

Abstract
Since the advent of the chromosome conformation capture technology, our understanding of the human genome 3D organization has grown rapidly and we now know that human interphase chromosomes are folded into multiple layers of hierarchical structures and each layer can play a critical role in transcriptional regulation. Alterations in any one of these finely-tuned layers can lead to unwanted cascade of molecular events and ultimately drive the manifestation of diseases and phenotypes. Here we discuss, starting from chromosome level organization going down to single nucleotide changes, recent studies linking diseases or phenotypes to changes in the 3D genome architecture.
Introduction
Control of gene expression is complex but vital and epigenetics play a critical role in modulating gene expression for normal cellular function. Abrogation of this epigenetic control is frequently associated with disease etiology and progression [1, 2]. Different epigenetic layers provide us context to understand the rules governing gene expression and chromatin stands out as front and center to all these layers. Chromatin is the biomolecular complex between the genomic DNA and histone proteins which allows the DNA to be folded and compacted by thousands of folds and get organized into cellular nuclei of 2–10 microns in size for human cells. Nucleosomes are the basic unit of chromatin, consisting of 146 bp of DNA wrapped around two copies of histone H2A, H2B, H3 and H4 proteins. Connecting the two nucleosomes is the linker DNA associated with histone H1 and H5 [3]. Although this initial folding and occupancy of nucleosomes along the DNA constitute a basic layer of gene regulatory system; the eukaryotic transcriptional mechanism (e.g., in human) is far from simple in nature [4]. The complexity of transcriptional machinery arises from a spatiotemporal coordination of many cis and trans acting genomic and proteomic elements that maintain the baseline gene expression profile in normal condition. Transcription is initiated by promoters, which first recruit the RNA polymerases and other necessary factors. Enhancers and insulators further fine tune the gene expression by recruiting transcriptional activators or suppressors in a tissue or lineage-dependent manner. Modulation of transcription through enhancers or insulators seems surprisingly difficult when the linear genomic distances between a transcription start site (TSS) and some of its distal regulatory elements are considered [5]. A simple linear map of histone modified chromatin is insufficient to fully explain cell type-and context-specific differences in gene expression and demands a detailed understanding of the three-dimensional organization of the chromosomes. To systematically elucidate the gene-regulatory programs in human or in other organisms and to decipher the complex circuit of connections between regulatory elements and genes, it is necessary to consider the chromatin in its folded state in 3D and in a tissue, lineage, and cell-type dependent manner.
The predominant method to study the 3D organization of genomes before the development of next generation sequencing (NGS)-based techniques was the fluorescence in situ hybridization (FISH) [6]. In recent years, chromosome conformation capture assays in combination with NGS technology successfully complemented imaging-based techniques to study the 3D organization of the genome. Chromosome conformation capture measures the pairwise contact frequency of genomic regions by a process that can be summarized as: crosslink, cut, label, re-ligate, shear, enrich and sequence. More specifically, chemically fixed cells with cross-links holding the spatially close DNA fragments together are subjected to chromatin digestion with a restriction enzyme (or enzymes) in order to create fragments, the ends of which are labeled with biotin and are allowed to re-ligate with the ends of nearby fragments that are cross-linked together. After shearing of the re-ligated circular DNA and potential steps for enrichment of newly created chimeric fragments (e.g., biotin pull down if ligation junctions are biotinylated) the resulting fragments are then quantified using PCR, microarrays or high throughput DNA sequencing [7–13]. Based on the number of interrogated genomic loci and enrichment of proteins, the conformation capture methods are subdivided into different categories such as 3C (one-to-one), 4C (one-to-all), 5C (many-to-many), Hi-C (all-to-all), ChIA-PET and HiChIP/PLAC-seq. In a typical 3C experiment, a pair of interacting genomic loci can be identified using PCR by targeting two known genomic loci with specific primers [14]. 4C experiments profile all interactions from a single genomic anchor point (i.e., bait region) [15–17]. Hi-C, on the other hand, measures all possible intra- and inter-chromosomal interactions in the genome in a single experiment [7]. In addition, finding genomic interactions in conjugate with different mediator proteins, especially that are associated with histones and transcription factors, several techniques have been developed adopting the chromatin immunoprecipitation (ChIP)-based enrichment methods [18–20]. Such methods provide high-resolution information about the presence of DNA-binding proteins on 3D organization of the genome and their implications on gene regulation. ChIA-PET was the first of such techniques [18], however, two other recent methods HiChIP [19] and PLAC-seq [20] overcome important limitations of ChIA-PET by reducing the input material requirement (i.e., number of cells) and increasing the sensitivity and robustness of the genome-wide interaction yield.
Since the advent of the chromosome conformation capture technology, our understanding of the human genome 3D organization has grown rapidly and we now know that human interphase chromosomes are folded into multiple layers of hierarchical structures (Figure 1) and each layer can play a critical role in transcriptional regulation [21, 22]. Alterations in any one of these finely-tuned layers can lead to unwanted cascade of molecular events and ultimately drive the manifestation of diseases and phenotypes (Figure 1, Table 1). Some examples include emergence or dissolution of compartments and chromosomal territories seen in several cancers (e.g. chromosomal translocations in breast cancer and prostate cancer) [23], dynamic arrangement of topologically associated domains (i.e., TADs) in some inherited diseases (e.g. F-syndrome, sex reversal) [24, 25], and formation or disappearance of chromatin-loops between enhancers and promoters in several types of cancers and other diseases (e.g. T cell acute lymphoblastic leukemia/T-ALL, asthma, heart-diseases) [26–28]. All these intricate layers of genome organization (e.g. chromatin loops, TAD arrangement, compartments) can be affected by changes in the DNA sequence or in the epigenetic landscape such as presence of single nucleotide polymorphism (SNPs) or mutations in enhancers and promoters, deletions, amplifications and translocations of genomic regions and binding of transcription factors. Such scenarios are increasingly evident from several recent studies where a strong relation is found between the changes in spatial organization of human genome due to chromosomal abrogation and the dysregulation of diseases related genes with intact DNA sequences. Our cumulative knowledge to perceive the missing links to connect human diseases with genome organization is ever increasing and summarizing the current understanding is of great importance to grasp the full picture and pave the way for the next wave of studies. There are already a number of excellent reviews that cover some of the published links between genome organization and diseases [21, 22, 29–31]. In this review, we take a top-down approach to discuss different layers of human genome organization, most of which are fully characterized only within the past decade, and we summarize the diseases or phenotypes linked to changes in the delicate fabric of the 3D genome architecture at each layer. We provide an up-to-date view of the literature and discuss potential future directions that are likely to transform the studies of 3D genome organization from discovery to translation.
Figure 1.
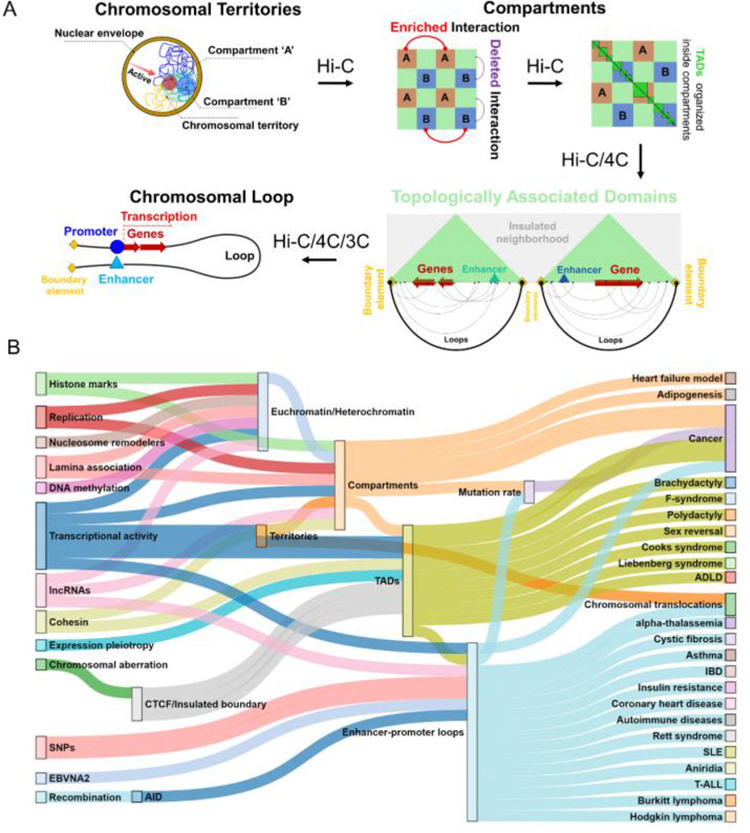
Panel A shows the overall hierarchical genome organization of human genome. Panel B shows the link between different genetic and epigenetic factors (left) and different human diseases (right) mediated through changes in different layers of the genome organization (mid).
Table 1.
The features of different layers of genome organization and diseases or phenotypes that are associated with changes in the corresponding layer. The last column summarizes possible mechanism/information about each listed 3D genome feature – disease link.
| Organizational Layer |
Features of the Layer | Associated Diseases/Phenotypes |
Mechanism/Available Information | |
|---|---|---|---|---|
| Chromosomal Territories | 1. Each chromosome in human genome occupies its own territory 2. Territories intermingle at inter-chromatin domains that are enriched in active genes 3. Territories play role in transcriptional activity and preferential positioning of loci in the nucleus |
Possibly various cancers [23] |
• Territories are dynamic in nature and intermingle significantly with each other • Intermingling between different chromosomes correlate with translocation frequencies in human cells |
|
| Compartments | A | 1. High gene density 2. Active histone marks 3. Early replication 4. Open chromatin 5. Low lamina association 6. High transcriptional activity 7. Low DNA methylation |
Breast cancer [47] |
• MCF-7 showed 12% compartment switching • Small chromosomes showed B to A transition and altered transcriptional activity • Activation of oncogenic pathways |
| Regional Variation in Mutational Rate (RViMR) [48, 50, 54] |
• Hetero/Euchromatin compartments play a major role and account for 55% of variation in mutational rates • Elevated somatic mutation in B compared to the A compartment in prostate cancer cell lines |
|||
| B | Opposite features compared to ‘A’ |
|||
| 1. Highly dynamic in nature and changes occur in accordance with lineage and cell- type specificity 2. Cohesin complex important for the formation and maintenance | ||||
| Adipogenesis [66] |
• lncRNA Firre plays a key role in adipogenesis • Firre forms a nuclear compartment with five distinct trans-chromosomal loci important for its function |
|||
| Heart failure model [57] |
• Altered transcriptional activity due to change in compartments |
|||
| Topologically Associated Domains | 1. Enriched intra-domain contact frequency compared to inter-domain 2. Highly dynamic in nature and changes occur in accordance with lineage and cell- type specificity 3. Boundaries are enriched in motifs and binding of insulator proteins (e.g. CTCF), transcriptional activity, housekeeping genes and SINE elements 4. Considered as a regulatory unit for transcriptional activity 5. CTCF and cohesin are two important factors in forming and regulating the TAD structure |
Brachydactyly [24] |
• Deletion of EPHA4 gene and its upstream eliminates TAD boundary • EPHA4 associated limb-enhancer now interacts with PAX3 genes and drives misexpression of PAX3 |
|
| F-syndrome [24] |
• In case of inversion, the IHH gene and TAD boundary including the limb-enhancer of EPHA4 gene gets inverted misplacing the enhancer close to WNT6 gene causing misexpression • In duplication, the WNT6, IHH and TAD boundary including the limb-enhancer is duplicated, which misplaces WNT6 gene closer to enhancer element causing misexpression |
|||
| Polydactyly [24] |
• Downstream deletion of EPHA4 TAD boundary causes reduction in linear distance between the limb-enhancer element and WNT6, IHH genes. This causes ectopic interactions between enhancer element and the said genes causing mis-expression |
|||
| Sex reversal [25] |
• An intra-TAD duplication event. • The duplication increases the interaction propensity among regulatory elements causing ectopic expression of SOX9 gene |
|||
| Cooks syndrome [25] |
• Inter-TAD duplication event. • The duplication results in formation of a new TAD involving KCNJ genes and regulatory elements previously associated with SOX9 gene. The new arrangement causes misexpression of KCNJ genes |
|||
| Liebenberg syndrome [82] |
• H1AFY gene upstream of PITX1 gene is an insulator element encoding gene and helps to insulate PITX1 from its neighborhood • PITX1 interacts with enhancer element from adjacent TAD upon H1AFy deletion causing ectopic expression of PITX1 |
|||
| Autosomal-dominant adult-onset demyelinating leukodystrophy (ADLD) [83] |
• Deletion upstream of Lamin B1 gene eliminates the TAD boundary causing an ectopic interaction between two merged TADs along with three enhancers with the Lamin B1 gene promoter |
|||
| T cell acute lymphoblastic leukemia (T-ALL) [26] |
• Microdeletions at CTCF boundaries eliminates insulated neighborhood containing proto-oncogenes causing aberrant expression of proto-oncogenes through ectopic interactions |
|||
| Chromosomal Loops | 1. Proximal in three-dimensional space as compared to their linear intervening nucleotide sequences 2. Can be of two types: Structural loops (e.g. CTCF-CTCF interaction to form TADs) and Functional loops (e.g. Enhancer-promoter loops to drive the expression of a gene) 3. Cell type and lineage specific interactions |
α-thalassemia [93] |
• Single nucleotide polymorphism creates a new enhancer-promoter link between alpha-globin genes leading to altered transcription initiation and reduced alpha-globin expression |
|
| Cystic Fibrosis [94] |
• Cystic fibrosis transmembrane conductance regulator (CFTR) gene regulate its own expression via a loop connecting the intronic-enhancer to the CFTR promoter • Common mutations abolish this intronic enhancer- promoter interaction leading to a reduced expression profile of CFTR gene |
|||
| Asthma [27] |
• SNPs overlap with immune cell enhancers. • CTCF binding sites are altered by SNPs • SNPs modify long-range ORMDL3 promoter- enhancer interaction |
|||
| Inflammatory bowel disease (IBD) [95] |
• Noteworthy IBD candidate genes shown to interact with enhancer region • eQTL level alters the interaction |
|||
| Insulin resistance, T2D, Coronary heart disease [28] |
• IRS1 enhancer harbours SNP causing more ectopic interaction with IRS1 promoter |
|||
| Autoimmune diseases [96] |
• SNPs associated to different autoimmune diseases physically interact with each other and regulate the associated genes with differing enhancer mechanisms |
|||
| Cardiac rhythm disorder [97] |
• SNP within SCN10A gene interacts with SCN5A promoter and alters expression |
|||
| Rett syndrome [98] |
• Mutation in MECP2 gene shown to abolish 11Kb chromatin loop around Dlx5/6 locus • Dlx5 regulates production of enzymes that synthesize gamma-aminobutyric acid (GABA). Absence of the loop in Dlx5/6 locus shown to alter GABAergic neuron activity in individuals |
|||
| Myeloproliferative disorders [99] |
• JAK2 gene promoter interacts with an enhancer 222kb away. The enhancer has a DHS peak and harbor a SNP. Presence of SNP within the DHS peak alters the transcription factor binding property of that region and thus causes reduced interaction with JAK2 promoter |
|||
| Systemic lupus erythematosus (SLE) [100] |
• Normal alleles control TNFAIP3 transcription through a long-range enhancer element and subsequent stimulation of A20 gene. • In risk allele the long-range enhancer-promoter interaction is hampered thus impaired expression of A20 takes place |
|||
| Aniridia [101] |
• Mutation in ultra-conserved PAX6 enhancer disrupts autoregulatory feedback loop |
|||
| Colorectal, Prostate and Breast cancer [103–106] |
• SNPs resides within the regulatory elements and/or enriched in transcriptional factor binding motifs in the non-coding region of the genome and exerts effects through long-range chromosomal interactions |
|||
| T cell acute lymphoblastic leukemia (T-ALL) [110] |
• IGF1R gene is activated by an aberrant interaction between LUNAR1 and the enhancer of IGF1R gene through Notch signaling. IGF1R plays a key role in the survival of the cancer cells |
|||
| Induced mutations and translocations [112] |
• Activation-induced cytidine deaminase (AID) target genes with specific features including super- enhancers and can physically interact with those genes to promote mutations and translocations |
|||
| Epstein-Barr virus (EBV) infection related cancers [113] |
• EBV nuclear antigen 2 exploits the transcriptional machinery of RCLs to its own favor by depleting repressive histone marks upstream of MYC gene and up-regulates MYC expression in RCLs though long-range enhancer-promoter interaction |
|||
Chromosomal Territories, Nuclear Compartments and Diseases
The concept of chromosomes occupying a defined space in the nucleus is as old as the study of chromosomes itself. The evidence of a territorial organization of chromosomes in animal cells was first furnished by Carl Rabl in 1885 [32] and since then numerous other studies [33–37] has backed this structural organization. Studies also demonstrated that chromosomal territories are segregated by inter-chromatin domain and according to this model the inter-chromatin domains are enriched in active genes and thus play a role in gene regulation [38]. Cremer & Cremer, 2001 [39] provided a good overview on the relationship between chromosomal territories and gene expression regulation mechanism. In general, chromosomal territories of different chromosomes exhibit several common features such as the preferential positioning in the nucleus depending on their genomic features [40], conserved nuclear localization after mitosis [41] and radial positioning that is correlated with the cell-type specific lamin association, replication timing and transcriptional activity [42, 43]. Further studies have also shown that these territories are dynamic in nature and shows a strong correlation between the frequency of chromosomal translocations and spatial proximity among them [23, 44]. Gene transcriptional machinery at the intermingling regions is also found to be a key factor in regulating the extent and the shape of the territories in a cell type-specific manner [23].
Analysis of the Hi-C data further refined these large-scale chromosomal territories into two sets of compartments. These compartments appear as a plaid, checkerboard-like pattern when a chromosomal Hi-C matrix is first transformed to an observed/expected ratio then to a correlation matrix and can be separated into two sets of megabase-sized regions called “A” and “B” compartments by eigenvalue decomposition [7]. The regions within a given compartment are enriched for interactions within the same compartment and depleted for those that are across. Compartment A is enriched in gene density, transcriptional activity, H3K36me3, early replication and higher DNA accessibility (e.g. DNAse), which all suggest an open chromatin state for the genomic regions in this category [7]. Compartment B regions are associated with lamina-associated domains, low transcriptional activity, late replication and, hence, are enriched in heterochromatin [7]. Compartments are highly dynamic in nature and changes occur in accordance with lineage and cell-type specificity. Recent studies suggest that about 36% and 59.6% of the genome is dynamically compartmentalized during stem cell differentiation and across different primary tissues, respectively [45, 46]. Altered compartmentalization has also been reported in cancer cells. A recent study from Barutcu et. al [47] comparing normal breast cells (MCF-10A) and its cancerous counterpart MCF-7 showed a homogeneous switching of 12% compartments. Even though the normal and cancerous cells showed a similar distribution of open (A) and closed (B) compartments across the genome, a significant number of B to A compartment transitions are observed within small chromosomes (chromosome 16–22) suggesting an overall increased open conformation in small chromosomes. RNAseq analysis from the same study revealed an altered gene expression pattern across the small chromosomes in MCF-7, related to the activation of oncogenic pathways such as WNT signaling. Previous studies have also demonstrated that chromatin organization plays a major role on mutational rates in human cancer cells [48]. It has long been known that the mutation rate varies over different scales across the genome with much of this variation largely unexplained [49]. This phenomenon of regional variation in mutation rates, RViMR, has been demonstrated for base-substitutions [50], small-indels [49, 51] and transposable elements [52]. Different genomic features such as GC content, SNP density, recombination hot spots and CpG islands have been shown to contribute towards RViMR [53]. Further studies performed by Schuster-Bockler and Lehner [48] showed that arrangement of the genome into heterochromatin and euchromatin like domains plays a major influence on regional mutation rate variation in human cancer cells and can account for 55% of variations observed across different cancer types. In another relevant study, Fortin and Hansen [54] demonstrated a relationship between A/B compartments and somatic mutations in prostate cancer cell lines. The analysis reconfirmed that similar to other cancer types, prostate cancers showed an elevated somatic mutation rate in closed or B compartments than in A. Compelling evidence [55] also suggests that replication timing, which is highly correlated with the organization of compartments [56]; is an effective predictor of mutational rate and subsequent somatic copy-number variations in several cancer types. Alteration in genomic compartmentalization are now being studied in other diseases such as heart failure models in mice [57]. Along with other findings, the Hi-C analysis also revealed an altered A/B compartmentalization with altered boundary strength upon CTCF depletion [57, 58].
Compartmentalization of the genome and different epigenetic mechanisms, which regulate the formation and stabilization these open and closed compartments play a dominant role in human disease. Few notable such epigenetic mechanisms include nucleosome remodelers [59], histone variants [60], post-translational modification of histones [61] and DNA methylation [62]. Interestingly, emerging evidence suggests that some long-noncoding RNAs (lncRNAs) act to establish genomic compartments [63, 64] and can shape the structure of chromosomes [10, 65]. A recent study by Hacisuleyman et. al. [66] showed that a lncRNA, Firre, which plays a key role in adipogenesis plays a role in forming a nuclear compartment with five distinct trans-chromosomal loci spatially proximal to its own loci on chromosome X and in modulating nuclear architecture across chromosomes. There is a plethora of other lncRNAs that are found to be associated with different diseases [67–70], and their roles in context to genomic architecture still needs to be explored.
Topologically Associated Domains and Diseases
In the study of human genome organization from Hi-C, topologically associated domains (TADs) emerged as a fundamental unit of the chromosomal structure since their first description in 2012 [71, 72]. These studies showed that human genome is segregated into megabase-sized topological domains and the genomic regions within a TAD have enriched intra-domain contact frequency compared to inter-domain. Although this spatial organization is found to be a general property of the interphase chromatin across different cell types in both human and mouse, further studies have suggested that TADs are not simple stable loops that are formed between two permanent genomic loci, rather they are dynamic in nature [10]. While mechanisms of TAD formation are still an open question, few studies have put forth some ideas to describe TAD formation including “strings and binders switch” [73], chromosomal supercoiling [74], block copolymer model [75] and loop extrusion model [76, 77]. Notably, the loop extrusion model is found to be generalizable at a genome-wide scale and it recapitulates experimental results in-silico by considering two important players, CTCF and cohesin, which have long been known to be critical in establishing and maintaining chromatin structure. In this model, the cis-acting loop-extruding factors such as cohesin rings form progressively larger loops until they are stalled by TAD boundary elements such as CTCF. More recently, Ganji et al provided an unambiguous evidence for Condensin-induced loop extrusion mechanism by directly visualizing the formation and processive extension of DNA loops in real time [78]. Irrespective of the TAD formation model, the importance of CTCF and cohesin in maintaining the boundaries and stable architecture of TADs are well-established [30]. CTCF, a highly conserved 11-zinc finger insulator protein that binds to a non-palindromic DNA motif and cohesin, a ring-like multiprotein complex that plays a role in DNA repair and segregation, are known to co-occupy different binding sites and CTCF has been shown to help cohesin in positioning to its target region [79]. Both are found to be very strongly enriched at the TAD boundaries [10, 30, 72] and depletion of either one has a significant impact on TAD organization and strength[58, 80].
The biological importance of TADs is highlighted by their potential to regulate the gene pleiotropy as suggested by studies performed on Hox locus [81] where TADs are found to evolve in multiple tissues with differences in enhancer-promoter interactions, which ultimately control the expression of genes at that locus. The relevance of TADs in human diseases has recently been shown also for the Epha4 locus [24]. Epha4 is an ephrin receptor and is involved in the formation of tissue boundaries and segmentation. Distinct structural variants involving a 1.7–1.9Mb heterozygous deletion, 1.1 Mb heterozygous inversion/or 1.4 Mb heterozygous duplication, and a 900 Kb duplication around this locus are associated with different types of limb malformations in human, namely Brachydactyly, F-syndrome, and Polydactyly diseases, respectively [24]. Lupianez et. al. [24] in their detailed study showed that all the disease-related structural aberrations disrupt the native TAD boundaries associated with this locus; resulting in altered promoter - limb-enhancer interactions and thus disrupts the normal Epha4 transcription regulation dysregulates expression profile of surrounding genes. In a separate study, Framke and Ibrahim et. al. [25] showed how genomic duplication events can affect TAD formation and lead to human disease etiology. They focused on the developmental transcription factor Sox9 locus and this locus contains two TADs, one including the Sox9 and the other includes potassium channel genes Kcnj2 and Kcnj16. The authors found that a duplication event within Sox9 TAD (intra-TAD duplication) leads to increased interaction propensity among the regulatory elements of Sox9 gene and in-effect causing female-to-male sex reversal phenotype, while an inter-TAD duplication resulted in a neo-TAD with new set of regulatory elements encompassing the Kcnj2 and Kcnj16 genes leads to Cooks syndrome, a limb malformation disease in humans. A third inter-TAD duplication event that extended within the Kcnj2 and Kcnj16 TAD but not the gene body did not show any phenotype. The results demonstrated how duplication events can affect higher-order genome structure and impact of TADs on regulating gene expression profiles. Liebenberg syndrome, in which arms of a patient acquire morphological characteristic similar to legs, linked to the deletion of H1afy gene 300 Kb upstream of Pitx1 gene. Pitx1 determines hindlimb identity and deregulation of Pitx1 in mice is associated with forelimb to hindlimb conversion. H1afy is a boundary element and in the normal scenario this helps to insulate the Pitx1 TAD from the neighborhood but when deleted an enhancer from adjacent TAD interacts with Pitx1 gene and thus misregulation of Pitx1 takes place causing Liebenberg syndrome [82]. In autosomal-dominant Adult-onset Demyelinating Leukodystrophy (ADLD) characterized by progressive demyelination of central nervous system through overexpression of Lamin B1 gene is also found to be linked to TAD boundary disruption. A deletion upstream of Lamin B1 gene eliminates the TAD boundary causing an ectopic interaction between two merged TADs along with three enhancers with the Lamin B1 gene promoter [83]. Importance of insulated neighborhood in the progression of cancer through activation of proto-oncogenes has also been studied [26, 84]. In one of such studies Hnisz and Weintraub et. al. [26] showed that in T-ALL cancer genome microdeletions are enriched at the CTCF boundary sites, thereby eliminates insulated neighborhood containing important T-ALL proto-oncogenes. With the loss of insulated neighborhood, aberrant activation of proto-oncogenes is possible via distal enhancer-promoter interactions normally located outside the neighborhood. The results from all these chromosome conformation capture studies confirm that TADs are a fundamental regulatory unit of our genome, which when disrupted lead to dysregulation and in turn disease phenotypes.
Chromosomal Loops and Diseases
Regulatory elements such as enhancers and insulators play a crucial role in controlling the gene expression profile of a cell in a context-dependent manner. Many human genes are regulated by at least one regulatory element positioned upstream or downstream relative to the gene promoter that influences the promoters transcriptional state. The detailed analysis of high-resolution chromosome conformation capture data strongly suggests the presence of regulatory chromosomal loops as a means of communication among the local and/or distal regulatory elements with promoters to control gene expression [28, 85]. A chromatin loop by definition is two genomic loci that are physically closer in the nucleus than their intervening sequences. Early studies used the 3C technique to investigate erythroid-specific mouse beta-globin gene and showed that its locus control regions (LCR) situated 50Kb apart is closer to the gene in proximity in fetal liver cells but not in fetal brain cells correlated with liver specific expression of beta-globin [86]. A classic example of long-range gene regulation involves the Shh gene, expression of which is regulated by a ~1Mb away enhancer element [87]. A more recent study of a GWAS SNP related to multiple vascular diseases clearly demonstrated the problems with “nearest gene” approaches by linking this putative causal variant to a more than 600kb away gene (EDN1) rather than the nearest gene that harbored the SNP in its intronic region (PHACTR1) [88]. Another recent study using super-resolution Hi-C data in human cells again provided compelling evidence on the importance of loops in regulating the gene expression through frequently linking the enhancer-promoter regions and in maintaining the TAD boundaries [10]. The mechanism of such loop formation is now gradually accepted [78] and through several loss-of-function experimental approaches it is now inferred that transcriptional factors like GAGA [89], GATA-1 [90], STAT6 [91] and architectural proteins especially CTCF, BEAF-32, Elba and Ibf1/2 helps to mediate and maintain long-range interactions across the genome [92]. As stated earlier in the previous section; the CTCF-cohesin based extrusion mechanism of TAD formation is also a leading model to describe how the chromosome loops may form in the genome [76, 77].
With the involvement of such large number of crucial transcription factors, chromatin modulators and their role in precise regulation in gene expression profile, it is not surprising that chromosomal loops are also found to play a dominant role in different human diseases. Early studies showed that illegitimate gene looping can contribute to a rare form of blood disorder alpha-thalassemia [93]. This study identified that in the diseased condition a gain-of-function SNP creates a new enhancer-promoter link between alpha-globin genes, leading to altered transcription initiation and reduced alpha-globin expression. In a separate study by Ott et. al. [94] investigating Cystic Fibrosis related markers, the authors showed that intronic enhancers within the cystic fibrosis transmembrane conductance regulator (CFTR) gene regulate its own expression via a loop connecting the intronic-enhancer to the CFTR promoter. Common mutations causing cystic fibrosis is found to abolish this intronic enhancer-promoter interaction leading to a reduced expression profile of CFTR gene in diseased condition. Further studies also demonstrated that a plethora of human diseases follow a similar mode of etiology where a SNP eliminates an enhancer-promoter loop, leading to mis-expression of a gene or a set of genes contributing to the development of the disease. Notable example includes Asthma [27], Inflammatory bowel disease [95], Insulin resistance, T2D, Coronary heart disease [28], autoimmune diseases [96], Cardiac rhythm disorder [97], Rett syndrome [98], Myeloproliferative disorders [99], systemic lupus erythematosus [100] and Aniridia [101]. In all of these referred human diseases, at least one enhancer-promoter interaction is lost due to a presence of a SNP either at the enhancer side or at the promoter loci causing an altered expression of the corresponding gene(s). The chromatin interaction model also offers an explanation to the effects of diseases-associated variants residing in the non-coding part of the genome. Combining chromosome conformation capture techniques with genome-wide association studies (GWAS) holds a great potential to identify new putative target genes/pathways for potentially causal SNPs/variants or revise existing connections made based on the nearest gene principle, thereby, significantly improving our understanding of human diseases including cancer.
A large number of common genetic variants in the human genome are associated with cancers and extensive studies have been performed nearly on all types of malignant cancers revealing novel target genes and pathways involved in carcinogenesis [102]. Recent studies involving capture Hi-C [103] and 3C techniques [104] in colorectal cancer risk loci also showed that most of the disease associated SNPs reside within the regulatory elements and/or enriched in transcriptional factor binding motifs in the non-coding region of the genome and exerts effects through long-range chromosomal interactions. Similar effects are also shown in cases of prostate cancer [105], breast cancer [106, 107], and in multiple other cancer systems [108]. Alteration of such 3D organization of regulatory elements has been suggested to drive the cancer development [109] and accumulating evidence suggests that chromosomal interactions also play a key role in the development of leukemia. The survival of T-ALL cells is dependent on an insulin-like growth factor receptor encoded by IGF1R gene and in T-ALL the IGF1R gene is found to be activated by an aberrant interaction between LUNAR1 (leukemia-induced noncoding activator RNA) and the enhancer of IGF1R gene [110] mediated through Notch signaling. In a separate example, it has been shown that AID (Activation-Induced cytidine Deaminase), a B-cell specific enzyme which helps in somatic hypermutation and class-switch recombination of immunoglobulin genes in mature B-cells, also induces mutations and translocations in genes not related to immunoglobulin class [111]. Recent study by Qian et al. showed that AID targeted genes share specific features including super-enhancers and multiple target genes that are megabases apart can physically interact with each other to promote mutations and translocations that are frequently observed in B cell lymphomas [112]. Another interesting case relates to Epstein-Barr virus (EBV) infection, which is prevalent in the human population and is a primary cause of Burkitt lymphoma, Hodgkin lymphoma development trough converting resting B lymphocytes (RBLs) to proliferating lymphoblastoid cells (LCLs). The conversion of RCLs to LCLs is mediated via EBV nuclear antigen 2 (EBVNA2) which upregulates MYC levels in RCLs. Zhao and Zou et al. [113] showed that EBV and particularly EBVNA2 evolved to exploit the transcriptional machinery of RCLs to its favor by depleting repressive histone marks upstream of MYC gene and thereby up-regulating MYC expression in RCLs though long-range enhancer-promoter interaction.
Future Directions
Chromosome conformation techniques have vastly improved our understanding towards the organization of the genome and further improvement in the methodology such as the ongoing development in the protocol to integrate the conjugate protein information with fewer input cells but better upshot in the resolution and throughput will be crucial to understand the biomolecular complexes that facilitate the interaction events. Acknowledging the current limitations to amalgamate the interaction data from other independent functional genomics experiments such as DNA-replication, DNA accessibility, RNA-seq, DNA-methylation and proteomics will provide us with better understanding of the structure-function relationship of disease-associated structural variants to the progress and subsequent pathology of the disease. Although common to many other sequencing-based investigations, most of our knowledge of genome organization is derived from the cell lines, which necessitates further analysis in primary cells coupled with relevant clinical information such as genotyping and copy-number variation (CNV). Studies of patient samples from large disease cohorts will be crucial in identifying the key factors controlling the integrity of the genome organization, which is likely disrupted in many diseases. Results from such studies will be of immense importance in addressing one of the long-standing challenging questions in epigenetic studies about the chromosomal changes that drive progression of cancer. Recent development in the CRISPR-Cas9 system provides an efficient molecular machinery to generate large-scale genomic deletions and other types of chromosomal rearrangements in a well-controlled biological system. The Chromatin Loop Reorganization using CRISPR-dCas9 or CLOuD9 system [114] now lets researcher form de-novo loops between specific genomic loci and provides a powerful hypothesis-driven framework to understand the role of genome organization in gene regulation, rather than depending on observational approaches. As we confront new challenges in biology, new tools such as CRISPR, CLOuD9 or other advanced synthetic biology techniques that allow us to control biochemical modifications of chromatin will be particularly important in characterizing molecular roles of factors that control several layers of the 3D genome architecture.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Jiang YH, Bressler J, Beaudet AL, Epigenetics and human disease, Annu Rev Genomics Hum Genet 5 (2004) 479–510. [DOI] [PubMed] [Google Scholar]
- [2].Brookes E, Shi Y, Diverse epigenetic mechanisms of human disease, Annu Rev Genet 48 (2014) 237–68. [DOI] [PubMed] [Google Scholar]
- [3].Li G, Zhu P, Structure and organization of chromatin fiber in the nucleus, FEBS Lett 589(20 Pt A) (2015) 2893–904. [DOI] [PubMed] [Google Scholar]
- [4].Kim TK, Shiekhattar R, Architectural and Functional Commonalities between Enhancers and Promoters, Cell 162(5) (2015) 948–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pennacchio LA, Bickmore W, Dean A, Nobrega MA, Bejerano G, Enhancers: five essential questions, Nat Rev Genet 14(4) (2013) 288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Casal J, Leptin M, Identification of novel genes in Drosophila reveals the complex regulation of early gene activity in the mesoderm, Proc Natl Acad Sci U S A 93(19) (1996) 10327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J, Comprehensive mapping of long-range interactions reveals folding principles of the human genome, Science 326(5950) (2009) 289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, Noble WS, A three-dimensional model of the yeast genome, Nature 465(7296) (2010) 363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kalhor R, Tjong H, Jayathilaka N, Alber F, Chen L, Genome architectures revealed by tethered chromosome conformation capture and population-based modeling, Nat Biotechnol 30(1) (2011) 90–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL, A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping, Cell 159(7) (2014) 1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P, Single-cell Hi-C reveals cell-to-cell variability in chromosome structure, Nature 502(7469) (2013) 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G, Three-dimensional folding and functional organization principles of the Drosophila genome, Cell 148(3) (2012) 458–72. [DOI] [PubMed] [Google Scholar]
- [13].Ay F, Vu TH, Zeitz MJ, Varoquaux N, Carette JE, Vert JP, Hoffman AR, Noble WS, Identifying multi-locus chromatin contacts in human cells using tethered multiple 3C, BMC Genomics 16 (2015) 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dekker J, Rippe K, Dekker M, Kleckner N, Capturing chromosome conformation, Science 295(5558) (2002) 1306–11. [DOI] [PubMed] [Google Scholar]
- [15].Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W, Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C), Nat Genet 38(11) (2006) 1348–54. [DOI] [PubMed] [Google Scholar]
- [16].Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, Pant V, Tiwari V, Kurukuti S, Ohlsson R, Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions, Nat Genet 38(11) (2006) 1341–7. [DOI] [PubMed] [Google Scholar]
- [17].van de Werken HJ, Landan G, Holwerda SJ, Hoichman M, Klous P, Chachik R, Splinter E, Valdes-Quezada C, Oz Y, Bouwman BA, Verstegen MJ, de Wit E, Tanay A, de Laat W, Robust 4C-seq data analysis to screen for regulatory DNA interactions, Nat Methods 9(10) (2012) 969–72. [DOI] [PubMed] [Google Scholar]
- [18].Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, Chew EG, Huang PY, Welboren WJ, Han Y, Ooi HS, Ariyaratne PN, Vega VB, Luo Y, Tan PY, Choy PY, Wansa KD, Zhao B, Lim KS, Leow SC, Yow JS, Joseph R, Li H, Desai KV, Thomsen JS, Lee YK, Karuturi RK, Herve T, Bourque G, Stunnenberg HG, Ruan X, Cacheux-Rataboul V, Sung WK, Liu ET, Wei CL, Cheung E, Ruan Y, An oestrogen-receptor-alpha-bound human chromatin interactome, Nature 462(7269) (2009) 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, Chang HY, HiChIP: efficient and sensitive analysis of protein-directed genome architecture, Nat Methods 13(11) (2016) 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD, Ren B, Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq, Cell Res 26(12) (2016) 1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Babu D, Fullwood MJ, 3D genome organization in health and disease: emerging opportunities in cancer translational medicine, Nucleus 6(5) (2015) 382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Krijger PH, de Laat W, Regulation of disease-associated gene expression in the 3D genome, Nat Rev Mol Cell Biol 17(12) (2016) 771–782. [DOI] [PubMed] [Google Scholar]
- [23].Branco MR, Pombo A, Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations, PLoS Biol 4(5) (2006) e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A, Mundlos S, Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions, Cell 161(5) (2015) 1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schopflin R, Kraft K, Kempfer R, Jerkovic I, Chan WL, Spielmann M, Timmermann B, Wittler L, Kurth I, Cambiaso P, Zuffardi O, Houge G, Lambie L, Brancati F, Pombo A, Vingron M, Spitz F, Mundlos S, Formation of new chromatin domains determines pathogenicity of genomic duplications, Nature 538(7624) (2016) 265–269. [DOI] [PubMed] [Google Scholar]
- [26].Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, Reddy J, Borges-Rivera D, Lee TI, Jaenisch R, Porteus MH, Dekker J, Young RA, Activation of proto-oncogenes by disruption of chromosome neighborhoods, Science 351(6280) (2016) 1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Schmiedel BJ, Seumois G, Samaniego-Castruita D, Cayford J, Schulten V, Chavez L, Ay F, Sette A, Peters B, Vijayanand P, 17q21 asthma-risk variants switch CTCF binding and regulate IL-2 production by T cells, Nat Commun 7 (2016) 13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, Sim HS, Peh SQ, Mulawadi FH, Ong CT, Orlov YL, Hong S, Zhang Z, Landt S, Raha D, Euskirchen G, Wei CL, Ge W, Wang H, Davis C, Fisher-Aylor KI, Mortazavi A, Gerstein M, Gingeras T, Wold B, Sun Y, Fullwood MJ, Cheung E, Liu E, Sung WK, Snyder M, Ruan Y, Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation, Cell 148(1–2) (2012) 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rajarajan P, Gil SE, Brennand KJ, Akbarian S, Spatial genome organization and cognition, Nat Rev Neurosci 17(11) (2016) 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Merkenschlager M, Nora EP, CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation, Annu Rev Genomics Hum Genet 17 (2016) 17–43. [DOI] [PubMed] [Google Scholar]
- [31].Bickmore WA, The spatial organization of the human genome, Annu Rev Genomics Hum Genet 14 (2013) 67–84. [DOI] [PubMed] [Google Scholar]
- [32].Rabl C, über Zellteilung, Morph Jb (10) (1885) 214–330. [Google Scholar]
- [33].Cremer T, Kurz A, Zirbel R, Dietzel S, Rinke B, Schrock E, Speicher MR, Mathieu U, Jauch A, Emmerich P, Scherthan H, Ried T, Cremer C, Lichter P, Role of chromosome territories in the functional compartmentalization of the cell nucleus, Cold Spring Harb Symp Quant Biol 58 (1993) 777–92. [DOI] [PubMed] [Google Scholar]
- [34].Manuelidis L, A view of interphase chromosomes, Science 250(4987) (1990) 1533–40. [DOI] [PubMed] [Google Scholar]
- [35].Boyle S, Gilchrist S, Bridger JM, Mahy NL, Ellis JA, Bickmore WA, The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells, Hum Mol Genet 10(3) (2001) 211–9. [DOI] [PubMed] [Google Scholar]
- [36].Albiez H, Cremer M, Tiberi C, Vecchio L, Schermelleh L, Dittrich S, Kupper K, Joffe B, Thormeyer T, von Hase J, Yang S, Rohr K, Leonhardt H, Solovei I, Cremer C, Fakan S, Cremer T, Chromatin domains and the interchromatin compartment form structurally defined and functionally interacting nuclear networks, Chromosome Res 14(7) (2006) 707–33. [DOI] [PubMed] [Google Scholar]
- [37].Cremer T, Cremer M, Chromosome territories, Cold Spring Harb Perspect Biol 2(3) (2010) a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cremer T, Kreth G, Koester H, Fink RH, Heintzmann R, Cremer M, Solovei I, Zink D, Cremer C, Chromosome territories, interchromatin domain compartment, and nuclear matrix: an integrated view of the functional nuclear architecture, Crit Rev Eukaryot Gene Expr 10(2) (2000) 179–212. [PubMed] [Google Scholar]
- [39].Cremer T, Cremer C, Chromosome territories, nuclear architecture and gene regulation in mammalian cells, Nat Rev Genet 2(4) (2001) 292–301. [DOI] [PubMed] [Google Scholar]
- [40].Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA, Differences in the localization and morphology of chromosomes in the human nucleus, J Cell Biol 145(6) (1999) 1119–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Parada LA, Roix JJ, Misteli T, An uncertainty principle in chromosome positioning, Trends Cell Biol 13(8) (2003) 393–6. [DOI] [PubMed] [Google Scholar]
- [42].Takizawa T, Meaburn KJ, Misteli T, The meaning of gene positioning, Cell 135(1) (2008) 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].van Steensel B, Belmont AS, Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression, Cell 169(5) (2017) 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Roukos V, Burman B, Misteli T, The cellular etiology of chromosome translocations, Curr Opin Cell Biol 25(3) (2013) 357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, Diao Y, Liang J, Zhao H, Lobanenkov VV, Ecker JR, Thomson JA, Ren B, Chromatin architecture reorganization during stem cell differentiation, Nature 518(7539) (2015) 331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schmitt AD, Hu M, Jung I, Xu Z, Qiu Y, Tan CL, Li Y, Lin S, Lin Y, Barr CL, Ren B, A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome, Cell Rep 17(8) (2016) 2042–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Barutcu AR, Lajoie BR, McCord RP, Tye CE, Hong D, Messier TL, Browne G, van Wijnen AJ, Lian JB, Stein JL, Dekker J, Imbalzano AN, Stein GS, Chromatin interaction analysis reveals changes in small chromosome and telomere clustering between epithelial and breast cancer cells, Genome Biol 16 (2015) 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schuster-Bockler B, Lehner B, Chromatin organization is a major influence on regional mutation rates in human cancer cells, Nature 488(7412) (2012) 504–7. [DOI] [PubMed] [Google Scholar]
- [49].Hodgkinson A, Eyre-Walker A, Variation in the mutation rate across mammalian genomes, Nat Rev Genet 12(11) (2011) 756–66. [DOI] [PubMed] [Google Scholar]
- [50].Ellegren H, Smith NG, Webster MT, Mutation rate variation in the mammalian genome, Curr Opin Genet Dev 13(6) (2003) 562–8. [DOI] [PubMed] [Google Scholar]
- [51].Chiaromonte F, Yang S, Elnitski L, Yap VB, Miller W, Hardison RC, Association between divergence and interspersed repeats in mammalian noncoding genomic DNA, Proc Natl Acad Sci U S A 98(25) (2001) 14503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kvikstad EM, Makova KD, The (r) evolution of SINE versus LINE distributions in primate genomes: sex chromosomes are important, Genome Res 20(5) (2010) 600–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Makova KD, Hardison RC, The effects of chromatin organization on variation in mutation rates in the genome, Nat Rev Genet 16(4) (2015) 213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fortin JP, Hansen KD, Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data, Genome Biol 16 (2015) 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].De S, Michor F, DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes, Nat Biotechnol 29(12) (2011) 1103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM, Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types, Genome Res 20(6) (2010) 761–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rosa-Garrido M, Chapski DJ, Schmitt AD, Kimball TH, Karbassi E, Monte E, Balderas E, Pellegrini M, Shih TT, Soehalim E, Liem D, Ping P, Galjart NJ, Ren S, Wang Y, Ren B, Vondriska TM, High-Resolution Mapping of Chromatin Conformation in Cardiac Myocytes Reveals Structural Remodeling of the Epigenome in Heart Failure, Circulation 136(17) (2017) 1613–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG, Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization, Cell 169(5) (2017) 930–944 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Korber P, Luckenbach T, Blaschke D, Horz W, Evidence for histone eviction in trans upon induction of the yeast PHO5 promoter, Mol Cell Biol 24(24) (2004) 10965–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mito Y, Henikoff JG, Henikoff S, Genome-scale profiling of histone H3.3 replacement patterns, Nat Genet 37(10) (2005) 1090–7. [DOI] [PubMed] [Google Scholar]
- [61].Lachner M, Sengupta R, Schotta G, Jenuwein T, Trilogies of histone lysine methylation as epigenetic landmarks of the eukaryotic genome, Cold Spring Harb Symp Quant Biol 69 (2004) 209–18. [DOI] [PubMed] [Google Scholar]
- [62].Sasai N, Defossez PA, Many paths to one goal? The proteins that recognize methylated DNA in eukaryotes, Int J Dev Biol 53(2–3) (2009) 323–34. [DOI] [PubMed] [Google Scholar]
- [63].Minajigi A, Froberg J, Wei C, Sunwoo H, Kesner B, Colognori D, Lessing D, Payer B, Boukhali M, Haas W, Lee JT, Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation, Science 349(6245) (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY, Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs, Cell 129(7) (2007) 1311–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Giorgetti L, Lajoie BR, Carter AC, Attia M, Zhan Y, Xu J, Chen CJ, Kaplan N, Chang HY, Heard E, Dekker J, Structural organization of the inactive X chromosome in the mouse, Nature 535(7613) (2016) 575–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hacisuleyman E, Goff LA, Trapnell C, Williams A, Henao-Mejia J, Sun L, McClanahan P, Hendrickson DG, Sauvageau M, Kelley DR, Morse M, Engreitz J, Lander ES, Guttman M, Lodish HF, Flavell R, Raj A, Rinn JL, Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre, Nat Struct Mol Biol 21(2) (2014) 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wapinski O, Chang HY, Long noncoding RNAs and human disease, Trends Cell Biol 21(6) (2011) 354–61. [DOI] [PubMed] [Google Scholar]
- [68].Ellis BC, Molloy PL, Graham LD, CRNDE: A Long Non-Coding RNA Involved in CanceR, Neurobiology, and DEvelopment, Front Genet 3 (2012) 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wang P, Ren Z, Sun P, Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation, J Cell Biochem 113(6) (2012) 1868–74. [DOI] [PubMed] [Google Scholar]
- [70].Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY, Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis, Nature 464(7291) (2010) 1071–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B, Topological domains in mammalian genomes identified by analysis of chromatin interactions, Nature 485(7398) (2012) 376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Bluthgen N, Dekker J, Heard E, Spatial partitioning of the regulatory landscape of the X-inactivation centre, Nature 485(7398) (2012) 381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Barbieri M, Chotalia M, Fraser J, Lavitas LM, Dostie J, Pombo A, Nicodemi M, Complexity of chromatin folding is captured by the strings and binders switch model, Proc Natl Acad Sci U S A 109(40) (2012) 16173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Benedetti F, Dorier J, Burnier Y, Stasiak A, Models that include supercoiling of topological domains reproduce several known features of interphase chromosomes, Nucleic Acids Res 42(5) (2014) 2848–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Jost D, Carrivain P, Cavalli G, Vaillant C, Modeling epigenome folding: formation and dynamics of topologically associated chromatin domains, Nucleic Acids Res 42(15) (2014) 9553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA, Formation of Chromosomal Domains by Loop Extrusion, Cell Rep 15(9) (2016) 2038–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, Geeting KP, Gnirke A, Melnikov A, McKenna D, Stamenova EK, Lander ES, Aiden EL, Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes, Proc Natl Acad Sci U S A 112(47) (2015) E6456–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ganji M, Shaltiel IA, Bisht S, Kim E, Kalichava A, Haering CH, Dekker C, Real-time imaging of DNA loop extrusion by condensin, Science 360(6384) (2018) 102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, Cobb BS, Yokomori K, Dillon N, Aragon L, Fisher AG, Merkenschlager M, Cohesins functionally associate with CTCF on mammalian chromosome arms, Cell 132(3) (2008) 422–33. [DOI] [PubMed] [Google Scholar]
- [80].Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, Huang X, Shamim MS, Shin J, Turner D, Ye Z, Omer AD, Robinson JT, Schlick T, Bernstein BE, Casellas R, Lander ES, Aiden EL, Cohesin Loss Eliminates All Loop Domains, Cell 171(2) (2017) 305–320 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lonfat N, Montavon T, Darbellay F, Gitto S, Duboule D, Convergent evolution of complex regulatory landscapes and pleiotropy at Hox loci, Science 346(6212) (2014) 1004–6. [DOI] [PubMed] [Google Scholar]
- [82].Spielmann M, Brancati F, Krawitz PM, Robinson PN, Ibrahim DM, Franke M, Hecht J, Lohan S, Dathe K, Nardone AM, Ferrari P, Landi A, Wittler L, Timmermann B, Chan D, Mennen U, Klopocki E, Mundlos S, Homeotic arm-to-leg transformation associated with genomic rearrangements at the PITX1 locus, Am J Hum Genet 91(4) (2012) 629–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Giorgio E, Robyr D, Spielmann M, Ferrero E, Di Gregorio E, Imperiale D, Vaula G, Stamoulis G, Santoni F, Atzori C, Gasparini L, Ferrera D, Canale C, Guipponi M, Pennacchio LA, Antonarakis SE, Brussino A, Brusco A, A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD), Hum Mol Genet 24(11) (2015) 3143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, Bernstein BE, Insulator dysfunction and oncogene activation in IDH mutant gliomas, Nature 529(7584) (2016) 110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Sanyal A, Lajoie BR, Jain G, Dekker J, The long-range interaction landscape of gene promoters, Nature 489(7414) (2012) 109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W, Looping and interaction between hypersensitive sites in the active beta-globin locus, Mol Cell 10(6) (2002) 1453–65. [DOI] [PubMed] [Google Scholar]
- [87].Amano T, Sagai T, Tanabe H, Mizushina Y, Nakazawa H, Shiroishi T, Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription, Dev Cell 16(1) (2009) 47–57. [DOI] [PubMed] [Google Scholar]
- [88].Gupta RM, Hadaya J, Trehan A, Zekavat SM, Roselli C, Klarin D, Emdin CA, Hilvering CRE, Bianchi V, Mueller C, Khera AV, Ryan RJH, Engreitz JM, Issner R, Shoresh N, Epstein CB, de Laat W, Brown JD, Schnabel RB, Bernstein BE, Kathiresan S, A Genetic Variant Associated with Five Vascular Diseases Is a Distal Regulator of Endothelin-1 Gene Expression, Cell 170(3) (2017) 522–533 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Mahmoudi T, Katsani KR, Verrijzer CP, GAGA can mediate enhancer function in trans by linking two separate DNA molecules, EMBO J 21(7) (2002) 1775–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Jing H, Vakoc CR, Ying L, Mandat S, Wang H, Zheng X, Blobel GA, Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus, Mol Cell 29(2) (2008) 232–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Spilianakis CG, Flavell RA, Long-range intrachromosomal interactions in the T helper type 2 cytokine locus, Nat Immunol 5(10) (2004) 1017–27. [DOI] [PubMed] [Google Scholar]
- [92].Cubenas-Potts C, Corces VG, Architectural proteins, transcription, and the three-dimensional organization of the genome, FEBS Lett 589(20 Pt A) (2015) 2923–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].De Gobbi M, Viprakasit V, Hughes JR, Fisher C, Buckle VJ, Ayyub H, Gibbons RJ, Vernimmen D, Yoshinaga Y, de Jong P, Cheng JF, Rubin EM, Wood WG, Bowden D, Higgs DR, A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter, Science 312(5777) (2006) 1215–7. [DOI] [PubMed] [Google Scholar]
- [94].Ott CJ, Blackledge NP, Kerschner JL, Leir SH, Crawford GE, Cotton CU, Harris A, Intronic enhancers coordinate epithelial-specific looping of the active CFTR locus, Proc Natl Acad Sci U S A 106(47) (2009) 19934–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Meddens CA, Harakalova M, van den Dungen NA, Foroughi Asl H, Hijma HJ, Cuppen EP, Bjorkegren JL, Asselbergs FW, Nieuwenhuis EE, Mokry M, Systematic analysis of chromatin interactions at disease associated loci links novel candidate genes to inflammatory bowel disease, Genome Biol 17(1) (2016) 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Martin P, McGovern A, Orozco G, Duffus K, Yarwood A, Schoenfelder S, Cooper NJ, Barton A, Wallace C, Fraser P, Worthington J, Eyre S, Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci, Nat Commun 6 (2015) 10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].van den Boogaard M, Smemo S, Burnicka-Turek O, Arnolds DE, van de Werken HJ, Klous P, McKean D, Muehlschlegel JD, Moosmann J, Toka O, Yang XH, Koopmann TT, Adriaens ME, Bezzina CR, de Laat W, Seidman C, Seidman JG, Christoffels VM, Nobrega MA, Barnett P, Moskowitz IP, A common genetic variant within SCN10A modulates cardiac SCN5A expression, J Clin Invest 124(4) (2014) 1844-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T, Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome, Nat Genet 37(1) (2005) 31–40. [DOI] [PubMed] [Google Scholar]
- [99].Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA, Systematic localization of common disease-associated variation in regulatory DNA, Science 337(6099) (2012) 1190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Wang S, Wen F, Wiley GB, Kinter MT, Gaffney PM, An enhancer element harboring variants associated with systemic lupus erythematosus engages the TNFAIP3 promoter to influence A20 expression, PLoS Genet 9(9) (2013) e1003750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Bhatia S, Bengani H, Fish M, Brown A, Divizia MT, de Marco R, Damante G, Grainger R, van Heyningen V, Kleinjan DA, Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia, Am J Hum Genet 93(6) (2013) 1126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Stadler ZK, Thom P, Robson ME, Weitzel JN, Kauff ND, Hurley KE, Devlin V, Gold B, Klein RJ, Offit K, Genome-wide association studies of cancer, J Clin Oncol 28(27) (2010) 4255–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Jager R, Migliorini G, Henrion M, Kandaswamy R, Speedy HE, Heindl A, Whiffin N, Carnicer MJ, Broome L, Dryden N, Nagano T, Schoenfelder S, Enge M, Yuan Y, Taipale J, Fraser P, Fletcher O, Houlston RS, Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci, Nat Commun 6 (2015) 6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Pittman AM, Naranjo S, Jalava SE, Twiss P, Ma Y, Olver B, Lloyd A, Vijayakrishnan J, Qureshi M, Broderick P, van Wezel T, Morreau H, Tuupanen S, Aaltonen LA, Alonso ME, Manzanares M, Gavilan A, Visakorpi T, Gomez-Skarmeta JL, Houlston RS, Allelic variation at the 8q23.3 colorectal cancer risk locus functions as a cis-acting regulator of EIF3H, PLoS Genet 6(9) (2010) e1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Meyer KB, Maia AT, O’Reilly M, Ghoussaini M, Prathalingam R, Porter-Gill P, Ambs S, Prokunina-Olsson L, Carroll J, Ponder BA, A functional variant at a prostate cancer predisposition locus at 8q24 is associated with PVT1 expression, PLoS Genet 7(7) (2011) e1002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Dryden NH, Broome LR, Dudbridge F, Johnson N, Orr N, Schoenfelder S, Nagano T, Andrews S, Wingett S, Kozarewa I, Assiotis I, Fenwick K, Maguire SL, Campbell J, Natrajan R, Lambros M, Perrakis E, Ashworth A, Fraser P, Fletcher O, Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C, Genome Res 24(11) (2014) 1854-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Zeitz MJ, Ay F, Heidmann JD, Lerner PL, Noble WS, Steelman BN, Hoffman AR, Genomic interaction profiles in breast cancer reveal altered chromatin architecture, PLoS One 8(9) (2013) e73974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Law PJ, Sud A, Mitchell JS, Henrion M, Orlando G, Lenive O, Broderick P, Speedy HE, Johnson DC,Kaiser M, Weinhold N, Cooke R, Sunter NJ, Jackson GH, Summerfield G, Harris RJ, Pettitt AR, Allsup DJ,Carmichael J, Bailey JR, Pratt G, Rahman T, Pepper C, Fegan C, von Strandmann EP, Engert A, Forsti A, Chen B, Filho MI, Thomsen H, Hoffmann P, Noethen MM, Eisele L, Jockel KH, Allan JM, Swerdlow AJ, Goldschmidt H, Catovsky D, Morgan GJ, Hemminki K, Houlston RS, Genome-wide association analysis of chronic lymphocytic leukaemia, Hodgkin lymphoma and multiple myeloma identifies pleiotropic risk loci, Sci Rep 7 (2017) 41071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Dekker J, Mirny L, The 3D Genome as Moderator of Chromosomal Communication, Cell 164(6) (2016) 1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Trimarchi T, Bilal E, Ntziachristos P, Fabbri G, Dalla-Favera R, Tsirigos A, Aifantis I, Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia, Cell 158(3) (2014) 593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Keim C, Kazadi D, Rothschild G, Basu U, Regulation of AID, the B-cell genome mutator, Genes Dev 27(1) (2013) 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, Liang G, Tang Z, Mathe E, Benner C, Dubois W, Nelson S, Vian L, Oliveira TY, Jankovic M, Hakim O, Gazumyan A, Pavri R, Awasthi P, Song B, Liu G, Chen L, Zhu S, Feigenbaum L, Staudt L, Murre C, Ruan Y, Robbiani DF, Pan-Hammarstrom Q, Nussenzweig MC, Casellas R, B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity, Cell 159(7) (2014) 1524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Zhao B, Zou J, Wang H, Johannsen E, Peng CW, Quackenbush J, Mar JC, Morton CC, Freedman ML, Blacklow SC, Aster JC, Bernstein BE, Kieff E, Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth, Proc Natl Acad Sci U S A 108(36) (2011) 14902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Morgan SL, Mariano NC, Bermudez A, Arruda NL, Wu F, Luo Y, Shankar G, Jia L, Chen H, Hu JF, Hoffman AR, Huang CC, Pitteri SJ, Wang KC, Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping, Nat Commun 8 (2017) 15993. [DOI] [PMC free article] [PubMed] [Google Scholar]