

Abstract
Purpose
Glioblastoma (GBM) is the most common and most lethal primary malignant brain tumor. The receptor tyrosine kinase MET is frequently upregulated or over activated in GBM. Although clinically applicable MET inhibitors have been developed, resistance to single modality anti-MET drugs frequently occurs, rendering these agents ineffective. We aimed to determine the mechanisms of MET inhibitor resistance in GBM and use the acquired information to develop novel therapeutic approaches to overcome resistance.
Experimental Design
We investigated two clinically applicable MET inhibitors: Crizotinib, an ATP-competitive small molecule inhibitor of MET, and Onartuzumab, a monovalent monoclonal antibody that binds to the extracellular domain of the MET receptor. We developed new MET inhibitor resistant cells lines and animal models and utilized reverse phase protein arrays (RPPA) and functional assays to uncover the compensatory pathways in MET inhibitor resistant GBM.
Results
We identified critical proteins that were altered in MET inhibitor resistant GBM including mTOR, FGFR1, EGFR, STAT3 and COX-2. Simultaneous inhibition of MET and one of these upregulated proteins led to increased cell death and inhibition of cell proliferation in resistant cells compared to either agent alone. Additionally, in vivo treatment of mice bearing MET resistant orthotopic xenografts with COX-2 or FGFR pharmacological inhibitors in combination with MET inhibitor restored sensitivity to MET inhibition and significantly inhibited tumor growth.
Conclusion
These data uncover the molecular basis of adaptive resistance to MET inhibitors and identifies new FDA-approved multi-drug therapeutic combinations that can overcome resistance.
Keywords: MET, Glioblastoma, Therapy Resistance, Crizotinib, Onartuzumab
Introduction
Glioblastoma (GBM) is the most common and most lethal primary malignant brain tumor1. Current standard of care includes surgical resection, radiotherapy and chemotherapy. Prognosis remains poor with a median survival of 15 months2. New therapies targeting common aberrations in signal transduction pathways in GBM are currently being investigated 3. Dysregulation of receptor tyrosine kinases (RTKs) have been found in approximately 90% of GBM 4–5. Consequently, tyrosine kinase inhibitors have been developed for anticancer therapy.
MET is a RTK that is essential for embryonic development and tissue repair 2, 6. Hepatocyte growth factor (HGF), the only known ligand for MET, activates MET and downstream signaling pathways including RAS/MAPK, PI3K/AKT and STAT7–8,9. MET is commonly dysregulated in GBM via various mechanisms including somatic mutations, rearrangement, amplification and overexpression of MET and HGF that leads to autocrine loop formation 10–12. Furthermore, MET expression inversely correlates with patient survival 12–13 and is upregulated in GBM 5, 14–15.
Several MET inhibitors are under investigation with mixed results in clinical trials 14. Crizotinib is a FDA-approved ATP-competitive small molecule inhibitor of MET. Approved for the treatment of advanced and metastatic ALK-positive non-small cell lung cancer (NSCLC), crizotinib is in phase II clinical trials for CNS and solid brain tumors 5, 16. In NSCLC patients, crizotinib displays initial potent anticancer activity. However, acquired resistance frequently ensues, rendering this drug ineffective as a monotherapy 17. Onartuzumab, is a monovalent monoclonal antibody that competes with HGF for binding to MET 18. Previous clinical trials involving Onartuzumab as a monotherapy in NSCLC patients have been disappointing, highlighting the importance of understanding resistance mechanisms to the drug.
Although MET inhibitors have displayed initial efficacy, acquired resistance to single agent modalities invariably occurs 19. The development of acquired resistance to monotherapy encompasses multiple mechanisms such as acquisition of secondary mutations in therapeutic targets, activation of bypass signaling pathways, or immune evasion 20. Deciphering the exact mechanism of acquired resistance would be advantageous to halt disease progression in GBM patients by using combinatorial therapies 20–22. This study had two aims: 1) to elucidate the bypass signaling pathways that are activated when GBM cells acquire MET inhibitor resistance; 2) to develop combinatorial drug therapies against MET inhibitor-resistant GBM. Our data uncovered a number of signaling molecules that are altered in MET inhibitor resistant cell lines compared to sensitive cell lines. Upregulation of RTKs such as fibroblast growth factor receptor (FGFR) and epidermal growth factor receptor (EGFR), signaling molecules such as mammalian target of rapamycin (mTOR) and signal transducer and activator of transcription 3 (STAT3), as well as elevation of cyclooxygenase 2 (COX-2) emphasized the extent of cross-talk between multiple signaling pathways in MET inhibitor resistant GBM. Multi-targeted combinational therapies against these molecules overcame single agent MET inhibitor resistance, paving the way for new therapeutic approaches that could be tested in clinical trials.
Methods
Cells and tumor specimens
Human GBM cell lines (U87 and U373) and stem cell line (GSC827) were used. U87 and U373 were from American Type Culture Collection (Manassas, VA) and were authenticated through short tandem repeat (STR) profiling. GSC827 was isolated from GBM specimens and characterized for tumorigenesis, pluripotency, self-renewal, stem cell markers, and neurosphere formation 23. The specimens were obtained with written informed consent by the patients and the studies conducted in accordance with recognized ethical guidelines and approved by the institutional review board of the Cleveland Clinic. All cell lines were tested for mycoplasma.
Developing MET inhibitor resistant cell lines
U87, U373 and GSC827 cells were exposed to increasing concentrations of either crizotinib (from 1 nM to 100 nM) or Onartuzumab (form 1 nM to 300 nM) over a period of 6 months. Cell lines were then tested for MET inhibitor resistance through trypan blue death assay as previously described 24. For this, cells were treated with MET inhibitor [crizotinib 100 nM or Onartuzumab 300 nM] for 48 hr, collected and stained with trypan blue reagent. MET inhibitor resistant cells were grown continually in the presence of either 100 nM crizotinib or 300 nM Onartuzumab.
Cell death and cell proliferation assays
Cell death was assessed by trypan blue assay as previously described 24. Cell proliferation was assessed by cell counting as previously described 25. All experiments were performed in triplicate. Apoptotic cell death was assessed by pre-treating cells with ZVAD (20 nM) followed by combinational drug treatment for 48 hr. Cell death was determined as described above.
Reverse Phase Protein Arrays
Proteomic screening was performed by reverse phase protein array (RPPA) as previously described 26–28. The cells were treated as described, in triplicates. Protein signaling analytes were chosen based on their previously described involvement in key aspects of tumor biology. Detection was performed using a fluorescence-based tyramide signal amplification strategy using Streptavidin-conjugated IRDye680 (LI-COR Biosciences, Lincoln NE) detection reagent. All antibodies were validated for single band specificity and for ligand-induction (for phospho-specific antibodies) by immunoblotting prior to use on the arrays as previously described 26–28. Each array was scanned using a TECAN LS (Vidar Systems Corporation, Herndon VA). Spot intensity was analysed, data were normalized to total protein and a standardized, single data value was generated for each sample on the array by MicroVigene software V2.999 (VigeneTech, North Billerica, MA).
Immunoblotting
Immunoblotting was performed as previously described 29. Antibodies used were p.FGFR1, FGFR1, p.ERK, ERK, p.AKT, AKT, p.MET, p.mTOR, mTOR, COX-2 and p.STAT3 (Cell Signaling Technology), MET and GAPDH (Santa Cruz Biotechnology).
Mechanistic studies of MET-inhibitor resistance
Functional rescue experiments were performed to determine whether molecules identified through RPPA screenings mediate resistance to MET inhibitors. Cells were pretreated with celecoxib (inhibits COX-2), debio-1347 (inhibits FGFR1), erlotinib (inhibits EGFR), rapamycin (inhibits mTOR) or STAT3 inhibitor as described for 2 hr then treated with either crizotinib or Onartuzumab for 48 hr. Cell death and proliferation was assessed as described above.
Cell transfections
U87 cells were transfected with either scrambled siRNA (control), si-FGFR1, si-COX2, si-RON, si-RET, si-Vimentin or si-ERBB3 (Thermo Fisher Scientific, Waltham, MA) then treated with crizotinib (100nM) for 48 hr. Knockdown was confirmed by immunoblotting. Cells death was determined as described above.
In vivo drug combination studies
Combination therapies to overcome resistance was assessed using an orthotopic xenograft mouse model. U87 cells (3 × 105) were stereotactically implanted into the right corpus striatum of immunodeficient mice (n = 10 per treatment group). Six days after implantation, the animals were treated with either control vehicle (DMSO), agent either alone [debio-1347 (25 mg/kg) or celecoxib (10 mg/kg), crizotinib (25 mg/kg)] or in combination [debio-1347+crizotinib or celecoxib+crizotinib] by oral gavage daily from day 6 post-tumor implantation for 7 days. Tumor volumes were visualized and quantified by MRI. These studies were approved by the University of Virginia Animal Care and Use Committee.
Statistical analyses
The continuous variable RPPA data generated were subjected to both unsupervised and supervised statistical analyses, as previously described 24. Statistical analyses were performed on final microarray intensity values obtained using R version 2.9.2 software (The R Foundation for Statistical Computing). If the distribution of variables for the analyzed groups were normal, a two-sample t-test was performed. If the variances of two groups were equal, two-sample t-test with a pooled variance procedure was used to compare the means of intensity between two groups. Otherwise, two-sample t-test without a pooled variance procedure was adopted. For non-normally distributed variables, the Wilcoxon rank sum test was used. Significance levels were set at p < 0.05. To evaluate the statistical significance of the difference between wildtype and resistant GBM cell proliferation and death, treated and control, we used a two sample t-test and significance levels were set at p < 0.05. To evaluate the statistical significance of the difference between each treated and control animal groups in vivo, we used both two-sample t-test and non-parametric Wilcoxen rank-sum test.
Results
Generation of MET inhibitor resistant GBM cell lines
Crizotinib-resistant and Onartuzumab-resistant GBM cell lines from U87, U373 and GSC827 were generated through dose escalation of each drug over 6 months until the cells were no longer sensitive to crizotinib at a concentration of 100 nM or Onartuzumab at a concentration of 300 nM (Figure 1A, B, C & D). When compared to the wildtype cells, the resistant cell lines exhibited no change in cell survival and proliferation when treated with either crizotinib or Onartuzumab for 48 hr. Treatment of wildtype GBM cells with crizotinib or Onartuzumab resulted in an antiproliferative effect and induction of apoptosis. Conversely, in crizotinib-resistant and Onartuzumab -resistant GBM cells, cell survival and proliferation remained unaffected.
Figure 1: Generation and testing of MET resistant GBM cells and GSCs.
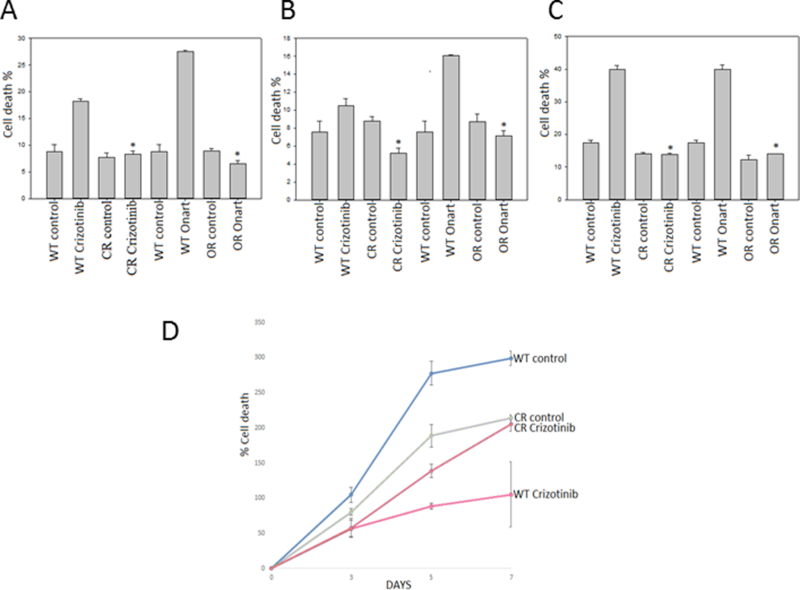
A). U87, B). U373 and C). GSC827 cells were exposed to increasing concentrations of either crizotinib or Onartuzumab (Onart) until resistance was confirmed at 100 nM for crizotinib-resistant (CR) and 300 nM for Onart-resistant (OR) respectively by trypan blue assay. The data confirm the generation of MET inhibitor resistant cells. D). U87 WT & CR cells were treated with crizotinib (100 nM) for 48 hr. Cell proliferation was assessed by cell counting over a period of 5 days and growth curves were established*, P < 0.05.
Discovery of bypass signaling pathways in resistant cells
To elucidate the mechanism(s) of resistance of GBM cells to crizotinib and Onartuzumab, we employed proteomic screening with RPPA to compare protein expression and activation changes between wildtype and resistant GBM cells when treated with the MET inhibitors for 48 hr. The experiment was performed in triplicate. The data revealed several upregulated-signaling pathways in the resistant cells. The most changed molecules were: p.EGFR, cABL, ALDH, Cyclin A and PDL1 increased 26.7, 4.3, 3.8, 3.3 and 3 fold respectively in crizotinib-resistant U373 cells; p.EGFR, p.ATPCL and PDL1 increased 7.7, 2.6, 2.3 fold respectively in Onartuzumab -resistant U373 cells; p.NFkB p65 and ERBB3 increased 2.7, 2 fold respectively in crizotinib-resistant U87 cells and COX-2, p.SRC and p.ERK increased 2.7, 2.4 and 2.3 fold respectively in Onartuzumab -resistant U87 cells compared to the wildtype after MET inhibitor treatment (Figure 2). Additional significantly changed molecules are shown in supplementary figures 2 & 3. In particular, MET inhibitor resistance resulted in the activation of the epidermal growth factor receptor (EGFR) pathway, the fibroblast growth factor receptor (FGFR) pathway, the mTOR pathway, STAT3 and COX-2. Increased activation of these molecules in resistant cells was further induced by additional treatment with MET inhibitors, forcing the resistant cells to upregulate existing bypass signaling pathways. These expression changes were validated by immunoblotting (Figure 2C). Several other changed molecules were also assessed but could either not be confirmed via immunoblotting, had no effect on the reversal of MET resistance, or had no available specific pharmacological inhibitors. These molecules were therefore not further investigated (Supplementary table. 1). Overall, the data demonstrate that MET inhibitor resistance is mediated through multiple key regulatory pathways that compensate for inhibition of the MET pathway (Figure 2A & B, supplementary figures 2 & 3). Celecoxib and debio-1347 were selected for evaluation in vivo, attributed to this, inhibition of COX-2 by celecoxib and FGFR by debio-1347, along with the effect on downstream pathways, was confirmed though immunoblotting (Supplementary figure 1).
Figure 2. MET inhibitor resistance is mediated by activation and expression changes in vital oncogenic molecules/pathways.
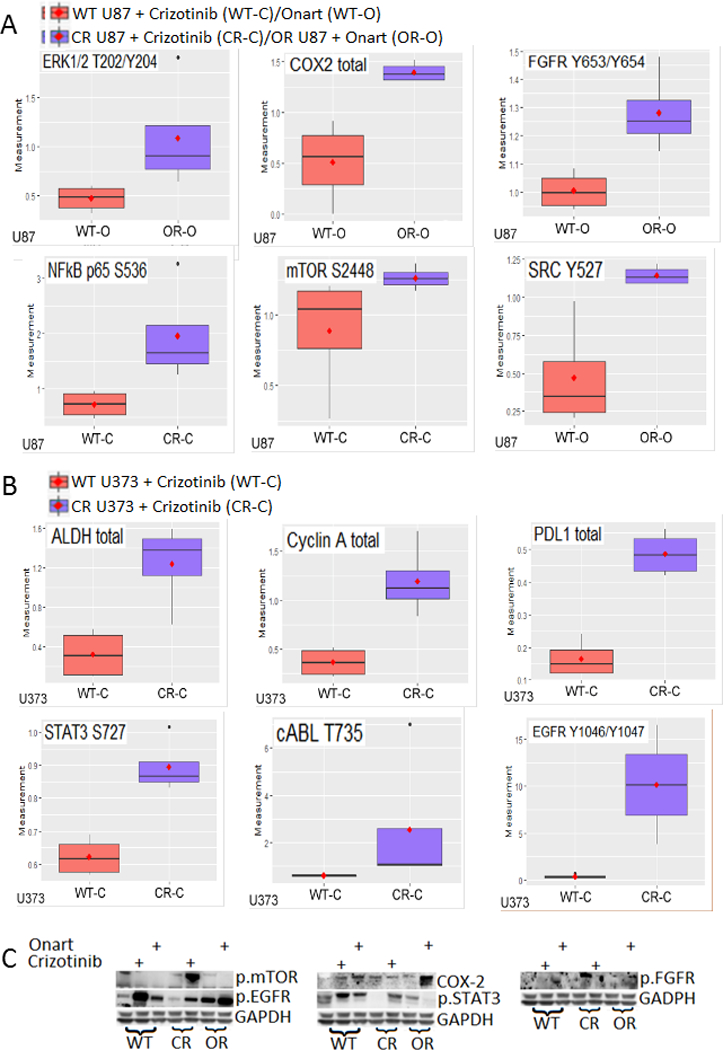
A). U87 wild type (WT), crizotinib-resistant (CR) and onartuzumab-resitant (OR) cells were treated with either control, crizotinib (100 nM) or Onartuzumab (Onart) (300 nM) for 48 hr and the cell lysate was subjected to RPPA. The most significantly changed molecules are shown in this figure. Among other, MET inhibitor resistance resulted in the upregulation of receptor tyrosine kinase FGFR1 and important survival signaling molecules mTOR and COX-2. B). U373 WT and CR cells were treated with either control or crizotinib (100 nM) for 48 hr and the cell lysate was subjected to RPPA. Notably, resistance resulted in the upregulation of EGFR and STAT3. C). RPPA data were verified by immunoblotting. These data show that MET inhibitor resistance is mediated via multiple signaling molecules.
Inhibition of bypass signaling pathways overcomes acquired resistance to MET inhibitor
To determine whether inhibition of select bypass-signaling pathways can re-sensitize the resistant cells to MET inhibitor, we treated the cells with MET inhibitors in combination with an inhibitor of one bypass signaling pathway and assessed the cell proliferation and death. A total of nine bypass signaling targets and their inhibitors were selected based on the availability of drugs that inhibit them. These targets included FGFR1, EGFR, mTOR, STAT3, COX-2, RON, RET, Vimentin and ERBB3. Wildtype and resistant U87, U373 and GSC827 cells were pretreated for 2 hr with A). COX-2 inhibitor, celecoxib (100 nM, 25 nM or 5 nM, respectively), B). FGFR1 inhibitor, debio-1347 (10 μM for U87 or 5 μM for U373 and GSC827), C). mTOR inhibitor, rapamycin (100 nM for U87 or 25 nM for U373 and GSC827), D). STAT3 inhibitor (STAT3i) (50 μΜ for U87 or 25 μΜ for U373 and GSC827) or E). EGFR inhibitor, erlotinib (100 nM for U87 or 25 nM for U373 and GSC827) then subjected to either crizotinib (100 nM) or Onartuzumab (300 nM) for 48 hr. When used alone, all five inhibitors decreased cell proliferation and increased cell death in wildtype cells but not in the resistant cells. However, when the resistant cells were treated with a MET inhibitor and one of the five inhibitors, we observed increased cell death (Figure 3A & B, 4A & B and 5A & B, supplemental figures 4A-C, 5A-B & 6A-E) and decreased proliferation (Figure 3C-F, 4C-F and 5C & D, supplemental figures 4D-I, 5C-F & 6F-O) indicating restored sensitivity to MET inhibitors. Inhibitions of RON, RET, Vimentin and ERBB3 either did not successfully restore sensitivity to MET inhibitors or clinically applicable inhibitors were not available for use (supplementary figure 1H - K). Additionally, to determine the predominant mode of cell death mediated by the combination of celecoxib and crizotinib, we pretreated U87 WT and CR cells with ZVAD for 30 min prior to treating with celecoxib followed by crizotinib for 48 hr then assessed the effect on cell death. We show that ZVAD reduced cell death caused by the combinational treatment, indicating that apoptosis as the preliminary mode of cell death (supplementary figure 4J & K). The above data demonstrate that when combined with either crizotinib or Onartuzumab, celecoxib, debio-1347, rapamycin, STAT3i and erlotinib are all partially but significantly effective at overcoming MET inhibitor resistance in GBM cells. P Values are stated in the figure legends.
Figure 3. Combination therapy with FGFR and COX-2 inhibitors restores sensitivity to MET inhibitors in resistant GBM cells.
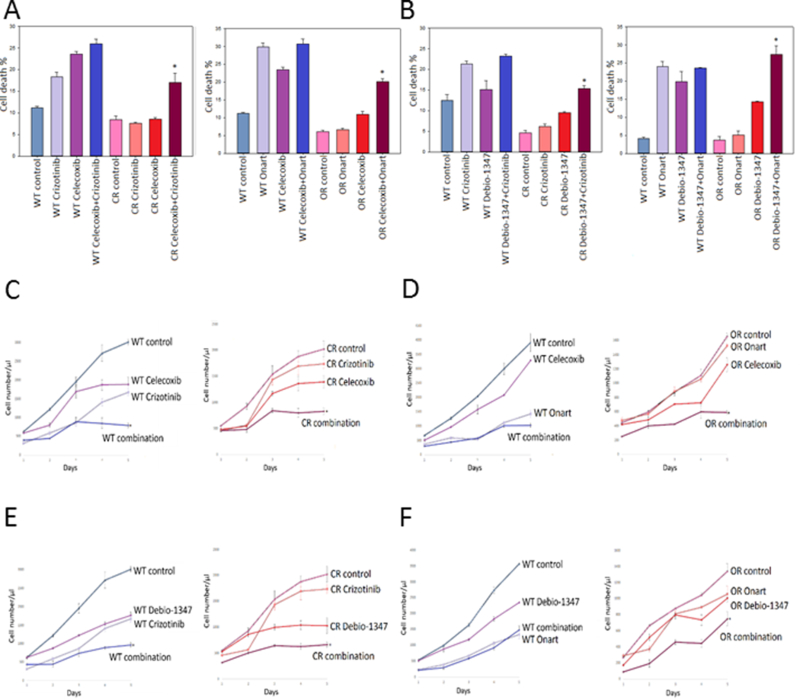
U87 wild type (WT), crizotinib-resistant (CR) & Onartuzumab-resistant (OR) cells were pretreated with A). COX-2 inhibitor celecoxib (100 nM) or B). FGFR1 inhibitor Debio-1347 (10 μM) for 2 hr then subsequently treated with either crizotinib (100 nM) or Onartuzumab (Onart) (300 nM) for 48 hr. Cell death was assessed via trypan blue assay. U87 WT, CR & OR cells were pretreated with C). & D). Celecoxib or E). & F). Debio-1347 for 2 hrs then subsequently treated with either crizotinib (100 nM) or Onart (300 nM) for 48 hr. Cell proliferation was assessed by cell counting over a period of 5 days and growth curves were established. *, P < 0.05.
Figure 4. Combination therapy with EGFR and mTOR inhibitors restores sensitivity to MET inhibitors in resistant GBM cells.

U373 wild type (WT), crizotinib-resistant (CR) & Onartuzumab-resistant (OR) cells were pretreated with A). EGFR inhibitor Erlotinib (25 nM) or B). mTOR inhibitor Rapamycin (25 μM) for 2 hr then subsequently treated with either crizotinib (100 nM) or (Onartuzumab) Onart (300 nM) for 48 hr. Cell death was assessed via trypan blue assay. U373 cells were pretreated with C). & D). Erlotinib or E). & F). Rapamycin for 2 hrs then subsequently treated with either crizotinib (100 nM) or Onart (300 nM) for 48 hr. Cell proliferation was assessed by cell counting over a period of 5 days and growth curves were established. *, P < 0.05.
Figure 5. Combination therapy with STAT3 inhibitor restores sensitivity to MET inhibitors in resistant GBM cells.
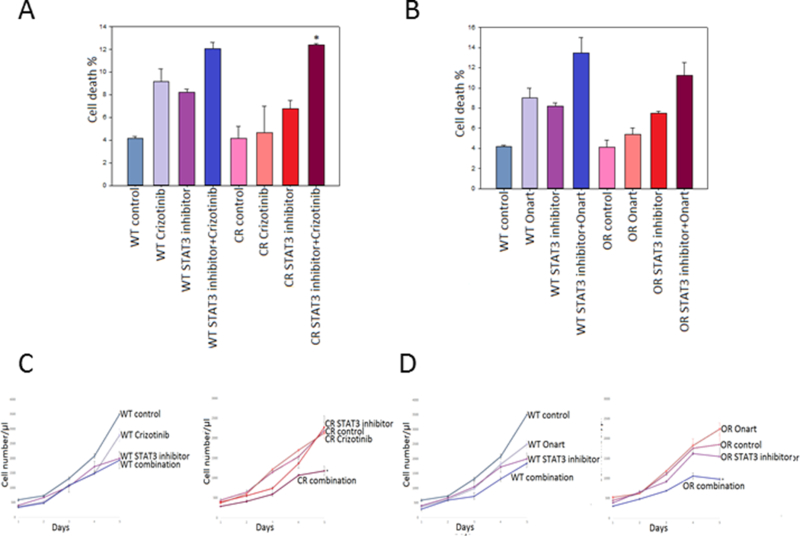
U373 wild type (WT), crizotinib-resistant (CR) & Onartuzumab-resistant (OR) cells were pretreated with a STAT3 inhibitor (25 μM) for 2 hr then subsequently treated with either A). crizotinib (100 nM) or B). Onartuzumab (Onart) (300 nM) for 48 hr. Cell death was assessed via trypan blue. U373 cells were pretreated with STAT3 inhibitor (25 μM) for 2 hrs then subsequently treated with either C). crizotinib (100 nM) or D). Onart (300 nM) for 48 hr. Cell proliferation was assessed by cell counting over a period of 5 days and growth curves were established. *, P < 0.05.
COX2 and FGFR1 siRNA-mediated knock-down induces cell death in crizotinib-resistant cells
To confirm that the cell death induced by celecoxib and debio-1347 was attributed to inhibition of COX2 and FGFR1 respectively, we silenced both COX2 and FGFR1 with siRNA and analyzed the effect on cell death. Cells were transfected with either si-control, si-COX2 or si-FGFR1 then treated with crizotinib for 48 h. siRNA-mediated knockdown of COX2 and FGFR1 was verified by immunobloting (Supplementary Fig. 1G). To assess whether COX2 or FGFR1 silencing sensitized resistant cells to MET inhibitors as celecoxib (inhibits COX2) and debio-1347 (inhibits FGFR1), cells were transfected as above and a trypan blue assay was performed. The data showed that silencing of COX2 and FGFR1 restored MET inhibitor sensitivity in crizotinib-resistant cells (Supplementary Fig. 1E & F). The above data show that silencing of either COX2 or FGFR1 expression leads to comparable anti-cancer effects on crizotinib-resistant U87 cells as celecoxib or debio-1347 treatment.
Combinational treatment is also effective against GSCs.
GSCs play an important role in mediating resistance to cytotoxic therapies 5. MET inhibition reduces GSC population sensitizing the tumor to therapies 5. With this in mind, we tested these combinational therapies on resistant and wildtype GSC827 and assessed the effect on cell death and proliferation. Although the concentration of all inhibitors (except MET inhibitors) had to be decreased due to toxicity, we observed a similar pattern as seen with U87 and U373 cells. All single agent treatments, although effective in wildtype GSC827 cells, were inadequate in MET inhibitor resistant GSC827 cells whilst simultaneous inhibition of a bypass signaling pathway (EGFR, FGFR, mTOR, STAT3 or COX-2) and MET inhibitor demonstrated significant induction of cell death and a dramatic suppression of cell proliferation. Importantly, combinational treatment with celecoxib and crizotinib decreased tumor cell proliferation and increased cell death significantly in crizotinib-resistant GSC827 cells. These data suggest that similar pathways are implicated in MET inhibitor resistance in GSCs and GBM cells indicating that these therapies may prove to be an extremely effective therapy for resistant GBM (Supplementary figure 6).
COX2 or FGFR1 inhibitions reverse resistance and cooperate with MET inhibitor to inhibit GBM xenograft growth in vivo
To examine whether these bypass signaling pathways can overcome MET inhibitor resistance in vivo, we assessed the effect of crizotinib alone and in combination with celecoxib and debio-1347 on tumor growth in immunodeficient mice bearing GBM xenografts. Celecoxib is an FDA-approved drug that demonstrates potent anti-cancer properties through the modulation of both the pro-survival BCL-2 30 family and COX-2, and is currently in clinical trials for the treatment of numerous neoplasms. Debio-1347 is in clinical trials for the treatment of advanced solid tumors and metastatic breast cancer with FGFR alterations. We injected either wildtype or resistant U87 cells into the striata of immunodeficient mice, six days after implantation the animals were treated with either control (DMSO), either agent alone [Debio-1347 (25 mg/kg), celecoxib (10 mg/kg) or crizotinib (25 mg/kg)] or in combination [Debio-1347+crizotinib or celecoxib+crizotinib] by oral gavage daily for 7 days. Tumors were visualized by MRI and volumes were quantified. The data show that both therapeutic combinations inhibited tumor growth in crizotinib-resistant mice significantly more than either agent alone (Figure. 6A & B). The resistant cells displayed significant tumor volume reduction when subjected to combinational treatment indicating restored sensitivity to MET inhibitors.
Figure 6. Combinational treatment inhibits MET inhibitor resistant xenograft growth.
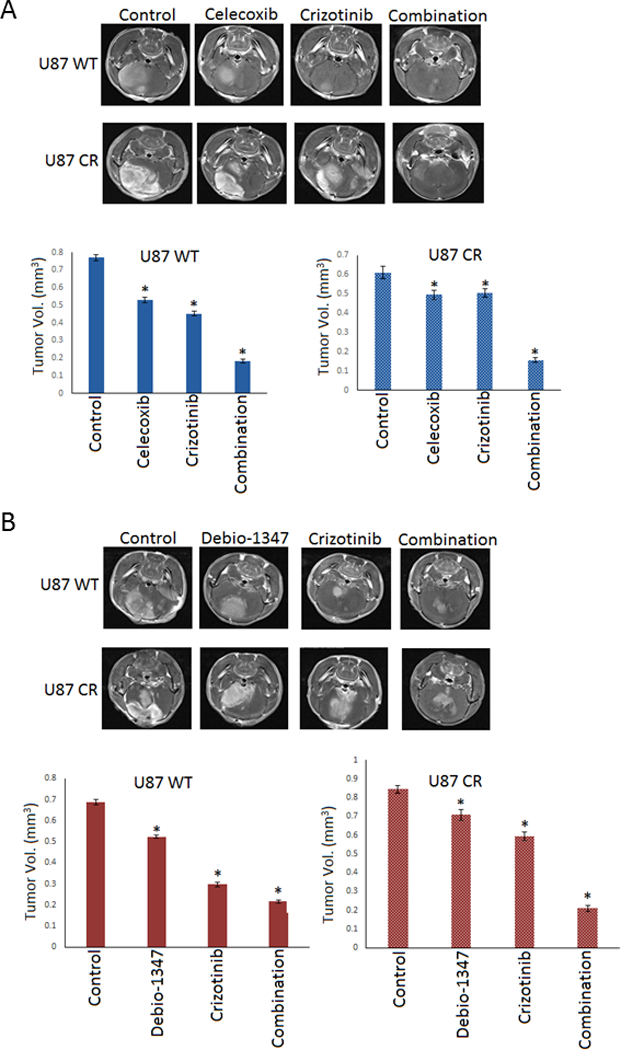
U87 wild type (WT) & crizotinib-resistant (CR) cells were stereotactically implanted in the striata of immunodeficient mice (n = 10). A). vehicle control, Celecoxib, crizotinib or the combination, B). Vehicle control, Debio-1347, crizotinib or the combination was administered daily by oral gavage starting 6 days after tumor implantation. The animals were subjected to MRI scan at 3 weeks after tumor implantation and tumor volumes were quantified. The data show that both celecoxib and debio-1347 significantly inhibit tumor growth and resensitize tumors to crizotinib treatment. *, P < 0.05 relative to control and single drug treatment.
Discussion
Although the MET pathway is often dysregulated in GBM, MET inhibitors have not been particularly effective in treating cancer patients due to acquired resistance. One mechanism of acquiring resistance against MET inhibitors is via activation of bypass pathways that compensate for the loss of survival signaling when MET is inhibited. Elucidating these bypass pathways offers the potential to develop combinatorial drug therapy to re-sensitize GBM cells to MET inhibitors. In this study, we developed GBM cell and animal models of resistance to MET inhibitors. Resistant cells revealed increased levels of active p.MET that could not be suppressed by the MET inhibitors, thus proving resistance and suggesting that MET receptor activation could contribute to this resistance. This finding differs from published mechanisms of resistance to EGFR inhibitors which involve loss of oncogenic mutant EGFRvIII 31. Using this model, we investigated the proteomic changes that occur when GBM cells become resistant to two clinically applicable MET inhibitors (crizotinib and Onartuzumab), uncovered several important bypass pathways that include mTOR, FGFR1, EGFR STAT3 and COX-2 and showed that targeting these pathways in combination with MET inhibitors, reverses resistance to the MET inhibitors.
The mTOR pathway is highly activated in GBM 32 and, although mTOR has been implicated in acquired resistance in small cell lung cancer 33, less is known about its role in GBM therapy resistance. MET inhibition leads to downregulation of PI3K signaling, which, in turn, leads to decreased activation of mTOR resulting in the induction of apoptosis and decreased cell proliferation 34. However, aberrant activation of PI3K/AKT signaling as a bypass mechanism results in increased mTOR activation that promotes cancer progression, metastasis and invasion 35. We demonstrate that mTOR phosphorylation is significantly increased in MET inhibitor resistant GBM cells, suggesting a role for the mTOR pathway in MET inhibitor resistance. Rapamycin, an FDA-approved inhibitor of mTOR, alone did not significantly enhance cell death but did have antiproliferative effects in MET inhibitor resistant GBM cells. Combinational treatment of resistant GBM cells with rapamycin and either MET inhibitor induced apoptosis and further suppressed cell proliferation indicating restored MET inhibitor sensitivity.
Aberrant activation of RTKs such as FGFR1 and EGFR is a recognized mechanism by which malignant cells acquire resistance to other RTK monotherapies 36. FGFR1 and EGFR compensate for the loss of MET-mediated survival signaling through reactivation of downstream PI3K and STAT signaling 37. We demonstrate that FGFR1 is implicated in MET inhibitor resistance in GBM. FGFR1 is upregulated in MET inhibitor resistant GBM cells and shows a trend towards correlation with GBM patient survival based on TCGA data analysis. Therefore, we assessed the effect of debio-1347, an FGFR1 inhibitor, in combination with crizotinib or Onartuzumab on MET inhibitor resistant GBM cells and we showed that FGFR1 inhibition can circumvent MET inhibitor resistance and simultaneous inhibition of both FGFR1 and MET is advantageous for reversal of MET inhibitor resistance 38.
EGFR and MET are frequently co-expressed in cancer and HGF can transactivate EGFR, which in turn, activates MET resulting in synergistic tumor growth 36–37. Furthermore, MET has been reported to play a role in acquired resistance to EGFR-targeted therapies in many cancers 17, 39. Attributed to this and the fact that cross-talk exists between MET and EGFR, MET inhibitor resistant GBM cells were subjected to combined treatment with the EGFR inhibitor, erlotinib, and either crizotinib or Onartuzumab. Erlotinib alone did not significantly induce apoptotic cell death in MET inhibitor resistant GBM cells however, concomitant treatment with erlotinib and either crizotinib or Onartuzumab restored MET inhibitor sensitivity leading to enhanced cell death and decreased cell proliferation.
Many growth factor receptors including MET activate STAT3 40, 32. STAT3 is elevated in GBM, driving tumor growth, angiogenesis and invasion 41 through the regulation of downstream targets including c-myc, Bcl-2 and Bcl-XL and is emerging as a drug resistance mechanism in GBM 42. Based on our screening data, we assessed the effect of STAT3 inhibition on MET inhibitor resistance and found that combinational treatment of MET inhibitor resistant GBM cells with a STAT3 inhibitor and MET inhibitor induced cell death and significantly inhibited cell proliferation indicating restored MET inhibitor sensitivity.
We also found that MET inhibitor resistance resulted in upregulation of COX-2, an inducible cyclooxygenase that is overexpressed GBM 43. Elevation of COX-2 stimulates increased angiogenesis and invasion of tumor cells and correlates with poor prognosis 44 although little is known about COX-2 and MET inhibitor resistance in GBM. Celecoxib, a COX-2 inhibitor, was used in conjunction with either crizotinib or Onartuzumab to assess the effect of COX-2 inhibition on MET inhibitor resistant GBM cells. MET inhibitor resistant GBM cells demonstrated significant toxicity to combination therapy. Although COX-2 inhibition is thought to be the primary mode of action, celecoxib has also been reported to act in a COX-2 independent manner 45. Further evaluation may be needed to completely elucidate the mechanism by which celecoxib reverts MET inhibitor resistance.
GSCs, a small subpopulation responsible for self-renewal, have been implicated in GBM relapse 46–47. Inhibition of MET, which is expressed in GSCs, halts GBM progression and decreases the expression of stem markers such as CD133 and Sox2. GSCs display enhanced sensitivity to MET inhibitors indicating a vital role for MET in GSC maintenance 48. Attributed to this, the development of anti-cancer therapies that target GSCs appears imperative for optimal GBM treatment 49. We show that treatment with MET inhibitor and simultaneous inhibition of one of the bypass proteins reverses MET resistance in GSCs. Interestingly, RPPA exposed considerable overlap in the bypass signaling involved in resistance to both MET inhibitors. These common up-regulated pathways are vital for cell survival and include important molecules such as EGFR, BCL-2 COX-2 and FGFR1. The ability to target a commonly altered pathway, that would be effective at reversing the effects of drug resistance to multiple inhibitors, is the ultimate goal for personalized therapy 50.
Our data identify the mechanisms of resistance to MET inhibitors in GBM and suggest new combination therapies that overcome resistance. Device-based and PDX model-based screens are currently being assessed for optimal, patient-specific oncogenic driver identification. Informed by our findings, individual patients that display drug resistance could be assessed for their unique oncogenic driver signature and receive the most effective combination therapy.
Supplementary Material
Translational relevance.
The receptor tyrosine kinase MET is frequently upregulated or over activated in many cancers including GBM. Consequently, several clinically applicable MET inhibitors have been developed. Although MET inhibitors initially display anticancer activity, resistance to the drugs frequently occurs, rendering these agents ineffective. Elucidating the mechanisms of acquired resistance to MET inhibitors is a challenge that must be overcome in order to halt disease progression. We used proteomic screenings to identify pathways altered in response to acquired MET inhibitor resistance in GBM. We uncovered several critical signaling molecules that mediated resistance to two clinically applicable anti-MET drugs that were previously tested in clinical trials. Inhibition of these molecules with clinically applicable drugs reversed resistance to MET inhibitors. Our data uncover the mechanisms of adaptive resistance to MET inhibitors and describe new combination therapies that overcome resistance and that could be tested in clinical trials.
Acknowledgments
Financial Support:
This work was supported by NIH grants RO1 NS045209 (Abounader) and UO1 CA220841 (Abounader).
Footnotes
Conflict of interest :
See Phan is the Associate Group Medical Director at Genentech Inc. that commercializes Onartuzumab.
References
- 1.Wen PY; Kesari S, Malignant gliomas in adults. New Engl J Med 2008, 359 (5), 492–507. [DOI] [PubMed] [Google Scholar]
- 2.Awad AJ; Burns TC; Zhang Y; Abounader R, Targeting MET for glioma therapy. Neurosurgical focus 2014, 37 (6), E10. [DOI] [PubMed] [Google Scholar]
- 3.Pearson JRD; Regad T, Targeting cellular pathways in glioblastoma multiforme. Signal Transduct Target Ther 2017, 2, 17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brennan CW; Verhaak RG; McKenna A; Campos B; Noushmehr H; Salama SR; Zheng S; Chakravarty D; Sanborn JZ; Berman SH; Beroukhim R; Bernard B; Wu CJ; Genovese G; Shmulevich I; Barnholtz-Sloan J; Zou L; Vegesna R; Shukla SA; Ciriello G; Yung WK; Zhang W; Sougnez C; Mikkelsen T; Aldape K; Bigner DD; Van Meir EG; Prados M; Sloan A; Black KL; Eschbacher J; Finocchiaro G; Friedman W; Andrews DW; Guha A; Iacocca M; O’Neill BP; Foltz G; Myers J; Weisenberger DJ; Penny R; Kucherlapati R; Perou CM; Hayes DN; Gibbs R; Marra M; Mills GB; Lander E; Spellman P; Wilson R; Sander C; Weinstein J; Meyerson M; Gabriel S; Laird PW; Haussler D; Getz G; Chin L; Network TR, The somatic genomic landscape of glioblastoma. Cell 2013, 155 (2), 462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cruickshanks N; Zhang Y; Yuan F; Pahuski M; Gibert M; Abounader R, Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma. Cancers 2017, 9 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cecchi F; Rabe DC; Bottaro DP, The Hepatocyte Growth Factor Receptor: Structure, Function and Pharmacological Targeting in Cancer. Curr Signal Transduct Ther 2011, 6 (2), 146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abounader R; Ranganathan S; Lal B; Fielding K; Book A; Dietz H; Burger P; Laterra J, Reversion of human glioblastoma malignancy by U1 small nuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c-met expression. J Natl Cancer Inst 1999, 91 (18), 1548–56. [DOI] [PubMed] [Google Scholar]
- 8.Abounader R; Laterra J, Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol 2005, 7 (4), 436–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y; Farenholtz K; Yang Y; Guessous F; Dipierro CG; Calvert V; Deng J; Schiff D; Xin W; Lee J; Purow BW; Christensen JG; Petricoin EF 3rd; Abounader R, Hepatocyte growth factor sensitizes brain tumors to c-Met kinase inhibition. Clin Cancer Res 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garajova I; Giovannetti E; Biasco G; Peters GJ, c-Met as a Target for Personalized Therapy. Transl Oncogenomics 2015, 7 (Suppl 1), 13–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwak Y; Kim SI; Park CK; Paek SH; Lee ST; Park SH, C-MET overexpression and amplification in gliomas. Int J Clin Exp Pathol 2015, 8 (11), 14932–8. [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y; Farenholtz KE; Yang Y; Guessous F; Dipierro CG; Calvert VS; Deng J; Schiff D; Xin W; Lee JK; Purow B; Christensen J; Petricoin E; Abounader R, Hepatocyte growth factor sensitizes brain tumors to c-MET kinase inhibition. Clin Cancer Res 2013, 19 (6), 1433–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guessous F; Zhang Y; diPierro C; Marcinkiewicz L; Sarkaria J; Schiff D; Buchanan S; Abounader R, An orally bioavailable c-Met kinase inhibitor potently inhibits brain tumor malignancy and growth. Anticancer Agents Med Chem 2010, 10 (1), 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nehoff H; Parayath NN; McConnell MJ; Taurin S; Greish K, A combination of tyrosine kinase inhibitors, crizotinib and dasatinib for the treatment of glioblastoma multiforme. Oncotarget 2015, 6 (35), 37948–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petterson SA; Dahlrot RH; Hermansen SK; S KAM; Gundesen MT; Wohlleben H; Rasmussen T; Beier CP; Hansen S; Kristensen BW, High levels of c-Met is associated with poor prognosis in glioblastoma. J Neurooncol 2015, 122 (3), 517–27. [DOI] [PubMed] [Google Scholar]
- 16.Kazandjian D; Blumenthal GM; Chen HY; He K; Patel M; Justice R; Keegan P; Pazdur R, FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. The oncologist 2014, 19 (10), e5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nanjo S; Yamada T; Nishihara H; Takeuchi S; Sano T; Nakagawa T; Ishikawa D; Zhao L; Ebi H; Yasumoto K; Matsumoto K; Yano S, Ability of the Met kinase inhibitor crizotinib and new generation EGFR inhibitors to overcome resistance to EGFR inhibitors. PLoS One 2013, 8 (12), e84700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morley R; Cardenas A; Hawkins P; Suzuki Y; Paton V; Phan SC; Merchant M; Hsu J; Yu W; Xia Q; Koralek D; Luhn P; Aldairy W, Safety of Onartuzumab in Patients with Solid Tumors: Experience to Date from the Onartuzumab Clinical Trial Program. PLoS One 2015, 10 (10), e0139679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heist RS; Sequist LV; Borger D; Gainor JF; Arellano RS; Le LP; Dias-Santagata D; Clark JW; Engelman JA; Shaw AT; Iafrate AJ, Acquired Resistance to Crizotinib in NSCLC with MET Exon 14 Skipping. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2016, 11 (8), 1242–1245. [DOI] [PubMed] [Google Scholar]
- 20.Ahronian LG; Corcoran RB, Strategies for monitoring and combating resistance to combination kinase inhibitors for cancer therapy. Genome medicine 2017, 9 (1), 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Botting GM; Rastogi I; Chhabra G; Nlend M; Puri N, Mechanism of Resistance and Novel Targets Mediating Resistance to EGFR and c-Met Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. PLoS One 2015, 10 (8), e0136155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engstrom LD; Aranda R; Lee M; Tovar EA; Essenburg CJ; Madaj Z; Chiang H; Briere D; Hallin J; Lopez-Casas PP; Banos N; Menendez C; Hidalgo M; Tassell V; Chao R; Chudova DI; Lanman RB; Olson P; Bazhenova L; Patel SP; Graveel C; Nishino M; Shapiro GI; Peled N; Awad MM; Janne PA; Christensen JG, Glesatinib Exhibits Antitumor Activity in Lung Cancer Models and Patients Harboring MET Exon 14 Mutations and Overcomes Mutation-mediated Resistance to Type I MET Inhibitors in Nonclinical Models. Clin Cancer Res 2017, 23 (21), 6661–6672. [DOI] [PubMed] [Google Scholar]
- 23.Kim E; Kim M; Woo DH; Shin Y; Shin J; Chang N; Oh YT; Kim H; Rheey J; Nakano I; Lee C; Joo KM; Rich JN; Nam DH; Lee J, Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23 (6), 839–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y; Cruickshanks N; Yuan F; Wang B; Pahuski M; Wulfkuhle J; Gallagher I; Koeppel AF; Hatef S; Papanicolas C; Lee J; Bar EE; Schiff D; Turner SD; Petricoin EF; Gray LS; Abounader R, Targetable T-type Calcium Channels Drive Glioblastoma. Cancer Res 2017, 77 (13), 3479–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y; Kim J; Mueller AC; Dey B; Yang Y; Lee DH; Hachmann J; Finderle S; Park DM; Christensen J; Schiff D; Purow B; Dutta A; Abounader R, Multiple receptor tyrosine kinases converge on microRNA-134 to control KRAS, STAT5B, and glioblastoma. Cell death and differentiation 2014, 21 (5), 720–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Einspahr JG; Calvert V; Alberts DS; Curiel-Lewandrowski C; Warneke J; Krouse R; Stratton SP; Liotta L; Longo C; Pellicani G; Prasad A; Sagerman P; Bermudez Y; Deng J; Bowden GT; Petricoin EF 3rd, Functional protein pathway activation mapping of the progression of normal skin to squamous cell carcinoma. Cancer Prev Res (Phila) 5 (3), 403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pierobon M; Vanmeter AJ; Moroni N; Galdi F; Petricoin EF 3rd, Reverse-phase protein microarrays. Methods Mol Biol 823, 215–35. [DOI] [PubMed] [Google Scholar]
- 28.Paweletz CP; Charboneau L; Bichsel VE; Simone NL; Chen T; Gillespie JW; Emmert-Buck MR; Roth MJ; Petricoin IE; Liotta LA, Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene 2001, 20 (16), 1981–9. [DOI] [PubMed] [Google Scholar]
- 29.Li Y; Guessous F; DiPierro C; Zhang Y; Mudrick T; Fuller L; Johnson E; Marcinkiewicz L; Engelhardt M; Kefas B; Schiff D; Kim J; Abounader R, Interactions between PTEN and the c-Met pathway in glioblastoma and implications for therapy. Mol Cancer Ther 2009, 8 (2), 376–85. [DOI] [PubMed] [Google Scholar]
- 30.Winfield LL; Payton-Stewart F, Celecoxib and Bcl-2: emerging possibilities for anticancer drug design. Future Med Chem 2012, 4 (3), 361–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nathanson DA; Gini B; Mottahedeh J; Visnyei K; Koga T; Gomez G; Eskin A; Hwang K; Wang J; Masui K; Paucar A; Yang H; Ohashi M; Zhu S; Wykosky J; Reed R; Nelson SF; Cloughesy TF; James CD; Rao PN; Kornblum HI; Heath JR; Cavenee WK; Furnari FB; Mischel PS, Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2014, 343 (6166), 72–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lo HW, EGFR-targeted therapy in malignant glioma: novel aspects and mechanisms of drug resistance. Curr Mol Pharmacol 2010, 3 (1), 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sen T; Tong P; Diao L; Li L; Fan Y; Hoff J; Heymach JV; Wang J; Byers LA, Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin Cancer Res 2017, 23 (20), 6239–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanteti R; Dhanasingh I; Kawada I; Lennon FE; Arif Q; Bueno R; Hasina R; Husain AN; Vigneswaran W; Seiwert T; Kindler HL; Salgia R, MET and PI3K/mTOR as a potential combinatorial therapeutic target in malignant pleural mesothelioma. PLoS One 2014, 9 (9), e105919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akhavan D; Cloughesy TF; Mischel PS, mTOR signaling in glioblastoma: lessons learned from bench to bedside. Neuro Oncol 2010, 12 (8), 882–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fong JT; Jacobs RJ; Moravec DN; Uppada SB; Botting GM; Nlend M; Puri N, Alternative signaling pathways as potential therapeutic targets for overcoming EGFR and c-Met inhibitor resistance in non-small cell lung cancer. PLoS One 2013, 8 (11), e78398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Velpula KK; Dasari VR; Asuthkar S; Gorantla B; Tsung AJ, EGFR and c-Met Cross Talk in Glioblastoma and Its Regulation by Human Cord Blood Stem Cells. Translational oncology 2012, 5 (5), 379–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim SM; Kim H; Yun MR; Kang HN; Pyo KH; Park HJ; Lee JM; Choi HM; Ellinghaus P; Ocker M; Paik S; Kim HR; Cho BC, Activation of the Met kinase confers acquired drug resistance in FGFR-targeted lung cancer therapy. Oncogenesis 2016, 5 (7), e241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hrustanovic G; Lee BJ; Bivona TG, Mechanisms of resistance to EGFR targeted therapies. Cancer Biol Ther 2013, 14 (4), 304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu H; Kortylewski M; Pardoll D, Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nature reviews. Immunology 2007, 7 (1), 41–51. [DOI] [PubMed] [Google Scholar]
- 41.Mukthavaram R; Ouyang X; Saklecha R; Jiang P; Nomura N; Pingle SC; Guo F; Makale M; Kesari S, Effect of the JAK2/STAT3 inhibitor SAR317461 on human glioblastoma tumorspheres. Journal of translational medicine 2015, 13, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gray GK; McFarland BC; Nozell SE; Benveniste EN, NF-kappaB and STAT3 in glioblastoma: therapeutic targets coming of age. Expert review of neurotherapeutics 2014, 14 (11), 1293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sminia P; Stoter TR; van der Valk P; Elkhuizen PH; Tadema TM; Kuipers GK; Vandertop WP; Lafleur MV; Slotman BJ, Expression of cyclooxygenase-2 and epidermal growth factor receptor in primary and recurrent glioblastoma multiforme. J Cancer Res Clin Oncol 2005, 131 (10), 653–61. [DOI] [PubMed] [Google Scholar]
- 44.Xu K; Wang L; Shu HK, COX-2 overexpression increases malignant potential of human glioma cells through Id1. Oncotarget 2014, 5 (5), 1241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Booth L; Roberts JL; Cruickshanks N; Tavallai S; Webb T; Samuel P; Conley A; Binion B; Young HF; Poklepovic A; Spiegel S; Dent P, PDE5 inhibitors enhance celecoxib killing in multiple tumor types. J Cell Physiol 2015, 230 (5), 1115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Auffinger B; Spencer D; Pytel P; Ahmed AU; Lesniak MS, The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert review of neurotherapeutics 2015, 15 (7), 741–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng L; Bao S; Rich JN, Potential therapeutic implications of cancer stem cells in glioblastoma. Biochem Pharmacol 2010, 80 (5), 654–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rath P; Lal B; Ajala O; Li Y; Xia S; Kim J; Laterra J, In Vivo c-Met Pathway Inhibition Depletes Human Glioma Xenografts of Tumor-Propagating Stem-Like Cells. Translational oncology 2013, 6 (2), 104–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang Z; Cheng L; Guryanova OA; Wu Q; Bao S, Cancer stem cells in glioblastoma--molecular signaling and therapeutic targeting. Protein Cell 2010, 1 (7), 638–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J; Di C; Mattox AK; Wu L; Adamson DC, The future role of personalized medicine in the treatment of glioblastoma multiforme. Pharmacogenomics and personalized medicine 2010, 3, 111–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.