle Jak2 mediates angiotensin II-induced hypertension via increased levels of reactive oxygen species. Cardiovasc Res 91: 171–179, 2011. doi: 10.1093/cvr/cvr059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Kishore R, Krishnamurthy P, Garikipati VN, Benedict C, Nickoloff E, Khan M, Johnson J, Gumpert AM, Koch WJ, Verma SK. Interleukin-10 inhibits chronic angiotensin II-induced pathological autophagy. J Mol Cell Cardiol 89, Pt B: 203–213, 2015. doi: 10.1016/j.yjmcc.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Klein JD, Murrell BP, Tucker S, Kim YH, Sands JM. Urea transporter UT-A1 and aquaporin-2 proteins decrease in response to angiotensin II or norepinephrine-induced acute hypertension. Am J Physiol Renal Physiol 291: F952–F959, 2006. doi: 10.1152/ajprenal.00173.2006. [DOI] [PubMed] [Google Scholar]
- 502.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 59: 251–287, 2007. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 503.Koga J, Egashira K, Matoba T, Kubo M, Ihara Y, Iwai M, Horiuchi M, Sunagawa K. Essential role of angiotensin II type 1a receptors in the host vascular wall, but not the bone marrow, in the pathogenesis of angiotensin II-induced atherosclerosis. Hypertens Res 31: 1791–1800, 2008. doi: 10.1291/hypres.31.1791. [DOI] [PubMed] [Google Scholar]
- 504.Kohlstedt K, Gershome C, Trouvain C, Hofmann WK, Fichtlscherer S, Fleming I. Angiotensin-converting enzyme (ACE) inhibitors modulate cellular retinol-binding protein 1 and adiponectin expression in adipocytes via the ACE-dependent signaling cascade. Mol Pharmacol 75: 685–692, 2009. doi: 10.1124/mol.108.051631. [DOI] [PubMed] [Google Scholar]
- 505.Kohlstedt K, Trouvain C, Namgaladze D, Fleming I. Adipocyte-derived lipids increase angiotensin-converting enzyme (ACE) expression and modulate macrophage phenotype. Basic Res Cardiol 106: 205–215, 2011. doi: 10.1007/s00395-010-0137-9. [DOI] [PubMed] [Google Scholar]
- 506.Kolwicz SC Jr, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res 111: 728–738, 2012. doi: 10.1161/CIRCRESAHA.112.268128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Kono S, Kurata T, Sato K, Omote Y, Hishikawa N, Yamashita T, Deguchi K, Abe K. Neurovascular protection by telmisartan via reducing neuroinflammation in stroke-resistant spontaneously hypertensive rat brain after ischemic stroke. J Stroke Cerebrovasc Dis 24: 537–547, 2015. doi: 10.1016/j.jstrokecerebrovasdis.2014.09.037. [DOI] [PubMed] [Google Scholar]
- 508.Konvalinka A, Zhou J, Dimitromanolakis A, Drabovich AP, Fang F, Gurley S, Coffman T, John R, Zhang SL, Diamandis EP, Scholey JW. Determination of an angiotensin II-regulated proteome in primary human kidney cells by stable isotope labeling of amino acids in cell culture (SILAC). J Biol Chem 288: 24834–24847, 2013. doi: 10.1074/jbc.M113.485326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Kopf PG, Campbell WB. Endothelial metabolism of angiotensin II to angiotensin III, not angiotensin (1-7), augments the vasorelaxation response in adrenal cortical arteries. Endocrinology 154: 4768–4776, 2013. doi: 10.1210/en.2013-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Kossmann S, Hu H, Steven S, Schönfelder T, Fraccarollo D, Mikhed Y, Brähler M, Knorr M, Brandt M, Karbach SH, Becker C, Oelze M, Bauersachs J, Widder J, Münzel T, Daiber A, Wenzel P. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J Biol Chem 289: 27540–27550, 2014. doi: 10.1074/jbc.M114.604231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M, Knorr M, Hu H, Kröller-Schön S, Schönfelder T, Grabbe S, Oelze M, Daiber A, Münzel T, Becker C, Wenzel P. Angiotensin II-induced vascular dysfunction depends on interferon-γ-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler Thromb Vasc Biol 33: 1313–1319, 2013. doi: 10.1161/ATVBAHA.113.301437. [DOI] [PubMed] [Google Scholar]
- 512.Kostenis E, Milligan G, Christopoulos A, Sanchez-Ferrer CF, Heringer-Walther S, Sexton PM, Gembardt F, Kellett E, Martini L, Vanderheyden P, Schultheiss HP, Walther T. G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation 111: 1806–1813, 2005. doi: 10.1161/01.CIR.0000160867.23556.7D. [DOI] [PubMed] [Google Scholar]
- 513.Koulis C, Chow BS, McKelvey M, Steckelings UM, Unger T, Thallas-Bonke V, Thomas MC, Cooper ME, Jandeleit-Dahm KA, Allen TJ. AT2R agonist, compound 21, is reno-protective against type 1 diabetic nephropathy. Hypertension 65: 1073–1081, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05204. [DOI] [PubMed] [Google Scholar]
- 514.Kourembanas S. Exosomes: vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu Rev Physiol 77: 13–27, 2015. doi: 10.1146/annurev-physiol-021014-071641. [DOI] [PubMed] [Google Scholar]
- 515.Krebs CF, Lange S, Niemann G, Rosendahl A, Lehners A, Meyer-Schwesinger C, Stahl RA, Benndorf RA, Velden J, Paust HJ, Panzer U, Ehmke H, Wenzel UO. Deficiency of the interleukin 17/23 axis accelerates renal injury in mice with deoxycorticosterone acetate+angiotensin ii-induced hypertension. Hypertension 63: 565–571, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02620. [DOI] [PubMed] [Google Scholar]
- 516.Krishna SM, Seto SW, Jose RJ, Li J, Morton SK, Biros E, Wang Y, Nsengiyumva V, Lindeman JH, Loots GG, Rush CM, Craig JM, Golledge J. Wnt Signaling Pathway Inhibitor Sclerostin Inhibits Angiotensin II-Induced Aortic Aneurysm and Atherosclerosis. Arterioscler Thromb Vasc Biol 37: 553–566, 2017. doi: 10.1161/ATVBAHA.116.308723. [DOI] [PubMed] [Google Scholar]
- 517.Kröller-Schön S, Jansen T, Schüler A, Oelze M, Wenzel P, Hausding M, Kerahrodi JG, Beisele M, Lackner KJ, Daiber A, Münzel T, Schulz E. Peroxisome proliferator-activated receptor γ, coactivator 1α deletion induces angiotensin II-associated vascular dysfunction by increasing mitochondrial oxidative stress and vascular inflammation. Arterioscler Thromb Vasc Biol 33: 1928–1935, 2013. doi: 10.1161/ATVBAHA.113.301717. [DOI] [PubMed] [Google Scholar]
- 518.Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, Satoh S, Nakano R, Ishii C, Sugiyama T, Eto K, Tsubamoto Y, Okuno A, Murakami K, Sekihara H, Hasegawa G, Naito M, Toyoshima Y, Tanaka S, Shiota K, Kitamura T, Fujita T, Ezaki O, Aizawa S, Kadowaki T, , et al. . PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell 4: 597–609, 1999. doi: 10.1016/S1097-2765(00)80210-5. [DOI] [PubMed] [Google Scholar]
- 519.Kumagai K, Nakashima H, Urata H, Gondo N, Arakawa K, Saku K. Effects of angiotensin II type 1 receptor antagonist on electrical and structural remodeling in atrial fibrillation. J Am Coll Cardiol 41: 2197–2204, 2003. doi: 10.1016/S0735-1097(03)00464-9. [DOI] [PubMed] [Google Scholar]
- 520.Kumar R, Thomas CM, Yong QC, Chen W, Baker KM. The intracrine renin-angiotensin system. Clin Sci (Lond) 123: 273–284, 2012. doi: 10.1042/CS20120089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Kumar R, Yong QC, Thomas CM, Baker KM. Intracardiac intracellular angiotensin system in diabetes. Am J Physiol Regul Integr Comp Physiol 302: R510–R517, 2012. doi: 10.1152/ajpregu.00512.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Kunieda T, Minamino T, Nishi J, Tateno K, Oyama T, Katsuno T, Miyauchi H, Orimo M, Okada S, Takamura M, Nagai T, Kaneko S, Komuro I. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation 114: 953–960, 2006. doi: 10.1161/CIRCULATIONAHA.106.626606. [DOI] [PubMed] [Google Scholar]
- 523.Kurdi M, Booz GW. New take on the role of angiotensin II in cardiac hypertrophy and fibrosis. Hypertension 57: 1034–1038, 2011. doi: 10.1161/HYPERTENSIONAHA.111.172700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Kurdi M, Booz GW. Three 4-letter words of hypertension-related cardiac hypertrophy: TRPC, mTOR, and HDAC. J Mol Cell Cardiol 50: 964–971, 2011. doi: 10.1016/j.yjmcc.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Kurihara T, Shimizu-Hirota R, Shimoda M, Adachi T, Shimizu H, Weiss SJ, Itoh H, Hori S, Aikawa N, Okada Y. Neutrophil-derived matrix metalloproteinase 9 triggers acute aortic dissection. Circulation 126: 3070–3080, 2012. doi: 10.1161/CIRCULATIONAHA.112.097097. [DOI] [PubMed] [Google Scholar]
- 526.Kurisu S, Ozono R, Oshima T, Kambe M, Ishida T, Sugino H, Matsuura H, Chayama K, Teranishi Y, Iba O, Amano K, Matsubara H. Cardiac angiotensin II type 2 receptor activates the kinin/NO system and inhibits fibrosis. Hypertension 41: 99–107, 2003. doi: 10.1161/01.HYP.0000050101.90932.14. [DOI] [PubMed] [Google Scholar]
- 527.Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M. Suppression of aging in mice by the hormone Klotho. Science 309: 1829–1833, 2005. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Kwon WY, Cha HN, Heo JY, Choi JH, Jang BI, Lee IK, Park SY. Interleukin-10 deficiency aggravates angiotensin II-induced cardiac remodeling in mice. Life Sci 146: 214–221, 2016. doi: 10.1016/j.lfs.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 529.Labeyrie PE, Goulay R, Martinez de Lizarrondo S, Hébert M, Gauberti M, Maubert E, Delaunay B, Gory B, Signorelli F, Turjman F, Touzé E, Courthéoux P, Vivien D, Orset C. Vascular Tissue-Type Plasminogen Activator Promotes Intracranial Aneurysm Formation. Stroke 48: 2574–2582, 2017. doi: 10.1161/STROKEAHA.117.017305. [DOI] [PubMed] [Google Scholar]
- 530.Labiós M, Martínez M, Gabriel F, Guiral V, Munoz A, Aznar J. Effect of eprosartan on cytoplasmic free calcium mobilization, platelet activation, and microparticle formation in hypertension. Am J Hypertens 17: 757–763, 2004. doi: 10.1016/j.amjhyper.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 531.Laczy B, Hill BG, Wang K, Paterson AJ, White CR, Xing D, Chen YF, Darley-Usmar V, Oparil S, Chatham JC. Protein O-GlcNAcylation: a new signaling paradigm for the cardiovascular system. Am J Physiol Heart Circ Physiol 296: H13–H28, 2009. doi: 10.1152/ajpheart.01056.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Lai EY, Solis G, Luo Z, Carlstrom M, Sandberg K, Holland S, Wellstein A, Welch WJ, Wilcox CS. p47(phox) is required for afferent arteriolar contractile responses to angiotensin II and perfusion pressure in mice. Hypertension 59: 415–420, 2012. doi: 10.1161/HYPERTENSIONAHA.111.184291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Lakshmikanthan S, Zieba BJ, Ge ZD, Momotani K, Zheng X, Lund H, Artamonov MV, Maas JE, Szabo A, Zhang DX, Auchampach JA, Mattson DL, Somlyo AV, Chrzanowska-Wodnicka M. Rap1b in smooth muscle and endothelium is required for maintenance of vascular tone and normal blood pressure. Arterioscler Thromb Vasc Biol 34: 1486–1494, 2014. doi: 10.1161/ATVBAHA.114.303678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, Hooper NM, Turner AJ. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem 280: 30113–30119, 2005. doi: 10.1074/jbc.M505111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 40: 511–515, 2002. doi: 10.1161/01.HYP.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 149: 274–293, 2012. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110: 1364–1390, 2012. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Lastra G, Dhuper S, Johnson MS, Sowers JR. Salt, aldosterone, and insulin resistance: impact on the cardiovascular system. Nat Rev Cardiol 7: 577–584, 2010. doi: 10.1038/nrcardio.2010.123. [DOI] [PubMed] [Google Scholar]
- 539.Lastra G, Sowers JR. Obesity and cardiovascular disease: role of adipose tissue, inflammation, and the renin-angiotensin-aldosterone system. Horm Mol Biol Clin Investig 15: 49–57, 2013. doi: 10.1515/hmbci-2013-0025. [DOI] [PubMed] [Google Scholar]
- 540.Lauer D, Slavic S, Sommerfeld M, Thöne-Reineke C, Sharkovska Y, Hallberg A, Dahlöf B, Kintscher U, Unger T, Steckelings UM, Kaschina E. Angiotensin type 2 receptor stimulation ameliorates left ventricular fibrosis and dysfunction via regulation of tissue inhibitor of matrix metalloproteinase 1/matrix metalloproteinase 9 axis and transforming growth factor β1 in the rat heart. Hypertension 63: e60–e67, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02522. [DOI] [PubMed] [Google Scholar]
- 541.Laurindo FR, Araujo TL, Abrahão TB. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid Redox Signal 20: 2755–2775, 2014. doi: 10.1089/ars.2013.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, Costa-Fraga F, Jankowski J, Jankowski V, Sousa F, Alzamora A, Soares E, Barbosa C, Kjeldsen F, Oliveira A, Braga J, Savergnini S, Maia G, Peluso AB, Passos-Silva D, Ferreira A, Alves F, Martins A, Raizada M, Paula R, Motta-Santos D, Klempin F, Pimenta A, Alenina N, Sinisterra R, Bader M, Campagnole-Santos MJ, Santos RA. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res 112: 1104–1111, 2013. doi: 10.1161/CIRCRESAHA.113.301077. [DOI] [PubMed] [Google Scholar]
- 543.Lavandero S, Chiong M, Rothermel BA, Hill JA. Autophagy in cardiovascular biology. J Clin Invest 125: 55–64, 2015. doi: 10.1172/JCI73943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Lavoie JL, Cassell MD, Gross KW, Sigmund CD. Adjacent expression of renin and angiotensinogen in the rostral ventrolateral medulla using a dual-reporter transgenic model. Hypertension 43: 1116–1119, 2004. doi: 10.1161/01.HYP.0000125143.73301.94. [DOI] [PubMed] [Google Scholar]
- 545.Lavoie JL, Cassell MD, Gross KW, Sigmund CD. Localization of renin expressing cells in the brain, by use of a REN-eGFP transgenic model. Physiol Genomics 16: 240–246, 2004. doi: 10.1152/physiolgenomics.00131.2003. [DOI] [PubMed] [Google Scholar]
- 546.Lavoie JL, Liu X, Bianco RA, Beltz TG, Johnson AK, Sigmund CD. Evidence supporting a functional role for intracellular renin in the brain. Hypertension 47: 461–466, 2006. doi: 10.1161/01.HYP.0000203308.52919.dc. [DOI] [PubMed] [Google Scholar]
- 547.Lavoie JL, Sigmund CD. Minireview: overview of the renin-angiotensin system–an endocrine and paracrine system. Endocrinology 144: 2179–2183, 2003. doi: 10.1210/en.2003-0150. [DOI] [PubMed] [Google Scholar]
- 548.Lavoz C, Rodrigues-Diez R, Benito-Martin A, Rayego-Mateos S, Rodrigues-Diez RR, Alique M, Ortiz A, Mezzano S, Egido J, Ruiz-Ortega M. Angiotensin II contributes to renal fibrosis independently of Notch pathway activation. PLoS One 7: e40490, 2012. doi: 10.1371/journal.pone.0040490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Leask A. Getting to the heart of the matter: new insights into cardiac fibrosis. Circ Res 116: 1269–1276, 2015. doi: 10.1161/CIRCRESAHA.116.305381. [DOI] [PubMed] [Google Scholar]
- 550.Leclerc PC, Auger-Messier M, Lanctot PM, Escher E, Leduc R, Guillemette G. A polyaromatic caveolin-binding-like motif in the cytoplasmic tail of the type 1 receptor for angiotensin II plays an important role in receptor trafficking and signaling. Endocrinology 143: 4702–4710, 2002. doi: 10.1210/en.2002-220679. [DOI] [PubMed] [Google Scholar]
- 551.Leclerc PC, Lanctot PM, Auger-Messier M, Escher E, Leduc R, Guillemette G. S-nitrosylation of cysteine 289 of the AT1 receptor decreases its binding affinity for angiotensin II. Br J Pharmacol 148: 306–313, 2006. doi: 10.1038/sj.bjp.0706725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Lee DY, Wauquier F, Eid AA, Roman LJ, Ghosh-Choudhury G, Khazim K, Block K, Gorin Y. Nox4 NADPH oxidase mediates peroxynitrite-dependent uncoupling of endothelial nitric-oxide synthase and fibronectin expression in response to angiotensin II: role of mitochondrial reactive oxygen species. J Biol Chem 288: 28668–28686, 2013. doi: 10.1074/jbc.M113.470971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Lee S, Kim IK, Ahn JS, Woo DC, Kim ST, Song S, Koh GY, Kim HS, Jeon BH, Kim I. Deficiency of endothelium-specific transcription factor Sox17 induces intracranial aneurysm. Circulation 131: 995–1005, 2015. doi: 10.1161/CIRCULATIONAHA.114.012568. [DOI] [PubMed] [Google Scholar]
- 554.Lee YJ, Song IK, Jang KJ, Nielsen J, Frøkiaer J, Nielsen S, Kwon TH. Increased AQP2 targeting in primary cultured IMCD cells in response to angiotensin II through AT1 receptor. Am J Physiol Renal Physiol 292: F340–F350, 2007. doi: 10.1152/ajprenal.00090.2006. [DOI] [PubMed] [Google Scholar]
- 555.Lee-Kirsch MA, Gaudet F, Cardoso MC, Lindpaintner K. Distinct renin isoforms generated by tissue-specific transcription initiation and alternative splicing. Circ Res 84: 240–246, 1999. doi: 10.1161/01.RES.84.2.240. [DOI] [PubMed] [Google Scholar]
- 556.Lei XF, Kim-Kaneyama JR, Arita-Okubo S, Offermanns S, Itabe H, Miyazaki T, Miyazaki A. Identification of Hic-5 as a novel scaffold for the MKK4/p54 JNK pathway in the development of abdominal aortic aneurysms. J Am Heart Assoc 3: e000747, 2014. doi: 10.1161/JAHA.113.000747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Letavernier E, Perez J, Bellocq A, Mesnard L, de Castro Keller A, Haymann JP, Baud L. Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ Res 102: 720–728, 2008. doi: 10.1161/CIRCRESAHA.107.160077. [DOI] [PubMed] [Google Scholar]
- 558.Leung A, Natarajan R. Noncoding RNAs in vascular disease. Curr Opin Cardiol 29: 199–206, 2014. doi: 10.1097/HCO.0000000000000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Leung A, Trac C, Jin W, Lanting L, Akbany A, Sætrom P, Schones DE, Natarajan R. Novel long noncoding RNAs are regulated by angiotensin II in vascular smooth muscle cells. Circ Res 113: 266–278, 2013. doi: 10.1161/CIRCRESAHA.112.300849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Li C, Wang W, Rivard CJ, Lanaspa MA, Summer S, Schrier RW. Molecular mechanisms of angiotensin II stimulation on aquaporin-2 expression and trafficking. Am J Physiol Renal Physiol 300: F1255–F1261, 2011. doi: 10.1152/ajprenal.00469.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Li CY, Chen YH, Wang Q, Hou JW, Wang H, Wang YP, Li YG. Partial inhibition of activin receptor-like kinase 4 attenuates pressure overload-induced cardiac fibrosis and improves cardiac function. J Hypertens 34: 1766–1777, 2016. doi: 10.1097/HJH.0000000000001020. [DOI] [PubMed] [Google Scholar]
- 562.Li CY, Zhou Q, Yang LC, Chen YH, Hou JW, Guo K, Wang YP, Li YG. Dual-specificity phosphatase 14 protects the heart from aortic banding-induced cardiac hypertrophy and dysfunction through inactivation of TAK1-P38MAPK/-JNK1/2 signaling pathway. Basic Res Cardiol 111: 19, 2016. doi: 10.1007/s00395-016-0536-7. [DOI] [PubMed] [Google Scholar]
- 563.Li DJ, Huang F, Ni M, Fu H, Zhang LS, Shen FM. α7 Nicotinic Acetylcholine Receptor Relieves Angiotensin II-Induced Senescence in Vascular Smooth Muscle Cells by Raising Nicotinamide Adenine Dinucleotide-Dependent SIRT1 Activity. Arterioscler Thromb Vasc Biol 36: 1566–1576, 2016. doi: 10.1161/ATVBAHA.116.307157. [DOI] [PubMed] [Google Scholar]
- 564.Li H, He C, Feng J, Zhang Y, Tang Q, Bian Z, Bai X, Zhou H, Jiang H, Heximer SP, Qin M, Huang H, Liu PP, Huang C. Regulator of G protein signaling 5 protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Proc Natl Acad Sci USA 107: 13818–13823, 2010. doi: 10.1073/pnas.1008397107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Li H, Li W, Gupta AK, Mohler PJ, Anderson ME, Grumbach IM. Calmodulin kinase II is required for angiotensin II-mediated vascular smooth muscle hypertrophy. Am J Physiol Heart Circ Physiol 298: H688–H698, 2010. doi: 10.1152/ajpheart.01014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Li H, Weatherford ET, Davis DR, Keen HL, Grobe JL, Daugherty A, Cassis LA, Allen AM, Sigmund CD. Renal proximal tubule angiotensin AT1A receptors regulate blood pressure. Am J Physiol Regul Integr Comp Physiol 301: R1067–R1077, 2011. doi: 10.1152/ajpregu.00124.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 40: 477–484, 2002. doi: 10.1161/01.HYP.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 568.Li L, Fan D, Wang C, Wang JY, Cui XB, Wu D, Zhou Y, Wu LL. Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-β1 pathways in cardiac fibroblasts. Cardiovasc Res 91: 80–89, 2011. doi: 10.1093/cvr/cvr067. [DOI] [PubMed] [Google Scholar]
- 569.Li L, Feng D, Luo Z, Welch WJ, Wilcox CS, Lai EY. Remodeling of Afferent Arterioles From Mice With Oxidative Stress Does Not Account for Increased Contractility but Does Limit Excessive Wall Stress. Hypertension 66: 550–556, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Li M, Wu P, Shao J, Ke Z, Li D, Wu J. Losartan Inhibits Vascular Calcification by Suppressing the BMP2 and Runx2 Expression in Rats In Vivo. Cardiovasc Toxicol 16: 172–181, 2016. doi: 10.1007/s12012-015-9326-y. [DOI] [PubMed] [Google Scholar]
- 571.Li N, Wang HX, Han QY, Li WJ, Zhang YL, Du J, Xia YL, Li HH. Activation of the cardiac proteasome promotes angiotension II-induced hypertrophy by down-regulation of ATRAP. J Mol Cell Cardiol 79: 303–314, 2015. doi: 10.1016/j.yjmcc.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 572.Li Q, Lin X, Yang X, Chang J. NFATc4 is negatively regulated in miR-133a-mediated cardiomyocyte hypertrophic repression. Am J Physiol Heart Circ Physiol 298: H1340–H1347, 2010. doi: 10.1152/ajpheart.00592.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Li Q, Shen P, Zeng S, Liu P. TIEG1 Inhibits Angiotensin II-induced Cardiomyocyte Hypertrophy by Inhibiting Transcription Factor GATA4. J Cardiovasc Pharmacol 66: 196–203, 2015. doi: 10.1097/FJC.0000000000000265. [DOI] [PubMed] [Google Scholar]
- 574.Li Q, Xie J, Wang B, Li R, Bai J, Ding L, Gu R, Wang L, Xu B. Overexpression of microRNA-99a Attenuates Cardiac Hypertrophy. PLoS One 11: e0148480, 2016. doi: 10.1371/journal.pone.0148480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest 124: 755–767, 2014. doi: 10.1172/JCI69942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Li W, Liu J, Hammond SL, Tjalkens RB, Saifudeen Z, Feng Y. Angiotensin II regulates brain (pro)renin receptor expression through activation of cAMP response element-binding protein. Am J Physiol Regul Integr Comp Physiol 309: R138–R147, 2015. doi: 10.1152/ajpregu.00319.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Li XC, Gu V, Miguel-Qin E, Zhuo JL. Role of caveolin 1 in AT1a receptor-mediated uptake of angiotensin II in the proximal tubule of the kidney. Am J Physiol Renal Physiol 307: F949–F961, 2014. doi: 10.1152/ajprenal.00199.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Li XC, Shao Y, Zhuo JL. AT1a receptor knockout in mice impairs urine concentration by reducing basal vasopressin levels and its receptor signaling proteins in the inner medulla. Kidney Int 76: 169–177, 2009. doi: 10.1038/ki.2009.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Li XC, Shull GE, Miguel-Qin E, Chen F, Zhuo JL. Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension in NHE3-deficient mice with transgenic rescue of NHE3 in small intestines. Physiol Rep 3: e12605, 2015. doi: 10.14814/phy2.12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Li XC, Shull GE, Miguel-Qin E, Zhuo JL. Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension. Physiol Genomics 47: 479–487, 2015. doi: 10.1152/physiolgenomics.00056.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Li XC, Zhuo JL. Phosphoproteomic analysis of AT1 receptor-mediated signaling responses in proximal tubules of angiotensin II-induced hypertensive rats. Kidney Int 80: 620–632, 2011. doi: 10.1038/ki.2011.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Li Y, Cai X, Guan Y, Wang L, Wang S, Li Y, Fu Y, Gao X, Su G. Adiponectin Upregulates MiR-133a in Cardiac Hypertrophy through AMPK Activation and Reduced ERK1/2 Phosphorylation. PLoS One 11: e0148482, 2016. doi: 10.1371/journal.pone.0148482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Li Y, Liu Y, Tian X, Zhang Y, Song H, Liu M, Zhang X, Liu H, Zhang J, Zhang Q, Liu D, Peng C, Yan C, Han Y. Cellular Repressor of E1A-Stimulated Genes Is a Critical Determinant of Vascular Remodeling in Response to Angiotensin II. Arterioscler Thromb Vasc Biol 37: 485–494, 2017. doi: 10.1161/ATVBAHA.116.308794. [DOI] [PubMed] [Google Scholar]
- 584.Li Y, Wu Y, Zhang C, Li P, Cui W, Hao J, Ma X, Yin Z, Du J. γδT Cell-derived interleukin-17A via an interleukin-1β-dependent mechanism mediates cardiac injury and fibrosis in hypertension. Hypertension 64: 305–314, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02604. [DOI] [PubMed] [Google Scholar]
- 585.Li Y, Zhang C, Wu Y, Han Y, Cui W, Jia L, Cai L, Cheng J, Li H, Du J. Interleukin-12p35 deletion promotes CD4 T-cell-dependent macrophage differentiation and enhances angiotensin II-Induced cardiac fibrosis. Arterioscler Thromb Vasc Biol 32: 1662–1674, 2012. doi: 10.1161/ATVBAHA.112.249706. [DOI] [PubMed] [Google Scholar]
- 586.Li Z, Mo N, Li L, Cao Y, Wang W, Liang Y, Deng H, Xing R, Yang L, Ni C, Chui D, Guo X. Surgery-Induced Hippocampal Angiotensin II Elevation Causes Blood-Brain Barrier Disruption via MMP/TIMP in Aged Rats. Front Cell Neurosci 10: 105, 2016. doi: 10.3389/fncel.2016.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Liang B, Wang S, Wang Q, Zhang W, Viollet B, Zhu Y, Zou MH. Aberrant endoplasmic reticulum stress in vascular smooth muscle increases vascular contractility and blood pressure in mice deficient of AMP-activated protein kinase-α2 in vivo. Arterioscler Thromb Vasc Biol 33: 595–604, 2013. doi: 10.1161/ATVBAHA.112.300606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Liao S, Miralles M, Kelley BJ, Curci JA, Borhani M, Thompson RW. Suppression of experimental abdominal aortic aneurysms in the rat by treatment with angiotensin-converting enzyme inhibitors. J Vasc Surg 33: 1057–1064, 2001. doi: 10.1067/mva.2001.112810. [DOI] [PubMed] [Google Scholar]
- 589.Liao TD, Yang XP, Liu YH, Shesely EG, Cavasin MA, Kuziel WA, Pagano PJ, Carretero OA. Role of inflammation in the development of renal damage and dysfunction in angiotensin II-induced hypertension. Hypertension 52: 256–263, 2008. doi: 10.1161/HYPERTENSIONAHA.108.112706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Lim S, Lee SY, Seo HH, Ham O, Lee C, Park JH, Lee J, Seung M, Yun I, Han SM, Lee S, Choi E, Hwang KC. Regulation of mitochondrial morphology by positive feedback interaction between PKCδ and Drp1 in vascular smooth muscle cell. J Cell Biochem 116: 648–660, 2015. doi: 10.1002/jcb.25016. [DOI] [PubMed] [Google Scholar]
- 591.Lin CX, Rhaleb NE, Yang XP, Liao TD, D’Ambrosio MA, Carretero OA. Prevention of aortic fibrosis by N-acetyl-seryl-aspartyl-lysyl-proline in angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol 295: H1253–H1261, 2008. doi: 10.1152/ajpheart.00481.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Lin L, Liu X, Xu J, Weng L, Ren J, Ge J, Zou Y. Mas receptor mediates cardioprotection of angiotensin-(1-7) against Angiotensin II-induced cardiomyocyte autophagy and cardiac remodelling through inhibition of oxidative stress. J Cell Mol Med 20: 48–57, 2016. doi: 10.1111/jcmm.12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Lin Q, Zhao G, Fang X, Peng X, Tang H, Wang H, Jing R, Liu J, Lederer WJ, Chen J, Ouyang K. IP3 receptors regulate vascular smooth muscle contractility and hypertension. JCI Insight 1: e89402, 2016. doi: 10.1172/jci.insight.89402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Linder AE, Thakali KM, Thompson JM, Watts SW, Webb RC, Leite R. Methyl-beta-cyclodextrin prevents angiotensin II-induced tachyphylactic contractile responses in rat aorta. J Pharmacol Exp Ther 323: 78–84, 2007. doi: 10.1124/jpet.107.123463. [DOI] [PubMed] [Google Scholar]
- 595.Littlejohn NK, Keen HL, Weidemann BJ, Claflin KE, Tobin KV, Markan KR, Park S, Naber MC, Gourronc FA, Pearson NA, Liu X, Morgan DA, Klingelhutz AJ, Potthoff MJ, Rahmouni K, Sigmund CD, Grobe JL. Suppression of Resting Metabolism by the Angiotensin AT2 Receptor. Cell Reports 16: 1548–1560, 2016. doi: 10.1016/j.celrep.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Liu C, Luo R, Elliott SE, Wang W, Parchim NF, Iriyama T, Daugherty PS, Blackwell SC, Sibai BM, Kellems RE, Xia Y. Elevated Transglutaminase Activity Triggers Angiotensin Receptor Activating Autoantibody Production and Pathophysiology of Preeclampsia. J Am Heart Assoc 4: e002323, 2015. doi: 10.1161/JAHA.115.002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Liu C, Wang W, Parchim N, Irani RA, Blackwell SC, Sibai B, Jin J, Kellems RE, Xia Y. Tissue transglutaminase contributes to the pathogenesis of preeclampsia and stabilizes placental angiotensin receptor type 1 by ubiquitination-preventing isopeptide modification. Hypertension 63: 353–361, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Liu J, Daugherty A, Lu H. Angiotensin II and Abdominal Aortic Aneurysms: An Update. Curr Pharm Des 21: 4035–4048, 2015. doi: 10.2174/1381612821666150826093318. [DOI] [PubMed] [Google Scholar]
- 599.Liu J, Liu S, Matsumoto Y, Murakami S, Sugakawa Y, Kami A, Tanabe C, Maeda T, Michikawa M, Komano H, Zou K. Angiotensin type 1a receptor deficiency decreases amyloid β-protein generation and ameliorates brain amyloid pathology. Sci Rep 5: 12059, 2015. doi: 10.1038/srep12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Liu J, Shimosawa T, Matsui H, Meng F, Supowit SC, DiPette DJ, Ando K, Fujita T. Adrenomedullin inhibits angiotensin II-induced oxidative stress via Csk-mediated inhibition of Src activity. Am J Physiol Heart Circ Physiol 292: H1714–H1721, 2007. doi: 10.1152/ajpheart.00486.2006. [DOI] [PubMed] [Google Scholar]
- 601.Liu R, Lo L, Lay AJ, Zhao Y, Ting KK, Robertson EN, Sherrah AG, Jarrah S, Li H, Zhou Z, Hambly BD, Richmond DR, Jeremy RW, Bannon PG, Vadas MA, Gamble JR. ARHGAP18 Protects Against Thoracic Aortic Aneurysm Formation by Mitigating the Synthetic and Proinflammatory Smooth Muscle Cell Phenotype. Circ Res 121: 512–524, 2017. doi: 10.1161/CIRCRESAHA.117.310692. [DOI] [PubMed] [Google Scholar]
- 602.Liu W, Zi M, Naumann R, Ulm S, Jin J, Taglieri DM, Prehar S, Gui J, Tsui H, Xiao R-P, Neyses L, Solaro RJ, Ke Y, Cartwright EJ, Lei M, Wang X. Pak1 as a novel therapeutic target for antihypertrophic treatment in the heart. Circulation 124: 2702–2715, 2011. doi: 10.1161/CIRCULATIONAHA.111.048785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.Liu Y, Huang H, Zhang Y, Zhu XY, Zhang R, Guan LH, Tang Q, Jiang H, Huang C. Regulator of G protein signaling 3 protects against cardiac hypertrophy in mice. J Cell Biochem 115: 977–986, 2014. doi: 10.1002/jcb.24741. [DOI] [PubMed] [Google Scholar]
- 604.Liu Y, Wang TT, Zhang R, Fu WY, Wang X, Wang F, Gao P, Ding YN, Xie Y, Hao DL, Chen HZ, Liu DP. Calorie restriction protects against experimental abdominal aortic aneurysms in mice. J Exp Med 213: 2473–2488, 2016. doi: 10.1084/jem.20151794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Liu Y, Wang Z, Xiao W. MicroRNA-26a protects against cardiac hypertrophy via inhibiting GATA4 in rat model and cultured cardiomyocytes. Mol Med Rep 14: 2860–2866, 2016. doi: 10.3892/mmr.2016.5574. [DOI] [PubMed] [Google Scholar]
- 606.Lob HE, Schultz D, Marvar PJ, Davisson RL, Harrison DG. Role of the NADPH oxidases in the subfornical organ in angiotensin II-induced hypertension. Hypertension 61: 382–387, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Lob HE, Vinh A, Li L, Blinder Y, Offermanns S, Harrison DG. Role of vascular extracellular superoxide dismutase in hypertension. Hypertension 58: 232–239, 2011. doi: 10.1161/HYPERTENSIONAHA.111.172718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 608.Lobysheva I, Rath G, Sekkali B, Bouzin C, Feron O, Gallez B, Dessy C, Balligand JL. Moderate caveolin-1 downregulation prevents NADPH oxidase-dependent endothelial nitric oxide synthase uncoupling by angiotensin II in endothelial cells. Arterioscler Thromb Vasc Biol 31: 2098–2105, 2011. doi: 10.1161/ATVBAHA.111.230623. [DOI] [PubMed] [Google Scholar]
- 609.Loot AE, Schreiber JG, Fisslthaler B, Fleming I. Angiotensin II impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J Exp Med 206: 2889–2896, 2009. doi: 10.1084/jem.20090449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Lopez-Ilasaca M, Liu X, Tamura K, Dzau VJ. The angiotensin II type I receptor-associated protein, ATRAP, is a transmembrane protein and a modulator of angiotensin II signaling. Mol Biol Cell 14: 5038–5050, 2003. doi: 10.1091/mbc.E03-06-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Lorenzen JM, Schauerte C, Hübner A, Kölling M, Martino F, Scherf K, Batkai S, Zimmer K, Foinquinos A, Kaucsar T, Fiedler J, Kumarswamy R, Bang C, Hartmann D, Gupta SK, Kielstein J, Jungmann A, Katus HA, Weidemann F, Müller OJ, Haller H, Thum T. Osteopontin is indispensible for AP1-mediated angiotensin II-related miR-21 transcription during cardiac fibrosis. Eur Heart J 36: 2184–2196, 2015. doi: 10.1093/eurheartj/ehv109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Lu H, Daugherty A. Atherosclerosis. Arterioscler Thromb Vasc Biol 35: 485–491, 2015. doi: 10.1161/ATVBAHA.115.305380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Lu H, Rateri DL, Bruemmer D, Cassis LA, Daugherty A. Involvement of the renin-angiotensin system in abdominal and thoracic aortic aneurysms. Clin Sci (Lond) 123: 531–543, 2012. doi: 10.1042/CS20120097. [DOI] [PubMed] [Google Scholar]
- 614.Lu T, Zhang DM, Wang XL, He T, Wang RX, Chai Q, Katusic ZS, Lee HC. Regulation of coronary arterial BK channels by caveolae-mediated angiotensin II signaling in diabetes mellitus. Circ Res 106: 1164–1173, 2010. doi: 10.1161/CIRCRESAHA.109.209767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Luo R, Zhang W, Zhao C, Zhang Y, Wu H, Jin J, Zhang W, Grenz A, Eltzschig HK, Tao L, Kellems RE, Xia Y. Elevated Endothelial Hypoxia-Inducible Factor-1α Contributes to Glomerular Injury and Promotes Hypertensive Chronic Kidney Disease. Hypertension 66: 75–84, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Luo YX, Tang X, An XZ, Xie XM, Chen XF, Zhao X, Hao DL, Chen HZ, Liu DP. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur Heart J 38: 1389–1398, 2017. doi: 10.1093/eurheartj/ehw138. [DOI] [PubMed] [Google Scholar]
- 617.Luther JM, Brown NJ. The renin-angiotensin-aldosterone system and glucose homeostasis. Trends Pharmacol Sci 32: 734–739, 2011. doi: 10.1016/j.tips.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Lv P, Miao SB, Shu YN, Dong LH, Liu G, Xie XL, Gao M, Wang YC, Yin YJ, Wang XJ, Han M. Phosphorylation of smooth muscle 22α facilitates angiotensin II-induced ROS production via activation of the PKCδ-P47phox axis through release of PKCδ and actin dynamics and is associated with hypertrophy and hyperplasia of vascular smooth muscle cells in vitro and in vivo. Circ Res 111: 697–707, 2012. doi: 10.1161/CIRCRESAHA.112.272013. [DOI] [PubMed] [Google Scholar]
- 619.Lymperopoulos A, Bathgate A. Arrestins in the cardiovascular system. Prog Mol Biol Transl Sci 118: 297–334, 2013. doi: 10.1016/B978-0-12-394440-5.00012-7. [DOI] [PubMed] [Google Scholar]
- 620.Lymperopoulos A, Sturchler E, Bathgate-Siryk A, Dabul S, Garcia D, Walklett K, Rengo G, McDonald P, Koch WJ. Different potencies of angiotensin receptor blockers at suppressing adrenal β-Arrestin1-dependent post-myocardial infarction hyperaldosteronism. J Am Coll Cardiol 64: 2805–2806, 2014. doi: 10.1016/j.jacc.2014.09.070. [DOI] [PubMed] [Google Scholar]
- 621.Lyu L, Wang H, Li B, Qin Q, Qi L, Nagarkatti M, Nagarkatti P, Janicki JS, Wang XL, Cui T. A critical role of cardiac fibroblast-derived exosomes in activating renin angiotensin system in cardiomyocytes. J Mol Cell Cardiol 89, Pt B: 268–279, 2015. doi: 10.1016/j.yjmcc.2015.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Ma CY, Yin L. Neuroprotective effect of angiotensin II type 2 receptor during cerebral ischemia/reperfusion. Neural Regen Res 11: 1102–1107, 2016. doi: 10.4103/1673-5374.187044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623.Ma D, Zheng B, Suzuki T, Zhang R, Jiang C, Bai D, Yin W, Yang Z, Zhang X, Hou L, Zhan H, Wen JK. Inhibition of KLF5-Myo9b-RhoA Pathway-Mediated Podosome Formation in Macrophages Ameliorates Abdominal Aortic Aneurysm. Circ Res 120: 799–815, 2017. doi: 10.1161/CIRCRESAHA.116.310367. [DOI] [PubMed] [Google Scholar]
- 624.Ma F, Feng J, Zhang C, Li Y, Qi G, Li H, Wu Y, Fu Y, Zhao Y, Chen H, Du J, Tang H. The requirement of CD8+ T cells to initiate and augment acute cardiac inflammatory response to high blood pressure. J Immunol 192: 3365–3373, 2014. doi: 10.4049/jimmunol.1301522. [DOI] [PubMed] [Google Scholar]
- 625.Ma F, Li Y, Jia L, Han Y, Cheng J, Li H, Qi Y, Du J. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS One 7: e35144, 2012. doi: 10.1371/journal.pone.0035144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626.Macconi D, Perico L, Longaretti L, Morigi M, Cassis P, Buelli S, Perico N, Remuzzi G, Benigni A. Sirtuin3 Dysfunction Is the Key Determinant of Skeletal Muscle Insulin Resistance by Angiotensin II. PLoS One 10: e0127172, 2015. doi: 10.1371/journal.pone.0127172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 17: 9–26, 2009. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Madamanchi NR, Runge MS. Redox signaling in cardiovascular health and disease. Free Radic Biol Med 61: 473–501, 2013. doi: 10.1016/j.freeradbiomed.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Madhur MS, Funt SA, Li L, Vinh A, Chen W, Lob HE, Iwakura Y, Blinder Y, Rahman A, Quyyumi AA, Harrison DG. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol 31: 1565–1572, 2011. doi: 10.1161/ATVBAHA.111.227629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55: 500–507, 2010. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Maegdefessel L, Azuma J, Toh R, Deng A, Merk DR, Raiesdana A, Leeper NJ, Raaz U, Schoelmerich AM, McConnell MV, Dalman RL, Spin JM, Tsao PS. MicroRNA-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Sci Transl Med 4: 122ra22, 2012. doi: 10.1126/scitranslmed.3003441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Maegdefessel L, Azuma J, Toh R, Merk DR, Deng A, Chin JT, Raaz U, Schoelmerich AM, Raiesdana A, Leeper NJ, McConnell MV, Dalman RL, Spin JM, Tsao PS. Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J Clin Invest 122: 497–506, 2012. doi: 10.1172/JCI61598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Mahmud A, Feely J. Arterial stiffness and the renin-angiotensin-aldosterone system. J Renin Angiotensin Aldosterone Syst 5: 102–108, 2004. doi: 10.3317/jraas.2004.025. [DOI] [PubMed] [Google Scholar]
- 634.Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res 75: 640–648, 2007. doi: 10.1016/j.cardiores.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635.Maiellaro-Rafferty K, Weiss D, Joseph G, Wan W, Gleason RL, Taylor WR. Catalase overexpression in aortic smooth muscle prevents pathological mechanical changes underlying abdominal aortic aneurysm formation. Am J Physiol Heart Circ Physiol 301: H355–H362, 2011. doi: 10.1152/ajpheart.00040.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Majeed B, Tawinwung S, Eberson LS, Secomb TW, Larmonier N, Larson DF. Interleukin-2/Anti-Interleukin-2 Immune Complex Expands Regulatory T Cells and Reduces Angiotensin II-Induced Aortic Stiffening. Int J Hypertens 2014: 126365, 2014. doi: 10.1155/2014/126365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Makhanova NA, Crowley SD, Griffiths RC, Coffman TM. Gene expression profiles linked to AT1 angiotensin receptors in the kidney. Physiol Genomics 42A: 211–218, 2010. doi: 10.1152/physiolgenomics.00063.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Makino H, Tada Y, Wada K, Liang EI, Chang M, Mobashery S, Kanematsu Y, Kurihara C, Palova E, Kanematsu M, Kitazato K, Hashimoto T. Pharmacological stabilization of intracranial aneurysms in mice: a feasibility study. Stroke 43: 2450–2456, 2012. doi: 10.1161/STROKEAHA.112.659821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639.Malekzadeh S, Fraga-Silva RA, Trachet B, Montecucco F, Mach F, Stergiopulos N. Role of the renin-angiotensin system on abdominal aortic aneurysms. Eur J Clin Invest 43: 1328–1338, 2013. doi: 10.1111/eci.12173. [DOI] [PubMed] [Google Scholar]
- 640.Mallat Z, Daugherty A. AT1 receptor antagonism to reduce aortic expansion in Marfan syndrome: lost in translation or in need of different interpretation? Arterioscler Thromb Vasc Biol 35: e10–e12, 2015. doi: 10.1161/ATVBAHA.114.305173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641.Maltese G, Psefteli PM, Rizzo B, Srivastava S, Gnudi L, Mann GE, Siow RC. The anti-ageing hormone klotho induces Nrf2-mediated antioxidant defences in human aortic smooth muscle cells. J Cell Mol Med 21: 621–627, 2017. doi: 10.1111/jcmm.12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Mamenko M, Zaika O, Ilatovskaya DV, Staruschenko A, Pochynyuk O. Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem 287: 660–671, 2012. doi: 10.1074/jbc.M111.298919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Mamenko M, Zaika O, Prieto MC, Jensen VB, Doris PA, Navar LG, Pochynyuk O. Chronic angiotensin II infusion drives extensive aldosterone-independent epithelial Na+ channel activation. Hypertension 62: 1111–1122, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Manea A, Manea SA, Gafencu AV, Raicu M, Simionescu M. AP-1-dependent transcriptional regulation of NADPH oxidase in human aortic smooth muscle cells: role of p22phox subunit. Arterioscler Thromb Vasc Biol 28: 878–885, 2008. doi: 10.1161/ATVBAHA.108.163592. [DOI] [PubMed] [Google Scholar]
- 645.Manhiani MM, Seth DM, Banes-Berceli AK, Satou R, Navar LG, Brands MW. The role of IL-6 in the physiologic versus hypertensive blood pressure actions of angiotensin II. Physiol Rep 3: e12595, 2015. doi: 10.14814/phy2.12595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646.Manolis AJ, Rosei EA, Coca A, Cifkova R, Erdine SE, Kjeldsen S, Lip GY, Narkiewicz K, Parati G, Redon J, Schmieder R, Tsioufis C, Mancia G. Hypertension and atrial fibrillation: diagnostic approach, prevention and treatment. Position paper of the Working Group ‘Hypertension Arrhythmias and Thrombosis’ of the European Society of Hypertension. J Hypertens 30: 239–252, 2012. doi: 10.1097/HJH.0b013e32834f03bf. [DOI] [PubMed] [Google Scholar]
- 647.Manrique C, Lastra G, Sowers JR. New insights into insulin action and resistance in the vasculature. Ann N Y Acad Sci 1311: 138–150, 2014. doi: 10.1111/nyas.12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Marchesi C, Paradis P, Schiffrin EL. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci 29: 367–374, 2008. doi: 10.1016/j.tips.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 649.Marchesi C, Rehman A, Rautureau Y, Kasal DA, Briet M, Leibowitz A, Simeone SM, Ebrahimian T, Neves MF, Offermanns S, Gonzalez FJ, Paradis P, Schiffrin EL. Protective role of vascular smooth muscle cell PPARγ in angiotensin II-induced vascular disease. Cardiovasc Res 97: 562–570, 2013. doi: 10.1093/cvr/cvs362. [DOI] [PubMed] [Google Scholar]
- 650.Marcus Y, Shefer G, Stern N. Adipose tissue renin-angiotensin-aldosterone system (RAAS) and progression of insulin resistance. Mol Cell Endocrinol 378: 1–14, 2013. doi: 10.1016/j.mce.2012.06.021. [DOI] [PubMed] [Google Scholar]
- 651.Markó L, Henke N, Park JK, Spallek B, Qadri F, Balogh A, Apel IJ, Oravecz-Wilson KI, Choi M, Przybyl L, Binger KJ, Haase N, Wilck N, Heuser A, Fokuhl V, Ruland J, Lucas PC, McAllister-Lucas LM, Luft FC, Dechend R, Müller DN. Bcl10 mediates angiotensin II-induced cardiac damage and electrical remodeling. Hypertension 64: 1032–1039, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 652.Markó L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, Bowman EP, Kleinewietfeld M, Fokuhl V, Dechend R, Müller DN. Interferon-γ signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension 60: 1430–1436, 2012. doi: 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- 653.Marrachelli VG, Mastronardi ML, Sarr M, Soleti R, Leonetti D, Martínez MC, Andriantsitohaina R. Sonic hedgehog carried by microparticles corrects angiotensin II-induced hypertension and endothelial dysfunction in mice. PLoS One 8: e72861, 2013. doi: 10.1371/journal.pone.0072861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 654.Marsh SA, Dell’Italia LJ, Chatham JC. Activation of the hexosamine biosynthesis pathway and protein O-GlcNAcylation modulate hypertrophic and cell signaling pathways in cardiomyocytes from diabetic mice. Amino Acids 40: 819–828, 2011. doi: 10.1007/s00726-010-0699-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Martín-Alonso M, García-Redondo AB, Guo D, Camafeita E, Martínez F, Alfranca A, Méndez-Barbero N, Pollán Á, Sánchez-Camacho C, Denhardt DT, Seiki M, Vázquez J, Salaices M, Redondo JM, Milewicz D, Arroyo AG. Deficiency of MMP17/MT4-MMP proteolytic activity predisposes to aortic aneurysm in mice. Circ Res 117: e13–e26, 2015. doi: 10.1161/CIRCRESAHA.117.305108. [DOI] [PubMed] [Google Scholar]
- 656.Martínez-Revelles S, García-Redondo AB, Avendaño MS, Varona S, Palao T, Orriols M, Roque FR, Fortuño A, Touyz RM, Martínez-González J, Salaices M, Rodríguez C, Briones AM. Lysyl Oxidase Induces Vascular Oxidative Stress and Contributes to Arterial Stiffness and Abnormal Elastin Structure in Hypertension: Role of p38MAPK. Antioxid Redox Signal 27: 379–397, 2017. doi: 10.1089/ars.2016.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657.Marzetti E, Calvani R, DuPree J, Lees HA, Giovannini S, Seo DO, Buford TW, Sweet K, Morgan D, Strehler KYE, Diz D, Borst SE, Moningka N, Krotova K, Carter CS. Late-life enalapril administration induces nitric oxide-dependent and independent metabolic adaptations in the rat skeletal muscle. Age (Dordr) 35: 1061–1075, 2013. doi: 10.1007/s11357-012-9428-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 658.Massiéra F, Bloch-Faure M, Ceiler D, Murakami K, Fukamizu A, Gasc JM, Quignard-Boulange A, Negrel R, Ailhaud G, Seydoux J, Meneton P, Teboul M. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J 15: 2727–2729, 2001. doi: 10.1096/fj.01-0457fje. [DOI] [PubMed] [Google Scholar]
- 659.Massiera F, Seydoux J, Geloen A, Quignard-Boulange A, Turban S, Saint-Marc P, Fukamizu A, Negrel R, Ailhaud G, Teboul M. Angiotensinogen-deficient mice exhibit impairment of diet-induced weight gain with alteration in adipose tissue development and increased locomotor activity. Endocrinology 142: 5220–5225, 2001. doi: 10.1210/endo.142.12.8556. [DOI] [PubMed] [Google Scholar]
- 660.Matavelli LC, Siragy HM. AT2 receptor activities and pathophysiological implications. J Cardiovasc Pharmacol 65: 226–232, 2015. doi: 10.1097/FJC.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661.Mateos L, Perez-Alvarez MJ, Wandosell F. Angiotensin II type-2 receptor stimulation induces neuronal VEGF synthesis after cerebral ischemia. Biochim Biophys Acta 1862: 1297–1308, 2016. doi: 10.1016/j.bbadis.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 662.Matsuda S, Umemoto S, Yoshimura K, Itoh S, Murata T, Fukai T, Matsuzaki M. Angiotensin II Activates MCP-1 and Induces Cardiac Hypertrophy and Dysfunction via Toll-like Receptor 4. J Atheroscler Thromb 22: 833–844, 2015. doi: 10.5551/jat.27292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Matsuda T, Hiyama TY, Niimura F, Matsusaka T, Fukamizu A, Kobayashi K, Kobayashi K, Noda M. Distinct neural mechanisms for the control of thirst and salt appetite in the subfornical organ. Nat Neurosci 20: 230–241, 2017. doi: 10.1038/nn.4463. [DOI] [PubMed] [Google Scholar]
- 664.Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest 115: 599–609, 2005. doi: 10.1172/JCI22304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665.Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, Takai S, Yamanishi K, Miyazaki M, Matsubara H, Yabe-Nishimura C. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation 112: 2677–2685, 2005. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- 666.Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K, Kinugawa S, Tsutsui H. Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension 47: 711–717, 2006. doi: 10.1161/01.HYP.0000208840.30778.00. [DOI] [PubMed] [Google Scholar]
- 667.Matsusaka T, Niimura F, Pastan I, Shintani A, Nishiyama A, Ichikawa I. Podocyte injury enhances filtration of liver-derived angiotensinogen and renal angiotensin II generation. Kidney Int 85: 1068–1077, 2014. doi: 10.1038/ki.2013.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668.Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H, Nishiyama A, Ichikawa I. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol 23: 1181–1189, 2012. doi: 10.1681/ASN.2011121159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Matsushita K, Wu Y, Pratt RE, Dzau VJ. Deletion of angiotensin II type 2 receptor accelerates adipogenesis in murine mesenchymal stem cells via Wnt10b/beta-catenin signaling. Lab Invest 96: 909–917, 2016. doi: 10.1038/labinvest.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 670.Matsuura H, Ichiki T, Ikeda J, Takeda K, Miyazaki R, Hashimoto T, Narabayashi E, Kitamoto S, Tokunou T, Sunagawa K. Inhibition of prolyl hydroxylase domain-containing protein downregulates vascular angiotensin II type 1 receptor. Hypertension 58: 386–393, 2011. doi: 10.1161/HYPERTENSIONAHA.110.167106. [DOI] [PubMed] [Google Scholar]
- 672.Mayr M, Duerrschmid C, Medrano G, Taffet GE, Wang Y, Entman ML, Haudek SB. TNF/Ang-II synergy is obligate for fibroinflammatory pathology, but not for changes in cardiorenal function. Physiol Rep 4: e12765, 2016. doi: 10.14814/phy2.12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673.McAllister-Lucas LM, Jin X, Gu S, Siu K, McDonnell S, Ruland J, Delekta PC, Van Beek M, Lucas PC. The CARMA3-Bcl10-MALT1 signalosome promotes angiotensin II-dependent vascular inflammation and atherogenesis. J Biol Chem 285: 25880–25884, 2010. doi: 10.1074/jbc.C110.109421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674.McAllister-Lucas LM, Ruland J, Siu K, Jin X, Gu S, Kim DS, Kuffa P, Kohrt D, Mak TW, Nuñez G, Lucas PC. CARMA3/Bcl10/MALT1-dependent NF-kappaB activation mediates angiotensin II-responsive inflammatory signaling in nonimmune cells. Proc Natl Acad Sci USA 104: 139–144, 2007. doi: 10.1073/pnas.0601947103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 675.McCarthy CA, Widdop RE, Denton KM, Jones ES. Update on the angiotensin AT(2) receptor. Curr Hypertens Rep 15: 25–30, 2013. doi: 10.1007/s11906-012-0321-4. [DOI] [PubMed] [Google Scholar]
- 676.McCollum LT, Gallagher PE, Tallant EA. Angiotensin-(1-7) attenuates angiotensin II-induced cardiac remodeling associated with upregulation of dual-specificity phosphatase 1. Am J Physiol Heart Circ Physiol 302: H801–H810, 2012. doi: 10.1152/ajpheart.00908.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677.McCurley A, McGraw A, Pruthi D, Jaffe IZ. Smooth muscle cell mineralocorticoid receptors: role in vascular function and contribution to cardiovascular disease. Pflugers Arch 465: 1661–1670, 2013. doi: 10.1007/s00424-013-1282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678.McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME, Jaffe IZ. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med 18: 1429–1433, 2012. doi: 10.1038/nm.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 679.McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res 116: 1022–1033, 2015. doi: 10.1161/CIRCRESAHA.116.303697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 680.Mederle K, Gess B, Pluteanu F, Plackic J, Tiefenbach KJ, Grill A, Kockskämper J, Castrop H. The angiotensin receptor-associated protein Atrap is a stimulator of the cardiac Ca2+-ATPase SERCA2a. Cardiovasc Res 110: 359–370, 2016. doi: 10.1093/cvr/cvw064. [DOI] [PubMed] [Google Scholar]
- 681.Mederos y Schnitzler M, Storch U, Meibers S, Nurwakagari P, Breit A, Essin K, Gollasch M, Gudermann T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J 27: 3092–3103, 2008. doi: 10.1038/emboj.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292: C82–C97, 2007. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 683.Meijles DN, Pagano PJ. Nox and Inflammation in the Vascular Adventitia. Hypertension 67: 14–19, 2016. doi: 10.1161/HYPERTENSIONAHA.115.03622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 684.Meissner A, Miro F, Jiménez-Altayó F, Jurado A, Vila E, Planas AM. Sphingosine-1-phosphate signalling-a key player in the pathogenesis of Angiotensin II-induced hypertension. Cardiovasc Res 113: 123–133, 2017. doi: 10.1093/cvr/cvw256. [DOI] [PubMed] [Google Scholar]
- 685.Mellak S, Ait-Oufella H, Esposito B, Loyer X, Poirier M, Tedder TF, Tedgui A, Mallat Z, Potteaux S. Angiotensin II mobilizes spleen monocytes to promote the development of abdominal aortic aneurysm in Apoe-/- mice. Arterioscler Thromb Vasc Biol 35: 378–388, 2015. doi: 10.1161/ATVBAHA.114.304389. [DOI] [PubMed] [Google Scholar]
- 686.Mendelowitz D. How Does Angiotensin Activate Hypothalamic Neurons Essential for Controlling Sympathetic Activity and Blood Pressure? Hypertension 68: 1340–1341, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07914. [DOI] [PubMed] [Google Scholar]
- 687.Méndez-Bolaina E, Sánchez-González J, Ramírez-Sánchez I, Ocharán-Hernández E, Núñez-Sánchez M, Meaney-Mendiolea E, Meaney A, Asbun-Bojalil J, Miliar-García A, Olivares-Corichi I, Ceballos-Reyes G. Effect of caveolin-1 scaffolding peptide and 17beta-estradiol on intracellular Ca2+ kinetics evoked by angiotensin II in human vascular smooth muscle cells. Am J Physiol Cell Physiol 293: C1953–C1961, 2007. doi: 10.1152/ajpcell.00519.2006. [DOI] [PubMed] [Google Scholar]
- 688.Meng X, Yang J, Zhang K, An G, Kong J, Jiang F, Zhang Y, Zhang C. Regulatory T cells prevent angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E knockout mice. Hypertension 64: 875–882, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03950. [DOI] [PubMed] [Google Scholar]
- 689.Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev 30: 1–17, 2016. doi: 10.1101/gad.274027.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690.Mennuni S, Rubattu S, Pierelli G, Tocci G, Fofi C, Volpe M. Hypertension and kidneys: unraveling complex molecular mechanisms underlying hypertensive renal damage. J Hum Hypertens 28: 74–79, 2014. doi: 10.1038/jhh.2013.55. [DOI] [PubMed] [Google Scholar]
- 691.Merino VF, Todiras M, Mori MA, Sales VM, Fonseca RG, Saul V, Tenner K, Bader M, Pesquero JB. Predisposition to atherosclerosis and aortic aneurysms in mice deficient in kinin B1 receptor and apolipoprotein E. J Mol Med (Berl) 87: 953–963, 2009. doi: 10.1007/s00109-009-0501-0. [DOI] [PubMed] [Google Scholar]
- 692.Mervaala E, Biala A, Merasto S, Lempiäinen J, Mattila I, Martonen E, Eriksson O, Louhelainen M, Finckenberg P, Kaheinen P, Muller DN, Luft FC, Lapatto R, Oresic M. Metabolomics in angiotensin II-induced cardiac hypertrophy. Hypertension 55: 508–515, 2010. doi: 10.1161/HYPERTENSIONAHA.109.145490. [DOI] [PubMed] [Google Scholar]
- 693.Meyer MR, Fredette NC, Daniel C, Sharma G, Amann K, Arterburn JB, Barton M, Prossnitz ER. Obligatory role for GPER in cardiovascular aging and disease. Sci Signal 9: ra105, 2016. doi: 10.1126/scisignal.aag0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 694.Miao SB, Xie XL, Yin YJ, Zhao LL, Zhang F, Shu YN, Chen R, Chen P, Dong LH, Lin YL, Lv P, Zhang DD, Nie X, Xue ZY, Han M. Accumulation of Smooth Muscle 22α Protein Accelerates Senescence of Vascular Smooth Muscle Cells via Stabilization of p53 In Vitro and In Vivo. Arterioscler Thromb Vasc Biol 37: 1849–1859, 2017. doi: 10.1161/ATVBAHA.117.309378. [DOI] [PubMed] [Google Scholar]
- 695.Miao XN, Siu KL, Cai H. Nifedipine attenuation of abdominal aortic aneurysm in hypertensive and non-hypertensive mice: mechanisms and implications. J Mol Cell Cardiol 87: 152–159, 2015. doi: 10.1016/j.yjmcc.2015.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696.Mieth A, Revermann M, Babelova A, Weigert A, Schermuly RT, Brandes RP. L-type calcium channel inhibitor diltiazem prevents aneurysm formation by blood pressure-independent anti-inflammatory effects. Hypertension 62: 1098–1104, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01986. [DOI] [PubMed] [Google Scholar]
- 697.Mifune M, Ohtsu H, Suzuki H, Nakashima H, Brailoiu E, Dun NJ, Frank GD, Inagami T, Higashiyama S, Thomas WG, Eckhart AD, Dempsey PJ, Eguchi S. G protein coupling and second messenger generation are indispensable for metalloprotease-dependent, heparin-binding epidermal growth factor shedding through angiotensin II type-1 receptor. J Biol Chem 280: 26592–26599, 2005. doi: 10.1074/jbc.M502906200. [DOI] [PubMed] [Google Scholar]
- 698.Mikolajczyk TP, Nosalski R, Szczepaniak P, Budzyn K, Osmenda G, Skiba D, Sagan A, Wu J, Vinh A, Marvar PJ, Guzik B, Podolec J, Drummond G, Lob HE, Harrison DG, Guzik TJ. Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J 30: 1987–1999, 2016. doi: 10.1096/fj.201500088R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 699.Min L-J, Mogi M, Iwanami J, Jing F, Tsukuda K, Ohshima K, Horiuchi M. Angiotensin II type 2 receptor-interacting protein prevents vascular senescence. J Am Soc Hypertens 6: 179–184, 2012. doi: 10.1016/j.jash.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 700.Min L-J, Mogi M, Tamura K, Iwanami J, Sakata A, Fujita T, Tsukuda K, Jing F, Iwai M, Horiuchi M. Angiotensin II type 1 receptor-associated protein prevents vascular smooth muscle cell senescence via inactivation of calcineurin/nuclear factor of activated T cells pathway. J Mol Cell Cardiol 47: 798–809, 2009. doi: 10.1016/j.yjmcc.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 701.Min LJ, Mogi M, Iwanami J, Li JM, Sakata A, Fujita T, Tsukuda K, Iwai M, Horiuchi M. Angiotensin II type 2 receptor deletion enhances vascular senescence by methyl methanesulfonate sensitive 2 inhibition. Hypertension 51: 1339–1344, 2008. doi: 10.1161/HYPERTENSIONAHA.107.105692. [DOI] [PubMed] [Google Scholar]
- 702.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res 107: 1071–1082, 2010. doi: 10.1161/CIRCRESAHA.110.227819. [DOI] [PubMed] [Google Scholar]
- 703.Miura S, Zhang J, Boros J, Karnik SS. TM2-TM7 interaction in coupling movement of transmembrane helices to activation of the angiotensin II type-1 receptor. J Biol Chem 278: 3720–3725, 2003. doi: 10.1074/jbc.M211338200. [DOI] [PubMed] [Google Scholar]
- 704.Miura S, Zhang J, Matsuo Y, Saku K, Karnik SS. Activation of extracellular signal-activated kinase by angiotensin II-induced Gq-independent epidermal growth factor receptor transactivation. Hypertens Res 27: 765–770, 2004. doi: 10.1291/hypres.27.765. [DOI] [PubMed] [Google Scholar]
- 705.Mogi M, Horiuchi M. Effect of angiotensin II type 2 receptor on stroke, cognitive impairment and neurodegenerative diseases. Geriatr Gerontol Int 13: 13–18, 2013. doi: 10.1111/j.1447-0594.2012.00900.x. [DOI] [PubMed] [Google Scholar]
- 706.Mogi M, Iwai M, Horiuchi M. Emerging concepts of regulation of angiotensin II receptors: new players and targets for traditional receptors. Arterioscler Thromb Vasc Biol 27: 2532–2539, 2007. doi: 10.1161/ATVBAHA.107.144154. [DOI] [PubMed] [Google Scholar]
- 707.Monasky MM, Taglieri DM, Henze M, Warren CM, Utter MS, Soergel DG, Violin JD, Solaro RJ. The β-arrestin-biased ligand TRV120023 inhibits angiotensin II-induced cardiac hypertrophy while preserving enhanced myofilament response to calcium. Am J Physiol Heart Circ Physiol 305: H856–H866, 2013. doi: 10.1152/ajpheart.00327.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 708.Mondorf UF, Geiger H, Herrero M, Zeuzem S, Piiper A. Involvement of the platelet-derived growth factor receptor in angiotensin II-induced activation of extracellular regulated kinases 1 and 2 in human mesangial cells. FEBS Lett 472: 129–132, 2000. doi: 10.1016/S0014-5793(00)01433-2. [DOI] [PubMed] [Google Scholar]
- 709.Montecucco F, Pende A, Mach F. The renin-angiotensin system modulates inflammatory processes in atherosclerosis: evidence from basic research and clinical studies. Mediators Inflamm 2009: 752406, 2009. doi: 10.1155/2009/752406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 710.Montezano AC, Burger D, Paravicini TM, Chignalia AZ, Yusuf H, Almasri M, He Y, Callera GE, He G, Krause KH, Lambeth D, Quinn MT, Touyz RM. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ Res 106: 1363–1373, 2010. doi: 10.1161/CIRCRESAHA.109.216036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 711.Montezano AC, Touyz RM. Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxid Redox Signal 20: 164–182, 2014. doi: 10.1089/ars.2013.5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 712.Moore-Morris T, Guimarães-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest 124: 2921–2934, 2014. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 713.Moore-Morris T, Guimarães-Camboa N, Yutzey KE, Pucéat M, Evans SM. Cardiac fibroblasts: from development to heart failure. J Mol Med (Berl) 93: 823–830, 2015. doi: 10.1007/s00109-015-1314-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 714.Moraes JA, Frony AC, Dias AM, Renovato-Martins M, Rodrigues G, Marcinkiewicz C, Assreuy J, Barja-Fidalgo C. Alpha1beta1 and integrin-linked kinase interact and modulate angiotensin II effects in vascular smooth muscle cells. Atherosclerosis 243: 477–485, 2015. doi: 10.1016/j.atherosclerosis.2015.09.026. [DOI] [PubMed] [Google Scholar]
- 715.Moran CS, Seto SW, Krishna SM, Sharma S, Jose RJ, Biros E, Wang Y, Morton SK, Golledge J. Parenteral administration of factor Xa/IIa inhibitors limits experimental aortic aneurysm and atherosclerosis. Sci Rep 7: 43079, 2017. doi: 10.1038/srep43079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 716.Mori J, Basu R, McLean BA, Das SK, Zhang L, Patel VB, Wagg CS, Kassiri Z, Lopaschuk GD, Oudit GY. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: a metabolic contribution to heart failure with normal ejection fraction. Circ Heart Fail 5: 493–503, 2012. doi: 10.1161/CIRCHEARTFAILURE.112.966705. [DOI] [PubMed] [Google Scholar]
- 717.Morla L, Edwards A, Crambert G. New insights into sodium transport regulation in the distal nephron: role of G-protein coupled receptors. World J Biol Chem 7: 44–63, 2016. doi: 10.4331/wjbc.v7.i1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717a.Morris EM, Fletcher JA, Thyfault JP, Rector RS. The role of angiotensin II in nonalcoholic steatohepatitis. Mol Cell Endocrinol 378: 29–40, 2013. doi: 10.1016/j.mce.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 718.Mount DB. Thick ascending limb of the loop of Henle. Clin J Am Soc Nephrol 9: 1974–1986, 2014. doi: 10.2215/CJN.04480413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 719.Mukohda M, Lu KT, Guo DF, Wu J, Keen HL, Liu X, Ketsawatsomkron P, Stump M, Rahmouni K, Quelle FW, Sigmund CD. Hypertension-Causing Mutation in Peroxisome Proliferator-Activated Receptor γ Impairs Nuclear Export of Nuclear Factor-κB p65 in Vascular Smooth Muscle. Hypertension 70: 174–182, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720.Mukoyama M, Nakajima M, Horiuchi M, Sasamura H, Pratt RE, Dzau VJ. Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J Biol Chem 268: 24539–24542, 1993. [PubMed] [Google Scholar]
- 721.Muniyappa R, Yavuz S. Metabolic actions of angiotensin II and insulin: a microvascular endothelial balancing act. Mol Cell Endocrinol 378: 59–69, 2013. doi: 10.1016/j.mce.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 722.Muñoz MC, Giani JF, Dominici FP, Turyn D, Toblli JE. Long-term treatment with an angiotensin II receptor blocker decreases adipocyte size and improves insulin signaling in obese Zucker rats. J Hypertens 27: 2409–2420, 2009. doi: 10.1097/HJH.0b013e3283310e1b. [DOI] [PubMed] [Google Scholar]
- 723.Muñoz-Durango N, Fuentes CA, Castillo AE, González-Gómez LM, Vecchiola A, Fardella CE, Kalergis AM. Role of the Renin-Angiotensin-Aldosterone System beyond Blood Pressure Regulation: Molecular and Cellular Mechanisms Involved in End-Organ Damage during Arterial Hypertension. Int J Mol Sci 17: 797, 2016. doi: 10.3390/ijms17070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 724.Muntner P, Carey RM, Gidding S, Jones DW, Taler SJ, Wright JT Jr, Whelton PK. Potential U.S. Population Impact of the 2017 American College of Cardiology/American Heart Association High Blood Pressure Guideline. J Am Coll Cardiol 71: 109–118, 2018. doi: 10.1016/j.jacc.2017.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 725.Murdoch CE, Alom-Ruiz SP, Wang M, Zhang M, Walker S, Yu B, Brewer A, Shah AM. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res Cardiol 106: 527–538, 2011. doi: 10.1007/s00395-011-0179-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 726.Murdoch CE, Chaubey S, Zeng L, Yu B, Ivetic A, Walker SJ, Vanhoutte D, Heymans S, Grieve DJ, Cave AC, Brewer AC, Zhang M, Shah AM. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J Am Coll Cardiol 63: 2734–2741, 2014. doi: 10.1016/j.jacc.2014.02.572. [DOI] [PubMed] [Google Scholar]
- 727.Murphy KT, Allen AM, Chee A, Naim T, Lynch GS. Disruption of muscle renin-angiotensin system in AT1a-/- mice enhances muscle function despite reducing muscle mass but compromises repair after injury. Am J Physiol Regul Integr Comp Physiol 303: R321–R331, 2012. doi: 10.1152/ajpregu.00007.2012. [DOI] [PubMed] [Google Scholar]
- 728.Murphy TJ, Alexander RW, Griendling KK, Runge MS, Bernstein KE. Isolation of a cDNA encoding the vascular type-1 angiotensin II receptor. Nature 351: 233–236, 1991. doi: 10.1038/351233a0. [DOI] [PubMed] [Google Scholar]
- 729.Muscogiuri G, Chavez AO, Gastaldelli A, Perego L, Tripathy D, Saad MJ, Velloso L, Folli F. The crosstalk between insulin and renin-angiotensin-aldosterone signaling systems and its effect on glucose metabolism and diabetes prevention. Curr Vasc Pharmacol 6: 301–312, 2008. doi: 10.2174/157016108785909715. [DOI] [PubMed] [Google Scholar]
- 730.Muta K, Morgan DA, Grobe JL, Sigmund CD, Rahmouni K. mTORC1 Signaling Contributes to Drinking But Not Blood Pressure Responses to Brain Angiotensin II. Endocrinology 157: 3140–3148, 2016. doi: 10.1210/en.2016-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 731.Nabah YN, Mateo T, Estellés R, Mata M, Zagorski J, Sarau H, Cortijo J, Morcillo EJ, Jose PJ, Sanz MJ. Angiotensin II induces neutrophil accumulation in vivo through generation and release of CXC chemokines. Circulation 110: 3581–3586, 2004. doi: 10.1161/01.CIR.0000148824.93600.F3. [DOI] [PubMed] [Google Scholar]
- 732.Nabeebaccus A, Zhang M, Shah AM. NADPH oxidases and cardiac remodelling. Heart Fail Rev 16: 5–12, 2011. doi: 10.1007/s10741-010-9186-2. [DOI] [PubMed] [Google Scholar]
- 733.Nagai M, Terao S, Vital SA, Rodrigues SF, Yilmaz G, Granger DN. Role of blood cell-associated angiotensin II type 1 receptors in the cerebral microvascular response to ischemic stroke during angiotensin-induced hypertension. Exp Transl Stroke Med 3: 15, 2011. doi: 10.1186/2040-7378-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 734.Nagata S, Kato J, Sasaki K, Minamino N, Eto T, Kitamura K. Isolation and identification of proangiotensin-12, a possible component of the renin-angiotensin system. Biochem Biophys Res Commun 350: 1026–1031, 2006. doi: 10.1016/j.bbrc.2006.09.146. [DOI] [PubMed] [Google Scholar]
- 735.Nagpal V, Rai R, Place AT, Murphy SB, Verma SK, Ghosh AK, Vaughan DE. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 133: 291–301, 2016. doi: 10.1161/CIRCULATIONAHA.116.022627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736.Nagy T, Champattanachai V, Marchase RB, Chatham JC. Glucosamine inhibits angiotensin II-induced cytoplasmic Ca2+ elevation in neonatal cardiomyocytes via protein-associated O-linked N-acetylglucosamine. Am J Physiol Cell Physiol 290: C57–C65, 2006. doi: 10.1152/ajpcell.00263.2005. [DOI] [PubMed] [Google Scholar]
- 737.Nakagawa P, Sigmund CD. How Is the Brain Renin-Angiotensin System Regulated? Hypertension 70: 10–18, 2017. doi: 10.1161/HYPERTENSIONAHA.117.08550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 738.Nakagawa T, Hasegawa Y, Uekawa K, Senju S, Nakagata N, Matsui K, Kim-Mitsuyama S. Transient Mild Cerebral Ischemia Significantly Deteriorated Cognitive Impairment in a Mouse Model of Alzheimer’s Disease via Angiotensin AT1 Receptor. Am J Hypertens 30: 141–150, 2017. doi: 10.1093/ajh/hpw099. [DOI] [PubMed] [Google Scholar]
- 739.Nakamura T, Kataoka K, Fukuda M, Nako H, Tokutomi Y, Dong YF, Ichijo H, Ogawa H, Kim-Mitsuyama S. Critical role of apoptosis signal-regulating kinase 1 in aldosterone/salt-induced cardiac inflammation and fibrosis. Hypertension 54: 544–551, 2009. doi: 10.1161/HYPERTENSIONAHA.109.135392. [DOI] [PubMed] [Google Scholar]
- 740.Nakashima H, Frank GD, Shirai H, Hinoki A, Higuchi S, Ohtsu H, Eguchi K, Sanjay A, Reyland ME, Dempsey PJ, Inagami T, Eguchi S. Novel role of protein kinase C-delta Tyr 311 phosphorylation in vascular smooth muscle cell hypertrophy by angiotensin II. Hypertension 51: 232–238, 2008. doi: 10.1161/HYPERTENSIONAHA.107.101253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 741.Nakashima H, Kumagai K, Urata H, Gondo N, Ideishi M, Arakawa K. Angiotensin II antagonist prevents electrical remodeling in atrial fibrillation. Circulation 101: 2612–2617, 2000. doi: 10.1161/01.CIR.101.22.2612. [DOI] [PubMed] [Google Scholar]
- 742.Nakayama H, Bodi I, Maillet M, DeSantiago J, Domeier TL, Mikoshiba K, Lorenz JN, Blatter LA, Bers DM, Molkentin JD. The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ Res 107: 659–666, 2010. doi: 10.1161/CIRCRESAHA.110.220038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 743.Nawano M, Anai M, Funaki M, Kobayashi H, Kanda A, Fukushima Y, Inukai K, Ogihara T, Sakoda H, Onishi Y, Kikuchi M, Yazaki Y, Oka Y, Asano T. Imidapril, an angiotensin-converting enzyme inhibitor, improves insulin sensitivity by enhancing signal transduction via insulin receptor substrate proteins and improving vascular resistance in the Zucker fatty rat. Metabolism 48: 1248–1255, 1999. doi: 10.1016/S0026-0495(99)90263-9. [DOI] [PubMed] [Google Scholar]
- 744.Nehme A, Cerutti C, Zibara K. Transcriptomic Analysis Reveals Novel Transcription Factors Associated With Renin-Angiotensin-Aldosterone System in Human Atheroma. Hypertension 68: 1375–1384, 2016. doi: 10.1161/HYPERTENSIONAHA.116.08070. [DOI] [PubMed] [Google Scholar]
- 745.Newgard CB, Sharpless NE. Coming of age: molecular drivers of aging and therapeutic opportunities. J Clin Invest 123: 946–950, 2013. doi: 10.1172/JCI68833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 746.Nguyen Dinh Cat A, Montezano AC, Burger D, Touyz RM. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid Redox Signal 19: 1110–1120, 2013. doi: 10.1089/ars.2012.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 747.Nguyen Dinh Cat A, Touyz RM. Cell signaling of angiotensin II on vascular tone: novel mechanisms. Curr Hypertens Rep 13: 122–128, 2011. doi: 10.1007/s11906-011-0187-x. [DOI] [PubMed] [Google Scholar]
- 748.Nguyen G. Renin, (pro)renin and receptor: an update. Clin Sci (Lond) 120: 169–178, 2011. doi: 10.1042/CS20100432. [DOI] [PubMed] [Google Scholar]
- 749.Nguyen MTX, Han J, Ralph DL, Veiras LC, McDonough AA. Short-term nonpressor angiotensin II infusion stimulates sodium transporters in proximal tubule and distal nephron. Physiol Rep 3: e12496, 2015. doi: 10.14814/phy2.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 750.Ni W, Zhan Y, He H, Maynard E, Balschi JA, Oettgen P. Ets-1 is a critical transcriptional regulator of reactive oxygen species and p47(phox) gene expression in response to angiotensin II. Circ Res 101: 985–994, 2007. doi: 10.1161/CIRCRESAHA.107.152439. [DOI] [PubMed] [Google Scholar]
- 751.Nishida M, Ogushi M, Suda R, Toyotaka M, Saiki S, Kitajima N, Nakaya M, Kim KM, Ide T, Sato Y, Inoue K, Kurose H. Heterologous down-regulation of angiotensin type 1 receptors by purinergic P2Y2 receptor stimulation through S-nitrosylation of NF-kappaB. Proc Natl Acad Sci USA 108: 6662–6667, 2011. doi: 10.1073/pnas.1017640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Nishimura A, Sunggip C, Tozaki-Saitoh H, Shimauchi T, Numaga-Tomita T, Hirano K, Ide T, Boeynaems JM, Kurose H, Tsuda M, Robaye B, Inoue K, Nishida M. Purinergic P2Y6 receptors heterodimerize with angiotensin AT1 receptors to promote angiotensin II-induced hypertension. Sci Signal 9: ra7, 2016. doi: 10.1126/scisignal.aac9187. [DOI] [PubMed] [Google Scholar]
- 753.Nishioka K, Nishida M, Ariyoshi M, Jian Z, Saiki S, Hirano M, Nakaya M, Sato Y, Kita S, Iwamoto T, Hirano K, Inoue R, Kurose H. Cilostazol suppresses angiotensin II-induced vasoconstriction via protein kinase A-mediated phosphorylation of the transient receptor potential canonical 6 channel. Arterioscler Thromb Vasc Biol 31: 2278–2286, 2011. doi: 10.1161/ATVBAHA.110.221010. [DOI] [PubMed] [Google Scholar]
- 754.Niu P, Shindo T, Iwata H, Iimuro S, Takeda N, Zhang Y, Ebihara A, Suematsu Y, Kangawa K, Hirata Y, Nagai R. Protective effects of endogenous adrenomedullin on cardiac hypertrophy, fibrosis, and renal damage. Circulation 109: 1789–1794, 2004. doi: 10.1161/01.CIR.0000118466.47982.CC. [DOI] [PubMed] [Google Scholar]
- 755.Nogueira EF, Bollag WB, Rainey WE. Angiotensin II regulation of adrenocortical gene transcription. Mol Cell Endocrinol 302: 230–236, 2009. doi: 10.1016/j.mce.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 756.Noll C, Labbé SM, Pinard S, Shum M, Bilodeau L, Chouinard L, Phoenix S, Lecomte R, Carpentier AC, Gallo-Payet N. Postprandial fatty acid uptake and adipocyte remodeling in angiotensin type 2 receptor-deficient mice fed a high-fat/high-fructose diet. Adipocyte 5: 43–52, 2016. doi: 10.1080/21623945.2015.1115582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 757.Nomura S, Shouzu A, Omoto S, Nishikawa M, Iwasaka T. Effects of losartan and simvastatin on monocyte-derived microparticles in hypertensive patients with and without type 2 diabetes mellitus. Clin Appl Thromb Hemost 10: 133–141, 2004. doi: 10.1177/107602960401000203. [DOI] [PubMed] [Google Scholar]
- 758.Norlander AE, Saleh MA, Pandey AK, Itani HA, Wu J, Xiao L, Kang J, Dale BL, Goleva SB, Laroumanie F, Du L, Harrison DG, Madhur MS. A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage. JCI Insight 2: e92801, 2017. doi: 10.1172/jci.insight.92801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 759.Nouet S, Amzallag N, Li JM, Louis S, Seitz I, Cui TX, Alleaume AM, Di Benedetto M, Boden C, Masson M, Strosberg AD, Horiuchi M, Couraud PO, Nahmias C. Trans-inactivation of receptor tyrosine kinases by novel angiotensin II AT2 receptor-interacting protein, ATIP. J Biol Chem 279: 28989–28997, 2004. doi: 10.1074/jbc.M403880200. [DOI] [PubMed] [Google Scholar]
- 760.Nuki Y, Tsou TL, Kurihara C, Kanematsu M, Kanematsu Y, Hashimoto T. Elastase-induced intracranial aneurysms in hypertensive mice. Hypertension 54: 1337–1344, 2009. doi: 10.1161/HYPERTENSIONAHA.109.138297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 761.Nussberger J, Brunner DB, Nyfeler JA, Linder L, Brunner HR. Measurement of immunoreactive angiotensin-(1-7) heptapeptide in human blood. Clin Chem 47: 726–729, 2001. [PubMed] [Google Scholar]
- 762.Nussenzweig SC, Verma S, Finkel T. The role of autophagy in vascular biology. Circ Res 116: 480–488, 2015. doi: 10.1161/CIRCRESAHA.116.303805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 763.O’Leary R, Penrose H, Miyata K, Satou R. Macrophage-derived IL-6 contributes to ANG II-mediated angiotensinogen stimulation in renal proximal tubular cells. Am J Physiol Renal Physiol 310: F1000–F1007, 2016. doi: 10.1152/ajprenal.00482.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 764.Obama T, Eguchi S. MicroRNA as a novel component of the tissue renin angiotensin system. J Mol Cell Cardiol 75: 98–99, 2014. doi: 10.1016/j.yjmcc.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 765.Obama T, Takayanagi T, Kobayashi T, Bourne AM, Elliott KJ, Charbonneau M, Dubois CM, Eguchi S. Vascular induction of a disintegrin and metalloprotease 17 by angiotensin II through hypoxia inducible factor 1α. Am J Hypertens 28: 10–14, 2015. doi: 10.1093/ajh/hpu094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 766.Obama T, Tsuji T, Kobayashi T, Fukuda Y, Takayanagi T, Taro Y, Kawai T, Forrester SJ, Elliott KJ, Choi E, Daugherty A, Rizzo V, Eguchi S. Epidermal growth factor receptor inhibitor protects against abdominal aortic aneurysm in a mouse model. Clin Sci (Lond) 128: 559–565, 2015. doi: 10.1042/CS20140696. [DOI] [PubMed] [Google Scholar]
- 767.Ocaranza MP, Michea L, Chiong M, Lagos CF, Lavandero S, Jalil JE. Recent insights and therapeutic perspectives of angiotensin-(1-9) in the cardiovascular system. Clin Sci (Lond) 127: 549–557, 2014. doi: 10.1042/CS20130449. [DOI] [PubMed] [Google Scholar]
- 768.Odenbach J, Wang X, Cooper S, Chow FL, Oka T, Lopaschuk G, Kassiri Z, Fernandez-Patron C. MMP-2 mediates angiotensin II-induced hypertension under the transcriptional control of MMP-7 and TACE. Hypertension 57: 123–130, 2011. doi: 10.1161/HYPERTENSIONAHA.110.159525. [DOI] [PubMed] [Google Scholar]
- 769.Ohsawa M, Tamura K, Wakui H, Maeda A, Dejima T, Kanaoka T, Azushima K, Uneda K, Tsurumi-Ikeya Y, Kobayashi R, Matsuda M, Uchida S, Toya Y, Kobori H, Nishiyama A, Yamashita A, Ishikawa Y, Umemura S. Deletion of the angiotensin II type 1 receptor-associated protein enhances renal sodium reabsorption and exacerbates angiotensin II-mediated hypertension. Kidney Int 86: 570–581, 2014. doi: 10.1038/ki.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 770.Ohshima K, Mogi M, Jing F, Iwanami J, Tsukuda K, Min LJ, Higaki J, Horiuchi M. Roles of interleukin 17 in angiotensin II type 1 receptor-mediated insulin resistance. Hypertension 59: 493–499, 2012. doi: 10.1161/HYPERTENSIONAHA.111.183178. [DOI] [PubMed] [Google Scholar]
- 771.Ohtsu H, Dempsey PJ, Frank GD, Brailoiu E, Higuchi S, Suzuki H, Nakashima H, Eguchi K, Eguchi S. ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler Thromb Vasc Biol 26: e133–e137, 2006. doi: 10.1161/01.ATV.0000236203.90331.d0. [DOI] [PubMed] [Google Scholar]
- 772.Ohtsu H, Higuchi S, Shirai H, Eguchi K, Suzuki H, Hinoki A, Brailoiu E, Eckhart AD, Frank GD, Eguchi S. Central role of Gq in the hypertrophic signal transduction of angiotensin II in vascular smooth muscle cells. Endocrinology 149: 3569–3575, 2008. doi: 10.1210/en.2007-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 773.Oka Y, Ye M, Zuker CS. Thirst driving and suppressing signals encoded by distinct neural populations in the brain. Nature 520: 349–352, 2015. doi: 10.1038/nature14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 774.Olivares-Reyes JA, Shah BH, Hernández-Aranda J, García-Caballero A, Farshori MP, García-Sáinz JA, Catt KJ. Agonist-induced interactions between angiotensin AT1 and epidermal growth factor receptors. Mol Pharmacol 68: 356–364, 2005. doi: 10.1124/mol.104.010637. [DOI] [PubMed] [Google Scholar]
- 775.Olson ER, Shamhart PE, Naugle JE, Meszaros JG. Angiotensin II-induced extracellular signal-regulated kinase 1/2 activation is mediated by protein kinase Cdelta and intracellular calcium in adult rat cardiac fibroblasts. Hypertension 51: 704–711, 2008. doi: 10.1161/HYPERTENSIONAHA.107.098459. [DOI] [PubMed] [Google Scholar]
- 776.Omura T, Yoshiyama M, Kim S, Matsumoto R, Nakamura Y, Izumi Y, Ichijo H, Sudo T, Akioka K, Iwao H, Takeuchi K, Yoshikawa J. Involvement of apoptosis signal-regulating kinase-1 on angiotensin II-induced monocyte chemoattractant protein-1 expression. Arterioscler Thromb Vasc Biol 24: 270–275, 2004. doi: 10.1161/01.ATV.0000112930.40564.89. [DOI] [PubMed] [Google Scholar]
- 777.Ongherth A, Pasch S, Wuertz CM, Nowak K, Kittana N, Weis CA, Jatho A, Vettel C, Tiburcy M, Toischer K, Hasenfuss G, Zimmermann WH, Wieland T, Lutz S. p63RhoGEF regulates auto- and paracrine signaling in cardiac fibroblasts. J Mol Cell Cardiol 88: 39–54, 2015. doi: 10.1016/j.yjmcc.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 778.Orecchia P, Conte R, Balza E, Castellani P, Borsi L, Zardi L, Mingari MC, Carnemolla B. Identification of a novel cell binding site of periostin involved in tumour growth. Eur J Cancer 47: 2221–2229, 2011. doi: 10.1016/j.ejca.2011.04.026. [DOI] [PubMed] [Google Scholar]
- 779.Orlov SN, Koltsova SV, Kapilevich LV, Gusakova SV, Dulin NO. NKCC1 and NKCC2: The pathogenetic role of cation-chloride cotransporters in hypertension. Genes Dis 2: 186–196, 2015. doi: 10.1016/j.gendis.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 780.Osako MK, Nakagami H, Shimamura M, Koriyama H, Nakagami F, Shimizu H, Miyake T, Yoshizumi M, Rakugi H, Morishita R. Cross-talk of receptor activator of nuclear factor-κB ligand signaling with renin-angiotensin system in vascular calcification. Arterioscler Thromb Vasc Biol 33: 1287–1296, 2013. doi: 10.1161/ATVBAHA.112.301099. [DOI] [PubMed] [Google Scholar]
- 781.Oshima Y, Morimoto S, Ichihara A. Roles of the (pro)renin receptor in the kidney. World J Nephrol 3: 302–307, 2014. doi: 10.5527/wjn.v3.i4.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 782.Oshita A, Iwai M, Chen R, Ide A, Okumura M, Fukunaga S, Yoshii T, Mogi M, Higaki J, Horiuchi M. Attenuation of inflammatory vascular remodeling by angiotensin II type 1 receptor-associated protein. Hypertension 48: 671–676, 2006. doi: 10.1161/01.HYP.0000238141.99816.47. [DOI] [PubMed] [Google Scholar]
- 783.Owens AP III, Rateri DL, Howatt DA, Moore KJ, Tobias PS, Curtiss LK, Lu H, Cassis LA, Daugherty A. MyD88 deficiency attenuates angiotensin II-induced abdominal aortic aneurysm formation independent of signaling through Toll-like receptors 2 and 4. Arterioscler Thromb Vasc Biol 31: 2813–2819, 2011. doi: 10.1161/ATVBAHA.111.238642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 784.Owens AP III, Subramanian V, Moorleghen JJ, Guo Z, McNamara CA, Cassis LA, Daugherty A. Angiotensin II induces a region-specific hyperplasia of the ascending aorta through regulation of inhibitor of differentiation 3. Circ Res 106: 611–619, 2010. doi: 10.1161/CIRCRESAHA.109.212837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 785.Ozasa Y, Akazawa H, Qin Y, Tateno K, Ito K, Kudo-Sakamoto Y, Yano M, Yabumoto C, Naito AT, Oka T, Lee JK, Minamino T, Nagai T, Kobayashi Y, Komuro I. Notch activation mediates angiotensin II-induced vascular remodeling by promoting the proliferation and migration of vascular smooth muscle cells. Hypertens Res 36: 859–865, 2013. doi: 10.1038/hr.2013.52. [DOI] [PubMed] [Google Scholar]
- 786.Ozawa Y, Kobori H. Crucial role of Rho-nuclear factor-kappaB axis in angiotensin II-induced renal injury. Am J Physiol Renal Physiol 293: F100–F109, 2007. doi: 10.1152/ajprenal.00520.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 787.Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol 292: F330–F339, 2007. doi: 10.1152/ajprenal.00059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 788.Ozcan L, Tabas I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu Rev Med 63: 317–328, 2012. doi: 10.1146/annurev-med-043010-144749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 789.Ozumi K, Sudhahar V, Kim HW, Chen GF, Kohno T, Finney L, Vogt S, McKinney RD, Ushio-Fukai M, Fukai T. Role of copper transport protein antioxidant 1 in angiotensin II-induced hypertension: a key regulator of extracellular superoxide dismutase. Hypertension 60: 476–486, 2012. doi: 10.1161/HYPERTENSIONAHA.111.189571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 790.Padia SH, Carey RM. AT2 receptors: beneficial counter-regulatory role in cardiovascular and renal function. Pflugers Arch 465: 99–110, 2013. doi: 10.1007/s00424-012-1146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 791.Padia SH, Kemp BA, Howell NL, Gildea JJ, Keller SR, Carey RM. Intrarenal angiotensin III infusion induces natriuresis and angiotensin type 2 receptor translocation in Wistar-Kyoto but not in spontaneously hypertensive rats. Hypertension 53: 338–343, 2009. doi: 10.1161/HYPERTENSIONAHA.108.124198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 792.Paek J, Kalocsay M, Staus DP, Wingler L, Pascolutti R, Paulo JA, Gygi SP, Kruse AC. Multidimensional Tracking of GPCR Signaling via Peroxidase-Catalyzed Proximity Labeling. Cell 169: 338–349.e11, 2017. doi: 10.1016/j.cell.2017.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 793.Pan L, Glenn ST, Jones CA, Gross KW. Activation of the rat renin promoter by HOXD10.PBX1b.PREP1, Ets-1, and the intracellular domain of notch. J Biol Chem 280: 20860–20866, 2005. doi: 10.1074/jbc.M414618200. [DOI] [PubMed] [Google Scholar]
- 794.Pan W, Zhong Y, Cheng C, Liu B, Wang L, Li A, Xiong L, Liu S. MiR-30-regulated autophagy mediates angiotensin II-induced myocardial hypertrophy. PLoS One 8: e53950, 2013. doi: 10.1371/journal.pone.0053950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 795.Pang J, Yan C, Natarajan K, Cavet ME, Massett MP, Yin G, Berk BC. GIT1 mediates HDAC5 activation by angiotensin II in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 28: 892–898, 2008. doi: 10.1161/ATVBAHA.107.161349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 796.Pang PS, Butler J, Collins SP, Cotter G, Davison BA, Ezekowitz JA, Filippatos G, Levy PD, Metra M, Ponikowski P, Teerlink JR, Voors AA, Bharucha D, Goin K, Soergel DG, Felker GM. Biased ligand of the angiotensin II type 1 receptor in patients with acute heart failure: a randomized, double-blind, placebo-controlled, phase IIB, dose ranging trial (BLAST-AHF). Eur Heart J 38: 2364–2373, 2017. doi: 10.1093/eurheartj/ehx196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 797.Pankratz F, Bemtgen X, Zeiser R, Leonhardt F, Kreuzaler S, Hilgendorf I, Smolka C, Helbing T, Hoefer I, Esser JS, Kustermann M, Moser M, Bode C, Grundmann S. MicroRNA-155 Exerts Cell-Specific Antiangiogenic but Proarteriogenic Effects During Adaptive Neovascularization. Circulation 131: 1575–1589, 2015. doi: 10.1161/CIRCULATIONAHA.114.014579. [DOI] [PubMed] [Google Scholar]
- 798.Park BM, Gao S, Cha SA, Park BH, Kim SH. Cardioprotective effects of angiotensin III against ischemic injury via the AT2 receptor and KATP channels. Physiol Rep 1: e00151, 2013. doi: 10.1002/phy2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 799.Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol 14: 98–112, 2013. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- 800.Passos-Silva DG, Verano-Braga T, Santos RA. Angiotensin-(1-7): beyond the cardio-renal actions. Clin Sci (Lond) 124: 443–456, 2013. doi: 10.1042/CS20120461. [DOI] [PubMed] [Google Scholar]
- 801.Patel J, McNeill E, Douglas G, Hale AB, de Bono J, Lee R, Iqbal AJ, Regan-Komito D, Stylianou E, Greaves DR, Channon KM. RGS1 regulates myeloid cell accumulation in atherosclerosis and aortic aneurysm rupture through altered chemokine signalling. Nat Commun 6: 6614, 2015. doi: 10.1038/ncomms7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 802.Patel VB, Bodiga S, Basu R, Das SK, Wang W, Wang Z, Lo J, Grant MB, Zhong J, Kassiri Z, Oudit GY. Loss of angiotensin-converting enzyme-2 exacerbates diabetic cardiovascular complications and leads to systolic and vascular dysfunction: a critical role of the angiotensin II/AT1 receptor axis. Circ Res 110: 1322–1335, 2012. doi: 10.1161/CIRCRESAHA.112.268029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 803.Patel VB, Clarke N, Wang Z, Fan D, Parajuli N, Basu R, Putko B, Kassiri Z, Turner AJ, Oudit GY. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol 66: 167–176, 2014. doi: 10.1016/j.yjmcc.2013.11.017. [DOI] [PubMed] [Google Scholar]
- 804.Patrushev N, Seidel-Rogol B, Salazar G. Angiotensin II requires zinc and downregulation of the zinc transporters ZnT3 and ZnT10 to induce senescence of vascular smooth muscle cells. PLoS One 7: e33211, 2012. doi: 10.1371/journal.pone.0033211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 805.Paukku K, Backlund M, De Boer RA, Kalkkinen N, Kontula KK, Lehtonen JY. Regulation of AT1R expression through HuR by insulin. Nucleic Acids Res 40: 5250–5261, 2012. doi: 10.1093/nar/gks170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 806.Paulis L, Becker ST, Lucht K, Schwengel K, Slavic S, Kaschina E, Thöne-Reineke C, Dahlöf B, Baulmann J, Unger T, Steckelings UM. Direct angiotensin II type 2 receptor stimulation in Nω-nitro-l-arginine-methyl ester-induced hypertension: the effect on pulse wave velocity and aortic remodeling. Hypertension 59: 485–492, 2012. doi: 10.1161/HYPERTENSIONAHA.111.185496. [DOI] [PubMed] [Google Scholar]
- 807.Pavlatou MG, Mastorakos G, Margeli A, Kouskouni E, Tentolouris N, Katsilambros N, Chrousos GP, Papassotiriou I. Angiotensin blockade in diabetic patients decreases insulin resistance-associated low-grade inflammation. Eur J Clin Invest 41: 652–658, 2011. doi: 10.1111/j.1365-2362.2010.02453.x. [DOI] [PubMed] [Google Scholar]
- 808.Pelham CJ, Keen HL, Lentz SR, Sigmund CD. Dominant negative PPARγ promotes atherosclerosis, vascular dysfunction, and hypertension through distinct effects in endothelium and vascular muscle. Am J Physiol Regul Integr Comp Physiol 304: R690–R701, 2013. doi: 10.1152/ajpregu.00607.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 809.Pellegrino PR, Schiller AM, Haack KK, Zucker IH. Central Angiotensin-II Increases Blood Pressure and Sympathetic Outflow via Rho Kinase Activation in Conscious Rabbits. Hypertension 68: 1271–1280, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 810.Pellieux C, Foletti A, Peduto G, Aubert JF, Nussberger J, Beermann F, Brunner HR, Pedrazzini T. Dilated cardiomyopathy and impaired cardiac hypertrophic response to angiotensin II in mice lacking FGF-2. J Clin Invest 108: 1843–1851, 2001. doi: 10.1172/JCI13627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 811.Pellman J, Zhang J, Sheikh F. Myocyte-fibroblast communication in cardiac fibrosis and arrhythmias: mechanisms and model systems. J Mol Cell Cardiol 94: 22–31, 2016. doi: 10.1016/j.yjmcc.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 812.Pena MJ, Heinzel A, Rossing P, Parving HH, Dallmann G, Rossing K, Andersen S, Mayer B, Heerspink HJ. Serum metabolites predict response to angiotensin II receptor blockers in patients with diabetes mellitus. J Transl Med 14: 203, 2016. doi: 10.1186/s12967-016-0960-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 813.Peña-Silva RA, Chalouhi N, Wegman-Points L, Ali M, Mitchell I, Pierce GL, Chu Y, Ballas ZK, Heistad D, Hasan D. Novel role for endogenous hepatocyte growth factor in the pathogenesis of intracranial aneurysms. Hypertension 65: 587–593, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 814.Peng H, Li W, Seth DM, Nair AR, Francis J, Feng Y. (Pro)renin receptor mediates both angiotensin II-dependent and -independent oxidative stress in neuronal cells. PLoS One 8: e58339, 2013. doi: 10.1371/journal.pone.0058339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 815.Peng H, Sarwar Z, Yang XP, Peterson EL, Xu J, Janic B, Rhaleb N, Carretero OA, Rhaleb NE. Profibrotic Role for Interleukin-4 in Cardiac Remodeling and Dysfunction. Hypertension 66: 582–589, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 816.Peng H, Yang XP, Carretero OA, Nakagawa P, D’Ambrosio M, Leung P, Xu J, Peterson EL, González GE, Harding P, Rhaleb NE. Angiotensin II-induced dilated cardiomyopathy in Balb/c but not C57BL/6J mice. Exp Physiol 96: 756–764, 2011. doi: 10.1113/expphysiol.2011.057612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 817.Peng J, Gurantz D, Tran V, Cowling RT, Greenberg BH. Tumor necrosis factor-alpha-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res 91: 1119–1126, 2002. doi: 10.1161/01.RES.0000047090.08299.D5. [DOI] [PubMed] [Google Scholar]
- 818.Peng K, Lu X, Wang F, Nau A, Chen R, Zhou SF, Yang T. Collecting duct (pro)renin receptor targets ENaC to mediate angiotensin II-induced hypertension. Am J Physiol Renal Physiol 312: F245–F253, 2017. doi: 10.1152/ajprenal.00178.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 819.Peng K, Tian X, Qian Y, Skibba M, Zou C, Liu Z, Wang J, Xu Z, Li X, Liang G. Novel EGFR inhibitors attenuate cardiac hypertrophy induced by angiotensin II. J Cell Mol Med 20: 482–494, 2016. doi: 10.1111/jcmm.12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 820.Pernomian L, do Prado AF, Gomes MS, Pernomian L, da Silva CH, Gerlach RF, de Oliveira AM. MAS receptors mediate vasoprotective and atheroprotective effects of candesartan upon the recovery of vascular angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS axis functionality. Eur J Pharmacol 764: 173–188, 2015. doi: 10.1016/j.ejphar.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 821.Peterson JR, Burmeister MA, Tian X, Zhou Y, Guruju MR, Stupinski JA, Sharma RV, Davisson RL. Genetic silencing of Nox2 and Nox4 reveals differential roles of these NADPH oxidase homologues in the vasopressor and dipsogenic effects of brain angiotensin II. Hypertension 54: 1106–1114, 2009. doi: 10.1161/HYPERTENSIONAHA.109.140087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 822.Pillai VB, Sundaresan NR, Kim G, Samant S, Moreno-Vinasco L, Garcia JG, Gupta MP. Nampt secreted from cardiomyocytes promotes development of cardiac hypertrophy and adverse ventricular remodeling. Am J Physiol Heart Circ Physiol 304: H415–H426, 2013. doi: 10.1152/ajpheart.00468.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 823.Pingili AK, Kara M, Khan NS, Estes AM, Lin Z, Li W, Gonzalez FJ, Malik KU. 6β-Hydroxytestosterone, a cytochrome P450 1B1 metabolite of testosterone, contributes to angiotensin II-induced hypertension and its pathogenesis in male mice. Hypertension 65: 1279–1287, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 824.Pironti G, Strachan RT, Abraham D, Mon-Wei Yu S, Chen M, Chen W, Hanada K, Mao L, Watson LJ, Rockman HA. Circulating Exosomes Induced by Cardiac Pressure Overload Contain Functional Angiotensin II Type 1 Receptors. Circulation 131: 2120–2130, 2015. doi: 10.1161/CIRCULATIONAHA.115.015687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 825.Poduri A, Rateri DL, Howatt DA, Balakrishnan A, Moorleghen JJ, Cassis LA, Daugherty A. Fibroblast Angiotensin II Type 1a Receptors Contribute to Angiotensin II-Induced Medial Hyperplasia in the Ascending Aorta. Arterioscler Thromb Vasc Biol 35: 1995–2002, 2015. doi: 10.1161/ATVBAHA.115.305995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 826.Pojoga LH, Romero JR, Yao TM, Loutraris P, Ricchiuti V, Coutinho P, Guo C, Lapointe N, Stone JR, Adler GK, Williams GH. Caveolin-1 ablation reduces the adverse cardiovascular effects of N-omega-nitro-l-arginine methyl ester and angiotensin II. Endocrinology 151: 1236–1246, 2010. doi: 10.1210/en.2009-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 827.Polito L, Kehoe PG, Davin A, Benussi L, Ghidoni R, Binetti G, Quadri P, Lucca U, Tettamanti M, Clerici F, Bagnoli S, Galimberti D, Nacmias B, Sorbi S, Guaita A, Scarpini E, Mariani C, Forloni G, Albani D. The SIRT2 polymorphism rs10410544 and risk of Alzheimer’s disease in two Caucasian case-control cohorts. Alzheimers Dement 9: 392–399, 2013. doi: 10.1016/j.jalz.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 828.Polito L, Kehoe PG, Forloni G, Albani D. The molecular genetics of sirtuins: association with human longevity and age-related diseases. Int J Mol Epidemiol Genet 1: 214–225, 2010. [PMC free article] [PubMed] [Google Scholar]
- 829.Pollow DP, Uhrlaub J, Romero-Aleshire M, Sandberg K, Nikolich-Zugich J, Brooks HL, Hay M. Sex differences in T-lymphocyte tissue infiltration and development of angiotensin II hypertension. Hypertension 64: 384–390, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 830.Ponnuswamy P, Joffre J, Herbin O, Esposito B, Laurans L, Binder CJ, Tedder TF, Zeboudj L, Loyer X, Giraud A, Zhang Y, Tedgui A, Mallat Z, Ait-Oufella H. Angiotensin II synergizes with BAFF to promote atheroprotective regulatory B cells. Sci Rep 7: 4111, 2017. doi: 10.1038/s41598-017-04438-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 831.Pontes RB, Crajoinas RO, Nishi EE, Oliveira-Sales EB, Girardi AC, Campos RR, Bergamaschi CT. Renal nerve stimulation leads to the activation of the Na+/H+ exchanger isoform 3 via angiotensin II type I receptor. Am J Physiol Renal Physiol 308: F848–F856, 2015. doi: 10.1152/ajprenal.00515.2014. [DOI] [PubMed] [Google Scholar]
- 832.Pontes RB, Girardi AC, Nishi EE, Campos RR, Bergamaschi CT. Crosstalk between the renal sympathetic nerve and intrarenal angiotensin II modulates proximal tubular sodium reabsorption. Exp Physiol 100: 502–506, 2015. doi: 10.1113/EP085075. [DOI] [PubMed] [Google Scholar]
- 833.Porrello ER, D’Amore A, Curl CL, Allen AM, Harrap SB, Thomas WG, Delbridge LM. Angiotensin II type 2 receptor antagonizes angiotensin II type 1 receptor-mediated cardiomyocyte autophagy. Hypertension 53: 1032–1040, 2009. doi: 10.1161/HYPERTENSIONAHA.108.128488. [DOI] [PubMed] [Google Scholar]
- 834.Porrello ER, Delbridge LM, Thomas WG. The angiotensin II type 2 (AT2) receptor: an enigmatic seven transmembrane receptor. Front Biosci (Landmark Ed) 14: 958–972, 2009. doi: 10.2741/3289. [DOI] [PubMed] [Google Scholar]
- 835.Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res 119: 91–112, 2016. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 836.Prasad AM, Morgan DA, Nuno DW, Ketsawatsomkron P, Bair TB, Venema AN, Dibbern ME, Kutschke WJ, Weiss RM, Lamping KG, Chapleau MW, Sigmund CD, Rahmouni K, Grumbach IM. Calcium/calmodulin-dependent kinase II inhibition in smooth muscle reduces angiotensin II-induced hypertension by controlling aortic remodeling and baroreceptor function. J Am Heart Assoc 4: e001949, 2015. doi: 10.1161/JAHA.115.001949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 837.Prasad AM, Nuno DW, Koval OM, Ketsawatsomkron P, Li W, Li H, Shen FY, Joiner ML, Kutschke W, Weiss RM, Sigmund CD, Anderson ME, Lamping KG, Grumbach IM. Differential Control of Calcium Homeostasis and Vascular Reactivity by CaMKII. Hypertension 62: 434–441, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 838.Pruthi D, McCurley A, Aronovitz M, Galayda C, Karumanchi SA, Jaffe IZ. Aldosterone promotes vascular remodeling by direct effects on smooth muscle cell mineralocorticoid receptors. Arterioscler Thromb Vasc Biol 34: 355–364, 2014. doi: 10.1161/ATVBAHA.113.302854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 839.Purohit A, Rokita AG, Guan X, Chen B, Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, Stefansdottir H, Behunin AC, Li N, El-Accaoui RN, Yang B, Swaminathan PD, Weiss RM, Wehrens XHT, Song L-S, Dobrev D, Maier LS, Anderson ME. Oxidized Ca2+/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 128: 1748–1757, 2013. doi: 10.1161/CIRCULATIONAHA.113.003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 840.Putnam K, Batifoulier-Yiannikouris F, Bharadwaj KG, Lewis E, Karounos M, Daugherty A, Cassis LA. Deficiency of angiotensin type 1a receptors in adipocytes reduces differentiation and promotes hypertrophy of adipocytes in lean mice. Endocrinology 153: 4677–4686, 2012. doi: 10.1210/en.2012-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 841.Putnam K, Shoemaker R, Yiannikouris F, Cassis LA. The renin-angiotensin system: a target of and contributor to dyslipidemias, altered glucose homeostasis, and hypertension of the metabolic syndrome. Am J Physiol Heart Circ Physiol 302: H1219–H1230, 2012. doi: 10.1152/ajpheart.00796.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 842.Qi G, Jia L, Li Y, Bian Y, Cheng J, Li H, Xiao C, Du J. Angiotensin II infusion-induced inflammation, monocytic fibroblast precursor infiltration, and cardiac fibrosis are pressure dependent. Cardiovasc Toxicol 11: 157–167, 2011. doi: 10.1007/s12012-011-9109-z. [DOI] [PubMed] [Google Scholar]
- 843.Qi GM, Jia LX, Li YL, Li HH, Du J. Adiponectin suppresses angiotensin II-induced inflammation and cardiac fibrosis through activation of macrophage autophagy. Endocrinology 155: 2254–2265, 2014. doi: 10.1210/en.2013-2011. [DOI] [PubMed] [Google Scholar]
- 844.Qi X, Disatnik MH, Shen N, Sobel RA, Mochly-Rosen D. Aberrant mitochondrial fission in neurons induced by protein kinase Cdelta under oxidative stress conditions in vivo. Mol Biol Cell 22: 256–265, 2011. doi: 10.1091/mbc.E10-06-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 845.Qian J, Fulton D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front Physiol 4: 347, 2013. doi: 10.3389/fphys.2013.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 846.Qiao YN, He WQ, Chen CP, Zhang CH, Zhao W, Wang P, Zhang L, Wu YZ, Yang X, Peng YJ, Gao JM, Kamm KE, Stull JT, Zhu MS. Myosin phosphatase target subunit 1 (MYPT1) regulates the contraction and relaxation of vascular smooth muscle and maintains blood pressure. J Biol Chem 289: 22512–22523, 2014. doi: 10.1074/jbc.M113.525444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 847.Qin B, Zhou J. Src Family Kinases (SFK) Mediate Angiotensin II-Induced Myosin Light Chain Phosphorylation and Hypertension. PLoS One 10: e0127891, 2015. doi: 10.1371/journal.pone.0127891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 848.Qin Y, Wang Y, Liu O, Jia L, Fang W, Du J, Wei Y. Tauroursodeoxycholic Acid Attenuates Angiotensin II Induced Abdominal Aortic Aneurysm Formation in Apolipoprotein E-deficient Mice by Inhibiting Endoplasmic Reticulum Stress. Eur J Vasc Endovasc Surg 53: 337–345, 2017. doi: 10.1016/j.ejvs.2016.10.026. [DOI] [PubMed] [Google Scholar]
- 849.Qin Z, Bagley J, Sukhova G, Baur WE, Park HJ, Beasley D, Libby P, Zhang Y, Galper JB. Angiotensin II-induced TLR4 mediated abdominal aortic aneurysm in apolipoprotein E knockout mice is dependent on STAT3. J Mol Cell Cardiol 87: 160–170, 2015. doi: 10.1016/j.yjmcc.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 850.Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol 75: 346–359, 2008. doi: 10.1016/j.bcp.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 851.Ragot H, Monfort A, Baudet M, Azibani F, Fazal L, Merval R, Polidano E, Cohen-Solal A, Delcayre C, Vodovar N, Chatziantoniou C, Samuel JL. Loss of Notch3 Signaling in Vascular Smooth Muscle Cells Promotes Severe Heart Failure Upon Hypertension. Hypertension 68: 392–400, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07694. [DOI] [PubMed] [Google Scholar]
- 852.Rahman A, Swärd K. The role of caveolin-1 in cardiovascular regulation. Acta Physiol (Oxf) 195: 231–245, 2009. doi: 10.1111/j.1748-1716.2008.01907.x. [DOI] [PubMed] [Google Scholar]
- 853.Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal 3: ra46, 2010. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 854.Ramchandran R, Takezako T, Saad Y, Stull L, Fink B, Yamada H, Dikalov S, Harrison DG, Moravec C, Karnik SS. Angiotensinergic stimulation of vascular endothelium in mice causes hypotension, bradycardia, and attenuated angiotensin response. Proc Natl Acad Sci USA 103: 19087–19092, 2006. doi: 10.1073/pnas.0602715103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 855.Ramkumar N, Kohan DE. The nephron (pro)renin receptor: function and significance. Am J Physiol Renal Physiol 311: F1145–F1148, 2016. doi: 10.1152/ajprenal.00476.2016. [DOI] [PubMed] [Google Scholar]
- 856.Ramseyer VD, Ortiz PA, Carretero OA, Garvin JL. Angiotensin II-mediated hypertension impairs nitric oxide-induced NKCC2 inhibition in thick ascending limbs. Am J Physiol Renal Physiol 310: F748–F754, 2016. doi: 10.1152/ajprenal.00473.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 857.Rateri DL, Moorleghen JJ, Balakrishnan A, Owens AP III, Howatt DA, Subramanian V, Poduri A, Charnigo R, Cassis LA, Daugherty A. Endothelial cell-specific deficiency of Ang II type 1a receptors attenuates Ang II-induced ascending aortic aneurysms in LDL receptor-/- mice. Circ Res 108: 574–581, 2011. doi: 10.1161/CIRCRESAHA.110.222844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 858.Rateri DL, Moorleghen JJ, Knight V, Balakrishnan A, Howatt DA, Cassis LA, Daugherty A. Depletion of endothelial or smooth muscle cell-specific angiotensin II type 1a receptors does not influence aortic aneurysms or atherosclerosis in LDL receptor deficient mice. PLoS One 7: e51483, 2012. doi: 10.1371/journal.pone.0051483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 859.Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol 13: 333–342, 2012. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 860.Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, Shah AM. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol 31: 1368–1376, 2011. doi: 10.1161/ATVBAHA.110.219238. [DOI] [PubMed] [Google Scholar]
- 861.Re RN. Mechanisms of disease: local renin-angiotensin-aldosterone systems and the pathogenesis and treatment of cardiovascular disease. Nat Clin Pract Cardiovasc Med 1: 42–47, 2004. doi: 10.1038/ncpcardio0012. [DOI] [PubMed] [Google Scholar]
- 862.Regan JA, Mauro AG, Carbone S, Marchetti C, Gill R, Mezzaroma E, Valle Raleigh J, Salloum FN, Van Tassell BW, Abbate A, Toldo S. A mouse model of heart failure with preserved ejection fraction due to chronic infusion of a low subpressor dose of angiotensin II. Am J Physiol Heart Circ Physiol 309: H771–H778, 2015. doi: 10.1152/ajpheart.00282.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 863.Regitz-Zagrosek V, Fielitz J, Fleck E. Myocardial angiotensin receptors in human hearts. Basic Res Cardiol 93, Suppl 2: 37–42, 1998. doi: 10.1007/s003950050207. [DOI] [PubMed] [Google Scholar]
- 864.Rehman A, Leibowitz A, Yamamoto N, Rautureau Y, Paradis P, Schiffrin EL. Angiotensin type 2 receptor agonist compound 21 reduces vascular injury and myocardial fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension 59: 291–299, 2012. doi: 10.1161/HYPERTENSIONAHA.111.180158. [DOI] [PubMed] [Google Scholar]
- 865.Ren Z, Liang W, Chen C, Yang H, Singhal PC, Ding G. Angiotensin II induces nephrin dephosphorylation and podocyte injury: role of caveolin-1. Cell Signal 24: 443–450, 2012. doi: 10.1016/j.cellsig.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 866.Reudelhuber TL, Bernstein KE, Delafontaine P. Is angiotensin II a direct mediator of left ventricular hypertrophy? Time for another look. Hypertension 49: 1196–1201, 2007. doi: 10.1161/HYPERTENSIONAHA.106.075085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 867.Rickard AJ, Morgan J, Tesch G, Funder JW, Fuller PJ, Young MJ. Deletion of mineralocorticoid receptors from macrophages protects against deoxycorticosterone/salt-induced cardiac fibrosis and increased blood pressure. Hypertension 54: 537–543, 2009. doi: 10.1161/HYPERTENSIONAHA.109.131110. [DOI] [PubMed] [Google Scholar]
- 868.Rider SA, Mullins LJ, Verdon RF, MacRae CA, Mullins JJ. Renin expression in developing zebrafish is associated with angiogenesis and requires the Notch pathway and endothelium. Am J Physiol Renal Physiol 309: F531–F539, 2015. doi: 10.1152/ajprenal.00247.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 869.Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, Liao JK. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/- haploinsufficient mice. Circulation 112: 2959–2965, 2005. doi: 10.1161/CIRCULATIONAHA.105.584623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 870.Riquier-Brison AD, Leong PK, Pihakaski-Maunsbach K, McDonough AA. Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli. Am J Physiol Renal Physiol 298: F177–F186, 2010. doi: 10.1152/ajprenal.00464.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 871.Rivera-Torres J, Guzmán-Martínez G, Villa-Bellosta R, Orbe J, González-Gómez C, Serrano M, Díez J, Andrés V, Maraver A. Targeting γ-secretases protect against angiotensin II-induced cardiac hypertrophy. J Hypertens 33: 843–850, 2015. doi: 10.1097/HJH.0000000000000463. [DOI] [PubMed] [Google Scholar]
- 872.Rodrigues-Ferreira S, le Rouzic E, Pawlowski T, Srivastava A, Margottin-Goguet F, Nahmias C. AT2 Receptor-Interacting Proteins ATIPs in the Brain. Int J Hypertens 2013: 513047, 2013. doi: 10.1155/2013/513047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 873.Rodrigues-Ferreira S, Nahmias C. An ATIPical family of angiotensin II AT2 receptor-interacting proteins. Trends Endocrinol Metab 21: 684–690, 2010. doi: 10.1016/j.tem.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 874.Rodriguez-Perez AI, Borrajo A, Rodriguez-Pallares J, Guerra MJ, Labandeira-Garcia JL. Interaction between NADPH-oxidase and Rho-kinase in angiotensin II-induced microglial activation. Glia 63: 466–482, 2015. doi: 10.1002/glia.22765. [DOI] [PubMed] [Google Scholar]
- 875.Roman RJ, Fan F, Zhuo JL. Intrarenal Renin-Angiotensin System: Locally Synthesized or Taken up Via Endocytosis? Hypertension 67: 831–833, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 876.Romero M, Jiménez R, Toral M, León-Gómez E, Gómez-Gúzman M, Sánchez M, Zarzuelo MJ, Rodríguez-Gómez I, Rath G, Tamargo J, Pérez-Vizcaíno F, Dessy C, Duarte J. Vascular and Central Activation of Peroxisome Proliferator-Activated Receptor-β Attenuates Angiotensin II-Induced Hypertension: Role of RGS-5. J Pharmacol Exp Ther 358: 151–163, 2016. doi: 10.1124/jpet.116.233106. [DOI] [PubMed] [Google Scholar]
- 877.Rosenthal T, Gavras I. Angiotensin inhibition and malignancies: a review. J Hum Hypertens 23: 623–635, 2009. doi: 10.1038/jhh.2009.21. [DOI] [PubMed] [Google Scholar]
- 878.Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, MacDougald OA. Inhibition of adipogenesis by Wnt signaling. Science 289: 950–953, 2000. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- 879.Rossing K, Mischak H, Parving HH, Christensen PK, Walden M, Hillmann M, Kaiser T. Impact of diabetic nephropathy and angiotensin II receptor blockade on urinary polypeptide patterns. Kidney Int 68: 193–205, 2005. doi: 10.1111/j.1523-1755.2005.00394.x. [DOI] [PubMed] [Google Scholar]
- 880.Roy A, Al-Qusairi L, Donnelly BF, Ronzaud C, Marciszyn AL, Gong F, Chang YP, Butterworth MB, Pastor-Soler NM, Hallows KR, Staub O, Subramanya AR. Alternatively spliced proline-rich cassettes link WNK1 to aldosterone action. J Clin Invest 125: 3433–3448, 2015. doi: 10.1172/JCI75245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 881.Rozansky DJ, Cornwall T, Subramanya AR, Rogers S, Yang YF, David LL, Zhu X, Yang CL, Ellison DH. Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J Clin Invest 119: 2601–2612, 2009. doi: 10.1172/JCI38323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 882.Rudemiller NP, Crowley SD. Interactions Between the Immune and the Renin-Angiotensin Systems in Hypertension. Hypertension 68: 289–296, 2016. doi: 10.1161/HYPERTENSIONAHA.116.06591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 883.Rudemiller NP, Patel MB, Zhang JD, Jeffs AD, Karlovich NS, Griffiths R, Kan MJ, Buckley AF, Gunn MD, Crowley SD. C-C Motif Chemokine 5 Attenuates Angiotensin II-Dependent Kidney Injury by Limiting Renal Macrophage Infiltration. Am J Pathol 186: 2846–2856, 2016. doi: 10.1016/j.ajpath.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 884.Rudolph TK, Ravekes T, Klinke A, Friedrichs K, Mollenhauer M, Pekarova M, Ambrozova G, Martiskova H, Kaur JJ, Matthes B, Schwoerer A, Woodcock SR, Kubala L, Freeman BA, Baldus S, Rudolph V. Nitrated fatty acids suppress angiotensin II-mediated fibrotic remodelling and atrial fibrillation. Cardiovasc Res 109: 174–184, 2016. doi: 10.1093/cvr/cvv254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 885.Rudolph V, Andrié RP, Rudolph TK, Friedrichs K, Klinke A, Hirsch-Hoffmann B, Schwoerer AP, Lau D, Fu X, Klingel K, Sydow K, Didié M, Seniuk A, von Leitner EC, Szoecs K, Schrickel JW, Treede H, Wenzel U, Lewalter T, Nickenig G, Zimmermann WH, Meinertz T, Böger RH, Reichenspurner H, Freeman BA, Eschenhagen T, Ehmke H, Hazen SL, Willems S, Baldus S. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat Med 16: 470–474, 2010. doi: 10.1038/nm.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 886.Ruiz-Villalba A, Simón AM, Pogontke C, Castillo MI, Abizanda G, Pelacho B, Sánchez-Domínguez R, Segovia JC, Prósper F, Pérez-Pomares JM. Interacting resident epicardium-derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J Am Coll Cardiol 65: 2057–2066, 2015. doi: 10.1016/j.jacc.2015.03.520. [DOI] [PubMed] [Google Scholar]
- 887.Rupérez M, Sánchez-López E, Blanco-Colio LM, Esteban V, Rodríguez-Vita J, Plaza JJ, Egido J, Ruiz-Ortega M. The Rho-kinase pathway regulates angiotensin II-induced renal damage. Kidney Int Suppl 68: S39–S45, 2005. doi: 10.1111/j.1523-1755.2005.09908.x. [DOI] [PubMed] [Google Scholar]
- 888.Rush C, Nyara M, Moxon JV, Trollope A, Cullen B, Golledge J. Whole genome expression analysis within the angiotensin II-apolipoprotein E deficient mouse model of abdominal aortic aneurysm. BMC Genomics 10: 298, 2009. doi: 10.1186/1471-2164-10-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 889.Rüster C, Wolf G. Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J Am Soc Nephrol 22: 1189–1199, 2011. doi: 10.1681/ASN.2010040384. [DOI] [PubMed] [Google Scholar]
- 890.Ryba DM, Li J, Cowan CL, Russell B, Wolska BM, Solaro RJ. Long-Term Biased β-Arrestin Signaling Improves Cardiac Structure and Function in Dilated Cardiomyopathy. Circulation 135: 1056–1070, 2017. doi: 10.1161/CIRCULATIONAHA.116.024482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 891.Rygiel K. Can angiotensin-converting enzyme inhibitors impact cognitive decline in early stages of Alzheimer’s disease? An overview of research evidence in the elderly patient population. J Postgrad Med 62: 242–248, 2016. doi: 10.4103/0022-3859.188553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 892.Saad S, Stanners SR, Yong R, Tang O, Pollock CA. Notch mediated epithelial to mesenchymal transformation is associated with increased expression of the Snail transcription factor. Int J Biochem Cell Biol 42: 1115–1122, 2010. doi: 10.1016/j.biocel.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 893.Saavedra JM. Brain angiotensin II: new developments, unanswered questions and therapeutic opportunities. Cell Mol Neurobiol 25: 485–512, 2005. doi: 10.1007/s10571-005-4011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 894.Saavedra JM. Evidence to Consider Angiotensin II Receptor Blockers for the Treatment of Early Alzheimer’s Disease. Cell Mol Neurobiol 36: 259–279, 2016. doi: 10.1007/s10571-015-0327-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 895.Sabuhi R, Ali Q, Asghar M, Al-Zamily NR, Hussain T. Role of the angiotensin II AT2 receptor in inflammation and oxidative stress: opposing effects in lean and obese Zucker rats. Am J Physiol Renal Physiol 300: F700–F706, 2011. doi: 10.1152/ajprenal.00616.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 896.Sabuhi R, Asghar M, Hussain T. Inhibition of NAD(P)H oxidase potentiates AT2 receptor agonist-induced natriuresis in Sprague-Dawley rats. Am J Physiol Renal Physiol 299: F815–F820, 2010. doi: 10.1152/ajprenal.00310.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 897.Sadoshima J, Izumo S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res 73: 413–423, 1993. doi: 10.1161/01.RES.73.3.413. [DOI] [PubMed] [Google Scholar]
- 898.Sag CM, Schnelle M, Zhang J, Murdoch CE, Kossmann S, Protti A, Santos CXC, Sawyer G, Zhang X, Mongue-Din H, Richards DA, Brewer AC, Prysyazhna O, Maier LS, Wenzel P, Eaton PJ, Shah AM. Distinct Regulatory Effects of Myeloid Cell and Endothelial Cell NAPDH Oxidase 2 on Blood Pressure. Circulation 135: 2163–2177, 2017. doi: 10.1161/CIRCULATIONAHA.116.023877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 899.Sahar S, Reddy MA, Wong C, Meng L, Wang M, Natarajan R. Cooperation of SRC-1 and p300 with NF-kappaB and CREB in angiotensin II-induced IL-6 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 27: 1528–1534, 2007. doi: 10.1161/ATVBAHA.107.145862. [DOI] [PubMed] [Google Scholar]
- 900.Saito T, Hasegawa Y, Ishigaki Y, Yamada T, Gao J, Imai J, Uno K, Kaneko K, Ogihara T, Shimosawa T, Asano T, Fujita T, Oka Y, Katagiri H. Importance of endothelial NF-κB signalling in vascular remodelling and aortic aneurysm formation. Cardiovasc Res 97: 106–114, 2013. doi: 10.1093/cvr/cvs298. [DOI] [PubMed] [Google Scholar]
- 901.Sakai K, Agassandian K, Morimoto S, Sinnayah P, Cassell MD, Davisson RL, Sigmund CD. Local production of angiotensin II in the subfornical organ causes elevated drinking. J Clin Invest 117: 1088–1095, 2007. doi: 10.1172/JCI31242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 902.Sakaue T, Suzuki J, Hamaguchi M, Suehiro C, Tanino A, Nagao T, Uetani T, Aono J, Nakaoka H, Kurata M, Sakaue T, Okura T, Yasugi T, Izutani H, Higaki J, Ikeda S. Perivascular Adipose Tissue Angiotensin II Type 1 Receptor Promotes Vascular Inflammation and Aneurysm Formation. Hypertension 70: 780–789, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09512. [DOI] [PubMed] [Google Scholar]
- 903.Sakima A, Averill DB, Kasper SO, Jackson L, Ganten D, Ferrario CM, Gallagher PE, Diz DI. Baroreceptor reflex regulation in anesthetized transgenic rats with low glia-derived angiotensinogen. Am J Physiol Heart Circ Physiol 292: H1412–H1419, 2007. doi: 10.1152/ajpheart.00984.2006. [DOI] [PubMed] [Google Scholar]
- 904.Salazar G, Huang J, Feresin RG, Zhao Y, Griendling KK. Zinc regulates Nox1 expression through a NF-κB and mitochondrial ROS dependent mechanism to induce senescence of vascular smooth muscle cells. Free Radic Biol Med 108: 225–235, 2017. doi: 10.1016/j.freeradbiomed.2017.03.032. [DOI] [PubMed] [Google Scholar]
- 905.Salmon M, Johnston WF, Woo A, Pope NH, Su G, Upchurch GR Jr, Owens GK, Ailawadi G. KLF4 regulates abdominal aortic aneurysm morphology and deletion attenuates aneurysm formation. Circulation 128, Suppl 1: S163–S174, 2013. doi: 10.1161/CIRCULATIONAHA.112.000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 906.Sampson LJ, Davies LM, Barrett-Jolley R, Standen NB, Dart C. Angiotensin II-activated protein kinase C targets caveolae to inhibit aortic ATP-sensitive potassium channels. Cardiovasc Res 76: 61–70, 2007. doi: 10.1016/j.cardiores.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 907.San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, Chari D, Kahle KT, Leng Q, Bobadilla NA, Hebert SC, Alessi DR, Lifton RP, Gamba G. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci USA 106: 4384–4389, 2009. doi: 10.1073/pnas.0813238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 908.Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest 117: 1538–1549, 2007. doi: 10.1172/JCI30634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 909.Sandset EC, Bath PM, Boysen G, Jatuzis D, Kõrv J, Lüders S, Murray GD, Richter PS, Roine RO, Terént A, Thijs V, Berge E; SCAST Study Group . The angiotensin-receptor blocker candesartan for treatment of acute stroke (SCAST): a randomised, placebo-controlled, double-blind trial. Lancet 377: 741–750, 2011. doi: 10.1016/S0140-6736(11)60104-9. [DOI] [PubMed] [Google Scholar]
- 910.Sano M, Fukuda K, Sato T, Kawaguchi H, Suematsu M, Matsuda S, Koyasu S, Matsui H, Yamauchi-Takihara K, Harada M, Saito Y, Ogawa S. ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res 89: 661–669, 2001. doi: 10.1161/hh2001.098873. [DOI] [PubMed] [Google Scholar]
- 911.Santillo M, Colantuoni A, Mondola P, Guida B, Damiano S. NOX signaling in molecular cardiovascular mechanisms involved in the blood pressure homeostasis. Front Physiol 6: 194, 2015. doi: 10.3389/fphys.2015.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 912.Santisteban MM, Qi Y, Zubcevic J, Kim S, Yang T, Shenoy V, Cole-Jeffrey CT, Lobaton GO, Stewart DC, Rubiano A, Simmons CS, Garcia-Pereira F, Johnson RD, Pepine CJ, Raizada MK. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ Res 120: 312–323, 2017. doi: 10.1161/CIRCRESAHA.116.309006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 913.Santos RA, Castro CH, Gava E, Pinheiro SV, Almeida AP, Paula RD, Cruz JS, Ramos AS, Rosa KT, Irigoyen MC, Bader M, Alenina N, Kitten GT, Ferreira AJ. Impairment of in vitro and in vivo heart function in angiotensin-(1-7) receptor MAS knockout mice. Hypertension 47: 996–1002, 2006. doi: 10.1161/01.HYP.0000215289.51180.5c. [DOI] [PubMed] [Google Scholar]
- 914.Santulli G. MicroRNAs and Endothelial (Dys) Function. J Cell Physiol 231: 1638–1644, 2016. doi: 10.1002/jcp.25276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 915.Saratzis A, Bown MJ. The genetic basis for aortic aneurysmal disease. Heart 100: 916–922, 2014. doi: 10.1136/heartjnl-2013-305130. [DOI] [PubMed] [Google Scholar]
- 916.Sasaki K, Yamano Y, Bardhan S, Iwai N, Murray JJ, Hasegawa M, Matsuda Y, Inagami T. Cloning and expression of a complementary DNA encoding a bovine adrenal angiotensin II type-1 receptor. Nature 351: 230–233, 1991. doi: 10.1038/351230a0. [DOI] [PubMed] [Google Scholar]
- 917.Satija NK, Gurudutta GU, Sharma S, Afrin F, Gupta P, Verma YK, Singh VK, Tripathi RP. Mesenchymal stem cells: molecular targets for tissue engineering. Stem Cells Dev 16: 7–24, 2007. doi: 10.1089/scd.2006.9998. [DOI] [PubMed] [Google Scholar]
- 918.Sato T, Sato C, Kadowaki A, Watanabe H, Ho L, Ishida J, Yamaguchi T, Kimura A, Fukamizu A, Penninger JM, Reversade B, Ito H, Imai Y, Kuba K. ELABELA-APJ axis protects from pressure overload heart failure and angiotensin II-induced cardiac damage. Cardiovasc Res 113: 760–769, 2017. doi: 10.1093/cvr/cvx061. [DOI] [PubMed] [Google Scholar]
- 919.Satou R, Shao W, Navar LG. Role of stimulated intrarenal angiotensinogen in hypertension. Ther Adv Cardiovasc Dis 9: 181–190, 2015. doi: 10.1177/1753944715585512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 920.Saulière A, Bellot M, Paris H, Denis C, Finana F, Hansen JT, Altié MF, Seguelas MH, Pathak A, Hansen JL, Sénard JM, Galés C. Deciphering biased-agonism complexity reveals a new active AT1 receptor entity. Nat Chem Biol 8: 622–630, 2012. doi: 10.1038/nchembio.961. [DOI] [PubMed] [Google Scholar]
- 921.Saxena A, Little JT, Nedungadi TP, Cunningham JT. Angiotensin II type 1a receptors in subfornical organ contribute towards chronic intermittent hypoxia-associated sustained increase in mean arterial pressure. Am J Physiol Heart Circ Physiol 308: H435–H446, 2015. doi: 10.1152/ajpheart.00747.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 922.Scalia R, Gong Y, Berzins B, Freund B, Feather D, Landesberg G, Mishra G. A novel role for calpain in the endothelial dysfunction induced by activation of angiotensin II type 1 receptor signaling. Circ Res 108: 1102–1111, 2011. doi: 10.1161/CIRCRESAHA.110.229393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 923.Schafer S, Viswanathan S, Widjaja AA, Lim WW, Moreno-Moral A, DeLaughter DM, Ng B, Patone G, Chow K, Khin E, Tan J, Chothani SP, Ye L, Rackham OJL, Ko NSJ, Sahib NE, Pua CJ, Zhen NTG, Xie C, Wang M, Maatz H, Lim S, Saar K, Blachut S, Petretto E, Schmidt S, Putoczki T, Guimarães-Camboa N, Wakimoto H, van Heesch S, Sigmundsson K, Lim SL, Soon JL, Chao VTT, Chua YL, Tan TE, Evans SM, Loh YJ, Jamal MH, Ong KK, Chua KC, Ong BH, Chakaramakkil MJ, Seidman JG, Seidman CE, Hubner N, Sin KYK, Cook SA. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 552: 110–115, 2017. doi: 10.1038/nature24676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 924.Schaich CL, Wellman TL, Koi B, Erdos B. BDNF acting in the hypothalamus induces acute pressor responses under permissive control of angiotensin II. Auton Neurosci 197: 1–8, 2016. doi: 10.1016/j.autneu.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 925.Schellings MW, Baumann M, van Leeuwen RE, Duisters RF, Janssen SH, Schroen B, Peutz-Kootstra CJ, Heymans S, Pinto YM. Imatinib attenuates end-organ damage in hypertensive homozygous TGR(mRen2)27 rats. Hypertension 47: 467–474, 2006. doi: 10.1161/01.HYP.0000202487.68969.f7. [DOI] [PubMed] [Google Scholar]
- 926.Schieffer B, Paxton WG, Marrero MB, Bernstein KE. Importance of tyrosine phosphorylation in angiotensin II type 1 receptor signaling. Hypertension 27: 476–480, 1996. doi: 10.1161/01.HYP.27.3.476. [DOI] [PubMed] [Google Scholar]
- 927.Schinke M, Baltatu O, Böhm M, Peters J, Rascher W, Bricca G, Lippoldt A, Ganten D, Bader M. Blood pressure reduction and diabetes insipidus in transgenic rats deficient in brain angiotensinogen. Proc Natl Acad Sci USA 96: 3975–3980, 1999. doi: 10.1073/pnas.96.7.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 928.Schleifenbaum J, Kassmann M, Szijártó IA, Hercule HC, Tano JY, Weinert S, Heidenreich M, Pathan AR, Anistan YM, Alenina N, Rusch NJ, Bader M, Jentsch TJ, Gollasch M. Stretch-activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ Res 115: 263–272, 2014. doi: 10.1161/CIRCRESAHA.115.302882. [DOI] [PubMed] [Google Scholar]
- 929.Schmieder RE, Kjeldsen SE, Julius S, McInnes GT, Zanchetti A, Hua TA; VALUE Trial Group . Reduced incidence of new-onset atrial fibrillation with angiotensin II receptor blockade: the VALUE trial. J Hypertens 26: 403–411, 2008. doi: 10.1097/HJH.0b013e3282f35c67. [DOI] [PubMed] [Google Scholar]
- 930.Schröder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 110: 1217–1225, 2012. doi: 10.1161/CIRCRESAHA.112.267054. [DOI] [PubMed] [Google Scholar]
- 931.Schuhmacher S, Foretz M, Knorr M, Jansen T, Hortmann M, Wenzel P, Oelze M, Kleschyov AL, Daiber A, Keaney JF Jr, Wegener G, Lackner K, Münzel T, Viollet B, Schulz E. α1AMP-activated protein kinase preserves endothelial function during chronic angiotensin II treatment by limiting Nox2 upregulation. Arterioscler Thromb Vasc Biol 31: 560–566, 2011. doi: 10.1161/ATVBAHA.110.219543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 932.Schwartz F, Duka A, Triantafyllidi E, Johns C, Duka I, Cui J, Gavras H. Serial analysis of gene expression in mouse kidney following angiotensin II administration. Physiol Genomics 16: 90–98, 2003. doi: 10.1152/physiolgenomics.00108.2003. [DOI] [PubMed] [Google Scholar]
- 933.Schwengel K, Namsolleck P, Lucht K, Clausen BH, Lambertsen KL, Valero-Esquitino V, Thöne-Reineke C, Müller S, Widdop RE, Denton KM, Horiuchi M, Iwai M, Boato F, Dahlöf B, Hallberg A, Unger T, Steckelings UM. Angiotensin AT2-receptor stimulation improves survival and neurological outcome after experimental stroke in mice. J Mol Med (Berl) 94: 957–966, 2016. doi: 10.1007/s00109-016-1406-3. [DOI] [PubMed] [Google Scholar]
- 934.Senbonmatsu T, Ichihara S, Price E Jr, Gaffney FA, Inagami T. Evidence for angiotensin II type 2 receptor-mediated cardiac myocyte enlargement during in vivo pressure overload. J Clin Invest 106: R25–R29, 2000. doi: 10.1172/JCI10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 935.Senbonmatsu T, Saito T, Landon EJ, Watanabe O, Price E Jr, Roberts RL, Imboden H, Fitzgerald TG, Gaffney FA, Inagami T. A novel angiotensin II type 2 receptor signaling pathway: possible role in cardiac hypertrophy. EMBO J 22: 6471–6482, 2003. doi: 10.1093/emboj/cdg637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 936.Senchenkova EY, Russell J, Kurmaeva E, Ostanin D, Granger DN. Role of T lymphocytes in angiotensin II-mediated microvascular thrombosis. Hypertension 58: 959–965, 2011. doi: 10.1161/HYPERTENSIONAHA.111.173856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 937.Sénémaud J, Caligiuri G, Etienne H, Delbosc S, Michel JB, Coscas R. Translational Relevance and Recent Advances of Animal Models of Abdominal Aortic Aneurysm. Arterioscler Thromb Vasc Biol 37: 401–410, 2017. doi: 10.1161/ATVBAHA.116.308534. [DOI] [PubMed] [Google Scholar]
- 938.Sevá Pessôa B, van der Lubbe N, Verdonk K, Roks AJ, Hoorn EJ, Danser AH. Key developments in renin-angiotensin-aldosterone system inhibition. Nat Rev Nephrol 9: 26–36, 2013. doi: 10.1038/nrneph.2012.249. [DOI] [PubMed] [Google Scholar]
- 939.Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol 18: 361–374, 2017. doi: 10.1038/nrm.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 940.Shah KH, Shi P, Giani JF, Janjulia T, Bernstein EA, Li Y, Zhao T, Harrison DG, Bernstein KE, Shen XZ. Myeloid Suppressor Cells Accumulate and Regulate Blood Pressure in Hypertension. Circ Res 117: 858–869, 2015. doi: 10.1161/CIRCRESAHA.115.306539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 941.Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA, Paulus WJ. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation 134: 73–90, 2016. doi: 10.1161/CIRCULATIONAHA.116.021884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 942.Shahid M, Spagnolli E, Ernande L, Thoonen R, Kolodziej SA, Leyton PA, Cheng J, Tainsh RE, Mayeur C, Rhee DK, Wu MX, Scherrer-Crosbie M, Buys ES, Zapol WM, Bloch KD, Bloch DB. BMP type I receptor ALK2 is required for angiotensin II-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol 310: H984–H994, 2016. doi: 10.1152/ajpheart.00879.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 943.Shamansurova Z, Tan P, Ahmed B, Pepin E, Seda O, Lavoie JL. Adipose tissue (P)RR regulates insulin sensitivity, fat mass and body weight. Mol Metab 5: 959–969, 2016. doi: 10.1016/j.molmet.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 944.Shanmugam P, Valente AJ, Prabhu SD, Venkatesan B, Yoshida T, Delafontaine P, Chandrasekar B. Angiotensin-II type 1 receptor and NOX2 mediate TCF/LEF and CREB dependent WISP1 induction and cardiomyocyte hypertrophy. J Mol Cell Cardiol 50: 928–938, 2011. doi: 10.1016/j.yjmcc.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 945.Shen M, Lee J, Basu R, Sakamuri SS, Wang X, Fan D, Kassiri Z. Divergent roles of matrix metalloproteinase 2 in pathogenesis of thoracic aortic aneurysm. Arterioscler Thromb Vasc Biol 35: 888–898, 2015. doi: 10.1161/ATVBAHA.114.305115. [DOI] [PubMed] [Google Scholar]
- 946.Shen YH, Zhang L, Ren P, Nguyen MT, Zou S, Wu D, Wang XL, Coselli JS, LeMaire SA. AKT2 confers protection against aortic aneurysms and dissections. Circ Res 112: 618–632, 2013. doi: 10.1161/CIRCRESAHA.112.300735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 947.Sheng L, Yang M, Ding W, Zhang M, Niu J, Qiao Z, Gu Y. Epidermal growth factor receptor signaling mediates aldosterone-induced profibrotic responses in kidney. Exp Cell Res 346: 99–110, 2016. doi: 10.1016/j.yexcr.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 948.Sherrod M, Davis DR, Zhou X, Cassell MD, Sigmund CD. Glial-specific ablation of angiotensinogen lowers arterial pressure in renin and angiotensinogen transgenic mice. Am J Physiol Regul Integr Comp Physiol 289: R1763–R1769, 2005. doi: 10.1152/ajpregu.00435.2005. [DOI] [PubMed] [Google Scholar]
- 949.Shibata S, Arroyo JP, Castañeda-Bueno M, Puthumana J, Zhang J, Uchida S, Stone KL, Lam TT, Lifton RP. Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc Natl Acad Sci USA 111: 15556–15561, 2014. doi: 10.1073/pnas.1418342111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 950.Shimada K, Furukawa H, Wada K, Wei Y, Tada Y, Kuwabara A, Shikata F, Kanematsu Y, Lawton MT, Kitazato KT, Nagahiro S, Hashimoto T. Angiotensin-(1-7) protects against the development of aneurysmal subarachnoid hemorrhage in mice. J Cereb Blood Flow Metab 35: 1163–1168, 2015. doi: 10.1038/jcbfm.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 951.Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol 97: 245–262, 2016. doi: 10.1016/j.yjmcc.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 952.Shimizu T, Narang N, Chen P, Yu B, Knapp M, Janardanan J, Blair J, Liao JK. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight 2: e93187, 2017. doi: 10.1172/jci.insight.93187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 953.Shimojo N, Hashizume R, Kanayama K, Hara M, Suzuki Y, Nishioka T, Hiroe M, Yoshida T, Imanaka-Yoshida K. Tenascin-C may accelerate cardiac fibrosis by activating macrophages via the integrin αVβ3/nuclear factor-κB/interleukin-6 axis. Hypertension 66: 757–766, 2015. doi: 10.1161/HYPERTENSIONAHA.115.06004. [DOI] [PubMed] [Google Scholar]
- 954.Shinohara K, Liu X, Morgan DA, Davis DR, Sequeira-Lopez ML, Cassell MD, Grobe JL, Rahmouni K, Sigmund CD. Selective Deletion of the Brain-Specific Isoform of Renin Causes Neurogenic Hypertension. Hypertension 68: 1385–1392, 2016. doi: 10.1161/HYPERTENSIONAHA.116.08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 955.Shinohara K, Nakagawa P, Gomez J, Morgan DA, Littlejohn NK, Folchert MD, Weidemann BJ, Liu X, Walsh SA, Ponto LL, Rahmouni K, Grobe JL, Sigmund CD. Selective Deletion of Renin-b in the Brain Alters Drinking and Metabolism. Hypertension 70: 990–997, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 956.Shinshi Y, Higashiura K, Yoshida D, Togashi N, Yoshida H, Miyazaki Y, Ura N, Shimamoto K. Angiotensin II inhibits glucose uptake of skeletal muscle via the adenosine monophosphate-activated protein kinase pathway. J Am Soc Hypertens 1: 251–255, 2007. doi: 10.1016/j.jash.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 957.Shirasuna K, Karasawa T, Usui F, Kobayashi M, Komada T, Kimura H, Kawashima A, Ohkuchi A, Taniguchi S, Takahashi M. NLRP3 Deficiency Improves Angiotensin II-Induced Hypertension But Not Fetal Growth Restriction During Pregnancy. Endocrinology 156: 4281–4292, 2015. doi: 10.1210/en.2015-1408. [DOI] [PubMed] [Google Scholar]
- 958.Shoja MM, Agutter PS, Tubbs RS, Payner TD, Ghabili K, Cohen-Gadol AA. The role of the renin–angiotensin system in the pathogenesis of intracranial aneurysms. J Renin Angiotensin Aldosterone Syst 12: 262–273, 2011. doi: 10.1177/1470320310387845. [DOI] [PubMed] [Google Scholar]
- 959.Shrikrishna D, Astin R, Kemp PR, Hopkinson NS. Renin-angiotensin system blockade: a novel therapeutic approach in chronic obstructive pulmonary disease. Clin Sci (Lond) 123: 487–498, 2012. doi: 10.1042/CS20120081. [DOI] [PubMed] [Google Scholar]
- 960.Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci 36: 457–469, 2011. doi: 10.1016/j.tibs.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 961.Shum M, Pinard S, Guimond MO, Labbé SM, Roberge C, Baillargeon JP, Langlois MF, Alterman M, Wallinder C, Hallberg A, Carpentier AC, Gallo-Payet N. Angiotensin II type 2 receptor promotes adipocyte differentiation and restores adipocyte size in high-fat/high-fructose diet-induced insulin resistance in rats. Am J Physiol Endocrinol Metab 304: E197–E210, 2013. doi: 10.1152/ajpendo.00149.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 962.Shyu KG, Cheng WP, Wang BW. Angiotensin II Downregulates MicroRNA-145 to Regulate Kruppel-like Factor 4 and Myocardin Expression in Human Coronary Arterial Smooth Muscle Cells under High Glucose Conditions. Mol Med 21: 616–625, 2015. doi: 10.2119/molmed.2015.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 963.Sica V, Galluzzi L, Bravo-San Pedro JM, Izzo V, Maiuri MC, Kroemer G. Organelle-Specific Initiation of Autophagy. Mol Cell 59: 522–539, 2015. doi: 10.1016/j.molcel.2015.07.021. [DOI] [PubMed] [Google Scholar]
- 964.Siddesha JM, Valente AJ, Sakamuri SS, Yoshida T, Gardner JD, Somanna N, Takahashi C, Noda M, Chandrasekar B. Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK. J Mol Cell Cardiol 65: 9–18, 2013. doi: 10.1016/j.yjmcc.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 965.Siddesha JM, Valente AJ, Yoshida T, Sakamuri SS, Delafontaine P, Iba H, Noda M, Chandrasekar B. Docosahexaenoic acid reverses angiotensin II-induced RECK suppression and cardiac fibroblast migration. Cell Signal 26: 933–941, 2014. doi: 10.1016/j.cellsig.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 966.Siddiquee K, Hampton J, McAnally D, May L, Smith L. The apelin receptor inhibits the angiotensin II type 1 receptor via allosteric trans-inhibition. Br J Pharmacol 168: 1104–1117, 2013. doi: 10.1111/j.1476-5381.2012.02192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 967.Siedlinski M, Nosalski R, Szczepaniak P, Ludwig-Gałęzowska AH, Mikołajczyk T, Filip M, Osmenda G, Wilk G, Nowak M, Wołkow P, Guzik TJ. Vascular transcriptome profiling identifies sphingosine kinase 1 as a modulator of angiotensin II-induced vascular dysfunction. Sci Rep 7: 44131, 2017. doi: 10.1038/srep44131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 968.Sigmund CD, Diz DI, Chappell MC. No Brain Renin-Angiotensin System: Déjà vu All Over Again? Hypertension 69: 1007–1010, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 969.Simões e Silva AC, Silveira KD, Ferreira AJ, Teixeira MM. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol 169: 477–492, 2013. doi: 10.1111/bph.12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 970.Singh KD, Karnik SS. Angiotensin Receptors: Structure, Function, Signaling and Clinical Applications. J Cell Signal 1: 111, 2016. doi: 10.4172/jcs.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 971.Singh MV, Cicha MZ, Meyerholz DK, Chapleau MW, Abboud FM. Dual Activation of TRIF and MyD88 Adaptor Proteins by Angiotensin II Evokes Opposing Effects on Pressure, Cardiac Hypertrophy, and Inflammatory Gene Expression. Hypertension 66: 647–656, 2015. doi: 10.1161/HYPERTENSIONAHA.115.06011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 972.Singh P, Bahrami L, Castillo A, Majid DS. TNF-α type 2 receptor mediates renal inflammatory response to chronic angiotensin II administration with high salt intake in mice. Am J Physiol Renal Physiol 304: F991–F999, 2013. doi: 10.1152/ajprenal.00525.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 973.Sinn PL, Sigmund CD. Identification of three human renin mRNA isoforms from alternative tissue-specific transcriptional initiation. Physiol Genomics 3: 25–31, 2000. doi: 10.1152/physiolgenomics.2000.3.1.25. [DOI] [PubMed] [Google Scholar]
- 974.Sinnett-Smith J, Ni Y, Wang J, Ming M, Young SH, Rozengurt E. Protein kinase D1 mediates class IIa histone deacetylase phosphorylation and nuclear extrusion in intestinal epithelial cells: role in mitogenic signaling. Am J Physiol Cell Physiol 306: C961–C971, 2014. doi: 10.1152/ajpcell.00048.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 975.Siu KL, Li Q, Zhang Y, Guo J, Youn JY, Du J, Cai H. NOX isoforms in the development of abdominal aortic aneurysm. Redox Biol 11: 118–125, 2017. doi: 10.1016/j.redox.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 976.Skorska A, von Haehling S, Ludwig M, Lux CA, Gaebel R, Kleiner G, Klopsch C, Dong J, Curato C, Altarche-Xifró W, Slavic S, Unger T, Steinhoff G, Li J, David R. The CD4(+) AT2R(+) T cell subpopulation improves post-infarction remodelling and restores cardiac function. J Cell Mol Med 19: 1975–1985, 2015. doi: 10.1111/jcmm.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 977.Smith PM, Chambers AP, Price CJ, Ho W, Hopf C, Sharkey KA, Ferguson AV. The subfornical organ: a central nervous system site for actions of circulating leptin. Am J Physiol Regul Integr Comp Physiol 296: R512–R520, 2009. doi: 10.1152/ajpregu.90858.2008. [DOI] [PubMed] [Google Scholar]
- 978.Soe NN, Sowden M, Baskaran P, Kim Y, Nigro P, Smolock EM, Berk BC. Acetylation of cyclophilin A is required for its secretion and vascular cell activation. Cardiovasc Res 101: 444–453, 2014. doi: 10.1093/cvr/cvt268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 979.Soe NN, Sowden M, Baskaran P, Smolock EM, Kim Y, Nigro P, Berk BC. Cyclophilin A is required for angiotensin II-induced p47phox translocation to caveolae in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 33: 2147–2153, 2013. doi: 10.1161/ATVBAHA.113.301894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 980.Somanna NK, Valente AJ, Krenz M, Fay WP, Delafontaine P, Chandrasekar B. The Nox1/4 Dual Inhibitor GKT137831 or Nox4 Knockdown Inhibits Angiotensin-II-Induced Adult Mouse Cardiac Fibroblast Proliferation and Migration. AT1 Physically Associates With Nox4. J Cell Physiol 231: 1130–1141, 2016. doi: 10.1002/jcp.25210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 981.Sopel MJ, Rosin NL, Lee TD, Légaré JF. Myocardial fibrosis in response to Angiotensin II is preceded by the recruitment of mesenchymal progenitor cells. Lab Invest 91: 565–578, 2011. doi: 10.1038/labinvest.2010.190. [DOI] [PubMed] [Google Scholar]
- 982.Sorci-Thomas MG, Thomas MJ. Microdomains, Inflammation, and Atherosclerosis. Circ Res 118: 679–691, 2016. doi: 10.1161/CIRCRESAHA.115.306246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 983.Souza ACP, Tsuji T, Baranova IN, Bocharov AV, Wilkins KJ, Street JM, Alvarez-Prats A, Hu X, Eggerman T, Yuen PST, Star RA. TLR4 mutant mice are protected from renal fibrosis and chronic kidney disease progression. Physiol Rep 3: e12558, 2015. doi: 10.14814/phy2.12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 984.Sovari AA, Iravanian S, Dolmatova E, Jiao Z, Liu H, Zandieh S, Kumar V, Wang K, Bernstein KE, Bonini MG, Duffy HS, Dudley SC. Inhibition of c-Src tyrosine kinase prevents angiotensin II-mediated connexin-43 remodeling and sudden cardiac death. J Am Coll Cardiol 58: 2332–2339, 2011. doi: 10.1016/j.jacc.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 985.Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM. Classical Renin-Angiotensin system in kidney physiology. Compr Physiol 4: 1201–1228, 2014. doi: 10.1002/cphy.c130040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 986.Sparks MA, Parsons KK, Stegbauer J, Gurley SB, Vivekanandan-Giri A, Fortner CN, Snouwaert J, Raasch EW, Griffiths RC, Haystead TA, Le TH, Pennathur S, Koller B, Coffman TM. Angiotensin II type 1A receptors in vascular smooth muscle cells do not influence aortic remodeling in hypertension. Hypertension 57: 577–585, 2011. doi: 10.1161/HYPERTENSIONAHA.110.165274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 987.Sparks MA, Stegbauer J, Chen D, Gomez JA, Griffiths RC, Azad HA, Herrera M, Gurley SB, Coffman TM. Vascular Type 1A Angiotensin II Receptors Control BP by Regulating Renal Blood Flow and Urinary Sodium Excretion. J Am Soc Nephrol 26: 2953–2962, 2015. doi: 10.1681/ASN.2014080816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 988.Spin JM, Hsu M, Azuma J, Tedesco MM, Deng A, Dyer JS, Maegdefessel L, Dalman RL, Tsao PS. Transcriptional profiling and network analysis of the murine angiotensin II-induced abdominal aortic aneurysm. Physiol Genomics 43: 993–1003, 2011. doi: 10.1152/physiolgenomics.00044.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 989.Spitler KM, Webb RC. Endoplasmic reticulum stress contributes to aortic stiffening via proapoptotic and fibrotic signaling mechanisms. Hypertension 63: e40–e45, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 990.Sriramula S, Francis J. Tumor Necrosis Factor - Alpha Is Essential for Angiotensin II-Induced Ventricular Remodeling: Role for Oxidative Stress. PLoS One 10: e0138372, 2015. doi: 10.1371/journal.pone.0138372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 991.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 51: 1345–1351, 2008. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 992.Stalker TJ, Gong Y, Scalia R. The calcium-dependent protease calpain causes endothelial dysfunction in type 2 diabetes. Diabetes 54: 1132–1140, 2005. doi: 10.2337/diabetes.54.4.1132. [DOI] [PubMed] [Google Scholar]
- 993.Stalker TJ, Skvarka CB, Scalia R. A novel role for calpains in the endothelial dysfunction of hyperglycemia. FASEB J 17: 1511–1513, 2003. doi: 10.1096/fj.02-1213fje. [DOI] [PubMed] [Google Scholar]
- 994.Starke RM, Chalouhi N, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Wada K, Shimada K, Hasan DM, Greig NH, Owens GK, Dumont AS. Critical role of TNF-α in cerebral aneurysm formation and progression to rupture. J Neuroinflammation 11: 77, 2014. doi: 10.1186/1742-2094-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 995.Stawski L, Haines P, Fine A, Rudnicka L, Trojanowska M. MMP-12 deficiency attenuates angiotensin II-induced vascular injury, M2 macrophage accumulation, and skin and heart fibrosis. PLoS One 9: e109763, 2014. doi: 10.1371/journal.pone.0109763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 996.Stec DE, Davisson RL, Haskell RE, Davidson BL, Sigmund CD. Efficient liver-specific deletion of a floxed human angiotensinogen transgene by adenoviral delivery of Cre recombinase in vivo. J Biol Chem 274: 21285–21290, 1999. doi: 10.1074/jbc.274.30.21285. [DOI] [PubMed] [Google Scholar]
- 997.Stec DE, Keen HL, Sigmund CD. Lower blood pressure in floxed angiotensinogen mice after adenoviral delivery of Cre-recombinase. Hypertension 39: 629–633, 2002. doi: 10.1161/hy0202.103418. [DOI] [PubMed] [Google Scholar]
- 998.Steckelings UM, Widdop RE, Paulis L, Unger T. The angiotensin AT2 receptor in left ventricular hypertrophy. J Hypertens 28, Suppl 1: S50–S55, 2010. doi: 10.1097/01.hjh.0000388495.66330.63. [DOI] [PubMed] [Google Scholar]
- 999.Stegbauer J, Chen D, Herrera M, Sparks MA, Yang T, Königshausen E, Gurley SB, Coffman TM. Resistance to hypertension mediated by intercalated cells of the collecting duct. JCI Insight 2: e92720, 2017. doi: 10.1172/jci.insight.92720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000.Stegbauer J, Gurley SB, Sparks MA, Woznowski M, Kohan DE, Yan M, Lehrich RW, Coffman TM. AT1 receptors in the collecting duct directly modulate the concentration of urine. J Am Soc Nephrol 22: 2237–2246, 2011. doi: 10.1681/ASN.2010101095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1001.Stern JE, Son S, Biancardi VC, Zheng H, Sharma N, Patel KP. Astrocytes Contribute to Angiotensin II Stimulation of Hypothalamic Neuronal Activity and Sympathetic Outflow. Hypertension 68: 1483–1493, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1002.Stornetta RL, Hawelu-Johnson CL, Guyenet PG, Lynch KR. Astrocytes synthesize angiotensinogen in brain. Science 242: 1444–1446, 1988. doi: 10.1126/science.3201232. [DOI] [PubMed] [Google Scholar]
- 1003.Su W, Xie Z, Liu S, Calderon LE, Guo Z, Gong MC. Smooth muscle-selective CPI-17 expression increases vascular smooth muscle contraction and blood pressure. Am J Physiol Heart Circ Physiol 305: H104–H113, 2013. doi: 10.1152/ajpheart.00597.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1004.Subramanian V, Moorleghen JJ, Balakrishnan A, Howatt DA, Chishti AH, Uchida HA. Calpain-2 compensation promotes angiotensin II-induced ascending and abdominal aortic aneurysms in calpain-1 deficient mice. PLoS One 8: e72214, 2013. doi: 10.1371/journal.pone.0072214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1005.Subramanian V, Uchida HA, Ijaz T, Moorleghen JJ, Howatt DA, Balakrishnan A. Calpain Inhibition Attenuates Angiotensin II-induced Abdominal Aortic Aneurysms and Atherosclerosis in Low-Density Lipoprotein Receptor Deficient Mice. J Cardiovasc Pharmacol 59: 66–76, 2012. doi: 10.1097/FJC.0b013e318235d5ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1006.Suen RS, Rampersad SN, Stewart DJ, Courtman DW. Differential roles of endothelin-1 in angiotensin II-induced atherosclerosis and aortic aneurysms in apolipoprotein E-null mice. Am J Pathol 179: 1549–1559, 2011. doi: 10.1016/j.ajpath.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1007.Sukhanov S, Semprun-Prieto L, Yoshida T, Michael Tabony A, Higashi Y, Galvez S, Delafontaine P. Angiotensin II, oxidative stress and skeletal muscle wasting. Am J Med Sci 342: 143–147, 2011. doi: 10.1097/MAJ.0b013e318222e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1008.Sullivan JC. Sex and the renin-angiotensin system: inequality between the sexes in response to RAS stimulation and inhibition. Am J Physiol Regul Integr Comp Physiol 294: R1220–R1226, 2008. doi: 10.1152/ajpregu.00864.2007. [DOI] [PubMed] [Google Scholar]
- 1009.Sullivan JC, Bhatia K, Yamamoto T, Elmarakby AA. Angiotensin (1-7) receptor antagonism equalizes angiotensin II-induced hypertension in male and female spontaneously hypertensive rats. Hypertension 56: 658–666, 2010. doi: 10.1161/HYPERTENSIONAHA.110.153668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1010.Sumida T, Naito AT, Nomura S, Nakagawa A, Higo T, Hashimoto A, Okada K, Sakai T, Ito M, Yamaguchi T, Oka T, Akazawa H, Lee JK, Minamino T, Offermanns S, Noda T, Botto M, Kobayashi Y, Morita H, Manabe I, Nagai T, Shiojima I, Komuro I. Complement C1q-induced activation of β-catenin signalling causes hypertensive arterial remodelling. Nat Commun 6: 6241, 2015. doi: 10.1038/ncomms7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1011.Sumners C, de Kloet AD, Krause EG, Unger T, Steckelings UM. Angiotensin type 2 receptors: blood pressure regulation and end organ damage. Curr Opin Pharmacol 21: 115–121, 2015. doi: 10.1016/j.coph.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1012.Sun B, Huo R, Sheng Y, Li Y, Xie X, Chen C, Liu HB, Li N, Li CB, Guo WT, Zhu JX, Yang BF, Dong DL. Bone morphogenetic protein-4 mediates cardiac hypertrophy, apoptosis, and fibrosis in experimentally pathological cardiac hypertrophy. Hypertension 61: 352–360, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00562. [DOI] [PubMed] [Google Scholar]
- 1013.Sun J, Luo J, Ruan Y, Xiu L, Fang B, Zhang H, Wang M, Chen H. Free Fatty Acids Activate Renin-Angiotensin System in 3T3-L1 Adipocytes through Nuclear Factor-kappa B Pathway. J Diabetes Res 2016: 1587594, 2016. doi: 10.1155/2016/1587594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1014.Sun P, Yue P, Wang WH. Angiotensin II stimulates epithelial sodium channels in the cortical collecting duct of the rat kidney. Am J Physiol Renal Physiol 302: F679–F687, 2012. doi: 10.1152/ajprenal.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1015.Sun X, Tommasi E, Molina D, Sah R, Brosnihan KB, Diz D, Petrovic S. Deletion of proton-sensing receptor GPR4 associates with lower blood pressure and lower binding of angiotensin II receptor in SFO. Am J Physiol Renal Physiol 311: F1260–F1266, 2016. doi: 10.1152/ajprenal.00410.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1016.Sun XN, Li C, Liu Y, Du LJ, Zeng MR, Zheng XJ, Zhang WC, Liu Y, Zhu M, Kong D, Zhou L, Lu L, Shen ZX, Yi Y, Du L, Qin M, Liu X, Hua Z, Sun S, Yin H, Zhou B, Yu Y, Zhang Z, Duan SZ. T-Cell Mineralocorticoid Receptor Controls Blood Pressure by Regulating Interferon-Gamma. Circ Res 120: 1584–1597, 2017. doi: 10.1161/CIRCRESAHA.116.310480. [DOI] [PubMed] [Google Scholar]
- 1017.Suzuki H, Eguchi K, Ohtsu H, Higuchi S, Dhobale S, Frank GD, Motley ED, Eguchi S. Activation of endothelial nitric oxide synthase by the angiotensin II type 1 receptor. Endocrinology 147: 5914–5920, 2006. doi: 10.1210/en.2006-0834. [DOI] [PubMed] [Google Scholar]
- 1018.Suzuki H, Kimura K, Shirai H, Eguchi K, Higuchi S, Hinoki A, Ishimaru K, Brailoiu E, Dhanasekaran DN, Stemmle LN, Fields TA, Frank GD, Autieri MV, Eguchi S. Endothelial nitric oxide synthase inhibits G12/13 and rho-kinase activated by the angiotensin II type-1 receptor: implication in vascular migration. Arterioscler Thromb Vasc Biol 29: 217–224, 2009. doi: 10.1161/ATVBAHA.108.181024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1019.Szakadáti G, Tóth AD, Oláh I, Erdélyi LS, Balla T, Várnai P, Hunyady L, Balla A. Investigation of the fate of type I angiotensin receptor after biased activation. Mol Pharmacol 87: 972–981, 2015. doi: 10.1124/mol.114.097030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1020.Szalai B, Barkai L, Turu G, Szidonya L, Várnai P, Hunyady L. Allosteric interactions within the AT1 angiotensin receptor homodimer: role of the conserved DRY motif. Biochem Pharmacol 84: 477–485, 2012. doi: 10.1016/j.bcp.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 1021.Tabas I, Bornfeldt KE. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ Res 118: 653–667, 2016. doi: 10.1161/CIRCRESAHA.115.306256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1022.Tabony AM, Yoshida T, Sukhanov S, Delafontaine P. Protein phosphatase 2C-alpha knockdown reduces angiotensin II-mediated skeletal muscle wasting via restoration of mitochondrial recycling and function. Skelet Muscle 4: 20, 2014. doi: 10.1186/2044-5040-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1023.Tadevosyan A, Létourneau M, Folch B, Doucet N, Villeneuve LR, Mamarbachi AM, Pétrin D, Hébert TE, Fournier A, Chatenet D, Allen BG, Nattel S. Photoreleasable ligands to study intracrine angiotensin II signalling. J Physiol 593: 521–539, 2015. doi: 10.1113/jphysiol.2014.279109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1024.Tadevosyan A, Xiao J, Surinkaew S, Naud P, Merlen C, Harada M, Qi X, Chatenet D, Fournier A, Allen BG, Nattel S. Intracellular Angiotensin-II Interacts With Nuclear Angiotensin Receptors in Cardiac Fibroblasts and Regulates RNA Synthesis, Cell Proliferation, and Collagen Secretion. J Am Heart Assoc 6: e004965, 2017. doi: 10.1161/JAHA.116.004965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1025.Taglieri DM, Johnson KR, Burmeister BT, Monasky MM, Spindler MJ, DeSantiago J, Banach K, Conklin BR, Carnegie GK. The C-terminus of the long AKAP13 isoform (AKAP-Lbc) is critical for development of compensatory cardiac hypertrophy. J Mol Cell Cardiol 66: 27–40, 2014. doi: 10.1016/j.yjmcc.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1026.Taguchi K, Hida M, Narimatsu H, Matsumoto T, Kobayashi T. Glucose and angiotensin II-derived endothelial extracellular vesicles regulate endothelial dysfunction via ERK1/2 activation. Pflugers Arch 469: 293–302, 2017. doi: 10.1007/s00424-016-1926-2. [DOI] [PubMed] [Google Scholar]
- 1027.Takaguri A, Shirai H, Kimura K, Hinoki A, Eguchi K, Carlile-Klusacek M, Yang B, Rizzo V, Eguchi S. Caveolin-1 negatively regulates a metalloprotease-dependent epidermal growth factor receptor transactivation by angiotensin II. J Mol Cell Cardiol 50: 545–551, 2011. doi: 10.1016/j.yjmcc.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1028.Takahashi M, Tanaka J. Noradrenaline receptor mechanisms modulate the angiotensin II-induced water intake in the subfornical organ in rats. Exp Brain Res 235: 833–839, 2017. doi: 10.1007/s00221-016-4844-9. [DOI] [PubMed] [Google Scholar]
- 1029.Takane K, Hasegawa Y, Lin B, Koibuchi N, Cao C, Yokoo T, Kim-Mitsuyama S. Detrimental Effects of Centrally Administered Angiotensin II are Enhanced in a Mouse Model of Alzheimer Disease Independently of Blood Pressure. J Am Heart Assoc 6: e004897, 2017. doi: 10.1161/JAHA.116.004897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1030.Takata Y, Liu J, Yin F, Collins AR, Lyon CJ, Lee CH, Atkins AR, Downes M, Barish GD, Evans RM, Hsueh WA, Tangirala RK. PPARdelta-mediated anti-inflammatory mechanisms inhibit angiotensin II-accelerated atherosclerosis. Proc Natl Acad Sci USA 105: 4277–4282, 2008. doi: 10.1073/pnas.0708647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1031.Takawale A, Zhang P, Patel VB, Wang X, Oudit G, Kassiri Z. Tissue Inhibitor of Matrix Metalloproteinase-1 Promotes Myocardial Fibrosis by Mediating CD63-Integrin β1 Interaction. Hypertension 69: 1092–1103, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09045. [DOI] [PubMed] [Google Scholar]
- 1032.Takayanagi T, Bourne AM, Kimura K, Takaguri A, Elliott KJ, Eguchi K, Eguchi S. Constitutive stimulation of vascular smooth muscle cells by angiotensin II derived from an adenovirus encoding a furin-cleavable fusion protein. Am J Hypertens 25: 280–283, 2012. doi: 10.1038/ajh.2011.221. [DOI] [PubMed] [Google Scholar]
- 1033.Takayanagi T, Crawford KJ, Kobayashi T, Obama T, Tsuji T, Elliott KJ, Hashimoto T, Rizzo V, Eguchi S. Caveolin 1 is critical for abdominal aortic aneurysm formation induced by angiotensin II and inhibition of lysyl oxidase. Clin Sci (Lond) 126: 785–800, 2014. doi: 10.1042/CS20130660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1034.Takayanagi T, Forrester SJ, Kawai T, Obama T, Tsuji T, Elliott KJ, Nuti E, Rossello A, Kwok HF, Scalia R, Rizzo V, Eguchi S. Vascular ADAM17 as a Novel Therapeutic Target in Mediating Cardiovascular Hypertrophy and Perivascular Fibrosis Induced by Angiotensin II. Hypertension 68: 949–955, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1035.Takayanagi T, Kawai T, Forrester SJ, Obama T, Tsuji T, Fukuda Y, Elliott KJ, Tilley DG, Davisson RL, Park JY, Eguchi S. Role of epidermal growth factor receptor and endoplasmic reticulum stress in vascular remodeling induced by angiotensin II. Hypertension 65: 1349–1355, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1036.Takefuji M, Wirth A, Lukasova M, Takefuji S, Boettger T, Braun T, Althoff T, Offermanns S, Wettschureck N. G(13)-mediated signaling pathway is required for pressure overload-induced cardiac remodeling and heart failure. Circulation 126: 1972–1982, 2012. doi: 10.1161/CIRCULATIONAHA.112.109256. [DOI] [PubMed] [Google Scholar]
- 1037.Tan CK, Tan EH, Luo B, Huang CL, Loo JS, Choong C, Tan NS. SMAD3 deficiency promotes inflammatory aortic aneurysms in angiotensin II-infused mice via activation of iNOS. J Am Heart Assoc 2: e000269, 2013. doi: 10.1161/JAHA.113.000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1038.Tan P, Blais C, Nguyen TM, Schiller PW, Gutkowska J, Lavoie JL. Prorenin/renin receptor blockade promotes a healthy fat distribution in obese mice. Obesity (Silver Spring) 24: 1946–1954, 2016. doi: 10.1002/oby.21592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1039.Tan P, Shamansurova Z, Bisotto S, Michel C, Gauthier MS, Rabasa-Lhoret R, Nguyen TM, Schiller PW, Gutkowska J, Lavoie JL. Impact of the prorenin/renin receptor on the development of obesity and associated cardiometabolic risk factors. Obesity (Silver Spring) 22: 2201–2209, 2014. doi: 10.1002/oby.20844. [DOI] [PubMed] [Google Scholar]
- 1040.Tanaka Y, Tamura K, Koide Y, Sakai M, Tsurumi Y, Noda Y, Umemura M, Ishigami T, Uchino K, Kimura K, Horiuchi M, Umemura S. The novel angiotensin II type 1 receptor (AT1R)-associated protein ATRAP downregulates AT1R and ameliorates cardiomyocyte hypertrophy. FEBS Lett 579: 1579–1586, 2005. doi: 10.1016/j.febslet.2005.01.068. [DOI] [PubMed] [Google Scholar]
- 1041.Tarigopula M, Davis RT III, Mungai PT, Ryba DM, Wieczorek DF, Cowan CL, Violin JD, Wolska BM, Solaro RJ. Cardiac myosin light chain phosphorylation and inotropic effects of a biased ligand, TRV120023, in a dilated cardiomyopathy model. Cardiovasc Res 107: 226–234, 2015. doi: 10.1093/cvr/cvv162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1042.Te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH. Hypertension: renin-angiotensin-aldosterone system alterations. Circ Res 116: 960–975, 2015. doi: 10.1161/CIRCRESAHA.116.303587. [DOI] [PubMed] [Google Scholar]
- 1043.Ter Horst P, Smits JF, Blankesteijn WM. The Wnt/Frizzled pathway as a therapeutic target for cardiac hypertrophy: where do we stand? Acta Physiol (Oxf) 204: 110–117, 2012. doi: 10.1111/j.1748-1716.2011.02309.x. [DOI] [PubMed] [Google Scholar]
- 1044.Tetzner A, Gebolys K, Meinert C, Klein S, Uhlich A, Trebicka J, Villacañas Ó, Walther T. G-Protein-Coupled Receptor MrgD Is a Receptor for Angiotensin-(1-7) Involving Adenylyl Cyclase, cAMP, and Phosphokinase A. Hypertension 68: 185–194, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07572. [DOI] [PubMed] [Google Scholar]
- 1045.Thakur S, Li L, Gupta S. NF-κB-mediated integrin-linked kinase regulation in angiotensin II-induced pro-fibrotic process in cardiac fibroblasts. Life Sci 107: 68–75, 2014. doi: 10.1016/j.lfs.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 1046.Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol 89: 1401–1438, 2015. doi: 10.1007/s00204-015-1477-x. [DOI] [PubMed] [Google Scholar]
- 1047.Than A, Leow MK, Chen P. Control of adipogenesis by the autocrine interplays between angiotensin 1-7/Mas receptor and angiotensin II/AT1 receptor signaling pathways. J Biol Chem 288: 15520–15531, 2013. doi: 10.1074/jbc.M113.459792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1048.Thomas M, Gavrila D, McCormick ML, Miller FJ Jr, Daugherty A, Cassis LA, Dellsperger KC, Weintraub NL. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Circulation 114: 404–413, 2006. doi: 10.1161/CIRCULATIONAHA.105.607168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1049.Thomas WG, Pipolo L, Qian H. Identification of a Ca2+/calmodulin-binding domain within the carboxyl-terminus of the angiotensin II (AT1A) receptor. FEBS Lett 455: 367–371, 1999. doi: 10.1016/S0014-5793(99)00904-7. [DOI] [PubMed] [Google Scholar]
- 1050.Thrumurthy SG, Karthikesalingam A, Patterson BO, Holt PJ, Thompson MM. The diagnosis and management of aortic dissection. BMJ 344, jan11 1: d8290, 2011. doi: 10.1136/bmj.d8290. [DOI] [PubMed] [Google Scholar]
- 1051.Tian F, Luo R, Zhao Z, Wu Y, Ban D. Blockade of the RAS increases plasma adiponectin in subjects with metabolic syndrome and enhances differentiation and adiponectin expression of human preadipocytes. Exp Clin Endocrinol Diabetes 118: 258–265, 2010. doi: 10.1055/s-0029-1237706. [DOI] [PubMed] [Google Scholar]
- 1052.Tilley DG. G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ Res 109: 217–230, 2011. doi: 10.1161/CIRCRESAHA.110.231225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1053.Tiruneh MA, Huang BS, Leenen FH. Role of angiotensin II type 1 receptors in the subfornical organ in the pressor responses to central sodium in rats. Brain Res 1527: 79–86, 2013. doi: 10.1016/j.brainres.2013.06.028. [DOI] [PubMed] [Google Scholar]
- 1054.Tirupula KC, Desnoyer R, Speth RC, Karnik SS. Atypical signaling and functional desensitization response of MAS receptor to peptide ligands. PLoS One 9: e103520, 2014. doi: 10.1371/journal.pone.0103520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1055.Tirupula KC, Ithychanda SS, Mohan ML, Naga Prasad SV, Qin J, Karnik SS. G protein-coupled receptors directly bind filamin A with high affinity and promote filamin phosphorylation. Biochemistry 54: 6673–6683, 2015. doi: 10.1021/acs.biochem.5b00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1056.Torika N, Asraf K, Danon A, Apte RN, Fleisher-Berkovich S. Telmisartan Modulates Glial Activation: In Vitro and In Vivo Studies. PLoS One 11: e0155823, 2016. doi: 10.1371/journal.pone.0155823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1057.Toth P, Csiszar A, Tucsek Z, Sosnowska D, Gautam T, Koller A, Schwartzman ML, Sonntag WE, Ungvari Z. Role of 20-HETE, TRPC channels, and BKCa in dysregulation of pressure-induced Ca2+ signaling and myogenic constriction of cerebral arteries in aged hypertensive mice. Am J Physiol Heart Circ Physiol 305: H1698–H1708, 2013. doi: 10.1152/ajpheart.00377.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1058.Touyz RM, Cruzado M, Tabet F, Yao G, Salomon S, Schiffrin EL. Redox-dependent MAP kinase signaling by Ang II in vascular smooth muscle cells: role of receptor tyrosine kinase transactivation. Can J Physiol Pharmacol 81: 159–167, 2003. doi: 10.1139/y02-164. [DOI] [PubMed] [Google Scholar]
- 1059.Touyz RM, Mercure C, He Y, Javeshghani D, Yao G, Callera GE, Yogi A, Lochard N, Reudelhuber TL. Angiotensin II-dependent chronic hypertension and cardiac hypertrophy are unaffected by gp91phox-containing NADPH oxidase. Hypertension 45: 530–537, 2005. doi: 10.1161/01.HYP.0000158845.49943.5e. [DOI] [PubMed] [Google Scholar]
- 1060.Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev 52: 639–672, 2000. [PubMed] [Google Scholar]
- 1061.Touyz RM, Wu XH, He G, Salomon S, Schiffrin EL. Increased angiotensin II-mediated Src signaling via epidermal growth factor receptor transactivation is associated with decreased C-terminal Src kinase activity in vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension 39: 479–485, 2002. doi: 10.1161/hy02t2.102909. [DOI] [PubMed] [Google Scholar]
- 1062.Touyz RM, Yao G, Schiffrin EL. c-Src induces phosphorylation and translocation of p47phox: role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 23: 981–987, 2003. doi: 10.1161/01.ATV.0000069236.27911.68. [DOI] [PubMed] [Google Scholar]
- 1063.Trachet B, Aslanidou L, Piersigilli A, Fraga-Silva RA, Sordet-Dessimoz J, Villanueva-Perez P, Stampanoni MFM, Stergiopulos N, Segers P. Angiotensin II infusion into ApoE-/- mice: a model for aortic dissection rather than abdominal aortic aneurysm? Cardiovasc Res 113: 1230–1242, 2017. doi: 10.1093/cvr/cvx128. [DOI] [PubMed] [Google Scholar]
- 1064.Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res 118: 1021–1040, 2016. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1065.Trivedi DB, Loftin CD, Clark J, Myers P, DeGraff LM, Cheng J, Zeldin DC, Langenbach R. β-Arrestin-2 deficiency attenuates abdominal aortic aneurysm formation in mice. Circ Res 112: 1219–1229, 2013. doi: 10.1161/CIRCRESAHA.112.280399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1066.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li CI, Shyr Y, Harrison DG. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension 64: 1108–1115, 2014. doi: 10.1161/HYPERTENSIONAHA.114.04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1067.Tsai CT, Lai LP, Kuo KT, Hwang JJ, Hsieh CS, Hsu KL, Tseng CD, Tseng YZ, Chiang FT, Lin JL. Angiotensin II activates signal transducer and activators of transcription 3 via Rac1 in atrial myocytes and fibroblasts: implication for the therapeutic effect of statin in atrial structural remodeling. Circulation 117: 344–355, 2008. doi: 10.1161/CIRCULATIONAHA.107.695346. [DOI] [PubMed] [Google Scholar]
- 1068.Tsai IC, Pan ZC, Cheng HP, Liu CH, Lin BT, Jiang MJ. Reactive oxygen species derived from NADPH oxidase 1 and mitochondria mediate angiotensin II-induced smooth muscle cell senescence. J Mol Cell Cardiol 98: 18–27, 2016. doi: 10.1016/j.yjmcc.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 1069.Tsukuda K, Mogi M, Iwanami J, Kanno H, Nakaoka H, Wang XL, Bai HY, Shan BS, Kukida M, Higaki A, Yamauchi T, Min LJ, Horiuchi M. Enhancement of Adipocyte Browning by Angiotensin II Type 1 Receptor Blockade. PLoS One 11: e0167704, 2016. doi: 10.1371/journal.pone.0167704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1070.Tsuruda T, Sekita-Hatakeyama Y, Hao Y, Sakamoto S, Kurogi S, Nakamura M, Udagawa N, Funamoto T, Sekimoto T, Hatakeyama K, Chosa E, Kato J, Asada Y, Kitamura K. Angiotensin II Stimulation of Cardiac Hypertrophy and Functional Decompensation in Osteoprotegerin-Deficient Mice. Hypertension 67: 848–856, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06689. [DOI] [PubMed] [Google Scholar]
- 1071.Tsurumi Y, Tamura K, Tanaka Y, Koide Y, Sakai M, Yabana M, Noda Y, Hashimoto T, Kihara M, Hirawa N, Toya Y, Kiuchi Y, Iwai M, Horiuchi M, Umemura S. Interacting molecule of AT1 receptor, ATRAP, is colocalized with AT1 receptor in the mouse renal tubules. Kidney Int 69: 488–494, 2006. doi: 10.1038/sj.ki.5000130. [DOI] [PubMed] [Google Scholar]
- 1072.Tsushima K, Osawa T, Yanai H, Nakajima A, Takaoka A, Manabe I, Ohba Y, Imai Y, Taniguchi T, Nagai R. IRF3 regulates cardiac fibrosis but not hypertrophy in mice during angiotensin II-induced hypertension. FASEB J 25: 1531–1543, 2011. doi: 10.1096/fj.10-174615. [DOI] [PubMed] [Google Scholar]
- 1073.Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S, Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, Miwa T, Kawada N, Mori Y, Shibasaki Y, Tanaka Y, Fujiyama S, Koyama Y, Fujiyama A, Takahashi H, Iwasaka T. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J Clin Invest 104: 925–935, 1999. doi: 10.1172/JCI7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1074.Uchida HA, Kristo F, Rateri DL, Lu H, Charnigo R, Cassis LA, Daugherty A. Total lymphocyte deficiency attenuates AngII-induced atherosclerosis in males but not abdominal aortic aneurysms in apoE deficient mice. Atherosclerosis 211: 399–403, 2010. doi: 10.1016/j.atherosclerosis.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1075.Uchida S, Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ Res 116: 737–750, 2015. doi: 10.1161/CIRCRESAHA.116.302521. [DOI] [PubMed] [Google Scholar]
- 1076.Uehara Y, Miura S, Yahiro E, Saku K. Non-ACE pathway-induced angiotensin II production. Curr Pharm Des 19: 3054–3059, 2013. doi: 10.2174/1381612811319170012. [DOI] [PubMed] [Google Scholar]
- 1077.Umesalma S, Houwen FK, Baumbach GL, Chan SL. Roles of Caveolin-1 in Angiotensin II-Induced Hypertrophy and Inward Remodeling of Cerebral Pial Arterioles. Hypertension 67: 623–629, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1078.Underwood PC, Adler GK. The renin angiotensin aldosterone system and insulin resistance in humans. Curr Hypertens Rep 15: 59–70, 2013. doi: 10.1007/s11906-012-0323-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1079.Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, Yoshimura K, Aoki H, Tsutsui H, Noda T, Sagara J, Taniguchi S, Takahashi M. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler Thromb Vasc Biol 35: 127–136, 2015. doi: 10.1161/ATVBAHA.114.303763. [DOI] [PubMed] [Google Scholar]
- 1080.Usui T, Okada M, Hara Y, Yamawaki H. Death-associated protein kinase 3 mediates vascular inflammation and development of hypertension in spontaneously hypertensive rats. Hypertension 60: 1031–1039, 2012. doi: 10.1161/HYPERTENSIONAHA.112.200337. [DOI] [PubMed] [Google Scholar]
- 1081.Vaccari CS, Lerakis S, Hammoud R, Khan BV. Mechanisms of benefit of angiotensin receptor blockers in coronary atherosclerosis. Am J Med Sci 336: 270–277, 2008. doi: 10.1097/MAJ.0b013e31816d1dc5. [DOI] [PubMed] [Google Scholar]
- 1082.Valente AJ, Clark RA, Siddesha JM, Siebenlist U, Chandrasekar B. CIKS (Act1 or TRAF3IP2) mediates Angiotensin-II-induced Interleukin-18 expression, and Nox2-dependent cardiomyocyte hypertrophy. J Mol Cell Cardiol 53: 113–124, 2012. doi: 10.1016/j.yjmcc.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1083.Van der Lubbe N, Lim CH, Fenton RA, Meima ME, Jan Danser AH, Zietse R, Hoorn EJ. Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int 79: 66–76, 2011. doi: 10.1038/ki.2010.290. [DOI] [PubMed] [Google Scholar]
- 1084.Van Esch JH, Gembardt F, Sterner-Kock A, Heringer-Walther S, Le TH, Lassner D, Stijnen T, Coffman TM, Schultheiss HP, Danser AH, Walther T. Cardiac phenotype and angiotensin II levels in AT1a, AT1b, and AT2 receptor single, double, and triple knockouts. Cardiovasc Res 86: 401–409, 2010. doi: 10.1093/cvr/cvq004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1085.Van Rooij E. The art of microRNA research. Circ Res 108: 219–234, 2011. doi: 10.1161/CIRCRESAHA.110.227496. [DOI] [PubMed] [Google Scholar]
- 1086.Van Thiel BS, Góes Martini A, Te Riet L, Severs D, Uijl E, Garrelds IM, Leijten FPJ, van der Pluijm I, Essers J, Qadri F, Alenina N, Bader M, Paulis L, Rajkovicova R, Domenig O, Poglitsch M, Danser AHJ. Brain Renin-Angiotensin System: Does It Exist? Hypertension 69: 1136–1144, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08922. [DOI] [PubMed] [Google Scholar]
- 1087.Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes 64: 2028–2041, 2015. doi: 10.2337/db14-1225. [DOI] [PubMed] [Google Scholar]
- 1088.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Müller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426: 620, 2003. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- 1089.Velloso LA, Folli F, Sun XJ, White MF, Saad MJ, Kahn CR. Cross-talk between the insulin and angiotensin signaling systems. Proc Natl Acad Sci USA 93: 12490–12495, 1996. doi: 10.1073/pnas.93.22.12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1090.Venema RC, Ju H, Venema VJ, Schieffer B, Harp JB, Ling BN, Eaton DC, Marrero MB. Angiotensin II-induced association of phospholipase Cgamma1 with the G-protein-coupled AT1 receptor. J Biol Chem 273: 7703–7708, 1998. doi: 10.1074/jbc.273.13.7703. [DOI] [PubMed] [Google Scholar]
- 1091.Verdonk K, Visser W, Van Den Meiracker AH, Danser AH. The renin-angiotensin-aldosterone system in pre-eclampsia: the delicate balance between good and bad. Clin Sci (Lond) 126: 537–544, 2014. doi: 10.1042/CS20130455. [DOI] [PubMed] [Google Scholar]
- 1092.Vidigal JA, Ventura A. The biological functions of miRNAs: lessons from in vivo studies. Trends Cell Biol 25: 137–147, 2015. doi: 10.1016/j.tcb.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1093.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation 122: 2529–2537, 2010. doi: 10.1161/CIRCULATIONAHA.109.930446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1094.Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, Whalen EJ, Gowen M, Lark MW. Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther 335: 572–579, 2010. doi: 10.1124/jpet.110.173005. [DOI] [PubMed] [Google Scholar]
- 1095.Vital SA, Terao S, Nagai M, Granger DN. Mechanisms underlying the cerebral microvascular responses to angiotensin II-induced hypertension. Microcirculation 17: 641–649, 2010. doi: 10.1111/j.1549-8719.2010.00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1096.Von Lueder TG, Wang BH, Kompa AR, Huang L, Webb R, Jordaan P, Atar D, Krum H. Angiotensin receptor neprilysin inhibitor LCZ696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ Heart Fail 8: 71–78, 2015. doi: 10.1161/CIRCHEARTFAILURE.114.001785. [DOI] [PubMed] [Google Scholar]
- 1097.Vorkapic E, Lundberg AM, Mäyränpää MI, Eriksson P, Wågsäter D. TRIF adaptor signaling is important in abdominal aortic aneurysm formation. Atherosclerosis 241: 561–568, 2015. doi: 10.1016/j.atherosclerosis.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 1098.Vukelic S, Griendling KK. Angiotensin II, from vasoconstrictor to growth factor: a paradigm shift. Circ Res 114: 754–757, 2014. doi: 10.1161/CIRCRESAHA.114.303045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1099.Wagner S, Dantz C, Flebbe H, Azizian A, Sag CM, Engels S, Möllencamp J, Dybkova N, Islam T, Shah AM, Maier LS. NADPH oxidase 2 mediates angiotensin II-dependent cellular arrhythmias via PKA and CaMKII. J Mol Cell Cardiol 75: 206–215, 2014. doi: 10.1016/j.yjmcc.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 1100.Wakayama K, Shimamura M, Suzuki JI, Watanabe R, Koriyama H, Akazawa H, Nakagami H, Mochizuki H, Isobe M, Morishita R. Angiotensin II Peptide Vaccine Protects Ischemic Brain Through Reducing Oxidative Stress. Stroke 48: 1362–1368, 2017. doi: 10.1161/STROKEAHA.116.016269. [DOI] [PubMed] [Google Scholar]
- 1101.Wakui H, Tamura K, Tanaka Y, Matsuda M, Bai Y, Dejima T, Masuda S, Shigenaga A, Maeda A, Mogi M, Ichihara N, Kobayashi Y, Hirawa N, Ishigami T, Toya Y, Yabana M, Horiuchi M, Minamisawa S, Umemura S. Cardiac-specific activation of angiotensin II type 1 receptor-associated protein completely suppresses cardiac hypertrophy in chronic angiotensin II-infused mice. Hypertension 55: 1157–1164, 2010. doi: 10.1161/HYPERTENSIONAHA.109.147207. [DOI] [PubMed] [Google Scholar]
- 1102.Walters PE, Gaspari TA, Widdop RE. Angiotensin-(1-7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension 45: 960–966, 2005. doi: 10.1161/01.HYP.0000160325.59323.b8. [DOI] [PubMed] [Google Scholar]
- 1103.Wang B, Li C, Huai R, Qu Z. Overexpression of ANO1/TMEM16A, an arterial Ca2+-activated Cl− channel, contributes to spontaneous hypertension. J Mol Cell Cardiol 82: 22–32, 2015. doi: 10.1016/j.yjmcc.2015.02.020. [DOI] [PubMed] [Google Scholar]
- 1104.Wang F, Lu X, Peng K, Zhou L, Li C, Wang W, Yu X, Kohan DE, Zhu S-F, Yang T. COX-2 mediates angiotensin II-induced (pro)renin receptor expression in the rat renal medulla. Am J Physiol Renal Physiol 307: F25–F32, 2014. doi: 10.1152/ajprenal.00548.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1105.Wang G, Coleman CG, Chan J, Faraco G, Marques-Lopes J, Milner TA, Guruju MR, Anrather J, Davisson RL, Iadecola C, Pickel VM. Angiotensin II slow-pressor hypertension enhances NMDA currents and NOX2-dependent superoxide production in hypothalamic paraventricular neurons. Am J Physiol Regul Integr Comp Physiol 304: R1096–R1106, 2013. doi: 10.1152/ajpregu.00367.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1106.Wang HW, Huang BS, White RA, Chen A, Ahmad M, Leenen FH. Mineralocorticoid and angiotensin II type 1 receptors in the subfornical organ mediate angiotensin II - induced hypothalamic reactive oxygen species and hypertension. Neuroscience 329: 112–121, 2016. doi: 10.1016/j.neuroscience.2016.04.050. [DOI] [PubMed] [Google Scholar]
- 1107.Wang HX, Yang H, Han QY, Li N, Jiang X, Tian C, Du J, Li HH. NADPH oxidases mediate a cellular “memory” of angiotensin II stress in hypertensive cardiac hypertrophy. Free Radic Biol Med 65: 897–907, 2013. doi: 10.1016/j.freeradbiomed.2013.08.179. [DOI] [PubMed] [Google Scholar]
- 1108.Wang J, Ma R, Ma W, Chen J, Yang J, Xi Y, Cui Q. LncDisease: a sequence based bioinformatics tool for predicting lncRNA-disease associations. Nucleic Acids Res 44: e90, 2016. doi: 10.1093/nar/gkw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1109.Wang K, Liu F, Zhou LY, Long B, Yuan SM, Wang Y, Liu CY, Sun T, Zhang XJ, Li PF. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res 114: 1377–1388, 2014. doi: 10.1161/CIRCRESAHA.114.302476. [DOI] [PubMed] [Google Scholar]
- 1110.Wang L, Li YL, Zhang CC, Cui W, Wang X, Xia Y, Du J, Li HH. Inhibition of Toll-like receptor 2 reduces cardiac fibrosis by attenuating macrophage-mediated inflammation. Cardiovasc Res 101: 383–392, 2014. doi: 10.1093/cvr/cvt258. [DOI] [PubMed] [Google Scholar]
- 1111.Wang LP, Fan SJ, Li SM, Wang XJ, Gao JL, Yang XH. Protective role of ACE2-Ang-(1-7)-Mas in myocardial fibrosis by downregulating KCa3.1 channel via ERK1/2 pathway. Pflugers Arch 468: 2041–2051, 2016. doi: 10.1007/s00424-016-1875-9. [DOI] [PubMed] [Google Scholar]
- 1112.Wang M, Kim SH, Monticone RE, Lakatta EG. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension 65: 698–703, 2015. doi: 10.1161/HYPERTENSIONAHA.114.03618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1113.Wang N, Frank GD, Ding R, Tan Z, Rachakonda A, Pandolfi PP, Senbonmatsu T, Landon EJ, Inagami T. Promyelocytic leukemia zinc finger protein activates GATA4 transcription and mediates cardiac hypertrophic signaling from angiotensin II receptor 2. PLoS One 7: e35632, 2012. doi: 10.1371/journal.pone.0035632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1114.Wang Q, Lin JL, Erives AJ, Lin CI, Lin JJ. New insights into the roles of Xin repeat-containing proteins in cardiac development, function, and disease. Int Rev Cell Mol Biol 310: 89–128, 2014. doi: 10.1016/B978-0-12-800180-6.00003-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1115.Wang RH, He JP, Su ML, Luo J, Xu M, Du XD, Chen HZ, Wang WJ, Wang Y, Zhang N, Zhao BX, Zhao WX, Shan ZG, Han J, Chang C, Wu Q. The orphan receptor TR3 participates in angiotensin II-induced cardiac hypertrophy by controlling mTOR signalling. EMBO Mol Med 5: 137–148, 2013. doi: 10.1002/emmm.201201369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1116.Wang S, Gong H, Jiang G, Ye Y, Wu J, You J, Zhang G, Sun A, Komuro I, Ge J, Zou Y. Src is required for mechanical stretch-induced cardiomyocyte hypertrophy through angiotensin II type 1 receptor-dependent β-arrestin2 pathways. PLoS One 9: e92926, 2014. doi: 10.1371/journal.pone.0092926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1117.Wang S, Zhang C, Zhang M, Liang B, Zhu H, Lee J, Viollet B, Xia L, Zhang Y, Zou M-H. Activation of AMP-activated protein kinase α2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med 18: 902–910, 2012. doi: 10.1038/nm.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1118.Wang TN, Chen X, Li R, Gao B, Mohammed-Ali Z, Lu C, Yum V, Dickhout JG, Krepinsky JC. SREBP-1 Mediates Angiotensin II-Induced TGF-β1 Upregulation and Glomerular Fibrosis. J Am Soc Nephrol 26: 1839–1854, 2015. doi: 10.1681/ASN.2013121332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1119.Wang W, Huang XR, Canlas E, Oka K, Truong LD, Deng C, Bhowmick NA, Ju W, Bottinger EP, Lan HY. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ Res 98: 1032–1039, 2006. doi: 10.1161/01.RES.0000218782.52610.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1120.Wang W-H, Giebisch G. Regulation of potassium (K) handling in the renal collecting duct. Pflugers Arch 458: 157–168, 2009. doi: 10.1007/s00424-008-0593-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1121.Wang X, Berry E, Hernandez-Anzaldo S, Takawale A, Kassiri Z, Fernandez-Patron C. Matrix metalloproteinase-2 mediates a mechanism of metabolic cardioprotection consisting of negative regulation of the sterol regulatory element-binding protein-2/3-hydroxy-3-methylglutaryl-CoA reductase pathway in the heart. Hypertension 65: 882–888, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04989. [DOI] [PubMed] [Google Scholar]
- 1122.Wang X, Dai Y, Ding Z, Khaidakov M, Mercanti F, Mehta JL. Regulation of autophagy and apoptosis in response to angiotensin II in HL-1 cardiomyocytes. Biochem Biophys Res Commun 440: 696–700, 2013. doi: 10.1016/j.bbrc.2013.09.131. [DOI] [PubMed] [Google Scholar]
- 1123.Wang X, Oka T, Chow FL, Cooper SB, Odenbach J, Lopaschuk GD, Kassiri Z, Fernandez-Patron C. Tumor necrosis factor-alpha-converting enzyme is a key regulator of agonist-induced cardiac hypertrophy and fibrosis. Hypertension 54: 575–582, 2009. doi: 10.1161/HYPERTENSIONAHA.108.127670. [DOI] [PubMed] [Google Scholar]
- 1124.Wang X, Wang HX, Li YL, Zhang CC, Zhou CY, Wang L, Xia YL, Du J, Li HH. MicroRNA Let-7i negatively regulates cardiac inflammation and fibrosis. Hypertension 66: 776–785, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05548. [DOI] [PubMed] [Google Scholar]
- 1125.Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, Huang J, Offenstadt G, Combadière C, Rénia L, Johnson JL, Tharaux PL, Tedgui A, Mallat Z. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest 120: 422–432, 2010. doi: 10.1172/JCI38136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1126.Wang Y, Emeto TI, Lee J, Marshman L, Moran C, Seto SW, Golledge J. Mouse models of intracranial aneurysm. Brain Pathol 25: 237–247, 2015. doi: 10.1111/bpa.12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1127.Wang Y, Kuro-o M, Sun Z. Klotho gene delivery suppresses Nox2 expression and attenuates oxidative stress in rat aortic smooth muscle cells via the cAMP-PKA pathway. Aging Cell 11: 410–417, 2012. doi: 10.1111/j.1474-9726.2012.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1128.Wassmann S, Czech T, van Eickels M, Fleming I, Böhm M, Nickenig G. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double-knockout mice. Circulation 110: 3062–3067, 2004. doi: 10.1161/01.CIR.0000137970.47771.AF. [DOI] [PubMed] [Google Scholar]
- 1129.Watson A, Nong Z, Yin H, O’Neil C, Fox S, Balint B, Guo L, Leo O, Chu MWA, Gros R, Pickering JG. Nicotinamide Phosphoribosyltransferase in Smooth Muscle Cells Maintains Genome Integrity, Resists Aortic Medial Degeneration, and Is Suppressed in Human Thoracic Aortic Aneurysm Disease. Circ Res 120: 1889–1902, 2017. doi: 10.1161/CIRCRESAHA.116.310022. [DOI] [PubMed] [Google Scholar]
- 1130.Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol 10: 15–26, 2013. doi: 10.1038/nrcardio.2012.158. [DOI] [PubMed] [Google Scholar]
- 1131.Wei C, Kim IK, Kumar S, Jayasinghe S, Hong N, Castoldi G, Catalucci D, Jones WK, Gupta S. NF-κB mediated miR-26a regulation in cardiac fibrosis. J Cell Physiol 228: 1433–1442, 2013. doi: 10.1002/jcp.24296. [DOI] [PubMed] [Google Scholar]
- 1132.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA 100: 10782–10787, 2003. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1133.Wei L, Yuan M, Zhou R, Bai Q, Zhang W, Zhang M, Huang Y, Shi L. MicroRNA-101 inhibits rat cardiac hypertrophy by targeting Rab1a. J Cardiovasc Pharmacol 65: 357–363, 2015. doi: 10.1097/FJC.0000000000000203. [DOI] [PubMed] [Google Scholar]
- 1134.Wei LH, Huang XR, Zhang Y, Li YQ, Chen HY, Yan BP, Yu CM, Lan HY. Smad7 inhibits angiotensin II-induced hypertensive cardiac remodelling. Cardiovasc Res 99: 665–673, 2013. doi: 10.1093/cvr/cvt151. [DOI] [PubMed] [Google Scholar]
- 1135.Wei T, Huang G, Gao J, Huang C, Sun M, Wu J, Bu J, Shen W. Sirtuin 3 Deficiency Accelerates Hypertensive Cardiac Remodeling by Impairing Angiogenesis. J Am Heart Assoc 6: e006114, 2017. doi: 10.1161/JAHA.117.006114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1136.Wei Y, Zavilowitz B, Satlin LM, Wang WH. Angiotensin II inhibits the ROMK-like small conductance K channel in renal cortical collecting duct during dietary potassium restriction. J Biol Chem 282: 6455–6462, 2007. doi: 10.1074/jbc.M607477200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1137.Weiss D, Taylor WR. Deoxycorticosterone acetate salt hypertension in apolipoprotein E-/- mice results in accelerated atherosclerosis: the role of angiotensin II. Hypertension 51: 218–224, 2008. doi: 10.1161/HYPERTENSIONAHA.107.095885. [DOI] [PubMed] [Google Scholar]
- 1138.Weng X, Yu L, Liang P, Chen D, Cheng X, Yang Y, Li L, Zhang T, Zhou B, Wu X, Xu H, Fang M, Gao Y, Chen Q, Xu Y. Endothelial MRTF-A mediates angiotensin II induced cardiac hypertrophy. J Mol Cell Cardiol 80: 23–33, 2015. doi: 10.1016/j.yjmcc.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 1139.Weng X, Yu L, Liang P, Li L, Dai X, Zhou B, Wu X, Xu H, Fang M, Chen Q, Xu Y. A crosstalk between chromatin remodeling and histone H3K4 methyltransferase complexes in endothelial cells regulates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol 82: 48–58, 2015. doi: 10.1016/j.yjmcc.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 1140.Wennmann DO, Vollenbröker B, Eckart AK, Bonse J, Erdmann F, Wolters DA, Schenk LK, Schulze U, Kremerskothen J, Weide T, Pavenstädt H. The Hippo pathway is controlled by Angiotensin II signaling and its reactivation induces apoptosis in podocytes. Cell Death Dis 5: e1519, 2014. doi: 10.1038/cddis.2014.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1141.Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, Oelze M, Grabbe S, Jonuleit H, Becker C, Daiber A, Waisman A, Münzel T. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 124: 1370–1381, 2011. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]
- 1142.Wenzel U, Turner JE, Krebs C, Kurts C, Harrison DG, Ehmke H. Immune Mechanisms in Arterial Hypertension. J Am Soc Nephrol 27: 677–686, 2016. doi: 10.1681/ASN.2015050562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1143.Wettschureck N, Rütten H, Zywietz A, Gehring D, Wilkie TM, Chen J, Chien KR, Offermanns S. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat Med 7: 1236–1240, 2001. doi: 10.1038/nm1101-1236. [DOI] [PubMed] [Google Scholar]
- 1144.Whaley-Connell A, Govindarajan G, Habibi J, Hayden MR, Cooper SA, Wei Y, Ma L, Qazi M, Link D, Karuparthi PR, Stump C, Ferrario C, Sowers JR. Angiotensin II-mediated oxidative stress promotes myocardial tissue remodeling in the transgenic (mRen2) 27 Ren2 rat. Am J Physiol Endocrinol Metab 293: E355–E363, 2007. doi: 10.1152/ajpendo.00632.2006. [DOI] [PubMed] [Google Scholar]
- 1145.Whaley-Connell A, Sowers JR. Oxidative stress in the cardiorenal metabolic syndrome. Curr Hypertens Rep 14: 360–365, 2012. doi: 10.1007/s11906-012-0279-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1146.Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA 104: 7682–7687, 2007. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1147.Widder JD, Fraccarollo D, Galuppo P, Hansen JM, Jones DP, Ertl G, Bauersachs J. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension 54: 338–344, 2009. doi: 10.1161/HYPERTENSIONAHA.108.127928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1148.Wilkinson-Berka JL, Rana I, Armani R, Agrotis A. Reactive oxygen species, Nox and angiotensin II in angiogenesis: implications for retinopathy. Clin Sci (Lond) 124: 597–615, 2013. doi: 10.1042/CS20120212. [DOI] [PubMed] [Google Scholar]
- 1149.Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, Demos-Davies K, Yeager ME, Stenmark KR, McKinsey TA. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J Mol Cell Cardiol 67: 112–125, 2014. doi: 10.1016/j.yjmcc.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1150.Willis LM, El-Remessy AB, Somanath PR, Deremer DL, Fagan SC. Angiotensin receptor blockers and angiogenesis: clinical and experimental evidence. Clin Sci (Lond) 120: 307–319, 2011. doi: 10.1042/CS20100389. [DOI] [PubMed] [Google Scholar]
- 1151.Wincewicz D, Juchniewicz A, Waszkiewicz N, Braszko JJ. Angiotensin II type 1 receptor blockade by telmisartan prevents stress-induced impairment of memory via HPA axis deactivation and up-regulation of brain-derived neurotrophic factor gene expression. Pharmacol Biochem Behav 148: 108–118, 2016. doi: 10.1016/j.pbb.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 1152.Wirth A, Benyó Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horváth B, Maser-Gluth C, Greiner E, Lemmer B, Schütz G, Gutkind JS, Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 14: 64–68, 2008. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 1153.Wollert KC, Drexler H. The renin-angiotensin system and experimental heart failure. Cardiovasc Res 43: 838–849, 1999. doi: 10.1016/S0008-6363(99)00145-5. [DOI] [PubMed] [Google Scholar]
- 1154.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 430: 686–689, 2004. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 1155.Wright JW, Harding JW. The brain RAS and Alzheimer’s disease. Exp Neurol 223: 326–333, 2010. doi: 10.1016/j.expneurol.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 1156.Wright JW, Harding JW. The brain renin-angiotensin system: a diversity of functions and implications for CNS diseases. Pflugers Arch 465: 133–151, 2013. doi: 10.1007/s00424-012-1102-2. [DOI] [PubMed] [Google Scholar]
- 1157.Wu CH, Mohammadmoradi S, Thompson J, Su W, Gong M, Nguyen G, Yiannikouris F. Adipocyte (Pro)Renin-Receptor Deficiency Induces Lipodystrophy, Liver Steatosis and Increases Blood Pressure in Male Mice. Hypertension 68: 213–219, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1158.Wu D, Ren P, Zheng Y, Zhang L, Xu G, Xie W, Lloyd EE, Zhang S, Zhang Q, Curci JA, Coselli JS, Milewicz DM, Shen YH, LeMaire SA. NLRP3 (Nucleotide Oligomerization Domain-Like Receptor Family, Pyrin Domain Containing 3)-Caspase-1 Inflammasome Degrades Contractile Proteins: Implications for Aortic Biomechanical Dysfunction and Aneurysm and Dissection Formation. Arterioscler Thromb Vasc Biol 37: 694–706, 2017. doi: 10.1161/ATVBAHA.116.307648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1159.Wu H, Chen L, Xie J, Li R, Li GN, Chen QH, Zhang XL, Kang LN, Xu B. Periostin expression induced by oxidative stress contributes to myocardial fibrosis in a rat model of high salt-induced hypertension. Mol Med Rep 14: 776–782, 2016. doi: 10.3892/mmr.2016.5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1160.Wu J, Montaniel KR, Saleh MA, Xiao L, Chen W, Owens GK, Humphrey JD, Majesky MW, Paik DT, Hatzopoulos AK, Madhur MS, Harrison DG. Origin of Matrix-Producing Cells That Contribute to Aortic Fibrosis in Hypertension. Hypertension 67: 461–468, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1161.Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W, Harrison DG. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res 114: 616–625, 2014. doi: 10.1161/CIRCRESAHA.114.302157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1162.Wu Y, Li Y, Zhang C, A X, Wang Y, Cui W, Li H, Du J. S100a8/a9 released by CD11b+Gr1+ neutrophils activates cardiac fibroblasts to initiate angiotensin II-Induced cardiac inflammation and injury. Hypertension 63: 1241–1250, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02843. [DOI] [PubMed] [Google Scholar]
- 1163.Wynne BM, Chiao CW, Webb RC. Vascular Smooth Muscle Cell Signaling Mechanisms for Contraction to Angiotensin II and Endothelin-1. J Am Soc Hypertens 3: 84–95, 2009. doi: 10.1016/j.jash.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1164.Wyse BD, Prior IA, Qian H, Morrow IC, Nixon S, Muncke C, Kurzchalia TV, Thomas WG, Parton RG, Hancock JF. Caveolin interacts with the angiotensin II type 1 receptor during exocytic transport but not at the plasma membrane. J Biol Chem 278: 23738–23746, 2003. doi: 10.1074/jbc.M212892200. [DOI] [PubMed] [Google Scholar]
- 1165.Wysocki J, Ye M, Batlle D. Plasma and Kidney Angiotensin Peptides: Importance of the Aminopeptidase A/Angiotensin III Axis. Am J Hypertens 28: 1418–1426, 2015. doi: 10.1093/ajh/hpv054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1166.Xanthoulea S, Thelen M, Pöttgens C, Gijbels MJ, Lutgens E, de Winther MP. Absence of p55 TNF receptor reduces atherosclerosis, but has no major effect on angiotensin II induced aneurysms in LDL receptor deficient mice. PLoS One 4: e6113, 2009. doi: 10.1371/journal.pone.0006113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1167.Xia H, Feng Y, Obr TD, Hickman PJ, Lazartigues E. Angiotensin II type 1 receptor-mediated reduction of angiotensin-converting enzyme 2 activity in the brain impairs baroreflex function in hypertensive mice. Hypertension 53: 210–216, 2009. doi: 10.1161/HYPERTENSIONAHA.108.123844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1168.Xia H, Sriramula S, Chhabra KH, Lazartigues E. Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ Res 113: 1087–1096, 2013. doi: 10.1161/CIRCRESAHA.113.301811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1169.Xia Y, Kellems RE. Angiotensin receptor agonistic autoantibodies and hypertension: preeclampsia and beyond. Circ Res 113: 78–87, 2013. doi: 10.1161/CIRCRESAHA.113.300752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1170.Xiao K, Sun J, Kim J, Rajagopal S, Zhai B, Villén J, Haas W, Kovacs JJ, Shukla AK, Hara MR, Hernandez M, Lachmann A, Zhao S, Lin Y, Cheng Y, Mizuno K, Ma’ayan A, Gygi SP, Lefkowitz RJ. Global phosphorylation analysis of beta-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR). Proc Natl Acad Sci USA 107: 15299–15304, 2010. doi: 10.1073/pnas.1008461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1171.Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, Takahashi T, Loperena R, Foss JD, Mernaugh RL, Chen W, Roberts J II, Osborn JW, Itani HA, Harrison DG. Renal Denervation Prevents Immune Cell Activation and Renal Inflammation in Angiotensin II-Induced Hypertension. Circ Res 117: 547–557, 2015. doi: 10.1161/CIRCRESAHA.115.306010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1172.Xie P, Joladarashi D, Dudeja P, Sun L, Kanwar YS. Modulation of angiotensin II-induced inflammatory cytokines by the Epac1-Rap1A-NHE3 pathway: implications in renal tubular pathobiology. Am J Physiol Renal Physiol 306: F1260–F1274, 2014. doi: 10.1152/ajprenal.00069.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1173.Xiong S, Salazar G, Patrushev N, Ma M, Forouzandeh F, Hilenski L, Alexander RW. Peroxisome proliferator-activated receptor γ coactivator-1α is a central negative regulator of vascular senescence. Arterioscler Thromb Vasc Biol 33: 988–998, 2013. doi: 10.1161/ATVBAHA.112.301019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1174.Xiong S, Salazar G, San Martin A, Ahmad M, Patrushev N, Hilenski L, Nazarewicz RR, Ma M, Ushio-Fukai M, Alexander RW. PGC-1 alpha serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J Biol Chem 285: 2474–2487, 2010. doi: 10.1074/jbc.M109.065235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1175.Xu D, Borges GR, Davis DR, Agassandian K, Sequeira Lopez ML, Gomez RA, Cassell MD, Grobe JL, Sigmund CD. Neuron- or glial-specific ablation of secreted renin does not affect renal renin, baseline arterial pressure, or metabolism. Physiol Genomics 43: 286–294, 2011. doi: 10.1152/physiolgenomics.00208.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1176.Xu J, Lin SC, Chen J, Miao Y, Taffet GE, Entman ML, Wang Y. CCR2 mediates the uptake of bone marrow-derived fibroblast precursors in angiotensin II-induced cardiac fibrosis. Am J Physiol Heart Circ Physiol 301: H538–H547, 2011. doi: 10.1152/ajpheart.01114.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1177.Xu J, Sriramula S, Xia H, Moreno-Walton L, Culicchia F, Domenig O, Poglitsch M, Lazartigues E. Clinical Relevance and Role of Neuronal AT1 Receptors in ADAM17-Mediated ACE2 Shedding in Neurogenic Hypertension. Circ Res 121: 43–55, 2017. doi: 10.1161/CIRCRESAHA.116.310509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1178.Xu J, Sun Y, Carretero OA, Zhu L, Harding P, Shesely EG, Dai X, Rhaleb NE, Peterson E, Yang XP. Effects of cardiac overexpression of the angiotensin II type 2 receptor on remodeling and dysfunction in mice post-myocardial infarction. Hypertension 63: 1251–1259, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1179.Xu Q, Jensen DD, Peng H, Feng Y. The critical role of the central nervous system (pro)renin receptor in regulating systemic blood pressure. Pharmacol Ther 164: 126–134, 2016. doi: 10.1016/j.pharmthera.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1180.Xu S, Zhi H, Hou X, Cohen RA, Jiang B. IκBβ attenuates angiotensin II-induced cardiovascular inflammation and fibrosis in mice. Hypertension 58: 310–316, 2011. doi: 10.1161/HYPERTENSIONAHA.111.172031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1181.Xu X, Ha CH, Wong C, Wang W, Hausser A, Pfizenmaier K, Olson EN, McKinsey TA, Jin ZG. Angiotensin II stimulates protein kinase D-dependent histone deacetylase 5 phosphorylation and nuclear export leading to vascular smooth muscle cell hypertrophy. Arterioscler Thromb Vasc Biol 27: 2355–2362, 2007. doi: 10.1161/ATVBAHA.107.151704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1182.Xuan H, Xu B, Wang W, Tanaka H, Fujimura N, Miyata M, Michie SA, Dalman RL. Inhibition or deletion of angiotensin II type 1 receptor suppresses elastase-induced experimental abdominal aortic aneurysms. J Vasc Surg 67: 573–584.e2, 2018. doi: 10.1016/j.jvs.2016.12.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1183.Yabumoto C, Akazawa H, Yamamoto R, Yano M, Kudo-Sakamoto Y, Sumida T, Kamo T, Yagi H, Shimizu Y, Saga-Kamo A, Naito AT, Oka T, Lee JK, Suzuki J, Sakata Y, Uejima E, Komuro I. Angiotensin II receptor blockade promotes repair of skeletal muscle through down-regulation of aging-promoting C1q expression. Sci Rep 5: 14453, 2015. doi: 10.1038/srep14453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1184.Yagi S, Akaike M, Aihara K, Ishikawa K, Iwase T, Ikeda Y, Soeki T, Yoshida S, Sumitomo-Ueda Y, Matsumoto T, Sata M. Endothelial nitric oxide synthase-independent protective action of statin against angiotensin II-induced atrial remodeling via reduced oxidant injury. Hypertension 55: 918–923, 2010. doi: 10.1161/HYPERTENSIONAHA.109.146076. [DOI] [PubMed] [Google Scholar]
- 1185.Yamamoto K, Kakino A, Takeshita H, Hayashi N, Li L, Nakano A, Hanasaki-Yamamoto H, Fujita Y, Imaizumi Y, Toyama-Yokoyama S, Nakama C, Kawai T, Takeda M, Hongyo K, Oguro R, Maekawa Y, Itoh N, Takami Y, Onishi M, Takeya Y, Sugimoto K, Kamide K, Nakagami H, Ohishi M, Kurtz TW, Sawamura T, Rakugi H. Oxidized LDL (oxLDL) activates the angiotensin II type 1 receptor by binding to the lectin-like oxLDL receptor. FASEB J 29: 3342–3356, 2015. doi: 10.1096/fj.15-271627. [DOI] [PubMed] [Google Scholar]
- 1186.Yamashiro Y, Papke CL, Kim J, Ringuette LJ, Zhang QJ, Liu ZP, Mirzaei H, Wagenseil JE, Davis EC, Yanagisawa H. Abnormal mechanosensing and cofilin activation promote the progression of ascending aortic aneurysms in mice. Sci Signal 8: ra105, 2015. doi: 10.1126/scisignal.aab3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1187.Yan M, Chen C, Gong W, Yin Z, Zhou L, Chaugai S, Wang DW. miR-21-3p regulates cardiac hypertrophic response by targeting histone deacetylase-8. Cardiovasc Res 105: 340–352, 2015. doi: 10.1093/cvr/cvu254. [DOI] [PubMed] [Google Scholar]
- 1188.Yan X, Price RL, Nakayama M, Ito K, Schuldt AJ, Manning WJ, Sanbe A, Borg TK, Robbins J, Lorell BH. Ventricular-specific expression of angiotensin II type 2 receptors causes dilated cardiomyopathy and heart failure in transgenic mice. Am J Physiol Heart Circ Physiol 285: H2179–H2187, 2003. doi: 10.1152/ajpheart.00361.2003. [DOI] [PubMed] [Google Scholar]
- 1189.Yan X, Schuldt AJ, Price RL, Amende I, Liu FF, Okoshi K, Ho KK, Pope AJ, Borg TK, Lorell BH, Morgan JP. Pressure overload-induced hypertrophy in transgenic mice selectively overexpressing AT2 receptors in ventricular myocytes. Am J Physiol Heart Circ Physiol 294: H1274–H1281, 2008. doi: 10.1152/ajpheart.00174.2006. [DOI] [PubMed] [Google Scholar]
- 1190.Yang G, Gray TS, Sigmund CD, Cassell MD. The angiotensinogen gene is expressed in both astrocytes and neurons in murine central nervous system. Brain Res 817: 123–131, 1999. doi: 10.1016/S0006-8993(98)01236-0. [DOI] [PubMed] [Google Scholar]
- 1191.Yang J, Feng X, Zhou Q, Cheng W, Shang C, Han P, Lin CH, Chen HS, Quertermous T, Chang CP. Pathological Ace2-to-Ace enzyme switch in the stressed heart is transcriptionally controlled by the endothelial Brg1-FoxM1 complex. Proc Natl Acad Sci USA 113: E5628–E5635, 2016. doi: 10.1073/pnas.1525078113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1192.Yang L, Du C, Chen T, Li S, Nie W, Zhu W, Fan F, Zhu J, Yan H. Distinct MAPK pathways are involved in IL-23 production in dendritic cells cocultured with NK cells in the absence or presence of angiotensin II. Mol Immunol 51: 51–56, 2012. doi: 10.1016/j.molimm.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 1193.Yang M, Zheng J, Miao Y, Wang Y, Cui W, Guo J, Qiu S, Han Y, Jia L, Li H, Cheng J, Du J. Serum-glucocorticoid regulated kinase 1 regulates alternatively activated macrophage polarization contributing to angiotensin II-induced inflammation and cardiac fibrosis. Arterioscler Thromb Vasc Biol 32: 1675–1686, 2012. doi: 10.1161/ATVBAHA.112.248732. [DOI] [PubMed] [Google Scholar]
- 1194.Yang P, Schmit BM, Fu C, DeSart K, Oh SP, Berceli SA, Jiang Z. Smooth muscle cell-specific Tgfbr1 deficiency promotes aortic aneurysm formation by stimulating multiple signaling events. Sci Rep 6: 35444, 2016. doi: 10.1038/srep35444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1195.Yang RZ, Lee MJ, Hu H, Pollin TI, Ryan AS, Nicklas BJ, Snitker S, Horenstein RB, Hull K, Goldberg NH, Goldberg AP, Shuldiner AR, Fried SK, Gong DW. Acute-phase serum amyloid A: an inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med 3: e287, 2006. doi: 10.1371/journal.pmed.0030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1196.Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, Zadeh M, Gong M, Qi Y, Zubcevic J, Sahay B, Pepine CJ, Raizada MK, Mohamadzadeh M. Gut dysbiosis is linked to hypertension. Hypertension 65: 1331–1340, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1197.Yang Y, Ago T, Zhai P, Abdellatif M, Sadoshima J. Thioredoxin 1 negatively regulates angiotensin II-induced cardiac hypertrophy through upregulation of miR-98/let-7. Circ Res 108: 305–313, 2011. doi: 10.1161/CIRCRESAHA.110.228437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1198.Yang Z, Bove CM, French BA, Epstein FH, Berr SS, DiMaria JM, Gibson JJ, Carey RM, Kramer CM. Angiotensin II type 2 receptor overexpression preserves left ventricular function after myocardial infarction. Circulation 106: 106–111, 2002. doi: 10.1161/01.CIR.0000020014.14176.6D. [DOI] [PubMed] [Google Scholar]
- 1199.Yasuno S, Kuwahara K, Kinoshita H, Yamada C, Nakagawa Y, Usami S, Kuwabara Y, Ueshima K, Harada M, Nishikimi T, Nakao K. Angiotensin II type 1a receptor signalling directly contributes to the increased arrhythmogenicity in cardiac hypertrophy. Br J Pharmacol 170: 1384–1395, 2013. doi: 10.1111/bph.12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1200.Ye J, Ji Q, Liu J, Liu L, Huang Y, Shi Y, Shi L, Wang M, Liu M, Feng Y, Jiang H, Xu Y, Wang Z, Song J, Lin Y, Wan J. Interleukin 22 Promotes Blood Pressure Elevation and Endothelial Dysfunction in Angiotensin II-Treated Mice. J Am Heart Assoc 6: e005875, 2017. doi: 10.1161/JAHA.117.005875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1201.Ye ZW, Wu XM, Zhang LJ, Huang ZL, Jiang JG. Knockdown of angiotensinogen by shRNA-mediated RNA interference inhibits human visceral preadipocytes differentiation. Int J Obes 34: 157–164, 2010. doi: 10.1038/ijo.2009.197. [DOI] [PubMed] [Google Scholar]
- 1202.Yiannikouris F, Gupte M, Putnam K, Thatcher S, Charnigo R, Rateri DL, Daugherty A, Cassis LA. Adipocyte deficiency of angiotensinogen prevents obesity-induced hypertension in male mice. Hypertension 60: 1524–1530, 2012. doi: 10.1161/HYPERTENSIONAHA.112.192690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1203.Yiannikouris F, Karounos M, Charnigo R, English VL, Rateri DL, Daugherty A, Cassis LA. Adipocyte-specific deficiency of angiotensinogen decreases plasma angiotensinogen concentration and systolic blood pressure in mice. Am J Physiol Regul Integr Comp Physiol 302: R244–R251, 2012. doi: 10.1152/ajpregu.00323.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1204.Yiannikouris F, Wang Y, Shoemaker R, Larian N, Thompson J, English VL, Charnigo R, Su W, Gong M, Cassis LA. Deficiency of angiotensinogen in hepatocytes markedly decreases blood pressure in lean and obese male mice. Hypertension 66: 836–842, 2015. doi: 10.1161/HYPERTENSIONAHA.115.06040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1205.Ying Z, do Carmo JM, Xiang L, da Silva AA, Chen M, Ryan MJ, Ostrowski M, Rajagopalan S, Hall JE. Inhibitor κB kinase 2 is a myosin light chain kinase in vascular smooth muscle. Circ Res 113: 562–570, 2013. doi: 10.1161/CIRCRESAHA.113.301510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1206.Ying Z, Giachini FR, Tostes RC, Webb RC. PYK2/PDZ-RhoGEF links Ca2+ signaling to RhoA. Arterioscler Thromb Vasc Biol 29: 1657–1663, 2009. doi: 10.1161/ATVBAHA.109.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1207.Yodoi K, Yamashita T, Sasaki N, Kasahara K, Emoto T, Matsumoto T, Kita T, Sasaki Y, Mizoguchi T, Sparwasser T, Hirata K. Foxp3+ regulatory T cells play a protective role in angiotensin II-induced aortic aneurysm formation in mice. Hypertension 65: 889–895, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04934. [DOI] [PubMed] [Google Scholar]
- 1208.Yogi A, Mercure C, Touyz J, Callera GE, Montezano AC, Aranha AB, Tostes RC, Reudelhuber T, Touyz RM. Renal redox-sensitive signaling, but not blood pressure, is attenuated by Nox1 knockout in angiotensin II-dependent chronic hypertension. Hypertension 51: 500–506, 2008. doi: 10.1161/HYPERTENSIONAHA.107.103192. [DOI] [PubMed] [Google Scholar]
- 1209.Yoshida T, Delafontaine P. An Intronic Enhancer Element Regulates Angiotensin II Type 2 Receptor Expression during Satellite Cell Differentiation, and Its Activity Is Suppressed in Congestive Heart Failure. J Biol Chem 291: 25578–25590, 2016. doi: 10.1074/jbc.M116.752501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1210.Yoshida T, Galvez S, Tiwari S, Rezk BM, Semprun-Prieto L, Higashi Y, Sukhanov S, Yablonka-Reuveni Z, Delafontaine P. Angiotensin II inhibits satellite cell proliferation and prevents skeletal muscle regeneration. J Biol Chem 288: 23823–23832, 2013. doi: 10.1074/jbc.M112.449074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1211.Yoshida T, Huq TS, Delafontaine P. Angiotensin type 2 receptor signaling in satellite cells potentiates skeletal muscle regeneration. J Biol Chem 289: 26239–26248, 2014. doi: 10.1074/jbc.M114.585521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1212.Yoshida T, Semprun-Prieto L, Sukhanov S, Delafontaine P. IGF-1 prevents ANG II-induced skeletal muscle atrophy via Akt- and Foxo-dependent inhibition of the ubiquitin ligase atrogin-1 expression. Am J Physiol Heart Circ Physiol 298: H1565–H1570, 2010. doi: 10.1152/ajpheart.00146.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1213.Yoshida T, Tabony AM, Galvez S, Mitch WE, Higashi Y, Sukhanov S, Delafontaine P. Molecular mechanisms and signaling pathways of angiotensin II-induced muscle wasting: potential therapeutic targets for cardiac cachexia. Int J Biochem Cell Biol 45: 2322–2332, 2013. doi: 10.1016/j.biocel.2013.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1214.You D, Loufrani L, Baron C, Levy BI, Widdop RE, Henrion D. High blood pressure reduction reverses angiotensin II type 2 receptor-mediated vasoconstriction into vasodilation in spontaneously hypertensive rats. Circulation 111: 1006–1011, 2005. doi: 10.1161/01.CIR.0000156503.62815.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1215.Youn J-Y, Zhang J, Zhang Y, Chen H, Liu D, Ping P, Weiss JN, Cai H. Oxidative stress in atrial fibrillation: an emerging role of NADPH oxidase. J Mol Cell Cardiol 62: 72–79, 2013. doi: 10.1016/j.yjmcc.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1216.Young CN, Cao X, Guruju MR, Pierce JP, Morgan DA, Wang G, Iadecola C, Mark AL, Davisson RL. ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J Clin Invest 122: 3960–3964, 2012. doi: 10.1172/JCI64583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1217.Young CN, Davisson RL. Angiotensin-II, the Brain, and Hypertension: An Update. Hypertension 66: 920–926, 2015. doi: 10.1161/HYPERTENSIONAHA.115.03624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1218.Young CN, Li A, Dong FN, Horwath JA, Clark CG, Davisson RL. Endoplasmic reticulum and oxidant stress mediate nuclear factor-κB activation in the subfornical organ during angiotensin II hypertension. Am J Physiol Cell Physiol 308: C803–C812, 2015. doi: 10.1152/ajpcell.00223.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1219.Young CN, Morgan DA, Butler SD, Rahmouni K, Gurley SB, Coffman TM, Mark AL, Davisson RL. Angiotensin type 1a receptors in the forebrain subfornical organ facilitate leptin-induced weight loss through brown adipose tissue thermogenesis. Mol Metab 4: 337–343, 2015. doi: 10.1016/j.molmet.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1220.Yu H, Guo Y, Mi L, Wang X, Li L, Gao W. Mitofusin 2 inhibits angiotensin II-induced myocardial hypertrophy. J Cardiovasc Pharmacol Ther 16: 205–211, 2011. doi: 10.1177/1074248410385683. [DOI] [PubMed] [Google Scholar]
- 1221.Yu KY, Wang YP, Wang LH, Jian Y, Zhao XD, Chen JW, Murao K, Zhu W, Dong L, Wang GQ, Zhang GX. Mitochondrial KATP channel involvement in angiotensin II-induced autophagy in vascular smooth muscle cells. Basic Res Cardiol 109: 416, 2014. doi: 10.1007/s00395-014-0416-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1222.Yu L, Yang G, Weng X, Liang P, Li L, Li J, Fan Z, Tian W, Wu X, Xu H, Fang M, Ji Y, Li Y, Chen Q, Xu Y. Histone Methyltransferase SET1 Mediates Angiotensin II-Induced Endothelin-1 Transcription and Cardiac Hypertrophy in Mice. Arterioscler Thromb Vasc Biol 35: 1207–1217, 2015. doi: 10.1161/ATVBAHA.115.305230. [DOI] [PubMed] [Google Scholar]
- 1223.Yu Y, Xue BJ, Wei SG, Zhang ZH, Beltz TG, Guo F, Johnson AK, Felder RB. Activation of central PPAR-γ attenuates angiotensin II-induced hypertension. Hypertension 66: 403–411, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1224.Yu Y-H, Hu Z-Y, Li M-H, Li B, Wang Z-M, Chen S-L. Cardiac hypertrophy is positively regulated by long non-coding RNA PVT1. Int J Clin Exp Pathol 8: 2582–2589, 2015. [PMC free article] [PubMed] [Google Scholar]
- 1225.Yuan Y, Yan L, Wu QQ, Zhou H, Jin YG, Bian ZY, Deng W, Yang Z, Shen DF, Zeng XF, Wang SS, Li H, Tang QZ. Mnk1 (Mitogen-Activated Protein Kinase-Interacting Kinase 1) Deficiency Aggravates Cardiac Remodeling in Mice. Hypertension 68: 1393–1399, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07906. [DOI] [PubMed] [Google Scholar]
- 1226.Yue H, Li W, Desnoyer R, Karnik SS. Role of nuclear unphosphorylated STAT3 in angiotensin II type 1 receptor-induced cardiac hypertrophy. Cardiovasc Res 85: 90–99, 2010. doi: 10.1093/cvr/cvp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1227.Yue P, Sun P, Lin DH, Pan C, Xing W, Wang W. Angiotensin II diminishes the effect of SGK1 on the WNK4-mediated inhibition of ROMK1 channels. Kidney Int 79: 423–431, 2011. doi: 10.1038/ki.2010.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1228.Yugandhar VG, Clark MA. Angiotensin III: a physiological relevant peptide of the renin angiotensin system. Peptides 46: 26–32, 2013. doi: 10.1016/j.peptides.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 1229.Yvan-Charvet L, Even P, Bloch-Faure M, Guerre-Millo M, Moustaid-Moussa N, Ferre P, Quignard-Boulange A. Deletion of the angiotensin type 2 receptor (AT2R) reduces adipose cell size and protects from diet-induced obesity and insulin resistance. Diabetes 54: 991–999, 2005. doi: 10.2337/diabetes.54.4.991. [DOI] [PubMed] [Google Scholar]
- 1230.Zahradka P, Litchie B, Storie B, Helwer G. Transactivation of the insulin-like growth factor-I receptor by angiotensin II mediates downstream signaling from the angiotensin II type 1 receptor to phosphatidylinositol 3-kinase. Endocrinology 145: 2978–2987, 2004. doi: 10.1210/en.2004-0029. [DOI] [PubMed] [Google Scholar]
- 1231.Zahradka P, Storie B, Wright B. IGF-1 receptor transactivation mediates Src-dependent cortactin phosphorylation in response to angiotensin II. Can J Physiol Pharmacol 87: 805–812, 2009. doi: 10.1139/Y09-052. [DOI] [PubMed] [Google Scholar]
- 1232.Zaika O, Mamenko M, Staruschenko A, Pochynyuk O. Direct activation of ENaC by angiotensin II: recent advances and new insights. Curr Hypertens Rep 15: 17–24, 2013. doi: 10.1007/s11906-012-0316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1233.Zampetaki A, Attia R, Mayr U, Gomes RS, Phinikaridou A, Yin X, Langley SR, Willeit P, Lu R, Fanshawe B, Fava M, Barallobre-Barreiro J, Molenaar C, So PW, Abbas A, Jahangiri M, Waltham M, Botnar R, Smith A, Mayr M. Role of miR-195 in aortic aneurysmal disease. Circ Res 115: 857–866, 2014. doi: 10.1161/CIRCRESAHA.115.304361. [DOI] [PubMed] [Google Scholar]
- 1234.Zeng C, Hopfer U, Asico LD, Eisner GM, Felder RA, Jose PA. Altered AT1 receptor regulation of ETB receptors in renal proximal tubule cells of spontaneously hypertensive rats. Hypertension 46: 926–931, 2005. doi: 10.1161/01.HYP.0000174595.41637.13. [DOI] [PubMed] [Google Scholar]
- 1235.Zeng C, Luo Y, Asico LD, Hopfer U, Eisner GM, Felder RA, Jose PA. Perturbation of D1 dopamine and AT1 receptor interaction in spontaneously hypertensive rats. Hypertension 42: 787–792, 2003. doi: 10.1161/01.HYP.0000085334.34963.4E. [DOI] [PubMed] [Google Scholar]
- 1236.Zeng SY, Chen X, Chen SR, Li Q, Wang YH, Zou J, Cao WW, Luo JN, Gao H, Liu PQ. Upregulation of Nox4 promotes angiotensin II-induced epidermal growth factor receptor activation and subsequent cardiac hypertrophy by increasing ADAM17 expression. Can J Cardiol 29: 1310–1319, 2013. doi: 10.1016/j.cjca.2013.04.026. [DOI] [PubMed] [Google Scholar]
- 1237.Zeniya M, Morimoto N, Takahashi D, Mori Y, Mori T, Ando F, Araki Y, Yoshizaki Y, Inoue Y, Isobe K, Nomura N, Oi K, Nishida H, Sasaki S, Sohara E, Rai T, Uchida S. Kelch-Like Protein 2 Mediates Angiotensin II-With No Lysine 3 Signaling in the Regulation of Vascular Tonus. J Am Soc Nephrol 26: 2129–2138, 2015. doi: 10.1681/ASN.2014070639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1238.Zeniya M, Sohara E, Kita S, Iwamoto T, Susa K, Mori T, Oi K, Chiga M, Takahashi D, Yang SS, Lin SH, Rai T, Sasaki S, Uchida S. Dietary salt intake regulates WNK3-SPAK-NKCC1 phosphorylation cascade in mouse aorta through angiotensin II. Hypertension 62: 872–878, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01543. [DOI] [PubMed] [Google Scholar]
- 1239.Zhan Y, Brown C, Maynard E, Anshelevich A, Ni W, Ho IC, Oettgen P. Ets-1 is a critical regulator of Ang II-mediated vascular inflammation and remodeling. J Clin Invest 115: 2508–2516, 2005. doi: 10.1172/JCI24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1240.Zhan Y, Yuan L, Kondo M, Oettgen P. The counter-regulatory effects of ESE-1 during angiotensin II-mediated vascular inflammation and remodeling. Am J Hypertens 23: 1312–1317, 2010. doi: 10.1038/ajh.2010.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1241.Zhang B, Jiang J, Yue Z, Liu S, Ma Y, Yu N, Gao Y, Sun S, Chen S, Liu P. Store-Operated Ca2+ Entry (SOCE) contributes to angiotensin II-induced cardiac fibrosis in cardiac fibroblasts. J Pharmacol Sci 132: 171–180, 2016. doi: 10.1016/j.jphs.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 1242.Zhang C, Li Y, Wang C, Wu Y, Cui W, Miwa T, Sato S, Li H, Song WC, Du J. Complement 5a receptor mediates angiotensin II-induced cardiac inflammation and remodeling. Arterioscler Thromb Vasc Biol 34: 1240–1248, 2014. doi: 10.1161/ATVBAHA.113.303120. [DOI] [PubMed] [Google Scholar]
- 1243.Zhang C, van der Voort D, Shi H, Zhang R, Qing Y, Hiraoka S, Takemoto M, Yokote K, Moxon JV, Norman P, Rittié L, Kuivaniemi H, Atkins GB, Gerson SL, Shi GP, Golledge J, Dong N, Perbal B, Prosdocimo DA, Lin Z. Matricellular protein CCN3 mitigates abdominal aortic aneurysm. J Clin Invest 126: 1282–1299, 2016. doi: 10.1172/JCI82337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1244.Zhang H, Han GW, Batyuk A, Ishchenko A, White KL, Patel N, Sadybekov A, Zamlynny B, Rudd MT, Hollenstein K, Tolstikova A, White TA, Hunter MS, Weierstall U, Liu W, Babaoglu K, Moore EL, Katz RD, Shipman JM, Garcia-Calvo M, Sharma S, Sheth P, Soisson SM, Stevens RC, Katritch V, Cherezov V. Structural basis for selectivity and diversity in angiotensin II receptors. Nature 544: 327–332, 2017. doi: 10.1038/nature22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1245.Zhang H, Unal H, Desnoyer R, Han GW, Patel N, Katritch V, Karnik SS, Cherezov V, Stevens RC. Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J Biol Chem 290: 29127–29139, 2015. doi: 10.1074/jbc.M115.689000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1246.Zhang H, Unal H, Gati C, Han GW, Liu W, Zatsepin NA, James D, Wang D, Nelson G, Weierstall U, Sawaya MR, Xu Q, Messerschmidt M, Williams GJ, Boutet S, Yefanov OM, White TA, Wang C, Ishchenko A, Tirupula KC, Desnoyer R, Coe J, Conrad CE, Fromme P, Stevens RC, Katritch V, Karnik SS, Cherezov V. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 161: 833–844, 2015. doi: 10.1016/j.cell.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1247.Zhang H, Wu J, Dong H, Khan SA, Chu ML, Tsuda T. Fibulin-2 deficiency attenuates angiotensin II-induced cardiac hypertrophy by reducing transforming growth factor-β signalling. Clin Sci (Lond) 126: 275–288, 2014. doi: 10.1042/CS20120636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1248.Zhang J, Patel MB, Griffiths R, Mao A, Song YS, Karlovich NS, Sparks MA, Jin H, Wu M, Lin EE, Crowley SD. Tumor necrosis factor-α produced in the kidney contributes to angiotensin II-dependent hypertension. Hypertension 64: 1275–1281, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1249.Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA, Griffiths R, Sparks MA, Jeffs AD, Crowley SD. Interleukin-1 Receptor Activation Potentiates Salt Reabsorption in Angiotensin II-Induced Hypertension via the NKCC2 Co-transporter in the Nephron. Cell Metab 23: 360–368, 2016. doi: 10.1016/j.cmet.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1250.Zhang JD, Patel MB, Griffiths R, Dolber PC, Ruiz P, Sparks MA, Stegbauer J, Jin H, Gomez JA, Buckley AF, Lefler WS, Chen D, Crowley SD. Type 1 angiotensin receptors on macrophages ameliorate IL-1 receptor-mediated kidney fibrosis. J Clin Invest 124: 2198–2203, 2014. doi: 10.1172/JCI61368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1251.Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, Sparks MA, Yan M, Howell DN, Gomez JA, Spurney RF, Coffman TM, Crowley SD. A novel role for type 1 angiotensin receptors on T lymphocytes to limit target organ damage in hypertension. Circ Res 110: 1604–1617, 2012. doi: 10.1161/CIRCRESAHA.111.261768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1252.Zhang L, Cheng J, Ma Y, Thomas W, Zhang J, Du J. Dual pathways for nuclear factor kappaB activation by angiotensin II in vascular smooth muscle: phosphorylation of p65 by IkappaB kinase and ribosomal kinase. Circ Res 97: 975–982, 2005. doi: 10.1161/01.RES.0000190589.52286.41. [DOI] [PubMed] [Google Scholar]
- 1253.Zhang L, Ren Z, Yang Q, Ding G. Csk regulates angiotensin II-induced podocyte apoptosis. Apoptosis 21: 846–855, 2016. doi: 10.1007/s10495-016-1256-z. [DOI] [PubMed] [Google Scholar]
- 1254.Zhang M, Prosser BL, Bamboye MA, Gondim ANS, Santos CX, Martin D, Ghigo A, Perino A, Brewer AC, Ward CW, Hirsch E, Lederer WJ, Shah AM. Contractile Function During Angiotensin-II Activation: Increased Nox2 Activity Modulates Cardiac Calcium Handling via Phospholamban Phosphorylation. J Am Coll Cardiol 66: 261–272, 2015. doi: 10.1016/j.jacc.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1255.Zhang R, Liu Z, Qu Y, Xu Y, Yang Q. Two distinct calmodulin binding sites in the third intracellular loop and carboxyl tail of angiotensin II (AT(1A)) receptor. PLoS One 8: e65266, 2013. doi: 10.1371/journal.pone.0065266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1256.Zhang X, Li ZL, Crane JA, Jordan KL, Pawar AS, Textor SC, Lerman A, Lerman LO. Valsartan regulates myocardial autophagy and mitochondrial turnover in experimental hypertension. Hypertension 64: 87–93, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1257.Zhang X, Meng F, Song J, Zhang L, Wang J, Li D, Li L, Dong P, Yang B, Chen Y. Pentoxifylline Ameliorates Cardiac Fibrosis, Pathological Hypertrophy, and Cardiac Dysfunction in Angiotensin II-induced Hypertensive Rats. J Cardiovasc Pharmacol 67: 76–85, 2016. doi: 10.1097/FJC.0000000000000316. [DOI] [PubMed] [Google Scholar]
- 1258.Zhang X, Wang H, Duvernay MT, Zhu S, Wu G. The angiotensin II type 1 receptor C-terminal Lys residues interact with tubulin and modulate receptor export trafficking. PLoS One 8: e57805, 2013. doi: 10.1371/journal.pone.0057805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1259.Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol Ther 22: 974–985, 2014. doi: 10.1038/mt.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1260.Zhang ZB, Ruan CC, Chen DR, Zhang K, Yan C, Gao PJ. Activating transcription factor 3 SUMOylation is involved in angiotensin II-induced endothelial cell inflammation and dysfunction. J Mol Cell Cardiol 92: 149–157, 2016. doi: 10.1016/j.yjmcc.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 1261.Zhang ZZ, Shang QH, Jin HY, Song B, Oudit GY, Lu L, Zhou T, Xu YL, Gao PJ, Zhu DL, Penninger JM, Zhong JC. Cardiac protective effects of irbesartan via the PPAR-gamma signaling pathway in angiotensin-converting enzyme 2-deficient mice. J Transl Med 11: 229, 2013. doi: 10.1186/1479-5876-11-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1262.Zhao N, Koenig SN, Trask AJ, Lin CH, Hans CP, Garg V, Lilly B. MicroRNA miR145 regulates TGFBR2 expression and matrix synthesis in vascular smooth muscle cells. Circ Res 116: 23–34, 2015. doi: 10.1161/CIRCRESAHA.115.303970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1263.Zhao Q, Ishibashi M, Hiasa K, Tan C, Takeshita A, Egashira K. Essential role of vascular endothelial growth factor in angiotensin II-induced vascular inflammation and remodeling. Hypertension 44: 264–270, 2004. doi: 10.1161/01.HYP.0000138688.78906.6b. [DOI] [PubMed] [Google Scholar]
- 1264.Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X, Bhandari B, Abboud HE. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation 131: 643–655, 2015. doi: 10.1161/CIRCULATIONAHA.114.011079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1265.Zhao W, Li Y, Jia L, Pan L, Li H, Du J. Atg5 deficiency-mediated mitophagy aggravates cardiac inflammation and injury in response to angiotensin II. Free Radic Biol Med 69: 108–115, 2014. doi: 10.1016/j.freeradbiomed.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 1266.Zhao YX, Yin HQ, Yu QT, Qiao Y, Dai HY, Zhang MX, Zhang L, Liu YF, Wang LC, Liu DS, Deng BP, Zhang YH, Pan CM, Song HD, Qu X, Jiang H, Liu CX, Lu XT, Liu B, Gao F, Dong B. ACE2 overexpression ameliorates left ventricular remodeling and dysfunction in a rat model of myocardial infarction. Hum Gene Ther 21: 1545–1554, 2010. doi: 10.1089/hum.2009.160. [DOI] [PubMed] [Google Scholar]
- 1267.Zheng YH, Li FD, Tian C, Ren HL, Du J, Li HH. Notch γ-secretase inhibitor dibenzazepine attenuates angiotensin II-induced abdominal aortic aneurysm in ApoE knockout mice by multiple mechanisms. PLoS One 8: e83310, 2013. doi: 10.1371/journal.pone.0083310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1268.Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M, Loibner H, Wang XH, Penninger JM, Kassiri Z, Oudit GY. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 122: 717–728, 2010. doi: 10.1161/CIRCULATIONAHA.110.955369. [DOI] [PubMed] [Google Scholar]
- 1269.Zhou L, Li Y, Hao S, Zhou D, Tan RJ, Nie J, Hou FF, Kahn M, Liu Y. Multiple genes of the renin-angiotensin system are novel targets of Wnt/β-catenin signaling. J Am Soc Nephrol 26: 107–120, 2015. doi: 10.1681/ASN.2014010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1270.Zhou L, Liu Y. Wnt/β-catenin signaling and renin-angiotensin system in chronic kidney disease. Curr Opin Nephrol Hypertens 25: 100–106, 2016. doi: 10.1097/MNH.0000000000000205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1271.Zhou MS, Schulman IH, Zeng Q. Link between the renin-angiotensin system and insulin resistance: implications for cardiovascular disease. Vasc Med 17: 330–341, 2012. doi: 10.1177/1358863X12450094. [DOI] [PubMed] [Google Scholar]
- 1272.Zhou Q, Wei SS, Wang H, Wang Q, Li W, Li G, Hou JW, Chen XM, Chen J, Xu WP, Li YG, Wang YP. Crucial Role of ROCK2-Mediated Phosphorylation and Upregulation of FHOD3 in the Pathogenesis of Angiotensin II-Induced Cardiac Hypertrophy. Hypertension 69: 1070–1083, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08662. [DOI] [PubMed] [Google Scholar]
- 1273.Zhu N, Zhang D, Chen S, Liu X, Lin L, Huang X, Guo Z, Liu J, Wang Y, Yuan W, Qin Y. Endothelial enriched microRNAs regulate angiotensin II-induced endothelial inflammation and migration. Atherosclerosis 215: 286–293, 2011. doi: 10.1016/j.atherosclerosis.2010.12.024. [DOI] [PubMed] [Google Scholar]
- 1274.Zhu Q, Wang Z, Xia M, Li PL, Van Tassell BW, Abbate A, Dhaduk R, Li N. Silencing of hypoxia-inducible factor-1α gene attenuated angiotensin II-induced renal injury in Sprague-Dawley rats. Hypertension 58: 657–664, 2011. doi: 10.1161/HYPERTENSIONAHA.111.177626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1275.Zhu S, Zhang M, Davis JE, Wu WH, Surrao K, Wang H, Wu G. A single mutation in helix 8 enhances the angiotensin II type 1a receptor transport and signaling. Cell Signal 27: 2371–2379, 2015. doi: 10.1016/j.cellsig.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1276.Zilberberg L, Phoon CK, Robertson I, Dabovic B, Ramirez F, Rifkin DB. Genetic analysis of the contribution of LTBP-3 to thoracic aneurysm in Marfan syndrome. Proc Natl Acad Sci USA 112: 14012–14017, 2015. doi: 10.1073/pnas.1507652112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1277.Zimmerman AW, Veerkamp JH. New insights into the structure and function of fatty acid-binding proteins. Cell Mol Life Sci 59: 1096–1116, 2002. doi: 10.1007/s00018-002-8490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1278.Zimmerman CA, Lin YC, Leib DE, Guo L, Huey EL, Daly GE, Chen Y, Knight ZA. Thirst neurons anticipate the homeostatic consequences of eating and drinking. Nature 537: 680–684, 2016. doi: 10.1038/nature18950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1279.Zorad S, Dou JT, Benicky J, Hutanu D, Tybitanclova K, Zhou J, Saavedra JM. Long-term angiotensin II AT1 receptor inhibition produces adipose tissue hypotrophy accompanied by increased expression of adiponectin and PPARgamma. Eur J Pharmacol 552: 112–122, 2006. doi: 10.1016/j.ejphar.2006.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1280.Zou LX, Imig JD, von Thun AM, Hymel A, Ono H, Navar LG. Receptor-mediated intrarenal angiotensin II augmentation in angiotensin II-infused rats. Hypertension 28: 669–677, 1996. doi: 10.1161/01.HYP.28.4.669. [DOI] [PubMed] [Google Scholar]
- 1281.Zou T, Yang W, Hou Z, Yang J. Homocysteine enhances cell proliferation in vascular smooth muscle cells: role of p38 MAPK and p47phox. Acta Biochim Biophys Sin (Shanghai) 42: 908–915, 2010. doi: 10.1093/abbs/gmq102. [DOI] [PubMed] [Google Scholar]
- 1282.Zouein FA, Altara R, Chen Q, Lesnefsky EJ, Kurdi M, Booz GW. Pivotal Importance of STAT3 in Protecting the Heart from Acute and Chronic Stress: New Advancement and Unresolved Issues. Front Cardiovasc Med 2: 36, 2015. doi: 10.3389/fcvm.2015.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1283.Zouein FA, Zgheib C, Hamza S, Fuseler JW, Hall JE, Soljancic A, Lopez-Ruiz A, Kurdi M, Booz GW. Role of STAT3 in angiotensin II-induced hypertension and cardiac remodeling revealed by mice lacking STAT3 serine 727 phosphorylation. Hypertens Res 36: 496–503, 2013. doi: 10.1038/hr.2012.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1284.Zucker IH, Xiao L, Haack KK. The central renin-angiotensin system and sympathetic nerve activity in chronic heart failure. Clin Sci (Lond) 126: 695–706, 2014. doi: 10.1042/CS20130294. [DOI] [PMC free article] [PubMed] [Google Scholar]