

Abstract
The functions of TGF-β in the liver vary depending on specific cell types and their temporal response to TGF-β during different stages of hepatocarcinogenesis. Through analysis of tumor tissues from hepatocellular carcinoma (HCC) patients, we were able to cluster hepatic epithelial cell-derived TGF-β gene signatures in association with distinct clinical prognoses. To delineate the role of hepatic epithelial TGF-β signaling in HCC development, we employed a novel experimental system in which tumor-initiating hepatocytes (TICs) were isolated from TGF-β receptor II floxed mice (Tgfbr2f/f) and transplanted into the syngeneic C57BL/6J mice via splenic injection. The recipient mice were then administered Cre-expressing adenovirus to inactivate Tgfbr2 in the transplanted TICs. After latency, the Tgfbr2-inactivated TICs formed larger and more tumor nodules in the recipient livers compared to TICs without Tgfbr2 inactivation. In vitro analyses revealed that treatment of cultured TICs with TGF-β inhibited the expression of progenitor cell factors (including Sox2). RNA-seq analysis identified H19 as one of the most upregulated long noncoding RNA (lncRNA) in association with Tgfbr2 inactivation in TICs. Tgfbr2 inactivation by Ad-Cre led to a 5-fold increase of H19 expression in TICs. Accordingly, TGF-β treatment reduced H19 expression. We observed that forced overexpression of Sox2 in TICs increased the transcription of H19, whereas knockdown of Sox2 decreased it. Furthermore, depletion of H19 reduced the progenitor property of TICs in vitro and decreased their tumorigenic potential in vivo. Finally, we observed low level of H19 mRNA expression in human HCC tissues from patients with the epithelial TGF-β gene signature in association with favorable prognosis. Conclusion: Our findings disclose a novel TGF-β and H19 signaling axis via Sox2 in TICs that importantly regulates hepatocarcinogenesis.
Keywords: Hepatocellular carcinoma, transforming growth factor-β receptor-2, hepatocarcinogenesis, long noncoding RNA, Sox2
Introduction
Transforming growth factor-β (TGF-β) is a multifunctional cytokine importantly implicated in the pathogenesis of chronic liver disease and cancer(1–3). The processes regulated by TGF-β in the liver are complex, as TGF-β signaling operates in different hepatic cell types and their activation status varies during multiple stages of hepatic carcinogenesis. Among its effect on liver non-parenchymal cells, TGF-β is well known to activate hepatic stellate cells and cause liver fibrosis, which is thought to contribute to hepatocarcinogenesis and tumor progression(4). However, progressive liver fibrosis does not appear to affect the prognosis of HCC patients until cirrhosis is reached(5). On the other hand, TGF-β signaling is known to provide a cytostatic signal that inhibits hepatic epithelial cell proliferation and induces their apoptosis(6, 7). Consistent with role of TGF-β signaling in cells of hepatic epithelial origin, TGF-β is importantly implicated in the regulation of hepatic progenitor cells and cancer stem cells(2, 8). An even more complicated situation arises when persistent cell stress and damage during liver injuries switch TGF-β effects on epithelial cells from mitoinhibition/apoptosis to proliferation/survival, which can lead to increased cell plasticity and expression of stemness factors(2). Intriguingly, whereas TGF-β potently inhibits the growth of primary hepatic epithelial cells(6, 7), neoplastic hepatic cells often become resistant to TGFβ-mediated mitoinhibition/apoptosis(9, 10). Thus, alteration of epithelial response to TGF-β during hepatic carcinogenesis may represent a key factor that determines hepatic cancer development and progression.
Consistent with its inhibitory effect on DNA synthesis in primary hepatocytes(6, 7), TGF-β is known to inhibit the early proliferative response to partial hepatectomy in rodents(9, 10). Besides its anti-proliferative effect, TGF-β is also known to induce apoptosis in cultured hepatocytes and in animal models of hepatic proliferation(11). Given its antiproliferative and proapoptotic role in the liver, it is not surprising that TGF-β has been shown to act as a tumor suppressor. Indeed, mice heterozygous for the deletion of TGF-β1 (TGF-β1+/−) or TGF-β receptor II (Tgfbr2+/−) were found to be more susceptible to diethylnitrosamine (DEN)-induced hepatocarcinogenesis when compared to their wild-type littermates(12, 13) (mice homozygous for the deletion of TGF-β1 or Tgfbr2 are lethal). Enhanced hepatocarcinogenesis is also observed in transgenic mice overexpressing a dominant negative Tgfbr2(14) or in mice heterozygous for deletion of the Smad adaptor protein, embryonic liver fodrin (ELF)(15). Consistent with these observations, forced overexpression of Smad3 in the liver has been shown to inhibit DEN-induced hepatocarcinogenesis(16). The TGF-β adaptor, β2-spectrin, is also known to reduce the development of hepatocellular cancer(17). On the other hand, there is also evidence suggesting that TGF-β may promote hepatic carcinogenesis(18, 19) and that late stage human liver cancers often show overexpression of TGF-β(20). Consistent with the latter view point, inhibition of TGF-β signaling has been shown to block hepatocellular carcinoma growth and progression in experimental models(21). Thus far, there are evidences in support of both tumor-suppressive and tumor-promoting actions of TGFβ in hepatic carcinogenesis.
The above seemingly opposing effects of TGF-β on hepatic carcinogenesis may be explained by the fact that TGF-βs function as tumor suppressors early in tumorigenesis when epithelial cell responsiveness to TGF-β is still relatively normal. It is possible that during multistage tumorigenesis, the mitoinhibitory/apoptotic effect of TGF-βs becomes lost, either through mutation of the TGF-β signaling molecules or by subversion of the normal signaling pathway due to activation of other molecules(3). Since mutation of TGF-β signaling molecules occurs only in a minority of human hepatocellular cancers, disruption of TGF-β- mediated inhibition of cell growth by other oncogenic molecules appears to be an important mechanism for regulation of cell growth and carcinogenesis. In this context, it is intriguing that a gene expression study describes two different TGF-β specific gene signatures - one termed “early TGF-β signature” which is associated with longer survival; and the other termed “late TGF-β signature” which is associated with shorter survival(22). In light of the proposed targeting of TGF-β signaling for HCC treatment(21), there is an urgent and practical need to further delineate the precise role of TGFβ in hepatic carcinogenesis before target therapy can be considered clinically.
It is estimated that >40% of HCCs are clonal and may arise from cancer stem cells(2, 23). Recent evidences suggest a potentially important role of TGFβ signaling for the regulation of hepatic progenitor and cancer stem cells(2, 8). However, the effect and mechanism of TGFβ in hepatic cancer initiating or stem cells remain to be further defined. In the present study, we employed a novel experimental system based on the transplantation of tumor-initiating hepatocytes (TICs) from Tgfbr2-floxed mice into the livers of the recipient C57BL/6J mice via splenic injection. The recipient mice were subsequently administered Cre-expressing adenovirus (Ad-Cre) to inactivate Tgfbr2 in the transplanted TICs. This system allowed us to assess the impact of TGF-β signaling on tumor-initiating hepatocytes without altering TGF-β responsiveness of background parenchymal and non-parenchymal cells, thus avoiding the drawback of existing mouse models of germline or liver specific deletion of TGF-β pathway molecules. Our studies unveil a novel TGFβ-regulated long noncoding RNA H19 signaling axis in tumor-initiating hepatocytes which controls hepatic cancer development.
Materials and Methods
Mouse strain B6.129S6-Tgfbr2tm1Hlm(24) was provided by Frederick Animal Facility of NCI. The experimental procedures for induction and isolation of TICs were based on previously described methodology with modification(25, 26). Briefly, 14-day-old Tgfbr2fl/fl male littermates were injected intraperitoneally with 25mg/kg DEN. Three months after DEN injection, mouse livers were perfused and TICs were isolated (see Supplementary Methods for details). The isolated TICs were transplanted (via splenic injection) to four-week-old male wild type C57BL/6J mice pre-treated with retrorsine. One week after transplantation of TICs, the recipient mice were treated with CCl4 (i.p.) once a week for 3 weeks, followed by one tail vein injection of GFP or Cre-expressed adenovirus (1011 plaque-forming unit/mouse). The recipient mice were monitored and sacrificed 12 months afterward to document HCC development and tumor burden.
For culture of TICs, the cells were maintained in collagen-coated Petri dishes; TICs within 8 passages were used for experiments. To inactivate Tgfbr2 in cultured TICs, the cells were incubated with Cre-expressed adenovirus (100 MOI) for 72 to 96 h. To knockdown H19, pGFP-V-RS based constructs expressing H19 specific shRNAs were transfected into TICs with Lipofectamin2000 (Invitrogen, Carlsbad, CA). To assess the effect of H19 on HCC tumorigenicity, TICs with or without H19 knockdown were transplanted via splenic injection into the livers of male C57BL /6J mice.
For Statistical Analysis, the data are presented as mean ± SD from a minimum of 3 replicates. Difference between groups was evaluated by SPSS 19.0 statistical software (IBM, Armonk, NY) with one-way analysis of variance, Mann-Whitney U test, chi square test, or repeated measure analysis of generalized linear model. P < 0.05 was considered as statistically significant.
Additional methods, including the isolation and transplantation of DEN-initiated hepatocytes, culture of DEN-initiated hepatocytes, imaging flow cytometry, chromatin immunoprecipitation, luciferase assay, cell proliferation and spheroid assay, immunofluorescence, TUNEL, qRT-PCR, Western blot and bioinformatics analysis are described in the Supplementary Information.
Results
A subset of hepatic epithelial cell-derived TGF-β gene signature has important prognostic relevance.
TGF-β responding genes in primary hepatocytes are temporal-dependent: the “early response genes” are characterized by transcriptional activation of inducers of cell cycle arrest and apoptosis (termed “early TGF-β signature”), whereas the “late response genes” are predominantly implicated in microenvironment modulation and epithelial-mesenchymal transition (EMT) (termed “late TGF-β signature”)(22). We had applied the above described TGF-β signatures to analyze human HCC patients from the TCGA database, which included a group of 371 HCC patients. Based on the expression levels of the TGF-β signature genes, the HCC patients were hierarchical clustered into three distinct subgroups: patients with early TGF-β signature (cluster 1, including 102 patients), patients with late TGF-β signature (cluster 3, including 75 patients), and other undefined patients (cluster 2, including 194 patients) (Fig 1A). To evaluate the clinical significance of the TGF-β signatures, we compared the distribution of several clinical and pathological parameters in HCCs harboring early, late and undefined TGF-β signatures. While the three TGF-β signature groups were similar with respect to the patients’ age, histological grade, vascular invasion, tumor recurrence, and the presence of fibrosis/cirrhosis in the background livers. However, the tumor stage, serum AFP and patient survival were significantly different among different TGF-β signature groups, with early TGF-β signature group in association with earlier tumor stage, lower serum AFP, and longer patient survival (Fig 1B and Supplementary Table 1).
Fig 1. Hepatic epithelial cell TGF-β gene expressing signatures define distinct subtypes of human HCCs with prognostic significance.
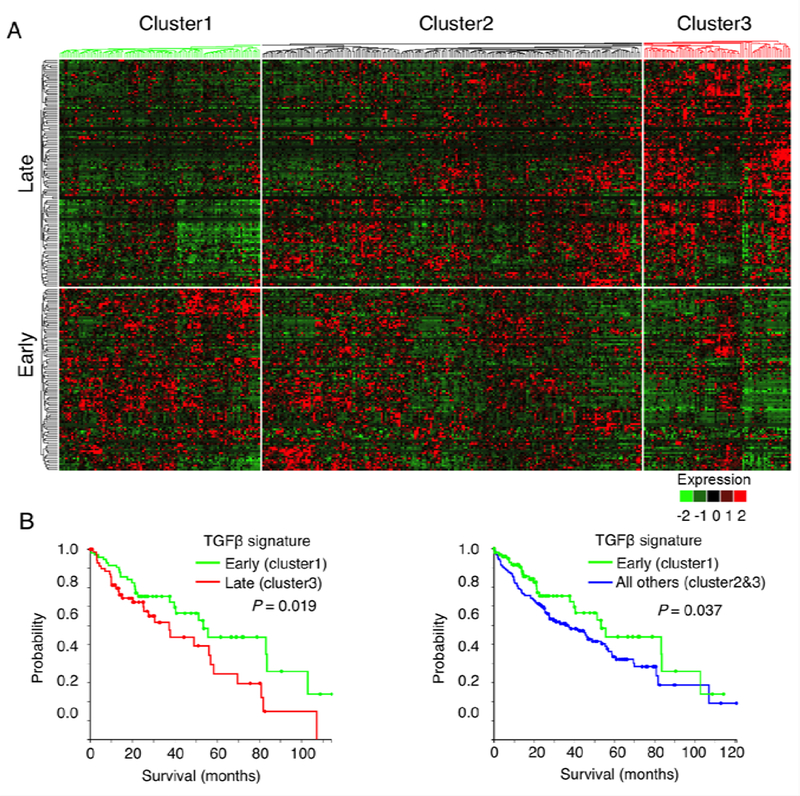
A. Dendrogram and heat-map overview of TGFβ-regulated epithelial cell gene expression in 371 cases of human HCC from TCGA. Based on the expression of hepatic TGF-β signature genes (which include 249 TGF-β early or late-response genes in hepatocyte)(22), the patients were divided into three subgroups: patients with high expression of TGF-β early-response genes (cluster 1), patients with high expression of TGF-β late-response genes (cluster 3), and the remaining undefined patients (cluster 2). The data were presented in a matrix format in which rows and columns represent genes and patients, respectively.B. Overall survival of TCGA HCC patients (Kaplan-Meier and log-rank test). The patients defined by TGF-β early-signatures (cluster 1, 102 patients) have longer overall survival when compared to patients defined by TGF-β late-signatures (cluster 3, 75 patients) (left). Similarly, the patients with TGF-β early-signatures (cluster 1) were also found to have longer overall survival when compared to the 269 patients in clusters 2 plus 3 (right).
Given that TGF-β is a well-characterized pro-fibrogenic cytokine in the liver(4), we further examined whether TGF-β-induced fibrogenic signatures would influence the clinical and pathological parameters in HCC patients. We applied a well characterized set of 202 TGF-β-induced gene signature derived from human hepatic stellate cells (HSCs)(27) to analyze the background liver tissues (available in 50 of 371 cases). Our analysis indicated that the patients can be clustered into two distinct subgroups according the 202 genes expression (Supplementary Fig S1A). These two groups showed similar clinical and pathological parameters, from the patients’ age, gender, histological grade, tumor stage, serum AFP level, vascular invasion, background liver fibrosis/cirrhosis, to patient survival and tumor recurrence (Supplementary Table 2, Fig S1B). Furthermore, we observed no difference in the overall survival between HCC patients with different fibrosis stages (Supplementary Fig S2). Taken together, the above-described bioinformatics analyses had enabled us to identify hepatic epithelial cell-derived TGF-β gene signatures in HCC which may have prognostic value. On the basis of these findings, we sought to further delineate the effect and mechanism of hepatic epithelial TGF-β signaling in hepatocarcinogenesis.
Inactivation of Tgfbr2 in tumor-initiating hepatocytes enhances hepatocellular cancer development.
Recent advances have enabled isolation of tumor-initiating hepatocytes (TICs) from mice treated with the hepatic carcinogen, DEN(25, 26). We adopted this strategy to isolate TICs from the Tgfbr2fl/fl mice. Specifically, male Tgfbr2fl/fl mice were treated intraperitoneally with a single dose of DEN (25mg/kg body-weight) at the age of 14 days and the animals were observed for three months to allow hepatocyte initiation and clonal expansion (Fig. 2A). As the initiated hepatocyte clones in DEN exposed liver usually exist as cell clusters in the hepatocyte population after the liver is routinely perfused and digested with collagenase(26), these collagenase-resistant hepatocyte clusters were isolated (Fig. 2B) and transplanted via splenic injection into the livers of male C57BL/6J mice (these animals were pretreated with 2 doses of retrorsine, a pyrrolizidine alkaloid that blocks the proliferation of endogenous hepatocytes, as described(25, 26). Following splenic injection of TICs, the recipient mice were treated with 3 weekly intraperitoneal injections of CCl4 (to induce compensatory hepatocyte proliferation). One week thereafter the recipient mice were injected once with Cre recombinase adenovirus (Ad-Cre) (to delete Tgfbr2 in transplanted cells) or Ad-GFP as control (1011 plaque-forming unit/mouse) (the experimental protocol is outlined in Fig. 2A). The animals were then monitored for liver tumor development. We observed that by 12 months after TIC transplantation, the recipient mice treated with Ad-Cre developed more and larger tumor nodules compared to the mice treated with Ad-GFP (Fig. 2C). Consistent with this observation, the Ad-Cre-treated mice showed significantly higher liver-to-body weight ratio compared to the Ad-GFP-treated group (Fig. 2C). Immunohistochemical analysis confirmed successful deletion of Tgfbr2 expression in hepatic tumors from the Ad-Cre treated mice (Fig. 2D). These findings demonstrate that deletion of Tgfbr2 in TICs enhances hepatocellular cancer development. Thus, TGF-β signaling in tumor-initiating hepatocytes appears to be tumor suppressive.
Fig 2. Tgfbr2 inactivation enhances the tumorigenic capacity of transplanted TICs in vivo.
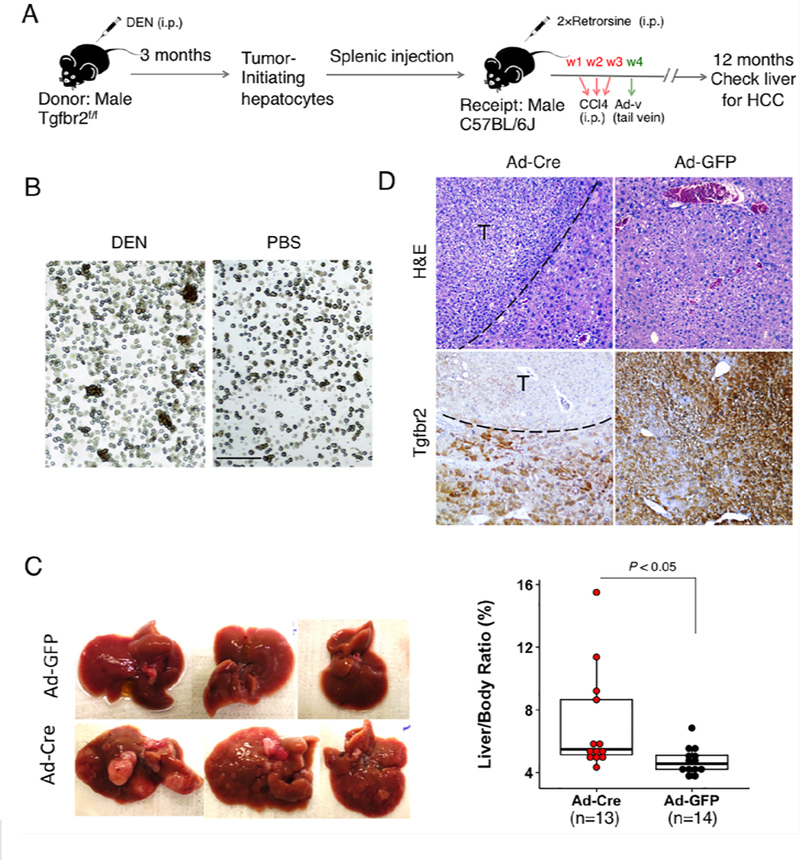
A. Outline of the experimental procedures. B. Hepatocytes isolated from mice with or without DEN injection. Note that hepatocyte clusters were seen from mice with DEN injection but not from mice without DEN injection. Scale bar, 150μm. C. Gross photos of representative livers from recipient C57BL/6J mice transplanted with Tgfbr2-floxed TICs and treated with Ad-Cre or Ad-GFP. The right graph showed the liver-body ratios of the two groups of mice. D. Sections of the liver tissues from the above groups were subjected to H&E and IHC staining with Tgfbr2 antibody (100×).
TGF-β signaling suppresses the progenitor property of tumor-initiating hepatocytes.
Given that CD44 is a marker for cancer stem/progenitor cells in HCC(26, 28), we performed immunofluorescence staining for CD44 in the isolated collagenase resistant-cell clusters. We observed that almost all of the cells in the freshly isolated clusters express CD44 (Fig. 3A). In fact, more than 99% of the isolated TICs continued to maintain CD44-positive status even after culture in vitro for a few days, as reflected by the Amins imaging flow cytometry results (Fig. 3B). However, prolonged culture in vitro (≥10 passages) resulted in reduction of CD44 expression in some cells. The latter phenomenon presented an opportunity to analyze the properties of cells with different CD44 expression levels after prolonged culture. We employed MACS technique with CD44-conjected magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany) to separate CD44high from CD44low cell populations. At 10th passage, approximately 80% of the cultured cells were allocated to CD44high group, while approximately 20% of the cells belonged to CD44low group. Immunofluorescence staining showed that the levels of other progenitor cell markers, including EpCam, Klf4, Oct4, Sox2 and c-Myc, were also higher in CD44high cells compared to CD44low cells (Fig. 3C). Based on these findings, we elected to use Tgfbr2-floxed TICs within 8 passages for subsequent studies to determine the functional impact of TGF-β signaling inactivation in these cells.
Fig 3. The tumor-initiating hepatocytes have progenitor cell characteristics.
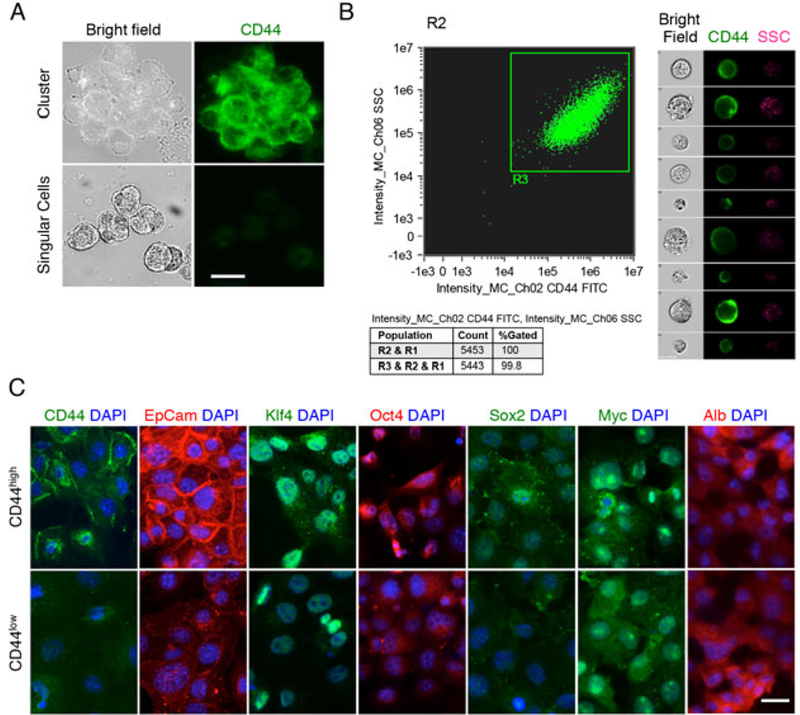
A. The cell clusters isolated from DEN treated mouse livers were composed of CD44+ cells, whereas the dispersed hepatocytes were CD44-. Scale bar, 20μm. B. 99.8% of primary clusters-derived cells were CD44+ cells, as determined by Imaging flow cytometry (left panel). Representative images of CD44+ cells are shown at the right panel (SSC: side scatter). C. The CD44high cells showed more progenitor cell characteristics than CD44low cells, as determined by IF staining with specific stem cell markers. Scale bar, 20μm.
We were able to successfully silence TGF-β singling in cultured TICs via Ad-Cre-mediated depletion of Tgfbr2 (successful reduction of Tgfbr2 was confirmed by immunoblotting) (Fig. 4A). As expected, Tgfbr2 depletion prevented TGFβ-induced Smad activation and morphological alteration (Fig. 4A-B). We observed that the cells with Tgfbr2 depletion expressed higher levels of progenitor markers including c-Myc, Klf4 and Sox2 (Fig. 4C, 3D) and showed increased capability to form spheroids (Fig. 4E). These findings indicate that TGF-β signaling suppresses the progenitor capability of TICs, in vitro. Intriguingly, Tgfbr2 depletion significantly increased the expression of the long noncoding RNA H19 (Fig. 4C).
Fig 4. Tgfbr2 deletion increased the expression of progenitor cell markers in tumor-initiating hepatocytes.
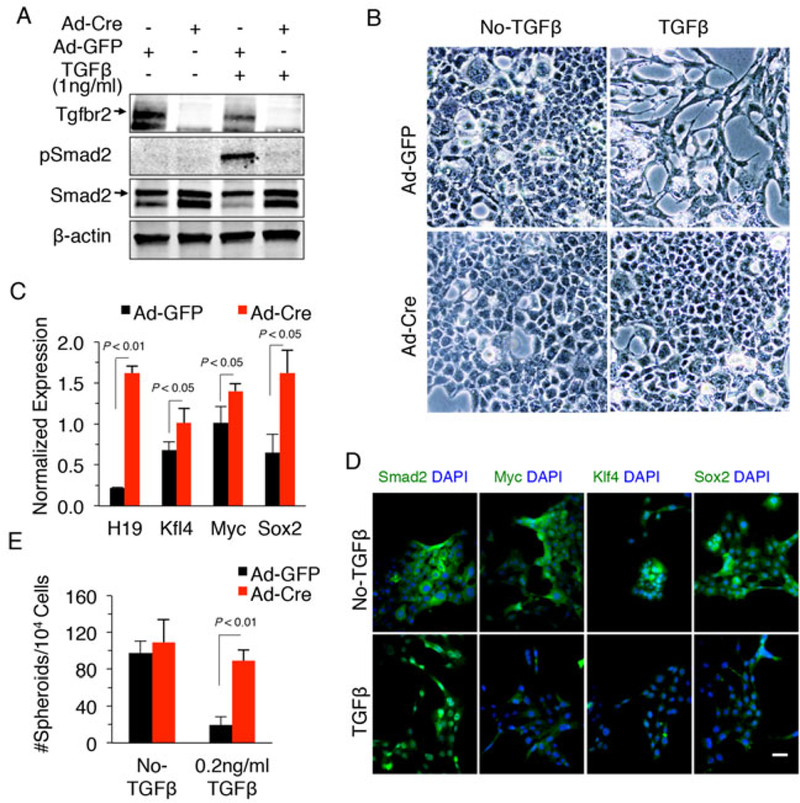
A. Cultured TICs were incubated with media containing Ad-Cre or Ad-GFP control at M0I>100 for 72–96 hours followed by treatment with 1ng/ml TGF-β or vehicle for 24 hours. The cells were then lysed and the cellular proteins were processed for immunoblotting to determine the levels of Tgfbr2, Smad2 and phospho-Smad2. B. The effects of TGF-β on cell morphology of TICs (the cells were treated as indicated in Panel A) (200x). C. Tumor-initiating hepatocytes infected with Ad-Cre or Ad-GFP were treated with 1ng/mL TGF-β for 48 hours. Total RNAs were isolated for qRT-PCR with primers of indicated genes. D. IF staining for Smad2, c-Myc, Klf4, and Sox2 in tumor-initiating hepatocytes. The cells were treated with 1ng/ml TGF-β or vehicle for 48 hours prior to IF. Scale bar, 20 μm. E. Spheroid formation of cultured tumor-initiating hepatocytes with indicated treatment.
We observed that the cultured TICs continued to maintain tumor-forming capability in vivo when transplanted into the livers of the recipient mice via splenic injection (see below). However, these cells did not have the capability to form tumors outside the liver, as subcutaneous inoculation of multiple batches of these cells into C57BL/6J mice (4~5×106 per injection, with or without Tgfbr2 depletion) failed to lead tumor formation (no tumor was observed after more than 35 injections). These observations suggest that the isolated TICs have not yet acquired all malignant characteristics as fully developed hepatocellular carcinoma cells.
TGF-β signaling inhibits the expression of lncRNA H19 in tumor-initiating hepatocytes.
Long noncoding RNAs (lncRNAs) are important players in diverse pathobiological processes, including the pathogenesis and progression of HCC(29). To identify lncRNAs which might be implicated in TGFβ-regulated tumorigenic process in TICs, we isolated total RNAs from TICs transduced with Ad-Cre or Ad-GFP and treated with TGF-β (1 ng/ml for 48 hours). The isolated RNAs were then subjected to sequencing analysis (procedure outlined in Fig. 5A). Gene Set Enrichment Analysis (GSEA) with RNA-seq data confirmed the lack of TGF-β response and the up-regulation of cell cycle related genes in Tgfbr2 depleted TICs (Fig. 5B). Tgfbr2-inactivation in TICs led to the identification of 1177 up-regulated and 822 down-regulated genes. Functional analyses with these differential expressed genes by DAVID annotation tool revealed that the up-regulated genes were allocated to Gene Ontology (GO) term of cell division and mitosis, while down-regulated genes to GO term of proteinaceous extracellular matrix, a type of cellular component increasingly produced by hepatocytes when undergoing EMT(30). These data validated the lack of TGF-β response in Tgfbr2-depleted cells. We next utilized the SAMMate 2.7.4 software to compare the expression abundance scores of annotated lncRNA genes to identify differentially expressed lncRNAs between TGFβ-inactivated and control cells. These analyses led to the identification of H19 as one of the most increased lncRNAs in Tgfrb2-depleted TICs (Fig. 5C). Quantitative RT-PCR assay further confirmed TGFβ-mediated inhibition of H19 expression in TICs (Fig. 5D). Together, these bioinformatics and experimental analyses provide novel evidence that TGF-β signaling inhibits the expression of lncRNA H19 in tumor-initiating hepatocytes.
Fig 5. H19 is the most up-regulated lncRNA in Tgfbr2-inactivated TICs.
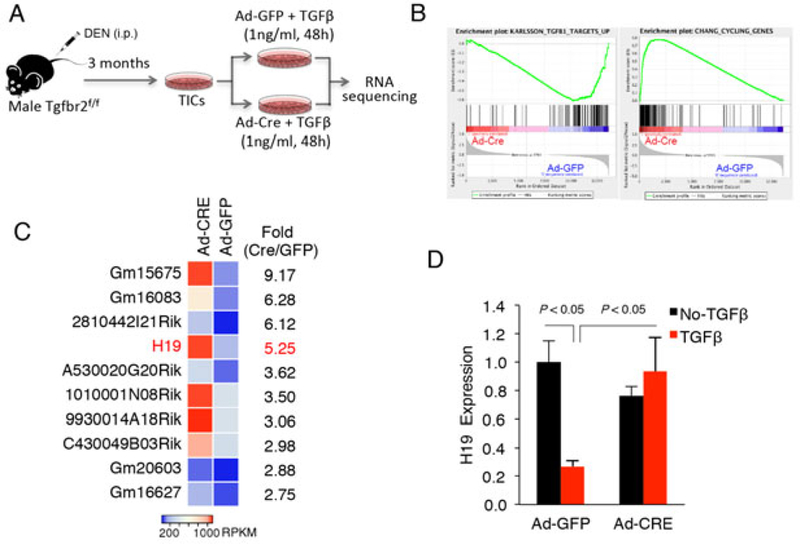
A. Outline of experimental protocol for RNA sequencing. B. GESA analysis using RNA-sequencing data of TICs. Ad-Cre-mediated Tgfbr2 deletion led to lower expression of TGFβ-stimulated genes and higher expression of cell cycling genes. C. Top 10 lncRNAs which are differentially expressed between Tgfbr2-inactivated and control TICs. The cells infected with Ad-Cre or Ad-GFP were treated with 1ng/ml TGF-β for 48 hours and the RNAs were isolated for sequencing analysis. D. qRT-PCR analysis for H19 in Tgfbr2-inactivated and control TICs with or without TGF-β treatment (1ng/ml).
TGF-β inhibits H19 gene transcription through Sox2 in tumor-initiating hepatocytes.
Our data showed that TGF-β treatment reduced the mRNA and protein levels of Sox2, c-Myc and Klf4 in TICs with intact TGF-β signaling, whereas this phenomenon was not observed in Tgfbr2-depleted cells (Fig. 4C, 4D and 6A). Given that Sox2 is an important transcription factor for maintenance of cancer stem cells in HCC(31), we further examined whether Sox2 might regulate H19 transcription. Bioinformatics analyses using PROMO algorithm with strict screening criteria revealed four putative Sox2 binding sites located at the upstream of the H19 gene. All these sites (site 1: TGACAAA; site 2: GGACAAA; site 3: ATACAAA; site 4: GGACAAT) were verified with chromatin immunoprecipitation (ChIP) assay by using anti-Sox2 antibody and site-specific primer pairs (Fig. 6B). We next performed qRT-PCR analysis to determine the effect of Sox2 on H19 expression in TICs. We observed that forced overexpression of Sox2 increased the level of H19 whereas Sox2 knockdown by siRNA decreased it (Fig. 6C). To further verify the role of Sox2 in H19 transcription, we amplified a 450bp DNA fragment upstream of H19 gene (−596 to −146, containing all of the four Sox2 binding sites) from the mouse genomic DNA by PCR; the amplified DNA fragment was purified and cloned into the multiple cloning sites of pGL4.21 vector to form the H19-promoter luciferase reporter construction. We then transfected the H19-promoter luciferase reporter construct to cultured TICs with renilla luciferase-expressed plasmid pRL-TK as internal control and the cell lysates were obtained for luciferase reporter activity assay. Our data showed that forced overexpression of Sox2 increased the H19-promoter luciferase reporter activity, while siRNA depletion of Sox2 reduced it (Fig. 6D). Moreover, we observed that TGF-β treatment significantly reduced the H19-promoter luciferase reporter activity in TICs (Fig. 6D). These findings implicate TGFβ-regulated Sox2 in H19 gene transcription. The results are further corroborated by TCGA analysis showing that HCCs with early TGF-β signature (when TGF-β still maintains its cytostasis inducing functions) exhibited significantly lower levels of Sox2 and H19 expression compared to those with late or undefined TGF-β signature (Supplementary Fig S3). Further TCGA analysis reveals that HCC patients with lower H19 level in the tumor tissues have longer disease-free survival when compared to patients with higher level of H19 (Supplementary Fig S4).
Fig 6. Sox2 mediates TGF-β induced inhibition of H19 expression in tumor-initiating hepatocytes.
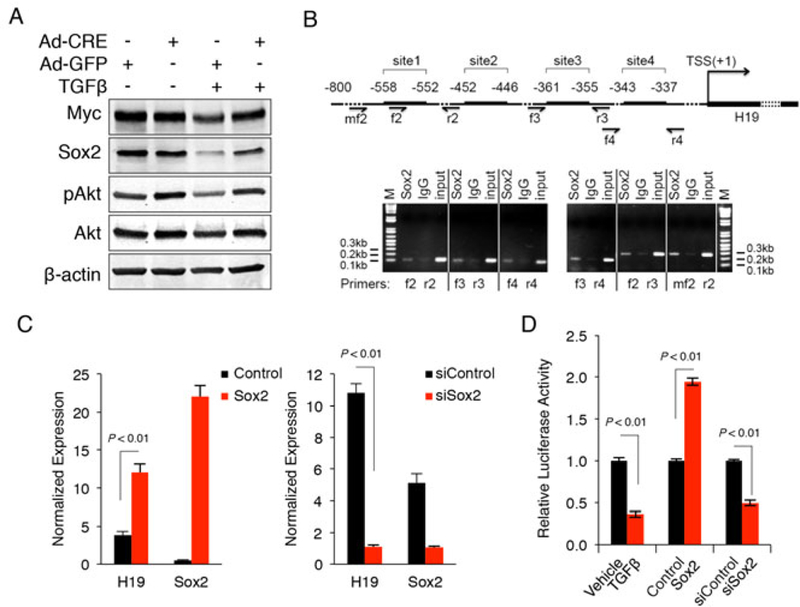
A. Protein levels of Myc, Sox2, Akt and phospho-Akt in Tgfbr2-inactivated and control tumor-initiating tumor cells, as detected by Western blotting. The cells infected with Ad-Cre or Ad-GFP were treated with 1ng/mL TGF-β or vehicle for 48 hours. B. ChIP assay in TICs with anti-Sox2 antibody or IgG as control. The PCR primers flanking the four different Sox2 binding sites in the H19 promoter were synthesized and utilized for ChIP analysis. C. qRT-PCR analysis for H19 in TICs transfected with Sox2 expression vector (left) or siRNA (right). D. H19-promoter activity assays. Tumor-initialing hepatocytes were transfected with H19-promoter luciferase reporter plasmid, followed by incubation with 1ng/mL TGF-β or cotransfection with Sox2 expression plasmid or siRNA. The cells were cultured in serumfree Opti-MEM medium for 24 hours, and then the cell lysates were obtained to measure the luciferase reporter activity with a luminometer.
H19 is implicated in TIC proliferation, survival and progenitor capacity.
To investigate the functional impact of H19 in tumor-initiating hepatocytes, we generated two H19 shRNA expressing constructs and these vectors were transfected into TICs individually to establish cell lines with stable knockdown of H19. Significant reduction of H19 levels by shRNA in the selected stable cell lines were verified by real time RT-PCR (Fig. 7A). We observed that H19 depletion significantly decreased the proliferation of TICs when compared to their corresponding controls (Fig. 7B). H19 knockdown also decreased the spheroid formation efficiency in TICs (Fig. 7C). TUNEL assays showed that H19 knockdown led to apoptosis of tumor-initiating hepatocytes in response to TGF-β treatment (Fig. 7D). These data demonstrate an important role of H19 in TIC proliferation, survival and progenitor capacity.
Fig 7. H19 depletion induces apoptosis of tumor-initiating hepatocytes, in vitro.
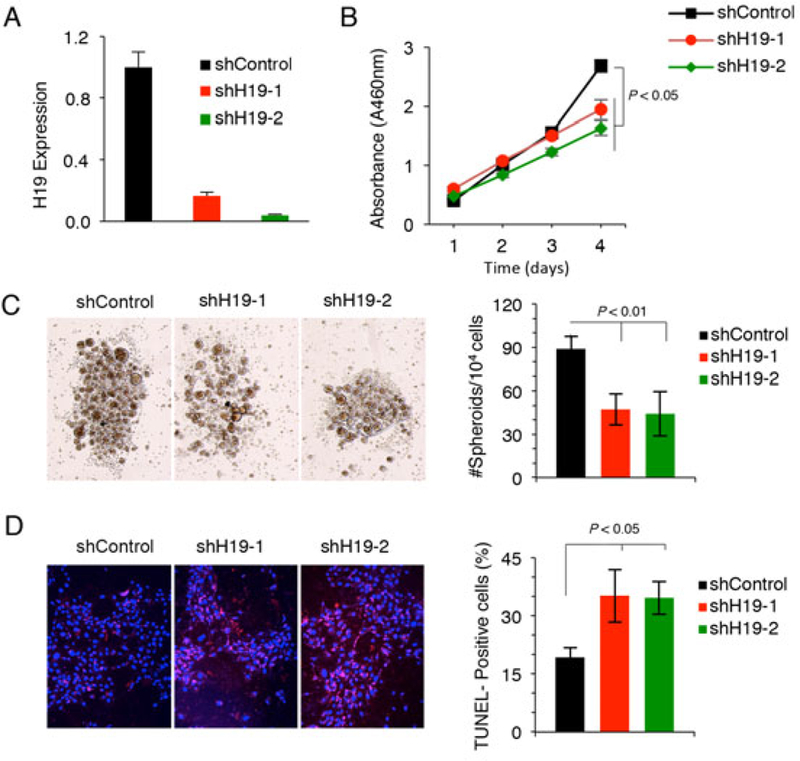
A. H19 level in tumor-initiating hepatocytes transfected with H19 specific shRNA plasmids or control vector, as measured by qRT-PCR. B. Cell proliferation assay (WST-1) of tumor-initiating hepatocytes transfected with the control or H19 shRNA plasmids. C. Spheroid formation of tumor-initiating hepatocytes with or without shRNA knockdown of H19. Representative spheroids for indicated treatments are shown at the left panel. The bar graph on the right represents quantitative results of 3 independent experiments. D. TUNEL analyses of tumor-initiating hepatocytes with or without H19 knockdown. The cells were treated with TGF-β (1ng/ml) for 24 hours prior to analyses. The data represent the results from 3 separate treatments.
We next performed RNA-seq analyses to determine the expression of mRNAs and miRNAs in TICs with or without H19 knockdown (Supplementary Table 3, 4, 5 and 6). GSEA analyses of the RNA-seq data showed that knockdown of H19 led to increased expression of TP53 and CDKN1A/p21 target genes (Supplementary Fig S5A). We observed that H19 knockdown counteracted Akt activation (as reflected by increased the expression of genes which are normally suppressed by Akt (Supplementary Fig S5B). Our further analysis indicated that H19 knockdown reduced the expression of Src target genes (Supplementary Fig S5C). These data suggest that H19 signaling targets several pathways (including TP53, Akt and Src) in TICs.
H19 knockdown attenuates Tgfbr2 deletion-induced tumorigenic capacity of TICs.
We sought to further investigate whether H19 depletion would impact TGFβ-regulated tumorigenic potential of TICs in vivo. For this purpose, 3×106 Tgfbr2-floxed TICs infected with Ad-Cre and transfected with H19 shRNA or control shRNA were inoculated into the livers of male C57BL/6J mice through splenic injection (the recipient mice were pretreated with retrorsine and received three doses of CCL4 i.p after splenic injection as described in the above section). We observed that the recipient mice receiving TICs treated with Ad-Cre plus H19 shRNA developed fewer tumor nodules compared to the mice receiving TICs exposed to Ad-Cre plus control shRNA (Fig. 8A). Consistent with these observations, the recipient mice receiving TICs treated with Ad-Cre plus H19 shRNA exhibited significantly lower liver-to-body ratio compared to the group receiving TICs treated with Ad-Cre plus control shRNA. Thus, H19 knockdown attenuated Tgfbr2 deletion-induced tumorigenic capacity of TICs, in vivo. We also examined H19 levels in the liver tumor tissues from mice transplanted with Tgfbr2 floxed TICs and treated with Ad-GFP or Ad-Cre (as described in Fig. 2C); these analyses revealed higher levels of H19 as well as Sox2 in Tgfbr2 depleted tumors compared to the control tumors (Fig. 8B). Taken together, our findings disclose a novel TGFβ-regulated H19 signaling axis via Sox2 in tumor-initiating hepatocytes which is crucial for liver cancer development (outlined in Fig. 8C).
Fig 8. H19 knockdown attenuates Tgfbr2 deletion-induced tumorigenic capacity of TICs in vivo.
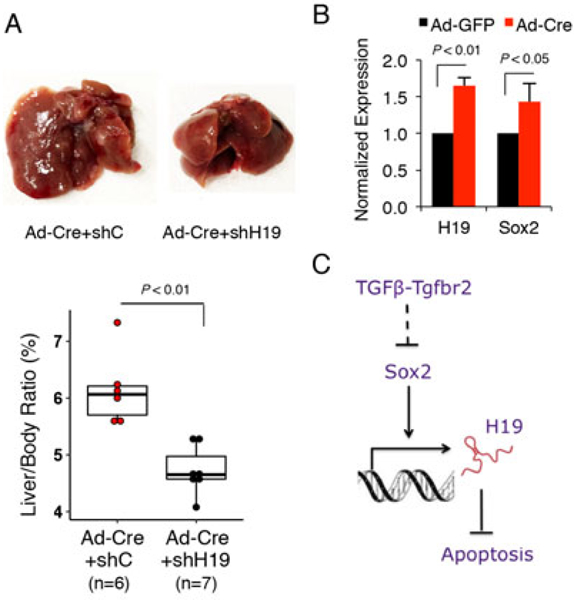
A. TICs infected with Ad-Cre were transfected with H19 shRNA or control vector respectively. The stably transfected cells (3×106) were inoculated into the livers of the C57BL/6J mice according to the protocol outlined in Figure 1A. Twelve months later, the livers were removed and the liver and body weights were recorded. Gross photos of representative livers are shown at the top panel. The liver-body weight ratios are presented at the graph below. B. Increased expression of H19 and Sox2 RNAs in Tgfbr2-deleted tumors. The levels of H19 and Sox2 RNAs were assessed by qRT-PCR in the tumor tissues with or without Tgfbr2 deletion as described in Figure 2C. C. Schematic illustration of TGFβ-regulated H19 signaling axis via Sox2 in tumor-initiating hepatocytes.
Discussion
Hepatocellular carcinoma initiation and progression are greatly affected by the liver milieus in which it arises (1, 4, 32). More than 80% of HCC develop on the basis of a chronic liver disease(4, 32). As results of the constant injuries, hepatocyte necrosis and high cellular turnover perpetuate error-prone chronic repair processes which exert selective pressure on the proliferating hepatocytes in the background liver. This pathological process not only causes hepatocyte genomic instability but also creates a pro-oncogenic hepatic microenvironment. Such combined effects importantly contribute to the clinically heterogeneous phenotypes and genotypes of HCC(32). Given the complex reciprocal signaling between proliferated hepatocytes and pro-oncogenic milieus, it is not surprising that the functional roles of genes in hepatocarcinogenesis collectively depend on the influences they brought to hepatocytes as well as on the microenvironment alterations resulting from the injured and/or proliferating hepatocytes. Such molecular intricacies of HCC development are attested by the paradox of conflicting roles of certain pro-oncogenic pathways or genes in chemically induced HCC mouse models, such as NF-kB, β-catenin, Jnk and Met(33). Those studies have revealed that either enhancing or reducing one of the aforementioned pro-oncogenic genes or pathways activity in hepatocyte could increase DEN-induced hepatocarcinogenesis in mouse. One possible explanation of this paradoxical phenomenon is that the loss of pro-survival signaling may trigger chronic hepatic injury, which in turn may induce compensatory hepatocyte propagation leading to neoplastic growth (so called “indirect effect”)(33). While this theory remains to be further verified experimentally, it cautions the limitations of the currently widely used hepatocyte-specific or liver-specific deletion of oncogenic molecules in experimental hepatocarcinogenesis. It remains to be determined whether such an “indirect effect” may also compromise experiments aimed to hepatocyte- or liver-specific deletion of other molecules that are known to have conflicting phonotypical changes in hepatic carcinogenesis, notably TGF-β and its receptors.
Previous studies have shown that germline or liver-specific deletion of TGF-β signaling molecules in mice variably influence hepatic carcinogeneic processes(2, 12–14, 21, 34–37). However, the results are often difficult to interpret and sometimes conflicting, partly due to alteration of TGF-β responsiveness in the background hepatic cells. While germline ablation of TGF-β signaling is known to affect liver parenchymal and non-parenchymal cells as well as the multiple reciprocal interactions in cellular niches, the more widely used hepatocyte-specific or liver-specific deletion also masquerades problems such as gene deletion-related cell injury and the resultant compensatory hepatocyte proliferation. In fact, the current available mouse models of liver carcinogenesis with liver- or hepatocyte- specific deletion of molecules are all associated with the drawback of indirect hepatocyte injury-associated carcinogenesis(33). To overcome these limitations, in the current study we created a new experimental system in which tumor-initiating hepatocytes were isolated from livers of DEN exposed Tgfbr2fl/fl mice, and the isolated TICs were transplanted via splenic injection into livers of wild type syngeneic mice followed by Tgfbr2 inactivation through tail vein injection with Cre-expressed adenovirus. This unique experimental system allowed us to precisely assess the impact of Tgfbr2-inactivated and Tgfbr2-intact tumor-initiating hepatocytes under the same background liver milieus. Our findings demonstrate a tumor-suppressive role of TGF-β signaling in tumor-initiating hepatocytes and uncover a novel TGFβ-regulated long noncoding RNA H19 signaling axis that is pivotal in hepatocellular cancer development.
Our findings in the current study suggest an important interaction between TGF-β signaling and Sox2 in tumor-initiating hepatocytes. We observed steady increase of Sox2 expression in Tgfbr2 depleted TICs and tumors; conversely, our data showed that TGF-β treatment of TICs decreased the level of Sox2. These findings indicated an inverse relationship between TGF-β signaling status and Sox2 level in tumor-initiating hepatocytes. Our finding are noteworthy, given that Sox2 is a critical factor sufficient to reprogram somatic cells and that TGF-β inhibition can replace Sox2 for cell reprogramming(38). These observations are further corroborated by patient-based studies that Sox2 is the most increased gene amongst core pluripotency factors in pre-malignant cirrhotic liver(39), where hepatocytes have already became resistant to TGFβ-induced mitoinhibition/apoptosis(40). In addition to being considered as a predictor for poor survival of HCC patients(41), Sox2 has also been shown to play an essential role in self-renewal and maintenance of cancer stem-like cell populations in HCC(42). Therefore, TGF-β signaling may counterbalance Sox2 in cancer initiating and progenitor cell differentiation.
LncRNAs, which are defined as a group of non-protein coding transcripts longer than 200 nucleotides, are now considered as substantial players in a broad spectrum of cellular activities(43). However, to date, the functions and mechanisms of lncRNAs in the liver remain largely unknown. Our experimental results in the current study disclose a novel TGFβ-regulated lncRNA H19 in tumor-initiating hepatocytes. We observed that Tgfbr2 depletion by Ad-Cre in TICs led to a 5-fold increase of H19 expression. Our data showed that H19 knockdown by shRNA significantly decreased the proliferation, survival and progenitor capacity of TICs in vitro and decreased their tumorigenic potential in association with TGF-β signaling inactivation in vivo. These results are highly significant, considering that lncRNA H19 was first discovered as a up-regulated gene by α-fetoprotein in the liver(44) and is now recognized as an important player in promoting cancer growth(45, 46).Furthermore, our findings reveal an important role of Sox2 for regulation of H19 gene expression. The latter assertion is based on the following observations: (i) ChIP assay showed Sox2 association with its consensus sites in the promoter region of H19 gene; (ii) Sox2 overexpression in TICs enhanced H19 promoter luciferase reporter activity and increased H19 RNA level; (iii) Sox2 knockdown in TICs decreased H19 promoter luciferase reporter activity and reduced H19 RNA level; (iv) TGF-β treatment reduced the H19-promoter luciferase reporter activity in TICs; (v) Tgfbr2-inactivated tumor-initiating cells and tissues showed simultaneous increase of Sox2 and H19 levels. Together, these data support Sox2-mediated activation of H19 gene transcription, although possible involvement of other transcription factors cannot be excluded.
In the current study, we discover that TGF-β inhibited the expression of the progenitor cell transcription factor, Sox2, in TICs. In this context, a recent study has reported that TGF-β regulates Sox4 in pancreatic ductal carcinoma cells(47). In that study, TGF-β was found to regulate the interaction of Sox4 with the gastrointestinal lineage-master regulator Klf5 through epithelial mesenchymal transition (EMT)-mediated disruption of a lineage-specific transcriptional network. It will be of further interest to determine whether TGF-β may also regulate other progenitor cell or lineage-specific transcription factors during hepatic carcinogenic processes.
A previous study(48) reported that H19 inhibited HCC metastasis in spite of its carcinogenic actions. Our data presented in the current manuscript provide novel evidence indicating an important role of H19 in TICs. Further studies are warranted to delineate the exact role of H19 in specific cell types in the HCC microenvironment.
In summary, our findings in the current study provide novel evidence that TGF-β signaling in tumor-initiating hepatocytes inhibits H19 expression via Sox2 and that this signaling axis is crucial for inhibition of HCC development. These findings not only help further understand the molecular mechanisms of hepatocarcinogenesis, but also provide important implications for stage-based target therapy against HCC.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by the National Institutes of Health grants DK077776 and CA102325.
List of abbreviations:
- HCC
hepatocellular carcinoma
- TICs
tumor-initiating hepatocytes
- lncRNA
long noncoding RNA
- TGF-β
transforming growth factor-β
- Sox2
SRY (sex determining region Y)-box 2
- Tgfbr2
transforming growth factor-β receptor-2
- ELF
embryonic liver fodrin
- EMT
epithelial-mesenchymal transition
- AFP
α- fetoprotein
- DEN
diethylnitrosamine
- CCl4
carbon tetrachloride
Footnotes
No conflicts of interest exist
References
- 1.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 2006;6:674–687. [DOI] [PubMed] [Google Scholar]
- 2.Majumdar A, Curley SA, Wu X, Brown P, Hwang JP, Shetty K, Yao ZX, et al. Hepatic stem cells and transforming growth factor beta in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2012;9:530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Massague J TGFbeta signalling in context. Nat Rev Mol Cell Biol 2012;13:616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013;144:512–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Q, Fiel MI, Blank S, Luan W, Kadri H, Kim KW, Manizate F, et al. Impact of liver fibrosis on prognosis following liver resection for hepatitis B-associated hepatocellular carcinoma. Br J Cancer 2013;109:573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carr BI, Hayashi I, Branum EL, Moses HL. Inhibition of DNA synthesis in rat hepatocytes by platelet-derived type beta transforming growth factor. Cancer Res 1986;46:2330–2334. [PubMed] [Google Scholar]
- 7.Han C, Bowen WC, Li G, Demetris AJ, Michalopoulos GK, Wu T. Cytosolic phospholipase A2alpha and peroxisome proliferator-activated receptor gamma signaling pathway counteracts transforming growth factor beta-mediated inhibition of primary and transformed hepatocyte growth. Hepatology 2010;52:644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Yao ZX, Chen JS, Gi YJ, Munoz NM, Kundra S, Herlong HF, et al. TGF- beta/beta2-spectrin/CTCF-regulated tumor suppression in human stem cell disorder Beckwith-Wiedemann syndrome. J Clin Invest 2016;126:527–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russell WE, Coffey RJ Jr., Ouellette AJ, Moses HL. Type beta transforming growth factor reversibly inhibits the early proliferative response to partial hepatectomy in the rat. Proc Natl Acad Sci U S A 1988;85:5126–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grasl-Kraupp B, Rossmanith W, Ruttkay-Nedecky B, Mullauer L, Kammerer B, Bursch W, Schulte-Hermann R. Levels of transforming growth factor beta and transforming growth factor beta receptors in rat liver during growth, regression by apoptosis and neoplasia. Hepatology 1998;28:717–726. [DOI] [PubMed] [Google Scholar]
- 11.Oberhammer FA, Pavelka M, Sharma S, Tiefenbacher R, Purchio AF, Bursch W, Schulte-Hermann R. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc Natl Acad Sci U S A 1992;89:5408–5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang B, Bottinger EP, Jakowlew SB, Bagnall KM, Mariano J, Anver MR, Letterio JJ, et al. Transforming growth factor-beta1 is a new form of tumor suppressor with true haploid insufficiency. Nat Med 1998;4:802–807. [DOI] [PubMed] [Google Scholar]
- 13.Im YH, Kim HT, Kim IY, Factor VM, Hahm KB, Anzano M, Jang JJ, et al. Heterozygous mice for the transforming growth factor-beta type II receptor gene have increased susceptibility to hepatocellular carcinogenesis. Cancer Res 2001;61:6665–6668. [PubMed] [Google Scholar]
- 14.Kanzler S, Meyer E, Lohse AW, Schirmacher P, Henninger J, Galle PR, Blessing M. Hepatocellular expression of a dominant-negative mutant TGF-beta type II receptor accelerates chemically induced hepatocarcinogenesis. Oncogene 2001;20:5015–5024. [DOI] [PubMed] [Google Scholar]
- 15.Kitisin K, Ganesan N, Tang Y, Jogunoori W, Volpe EA, Kim SS, Katuri V, et al. Disruption of transforming growth factor-beta signaling through beta-spectrin ELF leads to hepatocellular cancer through cyclin D1 activation. Oncogene 2007;26:7103–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang YA, Zhang GM, Feigenbaum L, Zhang YE. Smad3 reduces susceptibility to hepatocarcinoma by sensitizing hepatocytes to apoptosis through downregulation of Bcl-2. Cancer Cell 2006;9:445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baek HJ, Pishvaian MJ, Tang Y, Kim TH, Yang S, Zouhairi ME, Mendelson J, et al. Transforming growth factor-beta adaptor, beta2-spectrin, modulates cyclin dependent kinase 4 to reduce development of hepatocellular cancer. Hepatology 2011;53:1676–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Factor VM, Kao CY, Santoni-Rugiu E, Woitach JT, Jensen MR, Thorgeirsson SS. Constitutive expression of mature transforming growth factor beta1 in the liver accelerates hepatocarcinogenesis in transgenic mice. Cancer Res 1997;57:2089–2095. [PubMed] [Google Scholar]
- 19.Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, Roberts AB, et al. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci U S A 1995;92:2572–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito N, Kawata S, Tamura S, Takaishi K, Shirai Y, Kiso S, Yabuuchi I, et al. Elevated levels of transforming growth factor beta messenger RNA and its polypeptide in human hepatocellular carcinoma. Cancer Res 1991;51:4080–4083. [PubMed] [Google Scholar]
- 21.Giannelli G, Villa E, Lahn M. Transforming growth factor-beta as a therapeutic target in hepatocellular carcinoma. Cancer Res 2014;74:1890–1894. [DOI] [PubMed] [Google Scholar]
- 22.Coulouarn C, Factor VM, Thorgeirsson SS. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology 2008;47:2059–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rountree CB, Mishra L, Willenbring H. Stem cells in liver diseases and cancer: recent advances on the path to new therapies. Hepatology 2012;55:298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chytil A, Magnuson MA, Wright CV, Moses HL. Conditional inactivation of the TGF- beta type II receptor using Cre:Lox. Genesis 2002;32:73–75. [DOI] [PubMed] [Google Scholar]
- 25.He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, Sieghart W, et al. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010;17:286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, Shalapour S, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 2013;155:384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang LY, Heller M, Meng Z, Yu LR, Tang Y, Zhou M, Zhang YE. Transforming Growth Factor-beta (TGF-beta) Directly Activates the JAK1-STAT3 Axis to Induce Hepatic Fibrosis in Coordination with the SMAD Pathway. J Biol Chem 2017;292:4302–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Z, Hao X, Yan M, Yao M, Ge C, Gu J, Li J. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int J Cancer 2010;126:2067–2078. [DOI] [PubMed] [Google Scholar]
- 29.George J, Patel T. Noncoding RNA as therapeutic targets for hepatocellular carcinoma. Semin Liver Dis 2015;35:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wells RG. Cellular sources of extracellular matrix in hepatic fibrosis. Clin Liver Dis 2008;12:759–768, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest 2013;123:1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marquardt JU, Andersen JB, Thorgeirsson SS. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat Rev Cancer 2015;15:653–667. [DOI] [PubMed] [Google Scholar]
- 33.Feng GS. Conflicting roles of molecules in hepatocarcinogenesis: paradigm or paradox. Cancer Cell 2012;21:150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshida K, Murata M, Yamaguchi T, Matsuzaki K, Okazaki K. Reversible Human TGF-beta Signal Shifting between Tumor Suppression and Fibro-Carcinogenesis: Implications of Smad Phospho-Isoforms for Hepatic Epithelial-Mesenchymal Transitions. J Clin Med 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morris SM, Carter KT, Baek JY, Koszarek A, Yeh MM, Knoblaugh SE, Grady WM. TGF-beta signaling alters the pattern of liver tumorigenesis induced by Pten inactivation. Oncogene 2015;34:3273–3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris SM, Baek JY, Koszarek A, Kanngurn S, Knoblaugh SE, Grady WM. Transforming growth factor-beta signaling promotes hepatocarcinogenesis induced by p53 loss. Hepatology 2012;55:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mu X, Pradere JP, Affo S, Dapito DH, Friedman R, Lefkovitch JH, Schwabe RF. Epithelial Transforming Growth Factor-beta Signaling Does Not Contribute to Liver Fibrosis but Protects Mice From Cholangiocarcinoma. Gastroenterology 2016;150:720–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ichida JK, Blanchard J, Lam K, Son EY, Chung JE, Egli D, Loh KM, et al. A small- molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog. Cell Stem Cell 2009;5:491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toraih EA, Fawzy MS, El-Falouji AI, Hamed EO, Nemr NA, Hussein MH, Abd El Fadeal NM. Stemness-related transcriptional factors and homing gene expression profiles in hepatic differentiation and cancer. Mol Med 2016;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Black D, Bird MA, Samson CM, Lyman S, Lange PA, Schrum LW, Qian T, et al. Primary cirrhotic hepatocytes resist TGFbeta-induced apoptosis through a ROS- dependent mechanism. J Hepatol 2004;40:942–951. [DOI] [PubMed] [Google Scholar]
- 41.Sun C, Sun L, Li Y, Kang X, Zhang S, Liu Y. Sox2 expression predicts poor survival of hepatocellular carcinoma patients and it promotes liver cancer cell invasion by activating Slug. Med Oncol 2013;30:503. [DOI] [PubMed] [Google Scholar]
- 42.Liu L, Liu C, Zhang Q, Shen J, Zhang H, Shan J, Duan G, et al. SIRT1-mediated transcriptional regulation of SOX2 is important for self-renewal of liver cancer stem cells. Hepatology 2016;64:814–827. [DOI] [PubMed] [Google Scholar]
- 43.Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol 2013;14:699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pachnis V, Belayew A, Tilghman SM. Locus unlinked to alpha-fetoprotein under the control of the murine raf and Rif genes. Proc Natl Acad Sci U S A 1984;81:5523–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Conigliaro A, Costa V, Lo Dico A, Saieva L, Buccheri S, Dieli F, Manno M, et al. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol Cancer 2015;14:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raveh E, Matouk IJ, Gilon M, Hochberg A. The H19 Long non-coding RNA in cancer initiation, progression and metastasis - a proposed unifying theory. Mol Cancer 2015;14:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, et al. TGF-beta Tumor Suppression through a Lethal EMT. Cell 2016;164:1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L, Yang F, Yuan JH, Yuan SX, Zhou WP, Huo XS, Xu D, et al. Epigenetic activation of the MiR-200 family contributes to H19-mediated metastasis suppression in hepatocellular carcinoma. Carcinogenesis 2013;34:577–586. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.