

Abstract
Pericellular proteolysis provides a significant advantage to developing tumors through the ability to remodel the extracellular matrix, promote cell invasion and migration, and facilitate angiogenesis. Recent advances demonstrate that pericellular proteases can also communicate directly to cells by activation of a unique group of transmembrane G protein-coupled receptors (GPCRs) known as protease activated receptors (PARs). In this review we discuss the specific roles of one of four mammalian PARs, namely PAR-2, which is over-expressed in advanced stage tumors and is activated by trypsin-like serine proteases that are highly expressed or otherwise dysregulated in many cancers. We highlight recent insights into the ability of different protease agonists to bias PAR-2 signaling and the newly emerging evidence for an interplay between PAR-2 and membrane-anchored serine proteases, which may co-conspire to promote tumor progression and metastasis. Interfering with these pathways might provide unique opportunities for the development of new mechanism-based strategies for the treatment of advanced and metastatic cancers.
Keywords: membrane-anchored serine protease, TTSP, protease-activated receptor, biased signaling, cancer, metastasis
Introduction
G protein-coupled receptors (GPCRs) are a large family of cell surface receptors that react to extracellular molecules to activate internal signaling pathways, facilitating a wide range of physiological responses (1). Dysregulation of GPCR functions and their ligands are linked to tumorigenesis, angiogenesis and metastasis (2). A unique class of GPCRs, known as the protease activated receptors or PARs, sense and respond to active proteases in the cell microenvironment (3,4). Uniquely, the proteolytic nature of PAR activation is irreversible, distinct from many other GPCRs. The four PARs found in mammals are activated by various different protease agonists. PAR-1, PAR-3 and PAR-4 are main targets for the coagulation protease thrombin, orchestrating physiological responses to vascular injury, thrombosis and inflammation (5–9). PAR-2 on the other hand, is activated by trypsin, several trypsin-like serine proteases (3,10,11) and synthetic soluble PAR-2-activating peptides (12), signaling to various downstream pathways that modulate cell proliferation, migration and invasion, cytokine production, stimulation of angiogenesis and other functions promoting tumor development (2).
This review concerns the roles of PAR-2 and a network of membrane-anchored serine proteases in cancer. There are several excellent comprehensive reviews of PARs in cancer and other diseases (13–16), as well as reviews on membrane-anchored serine proteases in development, tissue homeostasis and tumor progression (17–21). Here we focus on recent evidence in support of an interplay between PAR-2 and membrane-anchored serine proteases in proximity on the tumor cell surface that could significantly modulate the local magnitude, duration, and nature of PAR-2 signaling, as well as restrict PAR-2 signaling to local membrane microdomains. Their overexpression and dysregulation in tumors have the potential to cooperate to promote aggressive disease through cell-surface interactions, integration of extracellular signals, and induction of intracellular signaling pathways.
Membrane-anchored serine proteases
Unlike trypsin and other secreted, soluble serine proteases, members of the family of membrane-anchored serine proteases are synthesized as catalytically inactive or near-inactive proenzymes (zymogens) that are converted into active serine proteases by proteolytic cleavage after an arginine or lysine amino acid residue that is positioned in a conserved activation motif within the catalytic domain (22). These proteases possess domains that tether the extracellular catalytic serine protease domain directly to the cell surface, allowing cleavage of cell surface and pericellular substrates (19,20,22–24) (Figure 1). The manner in which they are linked to the cell surface may be through type I or type II single pass transmembrane domains or linked via glycophosphatidylinositol (GPI)-anchors. The serine protease domains of these enzymes are structurally highly conserved and contain a triad of amino acids (serine, histidine, and aspartate) required for catalytic activity (25). Overexpression of many of the 20 human members of this family has been documented in many cancers, and several membrane-anchored serine proteases have been shown to promote experimental malignant transformation when aberrantly expressed in tumor cells or in in vivo tumor models (21,26). In this review, we will focus on those membrane-anchored serine proteases which have been identified to date to be associated with tumor biology and linked to the PAR-2 signaling axis; namely, matriptase, hepsin, prostasin, TMPRSS2, testisin, and the membrane-associated pathway triggered by tissue factor (TF), factor VIIa, and factor Xa (TF:FVIIa/FXa).
Figure 1. Activation of PAR-2 by membrane-anchored and secreted serine proteases and implications in cancer.
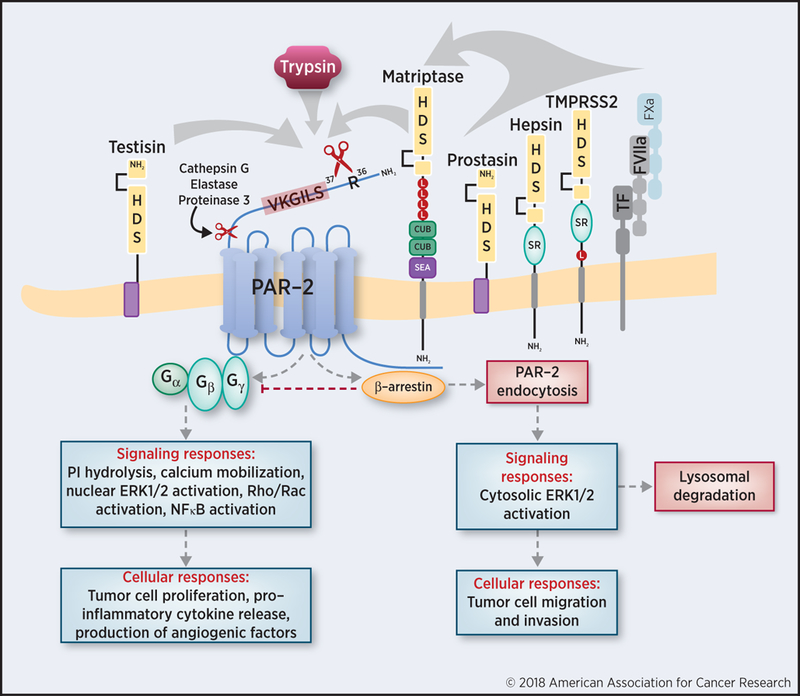
Human PAR-2 is cleaved by its various agonists on the cell surface at the canonical cleavage site, R36, revealing the S37LIGKV peptide sequence as a tethered ligand (in red text). Membrane-anchored serine proteases are illustrated with their conserved catalytic domains containing the serine (S), aspartate (D) and histidine (H) residues, their respective extracellular domains (low density lipoprotein (LDL) receptor class A domains (indicated by red circles labeled ‘L’), Cls/Clr, urchin embryonic growth factor and bone morphogenic protein 1 (CUB) domains, sea urchin sperm protein, enterokinase, agrin (SEA), and group A scavenger receptor (SR) domains), as well as their respective membrane-tethering regions. Testisin and matriptase cleave PAR-2 directly at the trypsin cleavage site, while prostasin, hepsin, TMPRSS2, and TF:FVIIa/Xa complex have been shown to activate matriptase and thus indirectly activate PAR-2. Upon proteolytic cleavage, PAR-2 can couple to various G proteins or once phosphorylated, bind to β-arrestin; both outcomes can activate subsequent signaling pathways and influencing tumor cell behavior. It is possible that various membrane-anchored serine proteases are capable of activating similar, overlapping, or distinct signaling responses to induce various cellular responses depending on the context.
PAR-2 signaling and cancer
In the majority of studies to date, PAR-2 has been reported to have oncogenic activities, functioning as a positive regulator of tumor growth and/or progression. Initial evidence that PAR-2 may drive tumorigenesis came from experimental studies showing that PAR-2 indirectly enhances thrombin-dependent tumor cell migration and metastasis (27). Increased PAR-2 expression has been reported in a diverse set of human cancers such as breast, ovarian, prostate and gastric cancer, when compared to normal patient tissue specimens (28–31). In addition, a recent survey of PAR family member expression in human tumor samples of various cancer types from the Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression projects (GTEx) reveals upregulated PAR-2 in 15 different cancer types compared with normal tissues (16). A global transcriptome array analysis of PAR expression in over 1,000 ovarian cancer and normal tissue samples showed that human epithelial ovarian cancers predominantly overexpress PAR-2, followed closely by PAR-1, with minimal detection of PAR3 and PAR4 (32). Consistent with this, increased PAR-2 is associated with poor prognosis and decreased progression-free and overall survival in ovarian, cervical, and hepatocellular carcinoma patients (30,33–35). Increased PAR-2 expression and activation is also correlated with the degree of invasiveness exhibited by both primary and metastatic tumors (29,30,36). Pro-tumorigenic activities attributed to PAR-2 signaling include chemokinesis, cell proliferation, invasion and migration, inflammatory signaling and increased angiogenesis (Figure 1) in several tumor types including breast, oral, renal, pancreatic, gastric, lung, and esophageal cancers (reviewed in (36–39)). PAR-2 may also modulate transactivation of other cell surface receptors (i.e. EGF, TGFβ, and Met tyrosine kinase receptors) that are frequently drivers of tumor progression (34,40–42). In contrast, a few studies have demonstrated tumor-suppressive functions of PAR-2 (43,44). In a DMBA-induced mouse model of skin carcinogenesis, PAR-2 deficient mice displayed increased tumor number and increased blood vessel infiltration, which was attributed to modulation of tumor-suppressing TGFβ-1 secretion (44). The specific protease agonists associated with the tumor suppressive and oncogenic roles of PAR-2 in various tumor types are poorly characterized.
Protease-stimulated PAR-2 activation and signaling
PARs are activated by a tethered ligand that is revealed by proteolytic cleavage of an N-terminal sequence and which can bind to an extracellular docking domain to cause a receptor conformational change that triggers intracellular signaling. There are several modes of regulation for PAR-2 activation and signaling. Different ligands can stabilize unique conformations of the cleaved PAR-2 that activate distinct signaling pathways, a phenomenon referred to as biased agonism or functional selectivity (45–47). A study of mutations within the tethered ligand sequence of PAR-2 (48) revealed that the nature of the tethered ligand sequence and the mode of its presentation to the receptor determine biased signaling by PAR-2. In addition, different proteases that cleave PAR-2 at distinct sites activate divergent patterns of receptor signaling and trafficking (reviewed in (42,47)). Signaling outcomes are diverse and can activate pathways leading to release of pro-inflammatory cytokines and angiogenic factors, increased cell motility and migration, and increased inflammatory responses (47,49) (Figure 1).
Activation of PAR-2 by trypsin and other soluble proteases has been most widely studied. Trypsin cleavage of PAR-2 involves hydrolysis at the canonical cleavage site R36↓S37, which reveals the tethered ligand SLIGKV (human) (50) or SLIGRL (mouse) (51). The exposed tethered ligand interacts with the second extracellular domain of the cleaved receptor and can trigger MAP kinase/ERK1/2 activation, calcium mobilization via Gαq activation, cAMP formation via Gαs activation, and Rho-kinase activity via Gα12/13 activation (47,49) (Figure 1). The binding of β-arrestin to phosphorylated residues on the PAR-2 C-terminal tail uncouples and terminates G protein signaling (52,53), results in endocytosis of the complex (52), and mediates early endosomal signaling via scaffolding complexes containing Raf1 and activated cytosolic ERK1/2 (54).
β-Arrestins are not only active participants in signaling by internalized PAR-2, they can also direct receptor trafficking to regulate the duration and magnitude of PAR-2 signaling. PAR-2 signal termination occurs by direct ubiquitination and trafficking of PAR-2 to lysosomes for degradation by distinct components of the ESCRT machinery, a process that is unique to PAR-2 within the PAR family (42,55,56). The pathways that regulate β-arrestin-mediated signaling versus signal termination are incompletely understood.
The specific signaling pathways activated by PAR-2 in the context of cancer in vivo have received limited attention and will likely depend on the protease activator(s) and the (patho)biological context. For example, in murine asthma models, disease-promoting PAR-2 pro-inflammatory signaling is dependent on β-arrestin-2, whereas G-protein-dependent signaling is beneficial (57,58). Several in vitro, in vivo, and human patient data suggest that dysregulation of β-arrestin expression, localization, and/or phosphorylation is associated with increased migration and invasion and ultimately poorer outcomes in various types of cancer (59,60). This may be attributed not only to direct tumorigenic signaling through β-arrestin but also other selective pathways of PAR-2 signaling. The contributions of various G-proteins and β-arrestin signaling downstream of membrane-anchored serine protease activation of PAR-2 in vivo are not yet well characterized.
PAR-2 activation by matriptase
Numerous in vitro and in vivo studies identify matriptase (encoded by ST14) to be a potent activator of PAR-2. Matriptase (Figure 1) is a type II single pass transmembrane serine protease with a unique extracellular stem region containing various conserved protein-binding domains (SEA, CUB, and LDLR-a repeats) which are involved in matriptase activation as well as interaction with its cognate inhibitor hepatocyte growth factor activator (HAI)-1 (61–66) and other proteins. Matriptase is expressed as a precursor or zymogen form that may be proteolytically processed first within the SEA domain, and then activated by further cleavage at a highly-conserved R↓VVGG motif by pericellular serine proteases or by autoactivation by matriptase itself (67). Interestingly, the zymogen form of matriptase, unusual among trypsin-like serine proteases, possesses measurable enzymatic activity and was recently shown to be capable of executing the in vivo developmental and homeostatic functions of the proteolytically-processed protease (68).
Matriptase is widely expressed in normal epithelial tissues where it plays a critical role in maintaining epithelial barrier homeostasis (69–72). Matriptase was first discovered in breast cancer cell lines (73) and its expression is associated with breast cancer progression (74,75). Matriptase is also upregulated in many other tumors of epithelial origin, namely prostate, cervical, gastric, esophageal, renal cell, skin, oral squamous cell, ovarian and cervical carcinomas (reviewed in (76,77)). Up-regulation of matriptase expression in many of these cancers is associated with poor outcomes (reviewed in (21)).
Substantial molecular and cellular data identifies matriptase to be a direct proteolytic activator of PAR-2. Early studies using the human HaCaT transformed keratinocyte cell line, which endogenously expresses PAR-2, showed that treatment with soluble recombinant matriptase (protease domain only) stimulates canonical PAR-2 cleavage (R36↓S37) and potent PI hydrolysis (78). In KOLF cells (which do not express endogenous PARs, matriptase, or hepsin), PI hydrolysis in response to recombinant matriptase was observed only upon PAR-2 transfection, indicating direct and specific PAR-2 activation (78). These findings have since been confirmed in several other in vitro studies using PAR-2 expressing KOLF or HEK293 cells and soluble or co-expressed matriptase (68,79–82).
In vivo studies using murine transgenic models of matriptase and PAR-2 deficiencies have provided compelling evidence for a matriptase-PAR-2 signaling axis, specifically during normal embryonic development and placental barrier function (78,83). Is it possible that a matriptase-PAR-2 signaling pathway regulates global epithelial integrity during homeostasis, and that this pathway becomes dysregulated in cancer? In support of this, PAR-2 has been shown to be critical for matriptase-mediated tumor progression using several in vivo tumor models in which matriptase is over-expressed. In a transgenic mouse model of squamous cell carcinoma (SCC) where matriptase is over-expressed in the epidermis via a keratin-5 promoter (K5-matriptase), animals developed spontaneous multistage SCCs (84) and displayed pro-tumorigenic inflammatory cytokine release that was PAR-2 dependent (81). The downstream effects of matriptase activation of PAR-2 were attributed to selective signaling through Gαi and NFκB directed cytokine release (81). In this model, matriptase also induced the activation of a HGF/c-Met dependent pathway, and both c-Met and PAR-2 signaling were independently required for tumor initiation (81).
Matriptase activity seems to be critical for the regulation of inflammatory signaling via the matriptase-PAR-2 axis. Matriptase expression and trafficking, activity and shedding are controlled by Kunitz-type serine protease inhibitors, specifically HAI-1/SPINT1 and HAI-2/SPINT2, and down-regulation of these endogenous matriptase inhibitors increases aberrant matriptase activity (62,85–88). Interestingly, the specific HAI required for proper matriptase trafficking is cell type dependent (reviewed in (89)). The ratio of matriptase to its inhibitors, or the protease-inhibitor balance, is important: loss of or decreased endogenous HAI-1 or HAI-2 enables increased matriptase activity and this has been shown to promote in vitro tumorigenesis in several studies (90–96). In human SCCs, increased matriptase expression is correlated with diminished expression of matriptase-HAI-1 complexes and with reduced PAR-2 expression (97), possibly due to PAR-2 over-activation induced by deregulated matriptase activity. The importance of protease-inhibitor balance has also been demonstrated in vivo in murine transgenic and xenograft models, where loss or decreased levels of endogenous HAI-1 or HAI-2 and increased matriptase activity promotes carcinogenesis, which can be effectively reversed by increased expression the inhibitor (84,98,99). Recent studies also suggest that matriptase can function in a paracrine manner to activate PAR-2 (100). Pericellular matriptase activity on the surface of oral SCC, caused by insufficient HAI-1, was shown to activate PAR-2 on the surface of cancer-associated fibroblasts (CAF), leading to enhanced CAF migration and infiltration (100).
In human colon cancer, matriptase was originally designated as a tumor suppressor gene due to its loss of heterozygosity (101). Consistent with this, mice with tissue specific deletion of matriptase in the GI tract show increased intestinal permeability, spontaneously develop chronic colitis and ultimately inflammation-induced colon cancer (102). These data suggest that complete loss of matriptase can lead to colon carcinogenesis in the context of inflammation. Although matriptase expression may be down-regulated in human colon cancer, additional studies using a specific antibody (A11) that targets active matriptase show that there is increased active matriptase in human colon cancer tissues and in a patient derived colon cancer xenograft model (92,103). The presence of increased active matriptase in human colon cancer is also supported by studies on the ratio of matriptase:HAI-1 levels, which suggest that while both matriptase and HAI-1 are down-regulated during carcinogenesis, the matriptase:HAI-1 ratio increases during cancer progression, resulting in a population of active matriptase on the cell surface (90,92). In support of this, the presence of HAI-1 in intestinal epithelium was shown to be protective in two murine models of intestinal carcinogenesis (104). Whether these sequelae are related to matriptase mediated-PAR-2 signaling is an unexplored area.
Activation of the matriptase-PAR-2 signaling axis
Several other members of the membrane-anchored serine protease family were originally reported to functionally activate PAR-2 signaling, namely prostasin (encoded by PRSS8), hepsin (encoded by HPN), and TMPRSS2 (encoded by TMPRSS2). Recent studies indicate that these proteases indirectly trigger PAR-2 activation through the matriptase-PAR-2 axis (Figure 1). Hepsin and TMPRSS2 are type-II transmembrane serine proteases (18), while prostasin is anchored to the plasma membrane via a GPI anchor (105).
Hepsin was shown to indirectly activate PAR-2 and trigger PI hydrolysis only in the presence of matriptase in HaCaT cells. This was attenuated by a specific matriptase blocking antibody, suggesting that hepsin is capable of activating the matriptase zymogen which can then activate PAR-2 (78). In the same study, PAR-2 activation induced by recombinant prostasin was only observed in cells that also expressed catalytically active matriptase, indicating that prostasin is capable of functioning as an indirect activator of PAR-2 signaling via activation of the matriptase zymogen (78). TMPRSS2 was originally thought to activate PAR-2 directly, resulting in calcium mobilization in prostate cancer cell lines (106). In a later study, stable overexpression of TMPRSS2 in a variety of prostate cancer cell lines was shown to induce matriptase activation and to increase the metastasis of orthotopic xenografts (107). These results identify matriptase as a possible substrate of TMPRSS2 and indicate that like hepsin, TMPRSS2 activates the matriptase-PAR-2 axis.
The functional interactions between prostasin, matriptase, and PAR-2 activation have been most extensively-studied, however these interactions have proved to be complex and are still incompletely understood. In normal tissues, matriptase and prostasin are ubiquitously co-expressed in epithelial cells, while during the progression of multi-stage epithelial carcinogenesis, they are found to be expressed in separate tumor cell compartments, possibly indicating altered regulation or activation requirements during tumor progression (108). Results from several studies highlight the importance of tissue distribution and subcellular localization for the function and regulation of these two membrane anchored proteases in other disease contexts (108–111). It is possible that interactions between their extracellular domains as well as tightly regulated membrane distribution and subcellular localization, all contribute to regulating the activation of the matriptase-PAR-2 axis as well. When ectopically expressed in the skin of transgenic mice, prostasin was shown to induce epidermal hyperplasia, ichthyosis and inflammation, phenotypes which are completely negated when superimposed on a PAR-2-null background, establishing PAR-2 as a pivotal downstream mediator of prostasin inflammatory activity (112). Matriptase may be involved in this activity, since matriptase and prostasin are found to be capable of forming a reciprocal zymogen activation complex stimulating the activation of the zymogen forms of each other (80). The matriptase zymogen, which has a low rate of catalytic activity, has been shown to be capable of activating prostasin (80). Utilizing detailed cell-based analyses and genetically modified animals, Friis et al (68) recently demonstrated that the matriptase zymogen can induce PAR-2 activation in the presence of prostasin, and that this activity requires catalytically active and membrane-anchored prostasin. This finding may indicate that matriptase zymogen-activated prostasin can execute the activation site cleavage of PAR-2 directly or that the intrinsic catalytic activity of the matriptase zymogen is stimulated effectively by catalytically active prostasin (68).
Hepsin, prostasin, and TMPRSS2 up-regulation in epithelial breast, prostate and ovarian cancer cell lines, mouse models, and patient samples are believed to contribute to increased proliferation, tumor growth, metastasis, ascites formation, and various other invasive processes (107,113–125). Overexpression of hepsin in prostate epithelium in a prostate tumor model (LPB-Tag mice) resulted in increased basement membrane disorganization and tumor metastasis to distant organs which did not occur in control mice, indicating that hepsin is capable of promoting prostate cancer metastatic processes (119). Expression of TMPRSS2 is also associated with prostate cancer progression, as knockdown of TMPRSS2 in prostate cancer cell lines results in decreased invasion, tumor size, and incidence in xenograft models (107). In the TRansgenic Adenocarcinoma Mouse Prostate (TRAMP) model of prostate cancer, mice with TMPRSS2 deficiency exhibited larger tumors but a lower incidence of distant metastasis (125). In contrast, prostasin, like matriptase, is reported to function as a tumor suppressor in colon cancer, with reduced expression correlating with more aggressive clinical stages and shorter patient survival time (114,116). In many of these cancer contexts, it is not yet known whether the tumorigenic processes attributed to protease expression or activity occur via PAR-2 activation.
In addition to these membrane-anchored serine proteases, the membrane-localized coagulation complex containing TF:FVIIa/FXa was also originally reported to activate PAR-2 directly (126), but was later shown to trigger the activation of matriptase zymogen, which mediates the cleavage and activation of PAR-2 (82) (Figure 1). In experimental studies using the spontaneous mammary tumor virus (MMTV) promoter-driven model of breast cancer in mice, the PAR-2 deficient phenotype was similar to that of mice with a truncated cytoplasmic domain of TF, suggesting an interplay between TF cytoplasmic domain signaling and PAR-2 in promoting breast cancer progression (127). Interestingly, in this study, PAR-2 deficiency led to a significant delay in the transition from adenomas to invasive carcinoma and was associated with less tumor vascularization and reduced immune cell infiltration. Reconstitution of the PAR-2-deficient tumor cells with PAR-2 mutated at the β-arrestin binding site restored proangiogenic chemokine induction, tumor growth and increased vessel density (127), demonstrating that the tumor promoting activities of PAR-2 activation in this model were due to G-protein, rather than β-arrestin signaling. The involvement of matriptase in this tumor-associated TF-PAR-2 signaling activity is not known.
Testisin
Testisin (encoded by PRSS21) is a GPI-anchored membrane serine protease (128–130), that has been found to induce PAR-2 activation (Figure 1). Exposure of PAR-2 over-expressing HeLa (human cervical carcinoma) cells to soluble recombinant testisin results in potent intracellular calcium mobilization, ERK1/2 phosphorylation, and NFκB activation (131). Further, co-expression of testisin and PAR-2 results in inflammatory cytokine (IL-8, IL-6) induction and a decrease in PAR-2 surface expression via receptor internalization (131). The data indicate that testisin is capable of PAR-2 cleavage and activation affecting signaling responses important for tumor cell motility, proliferation, and inflammation. The potential involvement of matriptase or the matriptase zymogen in testisin-mediated PAR-2 activation is not known. Interestingly, testisin shows very limited normal tissue distribution, but is overexpressed in human epithelial ovarian, cervical and lung carcinomas (132–134). Aberrant overexpression of testisin in epithelial ovarian tumor cells was shown to promote malignant transformation, increase tumor growth, and tumor formation in subcutaneous xenograft models (129). The involvement of PAR-2 activation in the in vivo tumor phenotypes is not known.
Other mechanisms of PAR-2 activation
Non-canonical PAR-2 cleavage.
Cleavage of the N-terminal PAR-2 sequence by membrane-anchored serine proteases at a non-canonical cleavage site has not yet been reported. However, several soluble secreted proteases alter PAR-2 signaling responses via cleavage at non-canonical sites. Cleavage of PAR-2 at residues C-terminal to the tethered ligand domain sequence, has the effect of removing the tethered ligand sequence and effectively ‘disarming’ PAR-2 to prevent its activation by trypsin-like proteases and membrane-anchored serine proteases (Figure 1). The neutrophil proteases cathepsin G and proteinase 3 cleave PAR-2 at F65↓S66 and V62↓D63 respectively, silencing trypsin-induced calcium mobilization and MAPK signaling, presumably by removing the tethered ligand sequence (135). Neutrophil elastase cleaves PAR-2 at S68↓V69, which, unlike trypsin, does not induce calcium mobilization via Gαq activation, recruitment of β−arrestin or receptor internalization (135). On the other hand, neutrophil elastase does activate MAPK signaling independent of the tethered ligand binding, suggesting that it instead stabilizes a unique receptor conformation that favors a distinct signaling profile (135). Thus in a protease rich environment, non-canonical PAR-2 cleavage adds another level of complexity in PAR-2 signaling that would be expected to impact the activities of membrane anchored serine proteases in the context of cancer. Whether non-canonical protease activators significantly impact biased signaling that influences tumor cell behavior is an interesting area for future study.
Activation of PAR-2 by PAR-1.
O’Brien et al. (136) were the first to show that the PAR-1 tethered ligand domain is capable of transactivating PAR-2, indicating a possible role for transactivation of PAR-2 induced by protease-mediated cleavage of PAR-1. Thrombin- and PAR-1-dependent migration of melanoma and prostate cancer cells was found to be dependent on indirect activation of PAR-2. This effect of thrombin on migration and chemokinesis required PAR-1 activation and transactivation of PAR-2 by the PAR-1 tethered ligand, independent of PAR-2 cleavage (27). In vivo, PAR-1 transactivation of PAR-2 was shown to contribute to altered responses in late stages of sepsis (137). This mechanism was further characterized and attributed to the formation of PAR-1-PAR-2 heterodimers. Thrombin was shown to cleave PAR-1 at its canonical site to reveal its tethered ligand, which can also bind to and activate PAR-2 (136). A PAR-1 cleavable but nonsignaling variant, with a mutation in the second extracellular ligand binding domain, was shown to donate its cleaved tethered ligand to wildtype PAR-2, which triggered thrombin-mediated signaling in COS-7 cells. This suggests that transactivation of PAR-2 by the PAR-1 tethered ligand facilitates PAR-1-associated signaling when present as a heterodimer (136). PAR-1-PAR-2 transactivation can furthermore recruit β-arrestin, co-internalize, and activate nuclear ERK1/2 signaling, all of which are different from the trafficking and signaling in response to PAR-1 activation alone (138). The impact and mechanisms by which PAR-1-PAR-2 heterodimers influence pro-tumorigenic signaling is an understudied area but provides yet another pathway of biased PAR-2 signaling in response to indirect activation by thrombin, and perhaps other PAR-1 activating proteases, such as the cancer associated protease plasmin and the coagulation protease FXa. To date, no membrane-anchored serine proteases are known to activate PAR-1 directly, but how the co-expression of PAR-2 activating membrane-anchored serine proteases may influence PAR-1-PAR-2 transactivation in these contexts has not been investigated.
Implications and future directions
While considerable progress has been made in our understanding of mechanisms by which proteases and synthetic agonists activate PARs, there is much to learn about the interactions between membrane anchored and soluble proteases, and specific protease-receptor interactions that influence PAR-2 signaling. This is important since PAR-2 is a signaling receptor with established links to cancer progression and metastasis, and whose protease-dependent activation can signal substantial changes in cell behavior. The emerging data implicates PAR-2 and membrane-anchored serine proteases in proximity on the tumor cell surface as co-conspirators in regulating the nature of PAR-2 signaling responses and as determinants of biased signaling in cancer.
Membrane anchored serine proteases may impact PAR-2 signaling bias in multiple and divergent ways. Biased PAR-2 signaling has mostly been studied with regard to cleavage at different sites by various protease agonists or antagonists. However while membrane-anchored serine protease activators of PAR-2 identified to date cleave PAR-2 at the trypsin canonical site, other mechanisms that modulate differential PAR-2 activities are likely. The extracellular domains of membrane-anchored serine proteases (Figure 1) offer unique opportunities for allosteric modulation of PAR-2, potentially by inducing conformational changes that can impart functional selectivity. It is also possible that cell surface and membrane localization may limit signaling responses to specific areas or local microdomains, whereas soluble proteases that cleave and activate PAR-2, such as trypsin, mast cell tryptase, kallikreins and gingipains, would induce transient activation independent of surface location. In nontransformed cells, matriptase is localized to cell-cell junctions and basolateral surfaces of polarized epithelium (139), prostasin is found on apical membranes (111) and hepsin is associated with desmosomes (121), while testisin is found in lipid rafts (130). The influence of these protease specific localizations on PAR-2 activation and signaling responses is currently unknown. Further, these distributions may be expected to significantly impact and be impacted by epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions associated with tumor progression and metastasis.
PAR-2 present on tumor cells could be considered to act as a ‘protease sensor’ of the tumor cell microenvironment, responding to circulating protease agonists that unmask the tethered ligand as well as circulating proteases that act indirectly as antagonists by ‘disarming’ the receptor. Since membrane-anchored serine proteases are overexpressed and their inhibitors are down-regulated in many cancers, these over-active proteases in direct contact with PAR-2 molecules on the cell membrane are poised to induce sustained signaling responses via continual surveillance of the cell membrane, activating PAR-2 present on the surface as well as newly synthesized receptors that repopulate the cell membrane in their vicinity. Such activities would not only affect the type, magnitude and duration of signaling responses, but would also be expected to modulate canonical activation by soluble proteases in the cell environment. A lack of protease-specific inhibitors, activity assays and other tools confounds studies aimed at teasing out these mechanistic details. This is further complicated by proteolytic cleavage or ‘shedding’ of protease domains from several of the membrane anchored serine proteases, potentially releasing protease activity in a soluble form. The emerging evidence for membrane serine protease zymogens with functional activities adds another level of complexity. Undoubtedly, it will be a challenge to understand the factors that determine which proteases cleave PAR-2 in a protease-rich environment, which may be disease or cell-type dependent and will also depend on the local milieu of protease inhibitors present. Membrane-anchored serine protease-mediated allosteric modulation of PAR-2 signaling has not yet been investigated, and could potentially facilitate tumor suppressive or oncogenic activities depending on the tumor cell context.
One approach to interfering with the pro-tumorigenic activities of membrane anchored serine proteases is the development of specific inhibitors. This strategy has had limited success, likely because there is redundancy and/or overlap in the catalytic specificities and substrates in physiological and pathological contexts. An early approach to the development of inhibitors that showed promise was the targeting of selective conformational changes required for protease activity. The human recombinant antibody termed A11, developed against a protease surface loop of matriptase that is less conserved amongst trypsin-fold proteases, was shown to selectively recognize the active form of matriptase over the zymogen form and is a specific inhibitor of its activity (103,140). Other strategies to inhibit matriptase activity include treatment with soluble recombinant HAI-1, synthetic small molecules, peptides, and monoclonal antibodies, all of which have been shown to inhibit matriptase to varying degrees in vitro but do not address therapeutic limitations of specificity and optimal serum half-lives in vivo (141–144). A recent approach for inhibition of matriptase consists of an engineered variant of natural HAI-1, utilizing a Kunitz-domain 1/Kunitz-domain 2 chimera to replace the less-specific Kunitz-domain 2 of HAI-1, fused to an antibody Fc domain to increase putative binding sites to matriptase (144). This fusion protein was shown to inhibit matriptase activation of pro-HGF and matriptase activity on the surface of cancer cells (144). The activities of membrane anchored serine proteases have also been exploited as functional biomarkers for imaging tumorigenesis. A11 has been used to visualize aberrant matriptase activity in epithelial tumors in vivo (92,140), demonstrating a potential application in non-invasive tumor imaging and monitoring of disease progression. Fluorescent nanoparticles targeted against hepsin allosteric binding peptides have also been shown to bind specifically to hepsin-expressing LNCaP xenografts (145). Such reagents hold promise for the diagnostic detection and experimental manipulation of protease activities that contribute to disease progression, with potential clinical applications.
An alternative strategy is to potentially manipulate the functional selectivity of PAR-2 to develop novel cancer therapies. In the broader context of GPCR signaling, therapeutic strategies are now being aimed at shifting the signal bias in the appropriate direction to mitigate disease progression (146,147). It is likely that membrane anchored serine proteases in proximity to PAR-2 will be a critical consideration in the development of such therapies. Many attempts to develop PAR-2 biased agonists (148) and antagonists (15,37) have been modeled on analogues of the PAR-2 activating peptide, some of which are capable of preferentially modulating Ca2+ or ERK1/2 signaling (148). Other small molecules and antibodies are being developed to target binding pockets and transmembrane regions, as well as allosteric sites on PAR-2, in an effort to prevent conformational changes required for receptor activation upon proteolytic cleavage or to prevent tethered ligand binding to the peptide binding site (149–151).
Several hallmarks of aggressive cancer are a direct result of proteolytic activity, including tumor cell invasion into the stroma, angiogenesis and metastasis (152), with the roles of proteases traditionally focused on protein degradation and extracellular matrix remodeling. Advances in our understanding of membrane-anchored serine proteases as modulators of PAR-2 activation and signaling are anticipated to uncover novel avenues for pharmaceutical intervention that could be used to selectively tune receptor activity for the treatment of cancer progression.
Acknowledgments
Financial Support: This work was supported by NIH Grants R01 CA196988 and R01 HL118390.
Footnotes
Potential Conflicts of Interest: none
REFERENCES
- 1.O’Hayre M, Degese MS, Gutkind JS. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr Opin Cell Biol 2014;27:126–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer 2007;7:79–94 [DOI] [PubMed] [Google Scholar]
- 3.Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R. Proteinase-activated receptors (PARs) - focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun Signal 2013;11:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, et al. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther 2011;130:248–82 [DOI] [PubMed] [Google Scholar]
- 5.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature 2000;407:258–64 [DOI] [PubMed] [Google Scholar]
- 6.O’Brien PJ, Molino M, Kahn M, Brass LF. Protease activated receptors: theme and variations. Oncogene 2001;20:1570–81 [DOI] [PubMed] [Google Scholar]
- 7.Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev 2004;84:579–621 [DOI] [PubMed] [Google Scholar]
- 8.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost 2005;3:1800–14 [DOI] [PubMed] [Google Scholar]
- 9.Ramachandran R, Hollenberg MD. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol 2008;153 Suppl 1:S263–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coughlin SR, Camerer E. PARticipation in inflammation. J Clin Invest 2003;111:25–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cottrell GS, Amadesi S, Schmidlin F, Bunnett N. Protease-activated receptor 2: activation, signalling and function. Biochem Soc Trans 2003;31:1191–7 [DOI] [PubMed] [Google Scholar]
- 12.Maryanoff BE, Santulli RJ, McComsey DF, Hoekstra WJ, Hoey K, Smith CE, et al. Protease-activated receptor-2 (PAR-2): structure-function study of receptor activation by diverse peptides related to tethered-ligand epitopes. Arch Biochem Biophys 2001;386:195–204 [DOI] [PubMed] [Google Scholar]
- 13.Arora P, Ricks TK, Trejo J. Protease-activated receptor signalling, endocytic sorting and dysregulation in cancer. J Cell Sci 2007;120:921–8 [DOI] [PubMed] [Google Scholar]
- 14.Schaffner F, Ruf W. Tissue factor and PAR2 signaling in the tumor microenvironment. Arterioscler Thromb Vasc Biol 2009;29:1999–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramachandran R, Noorbakhsh F, Defea K, Hollenberg MD. Targeting proteinase-activated receptors: therapeutic potential and challenges. Nat Rev Drug Discov 2012;11:69–86 [DOI] [PubMed] [Google Scholar]
- 16.Arakaki AKS, Pan WA, Trejo J. GPCRs in Cancer: Protease-Activated Receptors, Endocytic Adaptors and Signaling. Int J Mol Sci 2018;19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Netzel-Arnett S, Hooper JD, Szabo R, Madison EL, Quigley JP, Bugge TH, et al. Membrane anchored serine proteases: a rapidly expanding group of cell surface proteolytic enzymes with potential roles in cancer. Cancer Metastasis Rev 2003;22:237–58 [DOI] [PubMed] [Google Scholar]
- 18.Bugge TH, Antalis TM, Wu Q. Type II transmembrane serine proteases. J Biol Chem 2009;284:23177–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antalis TM, Bugge TH, Wu Q. Membrane-anchored serine proteases in health and disease. Prog Mol Biol Transl Sci 2011;99:1–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szabo R, Bugge TH. Membrane-anchored serine proteases in vertebrate cell and developmental biology. Annu Rev Cell Dev Biol 2011;27:213–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanabe LM, List K. The role of type II transmembrane serine protease-mediated signaling in cancer. FEBS J 2017;284:1421–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antalis TM, Buzza MS, Hodge KM, Hooper JD, Netzel-Arnett S. The cutting edge: membrane-anchored serine protease activities in the pericellular microenvironment. Biochem J 2010;428:325–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antalis TM, Conway GD, Peroutka RJ, Buzza MS. Membrane-anchored proteases in endothelial cell biology. Curr Opin Hematol 2016;23:243–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hooper JD, Clements JA, Quigley JP, Antalis TM. Type II transmembrane serine proteases. Insights into an emerging class of cell surface proteolytic enzymes. J Biol Chem 2001;276:857–60 [DOI] [PubMed] [Google Scholar]
- 25.Hedstrom L Serine protease mechanism and specificity. Chem Rev 2002;102:4501–24 [DOI] [PubMed] [Google Scholar]
- 26.Murray AS, Varela FA, List K. Type II transmembrane serine proteases as potential targets for cancer therapy. Biol Chem 2016;397:815–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi X, Gangadharan B, Brass LF, Ruf W, Mueller BM. Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Mol Cancer Res 2004;2:395–402 [PubMed] [Google Scholar]
- 28.Caruso R, Pallone F, Fina D, Gioia V, Peluso I, Caprioli F, et al. Protease-activated receptor-2 activation in gastric cancer cells promotes epidermal growth factor receptor trans-activation and proliferation. Am J Pathol 2006;169:268–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Black PC, Mize GJ, Karlin P, Greenberg DL, Hawley SJ, True LD, et al. Overexpression of protease-activated receptors-1,−2, and-4 (PAR-1, −2, and −4) in prostate cancer. Prostate 2007;67:743–56 [DOI] [PubMed] [Google Scholar]
- 30.Jahan I, Fujimoto J, Alam SM, Sato E, Sakaguchi H, Tamaya T. Role of protease activated receptor-2 in tumor advancement of ovarian cancers. Ann Oncol 2007;18:1506–12 [DOI] [PubMed] [Google Scholar]
- 31.Su S, Li Y, Luo Y, Sheng Y, Su Y, Padia RN, et al. Proteinase-activated receptor 2 expression in breast cancer and its role in breast cancer cell migration. Oncogene 2009;28:3047–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chanakira A, Westmark PR, Ong IM, Sheehan JP. Tissue factor-factor VIIa complex triggers protease activated receptor 2-dependent growth factor release and migration in ovarian cancer. Gynecol Oncol 2017;145:167–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aman M, Ohishi Y, Imamura H, Shinozaki T, Yasutake N, Kato K, et al. Expression of protease-activated receptor-2 (PAR-2) is related to advanced clinical stage and adverse prognosis in ovarian clear cell carcinoma. Hum Pathol 2017;64:156–63 [DOI] [PubMed] [Google Scholar]
- 34.Hugo de Almeida V, Guimaraes IDS, Almendra LR, Rondon AMR, Tilli TM, de Melo AC, et al. Positive crosstalk between EGFR and the TF-PAR2 pathway mediates resistance to cisplatin and poor survival in cervical cancer. Oncotarget 2018;9:30594–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun L, Li PB, Yao YF, Xiu AY, Peng Z, Bai YH, et al. Proteinase-activated receptor 2 promotes tumor cell proliferation and metastasis by inducing epithelial-mesenchymal transition and predicts poor prognosis in hepatocellular carcinoma. World J Gastroenterol 2018;24:1120–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wojtukiewicz MZ, Hempel D, Sierko E, Tucker SC, Honn KV. Protease-activated receptors (PARs)--biology and role in cancer invasion and metastasis. Cancer Metastasis Rev 2015;34:775–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ungefroren H, Witte D, Fiedler C, Gadeken T, Kaufmann R, Lehnert H, et al. The Role of PAR2 in TGF-beta1-Induced ERK Activation and Cell Motility. Int J Mol Sci 2017;18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kularathna PK, Pagel CN, Mackie EJ. Tumour progression and cancer-induced pain: a role for protease-activated receptor-2? Int J Biochem Cell Biol 2014;57:149–56 [DOI] [PubMed] [Google Scholar]
- 39.Sedda S, Marafini I, Caruso R, Pallone F, Monteleone G. Proteinase activated-receptors-associated signaling in the control of gastric cancer. World J Gastroenterol 2014;20:11977–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaufmann R, Oettel C, Horn A, Halbhuber KJ, Eitner A, Krieg R, et al. Met receptor tyrosine kinase transactivation is involved in proteinase-activated receptor-2-mediated hepatocellular carcinoma cell invasion. Carcinogenesis 2009;30:1487–96 [DOI] [PubMed] [Google Scholar]
- 41.Chung H, Ramachandran R, Hollenberg MD, Muruve DA. Proteinase-activated receptor-2 transactivation of epidermal growth factor receptor and transforming growth factor-beta receptor signaling pathways contributes to renal fibrosis. J Biol Chem 2013;288:37319–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soh UJ, Dores MR, Chen B, Trejo J. Signal transduction by protease-activated receptors. Br J Pharmacol 2010;160:191–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaufmann R, Schafberg H, Nowak G. Proteinase-activated receptor-2-mediated signaling and inhibition of DNA synthesis in human pancreatic cancer cells. Int J Pancreatol 1998;24:97–102 [DOI] [PubMed] [Google Scholar]
- 44.Rattenholl A, Seeliger S, Buddenkotte J, Schon M, Schon MP, Stander S, et al. Proteinase-activated receptor-2 (PAR2): a tumor suppressor in skin carcinogenesis. J Invest Dermatol 2007;127:2245–52 [DOI] [PubMed] [Google Scholar]
- 45.Galandrin S, Oligny-Longpre G, Bouvier M. The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci 2007;28:423–30 [DOI] [PubMed] [Google Scholar]
- 46.Hollenberg MD, Mihara K, Polley D, Suen JY, Han A, Fairlie DP, et al. Biased signalling and proteinase-activated receptors (PARs): targeting inflammatory disease. Br J Pharmacol 2014;171:1180–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao P, Metcalf M, Bunnett NW. Biased signaling of protease-activated receptors. Front Endocrinol (Lausanne) 2014;5:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, et al. Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol Pharmacol 2009;76:791–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rothmeier AS, Ruf W. Protease-activated receptor 2 signaling in inflammation. Semin Immunopathol 2012;34:133–49 [DOI] [PubMed] [Google Scholar]
- 50.Nystedt S, Emilsson K, Larsson AK, Strombeck B, Sundelin J. Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur J Biochem 1995;232:84–9 [DOI] [PubMed] [Google Scholar]
- 51.Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci U S A 1994;91:9208–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ricks TK, Trejo J. Phosphorylation of protease-activated receptor-2 differentially regulates desensitization and internalization. J Biol Chem 2009;284:34444–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jung SR, Seo JB, Deng Y, Asbury CL, Hille B, Koh DS. Contributions of protein kinases and beta-arrestin to termination of protease-activated receptor 2 signaling. J Gen Physiol 2016;147:255–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DeFea KA, Zalevsky J, Thoma MS, Dery O, Mullins RD, Bunnett NW. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol 2000;148:1267–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacob C, Cottrell GS, Gehringer D, Schmidlin F, Grady EF, Bunnett NW. c-Cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J Biol Chem 2005;280:16076–87 [DOI] [PubMed] [Google Scholar]
- 56.Hasdemir B, Murphy JE, Cottrell GS, Bunnett NW. Endosomal deubiquitinating enzymes control ubiquitination and down-regulation of protease-activated receptor 2. J Biol Chem 2009;284:28453–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nichols HL, Saffeddine M, Theriot BS, Hegde A, Polley D, El-Mays T, et al. beta-Arrestin-2 mediates the proinflammatory effects of proteinase-activated receptor-2 in the airway. Proc Natl Acad Sci U S A 2012;109:16660–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walker JK, DeFea KA. Role for beta-arrestin in mediating paradoxical beta2AR and PAR2 signaling in asthma. Curr Opin Pharmacol 2014;16:142–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sobolesky PM, Moussa O. The role of beta-arrestins in cancer. Prog Mol Biol Transl Sci 2013;118:395–411 [DOI] [PubMed] [Google Scholar]
- 60.Song Q, Ji Q, Li Q. The role and mechanism of betaarrestins in cancer invasion and metastasis (Review). Int J Mol Med 2018;41:631–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takeuchi T, Shuman MA, Craik CS. Reverse biochemistry: use of macromolecular protease inhibitors to dissect complex biological processes and identify a membrane-type serine protease in epithelial cancer and normal tissue. Proc Natl Acad Sci U S A 1999;96:11054–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oberst MD, Williams CA, Dickson RB, Johnson MD, Lin CY. The activation of matriptase requires its noncatalytic domains, serine protease domain, and its cognate inhibitor. J Biol Chem 2003;278:26773–9 [DOI] [PubMed] [Google Scholar]
- 63.Lee MS, Tseng IC, Wang Y, Kiyomiya K, Johnson MD, Dickson RB, et al. Autoactivation of matriptase in vitro: requirement for biomembrane and LDL receptor domain. Am J Physiol Cell Physiol 2007;293:C95–105 [DOI] [PubMed] [Google Scholar]
- 64.Kojima K, Tsuzuki S, Fushiki T, Inouye K. Roles of functional and structural domains of hepatocyte growth factor activator inhibitor type 1 in the inhibition of matriptase. J Biol Chem 2008;283:2478–87 [DOI] [PubMed] [Google Scholar]
- 65.Kojima K, Tsuzuki S, Fushiki T, Inouye K. Role of the stem domain of matriptase in the interaction with its physiological inhibitor, hepatocyte growth factor activator inhibitor type I. J Biochem 2009;145:783–90 [DOI] [PubMed] [Google Scholar]
- 66.Inouye K, Tomoishi M, Yasumoto M, Miyake Y, Kojima K, Tsuzuki S, et al. Roles of CUB and LDL receptor class A domain repeats of a transmembrane serine protease matriptase in its zymogen activation. J Biochem 2013;153:51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin CY, Tseng IC, Chou FP, Su SF, Chen YW, Johnson MD, et al. Zymogen activation, inhibition, and ectodomain shedding of matriptase. Front Biosci 2008;13:621–35 [DOI] [PubMed] [Google Scholar]
- 68.Friis S, Tadeo D, Le-Gall SM, Jurgensen HJ, Sales KU, Camerer E, et al. Matriptase zymogen supports epithelial development, homeostasis and regeneration. BMC Biol 2017;15:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.List K, Haudenschild CC, Szabo R, Chen W, Wahl SM, Swaim W, et al. Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene 2002;21:3765–79 [DOI] [PubMed] [Google Scholar]
- 70.List K, Kosa P, Szabo R, Bey AL, Wang CB, Molinolo A, et al. Epithelial integrity is maintained by a matriptase-dependent proteolytic pathway. Am J Pathol 2009;175:1453–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buzza MS, Netzel-Arnett S, Shea-Donohue T, Zhao A, Lin CY, List K, et al. Membrane-anchored serine protease matriptase regulates epithelial barrier formation and permeability in the intestine. Proc Natl Acad Sci U S A 2010;107:4200–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Netzel-Arnett S, Buzza MS, Shea-Donohue T, Desilets A, Leduc R, Fasano A, et al. Matriptase protects against experimental colitis and promotes intestinal barrier recovery. Inflamm Bowel Dis 2012;18:1303–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin CY, Wang JK, Torri J, Dou L, Sang QA, Dickson RB. Characterization of a novel, membrane-bound, 80-kDa matrix-degrading protease from human breast cancer cells. Monoclonal antibody production, isolation, and localization. J Biol Chem 1997;272:9147–52 [PubMed] [Google Scholar]
- 74.Zoratti GL, Tanabe LM, Varela FA, Murray AS, Bergum C, Colombo E, et al. Targeting matriptase in breast cancer abrogates tumour progression via impairment of stromal-epithelial growth factor signalling. Nat Commun 2015;6:6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zoratti GL, Tanabe LM, Hyland TE, Duhaime MJ, Colombo E, Leduc R, et al. Matriptase regulates c-Met mediated proliferation and invasion in inflammatory breast cancer. Oncotarget 2016;7:58162–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Uhland K. Matriptase and its putative role in cancer. Cell Mol Life Sci 2006;63:2968–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.List K Matriptase: a culprit in cancer? Future Oncol 2009;5:97–104 [DOI] [PubMed] [Google Scholar]
- 78.Camerer E, Barker A, Duong DN, Ganesan R, Kataoka H, Cornelissen I, et al. Local protease signaling contributes to neural tube closure in the mouse embryo. Dev Cell 2010;18:25–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Szabo R, Uzzun Sales K, Kosa P, Shylo NA, Godiksen S, Hansen KK, et al. Reduced prostasin (CAP1/PRSS8) activity eliminates HAI-1 and HAI-2 deficiency-associated developmental defects by preventing matriptase activation. PLoS Genet 2012;8:e1002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Friis S, Uzzun Sales K, Godiksen S, Peters DE, Lin CY, Vogel LK, et al. A matriptase-prostasin reciprocal zymogen activation complex with unique features: prostasin as a non-enzymatic co-factor for matriptase activation. J Biol Chem 2013;288:19028–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sales KU, Friis S, Konkel JE, Godiksen S, Hatakeyama M, Hansen KK, et al. Non-hematopoietic PAR-2 is essential for matriptase-driven pre-malignant progression and potentiation of ras-mediated squamous cell carcinogenesis. Oncogene 2015;34:346–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Le Gall SM, Szabo R, Lee M, Kirchhofer D, Craik CS, Bugge TH, et al. Matriptase activation connects tissue factor-dependent coagulation initiation to epithelial proteolysis and signaling. Blood 2016;127:3260–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Szabo R, Peters DE, Kosa P, Camerer E, Bugge TH. Regulation of feto-maternal barrier by matriptase- and PAR-2-mediated signaling is required for placental morphogenesis and mouse embryonic survival. PLoS Genet 2014;10:e1004470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.List K, Szabo R, Molinolo A, Sriuranpong V, Redeye V, Murdock T, et al. Deregulated matriptase causes ras-independent multistage carcinogenesis and promotes ras-mediated malignant transformation. Genes Dev 2005;19:1934–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lin CY, Anders J, Johnson M, Dickson RB. Purification and characterization of a complex containing matriptase and a Kunitz-type serine protease inhibitor from human milk. J Biol Chem 1999;274:18237–42 [DOI] [PubMed] [Google Scholar]
- 86.Szabo R, Hobson JP, List K, Molinolo A, Lin CY, Bugge TH. Potent inhibition and global co-localization implicate the transmembrane Kunitz-type serine protease inhibitor hepatocyte growth factor activator inhibitor-2 in the regulation of epithelial matriptase activity. J Biol Chem 2008;283:29495–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Friis S, Sales KU, Schafer JM, Vogel LK, Kataoka H, Bugge TH. The protease inhibitor HAI-2, but not HAI-1, regulates matriptase activation and shedding through prostasin. J Biol Chem 2014;289:22319–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oberst MD, Chen LY, Kiyomiya K, Williams CA, Lee MS, Johnson MD, et al. HAI-1 regulates activation and expression of matriptase, a membrane-bound serine protease. Am J Physiol Cell Physiol 2005;289:C462–70 [DOI] [PubMed] [Google Scholar]
- 89.Kataoka H, Kawaguchi M, Fukushima T, Shimomura T. Hepatocyte growth factor activator inhibitors (HAI-1 and HAI-2): Emerging key players in epithelial integrity and cancer. Pathol Int 2018;68:145–58 [DOI] [PubMed] [Google Scholar]
- 90.Vogel LK, Saebo M, Skjelbred CF, Abell K, Pedersen ED, Vogel U, et al. The ratio of Matriptase/HAI-1 mRNA is higher in colorectal cancer adenomas and carcinomas than corresponding tissue from control individuals. BMC Cancer 2006;6:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oberst MD, Johnson MD, Dickson RB, Lin CY, Singh B, Stewart M, et al. Expression of the serine protease matriptase and its inhibitor HAI-1 in epithelial ovarian cancer: correlation with clinical outcome and tumor clinicopathological parameters. Clin Cancer Res 2002;8:1101–7 [PubMed] [Google Scholar]
- 92.LeBeau AM, Lee M, Murphy ST, Hann BC, Warren RS, Delos Santos R, et al. Imaging a functional tumorigenic biomarker in the transformed epithelium. Proc Natl Acad Sci U S A 2013;110:93–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tsai CH, Teng CH, Tu YT, Cheng TS, Wu SR, Ko CJ, et al. HAI-2 suppresses the invasive growth and metastasis of prostate cancer through regulation of matriptase. Oncogene 2014;33:4643–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu SR, Teng CH, Tu YT, Ko CJ, Cheng TS, Lan SW, et al. The Kunitz Domain I of Hepatocyte Growth Factor Activator Inhibitor-2 Inhibits Matriptase Activity and Invasive Ability of Human Prostate Cancer Cells. Sci Rep 2017;7:15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nonboe AW, Krigslund O, Soendergaard C, Skovbjerg S, Friis S, Andersen MN, et al. HAI-2 stabilizes, inhibits and regulates SEA-cleavage-dependent secretory transport of matriptase. Traffic 2017;18:378–91 [DOI] [PubMed] [Google Scholar]
- 96.Sun P, Jiang Z, Chen X, Xue L, Mao X, Ruan G, et al. Decreasing the ratio of matriptase/HAI1 by downregulation of matriptase as a potential adjuvant therapy in ovarian cancer. Mol Med Rep 2016;14:1465–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bocheva G, Rattenholl A, Kempkes C, Goerge T, Lin CY, D’Andrea MR, et al. Role of matriptase and proteinase-activated receptor-2 in nonmelanoma skin cancer. J Invest Dermatol 2009;129:1816–23 [DOI] [PubMed] [Google Scholar]
- 98.Ye J, Kawaguchi M, Haruyama Y, Kanemaru A, Fukushima T, Yamamoto K, et al. Loss of hepatocyte growth factor activator inhibitor type 1 participates in metastatic spreading of human pancreatic cancer cells in a mouse orthotopic transplantation model. Cancer Sci 2014;105:44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sales KU, Friis S, Abusleme L, Moutsopoulos NM, Bugge TH. Matriptase promotes inflammatory cell accumulation and progression of established epidermal tumors. Oncogene 2015;34:4664–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kanemaru A, Yamamoto K, Kawaguchi M, Fukushima T, Lin CY, Johnson MD, et al. Deregulated matriptase activity in oral squamous cell carcinoma promotes the infiltration of cancer-associated fibroblasts by paracrine activation of protease-activated receptor 2. Int J Cancer 2017;140:130–41 [DOI] [PubMed] [Google Scholar]
- 101.Zhang Y, Cai X, Schlegelberger B, Zheng S. Assignment1 of human putative tumor suppressor genes ST13 (alias SNC6) and ST14 (alias SNC19) to human chromosome bands 22q13 and 11q24-->q25 by in situ hybridization. Cytogenet Cell Genet 1998;83:56–7 [DOI] [PubMed] [Google Scholar]
- 102.Kosa P, Szabo R, Molinolo AA, Bugge TH. Suppression of Tumorigenicity-14, encoding matriptase, is a critical suppressor of colitis and colitis-associated colon carcinogenesis. Oncogene 2012;31:3679–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schneider EL, Lee MS, Baharuddin A, Goetz DH, Farady CJ, Ward M, et al. A reverse binding motif that contributes to specific protease inhibition by antibodies. J Mol Biol 2012;415:699–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hoshiko S, Kawaguchi M, Fukushima T, Haruyama Y, Yorita K, Tanaka H, et al. Hepatocyte growth factor activator inhibitor type 1 is a suppressor of intestinal tumorigenesis. Cancer Res 2013;73:2659–70 [DOI] [PubMed] [Google Scholar]
- 105.Verghese GM, Gutknecht MF, Caughey GH. Prostasin regulates epithelial monolayer function: cell-specific Gpld1-mediated secretion and functional role for GPI anchor. Am J Physiol Cell Physiol 2006;291:C1258–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wilson S, Greer B, Hooper J, Zijlstra A, Walker B, Quigley J, et al. The membrane-anchored serine protease, TMPRSS2, activates PAR-2 in prostate cancer cells. Biochem J 2005;388:967–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ko CJ, Huang CC, Lin HY, Juan CP, Lan SW, Shyu HY, et al. Androgen-Induced TMPRSS2 Activates Matriptase and Promotes Extracellular Matrix Degradation, Prostate Cancer Cell Invasion, Tumor Growth, and Metastasis. Cancer Res 2015;75:2949–60 [DOI] [PubMed] [Google Scholar]
- 108.List K, Hobson JP, Molinolo A, Bugge TH. Co-localization of the channel activating protease prostasin/(CAP1/PRSS8) with its candidate activator, matriptase. J Cell Physiol 2007;213:237–45 [DOI] [PubMed] [Google Scholar]
- 109.Lee SP, Kao CY, Chang SC, Chiu YL, Chen YJ, Chen MG, et al. Tissue distribution and subcellular localizations determine in vivo functional relationship among prostasin, matriptase, HAI-1, and HAI-2 in human skin. PLoS One 2018;13:e0192632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lai CH, Chang SC, Chen YJ, Wang YJ, Lai YJ, Chang HD, et al. Matriptase and prostasin are expressed in human skin in an inverse trend over the course of differentiation and are targeted to different regions of the plasma membrane. Biol Open 2016;5:1380–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Friis S, Godiksen S, Bornholdt J, Selzer-Plon J, Rasmussen HB, Bugge TH, et al. Transport via the transcytotic pathway makes prostasin available as a substrate for matriptase. J Biol Chem 2011;286:5793–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Frateschi S, Camerer E, Crisante G, Rieser S, Membrez M, Charles RP, et al. PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin. Nat Commun 2011;2:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mok SC, Chao J, Skates S, Wong K, Yiu GK, Muto MG, et al. Prostasin, a potential serum marker for ovarian cancer: identification through microarray technology. J Natl Cancer Inst 2001;93:1458–64 [DOI] [PubMed] [Google Scholar]
- 114.Selzer-Plon J, Bornholdt J, Friis S, Bisgaard HC, Lothe IM, Tveit KM, et al. Expression of prostasin and its inhibitors during colorectal cancer carcinogenesis. BMC Cancer 2009;9:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yan BX, Ma JX, Zhang J, Guo Y, Mueller MD, Remick SC, et al. Prostasin may contribute to chemoresistance, repress cancer cells in ovarian cancer, and is involved in the signaling pathways of CASP/PAK2-p34/actin. Cell Death Dis 2014;5:e995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bao Y, Li K, Guo Y, Wang Q, Li Z, Yang Y, et al. Tumor suppressor PRSS8 targets Sphk1/S1P/Stat3/Akt signaling in colorectal cancer. Oncotarget 2016;7:26780–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tamir A, Gangadharan A, Balwani S, Tanaka T, Patel U, Hassan A, et al. The serine protease prostasin (PRSS8) is a potential biomarker for early detection of ovarian cancer. J Ovarian Res 2016;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tanimoto H, Yan Y, Clarke J, Korourian S, Shigemasa K, Parmley TH, et al. Hepsin, a cell surface serine protease identified in hepatoma cells, is overexpressed in ovarian cancer. Cancer Res 1997;57:2884–7 [PubMed] [Google Scholar]
- 119.Klezovitch O, Chevillet J, Mirosevich J, Roberts RL, Matusik RJ, Vasioukhin V. Hepsin promotes prostate cancer progression and metastasis. Cancer Cell 2004;6:185–95 [DOI] [PubMed] [Google Scholar]
- 120.Xuan JA, Schneider D, Toy P, Lin R, Newton A, Zhu Y, et al. Antibodies neutralizing hepsin protease activity do not impact cell growth but inhibit invasion of prostate and ovarian tumor cells in culture. Cancer Res 2006;66:3611–9 [DOI] [PubMed] [Google Scholar]
- 121.Miao J, Mu D, Ergel B, Singavarapu R, Duan Z, Powers S, et al. Hepsin colocalizes with desmosomes and induces progression of ovarian cancer in a mouse model. Int J Cancer 2008;123:2041–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xing P, Li JG, Jin F, Zhao TT, Liu Q, Dong HT, et al. Clinical and biological significance of hepsin overexpression in breast cancer. J Investig Med 2011;59:803–10 [DOI] [PubMed] [Google Scholar]
- 123.Tervonen TA, Belitskin D, Pant SM, Englund JI, Marques E, Ala-Hongisto H, et al. Deregulated hepsin protease activity confers oncogenicity by concomitantly augmenting HGF/MET signalling and disrupting epithelial cohesion. Oncogene 2016;35:1832–46 [DOI] [PubMed] [Google Scholar]
- 124.Vaarala MH, Porvari K, Kyllonen A, Lukkarinen O, Vihko P. The TMPRSS2 gene encoding transmembrane serine protease is overexpressed in a majority of prostate cancer patients: detection of mutated TMPRSS2 form in a case of aggressive disease. Int J Cancer 2001;94:705–10 [DOI] [PubMed] [Google Scholar]
- 125.Lucas JM, Heinlein C, Kim T, Hernandez SA, Malik MS, True LD, et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov 2014;4:1310–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci U S A 2000;97:5255–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schaffner F, Versteeg HH, Schillert A, Yokota N, Petersen LC, Mueller BM, et al. Cooperation of tissue factor cytoplasmic domain and PAR2 signaling in breast cancer development. Blood 2010;116:6106–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hooper JD, Nicol DL, Dickinson JL, Eyre HJ, Scarman AL, Normyle JF, et al. Testisin, a new human serine proteinase expressed by premeiotic testicular germ cells and lost in testicular germ cell tumors. Cancer Res 1999;59:3199–205 [PubMed] [Google Scholar]
- 129.Tang T, Kmet M, Corral L, Vartanian S, Tobler A, Papkoff J. Testisin, a glycosyl-phosphatidylinositol-linked serine protease, promotes malignant transformation in vitro and in vivo. Cancer Res 2005;65:868–78 [PubMed] [Google Scholar]
- 130.Honda A, Yamagata K, Sugiura S, Watanabe K, Baba T. A mouse serine protease TESP5 is selectively included into lipid rafts of sperm membrane presumably as a glycosylphosphatidylinositol-anchored protein. J Biol Chem 2002;277:16976–84 [DOI] [PubMed] [Google Scholar]
- 131.Driesbaugh KH, Buzza MS, Martin EW, Conway GD, Kao JP, Antalis TM. Proteolytic activation of the protease-activated receptor (PAR)-2 by the glycosylphosphatidylinositol-anchored serine protease testisin. J Biol Chem 2015;290:3529–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shigemasa K, Underwood LJ, Beard J, Tanimoto H, Ohama K, Parmley TH, et al. Overexpression of testisin, a serine protease expressed by testicular germ cells, in epithelial ovarian tumor cells. J Soc Gynecol Investig 2000;7:358–62 [PubMed] [Google Scholar]
- 133.Bignotti E, Tassi RA, Calza S, Ravaggi A, Bandiera E, Rossi E, et al. Gene expression profile of ovarian serous papillary carcinomas: identification of metastasis-associated genes. Am J Obstet Gynecol 2007;196:245 e1–11 [DOI] [PubMed] [Google Scholar]
- 134.Yeom SY, Jang HL, Lee SJ, Kim E, Son HJ, Kim BG, et al. Interaction of testisin with maspin and its impact on invasion and cell death resistance of cervical cancer cells. FEBS Lett 2010;584:1469–75 [DOI] [PubMed] [Google Scholar]
- 135.Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, et al. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J Biol Chem 2011;286:24638–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.O’Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, et al. Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J Biol Chem 2000;275:13502–9 [DOI] [PubMed] [Google Scholar]
- 137.Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, et al. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol 2007;8:1303–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lin H, Trejo J. Transactivation of the PAR1-PAR2 heterodimer by thrombin elicits beta-arrestin-mediated endosomal signaling. J Biol Chem 2013;288:11203–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hung RJ, Hsu Ia W, Dreiling JL, Lee MJ, Williams CA, Oberst MD, et al. Assembly of adherens junctions is required for sphingosine 1-phosphate-induced matriptase accumulation and activation at mammary epithelial cell-cell contacts. Am J Physiol Cell Physiol 2004;286:C1159–69 [DOI] [PubMed] [Google Scholar]
- 140.Darragh MR, Schneider EL, Lou J, Phojanakong PJ, Farady CJ, Marks JD, et al. Tumor detection by imaging proteolytic activity. Cancer Res 2010;70:1505–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Quimbar P, Malik U, Sommerhoff CP, Kaas Q, Chan LY, Huang YH, et al. High-affinity cyclic peptide matriptase inhibitors. J Biol Chem 2013;288:13885–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Owusu BY, Bansal N, Venukadasula PK, Ross LJ, Messick TE, Goel S, et al. Inhibition of pro-HGF activation by SRI31215, a novel approach to block oncogenic HGF/MET signaling. Oncotarget 2016;7:29492–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Colombo E, Desilets A, Duchene D, Chagnon F, Najmanovich R, Leduc R, et al. Design and synthesis of potent, selective inhibitors of matriptase. ACS Med Chem Lett 2012;3:530–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mitchell AC, Kannan D, Hunter SA, Parra Sperberg RA, Chang CH, Cochran JR. Engineering a potent inhibitor of matriptase from the natural hepatocyte growth factor activator inhibitor type-1 (HAI-1) protein. J Biol Chem 2018;293:4969–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kelly KA, Setlur SR, Ross R, Anbazhagan R, Waterman P, Rubin MA, et al. Detection of early prostate cancer using a hepsin-targeted imaging agent. Cancer Res 2008;68:2286–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rankovic Z, Brust TF, Bohn LM. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg Med Chem Lett 2016;26:241–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bologna Z, Teoh JP, Bayoumi AS, Tang Y, Kim IM. Biased G Protein-Coupled Receptor Signaling: New Player in Modulating Physiology and Pathology. Biomol Ther (Seoul) 2017;25:12–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jiang Y, Yau MK, Kok WM, Lim J, Wu KC, Liu L, et al. Biased Signaling by Agonists of Protease Activated Receptor 2. ACS Chem Biol 2017;12:1217–26 [DOI] [PubMed] [Google Scholar]
- 149.Cheng RKY, Fiez-Vandal C, Schlenker O, Edman K, Aggeler B, Brown DG, et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 2017;545:112–5 [DOI] [PubMed] [Google Scholar]
- 150.Yau MK, Liu L, Fairlie DP. Toward drugs for protease-activated receptor 2 (PAR2). J Med Chem 2013;56:7477–97 [DOI] [PubMed] [Google Scholar]
- 151.Yau MK, Liu L, Suen JY, Lim J, Lohman RJ, Jiang Y, et al. PAR2 Modulators Derived from GB88. ACS Med Chem Lett 2016;7:1179–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol 2011;21:228–37 [DOI] [PMC free article] [PubMed] [Google Scholar]