

SUMMARY.
Infants have higher risk of developing allergic asthma than adults. However, the underlying mechanism remains unknown. We show here that sensitization of mice with house-dust-mite (HDM) in the presence of low-dose lipopolysaccharide (LPS) prevented T helper-2 (Th2) cell-allergic responses in adult but not infant mice. Mechanistically, adult CD11b+migratory dendritic cells (mDCs) up-regulated the transcription factor T-bet in response to tumor necrosis factor-α (TNFα), which was rapidly induced after HDM+LPS sensitization. Consequently, adult CD11b+mDCs produced interleukin-12 (IL-12), which prevented Th2 cell development by promoting T-bet up-regulation in responding T cells. Conversely, infants failed to induce TNFα following HDM+LPS sensitization and therefore CD11b+mDCs failed to up-regulate T-bet, did not secrete IL-12 and Th2 cell responses normally developed in infant mice. Thus, the availability of TNFα dictates the ability of CD11b+mDCs to suppress allergic-Th2 cell responses upon dose-dependent endotoxin sensitization and is a key mediator governing susceptibility to allergic airway inflammation in infant mice.
Graphical Abstract
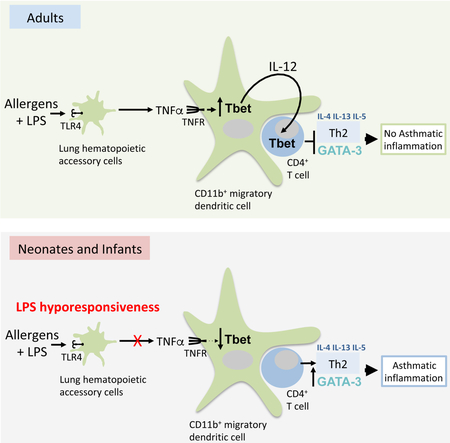
TOC blurb
Children are a higher risk of developing asthma in low-LPS “clean” environments. Bachus et al., demonstrate that infant mice require higher doses of LPS to prevent Th2-dependent allergic-responses due to the decreased ability to induce LPS-driven TNFα production and TNFR-mediated CD11b+ dendritic cell activation for Th2 cell suppression.
INTRODUCTION.
Asthma is the most common chronic disease of childhood, currently affecting ≈10% of school-aged children in U.S (Masoli et al., 2004). About 95% of those children begin developing asthma in very early childhood, normally before they turn five years of age (Masoli et al., 2004). Most cases of pediatric asthma are triggered by early sensitization to common environmental allergens, which ultimately lead to the activation of T-helper 2 cells (Th2) and the development of chronic Th2 cell-driven lung inflammation (Reynolds and Finlay, 2017). Despite the epidemiological and mechanistic studies, however, the underlying mechanisms for the high susceptibility to airway allergic inflammation and asthma development in infants remain elusive.
The incidence of allergic airway disease has increased over the past decades in industrialized countries (Masoli et al., 2004). Although numerous environmental factors associated to change of lifestyle may have contributed to this rise, the hygiene hypothesis proposes that the decreased exposure to microbial products is one of the main drivers. Supporting this idea, exposure to microbial products, such as lipopolysaccharide (LPS) protects from the development of experimental allergen-induced asthma (Daan de Boer et al., 2013; Schuijs et al., 2015). In addition, infants sensitized to common circulating allergens in a “clean” environment are at a higher risk of developing asthma later in life (Gereda et al., 2000; Reynolds and Finlay, 2017; Schuijs et al., 2015; Stein et al., 2016; Zhu et al., 2010); thus suggesting a particular requirement for high-endotoxin exposure during infancy for allergic asthma protection that is not fully understood.
Dendritic cells (DCs) are the main antigen-presenting cells for T cell activation and polarization. In lymph nodes (LN), DCs can be broadly divided into LN-resident DCs (rDCs) and migratory DCs (mDCs), which differentiate in peripheral tissues and migrate constantly through afferent lymphatics to the draining lymph node. Both rDCs and mDCs contain two major subsets: IRF8-dependent CD103+DCs (recently named cDC1s) and IRF4-dependent CD11b+DCs (recently named cDC2s) (Guilliams et al., 2014). Although the roles of the various DC subsets are not fully understood, CD11b+mDCs have been shown to be critical for the development of Th2 cell responses to common allergens (Leon, 2017; Plantinga et al., 2013). In contrast, CD103+mDCs are likely to prevent the development of allergic inflammation (Conejero et al., 2017). Whether alterations in individual DC subsets during infancy contribute to asthma susceptibility remains unexplored.
Here we show that CD11b+mDCs from adult mice up-regulated the transcription factor T-bet and produced interleukin-12 (IL-12) following house dust mite (HDM) sensitization in the presence of LPS. As a consequence, CD4+ T cells interacting with CD11b+mDCs in adult mice up-regulated T-bet, which precluded Th2 cell differentiation and subsequent pathogenic allergic responses to HDM. We found that the up-regulation of T-bet on CD11b+mDCs was dependent on TNFα production and TNFR signaling. Adults rapidly produced TNFα in response to low-dose LPS sensitization; however, infant mice had impaired ability to produce TNFα to LPS. As a result, CD11b+mDCs from infant mice failed to up-regulate T-bet and IL-12 in low-LPS conditions. Thus, CD4+ T cells interacting with CD11b+mDCs in infants failed to receive IL-12 signals, did not up-regulate T-bet and fully differentiated into pathogenic Th2 cells. Thus, whereas low LPS-exposure during HDM sensitization protected adult mice from developing Th2-driven allergic asthma, infant mice required higher doses of LPS. Collectively, our data demonstrated that LPS prevents Th2-dependent allergic-responses with different thresholds in adults and infants, and provide a plausible mechanism underlying the higher susceptibility to allergic airway inflammation observed in children.
RESULTS
LPS inhibits Th2 cell-mediated immunity to inhaled HDM with different thresholds in infant and adult mice.
To test whether LPS influenced Th2 cell response to HDM in infancy and adulthood, we intranasally (i.n.) sensitized adult (≥8 wk-old) and infant (18 d-old) IL-4 reporter B6.4get mice with HDM in the presence or absence of different amounts of LPS (sensitization phase, Fig. 1A). We then challenged them i.n. with HDM (challenge phase, Fig. 1A) and determined the frequency (Fig. 1B) and number (Fig. 1C) of GFP+CD44hiCD4+ T cells. GFP+CD4+ T cells similarly accumulated in the lungs of adult and infant mice that were sensitized with HDM (Fig. 1B-C), but failed to accumulate in adult mice that were sensitized with HDM in the presence of either low (5μg; LPSlo) or high doses (50μg; LPShi) of LPS (Fig. 1B-C). Similarly, LPShi significantly prevented the accumulation of GFP+CD4+ T cells in infant mice (Fig. 1B-C). Importantly, however, LPSlo exposure failed to prevent the expansion of GFP+CD4+ T cells in infant mice (Fig. 1B-C). As a result, GFP+CD4+ T cells largely accumulated in the lungs of HDM+LPSlo-sensitized infants relative to adult counterparts (Fig. 1C). We next sensitized and challenged adult and infant C57BL/6J (B6) mice as described in Fig. 1A and quantified IL-13+ and IFN+ CD4+ T cells in the lungs. As predicted, IL-13+CD4+ T cells were reduced in adults that were initially sensitized with HDM+LPSlo compared to HDM (Fig. 1D-E). Conversely, this effect was not observed in infants (Fig. 1D-E). LPSlo sensitization did not induce IFN-γ+CD4+ T cells in the lungs (Fig. 1D-E), but inhalation of LPShi results in IFN-γ+CD4+ T cells (Fig. S1A-B). These data show that relatively higher amounts of endotoxin exposure are required to prevent the accumulation of Th2 cells in infants. In agreement with these results, while LPSlo administration at the time of initial sensitization prevented eosinophilic airway inflammation (Fig. 1F and Fig. S1C-D), airway goblet cell hyperplasia (Fig. S1F) and accumulation of IgE+CD138+ antibody secreting cells (IgE+ASCs) in the lung-draining, mediastinal lymph node (mLN) (Fig. 1G and Fig. S1E) in adult challenged mice, it failed to do so in infants. Moreover, LPSlo sensitization in adults, but not in infant mice, reduced airway responsiveness to methacholine (Fig. 1H) and improved peripheral blood arterial oxygen saturation (SpO2) (Fig. 1I). Similar differences compared to adults were observed in neonatal (7 d-old), 14 d-old early infant and 18 d-old infant mice (Fig. S2A-J). Taken together, these data indicate that exposure to relatively low endotoxin amounts, in a defined time window from neonatal to infant period, fail to prevent the development of Th2 cell responses and allergic inflammation to inhaled HDM.
Figure 1. LPSlo sensitization in infants does not prevent HDM-induced Th2 cell priming and development of lung Th2 cell responses in adult life.

(A-I) B6.4get (A-C) or B6 (D-I) adults (>8wk-old) and infants (18d-old) were i.n treated with 50μg HDM plus different concentrations of LPS for 3 days. On day 20, mice were i.n challenged with 50μg HDM daily for 3 days and analyzed on day 25 (A). Frequencies and numbers of IL-4+ (EGFP+) (B-C), IL-13+ and IFNγ+ (D-E) CD4+ T cells in the lungs. Numbers of eosinophils, neutrophils and monocytes in the lungs (F). Numbers of IgE+ ACS in the mLN (G). Changes in lung resistance (cm H2O/ml/s) from baseline analyzed via mechanical ventilation using flexiVent system (H). Blood oxygen saturation (SpO2) determined by pulse-oximetry (I). (J-M) B6.4get adults and 18d-old infants were i.n treated with 50μg HDM+/−5μg LPS for 3 days and analyzed on day 6 (J). Frequencies (K-L) and numbers (M) of EGFP+CD4+ T cells in the mLN. (N-U) Adult B6 mice were adoptively transferred with 25×103 CD45.1+ OTII.4get cells from adult or infant donor mice (N-Q) or adult and 18d-old infant B6 mice were adoptively transferred with 25×103 CD45.1+ OTII.4get cells from naive adult donor mice (R-U). Recipients were i.n treated with 50μg HDM+5μg OVA+/−5μg LPS for 3 days and analyzed on day 6. Frequencies (O-P, S-T) and numbers (Q,U) of donor EGFP+OTII T cells in the mLN. *P < 0.05 **P < 0.01 ***P < 0.001 (unpaired Student’s t test). Data are representative of at least two independent experiments (mean and S.D. of 4-5 mice per group). Please also see Figure S1 and S2.
The environment of the infants is more resistant to LPS and more prone to prime HDM-specific Th2 cell responses.
The development of allergen-specific Th2 effector cells that migrate to the lungs requires T-cell priming in the mLN that occurs after initial sensitization (Ballesteros-Tato et al., 2016). Therefore, we next enumerated GFP+CD44hiCD4+ T cells in the mLN of adult and 18 d-old infant B6.4get mice during the sensitization phase (Fig. 1J). The frequency (Fig. 1K-L) and number (Fig. 1M) of GFP+CD4+ T cells were significantly reduced in HDM+LPSlo relative to HDM-sensitized adult mice. In contrast, LPSlo did not prevent the accumulation of GFP+CD4+ T cells in infants (Fig. 1K-M). Similar results were found in 7 and 14 d-old mice (Fig. S2K). Lung neutrophilia, was not examined since it is unlikely to significantly affect Th2 cell priming. These results suggested that low-dose LPS prevents Th2 cell priming in the mLN after HDM sensitization in adult but not in neonatal and infant mice. Next we tested whether differential capacity of low-dose LPS to prevent Th2 cell priming in adults and infants was dictated by intrinsic differences in the CD4+ T cell compartment (Fig. 1N-Q). We transferred 4get.OT-II TCR-transgenic CD4+ T cells from adult or infant mice into congenic adult recipients (Fig. 1N). Recipients were then i.n sensitized with HDM+endotoxin-free ovalbumin (HDM+OVA)+/−LPSlo and donor 4get.OT-II (CD45.1+) cells were analyzed for the expression of GFP-IL-4 in the mLN (Fig. 1O-Q). Data showed that the differential capacity of LPSlo to prevent Th2 cell development in adults and infants was not due to intrinsic differences in the CD4+ T cells. We next transferred 4get.OT-II cells from adult mice into adult or infant recipients (Fig. 1R). Those were then sensitized with HDM+OVA+−LPSlo and donor-derived GFP+ cells were analyzed (Fig. 1S-U). LPSlo failed to prevent the accumulation of donor-derived GFP+4get.OT-II cells when developing in infant but not in adult recipients (Fig. 1S-U). i.n. HDM+LPS delivery was similarly effective in adults and infants (Fig. S2L-N). Similar results were obtained when sensitization was given i.t. rather than i.n. (Fig. S2O-Q). Thus, whereas low-LPS prevents Th2 cell differentiation in adult mice following HDM sensitization, the environment of the infants is more resilient to LPS, hence infants are at higher risk for priming allergen-specific Th2 cell responses in the presence of low doses of endotoxin.
Infants do not induce T-bet expression in T cells under LPSlo sensitization.
T-bet suppress Th2 cell-associated program (Zhu et al., 2012). To test whether CD4+ T cells developing in an infant or adult environment differently up-regulated T-bet, we transferred 4get.OT-II cells from adult mice into either adult or infant recipients, sensitized them, and analyzed the dynamics of T-bet expression (Fig. 2A-C and S3A). As expected, LPSlo treatment prevented the accumulation of 4get.OT-II GFP+ cells in adult mice, but not infant mice, from day 5 (Fig. 2B). Importantly, 4get.OT-II cells expressed higher amounts of T-bet when developing in adults sensitized with HDM+OVA+LPSlo (Fig. 2C). Further, this up-regulation of T-bet occurred concomitant with the down-regulation of GATA-3 (Fig. S3B-D). In contrast, 4get.OT-II cells failed to up-regulate T-bet in HDM+OVA+LPSlo –sensitized infants (Fig. 2C). These results indicate that, while LPSlo promotes T-bet expression in CD4+ T cells differentiating in adult mice, CD4+ T cells fail to up-regulate T-bet when developing in infants. We next investigated whether T-bet up-regulation to LPS was required to prevent Th2 cell differentiation. Thus, we co-transferred WT (CD45.2+CD45.1+) and Tbx21−/− (CD45.2+) 4get.OT-II cells into CD45.1+ adult recipients, sensitized them, and analyzed the progeny from the WT and Tbx21−/− donors (Fig. 2D). As expected the frequency of GFP+ cells within the WT 4get.OT-II decreased in the mLN of HDM+OVA+LPSlo relative to HDM+OVA-sensitized mice (Fig. 2E-F). In contrast, the frequency of GFP+ cells within the Tbx21−/− 4get.OT-II was equivalent in both groups (Fig. 2E-F). We next analyzed the frequency of IL-13+ cells within the donor WT and Tbx21−/− OT-II cells in the lung following challenge (Fig. 2G). As predicted, the frequency of IL-13+WT OT-II cells was decreased in mice that were initially sensitized with HDM+OVA+LPSlo relative to HDM+OVA-sensitized mice (Fig. 2H-I). In contrast, the frequency of IL-13+Tbx21−/− OT-II resulted equivalent in both groups (Fig. 2H-I). Thus, expression of T-bet by CD4+ T cells in response to low-dose endotoxin is important to suppress the endogenous Th2 cell differentiation program to HDM.
Figure 2. T-bet induction by responding T cells is required to suppress endogenous Th2 cell differentiation program.

(A-C) Adult and 18d-old infant B6 mice were transferred with 25×103 CD45.1+ OTII.4get cells at day 0, i.n treated with 50μg HDM+5μg OVA+/−5μg LPS for 3 days starting on day 1 and analyzed on days 4,5 and 6. Frequencies of EGFP+ cells (B) and MFI of T-bet expression (C) in donor mLN OTII cells. (D-I) Adult CD45.1+B6 mice were co-transferred with 15×103 CD45.1+CD45.2+OTII.4get + 15×103 CD45.2+Tbx21−/− OTII.4get cells and treated as above described (D). Frequencies of donor EGFP+ WT and Tbx21−/− OTII cells in the mLN at day 6 (E-F). On day 10, recipients were i.n challenged with 50μg HDM+5μg OVA for 3 days and analyzed on day 15 (G). Frequencies of donor IL-13+ WT and Tbx21−/− OTII cells in the lungs (H-I). (J-M) Day 3 CD11c+ mLN DCs from HDM+OVA+/−LPS-treated adults and 18d-old infants were co-cultured for 3 days with CFSE-labeled OTII cells (J). Frequencies of CFSEloOTII cells (K), representative overlaid histogram of T-bet expression (L) and MFI of T-bet expression (M) in CFSEloOTII cells. *P < 0.05 **P < 0.01 ***P < 0.001 (unpaired Student’s t test). Data are representative of three independent experiments (mean and S.D. of 4 mice per group). Please also see Figure S3.
Lung mDCs from adult and infant mice differ in their ability to induce T-bet in responding T cells.
Lung mDCs are essential for priming Th2 cells to HDM (Leon, 2017). Thus, we tested whether LPSlo administration differentially affected adult and infant mDCs. Given that expression of T-bet by CD4+ T cells was important to suppress the Th2 cell development after HDM+LPSlo sensitization (Fig. 2A-I), we first tested the ability of infant and adult DCs to activate naive CD4+ T cells and promote T-bet expression. mLN CD11c+ DCs were co-cultured with CFSE-labeled naive OTII cells (Fig. 2J). OTII proliferated equally when primed with CD11c+ cells from adults or infants (Fig. 2K). OT-II cells primed by CD11c+ cells from HDM+OVA+LPSlo-sensitized adults up-regulated T-bet relative to OT-II cells primed by CD11c+ cells from HDM+OVA-sensitized adults (Fig. 2L-M). In contrast, OT-II cells primed by CD11c+ cells from HDM+OVA+LPSlo-sensitized infants failed to up-regulate T-bet (Fig. 2L-M). These results indicated that DCs from infant and adult mice differ in their ability to induce T-bet expression in T cells following HDM+LPSlo sensitization. We next characterized mLN mDC subsets in adult and infant mice. Frequency (Fig. S3E) and number (Fig. S3F) of CD103+mDCs decreased after HDM+LPSlo sensitization in adult mice. In contrast, CD11b+mDCs largely accumulated (Fig. S3E, G). Similar results were obtained in infants (Fig. S3E-G). We next assessed the capacity of DC subsets to capture and transport HDM-derived Ags into the mLN. Thus, we i.n sensitized adults and infants with Alexa-647 labeled-HDM+LPSlo and analyzed Alexa-647+CD11c+ DCs in the mLN (Fig. S3H-K). Although some CD103+mDCs were Alexa-647+, the vast majority of HDM-bearing, Alexa-647+CD11c+ DCs were CD11b+mDCs in both adults and infants (Fig. S3H-K). Moreover, the frequency (Fig. S3H) and number (Fig. S3I-K) of HDM-bearing, Alexa-647+ CD11b mDCs and CD103+mDCs resulted identical in infants and adults. Thus, the differences in Th2 cell responses between adults and infants following HDM+LPSlo sensitization was not due to differences in the number, nor the capacity of mDCs subsets to capture and transport HDM-derived Ags into the mLN, but to their functional capacity to induce T-bet expression in T cells.
T-bet expression by CD11b+mDCs is required to prevent Th2 cell responses to HDM.
Few reports have suggested that intrinsic T-bet expression might fine-tune the capacity of DCs to differentially induce Th1 and Th2 cells (Lipscomb et al., 2009; Lugo-Villarino et al., 2003). Thus, we next used T-bet-ZsGreen reporter mice (Zhu et al., 2012) to investigate whether infant and adult mDCs up-regulated T-bet in response to HDM+LPS sensitization (Fig. 3A-D). CD11b+mDCs from HDM+LPSlo-sensitized adult mice up-regulated T-bet expression after 48h compared to HDM-sensitized mice (Fig. 3A,C). In contrast, CD11b+mDCs from HDM+LPSlo-sensitized infants only modestly up-regulated T-bet expression (Fig. 3A,C). Notably, CD103+mDCs (Fig. 3B,D) failed to express T-bet. Similar results were obtained when T-bet protein expression was analyzed (Fig. 3E-F). Members of IFN regulatory factors (IRF) family play essential roles in the functional activation of DCs (Gao et al., 2013; Takaoka et al., 2005; Taki et al., 1997; Williams et al., 2013). Thus, we also investigated whether HDM+LPSlo sensitization differentially affected the expression of IRF members in infant and adult mDCs. Although CD11b+mDCs from HDM+LPSlo-sensitized adult and infant mice differentially up-regulated T-bet expression (Fig. 3J), no differences were observed in the expression of IRF4 (Fig. 3G), IRF1 (Fig. 3H) or IRF5 (Fig. 3I). Together, these data indicated that CD11b+mDCs from infant mice fail to up-regulate T-bet expression in response to HDM+LPSlo sensitization. To address whether T-bet expression by CD11b+mDCs was required for inducing T-bet expression in T cells, CD11c+ cells from the mLN of HDM+OVA+/−LPSlo-sensitized Tbx21−/− mice or adult and infant B6 mice were used to prime OTII cells in vitro (Fig. 4A). OTII cells proliferated equally well in all conditions (Fig. 4B). Compared to non-LPS sensitized adult-derived DCs, however, only CD11c+ cells from mLN of HDM+LPSlo-sensitized B6 adults, but not from B6 infants or Tbx21−/− mice induced significant T-bet expression (Fig. 4B-C). Correlating with this, OTII cells activated in the presence of CD11c+ cells from HDM+LPSlo-sensitized B6 adults expressed lower amounts of GATA-3 (Fig. 4B,D). These data suggested that T-bet expression by DCs is required to induce T-bet and prevent GATA3 expression in responding T cells. To address whether T-bet expression by DCs was required to prevent allergen-specific Th2 cell responses in vivo, we crossed Itgax-cre,-EGFP (Stranges et al., 2007) mice to Tbx21fl/fl mice (Intlekofer et al., 2008) to generate mice in which T-bet is conditionally deleted in CD11c+ cells (Itgaxcre-Tbx21fl/fl). Conditional deletion of T-bet in CD11c+ cells did not lead to defect in mDC subset development (Fig. S4A-B). Importantly, MHCIIhiCD11c+ DCs from the mLN of Itgaxcre-Tbx21fl/fl mice, but not CD11c− cells, expressed high amounts of GFP (Fig. S4C). All CD11chl DCs, including CD103+mDCs, CD11b+mDCs, and rDCs expressed GFP (Fig. S4D) and expected to have equimolar expression with Cre (Stranges et al., 2007). Plasmacytoid DCs expressed lower amounts of GFP (Fig. S4D) (Stranges et al., 2007). As predicted, mLN CD11b+mDCs of HDM+LPSlo-sensitized Itgaxcre-Tbx21fl/fl mice failed to express T-bet (Fig. S4E-F). Thus, we next transferred OT-II cells into either Itgaxcre-Tbx21fl/fl or control mice. Recipients were then sensitized and challenged and donor OTII cells were analyzed in the lungs (Fig. 4E). As expected, the frequency (Fig. 4F-G) and number (Fig. 4H) of IL-13+OTII cells were reduced in control LPSlo-sensitized mice compared to non-LPS-sensitized mice. Conversely, LPSlo sensitization failed to prevent the accumulation of IL-13+OTII cells in the lungs of Itgaxcre-Tbx21fl/fl mice (Fig. 4F-H). As a control, LPSlo sensitization did not induce IFNγ+CD4+ T cells (Fig. 4F-H). Likewise, HDM+LPSlo sensitization failed to prevent lung eosinophilia (Fig. S4G,I) or accumulation of mLN IgE+ASCs (Fig. S4H,J) in Itgaxcre-Tbx21fl/fl mice. Finally, 4get.OT-II cells were transferred into control or Itgaxcre-Tbx21fl/fl mice and the frequency of donor GFP+OTII were calculated in the mLN after sensitization (Fig. 4I). LPSlo sensitization prevented the accumulation of GFP+OT-II in the control recipients, but it failed to do so in Itgaxcre-Tbx21fl/fl mice (Fig. 4J-L). Correlating with these results, LPSlo sensitization induced T-bet up-regulation in control but not in Itgaxcre-Tbx21fl/fl mice (Fig. 4M). These results indicate that T-bet-expressing DCs are required for the suppression of Th2 cell responses to HDM in the presence of low doses of endotoxin. To further confirm these results, CD11c+ cells from lungs of HDM+/−LPSlo–sensitized adult B6 and Tbx21−/− mice were used to i.n. sensitize naive infant B6.4get mice (Fig. 4N). The frequency (Fig. 4O) and number (Fig. 4P) of GFP+CD69+CD44hiCD4+ responding T cells were determined in the mLN. As a control, some mice were also sensitized with infant DCs. Purified lung CD11c+ cells were enriched over 80% in mDCs (Fig. S4L) and were recovered in similar numbers in the mLN of all groups (Fig. S4M-N). Importantly, from the different donor mDC types, only the donor mDCs from HDM+LPS–sensitized adult mice were able to prevent the accumulation of activated GFP+CD4 T cells in the infant recipients (Fig. 4O-P). Collectively, these data suggested that up-regulation of T-bet in lung mDCs is required to prevent Th2 cell responses to low-dose endotoxin and HDM sensitization.
Figure 3. Infant CD11b+mDCs fail to up-regulate T-bet expression in response to LPSlo sensitization.

(A-D) T-bet-ZsGreen reporter adults and 18d-old infants were i.n treated with HDM+/−5μg LPS. Frequencies of T-bet-expressing (ZsG+) CD11b+mDCs (A,C) and CD103+mDCs (B,D). (E-J) B6 adult and 18d-old infant mice were i.n treated with HDM+5μg LPS. Frequencies of T-bet expression at 72h (E-F) and kinetics of IRF4 (G), IRF1 (H), IFR5 (I) and T-bet (J) expression in mLN CD11b+ and CD103+ mDCs. ***P < 0.001 (unpaired Student’s t test). Data are representative of at least two independent experiments (mean and S.D. of 3-4 mice per group).
Figure 4. T-bet expression by DCs is required to prevent allergen-specific Th2 cell responses upon LPSlo exposure.

(A-D) Day 3 CD11c+ mLN DCs from HDM+OVA+/−LPS-treated adult and 18d-old infant B6 mice or Tbx21−/− mice were co-cultured for 3 days with CFSE-labeled OTII cells (A). Frequencies of CFSEloOTII cells (B), frequencies of T-bet and GATA-3 expression (B) and MFI of T-bet (C) and GATA-3 (D) expression in CFSEloOTII cells. (E-H) Itgaxcre-Tbx21fl/fl and control mice were transferred with CD45.1+ OTII.4get cells and i.n treated with HDM+OVA+/−5μg LPS. On day 10, mice were i.n challenged with HDM+OVA and analyzed on day 15 (E). Frequencies of donor OTII cells (F) and frequencies (F-G) and numbers (H) of IL-13+ and IFNγ+ OTII cells from the lungs. (I-M) Mice sensitized as above described were analyzed at day 6 (I). Frequencies of donor OTII cells (J) and frequencies (J-K) and numbers (L) of EGFP+ cells and MFI of T-bet expression (M) in donor OTII cells from the mLN. (N-P) Day 3 CD11c+ lung mDCs from HDM+OVA+/−LPS-treated adult and 18d-old infant B6 mice or Tbx21−/− mice were i.n. transferred into B6.4get infant mice on days 0,1 and 2. Recipient mice were analyzed on day 5 (N). Frequencies (O) and numbers (P) of EGFP+ cells in CD69+CD4+ T cells from the mLN. **P < 0.01 ***P < 0.001 (unpaired Student’s t test). Data are representative of three independent experiments (mean and S.D. of 4-6 mice per group). Please also see Figure S4.
CD11b+mDCs from Tbx21−/− and infant mice are defective in producing IL-12 upon low-dose endotoxin sensitization.
We next made B6:Tbx21−/− mixed bone marrow (BM) chimeras to investigate the role of T-bet in DC function. Briefly, irradiated B6 (CD45.1+) recipient mice were reconstituted with a 50:50 combination of BM from B6 wild-type (WT) (CD45.1+) and Tbx21−/− (CD45.2+) donor mice (Fig. 5A). WT:Tbx21−/− mice were sensitized with HDM+/−LPSlo and mLN mDC were analyzed. mDC subsets derived equally from the WT and Tbx21−/− progenitors (Fig. S5A). Given than IL-12 production is up-regulated in DCs after LPS activation (Trinchieri, 2003), we next assessed IL-12 production by mDCs subsets. WT CD11b+mDCs largely produced IL-12p70 in day3-HDM+LPSlo-sensitized mice relative to HDM-sensitized or day1-HDM+LPSlo-sensitized mice (Fig. 5C and Fig. S5B). In contrast, sensitization in the presence of LPSlo failed to promote IL-12p70 production by Tbx21−/− CD11b+mDCs (Fig. 5C and Fig. S5B). Conversely, equivalent low numbers of IL-12+ WT and Tbx21−/− CD103+mDCs were detected in HDM and HDM+LPSlo-sensitized mice (Fig. 5C and Fig. S5C). These results indicated that, while T-bet is dispensable for the development of CD11b+mDCs, is intrinsically required for the capacity of these cells to produce sustained IL-12 in response to LPS. Corresponding with this, CD11b+mDCs from HDM-sensitized infant mice failed to up-regulate IL-12p70 production 3 days after LPSlo administration compared to adults (Fig. 5D-E and Fig. S5D). Although our data suggested that CD11b+mDCs up-regulate T-bet-dependent IL-12 production and suppress Th2 cell priming after HDM+LPS sensitization, a published report have suggested that CD103+mDCs have the ability to restrain Th2 cell responses to HDM through the production of IL-12 (Conejero et al., 2017). Thus, to test the role of CD103+mDCs during LPSlo-sensitization, we analyzed Batf3−/− mice that selectively lack the CD103+CD11b−DC subset (Edelson et al., 2010). We transferred 4get.OT-II CD4+ T cells into either WT or Batf3−/− recipients, sensitized them, and analyzed on day 6 (Fig. S5E). LPSlo sensitization abrogated GFP+ by donor OTII cells in both WT and Batf3−/− recipients (Fig. S5F-G), suggesting that CD103+mDCs are not necessary to suppress Th2 cell priming in response to low-dose endotoxin sensitization.
Figure 5. Infant and Tbx21−/− CD11b+mDCs are intrinsically impaired in their ability to produce IL-12 and to prevent allergen-specific Th2 cell responses upon LPSlo exposure.

(A-C) B6 (CD45.1+) mice were irradiated and reconstituted with 1:1 mixture of BM from B6 (CD45.1+) and Tbx21−/− (CD45.2+) donors (A). Numbers of IL-12p70+ WT and Tbx21−/− CD11b+mDCs (B) and CD103+mDCs (C) in the mLN of HDM+/−LPS-treated reconstituted chimeric mice. (D-E) Frequencies (D) and numbers (E) of IL-12p70+ CD11b+mDCs in the mLN of HDM+/−LPS-treated adult and 18d-old infant B6 mice. (F-I) Adult and 18d-old infant B6 mice or Itgaxcre-Tbx21fl/fl mice were transferred with CD45.1+ OTII.4get cells and i.n treated with HDM+OVA+/−LPS+/−150ng rIL-12p70 for 3 days. Frequencies of T-bet expression (F,H) and EGFP+ cells (G,I) in donor OTII cells from mLN on day 6. *P < 0.05 ***P < 0.001 (unpaired Student’s t test). Data are representative of two independent experiments (mean and S.D. of 3-6 mice per group). Please also see Figure S5.
Given that IL-12 induces T-bet expression in T cells (Zhu et al., 2012), we next tested whether recombinant IL-12 (rIL-12) administration during HDM+LPSlo sensitization restored the capacity of low-LPS treatment to abrogate Th2 cell development in infant and Itgaxcre-Tbx21fl/fl mice. Thus, we transferred 4get.OT-II CD4+ T cells into B6 adult, B6 infant and ItgaxcreTbx21 tfl/fl mice. Recipients were then sensitized with HDM+OVA+/−LPSlo, and PBS or rIL-12 was i.n co-administered (Fig. S5H). Expression of T-bet (Fig. 5F,H) and GFP (Fig. 5G,I) by donor 4get.OT-II cells was assessed in the mLN. As expected, sensitization in the presence of LPSlo promoted T-bet up-regulation (Fig. 5F,H), thereby preventing GFP expression (Fig. 5G,I) in B6 adults, but not in infant or Itgaxcre-Tbx21fl/fl mice. Importantly, however, rIL-12 administration restored T-bet expression in infant and Itgaxcre-Tbx21fl/fl mice sensitized in the presence of LPSlo (Fig. 5F,H). As a consequence, the frequency of donor GFP+OTII cells resulted similar in HDM+LPSlo-sensitized B6 adults and rIL-12-treated infant and Itgaxcre-Tbx21fl/fl mice (Fig. 5G,I). Administration of rIL-12, however, did not appear to affect T-bet or IL-12p70 expression by CD11b+mDCs (Fig. S5I-L). Collectively, our data indicate that adult CD11b+mDCs up-regulate T-bet after HDM+LPSlo sensitization, which promotes IL-12 production; thus preventing Th2 cell differentiation. In contrast, infant CD11b+mDC fail to up-regulate T-bet, which prevents optimal IL-12 production and subsequent Th2 cell inhibition.
TNFα-driven activation of CD11b+mDCs is required to prevent Th2 cell responses to HDM.
We next tested whether direct activation of CD11b+mDCs by LPS was required to induce T-bet expression after HDM+LPSlo sensitization. Thus, we made B6(CD45.1+):Tlr4−/− (CD45.2+) mixed BM chimeras. Chimeras were sensitized and the expression of T-bet in mLN CD11b+mDCs was determined. As expected, CD11b+mDCs from WT compartment up-regulated T-bet expression after HDM+LPSlo sensitization (Fig. 6A-B). Likewise, CD11b+mDCs from Tlr4−/− compartment also up-regulated T-bet expression (Fig. 6A-B), suggesting that LPS did not directly activate CD11b+mDCs to promote T-bet expression. LPS induces IL-1, which in turn activates IL-1R1-TLR4 signaling (Verstrepen et al., 2008), therefore we next tested whether indirect activation of CD11b+mDCs through the IL-1R1 was required to induce T-bet expression after HDM+LPSlo sensitization. B6(CD45.1+):Il1r1−/−(CD45.2+) mixed BM chimeras were analyzed and found that IL-1R1 signaling neither mediated T-bet up-regulation in CD11b+mDCs (Fig. 6A-B). To gain insight into the mechanisms underlying T-bet regulation in adult and infant CD11b+mDCs, we sorted CD11b+mDCs from mLN of adult and infant mice 24h after HDM+LPSlo sensitization and performed RNA sequencing (RNA-seq) and gene set enrichment analysis (GSEA) to identify hallmark-signaling pathways that were differentially regulated between adult and infant CD11b+mDCs. As a result, 173 differentially expressed genes were identified, 110 genes were down-regulated and 63 genes were up-regulated in infant CD11b+mDCs (Table S1). Notably, the pathway regulated by nuclear factor kappa B (NF-kB) in response to TNFα had the highest normalized enrichment score (NES = 2.64, FDR q-value= 1.28e−42) and contained the largest number of genes down-regulated in infant CD11b+mDCs (Fig. 6C, S6A-B and Tables S2-3). TNFα activates NF-kB and activator protein-1 (AP-1) transcription factors (Verstrepen et al., 2008). Gene expression of NF-kB-c-rel and the AP-1 family members c-Jun, JunB, c-Fos and FosB were decreased in infant CD11b+mDCs after HDM+LPSlo sensitization (Table S1). We next evaluated c-Rel and P-c-Jun protein expression in adult and infant mLN CD11b+mDCs. Adult CD11b+mDCs, but not infant CD11b+mDCs, induced c-Rel and P-c-Jun in response to HDM+LPSlo sensitization (Fig. S6C-E). We next tested whether LPS-induced TNFα played a role in the activation of NF-kB and AP-1 in CD11b+ mDCs. Therefore, we evaluated protein expression of NF-kB-c-Rel, the NF-kB early response protein IkBζ and P-c-Jun-AP-1 in adult mLN CD11b+mDCs after sensitization in presence or absence of TNFα neutralizing mAb or TNFα cytokine. As a control we also included the analysis of 18 d-old infant CD11b+mDCs. As expected, adult CD11b+mDCs, but not infant CD11b+mDCs, induced c-Rel, IkBζ and P-c-Jun in response to HDM+LPSlo sensitization (Fig. 6D-E). However, anti-TNFα treatment prevented these effects (Fig. 6D-E). Additionally, HDM sensitization in the presence of TNFα cytokine was able to induce c-Rel, IkBζ and P-c-Jun expression in CD11b+mDCs compared to HDM sensitization alone (Fig. 6D-E). These data suggested that the activation of NF-kB and AP-1 in CD11b+mDCs after HDM+LPSlo sensitization is mediated by TNFα signaling.
Figure 6. TNFα signaling is required to up-regulate T-bet in mCD11b+DCs and to prevent allergen-specific Th2 cell responses.

(A-B) B6 (CD45.1+) mice were irradiated and reconstituted with 1:1 mixture of BM from B6 (CD45.1+) and Tlr4−/− (CD45.2+) donors or from B6 (CD45.1+) and Il1r1−/− (CD45.2+) donors. Frequencies of T-bet+ WT, Tlr4−/− and Il1r1−/− CD11b+mDCs in the mLN of HDM+/−LPS-treated reconstituted chimeric mice on day 2. (C) mLN CD11b+mDCs from B6 adults and 18d-old infants were sorted 24h after i.n. HDM+5μg LPS and RNA-seq was performed. NES and number of down-regulated genes in infant CD11b+mDCs for each of the top ten hallmark signaling pathways resulting from GSEA (Broad institute) analysis (adjusted P value < 0.05, log2-fold > 0.58). (D-E) B6 adults and 18d-old infants were i.n treated with HDM+/−5μg LPS or HDM+/−30μg TNF. Some mice also receive 250μg anti-TNFα (i.p.). Expression c-Rel, IKBz and P-c-Jun in mLN CD11b+mDCs on day 1. (F-I) CD45.1+ mice were irradiated and reconstituted with 1:1 BM from B6 (CD45.1+) and Tnfr1&2−/− (CD45.2+) donors (F,H) or from B6 (CD45.1+) and Tlr4−/− (CD45.2+) donors (G,I). Frequencies of T-bet+ WT, Tnfr1&2−/− (F,H) and Tlr4−/− (G,I) CD11b+mDCs in the mLN of HDM+/−LPS+/−anti-TNFα-treated reconstituted mice on day 2. (J-M) Irradiated B6 mice were reconstituted with 80:20 BM mix of Itgax-DTR and B6 donors (DC-WT chimeras) or Itgax-DTR and Tnfr1&2−/− donors (DC-TNFR1&2 chimeras) (J). Chimeras were transferred with CD45.1+CD45.2+ OTII.4get cells, i.n treated with HDM+OVA+/−5μg LPS and i.p. treated with 60 ng DT (K). Frequencies of mLN donor EGFP+OTII cells on day 6 (L-M). *P < 0.05 **P < 0.01 ***P < 0.001 (unpaired Student’s t test). Data are representative of two independent experiments (mean and S.D. of 3-7 mice per group). Please also see Figure S6 and Tables S1-3.
We next examined whether the activation of CD11b+mDCs by TNFα signaling through TNF receptors (TNFR1 and TNFR2) was required to induce T-bet expression after HDM+LPSlo sensitization. As above, 50% CD45.1+WT:50% CD45.2+Tnfr1−/−Tnfr2−/− mixed BM chimeras were generated, sensitized, and the expression of T-bet in mLN CD11b+mDCs was determined. Whereas CD11b+mDCs from WT compartment up-regulated T-bet expression in HDM+LPSlo-sensitized chimeras, CD11b+mDCs from Tnfr1−/−Tnfr2−/− compartment failed to up-regulate T-bet (Fig. 6F,H), suggesting that TNFR signaling in CD11b+mDCs was required for the up-regulation of T-bet following HDM+LPSlo sensitization. To confirm these data we further determined the effect of TNFα neutralization after sensitization. Thus, we administered anti-TNFα mAb to 50% CD45.1+WT:50% CD45.2+Tlr4−/− mixed BM chimeras at the time of HDM+LPSlo sensitization. Anti-TNFα treatment prevented T-bet up-regulation in CD11b+mDCs from both WT and Tlr4−/− compartments (Fig. 6G,I). Together these data suggested that in adults, HDM+LPSlo sensitization induced activation of NF-kB and AP-1 and T-bet up-regulation in CD11b+mDCs by LPS-induced TNFα production and TNF-α-mediated signaling.
To test whether TNFα-mediated signaling in DCs was required to suppress Th2 cell priming in response to low-dose endotoxin sensitization, we generated BM chimeras that expressed TNFR normally (DC-WT) or that were selectively TNFR deficient in CD11c+ DCs after diphtheria toxin (DT) administration (DC-TNFR1&2) (Fig. 6J)(Leon et al., 2012). After reconstitution, we transferred 4get.OT-II CD4+ T cells into the chimeras, sensitized and treated them with DT to ablate CD11c+ cells derived from the Itgax-DTR BM, and analyzed GFP expression by donor 4get.OT-II cells in the mLN (Fig. 6K). Sensitization in the presence of LPSlo prevented GFP in DC-WT chimeras, but not in DC-TNFR1&2 chimeras (Fig. 6L-M). Thus, TNFR is required on CD11c+ DCs to suppress Th2 cell priming after HDM+LPSlo sensitization.
Impaired TNFα-driven activation precludes T-bet expression in infant CD11b+mDCs and their capability to suppress Th2 cell priming.
Our previous data show that in infants, HDM+LPSlo sensitization induced poor NF-kB and AP-1 activation and T-bet up-regulation in CD11b+mDCs, suggesting a defective TNFα-TNFR-mediated signaling. Thus, we next determined cytokine responses in bronchoalveolar lavage (BAL) fluid from adults and 18 d-old infants. HDM+LPSlo sensitization caused great increase in concentrations of TNFα in the BAL fluid of adult mice compared with moderate increase in infants (Fig. 7A). These differences were also observed between adults and 7-14 d-old mice (data not shown). No differences between adults and infants were found in the expression of other cytokines such as IL-1α, IL1β (Fig. 7A) or IL-6 (not shown). These data indicated that infants had a defective production of TNFα after HDM+LPSlo sensitization. To test whether defective T-bet up-regulation in infant CD11b+mDCs after HDM+LPSlo sensitization was due to defective TNFα production, 18 d-old T-bet-ZsGreen mice were sensitized with HDM+LPSlo in presence of different concentrations of TNFα and the expression of T-bet in mLN CD11b+mDCs was determined. As expected, HDM+LPSlo sensitization did not induce significant up-regulation of T-bet in CD11b+mDCs from infant mice (Fig. 7B-C). However, co-administration of TNFα induced T-bet up-regulation in infant CD11b+mDCs to amounts similar to those found in CD11b+mDCs from HDM+LPSlo-sensitized adults (Fig. 7B-C). Similar up-regulation of T-bet in CD11b+mDCs was seen from 7, 14 and 18 d-old mice after exogenous administration of TNF α (Fig. S7A-B). Thus, our data indicated that a defective TNFα production in infants prevented T-bet up-regulation in CD11b+mDCs after HDM+LPSlo sensitization. In addition, administration of TNFα during HDM+LPSlo sensitization recover the capacity of infant mLN CD11b+mDCs to produce IL-12 to amounts similar to adult CD11b+mDCs (Fig. 7D-E).
Figure 7. Defective TNFα signaling prevents T-bet up-regulation in mCD11b+DCs from infants and contributes toward the development of allergen-specific Th2 cell responses.

(A) Cytokines in BAL of day 1 HDM+/−LPS-treated B6 adults and 18d-old infants. (B-C) T-bet-ZsGreen reporter adults and 18d-old infants were i.n treated with HDM-/−5μg LPS+/−10ng or 30ng TNFα. Frequencies of ZsG+ mLN CD11b+ mDCs on day2. (D-E) B6 adults and 18d-old infants were i.n treated with HDM+/−5μg LPS+/−30ng TNFα. Frequencies of IL-12p70+ mLN CD11b+mDCs on day3. (F-G) B6 adults and 18d-old infants were transferred with CD45.1+ OTII.4get cells and i.n treated with HDM+OVA+/−5μg LPS. PBS or 30ng TNFα were co-administered on day 1. Frequencies of mLN EGFP+ donor OTII cells on day 6 (F-G). (H-L) B6 adults and 18d-old infants were i.n treated with HDM+/−5μg LPS+/−30μg TNFα. Some adults also received 250μg anti-TNFα (i.p.). On day 20, mice were i.n challenged with HDM and analyzed on day 25. Frequencies (H) and numbers (I) of IL-13+, IL-13-IL-5+ and IFNγ+ CD4+ T cells in the lungs. Numbers of lung eosinophils, neutrophils and monocytes (J). Numbers of mLN IgE+ ACS (K). SpO2 as determined by pulse-oximetry (L). (M-P) B6 (WT) and Tlr4−/− recipient mice were lethally irradiated and reconstituted with WT and Tlr4−/− BM. Chimeras were i.n treated with HDM+/−5μg LPS. Frequency of lung TNFα+ cells at 24h (M-N) and frequency of T-bet expression in mLN CD11b+mDCs at 72h (O-P). (Q-R) 1: 1 B6 (CD45.1+):Tlr4−/− (CD45.2+) and 1:1 B6 (CD45.1+):Il1r1−/− (CD45.2+) mixed BM chimeras were i.n treated with HDM+/−5μg LPS. TNFα expression 24h later in lung suspensions (Q). Frequencies of WT (CD45.1+) and KO (CD45.2+) cells within the TNFα+ and TNFα− gates (R). **P < 0.01 ***P < 0.001 (unpaired Student’s t test). Data are representative of two independent experiments (mean and S.D. of 3-5 mice per group). Please also see Figure S7.
Finally, we tested whether administration of TNFα during HDM+LPSlo sensitization restored the capacity of low-LPS treatment to abrogate Th2 cell development in infant mice. We transferred 4get.OT-II CD4+ T cells into B6 adults and 18 d-old infants, sensitized them in the presence of TNFα and assessed expression of GFP by donor 4get.OT-II cells (Fig. 7F-G). TNFα administration restored the capacity of LPSlo to prevent GFP expression in infant mice (Fig. 7F-G). We next sensitized adult and 18 d-old infant B6 mice as above, challenged them with HDM and quantified IL-13+, IL-5+ and IFNγ+ CD4+ T cells (Fig. 7H-I). As expected, HDM+LPSlo sensitization prevented the accumulation of IL-13+ and IL-13+IL-5+ CD4+ T cells in the lungs of adults but not in infants (Fig. 7H-I). Importantly, sensitization in the presence of TNF α restored the capacity of LPSlo to prevent the accumulation of IL-13+ and IL-13+IL-5+ CD4+ T cells in infants (Fig. 7H-I). Likewise, HDM+LPSlo sensitization in the presence of TNF α also prevented the accumulation of lung eosinophils (Fig. 7J and S7C-D), mLN IgE+ASCs (Fig. 7K and S7E), the induction of goblet cell hyperplasia (Fig. S7F) and improved SpO2 (Fig. 7L) in infant mice. Same results were seen when analyzing 10 d-old mice (data not shown). In contrast, TNF α neutralization after HDM+LPSlo sensitization in adult mice abolished the suppressive effect of LPS on asthma, including the accumulation of IL-13+CD4+ T cells (Fig. S7I-J) and eosinophils (Fig. S7G-H) in the lungs, the goblet cell hyperplasia (Fig. S7F) and the beneficial effect on SpO2 (Fig. 7L). Thus, our data indicated that a defective TNF α production prevents the suppression of Th2 cell responses to HDM in the presence of low doses of endotoxin.
To assess the cellular source of TNF α after HDM+LPSlo sensitization, we first evaluated whether LPS-driven TNF α production was mediated by radioresistant (including lung epithelium) or hematopoietic cells in the lungs. Therefore B6 (WT) or Tlr4−/− recipients were irradiated and reconstituted with BM from WT or Tlr4−/− mice. After reconstitution, TNF α production in the lungs was determined after HDM+LPSlo sensitization. Importantly, chimeric Tlr4−/− mice transplanted with WT BM, but not chimeric mice transplanted with Tlr4−/− BM, were able to produce WT amounts of TNF α + cells (Fig. 7M-N), suggesting that radiosensitive (hematopoietic) cells contained the principal source of TNF+ cells. Likewise, only mLN CD11b+mDCs of HDM+LPSlo-sensitized chimeric mice transplanted with WT BM, but not with Tlr4−/− BM, were able to up-regulate T-bet (Fig. 7O-P). Next, we evaluated whether hematopoietic cells directly responded to LPS to produce TNF α. Thus, WT:Tlr4−/− BM chimeras were sensitized and TNF α production in the lungs was determined. As expected, HDM+LPSlo sensitization induced TNF α production (Fig. 7Q), but importantly, TNF α+ cells were principally restricted to the WT phenotype (Fig. 7Q-R), suggesting that TNF α was produced by lung hematopoietic cells after direct recognition of LPS by TLR4. Conversely, the analysis of WT:Il1r1−/− BM chimeras showed that lung cells did not require to express IL-1R to produce TNF α in response to LPS (Fig. 7Q-R). These data suggested that TNF α -dependent up-regulation of T-bet in CD11b+mDCs was driven by hematopoietic cells that produce TNF α in response to LPS, but not by epithelial cells.
Collectively, our data indicated that adult CD11b+mDCs up-regulate T-bet in response to TNFα produced following HDM+LPSlo sensitization, which in turn promotes IL-12 production, thereby preventing Th2 cell differentiation by inducing T-bet expression in responding CD4+ T cells. In contrast, in early life and during a time window from d7 to d20 after birth, the CD11b+mDCs fail to up-regulate T-bet in response to low doses of LPS, due to impaired production of TNFα; which prevents optimal IL-12 production and subsequent Th2 cell inhibition despite the presence of LPS.
DISCUSSION.
Previous studies in mice have shown that during a time window from d7 to d20 after birth, the immune system is prone to allergic sensitization and Th2-driven allergic responses (de Kleer et al., 2016; Gollwitzer et al., 2014; Steer et al., 2017), suggesting that there is a “window of opportunity” in the postnatal period which influences the immune responses to allergens. Comparative immunological evaluations have demonstrated that the immunity in mice at 7 days is comparable to term human neonates, whereas mice <20 days old resemble infants (Siegrist, 2001). Thus, 1 to 3-wk-old mice yielded observations very similar to those of human neonates (0-2 months-old) and infants (under 1-2 years) (Siegrist, 2001). In correlation with animal studies, primary allergen sensitizations to environmental Ags gradually develop and peak in children at 2-3 years of age (Holt, 1996) and are associated with later development of asthma and allergic disease (Lodge et al., 2011). Therefore, both in human and mice there is a susceptibility window for inhalant allergen sensitization that covers the neonatal and infancy periods. Several studies in mice have evaluated this propensity to develop allergic sensitization during infancy (de Kleer et al., 2016; Steer et al., 2017). However, the understanding of the allergic sensitization process in the context of exposure to environmental endotoxin during infancy is still not clear. Previous studies show that LPS exposure prevents the development of allergic-Th2 cell responses (Daan de Boer et al., 2013; Gereda et al., 2000; Schuijs et al., 2015; Stein et al., 2016; Zhu et al., 2010). Our data demonstrated, however, that adults and infants respond to LPS with different thresholds. As a result, while relative low-dose LPS exposure normally prevents adults from developing allergic Th2 cell responses, infant mice require a relatively high-LPS environment to achieve the same prophylactic effect. The inability of low-LPS exposure to prevent Th2 cell differentiation in infants was attributed to the inability of responding infant CD4+ T cells to up-regulate T-bet, which we found was required to prevent Th2 cell development to HDM. Impaired T-bet expression, however, was not due to an intrinsic defect in the infant CD4+ T cells. Instead, we found that CD11b+mDCs from HDM+LPSlo sensitized infants poorly produce IL-12, thereby preventing IL-12-dependent T-bet up-regulation in responding CD4+ T cells. Mechanistically, we demonstrated that T-bet up-regulation in CD11b+mDCs is required for normal IL-12 production. However, while adult CD11b+mDCs normally up-regulate T-bet following HDM+LPSlo sensitization, infant CD11b+mDCs failed to express T-bet, which prevents CD11b+mDCs from secreting IL-12. Thus, the differential capacity to up-regulate T-bet in infant and adult CD11b+mDCs provides the underlying mechanism for how LPS prevents allergic-Th2 cell responses in adults and infants with different thresholds. Our data indicate that in addition of controlling important fate decisions in T cells (Szabo et al., 2000; Zhu et al., 2012), T-bet also critically regulates DC activity.
Importantly, our data show that T-bet up-regulation in CD11b+mDCs does not require direct recognition of LPS but instead it requires LPS-induced TNFα production and TNFα-TNFR-triggered signaling in CD11b+mDCs. Our data further show the requirement for a crosstalk between TLR4+ accessory hematopoietic cells producing TNFα and TNFR+CD11b+mDCs. Whereas adult hematopoietic cells produce large amounts of TNFα in response to HDM+LPS sensitization, infants have impaired TNFα production, which prevents TNFα-TNFR signaling and T-bet up-regulation in CD11b+mDCs. Therefore, the capacity of LPS to prevent allergic-Th2 cell responses relies in the capacity to induce increased amounts of TNF-α. Our data evidence a largely unexplored role of TNFα in mediating in vivo DC activation, in addition to its well-defined role in apoptosis and inflammation. In this way, TNFα act as a rheostat that can tune the activation of CD11b+mDCs and the subsequent balance of the T cell responses to inhaled Ags. Importantly, the regulatory activity of this rheostat is modulated by age.
Previous studies suggest that CD4+ T cells primed by CD103+mDCs preferentially differentiate into Th1 cells, while CD11b+mDCs promote Th2 cell differentiation (Conejero et al., 2017; Gao et al., 2013; Kumamoto et al., 2013; Leon, 2017; Plantinga et al., 2013). As such, CD103+mDCs produce IL-12p40 following HDM sensitization, thereby preventing Th2 cell differentiation (Conejero et al., 2017). Here, we found that LPS prevented Th2 cell differentiation in HDM-sensitized mice by an IL-12 dependent mechanism. We found, however, that CD103+mDCs are quickly depleted from the lung after HDM+LPS sensitization, possibly due to increased LPS-driven epithelial damage (Sung et al., 2006). In contrast, the number of CD11b+mDCs and their capacity to secrete IL-12 dramatically increases following HDM+LPS sensitization, thereby becoming the dominant IL-12+ DC subset in the mLN. In addition, CD11b+mDCs are the principal DC subset that capture and transport HDM-derived Ags from the lung into the mLN. Thus, given that the numbers of IL-12+CD11b+mDCs largely increase in HDM+LPS sensitized adult mice and that IL-12 prevents Th2 cell differentiation, we conclude that IL-12+CD11b+mDCs, and not CD103+ mDCs, are the main drivers preventing allergic Th2 cell responses in adult HDM+LPS-sensitized mice. Supporting this conclusion, LPS normally prevented Th2 cell differentiation in Batf3−/− mice despite the absence of CD103+mDCs. In contrast, LPS failed to suppress allergic-Th2 cell responses when CD11b+mDCs were unable to normally produce IL-12, such in ItgaxCRETbx21fl/fl or infant mice. Collectively, our results demonstrate that CD11b+mDCs have the ability to either promote or prevent Th2 cell responses depending on the amount of LPS. Therefore, the relative contribution of CD11b+ and CD103+mDCs to the development of allergic-Th2 cell responses is more complex than initially expected, as is highly influenced by the environmental amount of microbial products rather than being predetermined by the their intrinsic nature alone. Importantly, our data show that whereas IL-12 production by CD11b+mDCs required the up-regulation of T-bet, IL-12 production by CD103+mDCs is T-bet independent. However, the molecular mechanisms by which T-bet is only required in CD11b+mDCs is unclear. Various transcription factors, including IRF family members, have been implicated in the regulation of IL-12 production by DC subsets (Gao et al., 2013; Takaoka et al., 2005; Taki et al., 1997; Williams et al., 2013). Here, we found no differences in the expression of IRF1 or IRF5 between CD103+ and CD11b+mDCs from infant and adults. Importantly, the transcription factor IRF4 is highly expressed in CD11b+ but not in CD103+mDCs and essentially contributes to the differentiation and function of CD11b+mDCs. For example, IRF4 drives CD11b+mDCs to promote Th2 cell differentiation (Gao et al., 2013; Kumamoto et al., 2013; Williams et al., 2013), inhibits the capacity of DCs and macrophages to produce IL-12 (Akbari et al., 2014; Honma et al., 2005) and competes with IRF5 to inhibit IRF5-dependent up-regulation of pro-inflammatory cytokines, such as IL-12 and IL-6 (Negishi et al., 2005). These data suggest a role for IRF4 in the negative-feedback regulation of IL-12 production and since IRF4 is restricted to CD11b+mDCs, IRF4 may selectively control IL-12 gene regulation specifically in CD11b+mDCs. Therefore one could speculate that T-bet up-regulation, specifically in CD11b+mDCs is important to overcome the suppressive function of IRF4 in IL-12 production.
In summary, our comparative analysis between adult and neonatal-infant mice offers mechanistic insights into how priming of allergen-specific Th2 cell responses are differentially regulated during childhood and adulthood, suggests a perspective for the biological mechanism underlying the hygiene hypothesis and provides a plausible mechanism to explain higher susceptibility to allergic airway inflammation observed in children.
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the Lead Contact, Beatriz León. (bleon@uab.edu).
Experimental Model and Subject Details
Mice.
The mouse strains used in these experiments include: C57BL/6J (B6), B6.SJL-Ptprca Pepcb/BoyJ (CD45.1+ B6 congenic), C57BL/6-Tg(TcraTcrb)425Cbn/J (OTII), B6.129-Il4tm1Lky/J (B6.4get IL-4 reporter mice), B6.129S(C)-Batf3tm1Kmm/J (Baft−/−), B6.129-Tbx21tm2Srnr/J (Tbx21fl/fl), C57Bl/6J-Tg(Itgax-cre,-EGFP)4097Ach/J (Itgax-cre,-EGFP), B6.129S6-Tbx21tm1Glm/J (Tbx21−/−), B6(Cg)-Tlr4tm1.2Karp/J (Tlr4−/−), B6.129S7-Il1r1tm1Imx/J (Il1r1−/−), B6.129S-Tnfrsf1atm1Imx Tnfrsf1btm1Imx/J (Tnfr1−/−Tnfr2−/−) B6.FVB-Tg(Itgax-DTR/EGFP)57Lan/J (Itgax-DTR,-EGFP) and C57BL/6 T-bet-ZsGreen reporter (TBGR). B6.4get mice were originally obtained from Dr. M. Mohrs (Trudeau Institute). TBGR mice were developed and kindly provided by Dr. J. Zhu (NIAID, NIH). All other mice were originally obtained from Jackson Laboratory and were bred and housed in the University of Alabama at Birmingham animal facility under specific pathogen–free conditions. Experiments were equally performed with male and female mice. The University of Alabama at Birmingham Institutional Animal Care and Use Committee approved all procedures involving animals.
Method Details
Immunizations.
HDM (Dermatophagoides pteronyssinus and D. farinae) extracts were obtained from Greer laboratories (<12 EU/mg endotoxin). Mice were administered (i.n. or i.t.) with 50μg of HDM extract +/− 5μg of LPS-free EndoFit OVA (InvivoGen) +/− LPS from Escherichia coli 0111:B4 (Sigma-Aldrich) +/− 150 ng of recombinant IL-12 (PeproTech) +/− 10-30 ng of recombinant TNFα (R&D Systems) daily for 1-3 days and challenged (i.n.) with 50μg of HDM +/− 5μg of LPS-free EndoFit OVA. i.n. and i.t. administrations were given in 50μl of PBS for adults and 18-20d-old infant mice and in 10μl of PBS for 7d-old, 10d-old and 14d-old infant mice. In some experiments, mice were intraperitoneally administered (i.p.) with 250μg of anti-TNFα mAb (BioXCell) at the time of initial sensitization. To deplete CD11c+ cells, Itgax-DTR BM chimeras were i.p. treated with 60 ng DT (Sigma-Aldrich) on day 3 after sensitization. In some experiments HDM extract was labeled with AF647 labeling kit (Invitrogen) prior to administration to mice.
BM chimeras.
Recipient mice were irradiated with 950 Rads from a high-energy X-rays source delivered in a split dose and reconstituted with 107 total BM cells. Mice were allowed to reconstitute for at least 8-12 weeks before HDM treatment.
Cell preparation and flow cytometry.
Lungs were isolated, cut into small fragments and digested for 45 min at 37°C with 0.6 mg/ml collagenase A (Sigma) and 30 μg/ml DNAse I (Sigma) in RPMI-1640 medium (GIBCO). Digested lungs, mLN or spleens were mechanically disrupted by passage through a wire mesh. Red blood cells were lysed with 150 mM NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA. Fc receptors were blocked with anti-mouse CD16/32 (5 μg/ml; BioXCell5), followed by staining with fluorochrome-conjugated Ab. Fluorochrome-labeled anti-B220 (RA3-6B2), anti-CD4 (GK1.5), anti-CD11b (M1/70), anti-CD11c (HL3), anti-CD44 (IM7), anti-CD45.1 (A20), anti-CD45.2 (104), anti-CD69 (H1-2F3), anti-CD86 (GL1), anti-CD103 (M290), anti- CD138 (281-2), anti-IgE (R35-72y), anti-Ly6c (AL-21), anti-Siglec-F (E50-2440) and anti-Siglec-H (440c) were from BD Biosciences. Fluorochrome-labeled anti-CD64 (X54-5/7.1) and anti-MHC class II (M5/114.15.2) were from Biolegend.
For intracellular cytokine staining of T cells, cell suspensions were stimulated with PMA (20 ng/ml) plus Calcimycin (1μg/ml) in the presence of Brefeldin-A (BFA, 10 μg/ml) for 4h. Restimulated cells were surface stained, fixed in 4% paraformaldehyde, permeabilized with 0.1% saponin, and stained with Abs against IL-13 (13A; eBiosciences), IFNγ (XMG1.2; BD-Biosciences) and IL-5 (TRFK5; BioLegend). For cytokine staining of DCs, cell suspensions were surface stained, fixed, permeabilized, and stained with anti-IL-12 (C15.6; BD-Biosciences). For TNFα staining, lung cell suspensions were incubated in the presence of BFA for 4h, fixed, permeabilized, and stained with anti-TNFα (MP6-XT22; BD-Biosciences). T-bet, GATA-3, Foxp3, IRFs, c-Rel, P-c-Jun and IkBζ intracellular staining was performed using the Mouse regulatory T cell staining kit (eBioscience) and Abs against T-bet (4B10; biolegend), GATA-3 (TWAJ; eBioscience), Foxp3 (.FJK-16s; eBioscience), IRF1 (ab216114; Abcam), IRF4 (IRF4.3E4; BioLegend), IRF5 (ab212239; Abcam), c-Rel (sc-6955; Santa Cruz), P-c-Jun (D47G9; Cell Signaling Technology) and IkBζ (LK2NAP; eBioscience). Flow cytometry was performed on Attune NxT and FACSCanto II (BD-Biosciences) instruments.
Cell purifications, cell transfers and in vitro cultures.
CD4+ T cells were isolated by MACs (Miltenyi Biotec) from the spleens of naíve OTII mice. CD11c+ cells were isolated by MACs from pooled lungs or mLN of day 3 HDM+/−OVA+/−LPS-treated mice. All T and CD11c+ cell preparations were more than 95% pure. HDM+/−LPS sensitization greatly reduced the proportion of CD11c+ alveolar macrophages in the lungs (data not shown) and purification of CD11c+ cells from the lungs of sensitized mice resulted in more than 80-90% enrichment of CD11c+ Siglec-F− MHChi lung DCs. Equivalent numbers (25 × 103) of naíve OTII cells were transferred (i.v.) into naíve congenic recipients. Equivalent numbers (1 × 106) of CD11c+ lung cells were transferred (i.n.) into naíve congenic infant recipients daily for 3 days. For in vitro cultures, OTII cells were labeled for 10 min at 37°C with CellTrace™ CFSE (ThermoFisher). 2×104 DCs and 1×105 CFSE-labeled OTII cells were cultured in 200 μl of complete medium in round-bottomed 96-well plates for 72 h at 37°C. Complete medium included RPMI 1640 supplemented with sodium pyruvate, HEPES, non-essential amino acids, penicillin, streptomycin, 2-mercaptoethanol and 10% heat-inactivated FBS from Fisher Scientific.
BAL collection and measurement of cytokines.
BAL was collected using 0.5-1 ml sterilized PBS per mouse. The BAL fluids were centrifuged at 5,000íg for 10 min and the supernatants were frozen at − 80°C. Cytokine measurements were performed using ProcartaPlex assay (Life Technologies) and Luminex xMAP technology as per manufacturer's instructions.
Lung histology.
The lungs were fixed in buffered formalin (10%). Microtome sections of paraffin-embedded lungs were stained with periodic acid-Schiff (PAS) for visualization of goblet cells.
Pulmonary function assessment.
To measure airway responsiveness, mice on day 5 post-challenge were mechanically ventilated and challenged with increasing concentrations of methacholine, as described previously (Song et al., 2011). Briefly, mice were anesthetized, intubated, connected to a ventilator (FlexiVent; SCIREQ, Montreal, PQ, Canada), and ventilated at a rate of 160 breaths per minute. Airway responsiveness was recorded at baseline and after a linear dose response with methacholine challenge (10–75 mg/ml; Sigma-Aldrich). Data were normalized to basal airway resistance and expressed as changes induced by methacholine. To measure arterial O2 saturation (SpO2), the MouseOx™ Pulse-oximeter (Starr Life Sciences, Oakmont PA) was used on conscious mice 5 days-post-challenged, in accordance with manufacturer's instructions. Mice were shaved and the pulse-oximeter collar was placed behind the ears. To ensure accurate measurement, SpO2 data points were excluded from analysis if received an error code during measurement.
RNA-sequencing (RNA-seq).
mCD11b+DCs (B220−CD64−CD103− CD11c+MHCIIhiCD11b+) from adult (7wks-old) and infant (2-3wks-old) mice were sorted from the mLN 24h after HDM+LPS sensitization using a FACSAria (BD Biosciences) after positive selection with anti-CD11c MACS beads (Miltenyi Biotec). RNA was isolated from the sorted cells using the RNeasy Plus Micro RNA purification kit (QIAGEN). Three or four replicates from three or four independent experiments for each condition were analyzed with RNA-seq. Library preparation and RNA sequencing was conducted through Genewiz. Libraries were sequenced using a 1Å~ 50-bp single end rapid run on the HiSeq2500 platform. Sequence reads were trimmed to remove possible adaptor sequences and nucleotides with poor quality (error rate < 0.05) at the end. After trimming, sequence reads shorter than 30 nucleotides were discarded. Remaining sequence reads were mapped to the Mus musculus mm10 reference genome using CLC genomics workbench v. 9.0.1. Differential gene expression was determined using DESeq2. Genes with an adjusted P value < 0.05 and an absolute log1.5-fold change (ratio +/−0.58) were considered significantly differentially expressed genes between adult and infant mCD11b+DCs.
GSEA, hierarchical clustering and visualization of RNA sequencing results.
GSEA was performed using the Molecular Signatures Database on the publically available MIT BROAD Institute server. We ranked the 173 genes obtained from RNA sequencing (Genewiz) according to a logarithmic transformation of each gene’s P-value multiplied by the sign of the corresponding logarithmic fold change, and subsequently used these rank lists to perform a gene set enrichment analysis (Broad Institute’s GSEA Java app, version 3.0). Given gene sets can include both activated and repressed genes, we additionally performed an extended enrichment assessment of all identified gene sets. Separately, we performed hierarchical clustering analysis(Bar-Joseph et al., 2001; Eisen et al., 1998) of TNFα induced genes that are differentially expressed in adult vs infant mice, using Matlab (version R2017b). Differential clusters are presented in form of an annotated heatmap based on standardized expression values, along with the resulting hierarchical clustering dendogram.
Quantification and Statistical Analysis.
All plots and histograms were plotted in FlowJo v.9 software (Treestar). GraphPad Prism software (Version 7) was used for data and statistical analysis. Data were analyzed using the unpaired Student’s t test. Values of P < 0.05 were considered significant (*p < 0.05, **p < 0.01, ***p < 0.001).
Data and Software Availability
The accession number for the raw data files for the RNA-Seq analyses reported in this paper is GEO GSE110046.
Supplementary Material
Table S1. Differentially expressed genes in Infant CD11b+mDCs vs. Adult CD11b+mDCs. Related to Figure 6.
Table S2. Genes regulated by NF-kB in response to TNFα. Related to Figure 6.
Table S3. Genes of the Hallmark TNFA via NFKB signaling pathway down-regulated in Infant CD11b+mDCs vs. Adult CD11b+mDCs. Related to Figure 6.
Highlights.
Infant mice require high doses of LPS to suppress Th2-driven allergic asthma to HDM T-bet+ lung CD11b+DCs inhibit Th2 cell development after HDM+LPS sensitization T-bet expression on CD11b+DCs dependents on TNFα production and TNFR signaling. Infants have impaired ability to induce TNFα and T-bet+CD11b+DCs in response to LPS
ACKNOWLEDGEMENTS.
We thank Dr. J. Zhu (NIAID, NIH-Bethesda) for providing TBGR mice and Becca Burnham, Uma Mudunuru and Thomas S Simpler for animal husbandry. The work was supported by UAB and NIH grants R01 AI116584 to B.L., U01 ES026458 to S.M. and R01 AI110480 to A.B.T.
Footnotes
DECLARATION OF INTERESTS.
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Akbari M, Honma K, Kimura D, Miyakoda M, Kimura K, Matsuyama T, and Yui K (2014). IRF4 in dendritic cells inhibits IL-12 production and controls Th1 immune responses against Leishmania major. J Immunol 192, 2271–2279. [DOI] [PubMed] [Google Scholar]
- Ballesteros-Tato A, Leon B, Lund FE, and Randall TD (2010). Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8(+) T cell responses to influenza. Nat Immunol 11, 216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros-Tato A, Randall TD, Lund FE, Spolski R, Leonard WJ, and Leon B (2016). T Follicular Helper Cell Plasticity Shapes Pathogenic T Helper 2 Cell-Mediated Immunity to Inhaled House Dust Mite. Immunity 44, 259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Joseph Z, Gifford DK, and Jaakkola TS (2001). Fast optimal leaf ordering for hierarchical clustering. Bioinformatics 17 Suppl 1, S22–29. [DOI] [PubMed] [Google Scholar]
- Conejero L, Khouili SC, Martinez-Cano S, Izquierdo HM, Brandi P, and Sancho D (2017). Lung CD103+ dendritic cells restrain allergic airway inflammation through IL-12 production. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daan de Boer J, Roelofs JJ, de Vos AF, de Beer R, Schouten M, Hommes TJ, Hoogendijk AJ, de Boer OJ, Stroo I, van der Zee JS, et al. (2013). Lipopolysaccharide inhibits Th2 lung inflammation induced by house dust mite allergens in mice. Am J Respir Cell Mol Biol 48, 382–389. [DOI] [PubMed] [Google Scholar]
- de Kleer IM, Kool M, de Bruijn MJ, Willart M, van Moorleghem J, Schuijs MJ, Plantinga M, Beyaert R, Hams E, Fallon PG, et al. (2016). Perinatal Activation of the Interleukin-33 Pathway Promotes Type 2 Immunity in the Developing Lung. Immunity 45, 1285–1298. [DOI] [PubMed] [Google Scholar]
- Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG, et al. (2010). Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med 207, 823–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, and Botstein D (1998). Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Nish SA, Jiang R, Hou L, Licona-Limon P, Weinstein JS, Zhao H, and Medzhitov R (2013). Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity 39, 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gereda JE, Leung DY, Thatayatikom A, Streib JE, Price MR, Klinnert MD, and Liu AH (2000). Relation between house-dust endotoxin exposure, type 1 T-cell development, and allergen sensitisation in infants at high risk of asthma. Lancet 355, 1680–1683. [DOI] [PubMed] [Google Scholar]
- Gollwitzer ES, Saglani S, Trompette A, Yadava K, Sherburn R, McCoy KD, Nicod LP, Lloyd CM, and Marsland BJ (2014). Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med 20, 642–647. [DOI] [PubMed] [Google Scholar]
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, and Yona S (2014). Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 14, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt PG (1996). Primary allergic sensitization to environmental antigens: perinatal T cell priming as a determinant of responder phenotype in adulthood. J Exp Med 183, 1297–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma K, Udono H, Kohno T, Yamamoto K, Ogawa A, Takemori T, Kumatori A, Suzuki S, Matsuyama T, and Yui K (2005). Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc Natl Acad Sci U S A 102, 16001–16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Banerjee A, Takemoto N, Gordon SM, Dejong CS, Shin H, Hunter CA, Wherry EJ, Lindsten T, and Reiner SL (2008). Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science 321, 408–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, and Iwasaki A (2013). CD301b(+) dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity 39, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon B (2017). T Cells in Allergic Asthma: Key Players Beyond the Th2 Pathway. Curr Allergy Asthma Rep 17, 43. [DOI] [PubMed] [Google Scholar]
- Leon B, Ballesteros-Tato A, Browning JL, Dunn R, Randall TD, and Lund FE (2012). Regulation of T(H)2 development by CXCR5+ dendritic cells and lymphotoxin-expressing B cells. Nat Immunol 13, 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscomb MW, Chen L, Taylor JL, Goldbach C, Watkins SC, Kalinski P, Butterfield LH, Wesa AK, and Storkus WJ (2009). Ectopic T-bet expression licenses dendritic cells for IL-12-independent priming of type 1 T cells in vitro. J Immunol 183, 7250–7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge CJ, Lowe AJ, Gurrin LC, Hill DJ, Hosking CS, Khalafzai RU, Hopper JL, Matheson MC, Abramson MJ, Allen KJ, and Dharmage SC (2011). House dust mite sensitization in toddlers predicts current wheeze at age 12 years. J Allergy Clin Immunol 128, 782–788 e789. [DOI] [PubMed] [Google Scholar]
- Lugo-Villarino G, Maldonado-Lopez R, Possemato R, Penaranda C, and Glimcher LH (2003). T-bet is required for optimal production of IFN-gamma and antigen-specific T cell activation by dendritic cells. Proc Natl Acad Sci U S A 100, 7749–7754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoli M, Fabian D, Holt S, Beasley R, and Global Initiative for Asthma, P. (2004). The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy 59, 469–478. [DOI] [PubMed] [Google Scholar]
- Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, Matsuyama T, Taniguchi T, and Honda K (2005). Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A 102, 15989–15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, Vanhoutte L, Neyt K, Killeen N, Malissen B, et al. (2013). Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 38, 322–335. [DOI] [PubMed] [Google Scholar]
- Reynolds LA, and Finlay BB (2017). Early life factors that affect allergy development. Nat Rev Immunol. [DOI] [PubMed] [Google Scholar]
- Schuijs MJ, Willart MA, Vergote K, Gras D, Deswarte K, Ege MJ, Madeira FB, Beyaert R, van Loo G, Bracher F, et al. (2015). Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science 349, 1106–1110. [DOI] [PubMed] [Google Scholar]
- Siegrist CA (2001). Neonatal and early life vaccinology. Vaccine 19, 3331–3346. [DOI] [PubMed] [Google Scholar]
- Song W, Wei S, Liu G, Yu Z, Estell K, Yadav AK, Schwiebert LM, and Matalon S (2011). Postexposure administration of a {beta}2-agonist decreases chlorine-induced airway hyperreactivity in mice. Am J Respir Cell Mol Biol 45, 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steer CA, Martinez-Gonzalez I, Ghaedi M, Allinger P, Matha L, and Takei F (2017). Group 2 innate lymphoid cell activation in the neonatal lung drives type 2 immunity and allergen sensitization. J Allergy Clin Immunol 140, 593–595 e593. [DOI] [PubMed] [Google Scholar]
- Stein MM, Hrusch CL, Gozdz J, Igartua C, Pivniouk V, Murray SE, Ledford JG, Marques dos Santos M, Anderson RL, Metwali N, et al. (2016). Innate Immunity and Asthma Risk in Amish and Hutterite Farm Children. N Engl J Med 375, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, Hartig H, Sundberg JP, Servick S, Kaufmann G, et al. (2007). Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity 26, 629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung SS, Fu SM, Rose CE Jr., Gaskin F, Ju ST, and Beaty SR (2006). A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol 176, 2161–2172. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, and Glimcher LH (2000). A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak TW, and Taniguchi T (2005). Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434, 243–249. [DOI] [PubMed] [Google Scholar]
- Taki S, Sato T, Ogasawara K, Fukuda T, Sato M, Hida S, Suzuki G, Mitsuyama M, Shin EH, Kojima S, et al. (1997). Multistage regulation of Th1-type immune responses by the transcription factor IRF-1. Immunity 6, 673–679. [DOI] [PubMed] [Google Scholar]
- Trinchieri G (2003). Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 3, 133–146. [DOI] [PubMed] [Google Scholar]
- Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, and Beyaert R (2008). TLR-4, IL-1R and TNF-R signaling to NF-kappaB: variations on a common theme. Cell Mol Life Sci 65, 2964–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JW, Tjota MY, Clay BS, Vander Lugt B, Bandukwala HS, Hrusch CL, Decker DC, Blaine KM, Fixsen BR, Singh H, et al. (2013). Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Commun 4, 2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, Guo L, Yagi R, Yamane H, Punkosdy G, et al. (2012). The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity 37, 660–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Oh SY, Zheng T, and Kim YK (2010). Immunomodulating effects of endotoxin in mouse models of allergic asthma. Clin Exp Allergy 40, 536–546. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially expressed genes in Infant CD11b+mDCs vs. Adult CD11b+mDCs. Related to Figure 6.
Table S2. Genes regulated by NF-kB in response to TNFα. Related to Figure 6.
Table S3. Genes of the Hallmark TNFA via NFKB signaling pathway down-regulated in Infant CD11b+mDCs vs. Adult CD11b+mDCs. Related to Figure 6.
Data Availability Statement
The accession number for the raw data files for the RNA-Seq analyses reported in this paper is GEO GSE110046.