

Abstract
Background
Interferon-α favors a Th1 shift in immunity and combining with ipilimumab (ipi) at 3 or 10 mg/kg may downregulate CTLA4-mediated suppressive effects leading to more durable antitumor immune responses. A study of tremelimumab and high-dose interferon-α (HDI) showed promising efficacy supporting this hypothesis.
Methods
E3611 followed a 2 by 2 factorial design (A: ipi10+HDI; B: ipi10; C: ipi3+HDI; D: ipi3) to evaluate (i) no HDI versus HDI (across ipi doses) and (ii) ipi3 versus ipi10 (across HDI status). We hypothesized that median progression free survival (PFS) would improve from 3 to 6 months with HDI versus no HDI and with ipi10 versus ipi3.
Results
For eligible and treated patients (N=81) at a median follow-up time of 29.8 months, median PFS was 4.4 months (95%CI: 2.7–8.2) when ipilimumab was used alone and 7.5 months (95%CI: 5.1–11.0) when HDI was added. Median PFS was 3.8 months (95%CI: 2.6–7.5) with 3mg/kg ipilimumab and 6.5 months (95%CI: 5.1–13.5) with 10mg/kg. By study arm, median PFS was 8.0 months (95%CI: 2.8–20.2) in arm A, 6.2 months (95%CI: 2.7–25.7) in B, 5.7 months (95%CI: 1.5–11.1) in C and 2.8 months (95%CI: 2.6–5.7) in D. The differences in PFS and overall survival (OS) did not reach statistical significance. Adverse events were consistent with the known profiles of ipilimumab and HDI and significantly higher with HDI and ipi10.
Conclusions
While PFS was increased, the differences resulting from adding interferon-α or higher dose of ipilimumab did not reach statistical significance and do not outweigh the added toxicity risks.
Keywords: ipilimumab, high-dose interferon-α, Immunotherapy, Melanoma, Clinical trial
INTRODUCTION
An estimated 9,320 patients will die from metastatic melanoma in the United States in 2018 underscoring the need for new therapeutic approaches that may salvage patients not deriving benefits from existing treatment options, despite major recent advances [1]. Advances in the preceding few years have brought deepening and exciting understanding to the molecular biology of melanoma and the immune regulatory mechanisms that play an important role in the oncogenesis of this disease and the host immune tolerance conducive to disease progression. As a result, therapeutic options against melanoma have expanded to include several promising agents that exploit various targets the melanoma cell is dependent upon for survival [2, 3]. Recent advances at the molecular levels have been translated into the clinic with new molecularly targeted agents (BRAF and MEK kinase inhibitors) and immune checkpoint modulators (CTLA4 and PD-1 blocking antibodies) that led into significant benefits in tumor response and patient survival [4]. First line treatment for patients with metastatic melanoma currently consists of PD1 blockade as monotherapy or in combination with ipilimumab or targeted kinase inhibitors against BRAF and MEK in the presence of activating BRAF mutations. Second line treatment primarily consists of ipilimumab in ipilimumab-naïve patients, but, given the modest clinical activity observed with ipilimumab monotherapy, development of ipilimumab-based combinations is currently an active area of investigation [5].
Evidence suggests that patients with advanced melanoma display strong Th2-type polarization [6, 7]. Both CTLA-4 blockade and interferon-α (IFN) can up-regulate the pro-inflammatory cytokine response (Th1 polarization) [8, 9], and are associated with increased T cell and dendritic cell (DC) tumor infiltration [10–12]. The impact of IFN on DCs is well established, affecting stages of myeloid DC generation, maturation, differentiation and function [13]. In the immature state, IFN-treated myeloid DCs induce a Th1 ‘polarized’ cytokine response [14]. Similar to myeloid DCs, IFNs polarize lymphocytes toward a pro-inflammatory Th1 phenotype [15–17]. In the cytotoxic T cell compartment, type I IFNs induce antitumor cell-mediated cytotoxicity [18], and promote natural killer (NK) cell-mediated proliferation and cytotoxicity [19]. This Th1 shift in immunity induced by IFN can be countered by immune suppressive mechanisms (e.g. CTLA-4), likely contributing to the very limited clinical activity observed with IFN as monotherapy in metastatic melanoma. Combination with CTLA-4 blockade may alter this balance by down regulating the CTLA4 suppressive regulatory elements and possibly releasing inhibitory influences on activated CD4+ and CD8+ effector T cells.
We previously reported the results of a single arm phase II study testing the combination of the CTLA4 blocking monoclonal antibody tremelimumab and high dose IFN (HDI) in metastatic melanoma [20]. Tremelimumab was given at 15 mg/kg every 12 weeks. HDI was given concurrently. The tumor response rate by intention to treat was 24% (90% CI, 13% to 36%; four complete responses [CRs] and five partial responses [PRs] that lasted 6 to >37 months). Fourteen patients (38%) had stable disease (SD) that lasted 1.5 to 21 months. The median progression-free survival (PFS) was 6.4 months (95% CI, 3.3 to 12.1 months). The median overall survival (OS) was 21 months (95% CI, 9.5 to not reached). These results compared favorably to monotherapy studies testing IFN, tremelimumab [21], or ipilimumab [5], in metastatic melanoma.
Based on these data from our previously reported study with tremelimumab, we hypothesized that the combination of CTLA4 blockade and HDI would prove superior to single agent therapy in terms of efficacy and with acceptable toxicity. Given the interval FDA approval of ipilimumab in advanced melanoma, this study by extension argued for the evaluation of ipilimumab in combination with IFN in a randomized phase II design. Data had suggested a dose dependent effect of ipilimumab from 0.3 mg/kg to 10 mg/kg with increasing efficacy but also toxicity [22]. On the other hand, phase III studies demonstrated significant survival benefits from ipilimumab at 3 mg/kg and 10 mg/kg leading to approval of the 3 mg/kg dose in advanced melanoma. Therefore, we conducted a randomized phase II trial to test the combination of ipilimumab at 3 mg/kg and 10 mg/kg dose levels alone or in combination with IFN utilizing a unique 2 by 2 factorial design in order to simultaneously evaluate the four treatment options of interest.
PATIENTS AND METHODS
Eligibility
Eligibility criteria included patients with histologically confirmed inoperable Stage III or IV metastatic melanoma with measurable disease (RECIST v.1.1). Age ≥18; ECOG performance status of 0–1; adequate hematological and biochemical parameters; and no protocol specified limiting comorbidity or concurrent malignancy. Up to one prior regimen for metastatic melanoma and stable treated brain metastases were permitted.
Treatment
The study was approved by the institutional review board (IRB) of record at participating sites and was conducted in accordance with the Declaration of Helsinki. All patients provided an IRB approved written informed consent. Patients were randomly assigned to receive ipilimumab at 3 mg/kg or 10 mg/kg alone or in combination with IFN, stratified by the American Joint Committee on Cancer (AJCC) 7th edition stage (III/M1a, M1b, M1c). Ipilimumab was given by intravenous (IV) infusion every 3 weeks for up to 4 doses, then every 12 weeks, beginning at week 24, for a maximum of 4 doses. HDI was administered concurrently at 20 million units (MU)/m2;/day IV for 5 consecutive days every week for 4 weeks, followed by 10 MU/m2/day subcutaneously every other day, 3 times each week for up to 56 weeks. Patients who experienced toxicity were graded according to the National Cancer Institute Criteria (NCI) Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 and were managed in accordance with toxicity specific management and dose modification criteria provided in the study protocol.
Endpoints
The primary endpoint was the comparison of PFS between the combination and ipilimumab alone (across the 2 dose levels of ipilimumab). PFS was selected as a primary endpoint, primarily to provide a direct assessment of the durability of clinical benefits derived from the treatment and not potentially confounded by subsequent therapies. Secondary endpoints included the assessment of toxicity and PFS between the 10 mg/kg and 3 mg/kg dose levels of ipilimumab. Additional endpoints included response rate (RR) and overall survival (OS). PFS was defined as time from randomization to disease progression or death without progression. RR was assessed utilizing Response Evaluation Criteria In Solid Tumors (RECIST) criteria v.1.1 [23]. RR was further explored employing the immune-related response criteria (irRC) [24]. OS was defined as time from randomization to death from any cause. Adverse events (AEs) were coded and graded according to CTCAE version 4.0.
Statistical Design and Analysis
This was a phase II randomized study utilizing a 2 by 2 factorial design with (i) no HDI versus HDI (across ipilimumab treatment status) and (ii) ipilimumab 3 mg/kg versus ipilimumab 10 mg/kg (across HDI treatment status). We hypothesized that HDI combined with ipilimumab will lead to improved PFS in comparison to ipilimumab alone (primary hypothesis). To account for ineligible cases, 88 patients (for 80 eligible) were targeted to be accrued. Primarily, it was assumed that HDI would improve the median PFS from 3 to 6 months from no HDI. In addition, it was assumed 10 mg/kg ipilimumab would improve the median PFS from 3 to 6 months from 3 mg/kg ipilimumab. Based on the log-rank test, these comparisons had at least 82% power at a two-sided type I error rate of 0.10. The distribution of PFS was compared using the log-rank test. There was no planned interim analysis other than toxicity considerations. The study schema is shown in Figure 1.
Figure 1:

E3611: Study Schema, phase II randomized trial of of ipilimumab at 3 mg/kg or 10 mg/kg alone or in combination with high dose interferon-α in advanced melanoma
RESULTS
Efficacy
This United States (US) National Clinical Trials Network (NCTN) study was initiated by Eastern Cooperative Oncology Group (ECOG) and the American College of Radiology Imaging Network (ACRIN), with participation from multiple sites across the US. The study was open between December 27, 2012 and November 30, 2015 and had a total accrual of 88 patients. Of the 88 patients enrolled, 81 eligible and treated patients were included in the efficacy analysis and 83 treated patients were included for in the toxicity analysis. Patient disposition is described in the consort diagram (Figure 2). Baseline patient demographics and disease characteristics are listed in Table 1.
Figure 2:
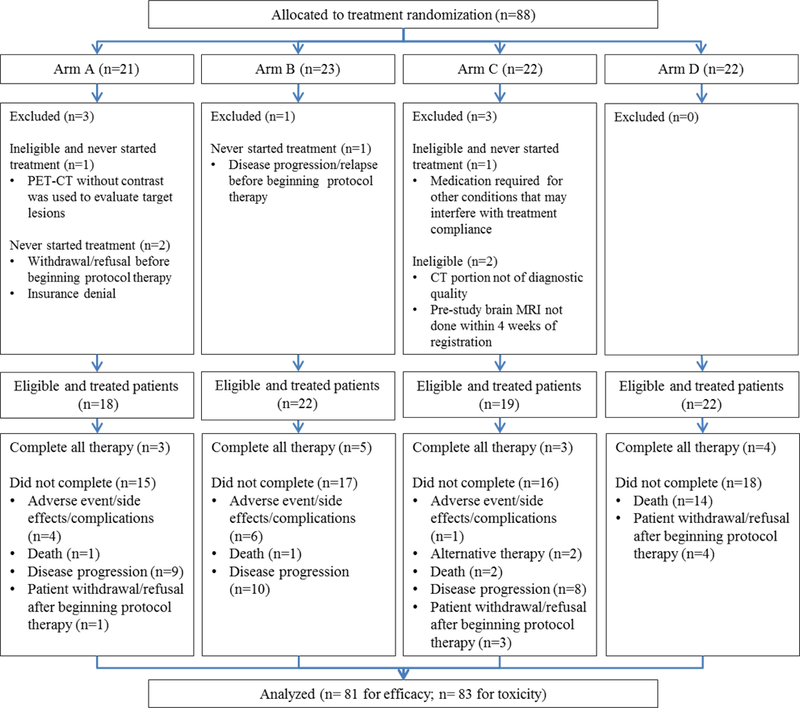
E3611 Consort diagram. The 2 additional patients analyzed for toxicity but not efficacy were enrolled and treated on Arm C, but later found to be ineligible
Table 1.
Patient Demographics and Baseline Disease Characteristics.
| Ipi10 + HDI (N = 18 ) |
Ipi10 (N = 22) |
Ipi3 + HDI (N = 19) |
Ipi3 (N = 22) |
|
|---|---|---|---|---|
| Age (median & range) | 60 (20–74) | 57 (27–83) | 65 (29–77) | 57 (26–73) |
| Gender | ||||
| Female | 10 (56%) | 8 (36%) | 6 (32%) | 9 (41%) |
| Male | 8 (44%) | 14 (64%) | 13 (68%) | 13 (59%) |
| Performance (ECOG) | ||||
| 0 | 9 (50%) | 11 (50%) | 8 (42%) | 15 (68%) |
| 1 | 9 (50%) | 11 (50%) | 11 (58%) | 7 (332%) |
| Primary | ||||
| Cutaneous | 16 (89%) | 16 (73%) | 14 (74%) | 17 (77%) |
| Unknown | 2 (11%) | 6 (27%) | 5 (26%) | 5 (23%) |
| Ulceration of primary | ||||
| Yes | 6 (33%) | 9 (41%) | 8 (42%) | 3 (14%) |
| No | 8 (44%) | 3 (14%) | 5 (26%) | 14 (64%) |
| Unknown | 4 (22%) | 10 (46%) | 6 (32%) | 5 (23%) |
| BRAF Status | ||||
| Mutant (E/K) | 5 (28%) | 11 (50%) | 5 (26%) | 8 (36%) |
| Wild type | 13 (72%) | 7 (32%) | 13 (68%) | 14 (64%) |
| Unknown | 0 | 4 (18.1%) | 1 (5%) | 0 |
| AJCC Stage | ||||
| III (N3)/M1a | 3 (17%) | 5 (23%) | 5 (26.3%) | 5 (22.7%) |
| M1b | 6 (33%) | 6 (27%) | 6 (31.6%) | 6 (27.3%) |
| M1c | 9 (50%) | 11 (50%) | 8 (42%) | 11 (50%) |
| Prior Treatment | 7 (39%) | 10 (45%) | 7 (37%) | 8 (36%) |
| Adjuvant IFN / peg-IFN | 5 (28%) | 4 (18%) | 4 (21%) | 7 (32%) |
| BRAF inhibitor | 0 | 3 (14%) | 2 (111%) | 0 |
| Interleukin-2 | 2 (11%) | 2 (9%) | 1 (5%) | 1 (5%) |
| PD1/PDL1 inhibitor | 0 | 1 (5%) | 0 | 0 |
A total of 88 patients were enrolled, but 7 who never started study treatment or found ineligible were excluded.
Arm A (ipilimumab 10 mg/kg [ipi10] + high-dose interferon-α [HDI])
Arm B (ipilimumab 10 mg/kg alone)
Arm C (ipilimumab 3 mg/kg [ipi3] + high-dose interferon-α)
Arm D (ipilimumab 3 mg/kg alone)
IFN: Interferon; peg-IFN: Pegylated-interferon
The data as of the cutoff date of August 8, 2017 was used, with a median follow-up time of 29.8 months (range: 1.46–47.9). For the 81 patients included in the efficacy analysis, median PFS was 8.0 months (95% CI: 2.8–20.2) in arm A (ipilimumab 10 mg/kg + HDI), 6.2 months (95% CI: 2.7–25.7) in arm B (ipilimumab 10 mg/kg alone), 5.7 months (95% CI: 1.5–11.1) in arm C (ipilimumab 3 mg/kg + HDI), and 2.8 months (95% CI: 2.6–5.7) in arm D (ipilimumab 3 mg/kg alone). The differences in PFS among treatment arms did not reach statistical significance. The median PFS was 4.4 months (95% CI: 2.7–8.2) when ipilimumab was used alone and 7.5 months (95% CI: 5.1–11.0) when IFN was used with ipilimumab. The median PFS was 3.8 months (95% CI: 2.6–7.5) with 3 mg ipilimumab and 6.5 months (95% CI: 5.1–13.5) with 10 mg ipilimumab. Here again, the differences in PFS by HDI or by ipilimumab dose did not reach statistical significance. PFS results are illustrated in Figure 3 and supplemental figure 1 (online only).
Figure 3a:
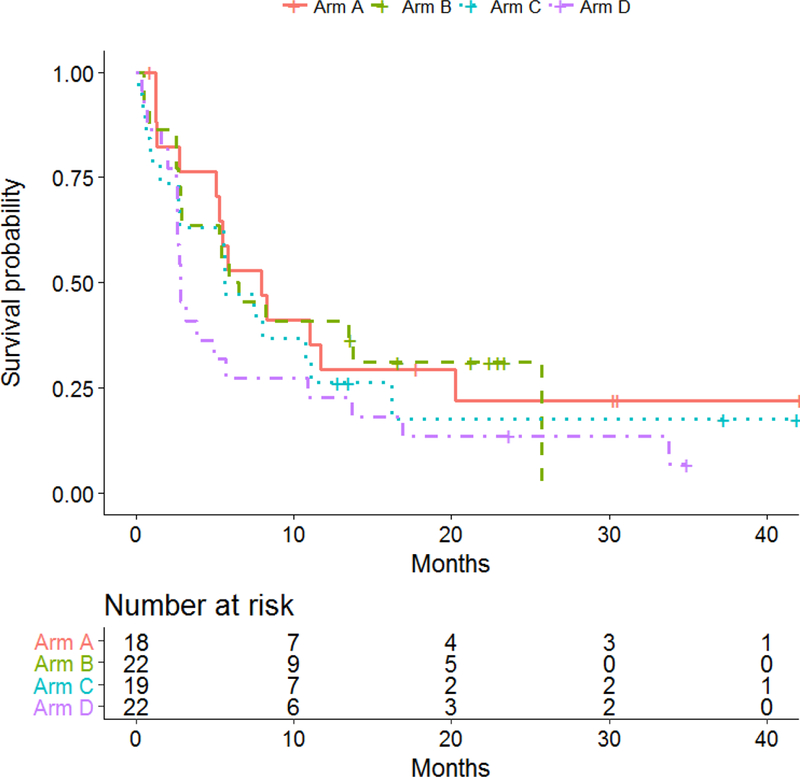
Kaplan-Meier plots of progression-free survival (PFS) by treatment arm
The median OS was 20.1 months (95% CI: 5.1-NA) in arm A, 19.6 months (95% CI: 6.5-NA) in arm B, 20.2 months (95% CI: 1.9-NA) in arm C, and 24.7 months (95% CI: 12.1-NA) months in arm D. The differences in OS were not significant among treatment arms. OS was not significantly different by interferon status or by ipilimumab dose. OS results are illustrated in Figure 4 and supplement figure 2 (online only).
Figure 4:
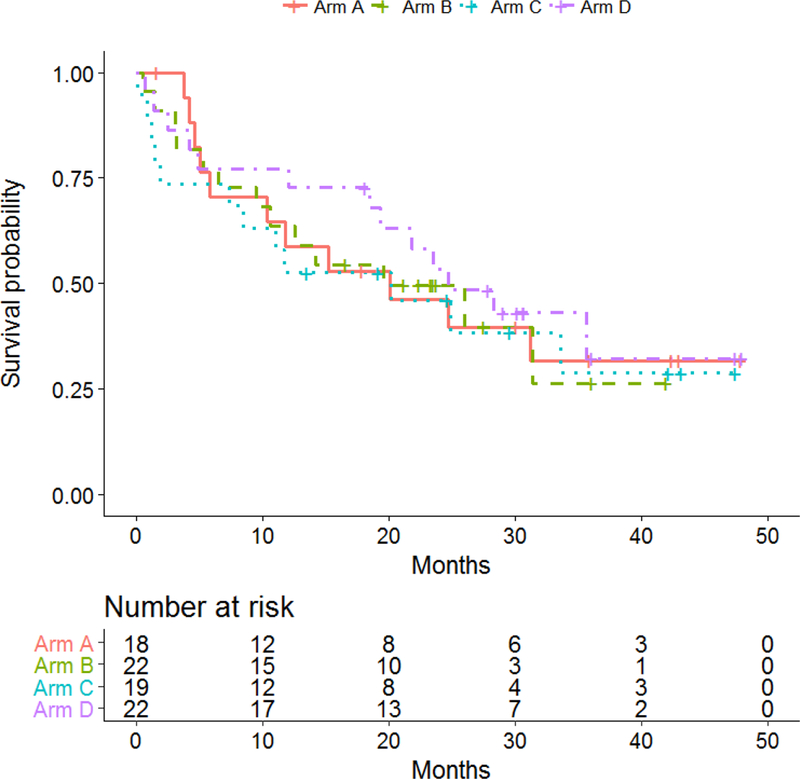
E3611: Kaplan-Meier plots of overall survival (OS) by treatment arm
The clinical response rate (RECIST v1.1) (CR+PR) was 28% (5/18, 95% CI: 10–54%) in arm A, 32% (7/22, 95% CI: 14–55%) in arm B, 16% (3/19, 95% CI: 3–40%) in arm C and 14% (3/22, 95% CI: 3–36%) in arm D. The response rates and durability are summarized in supplemental Table 1 (online only). The irRC response rate was 46% (6/13, 95% CI: 19–75%) in arm A, 38% (6/16, 95% CI: 15–65%) in arm B, 46% (5/11, 95% CI: 17–77%) in arm C and 27% (4/15, 95% CI: 8–55%) in arm D. The differences in clinical response rates or immune related response rates were not statistically significant by treatment arms.
Based on these analyses, there was no clear indication of whether the addition of IFN to ipilimumab or whether the higher dose of ipilimumab improves clinical outcome (PFS, OS, clinical response rates, immune-related response rates).
Treatment Details and Discontinuation
Treatment details by study arm and reasons for discontinuation are summarized in supplemental Table 2 (online only).
Toxicity
The toxicity rate for the worst degree grade 3 or higher adverse events was 94% (17/18, 95% CI: 72.7–99.9%) in arm A, 64% (14/22, 95% CI: 41–83%) in arm B, 76% (16/22, 95% CI: 50–89%) in arm C and 46% (10/22, 95% CI: 24%−68%) in arm D. The differences in worst degree of grade 3 or higher toxicity rates were significantly different by treatment arms (p =0.008). The immune-related toxicity (defined as AEs considered consistent with immune checkpoint inhibitors) incidence rate of grade 3 or higher adverse events was 39% (7/18, 95% CI: 17–64%) in arm A, 36% (8/22, 95% CI: 17–59%) in arm B, 33% (7/21, 95% CI: 15–57%) in arm C and 32% (7/22, 95% CI: 14–55%) in arm D. The incidence of immune-related toxicity rates was not significantly different by treatment arms. Table 2a shows the overall safety summary and Table 2b summarizes selected immune related adverse events. As expected, toxicities common with HDI were more frequent in the combination arms such as fatigue, anorexia, weight loss, fever, chills, flu-like symptoms, nausea, vomiting, liver function abnormalities, creatinine phosphokinase, neutropenia and thrombocytopenia. The use of corticosteroids to manage toxicities was higher in the ipi10 arms (67% in A and 59% in B) compared to the ipi3 arms (32% in C and 36% in D) and not significantly different by the addition of HDI. Only 2 patients required additional immunosuppressants and both were in Arm D (ipi3).
Table 2.
Treatment Related Toxicities. Table 2a shows the overall safety summary and Table 2b summarizes selected immune related adverse events.Table 2a. Safety summary by study arm (n=83). Table 2b.Selected Immune-Related Adverse Events (%)
| Ipi10 + HDI (n = 18) |
Ipi10 (n = 22) |
Ipi3 + HDI (n = 21) |
Ipi3 (n = 22) |
|||||
|---|---|---|---|---|---|---|---|---|
| Any Grade |
Grade 3/4/5 |
Any grade |
Grade 3/4/5 |
Any Grade |
Grade 3/4/5 |
Any Grade |
Grade 3/4/5 |
|
| Any AE, % | 100 | 94 | 100 | 63 | 100 | 76 | 100 | 46 |
| Any immune-related AE, % | 100 | 39 | 77 | 36 | 76 | 33 | 86.4 | 32 |
| Grade 5 events that were considered at least possibly related (n=3): • Suicide (Ipi10 + HDI) • Lung infection and haemorrhage (Ipi10) • Adult respiratory distress syndrome (Ipi3 + HDI) • An additional patient died of gastrointestinal bleed and cardiac arrest while on corticosteroids to treat temporal arteritis and vision loss (Ipi10) | ||||||||
| Ipi10 + HDI (n = 18) |
Ipi10 (n = 22) |
Ipi3 + HDI (n = 21) |
Ipi3 (n = 22) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Any Grade | Grade 3 | Grade 4 |
Any Grade | Grade 3 | Grade 4 |
Any Grade | Grade 3 |
Grade 4 |
Any Grade | Grade 3 |
Grade 4 |
|
| Dermatologic | ||||||||||||
| Rash | 83 | 11 | - | 64 | 5 | - | 29 | 5 | - | 50 | 5 | - |
| Gastrointestinal | ||||||||||||
| Diarrhea | 61 | 0 | 0 | 27 | 9 | 0 | 33 | 5 | - | 36 | 5 | - |
| Colitis | 6 | - | - | - | - | - | 5 | - | - | - | - | - |
| Pancreas | ||||||||||||
| Lipase increase | 39 | - | 6 | 9 | 9 | - | 14 | - | - | 9 | 5 | 5 |
| Endocrine | ||||||||||||
| Hypophysitis | 6 | 6 | - | - | - | - | - | - | - | 5 | - | 5 |
| Adrenal insuff. | 6 | 6 | - | 14 | - | - | 5 | - | - | 5 | - | - |
| Hypothyroid | 28 | - | - | 9 | - | - | 10 | - | - | 14 | - | - |
| Hepatic | ||||||||||||
| ALT/AST increase | 67 | 17 | - | 9 | 9 | - | 57 | 19 | - | 9 | - | 5 |
| Neurologic | ||||||||||||
| Peripheral sensory neuropathy | 6 | - | - | 5 | - | - | 5 | - | - | - | - | - |
| Other | ||||||||||||
| CPK increase | 11 | - | 11 | - | - | - | 10 | 5 | - | - | - | - |
Arm A (ipilimumab 10 mg/kg [ipi10] + high-dose interferon-α [HDI])
Arm B (ipilimumab 10 mg/kg alone)
Arm C (ipilimumab 3 mg/kg [ipi3] + high-dose interferon-α)
Arm D (ipilimumab 3 mg/kg alone)
IFN: Interferon; peg-IFN: Pegylated-interferon
DISCUSSION
In this study, our primary hypothesis was that the addition of IFN to ipilimumab would improve the clinical outcome as measured in terms of PFS. We also hypothesized that the clinical benefits of ipilimumab are dose dependent and the use of the higher dose of ipilimumab (10 mg/kg versus 3 mg/kg) would lead to an improvement in PFS. Overall, there were modest improvements in PFS supporting both hypotheses. However, the differences did not reach statistical significance based on our original study design and the sample size. Ipilimumab is active in patients with advanced stage melanoma providing overall survival benefit and durable objective responses as reported across the dose levels of 3 mg/kg and 10 mg/kg. There is evidence to support a modest clinical benefit in favor of the higher dose of ipilimumab as reported in a recent phase 3 trial (CA184–169) [25]. That study enrolled 727 patients and reported significantly longer OS with ipilimumab 10 mg/kg over ipilimumab 3 mg/kg (median OS of 15.7 months versus11.5 months; hazard ratio 0.84, 95% CI 0.70–0.99; p=0.04), although no significant differences in PFS, or distant metastasis-free survival (DMFS) were seen. Also, an increase in treatment-related adverse events was observed with the higher dosage of ipilimumab. Serious adverse events were seen in 37% and 18% of patients, respectively, including 4 (1%) versus 2 (<1%) of patients who died from treatment-related toxicity. These data are consistent with our findings and further solidify the notion that while limited OS benefits may be derived from the increased dose of ipilimumab, the differences are modest and do not outweigh the added toxicity. This is important since the currently approved dose of ipilimumab for the treatment of metastatic disease is 3 mg/kg based on the results of the MDX10–20 trial that tested ipilimumab at this lower dose level [26]. On the other hand, ipilimumab was approved at 10 mg/kg as adjuvant therapy in melanoma patients with surgically resected nodal metastasis, based on the results of the phase III EORTC 18071 trial which tested ipilimumab at 10 mg/kg versus placebo [27]. Furthermore, we recently reported the results of an interim analysis from the adjuvant E1609 trial testing ipilimumab at 3 and 10 mg/kg versus HDI, where no significant differences in PFS were seen between the 2 dose levels of ipilimumab in the adjuvant setting [28]. Toxicity in the combination arms was mostly additive and consistent with the known toxicity profiles of ipilimumab and HDI. Toxicity with ipilimumab was dose dependent, similar to the experience from the ipilimumab-anti-PD1 combination studies [29, 30].
IFN was the first recombinant cytokine to be investigated for the treatment of metastatic melanoma yielding response rates of about 16% [31]. However, the median duration of response was only a few months. Immunologically, IFNα is known to mount a potent pro-inflammatory (Th1 polarized) immune response that can be suppressed by host immune suppressive elements where CTLA-4 plays a critical role [32]. Our data appear to support the modest improvement in PFS with the addition of HDI to CTLA-4 blockade with ipilimumab, but the PFS difference did not reach statistical significance. On the other hand, toxicity was significantly increased with HDI. A previous phase IB study tested ipilimumab at 3 mg/kg dosing with peginterferon alfa-2b at 2 μg/kg/week for a total of 12 weeks in metastatic melanoma [33]. Grade 3 drug-related AEs were observed in 45% of patients. The response rate was reported as 40% by irRC, which is comparable to our findings in this study. Therefore, we conclude that further testing of the combination of ipilimumab and HDI in a larger study that may be better powered to detect a smaller difference in PFS or OS is not warranted. Furthermore, we conclude that our trial design and sample size as originally planned to compare the benefit-risk profiles of HDI versus no HDI and of ipilimumab 10 mg/kg versus 3 mg/kg was optimal and provided the needed answers with the least possible sample size. Adopting a standard approach to test our hypotheses utilizing a series of 2 clinical trials would have been more expensive to conduct and would have taken a longer time to execute and generate results. Methodological innovations in clinical trial design have become increasingly desired with an aim to answer more questions,with a smaller number of patients and in a shorter period of time [34]. Our study design provides a modest example of how to answer multiple clinical trial questions on a relatively small scale, but at the same time, we acknowledge the potential of treatment interaction as a confounder in our study design.
With the clear–cut advantage of anti-PD1 antibodies in the first line setting for metastatic melanoma, ipilimumab monotherapy has moved into the second line application. However, response rates with ipilimumab have been modest and appear to benefit only a subgroup of patients, supporting the need to investigate rationale combinations that may improve clinical outcomes. Combinations with cytokines such as interleukin-2 (IL2) are an attractive option, given the known pro-inflammatory (Th1 polarizing) impact on the immune response [35]. However, similar to our experience with IFN, there was no evidence to support added benefit while the added toxicity was significant [36]. Other studies pursued evidence suggesting enhanced antitumor activity by combining antiangiogenic agents with ipilimumab. This hypothesis was tested clinically in a phase I study combining bevacizumab and ipilimumab [37]. The response rate was approximately 20% and the median OS was 25.1 months, leading to an ongoing cooperative group randomized trial testing ipilimumab and bevacizumab versus ipilimumab alone (E3612; NCT01950390). Similarly, promising data have emerged from a study that tested intratumoral tilsotolimod (IMO-2125), a Toll-like receptor 9 agonist, in combination with ipilimumab in subjects with anti-PD-1 refractory melanoma [38]. Among 21 patients treated at the Recommended Phase 2 Dose (RP2D), 8 (38%) achieved an objective response, including one CR, and 15 of 21 (71%) had disease control (CR, PR, or SD ≥ 12 weeks). These data have led to an ongoing phase III study comparing tilsotolimod plus ipilimumab to ipilimumab alone in anti-PD-1 refractory melanoma patients. As a locally injectable oncolytic immunotherapy, talimogene laherparepvec in combination with ipilimumab demonstrated a tolerable safety profile and promising clinical activity [39]. Several other combination strategies, including radiation therapy have been reported with mixed results, overall [40]. However, continued development of CTLA4 blockade in combinations is warranted for the treatment of melanoma taking advantage of its potent effects in inducing lasting immune modulatory effects and clinical benefits [41].
Conclusions
Progression free survival was modestly increased with the addition of IFN to ipilimumab and with the use of ipilimumab at 10 mg/kg versus 3 mg/kg, but the differences did not reach statistical significance. Furthermore, there was significantly greater toxicity, eroding support for further development of the combination and the higher dose of ipilimumab.
Supplementary Material
Figure 3b:
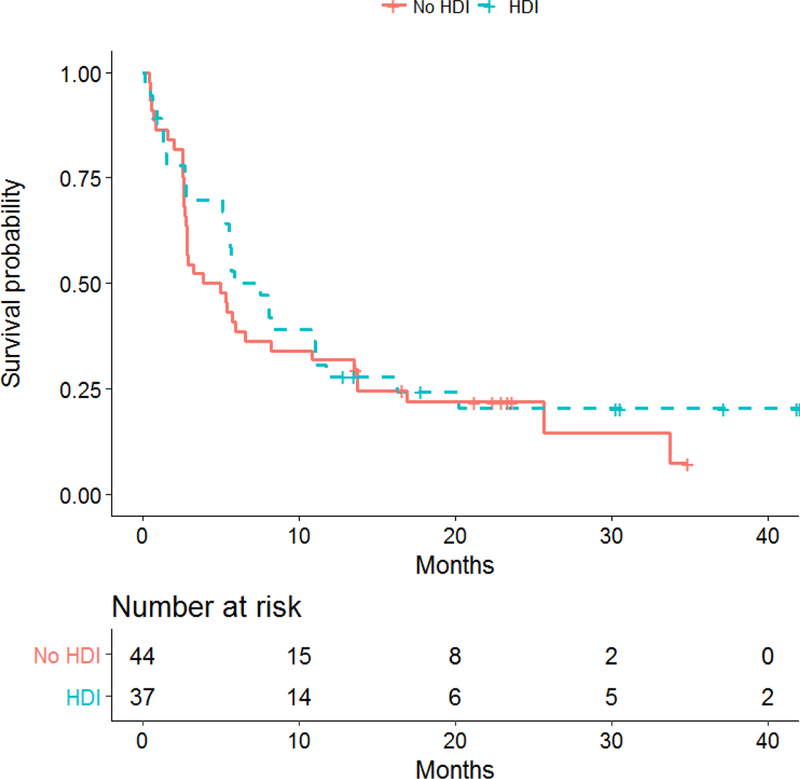
Kaplan-Meier plots of progression-free survival (PFS) by HDI treatment status
Figure 3c:
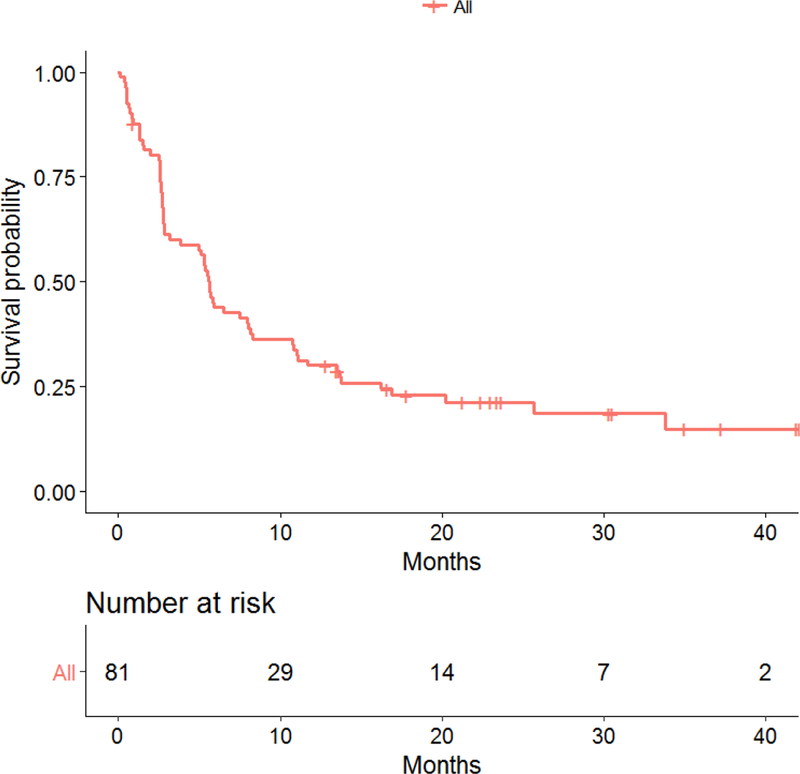
Kaplan-Meier plot of progression-free survival (PFS) all patients
Translational Relevance:
This was a phase II randomized study utilizing a 2 by 2 factorial design with (i) no high-dose interferon-α (HDI) versus HDI (across ipilimumab treatment status) and (ii) ipilimumab 3 mg/kg versus ipilimumab 10 mg/kg (across HDI treatment status). We tested the hypothesis that the addition of interferon-α to the anti-CTLA-4 antibody ipilimumab would lead to more durable antitumor response and improve the clinical outcome as measured in terms of progression free survival (PFS) based on possible synergistic mechanisms. A previous study of CTLA-4 blockade with tremelimumab and HDI showed promising antitumor efficacy supporting this hypothesis. We also hypothesized that the clinical benefits of ipilimumab are dose dependent and would lead to improved PFS with the higher dose of 10 mg/kg ipilimumab over 3 mg/kg. We observed that PFS was modestly increased with the addition of HDI to ipilimumab and with the use of ipilimumab at 10 mg/kg versus 3 mg/kg, but the differences did not reach statistical significance. Furthermore, there were significantly added toxicities denying support for further development of the combination and the higher dose of ipilimumab.
Acknowledgement:
This study was coordinated by the ECOG-ACRIN Cancer Research Group (Peter J. O’Dwyer, MD and Mitchell D. Schnall, MD, PhD, Group Co-Chairs) and supported by the National Cancer Institute of the National Institutes of Health under the following award numbers: CA180820, CA180794, CA180799, CA180844, CA180855, CA189863, CA189869, CA189954, CA189997. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Footnotes
Disclosure of Potential
Ahmad A Tarhini declared consulting or advisory role to BMS and Merck and research grant support from BMS and Merck. Robert M. Conry declared Speaker Bureau for Merck, Amgen, Novartis, BMS with honoraria as compensation. John M Kirkwood declared consulting or advisory role to BMS, Merck, Novartis, Roche, Genentech, EMD Serrono, Array Biopharma, Prometheus.
Conflicts of Interest:
The rest of the authors declare no potential conflicts of interest relevant to the study.
REFERENCES
- 1.Siegel RL, Miller KD, and Jemal A, Cancer statistics, 2018. CA Cancer J Clin, 2018. 68(1): p. 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Davies H, et al. , Mutations of the BRAF gene in human cancer. Nature, 2002. 417(6892): p. 949–54. [DOI] [PubMed] [Google Scholar]
- 3.Kirkwood JM, et al. , Immunotherapy of cancer in 2012. CA Cancer J Clin, 2012. 62(5): p. 309–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Achkar T and Tarhini AA, The use of immunotherapy in the treatment of melanoma. J Hematol Oncol, 2017. 10(1): p. 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodi FS, et al. , Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med, 2010. 363(8): p. 711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tatsumi T, et al. , Disease-associated bias in T helper type 1 (Th1)/Th2 CD4(+) T cell responses against MAGE-6 in HLA-DRB10401(+) patients with renal cell carcinoma or melanoma. J Exp Med, 2002. 196(5): p. 619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tatsumi T, et al. , Disease stage variation in CD4+ and CD8+ T-cell reactivity to the receptor tyrosine kinase EphA2 in patients with renal cell carcinoma. Cancer Res, 2003. 63(15): p. 4481–9. [PubMed] [Google Scholar]
- 8.Yurkovetsky ZR, et al. , Multiplex analysis of serum cytokines in melanoma patients treated with interferon-alpha2b. Clin Cancer Res, 2007. 13(8): p. 2422–8. [DOI] [PubMed] [Google Scholar]
- 9.Hanson DCCP, Primiano MJ, Donovan CB, Gardner JP, Natoli EJ, Morgan RW, Mather RJ, Singleton DH, Hermes PA, Neveu MJ, Andrews GC, Castro CD, Elliott EA, Bedian V Preclinical in vitro characterization of anti-CTLA4 therapeutic antibody CP-675,206. in PROC AM ASSOC CANCER RES 2004. 45 Abs 3802. [Google Scholar]
- 10.Moschos SJ, et al. , Neoadjuvant treatment of regional stage IIIB melanoma with high-dose interferon alfa-2b induces objective tumor regression in association with modulation of tumor infiltrating host cellular immune responses. J Clin Oncol, 2006. 24(19): p. 3164–71. [DOI] [PubMed] [Google Scholar]
- 11.Ribas A DT, Comin-Anduix B, de la Rocha P, Glaspy JA, Economou JS, Gomez-Navarro J, Cochran AJ Changes in intratumoral immune cell infiltrates, Foxp3 and indoleamine 2, 3-dioxygenase (IDO) expression with the CTLA4 blocking MAB CP-675,206. J IMMUNOTHER 2006. 29 (6 ): p. 636. [Google Scholar]
- 12.Hamid O, et al. Association of baseline and on-study tumor biopsy markers with clinical activity in patients (pts) with advanced melanoma treated with ipilimumab. J Clin Oncol 27:15s, 2009. (suppl; abstr 9008) 2009. [Google Scholar]
- 13.Paquette RL, Interferon-alpha and granulocyte-macrophage colony-stimulating factor differentiate peripheral blood monocytes into potent antigen-presenting cells. J Leukoc Biol, 1998. 64(3): p. 358–67. [DOI] [PubMed] [Google Scholar]
- 14.Parlato S, et al. , Expression of CCR-7, MIP-3beta, and Th-1 chemokines in type I IFN-induced monocyte-derived dendritic cells: importance for the rapid acquisition of potent migratory and functional activities. Blood, 2001. 98(10): p. 3022–9. [DOI] [PubMed] [Google Scholar]
- 15.Brinkmann V, et al. , Interferon alpha increases the frequency of interferon gamma-producing human CD4+ T cells. J Exp Med, 1993. 178(5): p. 1655–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wenner CA, et al. , Roles of IFN-gamma and IFN-alpha in IL-12-induced T helper cell-1 development. J Immunol, 1996. 156(4): p. 1442–7. [PubMed] [Google Scholar]
- 17.Rogge L, et al. , Selective expression of an interleukin-12 receptor component by human T helper 1 cells. J Exp Med, 1997. 185(5): p. 825–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palmer KJ, et al. , Interferon-alpha (IFN-alpha) stimulates anti-melanoma cytotoxic T lymphocyte (CTL) generation in mixed lymphocyte tumour cultures (MLTC). Clin Exp Immunol, 2000. 119(3): p. 412–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carballido JA, et al. , Interferon-alpha-2b enhances the natural killer activity of patients with transitional cell carcinoma of the bladder. Cancer, 1993. 72(5): p. 1743–8. [DOI] [PubMed] [Google Scholar]
- 20.Tarhini AA, et al. , Safety and efficacy of combination immunotherapy with interferon alfa-2b and tremelimumab in patients with stage IV melanoma. J Clin Oncol, 2012. 30(3): p. 322–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tarhini AA and Kirkwood JM, Tremelimumab (CP-675,206): a fully human anticytotoxic T lymphocyte-associated antigen 4 monoclonal antibody for treatment of patients with advanced cancers. Expert Opin Biol Ther, 2008. 8(10): p. 1583–93. [DOI] [PubMed] [Google Scholar]
- 22.Wolchok JD, et al. , Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol, 2010. 11(2): p. 155–64. [DOI] [PubMed] [Google Scholar]
- 23.Eisenhauer EA, et al. , New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer, 2009. 45(2): p. 228–47. [DOI] [PubMed] [Google Scholar]
- 24.Wolchok JD, et al. , Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res, 2009. 15(23): p. 7412–20. [DOI] [PubMed] [Google Scholar]
- 25.Ascierto PA, et al. , Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol, 2017. 18(5): p. 611–622. [DOI] [PubMed] [Google Scholar]
- 26.Hodi FS, et al. , Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncology, 2016. 17(11): p. 1558–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eggermont AM, et al. , Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. N Engl J Med, 2016. 375(19): p. 1845–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tarhini A, et al. , A phase III randomized study of adjuvant ipilimumab (3 or 10 mg/kg) versus high-dose interferon alfa-2b for resected high-risk melanoma (U.S. Intergroup E1609): preliminary safety and efficacy of the ipilimumab arms. Oral presentation at: 2017 American Society of Clinical Oncology Annual Meeting; June 2–6, 2017; Chicago, IL 2017. [Google Scholar]
- 29.Wolchok JD, et al. , Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Long GV, et al. , Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol, 2017. 18(9): p. 1202–1210. [DOI] [PubMed] [Google Scholar]
- 31.Tarhini AA and Kirkwood JM, Clinical and immunologic basis of interferon therapy in melanoma. Ann N Y Acad Sci, 2009. 1182: p. 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma P and Allison JP, The future of immune checkpoint therapy. Science, 2015. 348(6230): p. 56–61. [DOI] [PubMed] [Google Scholar]
- 33.Brohl AS, et al. , A phase IB study of ipilimumab with peginterferon alfa-2b in patients with unresectable melanoma. J Immunother Cancer, 2016. 4: p. 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woodcock J and LaVange LM, Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. N Engl J Med, 2017. 377(1): p. 62–70. [DOI] [PubMed] [Google Scholar]
- 35.Floros T and Tarhini AA, Anticancer Cytokines: Biology and Clinical Effects of Interferon-alpha2, Interleukin (IL)-2, IL-15, IL-21, and IL-12. Semin Oncol, 2015. 42(4): p. 539–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maker AV, et al. , Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2: a phase I/II study. Ann Surg Oncol, 2005. 12(12): p. 1005–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hodi FS, et al. , Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res, 2014. 2(7): p. 632–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diab A, et al. A Phase 1/2 trial of intratumoral (i.t.) IMO-2125 (IMO) in combination with checkpoint inhibitors (CPI) in PD-(L)1-refractory melanoma. ESMO 2017 Congress Abstract 1187P. 2017. [Google Scholar]
- 39.Chesney JA, et al. , Primary results from a randomized (1:1), open-label phase II study of talimogene laherparepvec (T) and ipilimumab (I) vs I alone in unresected stage IIIB- IV melanoma. Journal of Clinical Oncology, 2017. 35(15_suppl): p. 9509–9509. [Google Scholar]
- 40.Sundahl N, et al. , Phase 1 Dose Escalation Trial of Ipilimumab and Stereotactic Body Radiation Therapy in Metastatic Melanoma. Int J Radiat Oncol Biol Phys, 2018. 100(4): p. 906–915. [DOI] [PubMed] [Google Scholar]
- 41.Tarhini AA, Immune-Mediated Adverse Events Associated with Ipilimumab CTLA-4 Blockade Therapy: The Underlying Mechanisms and Clinical Management. Scientifica, 2013. 2013: p. pp 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.