

Abstract
Kidney-type glutaminase (GLS), the first enzyme in the glutaminolysis pathway, catalyzes the hydrolysis of glutamine to glutamate. GLS was found to be upregulated in many glutamine-dependent cancer cells. Therefore, selective inhibition of GLS has gained substantial interest as a therapeutic approach targeting cancer metabolism. Bis-2-[5-(phenylacetamido)-1,3,4-thiadiazol-2-yl]ethyl sulfide (BPTES), despite its poor physicochemical properties, has served as a key molecular template in subsequent efforts to identify more potent and drug-like allosteric GLS inhibitors. This review article provides an overview of the progress made to date in the development of GLS inhibitors and highlights the remarkable transformation of the unfavorable lead into “druglike” compounds guided by systematic SAR studies.
Graphical Abstract

1. INTRODUCTION
Glutaminase catalyzes the hydrolysis of glutamine into glutamate and ammonia in mitochondria. In mammalian cells, there exist two isoforms of glutaminase encoded by two paralogous genes located in distinct chromosomes.1 Kidney-type glutaminase (GLS) is widely distributed throughout extra-hepatic tissues, whereas liver-type glutaminase (GLS2) is predominantly found in the adult liver. The human GLS gene is located on chromosome 2 and spans 82 kb, and the human GLS2 gene spans 18 kb and is located on chromosome 12.2 Exons 3–17 of the GLS and GLS2 genes are identical in length and have 77% amino acid sequence identity.3 In contrast, the two isoforms differ significantly in their C-terminal sequences (33% identity), which may impart a distinct function to each isoform.
As one of the major sources of glutamate in nonhepatic tissues, GLS plays a range of critical roles in various biological pathways. For instance, GLS is a crucial enzyme in the glutamate-glutamine cycle designed to maintain an adequate supply of glutamate, a major excitatory neurotransmitter, in the brain.4 GLS is also the first enzyme of the glutaminolysis pathway, where glutamine serves as a precursor for a number of intermediates essential for other biosynthetic pathways and energy production.5 Importantly, GLS appears to be upregulated in many malignant cells in need of energy supply and nitrogen-rich substances.6 Two isoenzymes derived from the GLS gene by alternative splicing are known as kidney glutaminase (KGA) and glutaminase C (GAC), which share a common N-terminal sequence (1–550 derived from exons 1–14) but contain unique C-terminal segments (551–669 for KGA derived from exons 16–19 and 551–598 for GAC derived from exon 15).7 Although the functional differences between the two splicing variants of GLS have not yet been clearly understood, the GAC form of GLS seems to be playing a central role in the altered metabolic profile of many proliferating cells.8,9 Inhibition of GLS therefore has gained considerable attention as a new therapeutic approach for the treatment of cancer.
Despite the fact that glutaminase has been extensively studied as a key metabolic enzyme, until recently there have been limited molecular probes available for in-depth characterization of the enzyme. Although 6-diazo-5-oxo-L-norleucine 1 (DON, Figure 1),10 a conventional glutaminase inhibitor, has served as a valuable tool for deciphering the physiological roles of glutaminase, its lack of selectivity and weak potency has hampered its use in establishing the therapeutic benefit of selective GLS inhibition. The renewed interest in selective inhibition of GLS as a therapeutic approach has led to the search for new GLS inhibitors preferably more potent and selective for GLS. One of the most important breakthroughs in this regard was unarguably the discovery of bis-2-[5-(phenyl- acetamido)-1,3,4-thiadiazol-2-yl]ethyl sulfide 2 (BPTES, Figure 1), a truly novel GLS inhibitor structurally distinct from DON. Even though BPTES was first reported in 2002 in a patent application, its discovery went rather unnoticed until its therapeutic significance in cancer was realized years later.11 The past several years, however, have seen a wave of medicinal chemistry efforts in search of new GLS inhibitors mostly derived from BPTES. In fact, CB-839 3 (Figure 1), a GLS inhibitor discovered by Calithera Biosciences, entered clinical trials in 2014, underscoring the tremendous progress made toward the development of GLS inhibitors. The main objective of this review article is to provide an overview of the progress made to date in the area of GLS inhibitors subsequent to the discovery of BPTES. There are a number of well-written review articles dealing with the biochemistry and biology of GLS, including its potential as a therapeutic target for cancer treatment.7,12–16 Thus, we will focus primarily on the medicinal chemistry aspects of GLS inhibitors, specifically those derived from BPTES. In addition to GLS inhibitors reported in peer reviewed journals, an attempt was made to cover those found in the patent literature, where a wealth of new GLS inhibitors has been reported in recent years. It should be noted that a number of GLS inhibitors structurally distinct from BPTES have also been recently reported,17–20 but we have chosen not to cover these inhibitors as extensive SAR data are not available for these early stage compounds.
Figure 1.
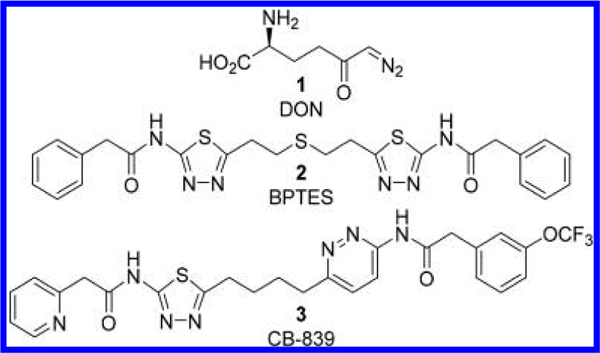
Structures of DON 1, BPTES 2, and CB-839 3.
2. GLUTAMINASE ASSAY METHODS
A variety of biochemical assay methods have been developed and are used to evaluate compounds for GLS inhibitory potency. These methods differ in many aspects including the concentrations of glutamine and/or phosphate used, the source of the enzyme, preincubation times, and glutamate (or ammonia) detection methods. Because BPTES and many of its derivatives are known to exhibit poor aqueous solubility, another source of variability might be introduced in glutaminase assays that use different dilution protocols. Additionally, minor differences in structure between BPTES analogues can lead to altered kinetics and inhibitory mechanisms of GLS. For instance, comparative kinetics studies conducted by Gross et al. at Calithera Biosciences indicate a predominantly uncompetitive mechanism displayed by BPTES but a primarily noncompetitive inhibition exhibited by CB-839.21 Thus, caution needs to be taken when comparing IC50 values of test compounds determined by different assay methods. Indeed, the consequences of the differences in assay conditions are apparent in the IC50 values reported for BPTES in the literature ranging from 0.1 to 3.3 μM. The most commonly used measurement of GLS activity has been performed by quantifying glutamate by coupling to glutamate dehydrogenase (GDH)-mediated NADH production. In some cases, NADH is further coupled to another detection system. Glutamate oxidase (GLOD) has also been utilized by coupling to horseradish peroxidase. When [3H]-glutamine is used as a substrate, [3H]-glutamate can be detected after separation from the unreacted [3H]-glutamine by ion-exchange resin.22 Although this radioactive assay is relatively low throughput compared to the enzyme-coupled assays, it has the advantage of producing fewer false positives due to assay interference. Different assay methods employed in assessing GLS inhibitory potency are summarized in Table 1 in an attempt to capture key elements in each method. When available, IC50 values determined for BPTES by the respective methods are also cited as a potential guide for comparing SAR data across different assay methods. It should be noted that some patent applications provided IC50 values as ranges and prohibited, to some extent, in-depth SAR data analysis.
Table 1.
Comparison of Assay Methods Used to Assess GLS Inhibitors
| compds tested | glutamine conc | phosphate conc | enzyme | preincubation time | detection method | IC50 (μM) for BPTES |
additional in vitro assays conducted | ref(s) |
|---|---|---|---|---|---|---|---|---|
| RPTES, 4-7 | 7 mM | 160 mM | hKGA1_669 | 20 min | GDH; formazan formation measured by absorbance at 570 nm | 0.19; 0.58 | 28 | |
| 8-10; 28; 29 | 2 mM | 45 mM | hKGA124-669 | 0 min | [3H]-glutamate | 3.3 | P493 proliferation | 22; 41 |
| CB-839, 11-25; 30; 31 | 10 mM | 150 mM | GAC | 0 or 60 min | GDH; NADH formation measured by absorbance at 340 nm | 0.1 (60 min) 0.2 (0 min) | P493/MDA-MB-231 /HCC1806 proliferation | 44 |
| 26; 27; 52-67 | 13 mM or 1.8 mM | 100 mM | GAC | 60 min | GDH-diaphorase; resorufin formation measured at ex544/em590 nm | 0.1-0.5 | 45; 50-52 | |
| 32-36; 68; 101-104 | 10 mM | unknown | mouse brain/kidney homogenate | 0 min | ammonia formation detected by Nessler s reagent by absorbance at 450 nm | not tested | MDA-MB-231 proliferation | 46; 56 |
| 69-72 | no biochemical GLS assay data reported | BT20 cellular glutaminase | 53 | |||||
| 37-51; 105-106 | 1 mM | 50 mM | GAC | 10 min | GLOD-horseradish peroxidase; resorufin formation measured at ex530/em590 nm | not tested | A549 proliferation | 47—49; 57 |
| 73-87 | 50 mM | 100 mM | hKGA63_669 | 15 min | GLOD-horseradish peroxidase; resorufin formation measured at ex535/em590 nm | not tested | PC3 cellular glutaminase; NCI-H1703 proliferation | 23-27 |
| 88-100 | 18 mM | 135 mM | GAC | unknown | GDH; NADH formation measured by absorbance at 340 nm | 0.4 | MDA-MB-231 proliferation | 54 |
In addition to these biochemical assays, some groups have reported IC50 values for cellular glutaminase and cancer cell proliferation assays. Figure 2 shows a correlation plot of IC50 values from biochemical GLS or cellular glutaminase assays versus IC50 values from the NCI-H1703 cell proliferation assay for more than 100 compounds collectively reported by the same group at AstraZeneca.23–27 It appears that IC50 values from the cellular glutaminase assay correlate better with those obtained from a NCI-H1703 cell proliferation assay, presumably due to the fact that only cell-permeable inhibitors are capable of suppressing cell proliferation. However, given that cellular glutaminase IC50 values are reported in a limited number of publications, IC50 values from biochemical assays are used as the primary source of information for SAR discussion in this review article.
Figure 2.
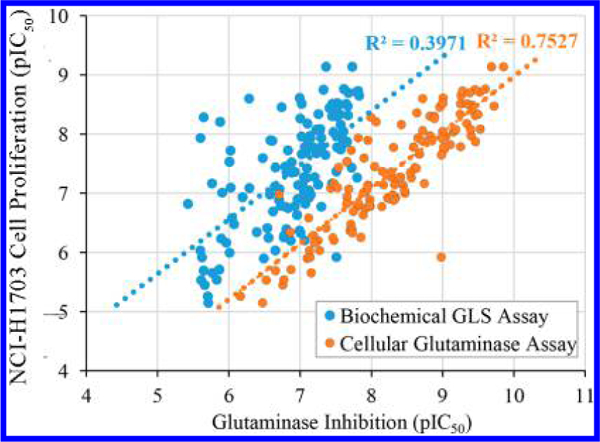
Correlation of inhibitory potency in biochemical GLS assay (blue) or cellular glutaminase assay (orange) with the NCI-H1703 cell proliferation assay.
3. BPTES
A US patent claiming BPTES 2 (termed SNX-1770) and its close analogues as GLS inhibitors filed by Newcomb at Elan Pharmaceuticals was issued in 2002.28 All but one of the compounds disclosed in this patent application are based on the symmetric bis-2-[5-(carboxamido)-1,3,4-thiadiazol-2-yl]-ethyl sulfide scaffold with variations in the two terminal carboxamide moieties (Figure 3). Although very limited SAR data were presented, changes in the carboxamide moiety appear to be tolerated by GLS considering that all compounds with this scaffold exhibited IC50 values below 5 μM. In addition to BPTES, which contains two phenylacetamide groups, benzamide derivatives 4 and 5 and 2-thiophenecarboxamide derivative 6 were reported to potently inhibit GLS. The least potent compound listed in this patent was compound 7 in which the sulfide group of 4 was replaced by an ether group.
Figure 3.
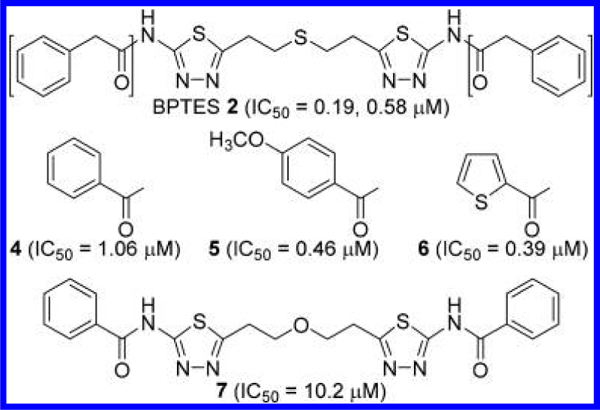
Structures of BPTES 2 and its close analogues 4—7.28
As described in more detail later, the discovery of BPTES and its analogues gave rise to a new generation of GLS inhibitors not only structurally but also pharmacologically distinct from DON in many aspects. Unlike DON, BPTES does not contain any reactive chemical groups that might form covalent bonds with the enzyme. In addition, BPTES bears no structural similarity to either glutamine or glutamate. This structural difference should minimize toxicological risk due to interaction with other enzymes, transporters, or receptors that recognize glutamine or glutamate as substrates. Indeed, BPTES was reported to selectively inhibit GLS over GLS2, glutamate dehydrogenase, and γ-glutamyl transpeptidase.29 This selectivity over other glutamine-related enzymes coupled with the lack of structural similarity to glutamine suggests that BPTES interacts with GLS at a site other than the active site where glutamine is being hydrolyzed. Indeed, in a kinetic study using rKGA128–674 (recombinant rat KGA lacking the sequence encoded by most of the first exon), increasing concentrations of BPTES produced a decrease in the apparent Vmax but had little effect on the Km, indicative of noncompetitive inhibition.29 GLS is known to show enhanced enzymatic activity (decreased Km value for glutamine) in the presence of increasing phosphate concentration. Although the precise mechanism by which phosphate activates GLS remains to be elucidated, phosphate-induced activation of GLS appears to involve facilitation of inactive dimers to form active tetramers or larger oligomers.30,31 BPTES, however, does not compete with phosphate but instead interferes with phosphate-induced activation by forming an inactive tetramer complex that has a reduced affinity for phosphate.29 Interestingly, a truncated form of human kidney-type glutaminase (hKGA124—551) was found to exist as a dimer even when activated by phosphate yet forms an inactive tetramer upon the addition of BPTES.32
In support of these findings, cocrystal structures of human GAC (hGAC)33 and hKGA34 in complex with BPTES uncovered the unique allosteric binding of two BPTES molecules per one GLS tetramer at the dimer—dimer interface region located ~18 Å away from the active site Ser286. As shown in Figure 4A, the hKGA cocrystal structure (PDB ID: 3VP1)34 reported by Thangavelu et al. at the National University of Singapore revealed that allosteric binding of BPTES induces a major conformational change of the Glu312- Pro329 loop, which plays a critical role in stabilization of the active site by forming a closed conformation in the absence of BPTES. BPTES, however, pulls the loop away from the active site region and stabilizes an open conformation, which renders the enzyme inactive. The thiadiazole group and the aliphatic linker of BPTES are encapsulated by a hydrophobic cluster formed by the side chains of Leu321, Phe322, and Leu323 from the Glu312-Pro329 loop as well as Tyr394 from the Asp386-Lys398 helix (Figure 4B). Interestingly, Phe322 is one of a few residues in the loop that is unique to GLS and its mutation to serine, the corresponding residue in GLS2, resulted in significant loss of sensitivity to BPTES. This finding provides the molecular basis for selectivity of BPTES for GLS over GLS2.34
Figure 4.
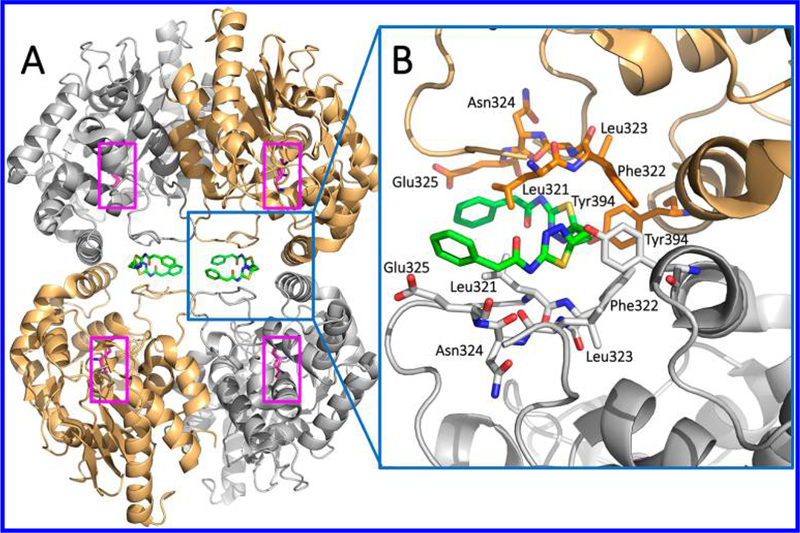
Crystal structure of hKGA in complex with BPTES (PDB ID: 3VP1). (A) GLS forms a tetramer with two molecules of BPTES (green), each of which binds to the dimer interface region. Ser286-Lys289 catalytic dyads are highlighted in pink. (B) Magnified view of the BPTES binding site.
Figure 5 compares GLS allosteric binding sites occupied by BPTES from two cocrystal structures: one reported by Thangavelu et al. (Figure 5A: 3VP1)34 and the other by DeLaBarre et al. at Agios (Figure 5B: 3UO9).33 As shown in Figure 5C, the key residues (Leu321, Phe322, Leu 323, and Tyr394) of the two structures are nearly superimposable. In both structures, the carbonyl oxygen of the Leu323 residue interacts with the NH group of the aminothiadiazole moiety. Another common feature between the two structures is the bent conformation adopted by the diethylsufide linker of BPTES, presumably to position the two thiadiazole rings for optimal interactions with the GLS allosteric site. This finding has formed the basis for the introduction of shorter and/or cyclic linkers as described later. One notable difference between the two cocrystal structures is the opposite orientations assumed by the thiadiazole rings of BPTES. In the 3VP1 structure, the OH group of Tyr394 and NH group of Phe322 appear to interact with one of the nitrogen atoms and the sulfur atom of the thiadiazole ring, respectively. On the other hand, in the 3UO9 structure, the OH group of Tyr394 interacts with the sulfur atom of the thiadiazole ring, whereas the two NH groups from Phe322 and Leu323 interact with the two nitrogen atoms of the thiadiazole ring. It should be noted that neither binding mode is exclusive to one of the two splicing variants hKGA and hGAC. For example, cocrystal structures of hKGA in complex with other BPTES-derived GLS inhibitors were found to adopt a binding mode similar to that of the 3UO9 structure obtained from hGAC as described later.35 It remains to be addressed whether these distinct binding modes reflect plasticity exhibited by the GLS allosteric binding site or an artifact of crystallization.
Figure 5.
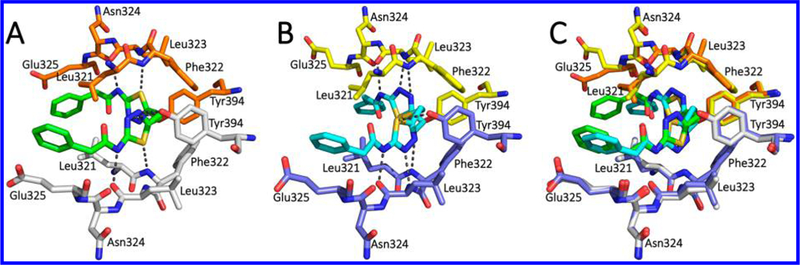
(A) Cocrystal structure of BPTES (green) bound to the allosteric site of hKGA (PDB ID: 3VP1). (B) Co-crystal structure of BPTES (cyan) bound to the allosteric site of hGAC (PDB ID: 3UO9). (C) Overlay of the two cocrystal structures (3VP1 and 3UO9).
More recently, using FRET assays, Stalnecker et al. at Cornell University showed that BPTES can stabilize the GAC tetramer containing 488 (FRET donor)-and QSY9 (FRET acceptor)-labeled GAC monomers.36 They also detected a dose-dependent quenching of tryptophan fluorescence in F327W GAC mutant upon binding of BPTES, most likely triggered by the conformational changes in the activation loop containing Phe327.
Although BPTES was originally intended for the treatment of CNS disorders, it has been most extensively utilized as a tool compound to study its effects on glutamine catabolism in cancer. In IMR90-ERMY cells with induced MYC activity, BPTES increased levels of glutamine and decreased the levels of glutamate and its downstream products including Krebs cycle intermediates such as fumarate and malate.37 Similarly, reduced incorporation of [U-13C]-glutamine-derived 13C into these products was observed in the presence of BPTES. In contrast, unlike DON, BPTES did not inhibit the incorporation of 15N2-glutamine-derived 15N into adenine nucleotide species presumably due to its inability to inhibit amido-transferase activities involved in nucleotide biosynthesis. In P493 cells, BPTES decreased ATP levels under both aerobic and hypoxic conditions, suggesting that glutamine metabolism is driving the Krebs cycle to maintain cellular energy production.11 Furthermore, in mice harboring P493 tumor xenografts, BPTES was found to reduce conversion of 13C-pyruvate to alanine by hyperpolarized 13C magnetic resonance spectroscopy as a result of the decline in the formation of glutamate, a substrate for the transamination of pyruvate to alanine.38 Collectively, these in vitro and in vivo findings provide molecular insights into the role played by GLS in glutamine metabolism and the consequence of its inhibition.
In addition to the molecular-level studies described above, BPTES has served as a tool compound to evaluate the therapeutic utility of GLS inhibition in many forms of cancer models, including lymphoma,11 glioma,39 breast,40,41 pancreatic,42,43 and nonsmall cell lung cancers.8 These studies with BPTES have firmly established the therapeutic potential of inhibiting GLS in various glutamine-dependent cancers. An indepth survey of these findings will not be provided herein as this review article is focused on the medicinal chemistry aspects of new GLS inhibitors derived from BPTES.
4. GLS INHIBITORS DERIVED FROM BPTES
Although it is undeniable that BPTES has played a key role in paving the way for the treatment of cancer using new pharmacological approaches targeting cellular metabolism, its poor druglike molecular properties, primarily its high degree of hydrophobicity, has presented a major setback. Indeed, BPTES has extremely poor aqueous solubility (<1 μg/mL) at any given pH, which has negated the feasibility of further development of BPTES as a therapeutic agent. To this end, tremendous efforts have been made to develop new allosteric GLS inhibitors with superior druglike properties. As outlined in Figure 6, structural evolution originating from BPTES has generated a number of new GLS inhibitors through systematic modifications carried out at various parts of the molecule, providing a high degree of structural diversity. Although it is impossible to present these inhibitors in the chronological order of discovery, herein we report the representative classes of BPTES-derived GLS inhibitors in a way that allows readers to appreciate how molecular structures have evolved over time as incremental changes have been made to improve potency and druglike properties.
Figure 6.
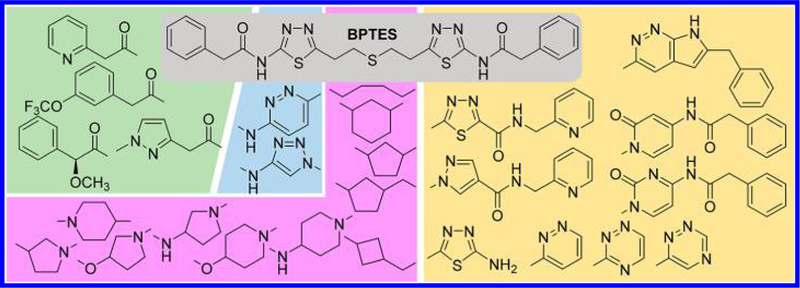
Iterative structural modifications systematically carried out on BPTES at various parts of the molecule. A number of linkers (purple background) were found to be effective as substitutes for the ethylsulfide moiety of BPTES. The phenylacetyl and 2-amino-1,3,4-thiadiazole moieties of BPTES were individually (green and cyan backgrounds, respectively) or collectively (beige background) altered, further expanding the structural diversity of GLS inhibitors derived from BPTES.
Shukla et al. at the Johns Hopkins University examined a number of truncated analogues of BPTES with the primary objective of improving solubility (Figure 7).22 They found that removal of one phenylacetyl group from BPTES (compound 8) results in little change in potency as compared to BPTES. Furthermore, deletion of the sulfide group from the middle linker (compound 9) was also tolerated by GLS. Although no substantial improvement in inhibitory potency was achieved by these modifications, the decreased molecular weight appears to contribute to the enhanced solubility. Compounds 8 and 9 displayed aqueous solubility of 13 and 3.4 μg/mL, respectively, as compared to 0.144 μg/mL for BPTES. Furthermore, 1,4-di(5-amino-1,3,4-thiadiazol-2-yl)butane of compound 9 also eliminated the metabolic liability associated with the sulfide oxidation reported for compound 8.22 The internal thiadiazole group of 9 was found to be essential as the removal of the N4 nitrogen from the ring of 9 led to the complete loss of inhibitory activity (compound 10), which underscores the crucial role played by the N4 of the thiadiazole ring.
Figure 7.
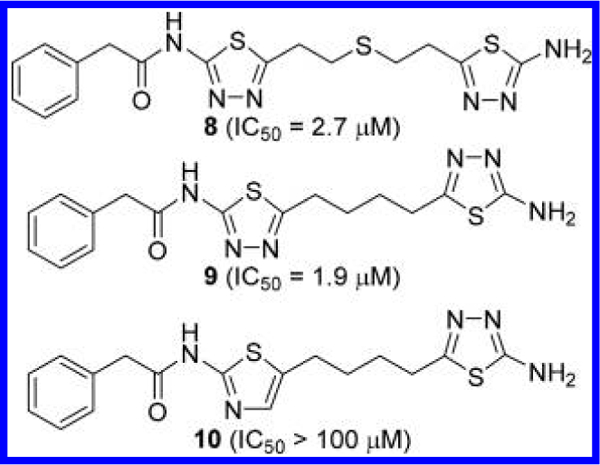
Truncated derivatives 8–10 of BPTES.22
Li et al. at Calithera Biosciences published a patent application covering a large number of BPTES analogues as GAC inhibitors (Figure 8).44 For the majority of compounds reported in this application, inhibitory activities were measured after preincubation of the GAC-compound mixture for a period of 60 min. Consistent with the findings from the Johns Hopkins group, compound 11 lacking the central sulfide group of BPTES was found to be as potent as BPTES. However, replacement of the two 1,3,4-thiadiazole rings in 11 with a 1,3,4-oxadiazole ring (compound 12) resulted in substantial loss of potency despite the structural similarity between the two heterocycles.
Figure 8.
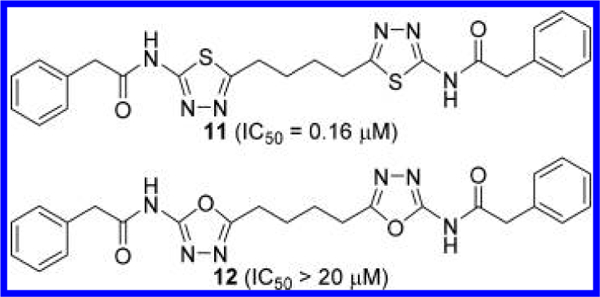
GLS inhibitors with 1,4-di(5-amino-1,3,4-thiadiazol-2- yl)butane scaffold.44
Various derivatives based on the 1,4-di(5-amino-1,3,4- thiadiazol-2-yl)butane scaffold were reported in Li’s application (Figure 9).44 The α-position of the phenylacetamide group can accommodate a hydroxyl (compounds 13 and 15) or methoxy (compounds 14 and 16) group without substantial loss of potency. Interestingly, two enantiomers 13 and 15 showed 5-fold difference in potency, whereas 14 and 16 displayed no difference. In contrast, disubstituted derivative 17 was substantially more potent than its enantiomer 18.
Figure 9.
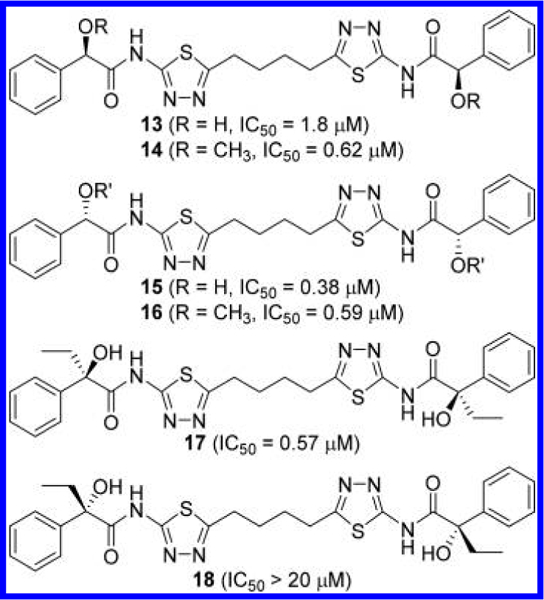
Structures of compounds 13–18 containing α-substituted phenylacetyl groups.44
A variety of substituents were also incorporated into the terminal phenyl groups of 11 (Figure 10). For example, 2-, 3-, and 4-methoxyphenyl derivatives 19–21 inhibited GLS with similar potency. More polar substituents were also explored, presumably in an attempt to improve aqueous solubility, including 4-amino (compound 22), 4-dimethylamino (compound 23), 3-(2-hydroxyethoxy) (compound 24), and 3-[2- (4-morpholinyl)ethoxy] (compound 25) groups. As is the case with compounds 19–21, these compounds exhibited comparable potency to that of compound 11.
Figure 10.
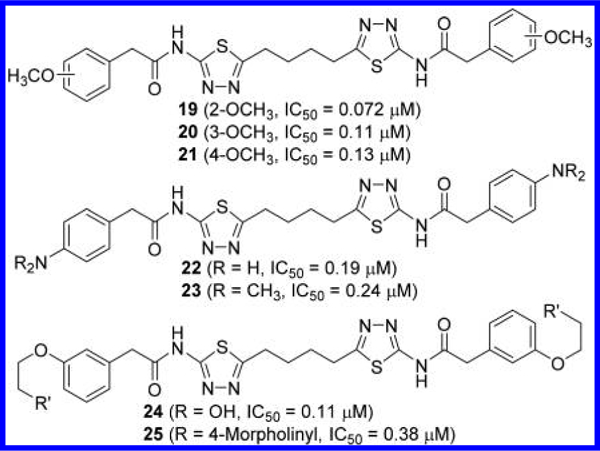
Structures of compounds 19–21 with substituted phenyl rings44
Lemieux et al. at Agios also disclosed a number of GLS inhibitors with the same scaffold (Figure 11).45 Some of their potent inhibitors (IC50 < 100 nM) possess bulky groups at both ends, such as compounds 26 and 27. These findings are consistent with Calithera’s findings and indicate the existence of a large space in the dimer interface that can accommodate sterically demanding side chains. Zimmermann et al. at Johns Hopkins University also reported GLS inhibitors on this scaffold in an attempt to improve potency and solubility.41 They found that replacement of one phenylacetyl group of 11 with a 4-hydroxyphenylacetyl group resulted in significant improvement in potency (compound 28), but replacing both of them did not enhance inhibition (compound 29). The role of the phenolic hydroxyl group in the improved potency is not well understood. It is speculated, however, that Arg317 and/or Glu325 residues located in the vicinity may play some role in the interaction with the phenolic moiety.
Figure 11.
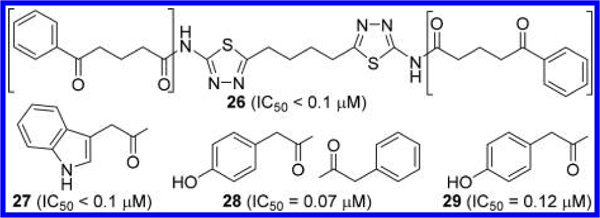
Structures of compounds 26–29 with substituted phenyl rings41,45
The most significant finding reported in Li’s application is, unarguably, the discovery of the pyridazine ring as an alternative to the thiadiazole ring (Figure 12).44 For instance, compound 30, in which one of the thiadiazole rings of compound 11 is replaced by a pyridazine ring, showed substantial improvement in potency. Furthermore, it was found that both thiadiazole rings can be replaced by a pyridazine ring as demonstrated by compound 31. One of the compounds disclosed in this patent application, CB-S39 3, is the first GLS inhibitor to enter clinical trials for the treatment of cancer. Direct comparison of inhibitory kinetics between BPTES and CB-839 revealed distinct modes of inhibition by the two GLS inhibitors.21 Although BPTES showed characteristics of an uncompetitive inhibitor supported by dose-dependent decreases in both Vmax and Km values for glutamine, CB-839 was found to primarily act as a noncompetitive inhibitor, resulting in lower Vmax values but largely unchanged Km values. In addition, inhibition of GAC by CB-839 was found to be time-dependent, although no reduction in IC50 values was observed after prolonged preincubation with BPTES. CB-839 possesses 2- pyridinylacetyl and 3-(trifluoromethoxy)phenyl)acetyl groups attached to the 2-amino-1,3,4-thiadiazole ring and the 3-aminopyridazine ring, respectively. It is not apparent why this particular compound has been chosen for clinical development from a number of potent compounds disclosed in this application, though many factors, including solubility, likely contributed to CB-839’s selection. Indeed, in a head-to-head comparison of solubility in citrate buffer, BPTES exhibited negligible solubility at all pH values tested (pH 2.3–5.4), whereas the solubility of CB-839 was 66 μg/mL in citrate buffer at pH 2.3 and was substantially enhanced (9.7 mg/mL) in citrate buffer containing 20% hydroxypropyl-β-cyclodextrin (HPBCD) compared to the 42 μg/mL reported for BPTES.44 As described later, many of the GLS inhibitors reported since the discovery of CB-839 incorporate the same acyl group combination, 2-pyridinylacetyl and 3-(trifluoromethoxy)phenyl)acetyl groups, while generating structural diversity at other parts of the molecules.
Figure 12.
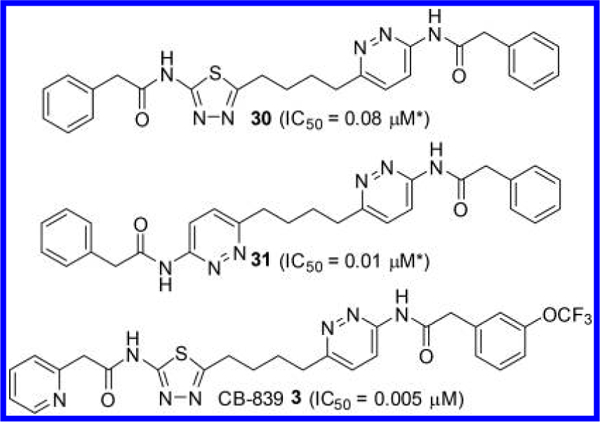
Structures of pyridazine-containing GLS inhibitors (*no preincubation).44
Ramachandran et al. at the National University of Singapore reported the crystal structure of hKGA221–533 in complex with CB-839 (PDB ID: 5JYO).35 As is the case with BPTES, two molecules of CB-839 were found to occupy the dimer—dimer interface of the GLS tetramer in a fashion similar to BPTES (Figure 13). Hydrogen bonds formed by the two nitrogen atoms of the pyridazine appear identical to those exhibited by the thiadiazole ring on the other side of the molecule.
Figure 13.
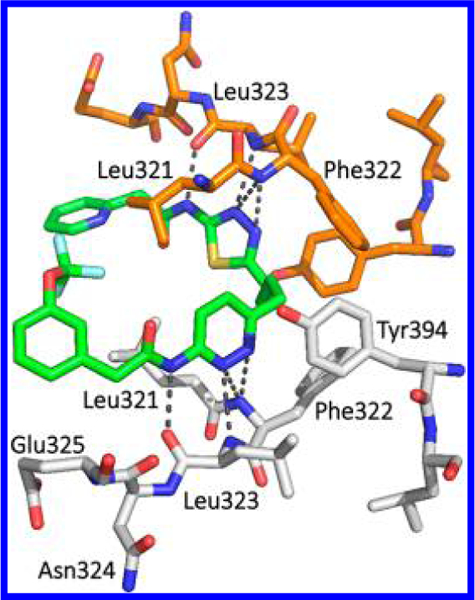
Cocrystal structure of CB-839 (green) bound to the allosteric site of hKGA (PDB ID: 5JYO).
Bhavar et al. at Incozen Therapeutics (in collaboration with Rhizen Pharmaceuticals) explored alternatives to the pyridazine of CB-839 in a systematic manner (Figure 14).46 Among their compounds 32—36, compound 32, in which the pyridazine ring of CB-839 was replaced by a pyridine ring, was reported to inhibit glutaminase prepared from mouse brain/kidney tissue homogenates with IC50 values in the range of 50—100 nM. This patent application, however, did not utilize CB-839 as a control for direct comparison, so one cannot assess the inhibitory potency of compound 32 relative to CB-839 given that their glutaminase assay conditions are very different from others.
Figure 14.

Structures of GLS inhibitors 32–36 containing a pyridazine replacement.46
Further exploration of alternatives to the 2-amino-1,3,4-thiadiazole ring moiety was conducted by Di Francesco et al. at the MD Anderson Cancer Center (Figure 15).47 As seen with compound 37, the 1,3,4-thiadiazole-2-amido moiety in CB-839 can be replaced by a 1,3,4-thiadiazole-2-carboxamide. This reverse amide strategy was successfully extended to 1,2,3-triazole derivative 38. The remaining N-pyridazinylamide in 38 can be further replaced by N-(1,3,4-thiadiazol-2-yl)amide 39 or 1,3,4-thiadiazole-2-carboxamide group 40. These findings enabled them to explore various combinations to identify additional scaffolds including compound 41, which bears two 1,2,3-triazole rings, one of which has a reverse amide group.
Figure 15.

Structures of reverse amide-containing GLS inhibitors 37–41 47
Di Francesco et al. also reported analogues of 11 where one of its 2-amino-1,3,4-thiadiazole rings was replaced by a 4- aminopyridin-2(1H)-one, as shown in compound 42 (Figure 16).48 As demonstrated by compound 43, the remaining 2-phenylacetamido-1,3,4-thiadiazole moiety can be substituted with an N-benzyl-1,2,3-triazole-4-carboxamide moiety. A number of compounds covered in this patent application contain a monofluorinated linker. For instance, compounds 44 and 45 are monofluorinated at the linker region of compound 43. A beneficial pharmacological effect of fluorination, however, is not entirely clear from the biological data.
Figure 16.
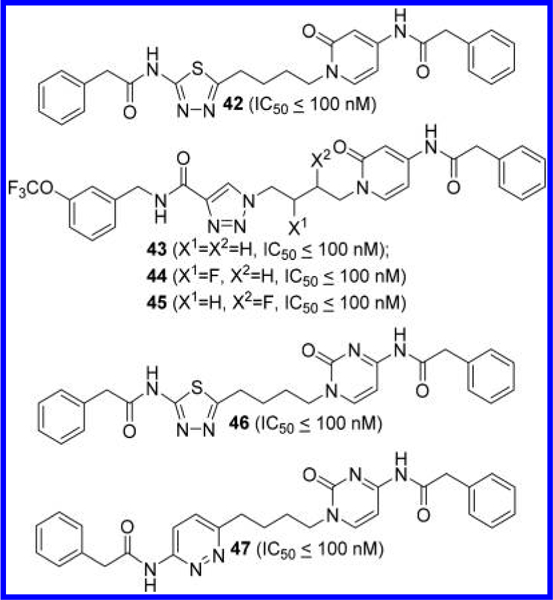
Structures of GLS inhibitors 42–45 containing a 4- aminopyridin-2(lH)-one ring and 46 and 47 containing a 4- aminopyrimidin-2(lH)-one ring.48
According to their patent application, the 2-amino-1,3,4-thiadiazole ring of 11 can also be replaced by a 4- aminopyrimidin-2(1H)-one ring, as exemplified by compound 46 (Figure 16). Further replacement of the thiadiazole ring with a pyridazine ring produced another potent GLS inhibitor 47.
In another patent application, Di Francesco et al. also disclosed GLS inhibitors containing a 7H-pyrrolo[2,3-c]-pyridazine scaffold, presumably acting as conformationally constrained derivatives of 3-acylamidopyridazine (Figure 17).49 In addition to the 2-phenylacetamido-1,3,4-thiadiazole moiety (compound 48), 4-phenylacetamidopyridin-2(1H)-one (compound 49) and N-benzyl-1,2,3-triazole-4-carboxamide (compound 50) can be introduced to the opposite side. Further structural modifications of 50 at both ends of the molecule gave rise to compound 51 containing a 2-fluorophenyl group attached to the 7H-pyrrolo[2,3-c]- pyridazine ring.
Figure 17.
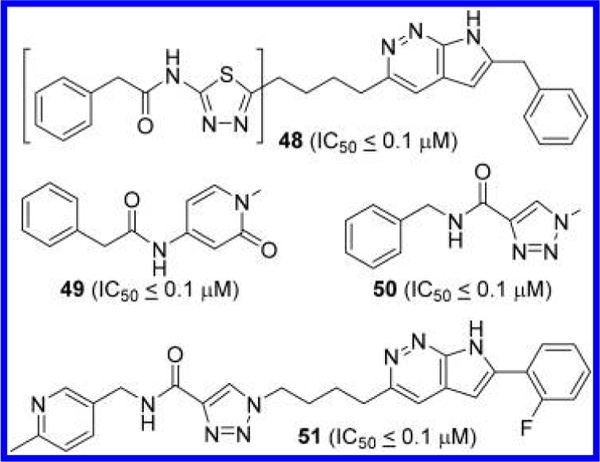
Structures of GLS inhibitors 48–51 containing a 7H- pyrrolo[2,3-c]pyridazine.49
Medicinal chemistry efforts on BPTES have also been expanded to more drastic modifications to the linker component. Lemieux et al. at Agios Pharmaceuticals disclosed BPTES derivatives where its diethyl sulfide portion was replaced with 3—7-membered cycloalkane-based linkers (Figure 18).50,51 Among these linkers, 1,3-disubstituted cyclohexane served as the most effective linker in terms of potency. Although cis-disubstituted analogue 52 and trans-(1R,3R) enantiomer 53 were not particularly potent, trans-(1S,3S)-enantiomer 54 was reported to potently inhibit GLS.
Figure 18.
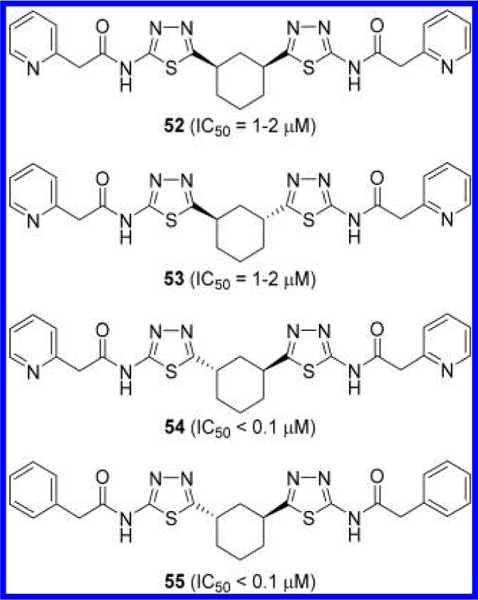
Structures of GLS inhibitors 52–55 containing a cyclohexane linker.50,51
Consistent with these findings, Ramachandran et al. reported that only (1S,3S) enantiomer 55 (Figure 18) was bound to GLS when a racemic mixture was used to obtain the crystal structure (PDB ID: 5JYP).35 The cocrystal structure in complex with 55 displayed the same network of hydrogen bonds as seen with BPTES (PDB ID: 3UO9) and CB-839 (PDB ID: 5JYO) bearing a noncyclic linker (Figure 19).
Figure 19.
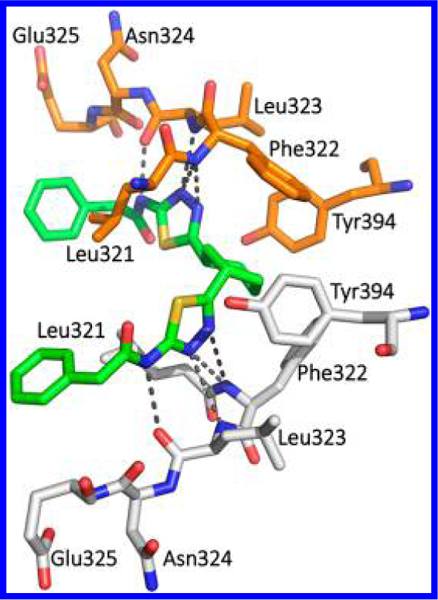
Cocrystal structure of compound 55 (green) bound to the allosteric site of hKGA (PDB ID: 5JYP).
Cianchetta et al. at Agios also replaced the thiadiazole rings of these cyclic GLS inhibitors with pyridazine rings (Figure 20),52 presumably inspired by CB-839. In contrast to compounds 52 and 54 (Figure 18), diacetyl derivative 56 with a cis-linker showed substantially better potency as compared to 58 containing a trans-(1S,3S) linker. In addition, both cis-derivative 57 and trans-(1S,3S) derivative 59 were found to be equally potent, presenting a sharp contrast to the trend of stereochemical preference in the thiadiazole series.
Figure 20.
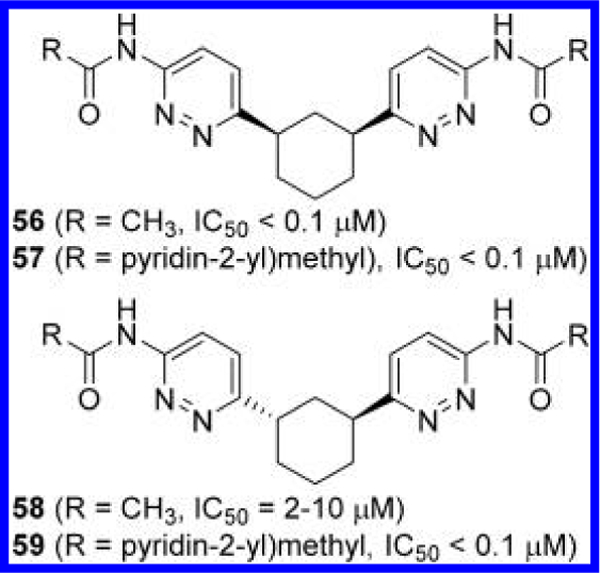
Structures of GLS inhibitors 56—59 containing a cyclohexane linker.52
Other linkers disclosed by the Agios team are summarized in Figure 21.51 A loss of potency was observed with trans-1,4-disubstitited cyclohexane-linked derivative 60 and racemic trans-1,2-dimethyl cyclopropane-linked derivative 61. In contrast to the marked difference in potency between the two enantiomers 53 and 54 (Figure 18), both enantiomers 62 and 63 containing a trans-1,3-disubstituted cyclopentane linker inhibited GLS with IC50 values below 100 nM. As in the case with compound 52, incorporation of cis-1,3-disubstituted cyclopentane (compound 64) resulted in weaker inhibitory potency. Insertion of an additional methylene unit onto each of the 1-and 3-positions of the cyclopentane linker also decreased potency (compound 65) as did the introduction of 1,3-dimethyl cyclobutane linker (compound 66) or a 1,3- disubstituted cylcoheptane linker (compound 67). These SAR findings collectively suggest that there is some degree of flexibility with regard to cyclic linkers, which has been further exploited by other groups as described later.
Figure 21.
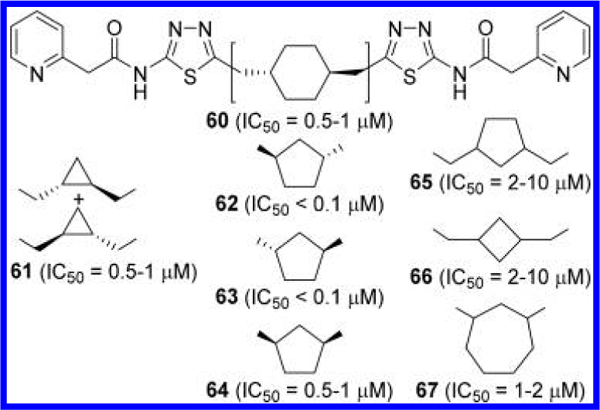
Structures of additional cycloalkane-linked GLS inhibitors 60–67.51
Other groups have also explored GLS inhibitors containing a cycloalkane linker (Figure 22). For example, Bhavar et al. reported compound 68, a cyclobutylmethyl linker-containing compound that inhibited mouse brain/kidney glutaminase with an IC50 value of less than 25 nM.46 Burns et al. at Pfizer reported a wide variety of cycloalkyl-linked GLS inhibitors including those containing cyclobutylmethyl (compound 69) and cyclopentylmethyl-based (compound 70) linkers.53 In addition, they reported spiro[3.3]heptane-linked GLS inhibitors such as 71 and 72. Another interesting element of these compounds is the 2-(l-methyl-1H-pyrazol-3-yl)acetyl group serving as an effective alternative to the phenylacetyl group. Although compounds 69–72 were reported to be highly potent, comparison to other GLS inhibitors should be made with caution as the IC50 values cited in Pfizer’s patent application were obtained from a cell-based assay (BT20) in which glutamate was quantified in the lysate.
Figure 22.
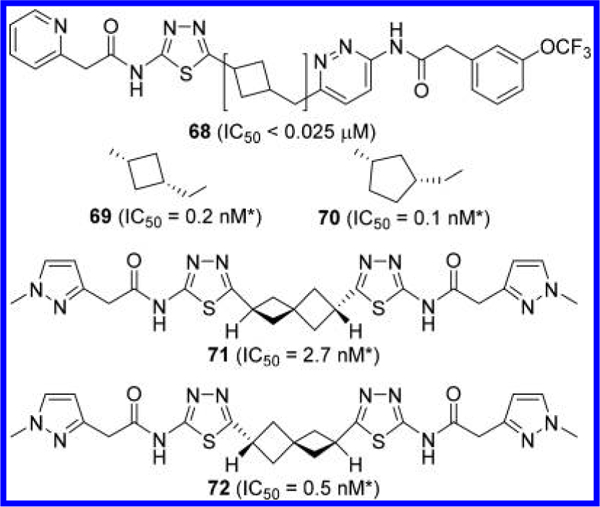
Structures of additional cycloalkane-linked GLS inhibitors 68–72 (*IC50 obtained from cell-based glutaminase assay).46,53
Finlay et al. at AstraZeneca disclosed GLS inhibitors containing a 3-aminopyrrolidine linker (Figure 23).23 Unique structural features of these compounds include the incorporation of an α-alkoxyphenylacetyl moiety to the amino-thiadiazole ring as well as a pyridazine or triazine ring lacking an acylamino group on the opposite side of the molecule. Although the effects of the alkoxy group cannot be assessed because of the lack of data for the nonsubstituted counterparts, its stereochemistry seems to have some impact on potency. In general, (2S) isomers such as compounds 73 and 74 are more potent than the corresponding (2R) derivatives such as compounds 75 and 76.
Figure 23.
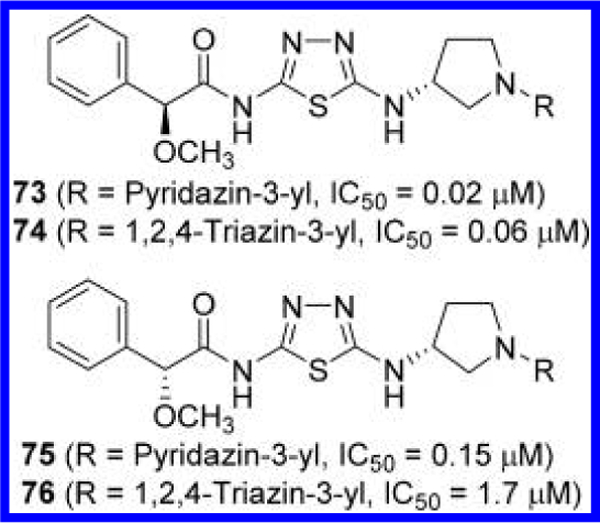
Structures GLS inhibitors 73–76 containing a 3- aminopyrrolidine linker.23
In subsequent applications,24,25 the AstraZeneca group disclosed additional analogues in which the phenylacetyl group and/or the pyridazine ring are further modified as represented by compounds 77–80 (Figure 24). Two diastereomers of 79 and 80, which differ in configuration at the α-carbon of the phenylacetyl group, were individually obtained, although the absolute stereochemistry at the α-carbon was not determined. Nevertheless, both diastereomers were found to be highly potent with IC50 values below 10 nM.
Figure 24.
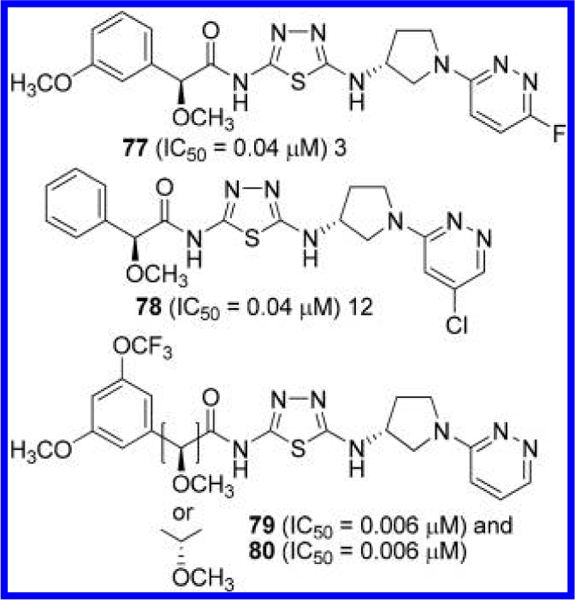
Structures of GLS inhibitors 77–80 containing a 3- aminopyrrolidine linker.24,25
Finlay et al. extended the above series by substituting a dialkylamino group for a methoxy group at the α-carbon of the phenylacetyl group (Figure 25).26 As is the case with the methoxy derivatives, (2S)-configuration at the α-carbon, as demonstrated by compounds 81 and 82, provided higher potency compared to those with (2R)-configuration (compounds 83 and 84). Other compounds disclosed in this application include azetidin-1-yl derivative 85 and 2-methyl-3,4-dihydro-1H-isoquinoline-containing analogue 86. It should be noted that the (2S)-configuration assigned to 85 and 86 in Figure 25 has been tentatively assigned based on their lower IC50 values compared to those of the other diastereomers. Finlay et al. also reported GLS inhibitors containing two pyridazine rings as represented by compound 87.27 In this series, the 2-aminopyrrolidine linker is reversed compared to the other compounds described earlier.
Figure 25.
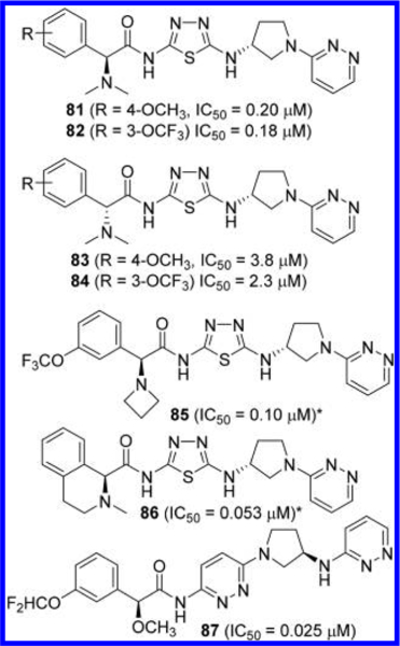
Structures of GLS inhibitors 81−87 containing a 3-aminopyrrolidine linker (*stereochemistry tentatively assigned).26,27
McDermott et al. at the University of Pittsburgh in collaboration with Cornell University conducted extensive linker optimization efforts prompted by the hypothesis that replacement of the flexible diethylsulfide linker of BPTES with small to medium size ring systems should result in not only a reduced number of rotatable bonds but also a reduction in the entropic penalty upon binding.54 As represented by compounds 88–90 (Figure 26), a variety of cyclic linkers were explored including 4-aminopiperidine (compound 88), 4-oxypiperidine (compound 89), and 1,3-diaminocyclopentane (compound 90). These compounds have reduced cLogP values, improved microsomal stability, and higher ligand efficiency when compared to BPTES and CB-839.
Figure 26.
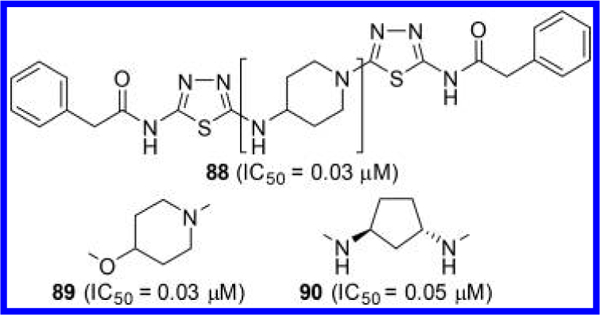
Structures of GLS inhibitors 88–90 each containing a 4- aminopiperidine, 4-oxypiperidine, or 1,3-diaminocyclopentane linker, respectively.54
Cocrystal structures of GAC complexed with compounds 88 (PDB ID: 5WJ6)55 and 89 (PDB ID: 5I94)54 were determined. As shown in Figure 27, compound 88 forms multiple interactions at the allosteric site of GLS. In particular, the terminal amino group of Lys320 from one GLS unit (orange) interacts with the carbonyl oxygen of the phenyl-acetyl group while the carbonyl oxygen of Lys320 from the other unit (white) interacts with the NH-bridge between the thiadiazole and piperidine rings. The contribution of these interactions to the binding of 88, however, may not be substantial as compound 89 exhibited similar potency despite the lack of these interactions.54
Figure 27.
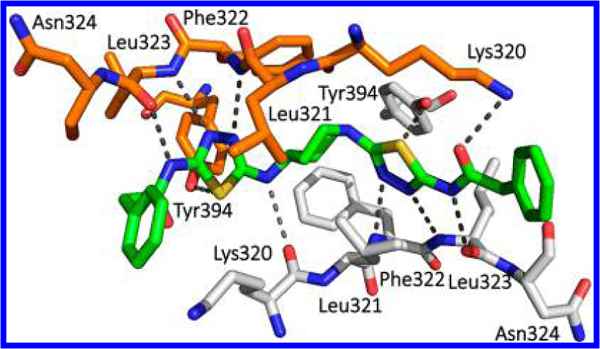
Cocrystal structure of compound 88 (green) bound to the allosteric site of hGAC (PDB ID: 5WJ6).
McDermott et al. also replaced the thiadiazole ring(s) of 89 with other heterocycles (Figure 28).54 Consistent with previous reports, derivatives in which one or both thiadiazole ring(s) were replaced by a pyridazine ring retained good inhibitory potency (compounds 91 and 92), whereas incorporation of pyridine and 1,4-pyrazine rings led to complete loss of potency (compounds 93–96). These findings reaffirm that two heterocyclic rings each containing two adjacent nitrogen atoms play an essential role in the majority of GLS inhibitors derived from BPTES.
Figure 28.

Structures of GLS inhibitors 91–96 containing a 4- oxypiperidine linker.54
In addition, McDermott et al. examined both enantiomers of additional GLS inhibitors containing a chiral cyclic linker (Figure 29).54 Interestingly, two enantiomers 97 and 98, containing a 3-aminopyrrolidine linker, displayed little difference in GLS inhibitory potency. A similar trend was seen for 3-oxypyrrolidine-linked 99 and its enantiomer 100. Indeed, crystal structures of GAC in complex with 97 (PDB ID: 5FI6) and 98 (PDB ID: 5FI2) revealed that both of these enantiomers can be well-accommodated in the GLS allosteric binding pocket.54 It should be noted that each of the two ligands within a GLS tetramer shows a distinct binding mode in these two cocrystal structures. For instance, in the 5FI2 tetramer structure (Figure 30), one molecule of 98 (green) binds to the allosteric site in a manner similar to BPTES in the 3VPI structure (Figure 5A), whereas the other molecule of 98 (cyan) adopts the binding mode of BPTES in the 3UO9 structure (Figure 5B).
Figure 29.
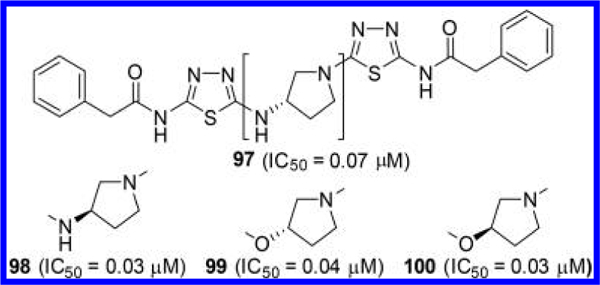
Structures of GLS inhibitors 97 and 98 containing a 3- aminopyrrolidine linker and 99 and 100 containing a 3-oxypyrrolidine linker.54
Figure 30.
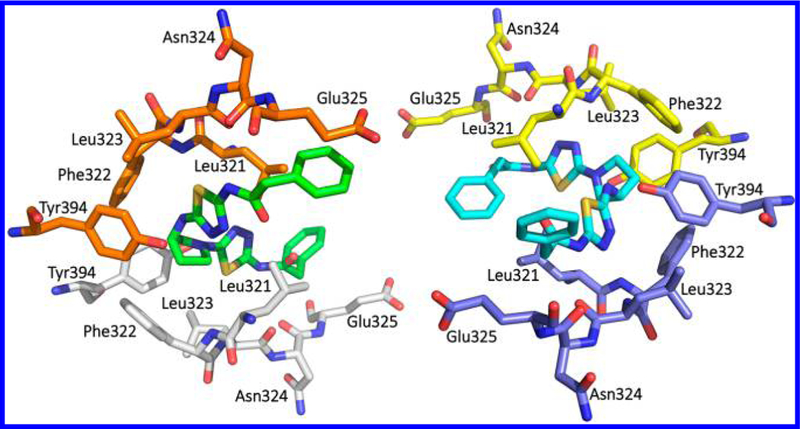
Cocrystal structure of compound 98 (green and cyan) bound to the allosteric site of hGAC (PDB ID: 5FI2).
Bhavar et al. published a number of GLS inhibitors with 1-(pyridazin-3-yl)piperidine scaffolds (Figure 31).56 The piperidine ring of these compounds also contains a 1,3,4-thiadiazole ring at its 3- or 4-position. Several 4-substituted derivatives including 101 displayed potent inhibitory activity (IC50 < 50 nM), whereas the corresponding 2-aminopyridin-5-yl derivative 102 showed substantial loss in potency. Swapping the position of the two terminal acyl groups of 101 produced another potent inhibitor 103. A good inhibitory potency was also seen by compound 104 containing a 1,3-linked piperidine.
Figure 31.
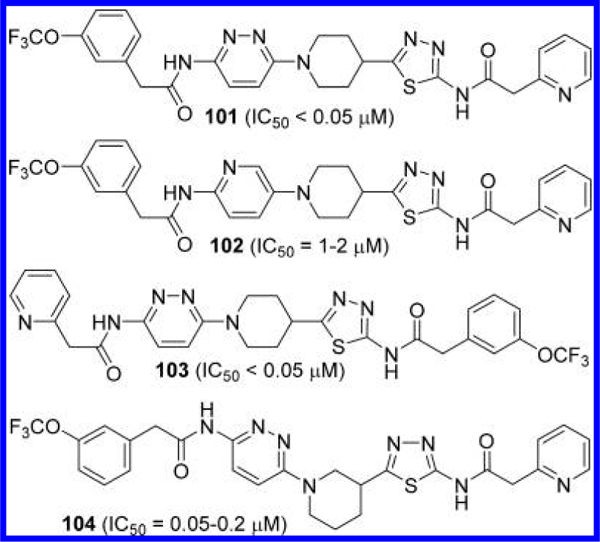
Structures of GLS inhibitors 101–104 containing a piperidine linker.56
Lewis et al. at the MD Anderson Cancer Center also disclosed pyrrolidine-linked GLS inhibitors 105 and 106,57 which only differ by the orientation of the pyrrolidine linker (Figure 32). The majority of the pyrrolidine-linked compounds in this application were made as racemates though two enantiomers 107 and 108 exhibited a slight difference in inhibitory potency.
Figure 32.
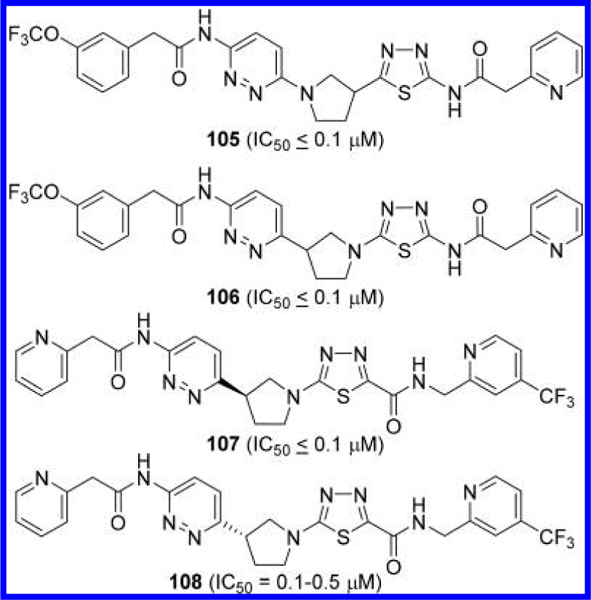
Structures of GLS inhibitors 105–108 containing a pyrrolidine linker.57
5. CONCLUSIONS
Over the past decade, tremendous progress has been made in the discovery and development of allosteric GLS inhibitors, particularly those derived from BPTES. The substantial efforts made by both industry and academic sectors are evident from the increasing number of GLS inhibitors appearing in journal and patent literature. Although BPTES was identified as a GLS inhibitor through a random screening, GLS inhibitors derived from BPTES undoubtedly provide an interesting case study, illuminating how an allosteric site can be methodically explored in pursuit of clinically viable inhibitors. The complex dynamics involved in the catalytic action of GLS is certainly intriguing, as is the mechanism by which BPTES exploits the oligomerization process, as an opportunity to selectively inhibit GLS over other glutamine-utilizing enzymes. This approach has more general implications beyond just GLS inhibition as a strategic way to target one isoform among those sharing a common substrate and to help mitigate the selectivity issues inherent to competitive inhibitors.
Although BPTES hardly possesses desirable “druglike” molecular properties, the structural evolution of GLS inhibitors has clearly shown that such a lead compound can be transformed into “druglike” compounds through systematic SAR studies. Analysis of the cumulative SAR data presented herein highlights some key structural features common to most BPTES-derived GLS inhibitors (with some exceptions): (i) two heterocycles with two adjacent nitrogen atoms are essential for interaction with the GLS allosteric site through interaction with Phe322 and Leu323 residues; (ii) the size and shape of the linkers can be flexible as long as they can be contained by the lipophilic pocket formed by Leu321, Phe322, and Tyr394 and can project the two heterocycles for optimal interactions with the GLS allosteric site; (iii) the NH groups of the terminal carboxamides play an important role in binding as hydrogen donors to the carbonyl oxygen of Leu323; (iv) the pockets that accommodate the two terminal carboxamide groups can tolerate a wide variety of substitutes; (v) one of the two carboxamide groups can be dispensable for GLS inhibition.
Development of CB-839, a first-in-class GLS inhibitor, unarguably represents an important milestone in the effort to translate GLS inhibitors into novel therapeutics. Outcomes of clinical studies of CB-839 should provide key insights into the patient populations that benefit most from GLS inhibition as well as the possibility of serving as an adjuvant therapy to the existing treatments for synergistic efficacy. The fact that the latest GLS inhibitors show little structural resemblance to BPTES is a testament to the incredible potential that lies in the hands of medicinal chemists. As some new GLS inhibitors have substantially lower molecular weight compared to BPTES, with a well thought-out strategy to optimize physicochemical properties for CNS permeability, one could explore the feasibility of developing brain-penetrant GLS inhibitors with potential therapeutic utility in neurological disorders where GLS-mediated formation of glutamate is believed to play a pathophysiological role.58–60
ACKNOWLEDGMENTS
The authors thank Dr. Norman P. Curthoys for his critical review of the manuscript and insightful discussions. The authors of this manuscript have been supported by NIH grants P30MH075673 (T.T.), R01CA193895 (T.T.), R21NS074151 (T.T.), and F32CA200278 (S.C.Z.). The authors are also grateful for the support provided by the Bloomberg-Kimmel Institute for Cancer Immunotherapy at Johns Hopkins.
ABBREVIATIONS USED
- KGA
kidney glutaminase
- GAC
glutaminase C
- DON
6-diazo-5-oxo-L-norleucine
- BPTES
bis-2-[5-(phenylacetami-do)-1,3,4-thiadiazol-2-yl]ethyl sulfide
- GDH
glutamate dehydrogenase
- GLOD
glutamate oxidase
- PDB
Protein Data Bank
Biographies
Sarah C. Zimmermann received her Ph.D. from the University of Maryland, Baltimore County. Her doctoral research, under the tutelage of Dr. Katherine L. Seley-Radtke, focused on the synthesis of carbocyclic nucleosides with flexible nucleobase motifs. Following her Ph.D., she was a Postdoctoral Fellow at the Johns Hopkins University with Dr. Takashi Tsukamoto, where she focused on the design and synthesis of GLS inhibitors and was awarded an NIH Ruth Kirschstein Postdoctoral Fellowship. Presently, Dr. Zimmermann is a chemist at the U.S. Food and Drug Administration.
Bridget R. Duvall earned her M.S. from the University of Maryland, College Park under the guidance of Philip DeShong and her thesis focused on the synthesis of (+)-pancratistatin featuring the chemistry of hypervalent silicates. Since graduation, she has worked as a medicinal chemist in both industry and academia. She is currently a member of Johns Hopkins Drug Discovery and involved in the design and synthesis of small molecules targeting a number of therapeutic targets including glutaminase.
Takashi Tsukamoto is an Associate Professor of Neurology at Johns Hopkins University and the Director of Medicinal Chemistry at Johns Hopkins Drug Discovery. He received his Ph.D. in chemistry from Tokyo Institute of Technology and pursued his postdoctoral studies at the University of Michigan. Prior to joining Johns Hopkins in 2009, Dr. Tsukamoto has held a variety of research positions in the pharmaceutical industry. During the course of his career, he has served as a lead medicinal chemist in a number of drug discovery projects exploring new therapeutics for neurological disorders and cancer.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Curthoys NP; Watford M Regulation of Glutaminase Activity and Glutamine Metabolism. Annu. Rev. Nutr. 1995, 15, 133–159. [DOI] [PubMed] [Google Scholar]
- (2).Aledo JC; Gomez-Fabre PM; Olalla L; Marquez J Identification of Two Human Glutaminase Loci and Tissue-Specific Expression of the Two Related Genes. Mamm. Genome 2000, 11, 1107–1110. [DOI] [PubMed] [Google Scholar]
- (3).Porter LD; Ibrahim H; Taylor L; Curthoys NP Complexity and Species Variation of the Kidney-Type Glutaminase Gene. Physiol. Genomics 2002, 9, 157–166. [DOI] [PubMed] [Google Scholar]
- (4).Sonnewald U; Schousboe A Introduction to the GlutamateGlutamine Cycle. Adv. Neurobiol 2016, 13, 1–7. [DOI] [PubMed] [Google Scholar]
- (5).Kovacevic Z; Mcgivan JD Mitochondrial Metabolism of Glutamine and Glutamate and Its Physiological Significance. Physiol. Rev. 1983, 63, 547–605. [DOI] [PubMed] [Google Scholar]
- (6).Gao P; Tchernyshyov I; Chang TC; Lee YS; Kita K; Ochi T; Zeller KI; De Marzo AM; Van Eyk JE; Mendell JT; Dang CV C-Myc Suppression of Mir-23a/B Enhances Mitochondrial Glutaminase Expression and Glutamine Metabolism. Nature 2009, 458, 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Mates JM; Segura JA; Martin-Rufian M; Campos-Sandoval JA; Alonso FJ; Marquez J Glutaminase Isoenzymes as Key Regulators in Metabolic and Oxidative Stress against Cancer. Curr. Mol. Med. 2013, 13, 514–534. [DOI] [PubMed] [Google Scholar]
- (8).Van Den Heuvel AP; Jing J; Wooster RF; Bachman KE Analysis of Glutamine Dependency in Non-Small Cell Lung Cancer: Gls1 Splice Variant Gac Is Essential for Cancer Cell Growth. Cancer Biol. Ther. 2012, 13, 1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cassago A; Ferreira AP; Ferreira IM; Fornezari C; Gomes ER; Greene KS; Pereira HM; Garratt RC; Dias SM; Ambrosio AL Mitochondrial Localization and Structure-Based Phosphate Activation Mechanism of Glutaminase C with Implications for Cancer Metabolism. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Shapiro RA; Clark VM; Curthoys NP Inactivation of Rat Renal Phosphate-Dependent Glutaminase with 6-Diazo-5-Oxo-L- Norleucine. Evidence for Interaction at the Glutamine Binding Site. J. Biol. Chem. 1979, 254, 2835–2838. [PubMed] [Google Scholar]
- (11).Le A; Lane AN; Hamaker M; Bose S; Gouw A; Barbi J; Tsukamoto T; Rojas CJ; Slusher BS; Zhang H; Zimmerman LJ; Liebler DC; Slebos RJ ; Lorkiewicz PK; Higashi RM; Fan TW; Dang CV Glucose-Independent Glutamine Metabolism Via Tca Cycling for Proliferation and Survival in B Cells. Cell Metab. 2012, 15, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Erickson JW; Cerione R A. Glutaminase: A Hot Spot for Regulation of Cancer Cell Metabolism? Oncotarget 2010, 1, 734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Katt WP; Cerione RA Glutaminase Regulation in Cancer Cells: A Druggable Chain of Events. Drug Discovery Today 2014, 19, 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lukey MJ; Wilson KF; Cerione R A. Therapeutic Strategies Impacting Cancer Cell Glutamine Metabolism. Future Med. Chem. 2013, 5, 1685—1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Altman BJ; Stine ZE; Dang CV From Krebs to Clinic: Glutamine Metabolism to Cancer Therapy. Nat. Rev. Cancer 2016, 16, 619—634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Katt WP; Lukey MJ; Cerione RA A Tale of Two Glutaminases: Homologous Enzymes with Distinct Roles in Tumori- genesis. Future Med. Chem. 2017, 9,223—243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang JB; Erickson JW; Fuji R; Ramachandran S; Gao P; Dinavahi R; Wilson KF; Ambrosio AL; Dias SM; Dang CV; Cerione R A. Targeting Mitochondrial Glutaminase Activity Inhibits Oncogenic Transformation. Cancer Cell 2010, 18, 207—219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Katt WP; Ramachandran S; Erickson JW; Cerione RA Dibenzophenanthridines as Inhibitors of Phosphate-Activated Glutaminase C and Cancer Cell Proliferation. Mol. Cancer Ther. 2012, 11, 1269—1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yeh TK; Kuo CC; Lee YZ; Ke YY; Chu KF; Hsu HY; Chang HY; Liu YW; Song JS; Yang CW; Lin LM; Sun M; Wu SH; Kuo PC; Shih C; Chen CT; Tsou LK; Lee SJ Design, Synthesis, and Evaluation of Thiazolidine-2,4-Dione Derivatives as a Novel Class of Glutaminase Inhibitors. J. Med. Chem. 2017, 60, 5599—5612. [DOI] [PubMed] [Google Scholar]
- (20).Cheng L; Wu CR; Zhu LH; Li H; Chen LX Physapubescin, a Natural Withanolide as a Kidney-Type Glutaminase (Kga) Inhibitor. Bioorg. Med. Chem. Lett. 2017, 27, 1243—1246. [DOI] [PubMed] [Google Scholar]
- (21).Gross MI; Demo SD; Dennison JB; Chen L; Chernov-Rogan T; Goyal B; Janes JR; Laidig GJ; Lewis ER; Li J; Mackinnon AL; Parlati F; Rodriguez ML; Shwonek PJ; Sjogren EB; Stanton TF; Wang T; Yang J; Zhao FY; Bennett MK Antitumor Activity of the Glutaminase Inhibitor Cb-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890—901. [DOI] [PubMed] [Google Scholar]
- (22).Shukla K; Ferraris DV; Thomas AG; Stathis M; Duvall B; Delahanty G; Alt J; Rais R; Rojas C; Gao P; Xiang Y; Dang V; Slusher BS; Tsukamoto T Design, Synthesis, and Pharmacological Evaluation of Bis-2-(5-Phenylacetamido-1,2,4-Thia-diazol-2-Yl)Ethyl Sulfide 3 (Bptes) Analogs as Glutaminase Inhibitors. J. Med. Chem. 2012, 55, 10551—10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Finlay MRV; Ekwuru CT; Charles MD; Raubo PA; Winter JJG; Nissink JWM 1,3,4-Thiadiazole Compounds and Their Use in Treating Cancer. PCT Int. Pat. Appl. WO 2015/181539, 2015. [Google Scholar]
- (24).Nissink JWM; Finlay MRV; Charles MD; Wood M 1,3,4-Thiadiazole Compounds and Their Use in Treating Cancer. U.S. Pat. Appl. 20170152254, 2017. [Google Scholar]
- (25).Nissink JWM; Finlay MRV; Charles MD 1,3,4-Thiadiazole Compounds and Their Use in Treating Cancer. U.S. Pat. Appl. 20170152255, 2017. [Google Scholar]
- (26).Finlay M; Verschoyle R; Perkins D; Nissink R; Wilhelmus J; Raubo M; Antoni P; Smith P; Bailey D; Cook A 1,3,4-Thiadiazole Compounds and Their Use in Treating Cancer. PCT Int. Pat. Appl. WO 2017/093301, 2017. [Google Scholar]
- (27).Finlay M, Verschoyle R Bis-Pyridazine Compounds and Their Use in Treating Cancer. PCT Int. Appl. WO 2017/089587, 2017. [Google Scholar]
- (28).Newcomb RW Selective Inhibition of Glutaminase by Bis-Thiadiazoles. US 6,451,828 B1, 2002.
- (29).Robinson MM; Mcbryant SJ; Tsukamoto T; Rojas C; Ferraris DV; Hamilton SK; Hansen JC; Curthoys NP Novel Mechanism of Inhibition of Rat Kidney-Type Glutaminase by Bis-2- (5-Phenylacetamido-1,2,4-Thiadiazol-2-Yl)Ethyl Sulfide (Bptes). Bio- chem. J. 2007, 406, 407—414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Godfrey S; Kuhlenschmidt T; Curthoys P Correlation between Activation and Dimer Formation of Rat Renal Phosphate- Dependent Glutaminase. J. Biol. Chem. 1977, 252, 1927—1931. [PubMed] [Google Scholar]
- (31).Morehouse RF; Curthoys NP Properties of Rat Renal Phosphate-Dependent Glutaminase Coupled to Sepharose. Evidence That Dimerization Is Essential for Activation. Biochem. J. 1981, 193, 709—716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hartwick EW; Curthoys NP Bptes Inhibition of Hga(124– 551), a Truncated Form of Human Kidney-Type Glutaminase. J. Enzvme Inhib. Med. Chem. 2012, 27, 861–867. [DOI] [PubMed] [Google Scholar]
- (33).Delabarre B; Gross S; Fang C; Gao Y; Jha A; Jiang F; Song JJ; Wei W; Hurov JB Full-Length Human Glutaminase in Complex with an Allosteric Inhibitor. Biochemistry 2011, 50, 10764–10770. [DOI] [PubMed] [Google Scholar]
- (34).Thangavelu K; Pan CQ; Karlberg T; Balaji G; Uttamchandani M; Suresh V; Schuler H; Low BC; Sivaraman J Structural Basis for the Allosteric Inhibitory Mechanism of Human Kidney-Type Glutaminase (Kga) and Its Regulation by Raf-Mek-Erk Signaling in Cancer Cell Metabolism. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 7705–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ramachandran S; Pan CQ; Zimmermann SC; Duvall B; Tsukamoto T; Low BC; Sivaraman J Structural Basis for Exploring the Allosteric Inhibition of Human Kidney Type Glutaminase. Oncotarget 2016, 7, 57943–57954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Stalnecker CA; Erickson JW; Cerione RA Conformational Changes in the Activation Loop of Mitochondrial Glutaminase C: A Direct Fluorescence Readout That Distinguishes the Binding of Allosteric Inhibitors from Activators. J. Biol. Chem. 2017, 292, 6095–6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Yuneva MO; Fan TW; Allen TD; Higashi RM; Ferraris DV; Tsukamoto T; Mates JM; Alonso FJ; Wang C; Seo Y; Chen X; Bishop JM The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Dutta P; Le A; Vander Jagt DL; Tsukamoto T; Martinez GV; Dang CV; Gillies RJ Evaluation of Ldh-a and Glutaminase Inhibition in Vivo by Hyperpolarized 13c-Pyruvate Magnetic Resonance Spectroscopy of Tumors. Cancer Res. 2013, 73, 4190–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Seltzer MJ; Bennett BD; Joshi AD; Gao P; Thomas AG; Ferraris DV; Tsukamoto T; Rojas CJ; Slusher BS; Rabinowitz JD; Dang CV; Riggins GJ Inhibition of Glutaminase Preferentially Slows Growth of Glioma Cells with Mutant Idh1. Cancer Res. 2010, 70, 8981–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Qie S; Chu C; Li W; Wang C; Sang N Erbb2 Activation Upregulates Glutaminase 1 Expression Which Promotes Breast Cancer Cell Proliferation. J. Cell. Biochem. 2014, 115, 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zimmermann SC; Wolf EF; Luu A; Thomas AG; Stathis M; Poore B; Nguyen C; Le A; Rojas C; Slusher BS; Tsukamoto T Allosteric Glutaminase Inhibitors Based on a 1,4-Di(5-Amino-1,3,4-Thiadiazol-2-Yl)Butane Scaffold. ACS Med. Chem. Lett. 2016. 7, 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Son J; Lyssiotis CA; Ying H; Wang X; Hua S; Ligorio M; Perera RM; Ferrone CR; Mullarky E; Shyh-Chang N; Kang Y; Fleming JB; Bardeesy N; Asara JM; Haigis MC; Depinho RA; Cantley LC; Kimmelman AC Glutamine Supports Pancreatic Cancer Growth through a Kras-Regulated Metabolic Pathway. Nature 2013, 496, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Elgogary A; Xu Q; Poore B; Alt J; Zimmermann SC; Zhao L; Fu J; Chen B; Xia S; Liu Y; Neisser M; Nguyen C; Lee R; Park JK; Reyes J; Hartung T; Rojas C; Rais R; Tsukamoto T; Semenza GL; Hanes J; Slusher BS; Le A Combination Therapy with Bptes Nanoparticles and Metformin Targets the Metabolic Heterogeneity of Pancreatic Cancer. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E5328–5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Li J; Chen L; Goyal B; Laidig G; Stanton TF; Sjogren EB Heterocyclic Inhibitors of Glutaminase. PCT Int. Pat. Appl. WO 2013/078123, 2013. [Google Scholar]
- (45).Lemieux RM; Popovici-Muller J; Salituro FG; Saunders JO; Travins J; Yan S Compounds and Their Methods of Use. U.S. Pat. Appl. 20140142146, 2014. [Google Scholar]
- (46).Bhavar PK; Vakkalanka SK; Viswanadha S; Swaroop MG; Babu G Novel Inhibitors of Glutaminase. PCT Int. Appl. WO 2015/101958, 2015. [Google Scholar]
- (47).Di Francesco ME; Jones P; Heffernan T; Hamilton MM; Kang Z; Soth MJ; Burke JP; Le K; Carroll CL; Palmer WS; Lewis R; Mcafoos T; Czako B; Liu G; Theroff J; Herrera Z; Yau A Glsl Inhibitors for Treating Disease. U.S. Pat. Appl. 20160009704, 2016. [Google Scholar]
- (48).Di Francesco ME; Jones P; Heffernan T; Hamilton MM; Kang Z; Soth MJ; Burke JP; Carroll CL; Mcafoos T; Palmer W; Herrera Z Glsl Inhibitors for Treating Disease. U.S. Pat. Appl. 20160002204, 2016. [Google Scholar]
- (49).Di Francesco ME; Heffernan T; Soth MJ; Le K; Carroll BL; Mcafoos T; Burke JP; Theroff J; Kang Z; Jones P Gls1 Inhibitors for Treating Disease. U.S. Pat. Appl. 20160002248, 2016. [Google Scholar]
- (50).Lemieux RM; Popovici-Muller J; Salituro FG; Saunders JO; Travins J; Chen Y Compounds and Their Methods of Use. U.S. Pat. Appl. 20140142081, 2014. [Google Scholar]
- (51).Lemieux RM; Popovici-Muller J; Salituro FG; Saunders JO; Travins J; Chen Y Compounds and Their Methods of Use. U.S. Pat. Appl. 20150291576, 2015. [Google Scholar]
- (52).Cianchetta G; Lemieux RM; Cao S; Ding Y; Ye Z Compounds and Their Methods of Use. PCT Int. Appl. WO 2015/143340, 2015. [Google Scholar]
- (53).Burns AC; Collins MR; Greasley SE; Hoffman RL; Huang Q; Kania RS; Kung P-P; Linton MA; Narasimhan LS; Richardson PF; Richter DT; Smith G Cycloalkyl-Linked Diheterocycle Derivatives. PCT Int. Appl. WO 2015/166373, 2015. [Google Scholar]
- (54).Mcdermott LA; Iyer P; Vernetti L; Rimer S; Sun J; Boby M; Yang T; Fioravanti M; O’neill J; Wang L; Drakes D; Katt W; Huang Q; Cerione R Design and Evaluation of Novel Glutaminase Inhibitors. Bioore. Med. Chem. 2016, 24, 1819–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Huang Q; Stalnecker C; Zhang C; Mcdermott LA; Iyer P; O’neill J; Reimer S; Cerione RA; Katt WP Characterization of the Interactions of Potent Allosteric Inhibitors with Glutaminase C, a Key Enzyme in Cancer Cell Glutamine Metabolism. J. Biol. Chem. 2018, 293, 3535–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Bhavar PK; Vakkalanka SKVS; Viswanadha S; Swaroop MG; Babu G Novel Glutaminase Inhibitors. PCT Int. Appl. WOw 2015/101957, 2015. [Google Scholar]
- (57).Lewis RT; Jones P; Petrocchi A; Reyna N; Hamilton M; Soth MJ; Heffernan T; Han M; Burke JP Gls1 Inhibitors for Treating Disease. U.S. Pat. Appl. 20170001996, 2017. [Google Scholar]
- (58).Yawata I; Takeuchi H; Doi Y; Liang J; Mizuno T; Suzumura A Macrophage-Induced Neurotoxicity Is Mediated by Glutamate and Attenuated by Glutaminase Inhibitors and Gap Junction Inhibitors. Life Sci. 2008, 82, 1111–1116. [DOI] [PubMed] [Google Scholar]
- (59).Potter MC; Figuera-Losada M; Rojas C; Slusher BS Targeting the Glutamatergic System for the Treatment of Hiv-Associated Neurocognitive Disorders. J. Neuroimmune Pharmacol 2013, 8, 594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Wang Y; Li Y; Zhao R; Wu B; Lanoha B; Tong Z; Peer J; Liu J; Xiong H; Huang Y; Zheng J Glutaminase C Overexpression in the Brain Induces Learning Deficits, Synaptic Dysfunctions, and Neuroinflammation in Mice. Brain Behav., Immun. 2017, 66, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]