

Abstract
Purpose:
We previously reported preventive and therapeutic effects of Smad7, a multifunctional protein, on radiation-induced mucositis in mice without promoting human oral cancer cell survival or migration in vitro. The current study aims to determine whether a Smad7-based biologic can treat existing oral mucositis during radiotherapy for oral cancer and whether this treatment compromises RT-induced cancer cell killing in neighboring oral cancer.
Experimental Design:
We transplanted human oral cancer cells into the tongues of mice and applied craniofacial irradiation to simultaneously kill tumor cells and induce oral mucositis, thus modeling RT and mucositis in oral cancer patients. We topically applied a recombinant human Smad7 protein fused with the cell-penetrating Tat tag (Tat-Smad7) to the oral mucosa of tumor-bearing mice post RT when oral mucositis began to develop.
Results:
Topically applied Tat-Smad7 penetrated cells in both the oral mucosa and oral cancer, attenuating TGFβ and NFκB signaling as well as inflammation at both sites. Tat-Smad7 treatment alleviated oral mucositis with reductions in DNA damage and apoptosis in keratinocytes, but increased keratinocyte proliferation compared to vehicle-treated mucositis lesions. In contrast, adjacent oral cancer exposed to Tat-Smad7 did not show alterations in proliferation or direct DNA damage, but showed increased oxidative stress damage and apoptosis compared to tumors treated with vehicle.
Conclusion:
Our results suggest that short-course Tat-Smad7 application to oral mucositis promotes its healing but does not compromise the cytotoxic effect of RT on oral cancer and has context-specific effects on oral mucosa vs. oral cancer.
Keywords: Oral ulcer, Head and neck neoplasm, Radiation toxicity, TGFbeta, NFkappaB
Introduction
Oral mucositis, painful ulceration of the oral mucosa, is a common toxic effect of radiation for bone marrow transplant, craniofacial RT for head and neck cancer (HNC), and chemo-radiotherapy for all cancer types (1). ~70% of HNC patients develop oral mucositis during treatment, which can be severe enough to cause reduction in oral intake or premature withdrawal from cancer treatment (1,2). New RT technologies such as intensity modulated RT (IMRT) and stereotactic body RT (SBRT) more precisely target cancer lesions and spare more normal tissue. However, these treatments do not reduce acute toxicity in the oral mucosa of HNC patients (3), because neighboring mucosa is still exposed to high intensity radiation, albeit at a lower dose than the cancer. Further, certain patients are at high risk for oral mucositis regardless of treatment regimen (4). Palifermin, recombinant human keratinocyte growth factor (KGF), is the only FDA approved targeted therapy for preventing oral mucositis in bone-marrow transplant patients (4% of the at-risk population), but it has no effect on existing mucositis (5). Palifermin clinical trials in oral cancer patients showed modest prevention of severe oral mucositis (6,7). A major challenge in treating oral mucositis is to repair ulcerated mucosa without promoting cancer, as growth factors (e.g., KGF) and their receptors are often overexpressed in cancer cells. To date, there is no FDA approved drug to treat oral mucositis in cancer patients. Clinical trials for GC4419, a superoxide dismutase mimetic, to treat oral mucositis in HNC patients revealed reductions in severe oral mucositis cases (8). GC4419 requires i.v. infusions one hour prior to each of 14 doses of radiation. Because it is difficult to predict which patients will develop severe oral mucositis, we sought to develop a therapeutic intervention that is topically applied to existing oral mucositis and targets multiple pathogenic processes of oral mucositis.
Oral mucositis develops as a consequence of complex molecular and cellular pathobiology processes, leading to apoptosis of basal epithelia cells, loss of epithelial renewal, atrophic damage, excessive inflammation and ulceration (9). Our previous study discovered that in addition to NFκB activation, activated transforming growth factor β (TGFβ) signaling contributes greatly to radiation-induced oral mucositis (10). TGFβ is a potent growth inhibitor and apoptosis inducer for epithelial cells and a pro-informatory cytokine in oral mucosa (11). To dampen both TGFβ and NFκB pathways to treat oral mucositis, we developed a recombinant Smad7 protein that contains human Smad7 fused to the HIV-1 Tat protein transduction domain. Tat-Smad7 protein rapidly penetrates cells upon contact (10). Local delivery of Tat-Smad7 to mouse oral mucosa shows prophylactic and therapeutic effects on radiation-induced oral mucositis (10). A remaining question is how to effectively utilize this therapeutic intervention without compromising RT-directed cancer cell killing. Smad7 can be either tumor suppressive or tumor promotive in different cancer types (12). For cancers outside the oral cavity, the concern for potential systemic tumor promoting effects of Tat-Smad7 is relatively minor because orally administered Tat-Smad7 protein will be degraded as it passes through the digestive tract. However, when RT-induced oral mucositis in oral cancer patients is treated with Tat-Smad7, the oral cancer will also be exposed to Tat-Smad7. Although increasing epithelial cell proliferation and reducing apoptosis in oral mucositis promotes wound healing, these effects could compromise RT-directed cancer cell death. We have previously shown that Tat-Smad7 increases survival of human oral keratinocytes but not HNSCC cells after RT. Similarly, Tat-Smad7 increases migration of normal keratinocytes but not HNSCC cells (10). These in vitro data indicate the necessity of in vivo testing of Tat-Smad7 using a model system mimicking RT in human oral cancer with a RT dose sufficient to induce oral mucositis. Although our previous study shows both preventive and therapeutic effect of Tat-Smad7 on oral mucositis (10), the current study focused on assessing the therapeutic effect of Tat-Smad7 on promoting healing of existing oral mucositis in a model of oral cancer. We chose two HNSCC cell lines: FaDu, with a SMAD4 deletion representing altered TGFβ signaling found in tobacco-associated oral cancer (13), and UM-SCC-1, with wildtype SMAD4, representing intact TGFβ signaling in tumor cells. We provide evidence that Tat-Smad7 application to RT-induced oral mucositis with neighboring oral cancer effectively promotes oral mucositis healing while paradoxically increasing tumor cell death in RT treated oral cancer.
Materials and Methods
Cell lines.
Two human oral cancer cell lines were used. FaDu was purchased from ATCC and UM-SCC-1 (14) was provided by MTA through University of Michigan. Cells were authenticated by STR profiling (University of Colorado Cancer Center Protein Production, Monoclonal Antibody, Tissue Culture Shared Resource) prior to our experiments. Cells were cultured in DMEM containing 10% FBS.
Generation and application of Tat-Smad7.
As previously described, we produced recombinant Tat-tagged, human Smad7 protein as a GST fusion protein from E. coli (10). Tat-Smad7 was cleaved from GST with PreScission protease followed by size exclusion chromatography (SEC) to purify Tat-Smad7 protein as a monomer (Supplementary Fig. 1). For in vivo treatment, 1 μg Tat-Smad7 (in 30 μL PBS containing 30% glycerol) was applied to mouse oral cavity; food/water was withdrawn for 1 hr post-treatment to minimize treatment disruption.
Orthotopic human oral cancer xenotransplantation.
Animal experiments were performed in accordance to an IACUC approved protocol. We used female 8- to 10-week-old athymic nude mice (Charles River Laboratories) as xenotransplantation recipients. Mice were anesthetized with 80 mg/kg ketamine, 12 mg/kg xylazine (i.p. injection). 105 oral cancer cells (FaDu or UM-SCC-1) were suspended in 20 μL of 50% PBS/50% Matrigel and injected directly into the anterior/middle tongue using a syringe with 30-gauge needle.
Craniofacial irradiation to oral tumor-bearing mice.
Mice with tongue tumors (7 days after injection of cancer cells) were cranially irradiated to induce oral mucositis using a RS2000 biological irradiator (Rad Source Technologies) as previously reported (10). Based on our previous observation that a single 18Gy dose has kinetics and severity of oral mucositis similar to 8Gy x3 fractionated irradiation (10), we chose 18Gy irradiation to reduce the death rate due to repeated anesthesia for fractionated irradiation to oral tumor-bearing mice that had deteriorating health. Each mouse was anesthetized with 80 mg/kg ketamine, 12 mg/kg xylazine and placed under a lead shield exposing only their head. The day of irradiation was designated day 1. All animals were provided soft food in addition to standard diet. On day 6, when mice began to lose weight due to oral ulcer-associated reduced food intake, mice were divided into 2 groups (of equal weight and tumor size) and treated daily with Tat-Smad7 or vehicle. 125 mg/kg BrdU was administered i.p. two hours before euthanasia. Mice were sacrificed and tongue samples collected on day 10 for pathological evaluation and immunostaining.
Pathological evaluation, immunostaining and TUNEL assay.
Tongue tissues were fixed in 10% formalin, embedded in paraffin, and cut in 5 μm sections. Tongue epithelium and tumor histology was evaluated using H&E stained slides. Open ulcer size, defined as complete loss of epithelium, was measured using NIS (Nikon Intensilight) Elements software by two independent investigators and the results averaged to determine ulcer size (mm). We performed immunohistochemical and immunofluorescent staining as previously described (10). The primary antibodies used were guinea pig anti-Keratin14 (1:200, Fitzgerald, 20R-CP200), chicken anti-Keratin5 (1:200, Biolegend, #905901), fluorescein isothiocyanate (FITC)-labeled antibody to BrdU (BD Bioscience, 347583), rat anti-mouse CD45 (1:50, BD Bioscience, 550539), rabbit anti-mouse F4/80 (1:400, Cell Signaling Technology, 70076), rat anti-mouse Ly6G (1A8, 1:3200, Biolegend #127602), rabbit anti-NFκB subunit p50 (1:100, Santa Cruz Biotechnology, SC-7178), rabbit anti-pSmad2 (1:200, Cell Signaling Technology, 3101), rabbit anti-pSmad3 (1:400, abcam, ab52903), rabbit anti-pH2AX (1:100, Cell Signaling Technology, 9718) and mouse anti-8-OHdG (1:100, Alpha Diagnostic, 8OHG11-M). Smad7 antibody was produced using human Smad7 recombinant protein to immunize rabbits. Specificity of Smad7 antiserum was confirmed by western blot (Supplementary Fig. 2). Secondary antibodies conjugated to Alexa Flour 594 (red) or 488 (green) were used (1:200 for all, Invitrogen). TUNEL (terminal deoxynucleotidyl transferase dUTP nick and labeling) staining was performed with a TUNEL kit (Promega) according to manufacturer directions to detect apoptotic cells. Slides were mounted with coverslips using Fluoromount-G or DAPI Fluoromount-G (SouthernBiotech).
Quantification of immunostaining.
In oral mucosa, we quantified BrdU-, pH2AX- or 8-OHdG-positive cells as cells per mm basement membrane length (including all epithelial cells), TUNEL-positive cells as cells per mm basement membrane length (including all epithelial layers and stroma above the muscle layer), CD45-positve cells as DAPI-positive cells per mm2 epithelial and stroma area above muscle layer, nuclear pSmad2- or NFκB p50-positive cells as the percent of positive cells per total epithelial cell count (excluding sloughed epithelial cells induced by irradiation). Sequential 20x images along the basement membrane were quantified and averaged per sample. In oral cancer regions, K14 or K5-positive cells were defined as tumor cells. All tumor cells were measured together as the area of tumor (mm2), excluding stromal cells. We quantified percentage of nuclear pSmad2- or NFκB p50-positive cells/total tumor (epithelial and stromal) cells. We quantified BrdU+, pH2AX+, 8-OHdG+, CD45+, and TUNEL+ cells as cells per mm2 tumor area. Ly6G+ or F4/80+ cells were quantified as cells per mm2 tumor area based upon staining intensity and morphology to distinguish them from non-specific staining of other cell types. For pH2AX staining, cells with more than three nuclear foci were defined as pH2AX-positive cells. Five random 20x tumor images were quantified and averaged per sample.
Statistical Analysis.
Statistical analyses were performed using GraphPad Prism software. Normality was determined by D’Agostino & Pearson normality test (or Shapiro-Wilk normality test for smaller samples size, n<8). Every dataset passed these normality tests; differences between two treatment groups was determined using Student’s t test and differences between more than two groups was determined by one way ANOVA with Tukey’s multiple comparison test. Data are presented as the mean ± SEM.
Results
Tat-Smad7 treatment reduced oral mucositis severity without protecting adjacent oral cancer.
To mimic RT-induced oral mucositis in oral cancer patients, we used a human oral cancer orthotopic tongue xenograft model in athymic nude mice. Bone marrow transplant patients are immune suppressed and experience severe radiation-induced oral mucositis (15) suggesting that T lymphocytes are not major contributors to mucositis. We transplanted two human oral cancer lines, FaDu or UM-SCC-1, into the tongues of athymic nude mice. The health of tumor-bearing mice rapidly deteriorated due to cancer burden and compromised oral intake. Therefore, after tumors were established, we irradiated mice with 18Gy cranial RT instead of fractionated RT to minimize death due to repeated doses of anesthesia required for fractionated RT. This irradiation dose induces oral mucositis with the kinetics and severity similar to 3×8Gy fractionated RT in mice with obvious mucosal damage starting on day 5 after RT (10). Six days after RT, when mice began to lose weight (10), we treated them with Tat-Smad7 produced as we previously described [(10), Supplementary Fig. 1]. We chose the dose and regimen of topical Tat-Smad7 application that effectively treated oral mucositis (1 μg/30 μL oral dose, daily). Vehicle treatment (30% glycerol in PBS) was used as control. Mice were sacrificed and tongues harvested ten days after RT. We assessed if Tat-Smad7 protein was delivered to both oral mucosa and oral cancer with immunofluorescent staining using a human Smad7 antibody that also cross-reacts with mouse Smad7 (Supplementary Fig. 2). Similar to our previous report (10), in the oral epithelium of Tat-Smad7-treated mice, Smad7 was present in both the cytoplasm and the nucleus, whereas endogenous Smad7 levels in vehicle-treated mice were low (Fig. 1A). In tumors, Tat-Smad7 was detected in the cytoplasm of both tumor epithelia and stroma (Fig. 1A), suggesting that TGFβ in these cells drives Smad7 cytoplasmic translocation to block TGFβ signaling (16). We measured tumor area microscopically and found that both FaDu and UM-SCC-1 tumor sizes were reduced in RT-treated mice and Tat-Smad7 treatment did not significantly affect tumor size compared to vehicle in this short-course treatment (Fig. 1B-C). Tongue mucosa adjacent to tumor showed RT damage and mucositis formation primarily at the posterior dorsal surface which has fewer cornified layers compared to tongue papillae at the tip of the tongue (Fig. 1F-G). Tat-Smad7 treated mice had significantly smaller RT-induced tongue ulcers compared to vehicle-treated mice (Fig. 1D-E).
Figure 1.
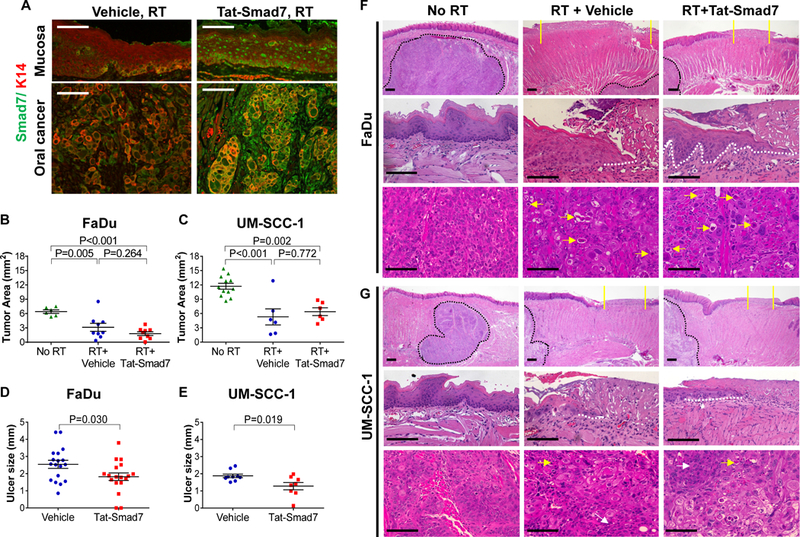
Oral Tat-Smad7 treatment alleviated radiation-induced oral mucositis but did not affect radiotherapy on neighboring oral cancer. (A) Representative immunofluorescent images using Smad7 antibody to detect endogenous Smad7 or Tat-Smad7 in irradiated mouse tongue mucosa and neighboring xenografted FaDu tumor (no endogenous Smad7). Weak endogenous Smad7 was detected in mouse tongue mucosa and oral cancer stromal cells of mouse origin. K14 antibody was used to counterstain epithelial cells. (B, C) Quantification of oral tumor size (mean ± SEM) 10 days after 18 Gy radiation in mice untreated (No RT), treated with RT and vehicle (RT+Vehicle) or RT and Tat-Smad7 (RT+Tat-Smad7). (D, E) Quantification of oral ulcer diameter (mean ± SEM) 10 days after 18 Gy radiation in mice treated as described in panels B and C. P values determined by Student’s t test (two groups) or one way ANOVA with Tukey’s multiple comparison test (more than two groups). (F, G) Representative H&E images of oral mucosa and oral cancer in tongue samples harvested from mice bearing FaDu (F) or UM-SCC-1 (G) tumors 10 days after 18 Gy radiation. Upper panels present low power images of tongue epithelium and tumor. Yellow solid lines define the ulcer boundary. Black dotted lines delineate the boundary of tongue tumor. Scale bars = 200 μm. Mid panels present high power images of oral epithelium from non-irradiated posterior dorsal tongue mucosa and irradiated mucosa from the same region with ulcer. White dotted lines delineate epithelial cells migrated underneath the ulcer. Scale bars = 100 μm. Lower panels present high power images of the oral cancer. Yellow arrows point to dying or apoptotic tumor cells. White arrows point to areas without obvious RT-induced damage. Scale bars = 100 μm.
Histopathological analysis revealed that irradiated epithelium exhibited atrophy, flattened tongue papillae, thinning cuticle and ulceration compared to untreated epithelium (Fig. 1F-G). Irradiated mucosa treated with Tat-Smad7 showed less epithelial atrophy, smaller ulcerated area and fewer immune cells compared to vehicle treated mucosa (Fig. 1F-G) (10). Compared to non-irradiated tumors, irradiated tumors near mucositis showed obvious cell damage with vacuolar degeneration and pyknotic nuclei or dysplastic cell shape with enlarged, smudged chromatin. Apoptotic cells, inflammation and interstitial fibrosis were obvious in irradiated tumors. FaDu tumors were more sensitive to irradiation as demonstrated by more damaged tumor cells than UM-SCC-1 tumors (Fig. 1F-G).
Tat-Smad7 reduced NFκB and TGFβ signaling in both oral mucositis and oral cancer.
To examine on-target effects of Tat-Smad7 on oral mucosa and cancer, we performed immunostaining for pSmad2 and pSmad3, markers of TGFβ pathway activation, and nuclear NFκBp50, a marker of NFκB pathway activation. Oral mucosa after RT had more nuclear pSmad3+ cells compared to non-irradiated mucosa (Supplementary Fig. 3), similar to changes in pSmad2+ cells as previously reported (10). Fewer nuclear pSmad2+ and pSmad3+ cells were observed in irradiated oral mucosa and adjacent oral cancer in Tat-Smad7 treated mice compared to vehicle treatment (Fig. 2A-B, E-F, I-J; Supplementary Fig. 3). In non-irradiated tongue tumors, both UM-SCC-1 and FaDu had more pSmad3+ cells than oral mucosa (Supplementary Fig. 3), indicating TGFβ-dependent Smad3 activation (even in Smad4-deficient FaDu tumors) as previously reported (13). Additionally, fewer nuclear NFκBp50+ cells were observed in both irradiated oral mucosa and adjacent oral cancer in Tat-Smad7 treated mice compared to vehicle treatment (Fig. 2C-D, G-H, K-L). These data demonstrate the on-target efficacy of Tat-Smad7 against known Smad7 targets in tongue mucosa with oral tumors.
Figure 2.

Oral Tat-Smad7 treatment attenuated nuclear pSmad2 and NFκB p50 in RT-induced oral mucositis and adjacent cancer. (A-D) Representative immunofluorescent staining of pSmad2 (A, B) and NFκB subunit p50 (C, D) in RT+Vehicle or RT+Tat-Smad7 treated oral mucosa and oral cancer (FaDu: A, C; UM-SCC-1: B, D) with K14 or K5 epithelial cell counterstain. Scale bars = 100 μm for all sections. (E-L) Quantifications of nuclear staining for pSmad2 or NFκB p50 (mean ± SEM). Nuclear pSmad2 and NFκB p50 positive cells were quantified as a percentage of total epithelial cells in oral mucosa (FaDu: E, G; UM-SCC-1: I, K) or in tumor cells (FaDu: F, H; UM-SCC-1: J, L). P values determined by Student’s t test.
Tat-Smad7 treatment alleviated inflammation and DNA damage-associated cell death in oral mucositis lesions of oral cancer-bearing mice.
We quantified leukocytes with CD45 staining and found Tat-Smad7 treated mucositis contained fewer CD45+ leukocytes (Fig. 3A-B). We next performed F4/80 and Ly6G staining to determine relative levels of F4/80+ macrophages and Ly6G+ cells [a marker of polymorphonuclear (PMN) neutrophils or PMN-myeloid derived suppressor cells (MDSC)]. RT-induced oral mucositis harbors primarily neutrophils and macrophages (9) that were obvious in oral mucositis lesions of tumor-bearing mice (Fig. 3A). Tat-Smad7 treated mucositis had fewer PMNs and macrophages than vehicle treated lesions (Fig. 3A), consistent with smaller ulcers and fewer inflammatory cells observed by H&E and CD45 staining. To examine epithelial slough due to DNA damage-induced cell death and cessation of proliferation, we first performed pH2AX staining as a marker of DNA damaged cells. Irradiated oral mucosa had numerous pH2AX+ cells and Tat-Smad7 treated oral mucosa had fewer pH2AX+ cells compared to vehicle-treated mucosa (Fig. 3A, C). Next, staining of nuclear 8-OHdG (8-hydroxy-2’-deoxyguanosine) was performed as a marker of oxidative DNA damage (17). There were fewer 8-OHdG+ cells in oral mucosa of Tat-Smad7 treated mice compared to vehicle treated mice (Fig. 3A, D). We performed TUNEL assays to quantify apoptosis and noted numerous apoptotic cells in the oral mucosa of irradiated mice. Tat-Smad7 treated mucosa contained fewer apoptotic cells than vehicle control (Fig. 3A, E). Together, these data demonstrate that Tat-Smad7 treatment reduces inflammation, DNA damage and apoptosis associated with RT-induced oral mucositis in tumor bearing mice.
Figure 3.
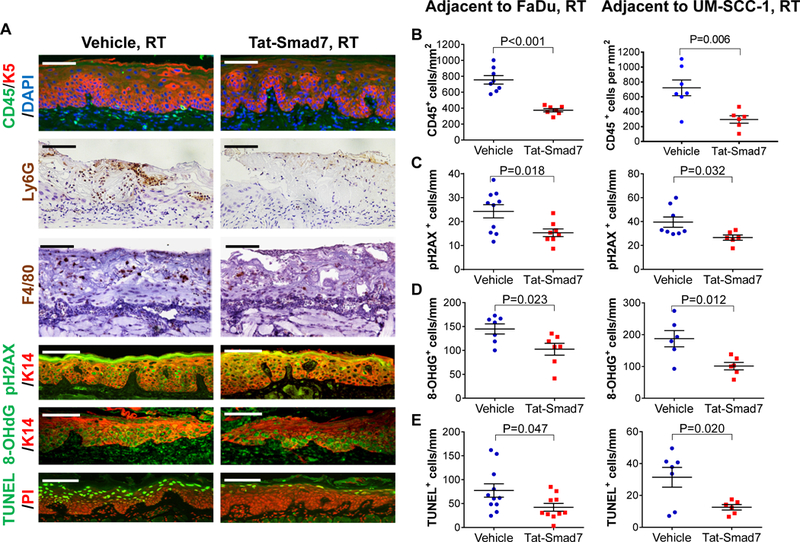
Oral Tat-Smad7 treatment mitigated inflammation, DNA damage and apoptosis in RT-induced oral mucositis. (A) Representative immunostaining of markers for immune cells (CD45, Ly6G, F4/80), DNA damage markers (pH2AX, 8-OHdG) and apoptosis (TUNEL) in RT+Vehicle or RT+Tat-Smad7 treated oral mucosa adjacent to irradiated FaDu tongue tumors. Similar staining patterns were also seen in irradiated mucosa adjacent to UM-SCC-1 tumors (not shown) and quantified in B-E. K5 or K14 antibody was used to counterstain epithelial cells. DAPI was used to counterstain nuclei for CD45 staining, propidium iodide (PI) was used to counterstain nuclei for TUNEL. Scale bars = 100 μm for all sections. (B-E): Quantification (mean ± SEM) of immunostaining markers in oral mucosa adjacent to irradiated FaDu and UM-SCC-1 shown in (A). CD45+ cells (B) were quantified based on mm2 epithelial and stromal areas above the muscle layer. Quantifications for pH2AX+ cells (C), 8-OHdG+ cells (D) and TUNEL+ cells (E) in keratinocytes per mm basement membrane length. P values were determined by Student’s t test.
Tat-Smad7 reduced inflammation in irradiated oral cancer
Unlike non-irradiated oral mucosa that had no obvious inflammation (Fig. 1), leukocyte infiltration was apparent in non-irradiated tumors that contained numerous F4/80+ macrophages and Ly6G+ cells (Supplementary Fig. 4). Very few infiltrated B cells were detected (data not shown). In irradiated FaDu tumors, F4/80+ cells were still prominent but Ly6G+ cells were largely diminished (Fig. 4A, C vs. Supplementary Fig. 4) even though Ly6G+ cells were numerous in adjacent mucositis (Fig. 3). Both F4/80+ cells and Ly6G+ cells were still pronounced in UM-SCC-1 irradiated tumors (Fig. 4D, F, G). Intriguingly, Tat-Smad7 reduced the number of CD45+ leukocytes and F4/80+ macrophages in both types of irradiated oral tumors and reduced Ly6G+ cells in irradiated UM-SCC-1 tumors (Fig. 4).
Figure 4.
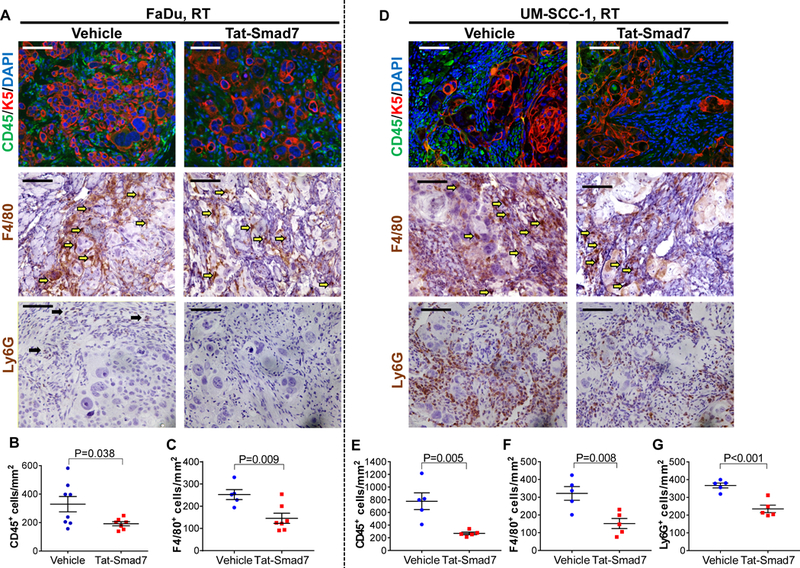
Oral Tat-Smad7 treatment reduced leukocyte infiltrate in irradiated tongue tumors. (A, D) Representative images of CD45, F4/80 and Ly6G staining in FaDu tumors (A) and UM-SCC-1 tumors (D). K5 antibody was used to counterstain epithelial cells and DAPI was used to counterstain nuclei for CD45 staining. Note that tumor cells after irradiation had more non-specific staining in F4/80 than in non-irradiated tumors (Supplementary Fig. 4). Quantification of F4/80+ macrophages in panels C and F is based upon staining intensity and morphology (indicated by yellow arrows, A and D). Black arrows in Ly6G vehicle panel in (A) point to a few remaining Ly6G+ cells which were absent in Tat-Smad7 treated tumor. Scale bars = 100 μm for all sections. (B, C) Quantifications of CD45+ and F4/80+ cells in FaDu tumors. (E-G) Quantifications of CD45+, F4/80+ and Ly6G+ cells in UM-SCC-1 tumors. Immune cells were quantified (mean ± SEM) per mm2 tumor area. P values were determined by Student’s t test.
Effects of Tat-Smad7 on irradiated oral cancer diverge from its effects on oral mucositis.
Similar to oral mucositis, irradiated tumors had numerous pH2AX+ cells that were significantly more abundant than in non-irradiated tumors (Fig. 5A, Supplementary Fig. 5). FaDu tumors harbored more pH2AX+ cells than UM-SCC-1 tumors (Fig. 5D-E, Supplementary Fig. 5). However, unlike oral mucositis, there was no difference in the number of pH2AX+ cells in oral cancers treated with Tat-Smad7 versus vehicle in both tumor types (Fig. 5D-E). 8-OHdG+ cells were hardly detectable in non-irradiated tumors (Supplementary Fig. 5) but were induced significantly by RT (Fig. 5B). In contrast to Tat-Smad7 effects on oral mucositis, 8-OHdG+ cells in oral cancer were significantly increased in Tat-Smad7 treated mice compared to vehicle (Fig. 5F-G). Basal levels of TUNEL+ apoptotic cells were present in non-irradiated tumors (Supplementary Fig. 5) and RT induced numerous apoptotic cells in both tumor types (Fig. 5C). Apoptotic cells in oral cancer were increased in Tat-Smad7 treated mice compared to vehicle-treated (Fig. 5H-I), opposite of what was observed in Tat-Smad7 treated mucositis. We performed BrdU staining to quantitate proliferating cells. Adjacent to ulceration, Tat-Smad7 treated oral mucosa showed more proliferative BrdU+ cells along the epithelial basement membrane than vehicle-treated oral mucosa (Fig. 6A-D). In contrast, Tat-Smad7 treated oral cancer had no difference in proliferation compared to vehicle-treated oral cancer (Fig. 6A-B, E-F). In summary, Tat-Smad7 treated mucositis lesions had reduced RT-induced DNA damage, inflammation and apoptosis and increased keratinocyte proliferation while adjacent irradiated tumors treated with Tat-Smad7 demonstrated no resolution to DNA damage, decreased inflammation and unaltered proliferation.
Figure 5.
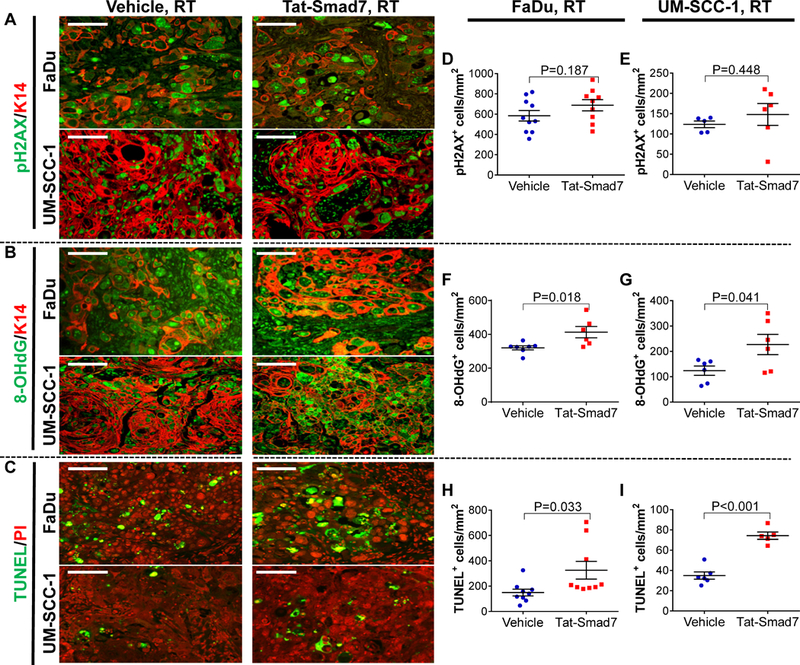
Tat-Smad7 did not reduce radiation-induced DNA damage but increased oxidative stress and apoptosis in irradiated tongue cancers. (A-C) Representative staining of pH2AX, 8-OHdG and TUNEL in RT+ vehicle and RT+ Tat-Smad7 treated tongue cancer. K14 antibody was used to counterstain epithelial cells or propidium iodide (PI) to counterstain nuclei. Scale bars = 100 μm for all sections. (D-I) Quantification (mean ± SEM) of pH2AX+, 8-OHdG+, and TUNEL+ tumor epithelial cells in cancer per mm2 tumor epithelial area (FaDu: D, F, H; UM-SCC 1: E, G, I). P values were determined by Student’s t test.
Figure 6.
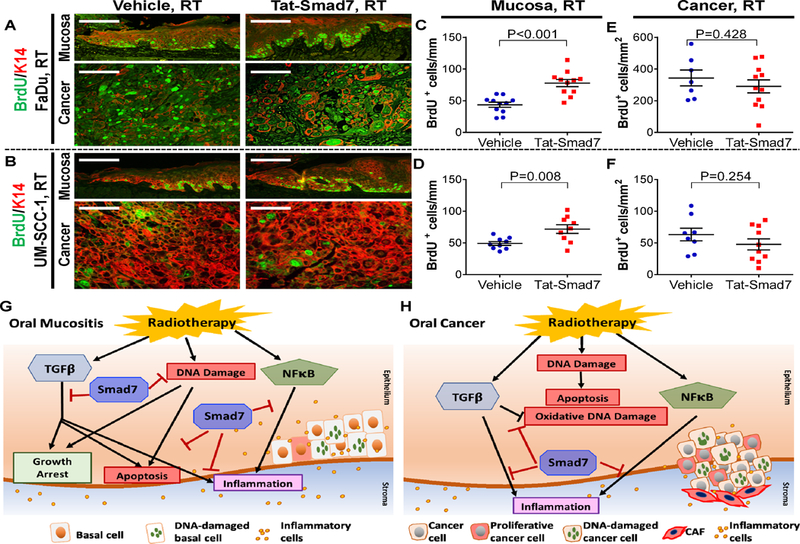
Tat-Smad7 increased proliferation in irradiated oral mucosa but not neighboring oral cancer and summary of Tat-Smad7 actions. (A, B) Representative staining of BrdU positive cells in irradiated mucosa and neighboring oral cancer (FaDu: A, UM-SCC-1: B). K14 antibody was used as counterstain epithelial cells. Scale bars = 100 μm. (C-F) Quantification (mean ± SEM) of BrdU+ cells in oral mucosal keratinocytes per mm basement membrane length (adjacent to FaDu: C; adjacent to UM-SCC-1: D) and in cancer epithelial cells per mm2 tumor area (FaDu: E; UM-SCC-1: F). P values were determined by Student’s t test. (G) Smad7 functional mechanisms in oral mucositis with neighboring irradiated oral cancer. RT directly activates TGFβ, NFκB and induces DNA damage. Smad7 alleviates RT-induced DNA damage (including DNA strand breaks and oxidative damage), attenuates TGFβ induced growth arrest, attenuates apoptosis, and blocks inflammation induced by TGFβ and NFκB, further promoting oral mucositis healing. (H) Smad7 functional mechanisms in irradiated oral cancer. RT directly activates TGFβ and NFκB. Tumor cells harbor intrinsically damaged DNA and are thus more sensitive to RT-induced apoptosis. Smad7 enhances RT-induced oxidative damage that consequently increases apoptosis. Smad7 also attenuates TGFβ and NFκB activation-induced inflammation.
Discussion
One of the major challenges for treating oral mucositis in cancer patients is that potential therapies reviving normal mucosal regeneration could also pose risk by protecting cancer cells. We show here that Tat-Smad7 topical application reduced activation of NFκB and TGFβ signaling and reduced inflammation in both oral mucosa and cancer, but differentially affected normal mucosa and oral cancer with respect to DNA damage and associated cell death, and cell proliferation.
Our current study confirms that in oral mucositis-induced acute inflammation associated with NFκB and TGFβ activation (10,18), macrophages and neutrophils are predominant leukocyte types (9). Fewer macrophages and neutrophils in Tat-Smad7 treated mucositis could reflect less damage (smaller ulcers and fewer dead cells) or a quicker resolution of inflammation. Oral cancer without RT showed chronic inflammation in the tumor microenvironment that harbored numerous F4/80+ putative M2 macrophages and Ly6G+ putative tumor-associated neutrophils or PMN-MDSCs; both are known to be tumor-promoting leukocytes attracted by TGFβ1 and also a major source of TGFβ1 (19). With RT, F4/80+ and Ly6G+ cells remained plentiful in UM-SCC-1 tumors and Tat-Smad7 reduced numbers of these cells similar to TGFβ inhibitors and NFκB inhibitors used in clinical trials of metastatic cancer, which has an inflammatory tumor microenvironment (20,21). The subtypes of these leukocytes in irradiated tumors requires further study, e.g., if they have shifted from PMN-MDSCs to neutrophils as part of acute inflammation induced by RT. Interestingly, Ly6G+ leukocytes were largely diminished in irradiated FaDu tumors despite their presence in the adjacent mucosa. Because FaDu cells lack Smad4 but retain nuclear pSmad2+ and pSmad3+, it is unclear if this altered TGFβ signaling affected RT response-associated changes in Ly6G+ cell infiltration or depletion in tumors. In contrast, Tat-Smad7 still reduced F4/80+ macrophages in FaDu tumors, possibly by blocking RT-induced TGFβ signaling through Smad2/Smad3 and by direct blocking of non-Smad-mediated NFκB activation by TGFβ (22). These data highlight the complex biology and heterogeneity of tumors in response to RT. The anti-TGFβ/NFκB effects of Smad7 could have a cascade effect on cytokine production directly or indirectly due to reduced leukocyte numbers. Therefore, future in-depth studies are needed to dissect subtypes of immune cells influenced by Smad7 as well as the production of cytokines by these cells.
Because reducing inflammation is insufficient to alleviate oral mucositis, other pathogenic processes must also be targeted for effective treatment. Among these processes, RT-induced DNA damage kills tumor cells but also initiates oral mucositis (1, 23). Therefore, reducing DNA damage or accelerating repair is key for oral mucositis healing, but such an effect is undesirable for eradicating tumor cells. The selective reduction of radiation-induced DNA damage by Smad7 in oral mucosa (Fig. 3) might be explained by its ability to facilitate DNA damage repair through protein-protein interactions with the DNA repair protein complex containing ATM and Mre11-Rad50-Nbs1 proteins (24). However, Tat-Smad7 did not reduce RT-induced DNA damage in tumor cells (pH2AX+ cells). This may be part of the reason that intrinsic DNA repair defects in tumor cells (evident in non-irradiated tumors) cannot be reversed by Tat-Smad7. Additionally, TGFβ-mediated DNA repair is an important mechanism of radioresistance in cancer treatment (25–29). In this context, blocking TGFβ signaling by Tat-Smad7 would enhance DNA damage in cancer cells. Further, RT-induced oxidative stress is required to amplify DNA damage, causing collateral damage leading to cancer cell death (30,31) and oral mucositis (1,23). Reduced oxidative stress marker 8-OHdG with Tat-Smad7 treatment in oral mucositis is consistent with previous reports that both TGFβ and NFκB pathways can induce oxidative stress (32–35). Paradoxically, TGFβ is reported to reduce oxidative stress in a skin SCC model (36). This could explain why Tat-Smad7 treatment increased 8-OHdG+ cells in oral cancer. Given these effects, it is not surprising that DNA damage-associated apoptosis was reduced by Tat-Smad7 in oral mucositis but increased in adjacent oral cancer. The effect of Tat-Smad7 on apoptosis is also consistent with previous reports showing Smad7 reduced apoptosis in normal epithelia (10,37) but enhanced apoptosis in cancer (38).
In a subset of HNSCCs, endogenous Smad7 is activated by TGFβ and NFκB, which transcriptionally suppress Smad target genes that could either be tumor suppressive or promotive (22). Therefore, pharmacological dosage of a Smad7-based biologic needs to be carefully assessed. Our study revealed that HNSCCs derived from SMAD4 mutant (FaDu) and SMAD4 wildtype (UM-SCC-1) cells responded to Tat-Smad7 similarly. This can be explained by the fact that the majority of human cancers escape Smad-dependent TGFβ growth inhibition, which is also supported by Tat-Smad7 not affecting cancer cell proliferation in our model. Additionally, we have shown Smad4 mutant epithelial cells rely primarily on Smad3-dependent signaling to mediate TGFβ1-induced inflammation (13). Therefore, the use of Tat-Smad7 in Smad4 mutant cancer could follow the same principle as TGFβ inhibitor in treating Smad4 mutant metastatic cancer including pancreatic cancer that has a high rate of Smad4 mutation (39).
In summary, we provide evidence that local short-term Tat-Smad7 protein delivery alleviated radiation-induced oral mucositis without compromising radiation-induced killing of neighboring oral cancer. Potential mechanisms for the context-specific effects of Smad7 are as follows: In oral mucositis, Tat-Smad7 attenuates TGFβ-mediated growth arrest and apoptosis as well as TGFβ/NFκB-mediated inflammation. Smad7 could also directly reduce DNA damage or accelerate repair (Fig. 6G). In neighboring oral cancer, Smad7 still attenuates TGFβ/NFκB-mediated inflammation, but is insufficient to reduce DNA damage (Fig. 6H). Additionally, Smad7 could block TGFβ-mediated suppression of oxidative damage and consequently induce apoptosis in tumor cells (Fig. 6H). Because high intensity/hypo-fractionated SBRT is increasingly used to treat oral cancers resistant to traditional low dose fractionated RT, future studies that more closely mimic SBRT will be needed to assess therapeutic efficacy of Tat-Smad7 in oral mucositis treatment. Additionally, HNSCC mouse models with an intact immune system (13,40) will further determine if effects of Tat-Smad7 are influenced by T cell-mediated tumor immunity. Future studies should carefully assess Tat-Smad7 dose-dependent efficacy to effectively treat oral mucositis but avoid potential oncogenic effects on neighboring oral cancer, and assess the effect of long-term Tat-Smad7 use on oral cancer progression with an intact immune microenvironment.
Supplementary Material
Translational Relevance.
Oral mucositis, painful oral ulcerations, is one of the most common toxic effects of radiotherapy (RT) and chemotherapy in cancer patients and can lead to therapy withdrawal or dose reduction. In head and neck cancer (HNC) patients, a major challenge in treating oral mucositis is to repair ulcerated mucosa without promoting neighboring cancer growth. To date, there is no FDA approved drug to treat oral mucositis in HNC patients. We studied a novel biologic agent for treating oral mucositis in a mouse model mimicking RT-induced oral mucositis adjacent to oral cancer. Our preclinical data demonstrate the feasibility of a novel therapeutic approach for treating existing oral mucositis without compromising radiotherapy in neighboring cancer.
Acknowledgements
We thank Drs. Yosef Refaeli, Brian Turner, Steve Sonis and Thomas Carey for advice and an anonymous donation for support of this work. D. Raben is supported by the Marsico Endowment fund for research.
Financial support: This work was supported by NIH grant DE024659 to CDY and XJW, and DE015953 to XJW. XJW is also a Research Biologist in Department of Veterans Affairs. JL and FW were supported by the National Nature Science Foundation of China No. 81772898, and the State Scholarship Fund of China Scholarship Council No. 201506240122 and No. 201606240201. MAB was supported by T32 CA174648.
Footnotes
Conflict of interest: XJW is an inventor of the patent filed by the University of Colorado for using Tat-Smad7 to treat oral mucositis. Allander Biotechnologies, LLC. has the exclusive license and commercial interests in developing Tat-Smad7-based therapy. Other authors have no conflicts of interest to disclose.
References
- 1.Sonis ST. Oral mucositis in head and neck cancer: risk, biology, and management. American Society of Clinical Oncology educational book / ASCO American Society of Clinical Oncology Meeting 2013;2013:236–40. [DOI] [PubMed] [Google Scholar]
- 2.Villa A, Sonis ST. Mucositis: pathobiology and management. Curr Opin Oncol 2015;27(3):159–64. [DOI] [PubMed] [Google Scholar]
- 3.Khuntia D, Harris J, Bentzen SM, Kies MS, Meyers JN, Foote RL, et al. Increased Oral Mucositis after IMRT versus Non-IMRT when Combined with Cetuximab and Cisplatin or Docetaxel for Head and Neck Cancer: Preliminary Results of RTOG 0234. International Journal of Radiation Oncology 2008;72(1). [Google Scholar]
- 4.Al-Dasooqi N, Sonis ST, Bowen JM, Bateman E, Blijlevens N, Gibson RJ, et al. Emerging evidence on the pathobiology of mucositis. Supportive care in cancer : official journal of the Multinational Association of Supportive Care in Cancer 2013. [DOI] [PubMed] [Google Scholar]
- 5.Sonis ST. Efficacy of palifermin (keratinocyte growth factor-1) in the amelioration of oral mucositis. Core Evid 2010;4:199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henke M, Alfonsi M, Foa P, Giralt J, Bardet E, Cerezo L, et al. Palifermin decreases severe oral mucositis of patients undergoing postoperative radiochemotherapy for head and neck cancer: a randomized, placebo-controlled trial. J Clin Oncol 2011;29(20):2815–20. [DOI] [PubMed] [Google Scholar]
- 7.Le QT, Kim HE, Schneider CJ, Murakozy G, Skladowski K, Reinisch S, et al. Palifermin reduces severe mucositis in definitive chemoradiotherapy of locally advanced head and neck cancer: a randomized, placebo-controlled study. J Clin Oncol 2011;29(20):2808–14. [DOI] [PubMed] [Google Scholar]
- 8.Anderson CM, Sonis ST, Lee CM, Adkins D, Allen BG, Sun W, et al. Phase 1b/2a Trial of the Superoxide Dismutase Mimetic GC4419 to Reduce Chemoradiotherapy-Induced Oral Mucositis in Patients With Oral Cavity or Oropharyngeal Carcinoma. Int J Radiat Oncol Biol Phys 2018;100(2):427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonis ST. The pathobiology of mucositis. Nat Rev Cancer 2004;4(4):277–84. [DOI] [PubMed] [Google Scholar]
- 10.Han G, Bian L, Li F, Cotrim A, Wang D, Lu J, et al. Preventive and therapeutic effects of Smad7 on radiation-induced oral mucositis. Nat Med 2013;19(4):421–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu SL, Reh D, Li AG, Woods J, Corless CL, Kulesz-Martin M, et al. Overexpression of transforming growth factor beta1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Res 2004;64(13):4405–10. [DOI] [PubMed] [Google Scholar]
- 12.Bian L, Han G, Zhao CW, Garl PJ, Wang XJ. The role of Smad7 in oral mucositis. Protein Cell 2015;6(3):160–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bornstein S, White R, Malkoski S, Oka M, Han G, Cleaver T, et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. JClinInvest 2009;119:3408–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brenner JC, Graham MP, Kumar B, Saunders LM, Kupfer R, Lyons RH, et al. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck 2010;32(4):417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gabriel DA, Shea T, Olajida O, Serody JS, Comeau T. The effect of oral mucositis on morbidity and mortality in bone marrow transplant. Semin Oncol 2003;30(6 Suppl 18):76–83. [DOI] [PubMed] [Google Scholar]
- 16.Itoh S, Landstrom M, Hermansson A, Itoh F, Heldin CH, Heldin NE, et al. Transforming growth factor beta1 induces nuclear export of inhibitory Smad7. J Biol Chem 1998;273(44):29195–201. [DOI] [PubMed] [Google Scholar]
- 17.Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2’ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 2009;27(2):120–39. [DOI] [PubMed] [Google Scholar]
- 18.Sonis ST. Pathobiology of oral mucositis: novel insights and opportunities. J Support Oncol 2007;5(9 Suppl 4):3–11. [PubMed] [Google Scholar]
- 19.Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Science translational medicine 2018;10(424). [DOI] [PubMed] [Google Scholar]
- 20.Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov 2004;3(12):1011–22. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J Clin Invest 2001;107(2):135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freudlsperger C, Bian Y, Contag Wise S, Burnett J, Coupar J, Yang X, et al. TGF-beta and NF-kappaB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene 2013;32(12):1549–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Treister N, Sonis S. Mucositis: biology and management. Curr Opin Otolaryngol Head Neck Surg 2007;15(2):123–9. [DOI] [PubMed] [Google Scholar]
- 24.Park S, Kang JM, Kim SJ, Kim H, Hong S, Lee YJ, et al. Smad7 enhances ATM activity by facilitating the interaction between ATM and Mre11-Rad50-Nbs1 complex in DNA double-strand break repair. Cell Mol Life Sci 2015;72(3):583–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirshner J, Jobling MF, Pajares MJ, Ravani SA, Glick AB, Lavin MJ, et al. Inhibition of transforming growth factor-beta1 signaling attenuates ataxia telangiectasia mutated activity in response to genotoxic stress. Cancer Res 2006;66(22):10861–9. [DOI] [PubMed] [Google Scholar]
- 26.Ewan KB, Henshall-Powell RL, Ravani SA, Pajares MJ, Arteaga C, Warters R, et al. Transforming growth factor-beta1 mediates cellular response to DNA damage in situ. Cancer Research 2002;62(20):5627–31. [PubMed] [Google Scholar]
- 27.Glick AB, Weinberg WC, Wu IH, Quan W, Yuspa SH. Transforming growth factor beta 1 suppresses genomic instability independent of a G1 arrest, p53, and Rb [published erratum appears in Cancer Res 1997 May 15;57(10):2079]. Cancer Research 1996;56(16):3645–50. [PubMed] [Google Scholar]
- 28.Wang J, Jacob NK, Ladner KJ, Beg A, Perko JD, Tanner SM, et al. RelA/p65 functions to maintain cellular senescence by regulating genomic stability and DNA repair. EMBO reports 2009;10(11):1272–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardee ME, Marciscano AE, Medina-Ramirez CM, Zagzag D, Narayana A, Lonning SM, et al. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-beta. Cancer Res 2012;72(16):4119–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dayal R, Singh A, Pandey A, Mishra KP. Reactive oxygen species as mediator of tumor radiosensitivity. J Cancer Res Ther 2014;10(4):811–18. [DOI] [PubMed] [Google Scholar]
- 31.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 2009;8(7):579–91. [DOI] [PubMed] [Google Scholar]
- 32.Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol 1996;10(9):1077–83. [DOI] [PubMed] [Google Scholar]
- 33.Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free radical biology & medicine 2010;48(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li N, Karin M. Ionizing radiation and short wavelength UV activate NF-kappaB through two distinct mechanisms. Proceedings of the National Academy of Sciences of the United States of America 1998;95(22):13012–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell research 2011;21(1):103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oshimori N, Oristian D, Fuchs E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015;160(5):963–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He W, Li AG, Wang D, Han S, Zheng B, Goumans MJ, et al. Overexpression of Smad7 results in severe pathological alterations in multiple epithelial tissues. EMBO J 2002;21(11):2580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landstrom M, Heldin NE, Bu S, Hermansson A, Itoh S, ten Dijke P, et al. Smad7 mediates apoptosis induced by transforming growth factor beta in prostatic carcinoma cells. Curr Biol 2000;10(9):535–8. [DOI] [PubMed] [Google Scholar]
- 39.Malkoski SP, Wang XJ. Two sides of the story? Smad4 loss in pancreatic cancer versus head-and-neck cancer. FEBS letters 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mishra AK, Kadoishi T, Wang X, Driver E, Chen Z, Wang XJ, et al. Squamous cell carcinomas escape immune surveillance via inducing chronic activation and exhaustion of CD8+ T Cells co-expressing PD-1 and LAG-3 inhibitory receptors. Oncotarget 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.