

Abstract
PURPOSE
Thyroid cancer is frequently difficult to diagnose due to an overlap of cytological features between malignant and benign nodules. This overlap leads to unnecessary removal of the thyroid in patients without cancer. While providing some improvement over cytopathologic diagnostics, molecular methods frequently fail to provide a correct diagnosis for thyroid nodules. These approaches are based on the difference between cancer and adjacent thyroid tissue and assume that adjacent tissues are the same as benign nodules. However, in contrast to adjacent tissues, benign thyroid nodules can contain genetic alterations that can be found in cancer.
PATIENTS AND METHODS
For the development of a new molecular diagnostic test for thyroid cancer, we evaluated DNA methylation in 109 thyroid tissues by using genome wide single base resolution DNA methylation analysis. The test was validated in a retrospective cohort containing 65 thyroid nodules.
RESULTS
By conducting Reduced Representation Bisulfite Sequencing in 109 thyroid specimens, we found significant differences between adjacent tissue, benign nodules, and cancer. These tissue specific signatures are strongly linked to active enhancers and cancer associated genes. Based on these signatures, we developed a new epigenetic approach for thyroid diagnostics. According to the validation cohort, our test has an estimated specificity of 97% (95% CI, 81 to 100), sensitivity of 100% (95% CI, 87 to 100), PPV of 97% (95% CI, 83-100), and NPV of 100% (95% CI, 86 to 100).
CONCLUSION
These data show that epigenetic testing can provide outstanding diagnostic accuracy for thyroid nodules.
Introduction
Every year, as many as 50,000 patients in the United States receive unnecessary thyroidectomies1. These unnecessary thyroidectomies occur due to the difficulty in pre-operatively distinguishing benign thyroid nodules from thyroid cancers. While some thyroidectomies are required for patients with thyroid nodules for reasons other than cancer, it has been estimated that as many as 70% of thyroidectomies are unnecessary. Surgical complications from thyroidectomy include damage to the recurrent laryngeal nerves which results in a permanently hoarse voice, or in severe cases, tracheostomy2,3. Thyroidectomy may cause damage or removal of the parathyroids which may result in a lifelong need for calcium supplementation. Lifelong thyroid hormone replacement is necessary when the whole thyroid is removed and does occur in a significant percentage of patients when only part of the thyroid is removed.
Papillary thyroid cancer, (PTC) represents about 75-80% of all thyroid cancers4. The diagnosis of thyroid cancer is established via fine needle aspiration (FNA) biopsy of thyroid nodules. In contrast to many other cancers, diagnosis of well-differentiated thyroid cancer is challenging due to frequent overlap of cytologic features between malignant and benign nodules from FNA. Many institutions have introduced the use of molecular tests for thyroid nodules with indeterminate cytology, which are observed in up to 40% of thyroid nodules5. The most widely used molecular test for thyroid nodules, the Afirma® Gene Expression Classifier is associated with a high rate of overdiagnosis due to low positive predictive value (PPV) (47%) and can be performed only on fresh aspirates due to the instability of the RNA used in the test6. Another diagnostic approach, ThyroSeqv2, is based on genetic alterations frequently found in benign nodules including H/K/NRAS mutations and RET/PTC rearrangements and is associated with a low PPV (42%-77%)7–13. These facts suggest that there is a need for a highly accurate thyroid nodule diagnostic test which uses time insensitive material.
DNA methylation is a stable epigenetic modification, which plays an important role in cancer initiation and progression. Studies comparing thyroid cancer versus adjacent thyroid tissues revealed that DNA methylation patterns in PTC are strongly associated with mRNA and miRNA signatures, BRAFV600E and H/K/NRAS mutations, and frequently reflect a histological tumor type14–17. However, use of this information for development of thyroid cancer testing can be problematic since (1) it is not adjacent thyroid tissue but benign nodules that undergo cancer diagnostic testing; and (2) there are potential differences between benign nodules and adjacent thyroid tissues. For instance, in contrast to adjacent thyroid tissues, some benign nodules have genetic alterations frequently found in cancer including H/K/NRAS mutations and RET/PTC rearrangements7–11. Thus comparison of malignant to benign nodules rather than adjacent thyroid tissue should be used for the development of thyroid nodule diagnostics.
Here, we demonstrate that benign thyroid nodules have a unique DNA methylation pattern different from normal thyroid tissues and malignant nodules with PTC. Based on this epigenetic signature specific for benign thyroid nodules and a DNA methylation signature specific for PTC, we have developed a highly accurate Diagnostic DNA Methylation Signature approach (DDMS) for thyroid nodule diagnostics. Our study suggests that DNA methylation sites analyzed by DDMS potentially plays a functional role in thyroid cancer development since these sites frequently co-localize with active enhancers in thyroid tissues and are associated with genes playing an important role in cancer development.
Material and Methods
Human tissue collection
Developmental and validation retrospective cohorts were represented by surgical leftover thyroid nodule samples which were obtained from the City of Hope Pathology Research Services Core – Biorepository according to the IRB protocols #97134 (J.H.Y.) and #04069 (P.C.). IRB protocol #97134 (de-identified retrospective leftover discard specimens) falls under non-human subject research criteria according to City of Hope IRB guidelines and does not require patient consent. For IRB protocol #04609 (discard leftover specimens), a waiver of informed consent and HIPAA authorization was issued by the City of Hope IRB. Both studies were conducted in accordance with recognized ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, U.S. Common Rule) and were approved by the City of Hope IRB. Tissue H&E slides were blindly evaluated by two pathologists (S.-W.T.T. and S.C.). Specimens were included in the study if the pathologists agreed with the diagnosis (benign versus malignant); and their diagnosis did not conflict with the original diagnosis of the nodule. Disputed cases were included and considered as malignant if they had BRAFV600E mutation or had metastasis.
DNA methylation profiling and mutation screening
DNA from fresh frozen samples was isolated by using the phenol/chloroform extraction approach. For DNA isolation from FFPE samples, QIAamp DNA FFPE Tissue Kit was used (Qiagen Inc., Valencia, CA). The RRBS procedure was previously described (Hahn et. al., 2015). DNA sequencing was performed on an HiSeq2500 Illumina instrument and aligned to the human genome at the City of Hope Integrative Genomics Core (GEO submission: GSE107738). The analysis pipelines were implemented using R statistical language. Refseq genes were downloaded from the UCSC HG19 genome annotation database. For the DDMS diagnostics, since information for all 373 DDMS sites was sometimes not available for each specimen, the benign and malignant scores were calculated based on only available information.
For BRAFV600E mutation screening, BRAF exon 15 was amplified by using primers (5’ GGGCCAAAAATTTAATCAG and 5’ TTCCTTTACTTACTACACCTCAGATA). Sequences of PCR products were evaluated by Sanger sequencing performed by the City of Hope Integrative Genomics Core.
Chromatin Immunoprecipitation Sequencing (ChIP-Seq)
Chromatin Immunopreciptation (ChIP) with fresh frozen tissue was previously described18. Enriched thyroid DNA fraction was repaired and linker ligated by using by using KAPA Hyper Prep Kit (KAPA biosystems). Further, DNA amplified by using Phusion® High-Fidelity DNA Polymerase (NEB), sequenced on an HiSeq2500 Illumina instrument and aligned to the human genome at the City of Hope Integrative Genomics Core (GEO: submission: GSE107738).
Results
Benign nodules differ from thyroid cancer and adjacent thyroid tissues
We characterized DNA methylation patterns in 109 surgical thyroid specimens including 28 benign nodules, 39 PTCs and 42 adjacent normal thyroid tissues (Table 1, Table S1).
Table 1.
Patient data for the developing and testing cohorts.
| Developing cohort | Training cohort | ||||
|---|---|---|---|---|---|
| Patients | Age | Patients | Age | ||
| Benign | Females | 25 | 49.64 +/−17 | 28 | 49.93 +/−15.61 |
| Males | 3 | 69 +/−11.36 | 9 | 57.33 +/−15.81 | |
| Total | 28 | 51.71 +/−17.43 | 37 | 51.73 +/−15.78 | |
| Malignant | Females | 29 | 45.86 +/−16.35 | 29 | 45.55 +/−20.18 |
| Males | 10 | 62.1 +/−10.16 | 12 | 52.17 +/−12.42 | |
| Total | 39 | 50.03 +/−16.52 | 41 | 47.49 +/−18.35 | |
For DNA methylation profiling, we applied the Reduced Representation Bisulfite Sequencing (RRBS) approach, which provides single base resolution for DNA methylation information for over 6 million cytosines genome wide (after generating at least 20 million aligned sequencing reads)19. Using this data we searched for DNA methylation sites, which are characterized by tissue specific DNA methylation patterns. We performed the following comparisons: malignant nodules versus adjacent thyroid tissues, malignant nodules versus benign nodules, benign nodules versus adjacent tissues and benign nodules versus malignant nodules and adjacent thyroid tissues. We identified 18,593 cytosine locations which demonstrated a significant difference (adjusted FDR<10E-2) between analyzed tissue groups (Figure 1). Based on the clustering analysis of these locations, PTC is associated with unique DNA methylation patterns. At the same time, benign nodules are frequently characterized by an intermediate epigenetic pattern between thyroid tissues and malignant nodules (Figure 1; see intermediate clusters #1 and #2). However, 4,575 cytosines demonstrated a unique DNA methylation pattern specific to benign nodules. These DNA locations were associated with intensive DNA hypermethylation in benign nodules compared to malignant nodules and adjacent thyroid tissues. These data suggest that the DNA methylome of benign nodules differs from methylomes of adjacent thyroid tissue and malignant nodules; and this fact should be taken into account for development of diagnostics for thyroid nodules. In this light, it is very important to note that some adjacent thyroid tissues demonstrated a very similar pattern to the adjacent nodules which suggests a field cancerization effect in thyroid cancer (Figure 1).
Figure 1.
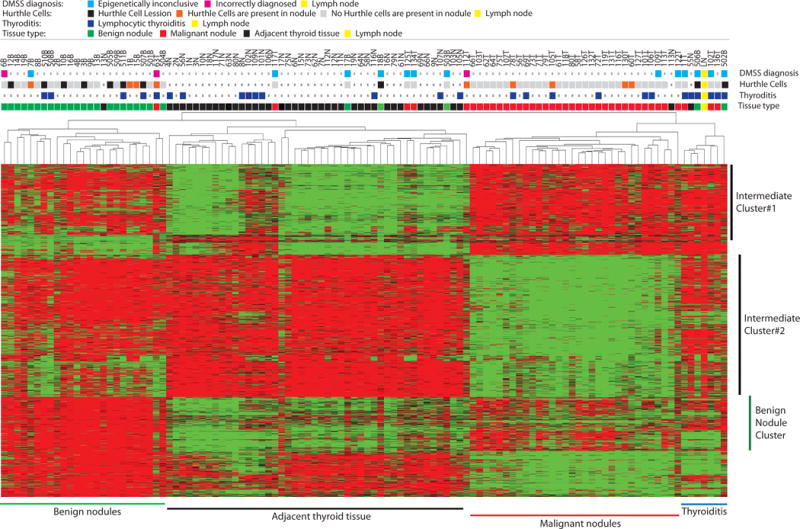
Differential DNA methylation in adjacent thyroid tissues, benign and malignant nodules. Centered clustering analysis of DNA methylation in normal thyroid tissues, benign and malignant nodules at 18,593 sites which demonstrated FDR<0.01 in comparisons: (i) malignant nodules versus adjacent thyroid tissues, (ii) malignant nodules versus benign nodules, (iii) benign nodules versus adjacent tissues and (iv) benign nodules versus malignant nodules and adjacent thyroid tissues. Each row represents DNA regions. Each column represents tissue specimen. Tissue status and lymphocytic thyroiditis are indicated. Red color indicates a high level of DNA methylation, green color reflects a low level DNA methylation while black color marks a medium level of DNA methylation. Different tissue clusters and cytosine groups are indicated.
We observed that a group of specimens with lymphocytic thyroiditis is clustered together and characterized by unique DNA methylation patterns (Figure 1). In order to evaluate if a high lymphocyte presence is responsible for the DNA methylation patterns in this “thyroiditis” tissue cluster, we profiled the DNA methylation pattern in lymph nodes as a tissue highly enriched with lymphocytes. Clustering analysis revealed that the lymph node had a very similar DNA methylation pattern to the “thyroiditis” cluster (Figure 1). These data suggest that a high presence of cells other than follicular cells in the analyzed thyroid tissue may interfere with DNA methylation pattern diagnostics.
DNA methylation tissue specific pattern frequently affects enhancers and cancer associated genes
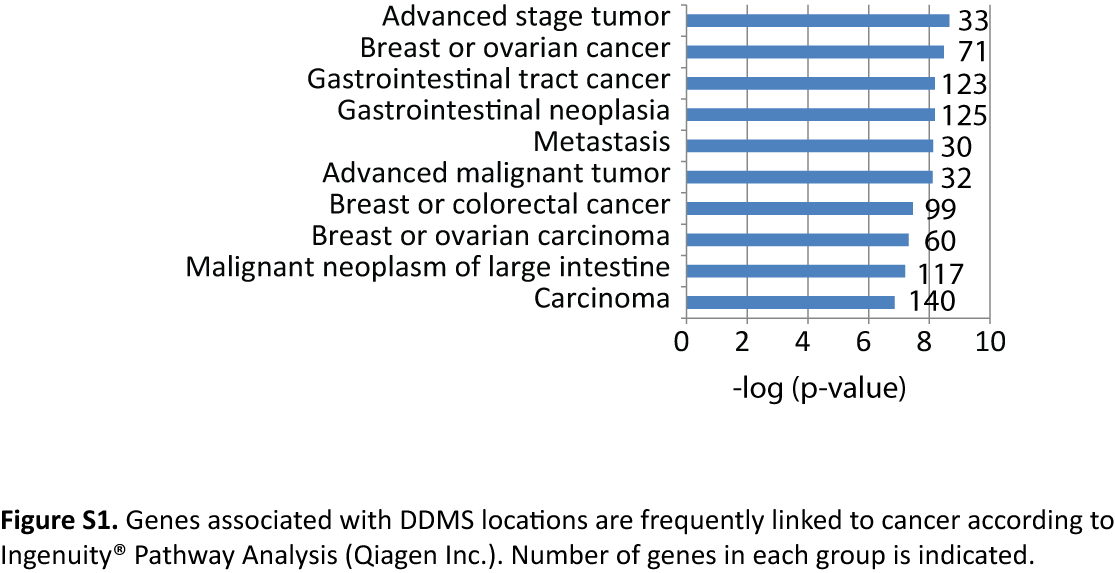
By using DNA methylation data for 109 specimens, we evaluated the presence of thyroid cancer-specific and benign nodule-specific patterns. We detected 373 genomic locations with tissue specific epigenetic patterns in benign and malignant nodules (Table S2). Based on these 373 differentially methylated genomic locations, we developed a Diagnostic DNA Methylation Signature approach (DDMS). According to our study, 62% (234 out 373) of DDMS locations were linked to gene bodies or gene promoters (Figure 2A). We identified 236 locations which are associated with the cancer-specific pattern and co-localized with 93 genes (Table S2). At the same time, 137 DDMS locations were associated with the benign-specific pattern and co-localized with 64 genes. According to Ingenuity® Pathway Analysis (Qiagen Inc.), genes associated with tissue specific signatures are linked to cancer and involved in cell proliferation, invasion and migration (Figure 2B, S1 and Table S3). Genes associated with tissue specific epigenetic signatures include GRP (gastrin releasing peptide), HMGA1 (high mobility group AT-hook 1), THY1 (Thy-1 cell surface antigen), TYK2 (tyrosine kinase 2) and GATA6 (GATA binding protein 6). Further, we investigated whether tissue specific DNA methylation signatures are associated with gene regulatory elements. We performed genome wide profiling of a chromatin mark of active enhancers, H3 lysine 27 acetylation (H3K27Ac) by using ChIP-sequencing with 2 normal adjacent thyroid tissues. A representative example of H3K27Ac pattern in adjacent thyroid tissues is shown (Figure S2A). Our data revealed that approximately 35% of DDMS locations are associated with H3K27Ac enriched regions in at least one of the adjacent tissues samples (Figure 2C and Table S2). Examples of co-localization of active enhancers with DDMS locations are shown for the genes ANGPTL4 (angiopoietin like 4), MUC1 (mucin 1, cell surface associated), NFIC (nuclear factor I C) and EPHA2 (EPH receptor A2) (Figure 2D, E and S1B, C). At the same time, approximately 54% of DDMS locations are located within H3K27Ac peaks or located at least within a 1 kb area surrounding H3K27Ac peaks, so-called “active enhancer shores”. These data suggest that DNA methylation at DDMS locations potentially play a regulatory role in thyroid cancer development.
Figure 2.
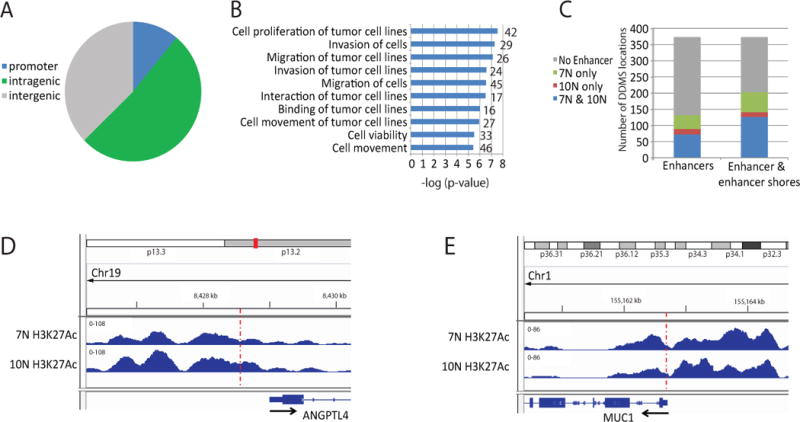
DNA locations associated with thyroid tissue specific signatures (DDMS locations) are linked to cancer associated genes and active enhancers. A. Distribution in genome of DDMS locations. B. Genes co-localized with DDMS locations frequently have cancer associated functions according to Ingenuity® Pathway Analysis (Qiagen Inc.). Number of genes in each group is indicated. C. Distribution of DDMS locations relative to active enhancers in adjacent thyroid tissues. A number of DDMS locations without active enhancers, with active enhancers in one out two adjacent thyroid tissue and active enhancers in both tissues are shown. Enhancer shores were defined as 1 kb areas surrounding H3K27Ac peaks. D. Representative snap-shot of co-localization DDMS site with H3K27Ac enrichment at the ANGPTL4 promoter in two analyzed adjacent thyroid tissues (7N and 10N). The snap-shot indicates the normalized number of reads. The red dotted line indicates the position of the DDMS location. Transcriptional start site and transcript direction are shown. E. Representative snap-shot of co-localization DDMS site with H3K27Ac enrichment at the MUC1 promoter in two analyzed adjacent thyroid tissues (7N and 10N). The snap-shot indicate the normalized number of reads. The red dotted line indicates the position of the DDMS location. Transcriptional start site and transcript direction are shown.
Validation of DDMS by using the leave-one-out cross-validation approach in the development cohort
We proposed that thyroid cancer diagnosis can be accomplished by a combination of both benign and thyroid cancer patterns/signatures; where each individual thyroid nodule is characterized by both the number of epigenetic changes specific to benign nodules and the number of epigenetic alterations specific to malignant nodules.
In order to validate our DDMS approach, we performed a statistical analysis based on the leave-one-out cross-validation technique20. In order to predict benign and cancer scores for 67 nodules, 67 benign and cancer unique predictive signatures were developed by using 109 specimens.
The leave-one-out cross-validation revealed that 11 of 67 analyzed specimens are associated with fewer than 5 out of the 137 epigenetic changes specific to benign nodules and fewer than 5 out of the 236 epigenetic changes specific to PTC (Table S1). We classified these specimens as un-diagnosable since (1) our approach is based on the identification of the tissue specific epigenetic signatures and (2) un-diagnosable tissues are missing tissue specific epigenetic alterations/signatures. These un-diagnosable specimens were excluded from further analysis.
The leave-one-out cross-validation indicated that some specimens are associated with the presence of both epigenetic scores, benign and malignant (Table S1). In order to diagnose these types of specimens, we introduced a cancer risk score which is defined as the ratio of the cancer score and the benign score [cancer risk score = (malignant score + 1)/(benign score + 1)]. We classified nodules with a cancer risk score above or equal to 1.0 as thyroid cancer, whereas the specimens with a cancer score below 1.0 were classified as benign nodules (Figure 3A). Based on these criteria, 23 out of 24 benign nodules were classified as benign; and 30 out of 32 malignancies were classified as cancer (Figure 3B). Both wrongly diagnosed malignant nodules (#63T and #112T) were Hurthle cell lesions but had more than 75% of adjacent thyroid tissue in the specimens according to H&E staining analysis (Table S1). Since (i) sensitivity is defined as the proportion of true positive in all positively classified samples; and (ii) we classify thyroid nodules as malignant when a specimen is characterized by at least one epigenetic signature and cancer risk score equal to 1.0 or more, the estimated sensitivity of DDMS is 94% (Figure 3C). On the other hand, since specificity is determined as a proportion of true negative in all negatively classified samples; and (ii) we classify benign nodules as nodules with at least one epigenetic signature and cancer risk score less than 1.0, the estimated specificity of our test is 95% (Figure 3C). Thus, based on the leave-one-out cross-validation technique in the developing nodule cohort, DDMS has an estimated specificity of 96%, sensitivity of 94%, PPV of 97%, NPV of 92% and accuracy of 95% (Figure 3C).
Figure 3.
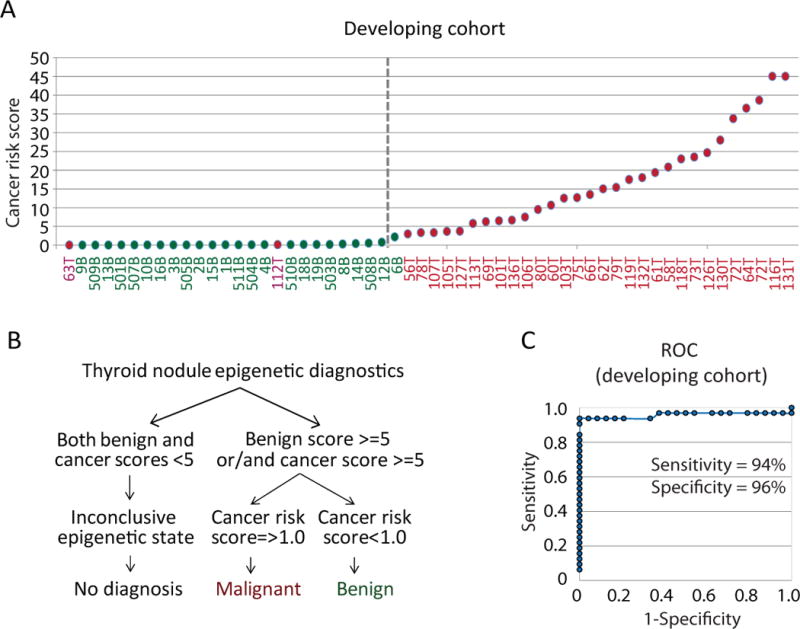
Diagnostic DNA Methylation Signature approach (DDMS). A. Cancer risk scores for specimens with conclusive epigenetic state according to leave-one-out cross-validation technique in the developing cohort. The threshold of cancer risk score of 1.0 is indicated. The values of cancer risk score for epigenetically conclusive thyroid nodules are shown. Abbreviation “B” is for thyroid benign nodule and “T” is for thyroid cancer. B. Diagnostics of thyroid nodules based on DDMS epigenetic signatures. C. Receiver operating characteristic for DDMS based on leave-one-out cross-validation technique results in the developing cohort.
Un-diagnosable specimens are frequently contaminated
In order to evaluate mechanisms responsible for an absence of epigenetic signatures in un-diagnosable specimens, we investigated an association between lymphocytic thyroiditis and un-diagnosable specimens (Figure 1). We found that all nodules associated with the “thyroiditis” tissue cluster were un-diagnosable according to DDMS; and 4 out of 11 un-diagnosable nodules were associated with thyroiditis (Figure 1).
Further we evaluated the potential impact of Hurtle cell presence on the DDMS epigenetic signatures (Figure 1 and Table S1). According to the pathological evaluation, the development cohort contained 9 nodules with some presence of Hurthle cells and 10 Hurthle cell lesions. From 10 Hurthle cell lesions, only 2 nodules were un-diagnosable. Both nodules were associated with the “thyroiditis” tissue cluster and had thyroiditis (Figure 1). At the same time, only one of 9 nodules with some presence of Hurthle cells was un-diagnosable.
Since some FNAs are falsely acquired from adjacent thyroid tissue we evaluated DDMS performance in adjacent thyroid tissue. We performed the leave-one-out cross-validation for 42 adjacent thyroid samples by using our 109-specimen cohort (Table S4). According to our study, only 38% (16 out 42) of adjacent tissues had epigenetic signatures. Fourteen out of these 16 adjacent tissues were classified as benign, when two adjacent tissues were classified as malignant. One wrongly classified adjacent tissue (113N) was associated with a very similar DNA methylation pattern to the adjacent malignant nodule (Fig. 1). This similarity can be potentially explained by a field cancerization effect. For another wrongly diagnosed normal adjacent tissue sample (55N), it is not clear whether the wrong diagnosis is associated with a field cancerization effect. The status of the nodule #55 located next to this normal adjacent tissue 55N is unknown due to a disagreement of the participating pathologists regarding the diagnosis of the nodule.
Further, we asked whether a high level of adjacent normal tissue in a biopsy might affect a DDMS diagnostic result. First, we determined the proportion of cancer cells in each malignant tissue by using H&E staining. We found 5 specimens 63T, 102T, 112T, 117T and 119T with approximately 25% or less of malignant cells in the specimen. Two (102T and 117T) out of five of these specimens were un-diagnosable when the other two (#63T and #112T) were misdiagnosed as benign. Thus, un-diagnosable specimens according to DDMS are frequently associated with a high presence of non-nodule cells including lymphocytes and adjacent tissues.
Validation of DDMS performance in a testing cohort
We performed validation of DDMS performance in a testing cohort by using RRBS. Our testing cohort contained 78 post-surgical thyroid nodules: 41 malignant and 37 benign thyroid nodules (Table 1, Table S1). We observed DNA methylation signatures in 34 malignant and 31 benign nodules. It is important to note that 8 out 13 of un-diagnosable specimens had areas of lymphocytic thyroiditis (Table S1). At the same time, only 3 out of 13 un-diagnosable nodules were associated with Hurthle cells; whereas 2 out of these 3 nodules had thyroiditis. All 34 cancers with epigenetic signatures were classified as malignant according to DDMS; and 30 out of 31 benign nodules were classified as benign (Figure 4A, Table S1). These data suggest that DDMS has an estimated specificity of 97% (95% CI, 80 to 100), sensitivity of 100% (95% CI, 86 to 100), PPV of 97% (95% CI, 82-100), NPV of 100% (95%, 85 to 100) (Figure 4B). Therefore, DDMS can serve as a potential diagnostic tool for thyroid cancer in specimens with epigenetic signatures.
Figure 4.
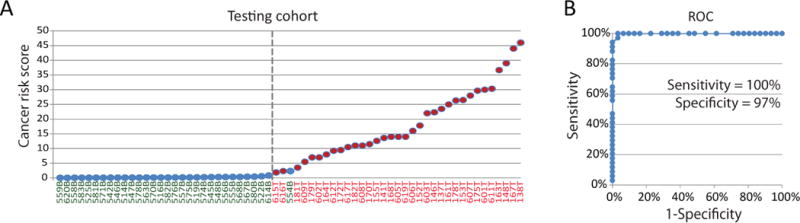
DDMS validation in the testing cohort. A. Cancer risk scores for specimens with conclusive epigenetic state in the developing cohort. The values of cancer risk score for epigenetically conclusive thyroid nodules are shown. Abbreviation “B” is for thyroid benign nodule and “T” is for thyroid cancer. The threshold of cancer risk score of 1.0 is indicated. B. Receiver operating characteristics for DDMS in the testing cohort.
Development of a Multiplex Bisulfite PCR (MB-PCR) approach for thyroid nodule diagnostics
In order to reduce costs, we developed an MB-PCR approach for the DDMS validation in thyroid nodules (Figure 5). This method is based on multiplex PCR with bisulfite converted DNA followed by ligation with a linker (a short double stranded oligonucleotide) and next generation sequencing (Figure 5A). MB-PCR demonstrated high reproducibility (R=0.97, p<0.0001) for the lowest range of DNA amount obtained by FNA, 100 ng (Figure 5B)21. The DNA methylation status of cytosines from the DDMS panel strongly correlates between RRBS and the newly developed MB-PCR for thyroid nodules with an average of correlation coefficient, R=0.93, P<0.0001 (Figure 5C). Thus, this inexpensive MB-PCR approach (under $100 per sample) based on DDMS can be used for thyroid nodule diagnostics.
Figure 5.
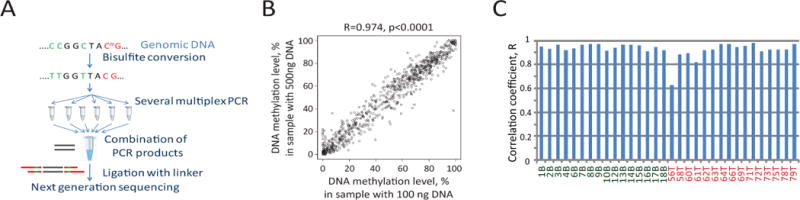
Multiplex bisulfite PCR assay (MB-PCR) for DNA methylation analysis in thyroid nodules. A. Flowchart of the multiplex bisulfite PCR assay combined with NextGen sequencing for DNA methylation analysis. B. Correlation between DNA methylation patterns from two technical replicates, one with 100 ng starting material and another 500 ng starting material. C. Correlation between RRBS and multiplex bisulfite PCR for 32 thyroid nodules. “B” is used for benign thyroid nodule and “T” is used for PTC.
Discussion
The evaluation of thyroid cancer has been very problematic due to a frequent overlap of cytologic features in benign and malignant nodules. Though current molecular testing methods have improved the accuracy of evaluation, their performance still has the potential for improvement. For instance, a classification of “suspicious” from the most widely used molecular test, the Afirma® Gene Expression Classifier, is seen in up to 60% of benign thyroid nodules6. ThyroSeqv2, which is based on known gene mutations and translocations, deems up to 58% of benign thyroid nodules as “suspicious”12,13. By contrast, our test is associated with PPV of 97% (95% CI, 82-100) according to the validation study.
According to our analysis, DDMS locations are strongly linked to genes involved in cancer development; and a third of DDMS locations are co-localized with active enhancers, gene regulatory elements (Figure 2, Table S2). These data suggest that DDMS locations play a role in functional cancer development.
In our study, DNA methylation is used for thyroid nodule diagnostics. Since DNA methylation is a very stable epigenetic mark, DNA methylation based testing does not require special handling and can be performed on FFPE preserved samples like cell blocks and even on FNA slides. Sometimes special sample handling is impossible when an unexpected incident, such as bleeding, occurs during a biopsy procedure. In this light, DDMS stands out in comparison to the RNA based test, Afirma® Gene Expression Classifier, that requires special tissue handling due to RNA instability.
Our approach is very different from standard cancer diagnostics. The standard diagnostics searches for cancer-associated alterations and classifies specimen as cancer negative in an absence of these characteristics. By contrast, our test defines both cancer negative and cancer positive states according to the nodule specific signatures. Therefore, an absence of those epigenetic changes makes the thyroid tissues un-diagnosable.
We found that 17% of thyroid nodules are missing DNA methylation changes specific for malignant or benign nodules. We observed that the absence of epigenetic signatures in thyroid nodules is frequently linked to lymphocytic thyroiditis. In fact, a group of thyroid nodules with thyroiditis had a very similar DNA methylation pattern to lymph node (Figure 1). At the same time, 62% of adjacent thyroid tissues and 40% (two out five) of malignant specimens with high levels (75% or more) of adjacent thyroid tissues were un-diagnosable according to DDMS. These data suggest that the presence (contamination) of cells other than follicular epithelial cells from a nodule in a specimen may lead to the absence of thyroid nodule epigenetic signatures. Such contamination is frequently found in FNA biopsies of thyroid nodules. Contaminating cells may include white blood cells, skeletal muscle, blood vessels or adjacent normal tissue. In order to overcome diagnostic difficulties associated with non-follicular cell contamination, current molecular thyroid nodule diagnostics such as ThyroSeqV2 perform an “assessment of sample adequacy for molecular testing”. The assessment is completed by analyzing an expression activity of a gene specifically expressed in thyroid epithelium, KRT71.
Regarding un-diagnosable samples which are not linked to thyroiditis, at this point, it is very difficult to determine whether these specimens have their own epigenetic signature since benign and malignant groups with the un-diagnosable specimens are fairly small. To solve this, a potential combination of DDMS and BRAFV600E genetic testing can reduce the number of undiagnosed patients. The BRAFV600E genetic screening is currently incorporated in Afirma® Gene Expression Classifier and ThyroSeqv213,22. According to developing and testing cohort data, the incorporation of BRAFV600E in DDMS resulted in 86% of specimens with a conclusive epigenetic state (Table S1). The positive side of the un-diagnosable classification is that it does not incorrectly diagnose but rather gives room for other testing approaches. Alternatively, microdissection such as laser capture microdissection may allow for an enrichment of the sample for non-contaminating cells.
In our study, the DNA methylation patterns specific to benign nodules were used not only for a better characterization of cancer specific alterations but also for the development of a benign specific signature. In fact, if we had not used the benign signature, the malignant un-diagnosable specimens would have been erroneously diagnosed as benign. Therefore, the introduction of the benign signature dramatically improves DDMS accuracy.
In contrast to current molecular thyroid nodule diagnostics, our test is based on differences between benign and malignant nodules. Our study suggests that benign nodules are distinct from adjacent thyroid tissues and malignant tissues. Therefore, not only adjacent tissues but the benign nodules themselves should be used for the development of thyroid nodule diagnostics. Furthermore, according to our study, some adjacent thyroid tissues have DNA methylation patterns (13N, 113N) that are very similar to the thyroid nodules to which these “normal tissues” are adjacent (Figure 1). This fact can be explained by a field cancerization effect which is observed in many types of cancer23–26. Field cancerization also explains classification of 113N thyroid tissues adjacent to a malignant nodule as malignant. Thus adjacent thyroid tissues may carry cancer associated alterations due to a field cancerization effect and have different DNA methylation compared to benign nodules. Our data shifts the emphasis of the research paradigm to compare the benign thyroid nodule instead of any thyroid tissue versus the cancer for the development of thyroid cancer diagnostics.
Taken together, we have developed a new, inexpensive, high precision diagnostic test based on DNA methylation patterns of benign and malignant thyroid nodules. We plan on performing a multicenter clinical trial to determine the performance of this test in FNAs in the clinic.
Supplementary Material
{kind=link}
{kind=link}
Translational Relevance Statement.
Up to 50,000 unnecessary thyroidectomies are performed annually in the United States due to the inability to diagnose benign versus cancerous thyroid nodules. These difficulties occur due to overlapping cytologic features between benign and malignant thyroid nodules. Molecular diagnostics for thyroid nodules has been introduced, but frequently fails to provide a correct diagnosis, and their routine use for thyroid nodule diagnostics is under debate.
By analyzing DNA methylation patterns from benign and malignant nodules, we developed a new method for thyroid nodule molecular diagnostics, the Diagnostic DNA Methylation Signature approach (DDMS), with an estimated specificity of 97%, sensitivity of 100%, PPV of 97%, and NPV of 100%. Thus, DDMS has the potential to provide standard of care thyroid cancer molecular diagnostics. Introduction of more accurate thyroid cancer diagnostics to the clinic will decrease the morbidity and even mortality of overdiagnosis and reduce health care costs associated with unnecessary thyroidectomies.
Acknowledgments
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award numbers R21CA223367 to M.A.H and J.H.Y. and P30CA033572 to City of Hope. Research reported in this publication included work performed in the Pathology Research Services Core – Biorepository and the City of Hope Integrative Genomics Core. Both cores are supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We thank Karen Miller, Michael Lewallen, and Maria De Lao for their support and outstanding work with patient specimens.
Footnotes
Presented, in part, at the 2016 AACR Annual Meeting; April 16-20, 2016, New Orleans, Louisiana
Conflict of interest: Audrey H. Choi, Hanjun Qin, Sue Chang, Sun-Wing T. Tong, Peiguo Chu, Byung-Wook Kim, Daniel Schmolze, Ryan Lew, Yasmine Ibrahim, Valeriy A. Poroyko, Sylvana Salvatierra, Alysha Baker, Jinhui Wang, Gerd P. Pfeifer declare no potential conflicts of interest.
John H. Yim, Arthur X. Li, Xiwei Wu, Yuman Fong and Maria A. Hahn are authors of a pending patent “METHODS, TREATMENT, AND COMPOSITIONS FOR CHARACTERIZING THYROID NODULE” (Publication number: 20170022570).
References
- 1.Nikiforov YE, Ohori NP, Hodak SP, et al. Impact of mutational testing on the diagnosis and management of patients with cytologically indeterminate thyroid nodules: a prospective analysis of 1056 FNA samples. J Clin Endocrinol Metab. 2011;96:3390–7. doi: 10.1210/jc.2011-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan AC, Lang BH, Wong KP. The pros and cons of routine central compartment neck dissection for clinically nodal negative (cN0) papillary thyroid cancer. Gland Surg. 2013;2:186–95. doi: 10.3978/j.issn.2227-684X.2013.10.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hambleton C, Kandil E. Appropriate and accurate diagnosis of thyroid nodules: a review of thyroid fine-needle aspiration. Int J Clin Exp Med. 2013;6:413–22. [PMC free article] [PubMed] [Google Scholar]
- 4.Sherman SI. Thyroid carcinoma. Lancet. 2003;361:501–11. doi: 10.1016/s0140-6736(03)12488-9. [DOI] [PubMed] [Google Scholar]
- 5.Prades JM, Querat C, Dumollard JM, et al. Thyroid nodule surgery: predictive diagnostic value of fine-needle aspiration cytology and frozen section. Eur Ann Otorhinolaryngol Head Neck Dis. 2013;130:195–9. doi: 10.1016/j.anorl.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Alexander EK, Kennedy GC, Baloch ZW, et al. Preoperative diagnosis of benign thyroid nodules with indeterminate cytology. N Engl J Med. 2012;367:705–15. doi: 10.1056/NEJMoa1203208. [DOI] [PubMed] [Google Scholar]
- 7.Puzziello A, Guerra A, Murino A, et al. Benign thyroid nodules with RAS mutation grow faster. Clin Endocrinol (Oxf) 2015 doi: 10.1111/cen.12875. [DOI] [PubMed] [Google Scholar]
- 8.Rossi M, Buratto M, Tagliati F, et al. Relevance of BRAF(V600E) mutation testing versus RAS point mutations and RET/PTC rearrangements evaluation in the diagnosis of thyroid cancer. Thyroid. 2015;25:221–8. doi: 10.1089/thy.2014.0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eszlinger M, Krogdahl A, Munz S, et al. Impact of molecular screening for point mutations and rearrangements in routine air-dried fine-needle aspiration samples of thyroid nodules. Thyroid. 2014;24:305–13. doi: 10.1089/thy.2013.0278. [DOI] [PubMed] [Google Scholar]
- 10.Beaudenon-Huibregtse S, Alexander EK, Guttler RB, et al. Centralized molecular testing for oncogenic gene mutations complements the local cytopathologic diagnosis of thyroid nodules. Thyroid. 2014;24:1479–87. doi: 10.1089/thy.2013.0640. [DOI] [PubMed] [Google Scholar]
- 11.Guerra A, Carrano M, Angrisani E, et al. Detection of RAS mutation by pyrosequencing in thyroid cytology samples. Int J Surg. 2014;12(Suppl 1):S91–4. doi: 10.1016/j.ijsu.2014.05.045. [DOI] [PubMed] [Google Scholar]
- 12.Valderrabano P, Khazai L, Leon ME, et al. Evaluation of ThyroSeq v2 performance in thyroid nodules with indeterminate cytology. Endocr Relat Cancer. 2017;24:127–136. doi: 10.1530/ERC-16-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikiforov YE, Carty SE, Chiosea SI, et al. Impact of the Multi-Gene ThyroSeq Next-Generation Sequencing Assay on Cancer Diagnosis in Thyroid Nodules with Atypia of Undetermined Significance/Follicular Lesion of Undetermined Significance Cytology. Thyroid. 2015;25:1217–23. doi: 10.1089/thy.2015.0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellis RJ, Wang Y, Stevenson HS, et al. Genome-wide methylation patterns in papillary thyroid cancer are distinct based on histological subtype and tumor genotype. J Clin Endocrinol Metab. 2014;99:E329–37. doi: 10.1210/jc.2013-2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mancikova V, Buj R, Castelblanco E, et al. DNA methylation profiling of well-differentiated thyroid cancer uncovers markers of recurrence free survival. Int J Cancer. 2014;135:598–610. doi: 10.1002/ijc.28703. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez-Rodero S, Fernandez AF, Fernandez-Morera JL, et al. DNA methylation signatures identify biologically distinct thyroid cancer subtypes. J Clin Endocrinol Metab. 2013;98:2811–21. doi: 10.1210/jc.2012-3566. [DOI] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research N. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676–90. doi: 10.1016/j.cell.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hahn MA, Li AX, Wu X, et al. Loss of the polycomb mark from bivalent promoters leads to activation of cancer-promoting genes in colorectal tumors. Cancer Res. 2014;74:3617–3629. doi: 10.1158/0008-5472.CAN-13-3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahn MA, Li AX, Wu X, et al. Single base resolution analysis of 5-methylcytosine and 5-hydroxymethylcytosine by RRBS and TAB-RRBS. Methods Mol Biol. 2015;1238:273–87. doi: 10.1007/978-1-4939-1804-1_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohavi R. A study of cross-validation and bootstrap for accuracy estimation and model selection. IJCAI’95 Proceedings of the 14th International Joint Conference on Artificial Intelligence. 1995;2:1137–1143. [Google Scholar]
- 21.Dyhdalo K, Macnamara S, Brainard J, et al. Assessment of cellularity, genomic DNA yields, and technical platforms for BRAF mutational testing in thyroid fine-needle aspirate samples. Cancer Cytopathol. 2014;122:114–22. doi: 10.1002/cncy.21356. [DOI] [PubMed] [Google Scholar]
- 22.Diggans J, Kim SY, Hu Z, et al. Machine learning from concept to clinic: reliable detection of BRAF V600E DNA mutations in thyroid nodules using high-dimensional RNA expression data. Pac Symp Biocomput. 2015:371–82. [PubMed] [Google Scholar]
- 23.Baba Y, Ishimoto T, Kurashige J, et al. Epigenetic field cancerization in gastrointestinal cancers. Cancer Lett. 2016;375:360–6. doi: 10.1016/j.canlet.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 24.Utsunomiya T, Shimada M, Morine Y, et al. Specific molecular signatures of non-tumor liver tissue may predict a risk of hepatocarcinogenesis. Cancer Sci. 2014;105:749–54. doi: 10.1111/cas.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nonn L, Ananthanarayanan V, Gann PH. Evidence for field cancerization of the prostate. Prostate. 2009;69:1470–9. doi: 10.1002/pros.20983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dworkin AM, Huang TH, Toland AE. Epigenetic alterations in the breast: Implications for breast cancer detection, prognosis and treatment. Semin Cancer Biol. 2009;19:165–71. doi: 10.1016/j.semcancer.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.