

Abstract
There is an urgent need for strategies to combat dengue and other emerging viral infections. We reported that cyclin G-associated kinase (GAK), a cellular regulator of the clathrin-associated host adaptor proteins AP-1 and AP-2, regulates intracellular trafficking of multiple unrelated RNA viruses during early and late stages of the viral lifecycle. We also reported the discovery of potent, selective GAK inhibitors based on an isothiazolo[4,3-b]pyridine scaffold, albeit with moderate antiviral activity. Here, we describe our efforts leading to the discovery of novel isothiazolo[4,3-b]pyridines that maintain high GAK affinity and selectivity. These compounds demonstrate improved in vitro activity against dengue virus, including in human primary dendritic cells, and efficacy against the unrelated Ebola and chikungunya viruses. Moreover, inhibition of GAK activity was validated as an important mechanism of antiviral action of these compounds. These findings demonstrate the potential utility of a GAK-targeted broad-spectrum approach for combating currently untreatable emerging viral infections.
Table of Contents graphic
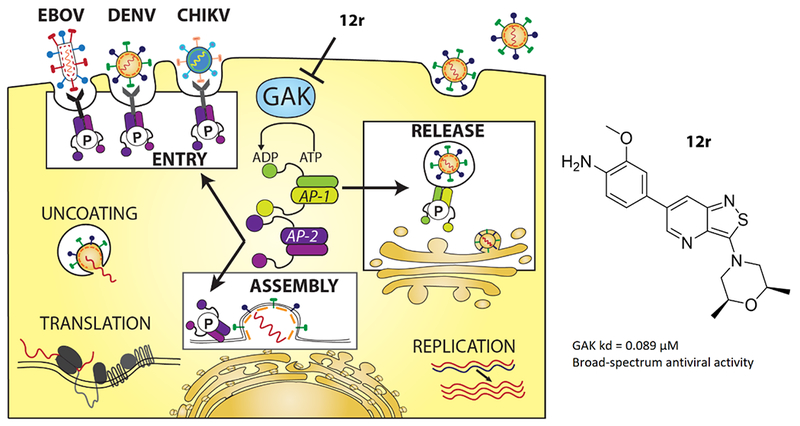
Introduction
Emerging viral infections, such as those caused by dengue (DENV), Ebola (EBOV) and chikungunya (CHIKV) viruses, represent major threats to global health. DENV is estimated to infect 390 million people annually in over 100 countries.1 The majority of individuals infected with any of the four DENV serotypes remain asymptomatic or present with acute dengue fever.2 A fraction (~5-20%) of dengue patients, particularly those secondarily infected with a heterologous DENV serotype, will progress to severe dengue, manifested by bleeding, plasma leakage, shock, organ failure, and death. The development of an effective vaccine for DENV has been hampered by the need to generate simultaneous protection against the four distinct DENV serotypes to avoid antibody-dependent enhancement (ADE), with recent data indicating an increase in dengue severity requiring hospitalization in vaccinated children.3 EBOV is the causative agent of a severe and often fatal hemorrhagic disease.4–6 The unprecedented scope of the 2013-2016 Ebola virus disease (EVD) epidemic in western Africa highlighted the need for effective medical countermeasures against this emerging infectious disease.7 CHIKV is a re-emerging alphavirus that has been causing massive outbreaks in various parts of Africa, Asia and more recently in Central and South America.8 There are currently no vaccines available for the prevention of CHIKV infection. While an EBOV vaccine has shown promise recently,9 it is not yet approved. Importantly, no effective antiviral treatment is available against DENV, EBOV, CHIKV, and most other emerging viral pathogens.
The majority of the currently approved antiviral drugs target viral enzymatic functions and thus typically have a narrow spectrum of coverage and a low genetic barrier to resistance. An attractive approach to overcome these limitations is to develop compounds that target host factors broadly required for the effective replication of multiple viral pathogens.10 Such a host-targeted broad-spectrum approach is more scalable to address the large unmet clinical need and is particularly attractive for the treatment of emerging viral infections lacking any treatment.10
Intracellular membrane trafficking is one of multiple cellular processes usurped by viruses. Cyclin G-associated kinase (GAK) is a ubiquitously expressed host cell kinase that regulates clathrin-mediated intracellular trafficking of cellular cargo proteins.11 GAK is a 160 kDa serine/threonine kinase belonging to the numb-associated kinase (NAK) family, which also includes adaptor-associated kinase 1 (AAK1), BMP-2-inducible kinase (BIKE/BMP2K) and myristoylated and palmitoylated serine/threonine kinase 1 (MPSK1/STK16). Clathrin-mediated membrane trafficking is dependent on the action of oligomeric clathrin and adaptor protein complexes (APs) that coordinate the specific recruitment and assembly of clathrin into clathrin-coated vesicles (CCVs) as well as its coupling to endocytic cargo.12–14 The heterotetrametic AP-1 and AP-2 complexes are major components of CCVs, responsible for vesicle formation in the trans-Golgi network (TGN) and plasma membrane, respectively.12,15 The μ subunit of these AP complexes recognizes and binds sorting signals in the form of tyrosine- or dileucine-based motifs present on cargo molecules. The efficiency of cargo protein binding to APs is regulated via phosphorylation of a threonine residue within their μ subunit (Thr 144 in AP-1 and Thr 156 in AP-2), which induces a conformational change.12,16,17 Both GAK and AAK1 phosphorylate these Thr residues, thereby enhancing cargo recruitment and vesicle assembly.18–21 Additionally, these kinases regulate the recruitment of clathrin and AP-2 to the plasma membrane, and GAK also controls the uncoating of CCVs, enabling recycling of clathrin back to the cell surface.12,14
We have previously demonstrated that depletion of GAK by siRNAs or ectopic expression of phosphorylation mutants of AP-1 and AP-2 are dispensable for RNA replication of hepatitis C virus (HCV), but significantly inhibit four temporally distinct steps of the HCV lifecycle, namely entry (AP-2), assembly (AP-2), release of cell-free virus and direct cell-to-cell spread (AP-1).14–16,22 Moreover, we discovered a requirement for GAK in early and late stages of the lifecycle of other viruses, including DENV and EBOV, thereby validating GAK as a potential cellular target for the development of broad-spectrum antiviral agents.15 Indeed, erlotinib (compound 1, Figure 1), an approved anticancer drug with potent, yet non-selective, anti-GAK activity (dissociation constant (Kd) value of 3.1 nM) inhibits replication of multiple viruses from six unrelated viral families in cell culture.15 Combinations of erlotinib and sunitinib, an approved anticancer drug with potent anti-AAK1 activity, protected mice from morbidity and mortality associated with DENV and EBOV infections.15 These data provide a proof-of-concept that small-molecule inhibitors of GAK and AAK1 are useful candidates for broad-spectrum antiviral therapy. The safety and efficacy of erlotinib and/or sunitinib will be evaluated in DENV infected patients in the near future and potentially in patients with EVD in future outbreaks (ClinicalTrials.gov NCT02380625). Nevertheless, erlotinib is a potent, yet non-selective inhibitor whose side effects result not only from on target inhibition of EGFR but also from inhibition of other cellular kinases and non-kinase proteins.23–25 A particularly relevant side effect that often complicates combination regimens of erlotinib with sunitinib is diarrhea, attributed in part to inhibition erlotinib’s cancer target, EGFR.26 This side effect may limit erlotinib’s use in patients with EVD, who often present with diarrhea.27 To overcome this challenge and avoid other potential toxicity, we have sought to develop novel, chemically distinct and more selective inhibitors of GAK.
Figure 1.
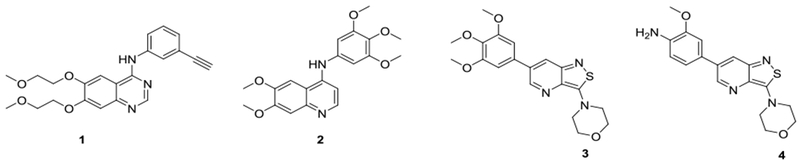
Known GAK inhibitors
To the best of our knowledge, only two series of potent and selective GAK inhibitors have been reported to date. The first, represented by 4-anilinoquinoline analogue 2 (Figure 1), displays low nM GAK inhibition, has over 50,000 fold greater selectivity to GAK than to other members of the NAK subfamily of kinases, and shows selective GAK inhibition when tested against a large panel of kinases.28 Nevertheless, this series has not been tested for its antiviral activity. We have previously reported the discovery and synthesis of isothiazolo[4,3-b]pyridines, a second, structurally distinct class of selective GAK inhibitors.29 The two most prominent derivatives within this series (compounds 3 and 4, Figure 1) have a high GAK affinity (Kd = 8 nM).29 We reported that these isothiazolo[4,3-b]pyridines have antiviral activity against both HCV and DENV, yet the measured 50% effective concentration (EC50) values were at a micromolar range.15,29 Here, we describe our efforts to optimize the antiviral activity of these isothiazolo[4,3-b]pyridines, while maintaining their potency and selectivity to GAK.
Results and discussion
Synthesis of isothiazolo[4,3-b]pyridines
To synthesize isothiazolo[4,3-b]pyridine derivatives, a known procedure was followed with minor modifications (Scheme 1).29,30 First, the nitro group of 3-nitro-5-bromopyridine-2-carbonitrile 5 was reduced by treatment with iron under acidic conditions, generating the desired aniline 6 as the major product. However, due to the acid hydrolysis of the cyano group, a substantial amount (12%) of 3-amino-5-bromopicolinamide was also formed. These two compounds were easily separated by silica gel flash chromatography. Thionation of 6 using phosphorus pentasulfide yielded thioamide 7 in moderate yield. Although this procedure was successful on a small scale (< 100 mg), upon upscaling, the starting material 6 was not completely consumed, drastically reducing the reaction yield. Substituting phosphorus pentasulfide by Lawesson’s reagent allowed isolation of 3-amino-5-bromopyridine-2-carbothioamide 7 in good yield at a larger (gram) scale. The isothiazole moiety was formed by an oxidative ring closure using hydrogen peroxide in methanol, generating 3-amino-6-bromo-isothiazolo[4,3-b]pyridine 8. The subsequent Sandmeyer reaction of the exocyclic amino group with sodium nitrite, hydrogen bromide and CuBr yielded the key intermediate 3,6-dibromo-isothiazolo[4,3-b]pyridine 9 in moderate yield. Work-up of this reaction was initially done by careful addition of solid K2CO3 to the reaction mixture. Since time consuming on a larger scale, this step was substituted with neutralization with a 30% aqueous NaOH solution. Various nitrogen-nucleophiles were introduced at position 3 of the isothiazolo[4,3-b]pyridine scaffold, generating compounds 10a-t in yields ranging from 25 to 97%. Subsequently, Suzuki coupling of compounds 10a-s using 4-amino-3-methoxyphenylboronic acid pinacol ester, potassium carbonate as a base and tetrakis(triphenylphosphine)palladium(0) as a catalyst in a mixture of dioxane/water yielded compound 11 and a series of isothiazolo[4,3-b]pyridines 12a-s. Finally, acidic cleavage of the Boc protecting group of compound 11 furnished compound 12t, giving rise to a small library of isothiazolo[4,3-b]pyridines 12a-t (Table 1), which was then evaluated biologically.
Scheme 1.
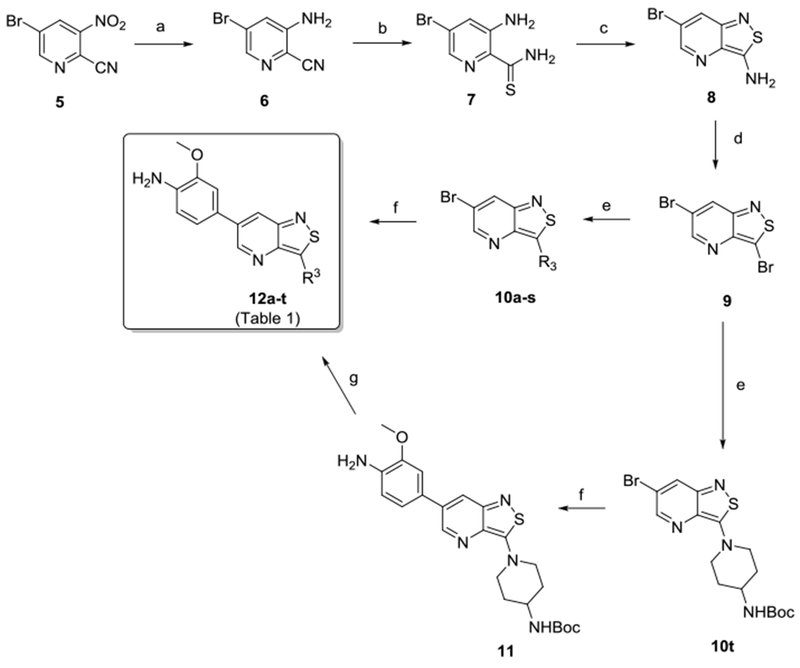
Reagents and conditions: a) Fe, CH3COOH, rt; b) Lawesson’s reagent, EtOH, reflux; c) 30% aq. H2O2, MeOH, 0 °C; d) NaNO2, HBr, CuBr, H2O, 0 °C to rt; e) R3H, EtOH or n-BuOH, reflux; f) 4-amino-3-methoxyphenylboronic acid pinacol ester, K2CO3, Pd(PPh3)4, dioxane/water, 100 °C; g) 37% HCl, dioxane, rt.
Table 1.
SAR at position 3 of isothiazolo[4,3-b]pyridines
| Compound# | R3 | GAK affinity | DENV-2 antiviral activity | Cytotox | |
|---|---|---|---|---|---|
| Kd (μM)a | EC50 (μM)b | EC90 (μM)c | CC50 (μM)d | ||
| 4 | 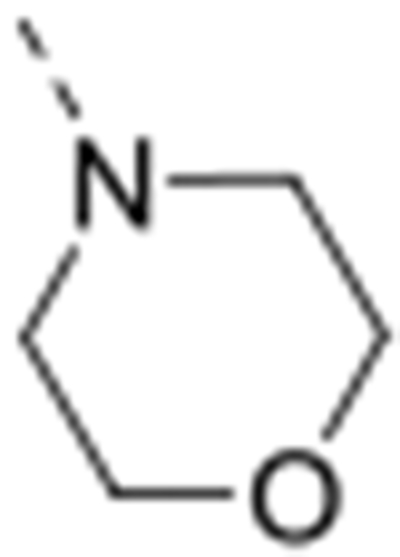 |
0.0089 | 1.844 | 8.05 | 17 |
| 12a | 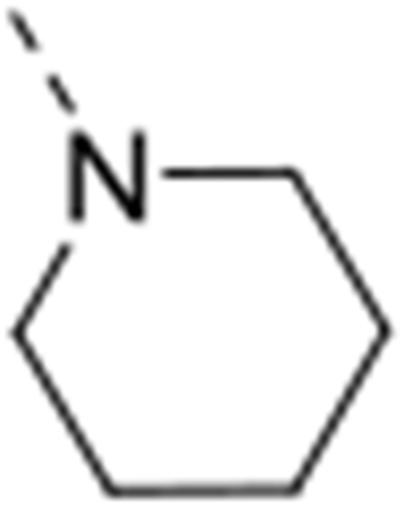 |
0.97 | 5.72 | >10 | >10 |
| 12b | 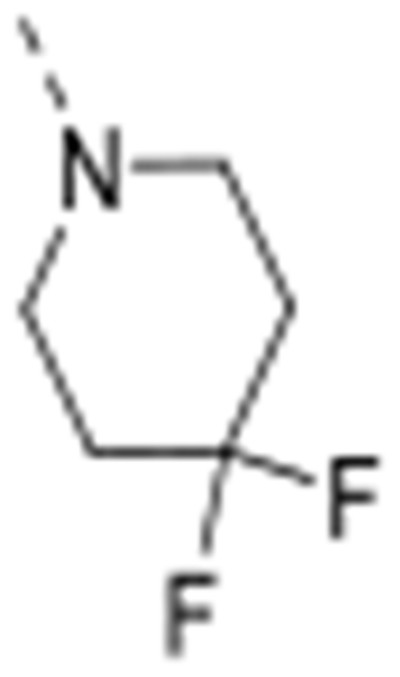 |
4.3 | >10 | >10 | >10 |
| 12t | 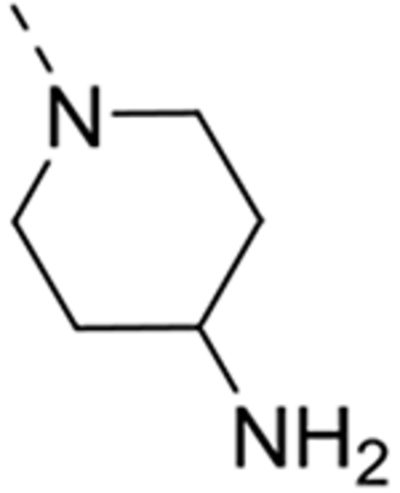 |
1.6 | 6.38 | >10 | >10 |
| 12c | 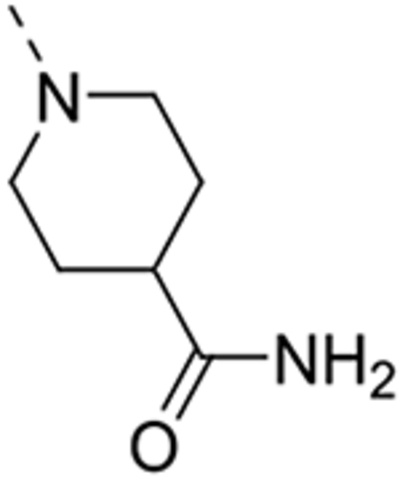 |
7.2 | >10 | >10 | >10 |
| 12d | 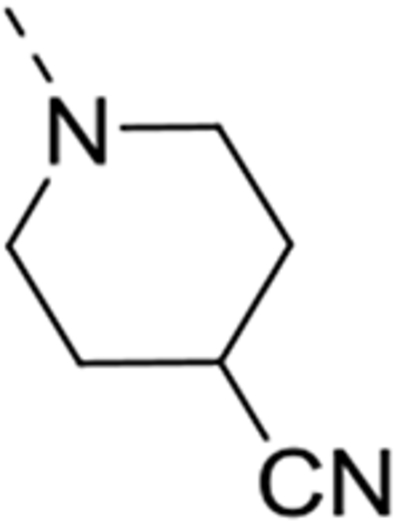 |
0.25 | 0.18 | 0.56 | 2.09 |
| 12e |  |
0.16 | 5.28 | >10 | >10 |
| 12f | 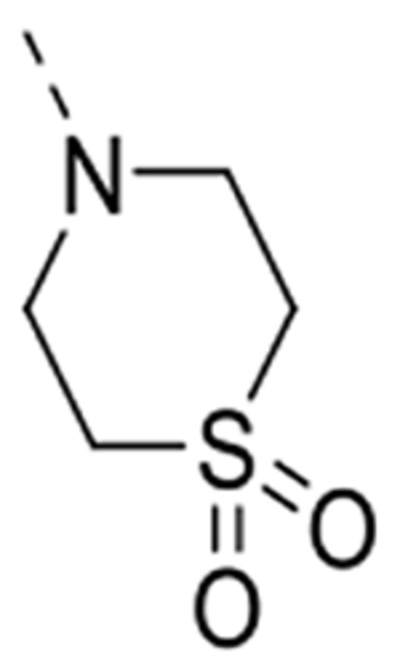 |
0.21 | 5.305 | >10 | >10 |
| 12g | 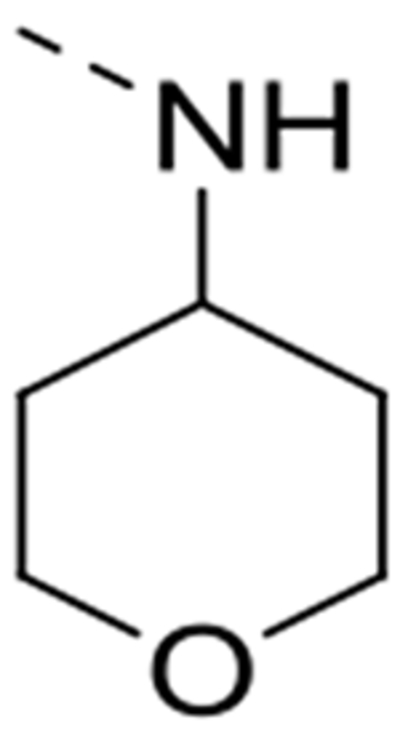 |
0.08 | >20 | >20 | >20 |
| 12h |  |
0.052 | 4.03 | >10 | >10 |
| 12i | 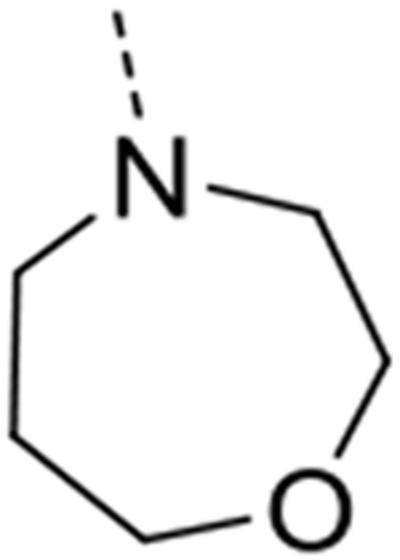 |
0.19 | 2 | 4.15 | >10 |
| 12j | 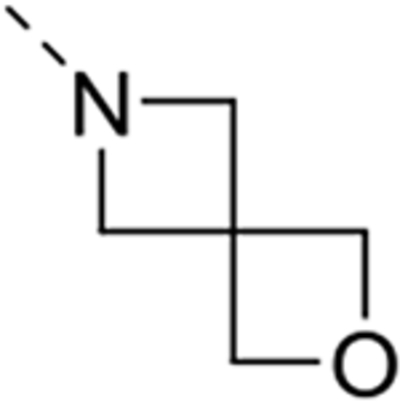 |
0.17 | 7.12 | >10 | >10 |
| 12k | 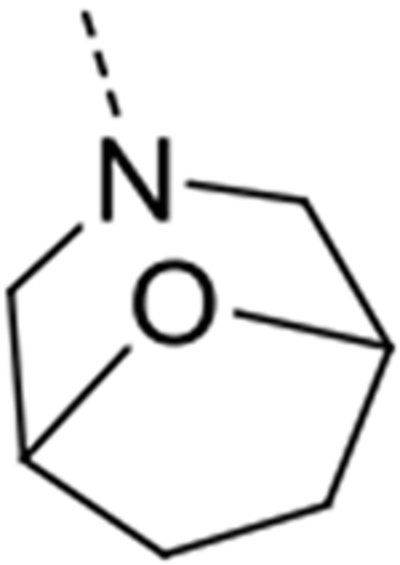 |
0.061 | 2.735 | >10 | >10 |
| 12l | 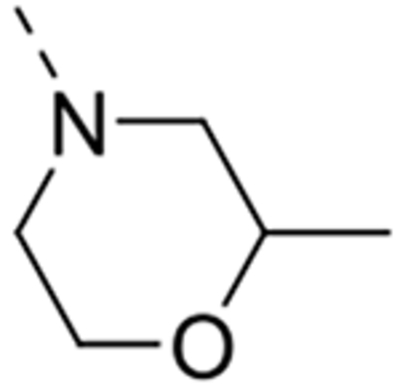 |
0.046 | 0.76 | 2.74 | 21.5 |
| 12m | 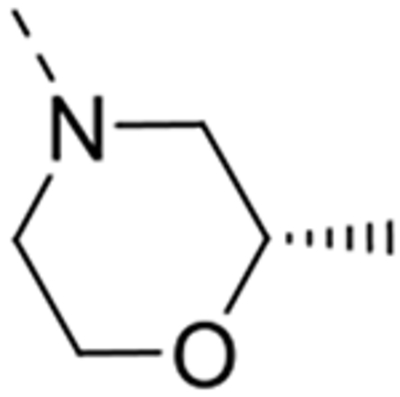 |
0.026 | 1.1 | 2.9 | >25 |
| 12n | 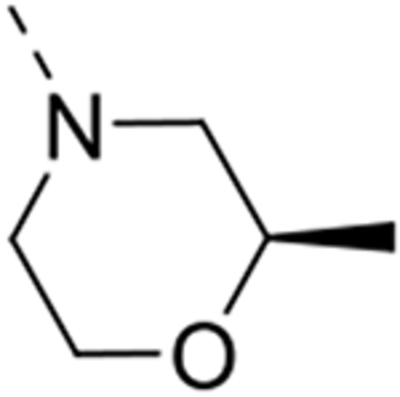 |
0.019 | 2.09 | 6.5 | 20.68 |
| 12o | 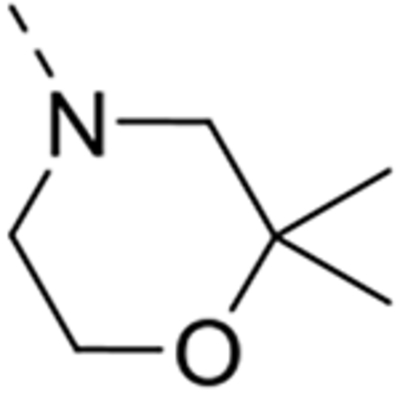 |
0.035 | 0.8416 | 3.92 | 4.14 |
| 12p | 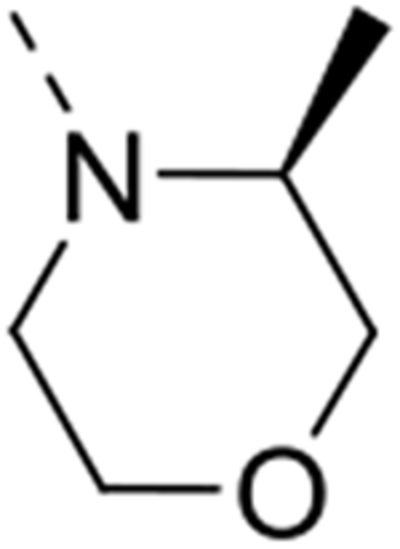 |
0.018 | 0.47 | 1.34 | 6.32 |
| 12q | 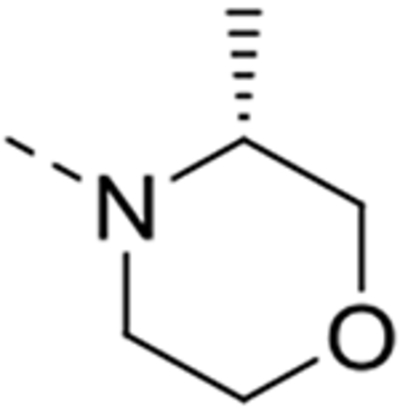 |
0.8 | >10 | >10 | >10 |
| 12r | 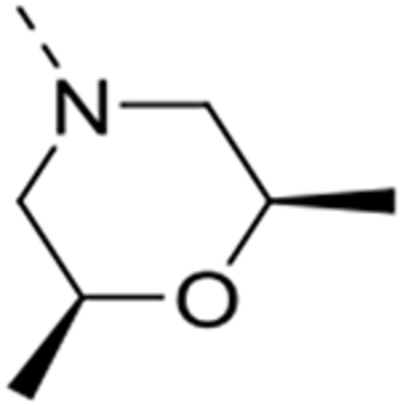 |
0.089 | 0.82 | 1.76 | >25 |
| 12s | 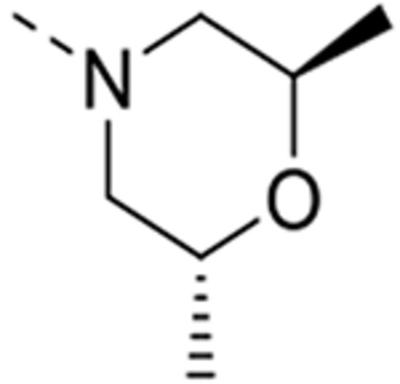 |
0.011 | 0.70 | 2.15 | >10 |
Kd = dissociation constant. Values represent the average of two independent experiments.
EC50 = half-maximal effective concentration.
EC90 = 90% effective concentration.
CC50 = half-maximal cytotoxic concentration.
GAK binding affinity studies
All compounds were tested for GAK binding affinity using the KINOMEscan™ platform, which quantitatively measures the ability of a compound to compete with an immobilized active-site-directed ligand.31 Compound 4 was used as a positive control, as it has a potent GAK affinity (Kd = 8 nM).29 Prior work revealed that position 3 of the isothiazolo[4,3-b]pyridine scaffold can tolerate structural modifications without impairing GAK affinity. Various amines29, alkoxy30 and carboxamide30 groups have thus been inserted at this position. Indeed, many of these molecules demonstrated high affinity to GAK (low μM range), with 3-N-morpholino-isothiazolo[4,3-b]pyridines emerging as the most potent derivatives within this series. In combination with the appropriate aryl substituent (e.g. 3,4,5-trimethoxyphenyl, 3-methoxy-4-aminophenyl) at position 6, these compounds demonstrated Kd values in a low nM range. We therefore focused on the synthesis and biological evaluation of isothiazolo[4,3-b]pyridine analogues bearing substituents at position 3 that closely resemble morpholine. The aryl moiety at position 6 was fixed as a 4-amino-3-methoxyphenyl residue, since this was shown to be optimal for GAK affinity and antiviral activity.
The isosteric replacement of the oxygen atom of the morpholino ring of compound 4 by a carbon gave rise to the 3-N-piperidine analogue 12a. Compound 12a displayed reasonable affinity to GAK (Kd = 0.97 μM), albeit 100 fold lower than that of compound 4 (Kd = 0.0089 μM). A number of substituents at position 4 of the piperidinyl moiety were introduced. Whereas the 4,4-difluoropiperidine (compound 12b), 4-aminopiperidine (compound 12t) and 4-carboxamidepiperidine (compound 12c) derivatives exhibit GAK Kd values exceeding 1 μM, the 4-cyano analog (compound 12d) has a quite potent GAK affinity (Kd = 0.25 μM). Remarkably, the 3-carboxamide congener (compound 12e) demonstrates potent affinity for GAK (Kd = 0.16 μM), yet its 4-substituted congener 12c is much less active. Replacing the morpholino oxygen with a sulphon moiety yielded compound 12f, which is 20-fold less active as a GAK ligand relative to the morpholino containing derivative 4. We have previously demonstrated that the presence of a piperazine ring in place of morpholine reduced GAK affinity, suggesting that a basic nitrogen atom is not tolerated at this position.29 A lactam containing analogue (compound 12h) was therefore prepared, with potent binding affinity to GAK (Kd = 0.052 μM). Since the morpholine oxygen seems to play a crucial role in mediating GAK affinity, it was kept fixed, whereas the morpholine nitrogen was changed to an externally secondary amine group. The resulting compound 12g bound GAK with a Kd of 80 nM.
Expansion of the six-membered morpholino moiety towards a seven-membered homomorpholino derivative yielded compound 12i. This 1,4-oxazepine derivative is 20-fold less potent as a GAK ligand than the morpholine analogue (compound 4). Additionally, using a spirocyclic oxetane, a well-known isoster for morpholine, compound 12j was obtained, with a GAK Kd value of 0.17 μM. Surprisingly, the bridged morpholine derivative 12k (with an 8-oxa-3-aza-bicyclo[3.2.1]octane moiety) displayed potent GAK affinity (Kd = 61 nM). Since this suggested a positive effect of aliphatic substituents, the SAR of the morpholine ring was further explored by introducing methyl groups at various positions of the morpholine moiety. Introducing a single methyl group at position 2 yielded compound 12l as a racemic mixture. This compound demonstrated potent GAK affinity (Kd = 46 nM). The enantiopure compounds 12m and 12n were thus prepared using optically pure building blocks. The two enantiomers demonstrated very similar Kd values (26 nM and 19 nM, respectively). To avoid the racemic mixtures and the high cost of enantiopure building blocks, a geminal dimethyl group was introduced at position 2 of the morpholine ring, yielding compound 12o as a potent GAK ligand (Kd = 35 nM). The stereochemistry of the 3-methyl substituted morpholine analogues demonstrated a strong impact on GAK binding, with the S-3-methylmorpholine derivative (compound 12p) showing a Kd value of 18 nM, and the corresponding R-isomer (compound 12q) demonstrating a 40-fold reduction in GAK affinity. To expand the SAR at position 2 of the morpholine moiety, two 2,6-dimethyl-substituted morpholine derivatives were prepared. Both the cis (compound 12r) and trans (compound 12s) diastereoisomers demonstrated potent GAK affinity (Kd values of 89 nM and 11 nM, respectively). X-ray crystallography has previously demonstrated that compound 4 bound to the ATP binding site of GAK according to a ‘type I’ binding mode.29 Given the close structural similarity between the most potent congeners of the current series and compound 4, we predict that their mode of binding to GAK is similar.
Anti-DENV activity of isothiazolo[4,3-b]pyridines
All the synthesized derivatives were tested for their activity against DENV, independently of their affinity to GAK. Human hepatoma (Huh7) cells infected with DENV2 (New Guinea C strain) harboring a luciferase reporter32,33 were treated with the individual compounds for 48 hours. Antiviral activity (EC50 and EC90) was measured via luciferase assays. Cytotoxicity (CC50) was measured in the same cell culture wells via AlamarBlue assays (Table 1). In general, isothiazolo[4,3-b]pyridines demonstrating GAK binding displayed a dose-dependent inhibition of DENV infection. The 3-N-piperidinyl isothiazolo[4,3-b]pyridine analogues, which displayed GAK Kd values in the range of 0.16 – 1.6 μM (compounds 12a, 12t, 12d and 12e), inhibited DENV with EC50 values ranging from 0.18 to 6.38 μM, albeit compound 12d was more cytotoxic. As predicted, inactive GAK ligands with substitutions at the piperidinyl ring (compounds 12b, 12c) demonstrated no antiviral activity. Nevertheless, the introduction of other saturated heterocycles at position 3, such as a tetrahydro-2H-pyran-4-amine (compound 12g) and a piperazinyl-2-one (compound 12h), yielded analogues with potent GAK affinity (Kds<100 nM), yet with limited antiviral activity (EC50> 20 μM and 4.03 μM, respectively). Among the series of isothiazolo[4,3-b]pyridines, which bear a closely related analogue of morpholine as substituent at position 3, the homomorpholino (compound 12i) and 2-oxa-6-azaspiro[3.3]heptane (compound 12j) analogues are the least active GAK binders. Accordingly, these compounds demonstrated a moderate antiviral activity (EC50 values of 2 μM and 7.12 μM, respectively). The majority of the 3-N-morpholinyl-isothiazolo[4,3-b]pyridines (compounds 12k-p and 12r-s), which demonstrated potent affinity to GAK, displayed potent antiviral activity, with EC50 values of ~1 μM. Notably, these new analogues also displayed significantly lower EC90 values (at a low μM range) and reduced cytotoxicity relative to the original lead compound 4. A comparison of the dose-response curves of compound 12r, the most promising compound in this series, and the parental compound 4 reveals the improved antiviral activity and toxicity profiles of 12r (Figure 2). Several derivatives within this series, such as the geminal 2,2-dimethylmorpholino (compound 12o) and the 3-S-methylmorpholino (compound 12p), demonstrated no antiviral activity beyond toxicity. The 3-R-methylmorpholine analogue 12q, which had poor GAK affinity, also lacked antiviral activity (Table 1).
Figure 2. Compound 12r suppresses DENV infection more effectively than compound 4.
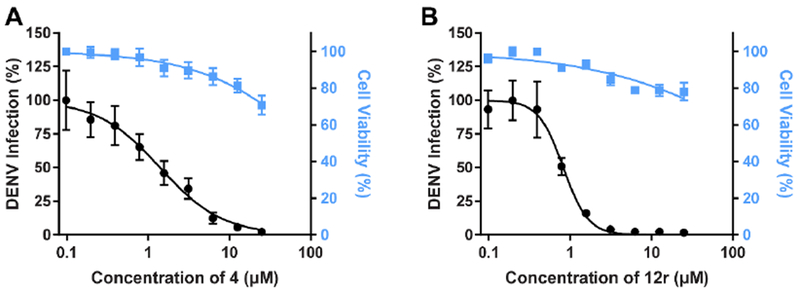
Cell viability (blue) and dose response of DENV infection (black) to compound 4 (A) and 12r (B) measured by luciferase and alamarBlue assays, respectively, 48 hours after infection. Data are plotted relative to vehicle control. Shown are representative experiments from at least 2 conducted, each with 6 biological replicates; shown are means ± SD.
Compound 12r inhibits DENV infection in human primary monocyte-derived dendritic cells.
To further determine the therapeutic potential of compound 12r as an anti-DENV compound, we studied its antiviral effect in human primary monocyte-derived dendritic cells (MDDCs); an established ex vivo model system for DENV.34 We measured a dose-dependent inhibition of DENV infection with minimal cytotoxicity following a 3-day compound treatment with an EC50 of 3.537 μM and CC50 > 20 μM by plaque assays and alamarBlue assays, respectively (Figure 3). Dendritic cells represent the primary target of DENV in humans.35 Moreover, primary cells model human physiology and disease better than immortalized cell lines.35 Our finding that 12r treatment exhibits antiviral efficacy in MDDCs may therefore more accurately reflect the dependence of DENV on GAK during human infection and support the biological relevance of this approach.
Figure 3. Ex vivo antiviral activity of 12r in human primary dendritic cells.
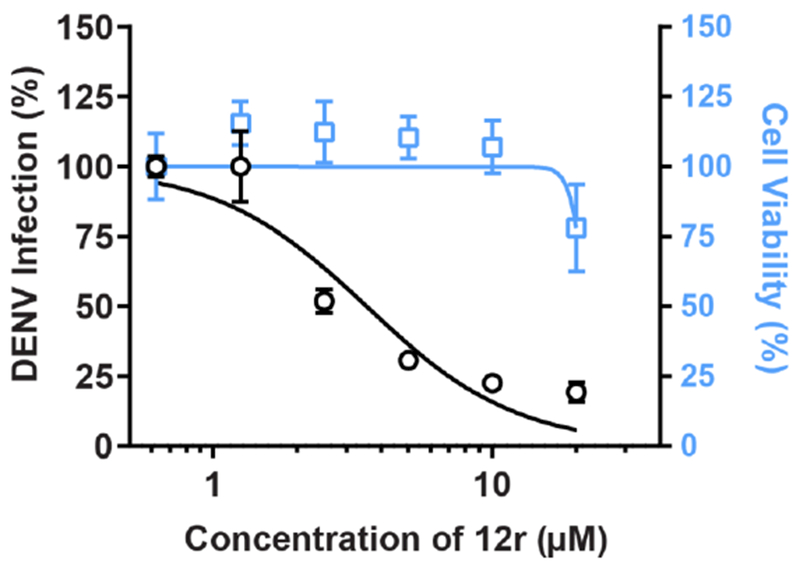
Cell viability (blue) and dose response of DENV infection (black) to 12r measured by plague assays and alamarBlue assays, respectively, 72 hours after infection of primary human monocyte-derived dendritic cells (MDDCs). Data are plotted relative to vehicle control. Shown is a representative experiment with cells from a single donor, out of 2 independent experiments conducted with cells derived from 2 donors, each with 5 biological replicates; shown are means ± SD.
Broad-spectrum antiviral activity of 3-N-morpholinyl-isothiazolo[4,3-b]pyridine analogues
To explore the spectrum of coverage of the new 3-N-morpholinyl-isothiazolo[4,3-b]pyridine analogues beyond DENV, we studied their antiviral effects against two unrelated viruses: EBOV (Filoviridae family), which was previously shown to hijack GAK15, and CHIKV (Togaviridae family). Huh7 cells were infected with authentic EBOV and treated for 48 hours with individual isothiazolo[4,3-b]pyridine derivatives. Antiviral activity was measured via an immunofluorescence assay and cytotoxicity via a Cell TiterGlo assay (Table 2). Whereas treatment with compound 4 showed no anti-EBOV activity beyond cytotoxicity, treatment with 12r and 3-N-morpholinyl-isothiazolo[4,3-b]pyridine analogues (compounds 12l, 12m and 12n) resulted in a dose-dependent inhibition of EBOV infection, with EC50 values of ~2 μM and CC50 > 10 μM (Figure 4A, Table 2). Additionally, treatment of Vero cells with 12r commencing 48 hours before infection resulted in a dose-dependent decrease in CHIKV infection measured by plaque assay 48 hours following infection (Figure 4B). These data expand the possible future indications of these compounds as antiviral agents beyond Flaviviridae infections to other emerging RNA viral infections.
Table 2.
Activity against EBOV
| Compound# | GAK affinity | EBOV antiviral activity | Cytotox | |
|---|---|---|---|---|
| Kd (μM)a | EC50 (μM)b | EC90 (μM)c | CC50 (μM)d | |
| 4 | 0.0089 | 11.86 | 21.91 | 11 |
| 12r | 0.089 | 2.436 | 7.732 | >10 |
| 12l | 0.046 | 2.001 | 7.865 | 9.398 |
| 12m | 0.026 | 2.728 | 8.062 | >10 |
| 12n | 0.019 | 4.345 | >10 | >10 |
Kd = dissociation constant. Values represent the average of two independent experiments.
EC50 = half-maximal effective concentration.
EC90 = 90% effective concentration.
CC50 = half-maximal cytotoxic concentration.
Figure 4: Compound 12r suppresses EBOV and CHIKV infections.
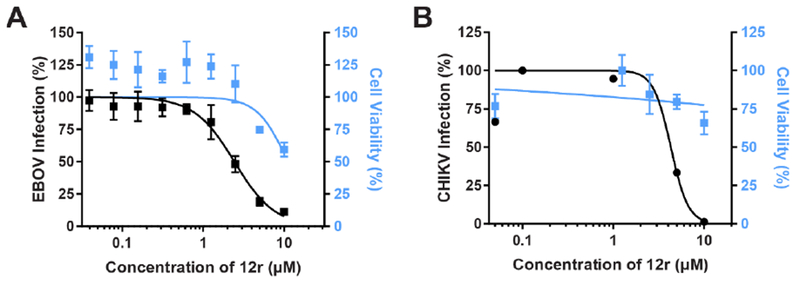
Cell viability (blue) and dose response of EBOV (A) or CHIKV (B) infection (black) to compound 12r measured by immunofluorescence (A) or plaque (B) assays in Huh7 (A) or Vero (B) cells 48 hours after infection. Data are plotted relative to vehicle control. Shown are representative experiments from at least 2 conducted, each with 6 biological replicates; shown are means ± SD.
The antiviral effect of compound 12r correlates with functional inhibition of GAK
To confirm that the observed antiviral activity is correlated with functional inhibition of GAK activity, we measured levels of the phosphorylated form of the μ subunit of the AP-2 complex, AP2M1, upon treatment with compound 12r. Since AP2M1 phosphorylation is transient (due to phosphatase PP2A activity)27, to allow capturing of the phosphorylated state, Huh7 cells were incubated for 30 minutes in the presence of the PP2A inhibitor calyculin A prior to lysis. Treatment with compound 12r reduced AP2M1 phosphorylation (Figure 5), indicating modulation of AP2M1 phosphorylation via GAK inhibition.
Figure 5. Antiviral effect of compound 12r correlates with functional inhibition of GAK.
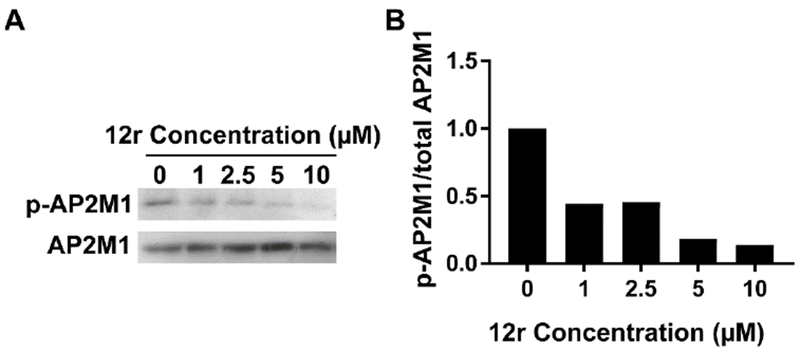
The effect of 12r on AP2M1 phosphorylation by Western analysis in lysates derived from Huh7 cells. Shown are a representative membrane (from two independent experiments) blotted with anti-phospho-AP2M1 (p-AP2M1) and anti-AP2M1 antibodies and quantitative data of p-AP2M1/total AP2M1 protein ratio normalized to DMSO controls.
GAK is a molecular target underlying the antiviral effect of compound 12r
Next, we conducted gain-of-function assays, to confirm that inhibition of GAK is a mechanism underlying the anti-DENV effect of compound 12r. A doxycycline-inducible cell line was established to overexpress GAK using the Flp-In™ recombination system in T-REx 293 cells.36 Following doxycycline-mediated induction of GAK expression, these cells were infected with a luciferase reporter DENV and treated with compound 12r for 72 hours prior to luciferase and viability assays. Doxycycline-induced GAK expression either partially or completely reversed the antiviral effect of compound 12r relative to uninduced cells (Figure 6). These results validate GAK as a key mediator of the anti-DENV effect of compound 12r.
Figure 6. GAK is a molecular target underlying the antiviral effect of compound 12r.
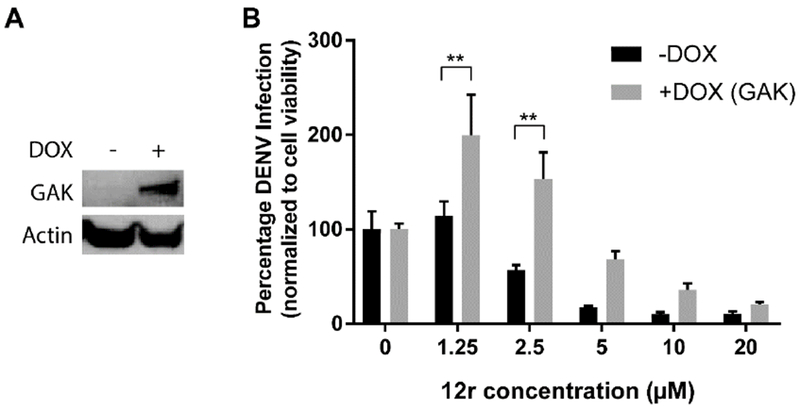
A. Expression of GAK in T-REx 293 cells induced with doxycycline and uninduced control cells. B. DENV infection normalized to cellular viability in the induced cells (+DOX, grey) or uninduced control cells (-DOX, black) 72 hours following infection with a luciferase reporter DENV and 12r treatment, via luciferase and alamarBlue assays, respectively. Shown is a representative experiment out of two conducted, each with 5 replicates. **P<0.01 relative to induced or uninduced cells treated with DMSO by one-way ANOVA with Dunnett’s multiple comparisons test. Means ± SD are shown.
Kinase selectivity profile of compound 12r
Due to its excellent GAK affinity, good antiviral activity, and relatively low cytotoxicity, compound 12r was selected as a lead compound for further analysis. While our data provide evidence that GAK is an important mediator of the observed antiviral effect of compound 12r, additional cellular kinases may mediate its antiviral activity. This is particularly relevant as X-ray crystallography has previously demonstrated targeting of the ATP binding site of GAK by compound 3, thereby classifying it as a classical type I kinase inhibitor.29 Since the ATP binding site is highly conserved across the kinome, achieving high selectivity of kinase inhibitors is quite challenging.31 To determine whether the improved antiviral activity of compound 12r resulted from reduced selectivity, we screened it against a diverse panel of 468 kinases (74 mutant and 394 wild-type kinases) using an in vitro ATP-site competition binding assay (DiscoveRX, KinomeScan). This assay measures binding and not functional activity, is conducted in the absence of ATP and is quite heterogeneous, yet it is commonly used for selectivity assessment. Overall, the selectivity profile of compounds 3 and 12r is comparable, as illustrated by their kinome trees (Figure 7), with selectivity indices S(35) of 0.033 and 0.057, respectively. Beyond GAK, at a concentration of 10 μM, compound 12r targets only 8 other kinases with greater than 90% inhibition (<10% control): ABL1, AURKC, EPHA7, FLT3, KIT, MAP2K5, MAPK14 and PDGRFB. ABL1 (Abelson Proto-Oncogene 1), a member of the Abl family of non-receptor tyrosine kinases, is implicated in the lifecycle of both DENV37 and EBOV38, and thus represents another target of 12r that may be contributing to the observed antiviral effect. We previously reported that siRNA-mediated silencing of KIT (KIT Proto-Oncogene Receptor Tyrosine Kinase) reduced DENV infection.15 Nevertheless, KIT’s role in DENV infection remained questionable since its suppression substantially reduced cellular viability (likely due to its role in cell survival and proliferation).39,15 Moreover, the anti-DENV activity of the tested derivatives does not correlate with the binding affinity to KIT. Compound 4 exhibits potent KIT affinity (Kd = 0.0045 μM), yet only moderate antiviral activity, whereas compound 12r is 300-fold less potent as a KIT inhibitor (Kd = 1.3 μM), but displays an improved antiviral activity. These data suggest that KIT is unlikely to be an important molecular target underlying the antiviral effect of these compounds.
Figure 7. Kinase selectivity of compound 12r.
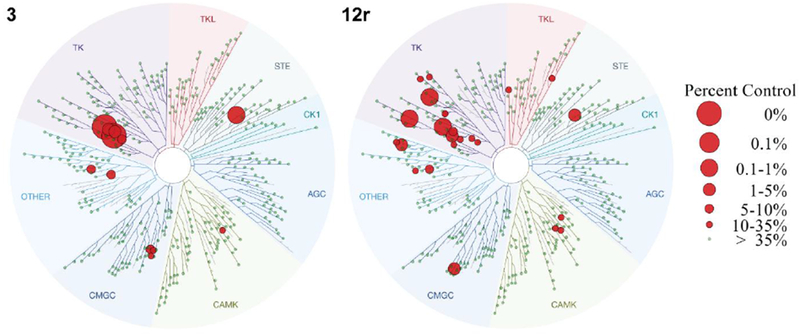
Kinome tree comparison of 3 (left) and 12r (right). Red circles indicate kinases inhibited by more than 90% at a concentration of 10 μM of the indicated compounds.
ADME profiling
Compounds 4 and 12h exhibit a large (200 fold) or moderate (75 fold) difference between their GAK affinity and anti-DENV cellular activity, respectively (Table 1). In contrast, the antiviral activity of the structurally related isothiazolo[4,3-b]pyridine 12r is only 10 fold lower than its GAK affinity (Table 1). We thus tested our hypothesis that the aqueous solubility and/or cellular permeability of these compounds account for these observed differences. We measured the solubility of compounds 4, 12h and 12r, as well as of three reference compounds (propranolol, ketoconazole and tamoxifene) in phosphate buffered saline at a pH of 7.4 (Table 3). Compound 12r exhibited very poor aqueous solubility yet potent antiviral activity, compound 4 demonstrated low solubility and intermediate antiviral activity, whereas compound 12h demonstrated high solubility but reduced antiviral activity relative to compounds 4 and 12r (Table 3). The membrane permeability properties of these compounds was then measured via MDR1-MDCK permeability assays. The permeability measured in the apical to basolateral (A-to-B) direction was higher (compound 4) or slightly lower (compounds 12h, 12r) than that of metoprolol (a highly permeable compound) and much higher than that of the low permeable control, atenolol. In the basolateral to apical (B-to-A) direction, the permeability of compound 4 was comparable to that of metoprolol, whereas the permeability of compound 12r was lower than metoprolol’s. Notably, compound 12h exhibited permeability in the B-A direction that was higher than that of quinidine’s, a compound that is subjected to efflux (Table 3). Accordingly, the calculated efflux ratios (B-to-A/A-to-B) (Table 3) suggest that compound 12h is likely subjected to efflux, which may explain in part its diminished anti-DENV activity relative to compounds 4 and 12r. In contrast, compound 12r has reduced efflux compared to compounds 4 and 12h, suggesting that it may achieve a higher intracellular concentration, contributing to its enhanced antiviral activity.
Table 3:
Solubility (in PBS) and Permeability values of compounds 4, 12h and 12r
| Compound | Solubility (μM) | Papp × 106 (cm/s) | Efflux ratioc | |
|---|---|---|---|---|
| 4 | 3.44 ± 0.03 | A-Ba | 45.45 ± 0.07 | 0.68 |
| B-Ab | 30.80 ± 0.08 | |||
| 12h | 51.30 ± 0.02 | A-Ba | 30.32 ± 0.01 | 1.43 |
| B-Ab | 43.48 ± 0.02 | |||
| 12r | 0.52 ± 0.23 | A-Ba | 31.33 ± 0.03 | 0.58 |
| B-Ab | 18.28 ± 0.26 | |||
| Propanolol | 117 ± 0.15 | NDd | ||
| Ketoconazole | 32.00 ± 0.03 | NDd | ||
| Tamoxifene | 1.32 ± 0.09 | NDd | ||
| Metoprolol | NDd | A-Ba | 39.29 ± 0.04 | 0.78 |
| B-Ab | 30.66 ± 0.00 | |||
| Atenolol | NDd | A-Ba | 1.21 ± 0.00 | 1.03 |
| B-Ab | 1.25 ± 0.00 | |||
| Quinidine | NDd | A-Ba | 13.03 ± 0.01 | 3.22 |
| B-Ab | 41.99 ± 0.01 |
A-B: Apical to Basolateral
B-A: Basolateral to Apical
Efflux ratio: B-A/A-B
ND: not determined
Taken together, these data suggest that differences in the efflux ratio of these compounds, but not their aqueous solubility, may in part account for the measured differences between their GAK affinity and antiviral activity. Additionally, GAK binding rather than enzymatic activity was measured. It is therefore possible that the degree of the functional inhibition of GAK activity by these compounds is different from their GAK affinity, particularly in a cellular environment where high ATP concentrations and other cellular proteins are present.
Conclusions
Based on our earlier discovery of isothiazolo[4,3-b]pyridine (compound 4) as a potent and selective GAK inhibitor with moderate antiviral activity, we embarked on an optimization campaign aimed at improving the antiviral activity while maintaining potent and selective GAK inhibition. Synthesis of novel isothiazolo[4,3-b]pyridines with structural variation at position 3 of the scaffold led to the discovery of 3-(cis-2,6-dimethylmorpholino)-6-(4-amino-3-methoxypheny)isothiazolo[4,3-b]pyridine 12r. Compound 12r maintains potent activity and high selectivity to GAK and displays significantly improved antiviral activity against DENV with reduced cellular toxicity relative to the parental compound 4. Our gain-of-function assays provide evidence that GAK is an important molecular target underlying the antiviral effect of compound 12r. Compound 12r treatment exhibits antiviral efficacy in human primary dendritic cells with minimal toxicity to host cells, further supporting the biological relevance of this approach. Lastly, our finding that compound 12r also demonstrates antiviral activity against EBOV and CHIKV establishes the broad-spectrum potential of this and related GAK inhibitors.
Experimental section
Chemistry
For all reactions, analytical grade solvents were used. All moisture-sensitive reactions were carried out in oven-dried glassware (125 °C). 1H and 13C NMR spectra were recorded on a Bruker Avance 300 MHz instrument (1H NMR, 300 MHz; 13C NMR, 75 MHz), 500 MHz instrument (1H NMR, 500 MHz; 13C NMR, 125 MHz) or a 600 MHz instrument (1H NMR, 600 MHz; 13C NMR, 150 MHz), using tetramethylsilane as internal standard for 1H NMR spectra and DMSO-d6 (39.5 ppm) or CDCl3 (77.2 ppm) for 13C NMR spectra. Abbreviations used are s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, b = broad. Coupling constants are expressed in Hz. High resolution mass spectra were acquired on a quadrupole orthogonal acceleration time-of-flight mass spectrometer (Synapt G2 HDMS, Waters, Milford, MA). Samples were infused at 3uL/min and spectra were obtained in positive or negative ionization mode with a resolution of 15000 (FWHM) using leucine enkephalin as lock mass. Precoated aluminum sheets (Fluka silica gel/TLC-cards, 254 nm) were used for TLC. Column chromatography was performed on silica gel 0.060 – 0.200 mm, 60 Å (Acros Organics). Purity of final compounds was verified to be >95% by HPLC analysis. HPLC conditions to asses purity were as follows: Shimadzu HPLC equipped with a LC-20AT pump, DGU-20A5 degasser, and a SPD-20A UV-VIS detector; Symmetry C18 column (5 μm, 4.6 mm × 150 mm); gradient elution of H2O/CH3CN from 95/5 or 70/30 to 5/95 over 25 minutes; flow rate 1 mL/min; wavelength, UV 254 nm. Preparative HPLC purifications were performed using a Phenomenex Gemini 110A column (C18, 10 μM, 21.2 mm × 250 mm).
3-Amino-5-bromopyridine-2-carbonitrile (6)
To a stirred solution of iron powder (4.900 g, 87.7 mmol) in acetic acid (50 mL) at room temperature, was added 5-bromo-3-nitropyridine-2-carbonitrile 5 (10.00 g, 43.9 mmol) in one portion. The resulting reaction mixture was stirred for 3 hours. After completion, ethyl acetate was added and the reaction mixture was filtered through a paper filter. The filtered cake was washed thoroughly with ethyl acetate and the filtrate was evaporated under reduced pressure. The crude residue was purified by silica gel flash column chromatography (using dichloromethane as mobile phase) to yield the title compound (4.845 g, 56 %). 1H NMR (300 MHz, DMSO-d6): δ = 8.08 – 7.66 (m, 1H), 7.55 – 7.26 (m, 1H), 6.58 (bs, 2H) ppm. HRMS m/z [M+H]+ calcd for C6H4BrN3: 197.96618, found 197.9656.
3-Amino-5-bromo-2-pyridinecarbothioamide (7)
To a solution of 3-amino-5-bromopyridine-2-carbonitrile 6 (5.395 g, 25.00 mmol) in absolute ethanol (50 mL) was added Lawesson’s reagent (20.22 g, 50.0 mmol). The mixture was refluxed for 24 hours. When the starting material was completely consumed according to TLC analysis, the solvent was evaporated under reduced pressure. The residue was redissolved in water and extracted three times with dichloromethane. The combined organic layers were evaporated under reduced pressure. The crude residue was purified by silica gel flash column chromatography (using a mixture of heptane/acetone in a ratio of 60:40 as mobile phase) yielding the title compound (4.513 g, 78 %). 1H NMR (300 MHz, DMSO) δ 9.64 (bs, 1H), 9.60 (bs, 1H), 7.83 (d, J = 2.0 Hz, 1H), 7.81 (s, 1H), 7.50 (d, J = 2.0 Hz, 1H) ppm.
3-Amino-6-bromoisothiazolo[4,3-b]pyridine (8)
To a solution of 3-amino-5-bromopyridine-2-carbothioamide 7 (2.0 g, 8.62 mmol) in methanol (30 mL) at 0 °C was added dropwise a 30% aqueous H2O2 solution (2.22 mL, 21.54 mmol). The mixture was stirred overnight at room temperature. After disappearance of the starting material, the mixture was cooled to 0 °C. The precipitate was filtered off and washed with cold methanol to yield the title compound (1.0 g, 50 %). 1H NMR (300 MHz, DMSO) δ 8.29 (d, J = 2.0 Hz, 1H), 8.07 (s, 2H), 8.03 (d, J = 2.0 Hz, 1H) ppm. HRMS m/z [M+H]+ calcd for C6H6BrN3S: 229.9383, found 229.9380.
3,6-Dibromoisothiazolo[4,3-b]pyridine (9)
A solution of 3-amino-6-bromoisothiazolo[4,3-b]pyridine 8 (1.1 g, 4.78 mmol) in HBr (50 mL) was stirred for 10 min at room temperature, and then CuBr was added in one portion (1.37 g, 9.56 mmol, 2.0 equiv). The resulting mixture was cooled to 0 °C. A solution of sodium nitrite (0.99 g, 14.34 mmol, 3.0 equiv) in H2O (15 mL) was added dropwise over a period of 30 min. The reaction mixture was stirred for 2 h at 0 °C and overnight at room temperature. The mixture was cooled to 0 °C and carefully neutralized with a 30% aqueous NaOH solution. The formed precipitate was filtered over Celite and thoroughly washed with dichloromethane and methanol. The filtrate was evaporated in vacuo and the crude residue was purified by silica gel flash chromatography (using a mixture of cyclohexane and ethylacetate in a ratio of 95:5 as mobile phase), yielding the title compound (1.1 g, 78%). 1H NMR (300 MHz, DMSO-d6): δ 8.92 (d, J = 2.04 Hz, 1H), 8.70 (d, J = 2.01 Hz, 1H) ppm. HRMS m/z [M+H]+ calcd for C6H2Br2N2S: 292.8379, found 292.8373.
Synthesis of 6-bromo-3-substituted-isothiazolo[4,3-b]pyridines (10a-t)
General procedure
To a solution of 3,6-dibromoisothiazolo[4,3-b]pyridine 9 in ethanol or as specified was added an appropriate nitrogen nucleophile (3 equivalents or specified otherwise). The reaction was stirred at reflux overnight or as specified. When TLC analysis indicated complete disappearance of the starting material, the solvent was evaporated in vacuo and the crude residue was purified either by silica gel flash chromatography or extraction with dichloromethane yielding the 3-substituted-6-bromoisothiazolo[4,3-b]pyridine derivatives. The following compounds were made according to this procedure:
6-Bromo-3-(piperidin-1-yl)isothiazolo[4,3-b]pyridine (10a)
This compound was prepared from compound 9 (0.68 mmol, 200 mg) and piperidine (2.04 mmol, 174 mg). The product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/ethyl acetate in a ratio of 90:10 as mobile phase) yielding the title compound (180 mg, 60%). 1H NMR (300 MHz, DMSO-d6): δ = 8.32 (d, J = 2.0 Hz, 1H), 8.07 (d, J = 2.0 Hz, 1H), 4.00 – 3.70 (m, 4H), 1.74 – 1.62 (m, 6H) ppm. HRMS m/z [M+H]+ calcd for C11H12BrN3S: 298.0009, found 298.0014.
6-Bromo-3-(4,4-difluoropiperidin-1-yl)isothiazolo[4,3-b]pyridine (10b)
This compound was prepared from compound 9 (0.34 mmol, 100 mg), 4,4-difluoropiperidine hydrochloride (1.02 mmol, 161 mg) and DIPEA (1.70 mmol, 296 μL). The product was purified by silica gel flash column chromatography (using a mixture of heptane/ethyl acetate in a ratio of 60:40 as mobile phase) yielding the title compound (110 mg, 97%). 1H NMR (300 MHz, DMSO-d6): δ = 8.40 (d, J = 1.4 Hz, 1H), 8.16 (d, J = 1.4 Hz, 1H), 4.20 – 3.98 (m, 4H), 2.32 – 2.16 (m, 4H) ppm. HRMS m/z [M+H]+ calcd for C11H12BrN3S: 333.9820, found 333.9823.
1-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)piperidine-4-carboxamide (10c)
This compound was prepared from compound 9 (0.68 mmol, 200 mg) and piperidine-4-carboxamide (1.36 mmol, 174 mg, 2 eq). The crude product was resuspended in water and washed three times with dichloromethane. The combined organic layers evaporated in vacuo yielding the title compound (160 mg, 69%). The compound was used as such for further reaction.
1-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)piperidine-4-carbonitrile (10d)
This compound was prepared from compound 9 (0.68 mmol, 200 mg) and piperidine-4-carbonitrile (1.36 mmol, 150 mg, 2 eq). The crude product was resuspended in water and washed with dichloromethane three times. The combined organic phases were evaporated in vacuo to yield the title compound (190 mg, 86%). 1H NMR (300 MHz, DMSO-d6): δ = 8.37 (d, J = 2.0 Hz, 1H), 8.13 (d, J = 2.0 Hz, 1H), 4.29 – 4.02 (m, 2H), 3.86 – 3.58 (m, 2H), 3.29 – 3.08 (m, 1H), 2.15 – 2.00 (m, 2H), 2.00 – 1.85 (m, 2H) ppm. HRMS m/z [M+H]+ calcd for C12H11BrN4S: 322.9961, found 322.9958.
1-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)piperidine-3-carboxamide (10e)
This compound was prepared from compound 9 (0.68 mmol, 200 mg) and piperidine-3-carboxamide (1.36 mmol, 174 mg, 2 eq). The crude product was resuspended in water and washed three times with dichloromethane. The combined organic layers were evaporated in vacuo to yield the title compound (150 mg, 65%). 1H NMR (300 MHz, DMSO-d6): δ = 8.36 (d, J = 2.0 Hz, 1H), 8.10 (d, J = 2.0 Hz, 1H), 7.44 (bs, 1H), 6.99 (bs, 1H), 4.51 (dd, J = 60.1, 11.6 Hz, 2H), 3.51 – 3.12 (m, 2H), 2.00 – 1.79 (m, 2H), 1.77 – 1.58 (m, 2H) ppm. HRMS m/z [M+H]+ calcd for C12H13BrN4OS: 341.0067, found 341.0064.
4-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)thiomorpholine 1,1-dioxide (10f)
This compound was prepared from compound 9 (1.02 mmol, 300 mg) and thiomorpholine-1,1-dioxide (3.06 mmol, 414 mg). The product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio 95:5 as mobile phase) generating the title compound (90 mg, 25%). 1H NMR (300 MHz, DMSO-d6): δ = 8.44 (d, J = 2.1 Hz, 1H), 8.21 (d, J = 2.1 Hz, 1H), 4.42 (m, 4H), 3.42 (m, 4H) ppm. HRMS m/z [M+H]+ calcd for C10H10BrN3O2S2: 347.9471, found 347.9480.
6-Bromo-N-(tetrahydro-2H-pyran-4-yl)isothiazolo[4,3-b]pyridin-3-amine (10g)
This compound was prepared from compound 9 (0.68 mmol, 200 mg), 4-aminotetrahydro-2H-pyran hydrochloride (3.4 mmol, 468 mg) and DIPEA (3.4 mmol, 595 μL). The product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio 95:5 as mobile phase) yielding the title compound (90 mg, 42%). 1H NMR (300 MHz, DMSO-d6): δ = 8.81 (d, J = 8.07 Hz, 1H), 8.30 (d, J = 1.98 Hz, 1H), 8.05 (d, J = 1.86 Hz, 1H), 3.92 (dd, J = 1.86 Hz, J = 10.08 Hz, 2H), 3.58 (m, 1H), 3.42 (td, J = 2.1 Hz, J = 11.78 Hz, 2H), 1.96 (dd, J = 12.7 Hz, J = 2.2 Hz, 2H), 1.68 (m, 2H) ppm. HRMS m/z [M+H]+ calcd for C11H12BrN3OS: 313.9958, found 313.9954.
4-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)piperazin-2-one (10h)
This compound was prepared from compound 9 (0.68 mmol, 200 mg) and 2-piperazinone (2.06 mmol, 206 mg) and stirred for 7 days. The crude product was purified by silica gel flash column chromatography (eluting with a mixture of dichloromethane/MeOH in a ratio of 95:5 as mobile phase) yielding the title compound (150 mg, 70%). 1H NMR (300 MHz, DMSO-d6): δ = 8.40 – 8.33 (m, 1H), 8.22 – 8.04 (m, 1H), 4.42 – 4.29 (m, 2H), 4.24 – 3.94 (m, 2H), 3.65 – 3.49 (m, 2H) ppm.
4-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)-1,4-oxazepane (10i)
This compound was prepared from compound 9 (0.34 mmol, 100 mg) and 1,4-oxazepane (1.02 mmol, 103 mg). The product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio 95:5 as mobile phase) affording the title compound (100 mg, 96%). 1H NMR (300 MHz, DMSO-d6): δ = 8.27 (s, 1H), 8.04 (s, 1H), 4.18 – 4.12 (m, 2H), 4.069 – 4.0 (m, 2H), 3.87 (d, J = 3.9 Hz, 2H), 3.76 – 3.68 (m, 2H), 2.08 – 1.91 (m, 2H) ppm. HRMS m/z [M+H]+ calcd for C11H12BrN3OS: 313.9957, found 313.9959.
6-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)-2-oxa-6-azaspiro[3.3]heptane (10j)
This compound was prepared from compound 9 (0.34 mmol, 100 mg) and 2-oxa-6-azaspiro[3.3]heptane (1.02 mmol, 206 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/ethyl acetate in a ratio of 90:10 as mobile phase) yielding the title compound (90 mg, 85%). 1H NMR (300 MHz, CDCl3): δ = 8.29 – 8.16 (m, 1H), 7.98 – 7.81 (m, 1H), 4.86 (s, 4H), 4.61 – 4.47 (m, 4H) ppm. HRMS m/z [M+H]+ calcd for C11H10BrN3OS: 311.9801, found 311.9815.
3-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)-8-oxa-3-azabicyclo[3.2.1]octane (10k)
This compound was prepared from compound 9 (0.68 mmol, 200 mg) and 8-oxa-3-azabicyclo[3.2.1]octane (1.36 mmol, 154 mg, 2 eq). The crude product was purified by silica gel flash column chromatography (using a mixture of heptane/acetone in a ratio 8:2 as mobile phase) yielding the title compound (155 mg, 70%). 1H NMR (300 MHz, DMSO-d6): δ = 8.35 (d, J = 2.1 Hz, 1H), 8.13 (d, 1H), 4.51 (s, 2H), 4.20 (d, J = 12.4 Hz, 2H), 3.43 (dd, J = 12.4, 2.1 Hz, 2H), 2.05 – 1.78 (m, 4H) ppm. HRMS m/z [M+H]+ calcd for C12H12BrN3OS: 325.9957, found 325.9964.
4-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)-2-methylmorpholine (10l)
This compound was prepared from compound 9 (0.34 mmol, 100 mg) and 2-methylmorpholine (1.02 mmol, 103 μL). The product was purified by silica gel flash column chromatography (eluting with a mixture of dichloromethane/acetone in a ratio 95:5), affording the title compound (93 mg, 87%). 1H NMR (300 MHz, DMSO-d6): δ = 8.37 (d, 1H), 8.14 (d, J = 2.1 Hz, 1H), 4.47 – 4.433 (m, 2H), 4.01 – 3.96 (m, 1H), 3.83 – 3.68 (m, 2H), 3.36 – 3.23 (m, 2H), 3.00 (dd, J = 12.4, 10.5 Hz, 1H), 1.18 (d, J = 6.2 Hz, 3H) ppm.
(S)-4-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)-2-methylmorpholine (10m)
This compound was prepared from compound 9 (0.68 mmol, 200 mg), (S)-2-methylmorpholine hydrochloride (2.04 mmol, 281 mg) and DIPEA (2.04 mmol, 264 mg). The crude residue was purified by silica gel flash column chromatography (using a mixture of heptane/ethyl acetate in a ratio 8:2 as mobile phase) yielding the title compound (200 mg, 93 %). 1H NMR (300 MHz, DMSO-d6) δ 8.39 (d, J = 2.1 Hz, 1H), 8.15 (d, J = 2.1 Hz, 1H), 4.53 – 4.25 (m, J = 29.3, 12.6 Hz, 2H), 4.06 – 3.92 (m, 1H), 3.88 – 3.66 (m, 2H), 3.37 – 3.24 (m, 1H), 3.02 (dd, J = 12.5, 10.5 Hz, 1H), 1.18 (d, J = 6.2 Hz, 3H) ppm. HRMS m/z [M+H]+ calcd for C11H12BrN3OS: 313.9958, found 313.9961.
(R)-4-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)-2-methylmorpholine (10n)
This compound was prepared from compound 9 (0.68 mmol, 200 mg) and (R)-2-methylmorpholine (2.04 mmol, 206 mg). The crude residue was purified by silica gel flash column chromatography (using a mixture of heptane/ethyl acetate in a ratio 8:2 as mobile phase), yielding the title compound (190 mg, 89%). 1H NMR (300 MHz, DMSO-d6): δ = 8.36 (d, J = 2.1 Hz, 1H), 8.13 (d, J = 2.1 Hz, 1H), 4.52 – 4.28 (m, 2H), 4.04 – 3.92 (m, 1H), 3.83 – 3.68 (m, 2H), 3.35 – 3.24 (m, 1H), 3.00 (dd, J = 12.4, 10.5 Hz, 1H), 1.18 (d, J = 6.2 Hz, 3H) ppm. HRMS m/z [M+H]+ calcd for C11H12BrN3OS: 313.9958, found 313.9954.
4-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)-2,2-dimethylmorpholine (10o)
This compound was prepared from compound 9 (0.68 mmol, 200 mg) and 3,3-dimethylmorpholine (2.04 mmol, 235 mg) in EtOH (5 mL). The crude product was purified by silica gel flash column chromatography (using a mixture of heptane/acetone in a ratio 80:20 as mobile phase) affording the title compound (200 mg, 90%). 1H NMR (300 MHz, CDCl3): δ = 8.24 (d, J = 1.9 Hz, 1H), 7.89 (d, J = 2.0 Hz, 1H), 4.02 – 3.79 (m, 4H), 3.75 (s, 2H), 1.32 (s, 6H) ppm. HRMS m/z [M+H]+ calcd for C12H14BrN3OS: 328.0114, found 328.0110.
(S)-4-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)-3-methylmorpholine (10p)
This compound was prepared from compound 9 (0.68 mmol, 200 mg) and (S)-3-methylmorpholine (2.04 mmol, 107 mg) in t-BuOH (10 mL). The crude residue was washed three times with a mixture of dichloromethane/water to yield the title compound (246 mg, 77%).
1H NMR (300 MHz, DMSO-d6): δ = 8.37 (d, J = 2.1 Hz, 1H), 8.14 (d, J = 2.1 Hz, 1H), 4.93 – 4.85 (m, 1H), 4.02 – 3.95 (m, 2H), 3.85 – 3.77 (m, 2H), 3.75 – 3.65 (m, 1H), 3.60 – 3.49 (m, 1H), 1.30 (d, J = 6.7 Hz, 3H) ppm. HRMS m/z [M+H]+ calcd for C11H12BrN3OS: 313.9958, found 313.9960.
(R)-4-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)-3-methylmorpholine (10q)
This compound was prepared from compound 9 (1.02 mmol, 300 mg) and (R)-3-methylmorpholine (3.06 mmol, 161 mg). The product was purified by silica gel flash column chromatography (using a mixture of heptane/ethyl acetate in a ratio of 60:40 as mobile phase) yielding the title compound (110 mg, 97%). 1H NMR (300 MHz, DMSO-d6): δ = 8.36 (d, J = 2.0 Hz, 1H), 8.14 (d, J = 2.0 Hz, 1H), 4.93 – 4.85 (m, 1H), 4.03 – 3.94 (m, 2H), 3.85 – 3.77 (m, 2H), 3.76 – 3.65 (m, 1H), 3.61 – 3.49 (m, 1H), 1.30 (d, J = 6.7 Hz, 3H) ppm.
6-Bromo-3-cis-2,6-dimethylmorpholinoisothiazolo[4,3-b]pyridine (10r)
This compound was prepared from compound 9 (1.02 mmol, 300 mg) and cis-2,6-dimethylmorpholine (3.06 mmol, 352 mg). The product was purified by silica gel flash column chromatography (using a mixture of heptane/ethyl acetate in a ratio of 90:10 as mobile phase) yielding the title compound (290 mg, 87%). 1H NMR (300 MHz, DMSO-d6) δ = 8.38 (d, J = 2.1 Hz, 1H), 8.14 (d, J = 2.1 Hz, 1H), 4.50 – 4.34 (m, 2H), 4.03 – 3.73 (m, 2H), 2.91 (dd, J = 12.5, 10.8 Hz, 2H), 1.20 (s, 3H), 1.17 (s, 3H) ppm. HRMS m/z [M+H]+ calcd for C12H14BrN3OS: 328.0114, found 328.0105.
trans-4-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)-2,6-dimethylmorpholine (10s)
This compound was prepared from compound 9 (0.51 mmol, 150 mg) and trans-2,6-dimethylmorpholine (1.53 mmol, 176 mg). The crude product was resuspended in water and washed three times with dichloromethane. The combined organic phases were evaporated in vacuo, yielding the title compound (159 mg, 95%). 1H NMR (300 MHz, CDCl3): δ = 8.26 (d, J = 1.9 Hz, 1H), 7.90 (d, J = 1.9 Hz, 1H), 4.32 – 4.10 (m, 2H), 3.95 (dd, J = 12.6, 3.3 Hz, 2H), 3.70 (dd, J = 12.6, 6.3 Hz, 3H), 1.33 (s, 3H), 1.30 (s, 3H) ppm. HRMS m/z [M+H]+ calcd for C12H14BrN3OS: 328.0114, found 328.0015.
tert-Butyl (1-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)piperidin-4-yl)carbamate (10t)
This compound was prepared from compound 9 (0.34 mmol, 100 mg) and tert-butyl piperidin-4-ylcarbamate (1.02 mmol, 206 mg). The crude product was purified by silica gel flash column chromatography (eluting with a mixture of heptane/acetone in a ratio of 80:20 as mobile phase), yielding the title compound (120 mg, 85%). 1H NMR (300 MHz, DMSO-d6): δ = 8.35 (d, J = 2.1 Hz, 1H), 8.10 (d, J = 2.1 Hz, 1H), 6.95 (bs, 1H), 4.47 (d, J = 13.3 Hz, 2H), 3.67 – 3.54 (m, 1H), 3.50 – 3.36 (m, 2H), 2.01 – 1.85 (m, 2H), 1.67 – 1.49 (m, 2H), 1.40 (s, 9H) ppm. HRMS m/z [M+H]+ calcd for C16H21BrN4O2S: 356.1176, found 356.1172.
Synthesis of 3-substituted-6-(4-amino-3-methoxyphenyl)isothiazolo[4,3-b]pyridines (11 and 12a-s)
General procedure
To a mixture of dioxane/water (in a ratio of 4:1) were added the appropriate 3-substituted-6-bromoisothiazolo[4,3-b]pyridine analogue 10a-t, 4-amino-3-methoxyphenylboronic acid pinacol ester (1.2 equiv) and potassium carbonate (2 equiv). The mixture was degassed and filled with nitrogen. Subsequently, Pd(PPh3)4 (1 mol % - 10 mol%) was added. The mixture was degassed a second time, filled with nitrogen and stirred at 100 °C. After completion of the reaction, solvents were evaporated. The crude residue was purified by silica gel flash chromatography and when needed further purified using RP-HPLC yielding the title compounds. All compounds submitted for biological assays had a purity of at least 95%. The following compounds were made according to this procedure:
tert-Butyl (1-(6-(4-amino-3-methoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)piperidin-4-yl)carbamate (11)
This compound was prepared from tert-butyl (1-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)piperidin-4-yl)carbamate 11 (0.25 mmol, 110 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio of 90:10 as mobile phase), yielding the title compound (90 mg, 79%). 1H NMR (300 MHz, CDCl3): δ = 8.63 (d, J = 2.1 Hz, 1H), 7.81 (d, J = 2.1 Hz, 1H), 7.13 (dd, J = 8.0, 1.9 Hz, 1H), 7.09 (d, J = 1.9 Hz, 1H), 6.81 (bs, 1H), 4.72 – 4.56 (m, 2H), 4.00 – 3.96 (m, 1H), 3.93 (s, 3H), 3.42 – 3.30 (m, 2H), 2.22 – 2.07 (m, 2H), 1.75 – 1.64 (m, 2H), 1.47 (s, 9H) ppm. HRMS m/z [M+H]+ calcd for C23H29N5O3S: 456.2064, found 456.2054.
2-Methoxy-4-(3-(piperidin-1-yl)isothiazolo[4,3-b]pyridin-6-yl)aniline (12a)
The title compound was prepared from 6-bromo-3-(piperidin-1-yl)isothiazolo[4,3-b]pyridine 10a (0.34 mmol, 100 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/ethyl acetate in a ratio ranging from 95:5 to 90:10 as mobile phase), yielding the title compound (60 mg, 52 %). 1H NMR (300 MHz, DMSO-d6): δ = 8.70 (d, J = 2.1 Hz, 1H), 7.84 (d, J = 2.1 Hz, 1H), 7.25 (d, J = 1.8 Hz, 1H), 7.20 (dd, J = 8.1, 1.9 Hz, 1H), 6.74 (d, J = 8.1 Hz, 1H), 5.07 (s, 2H), 3.94 – 3.89 (m, 4H), 3.89 (s, 3H), 1.77 – 1.66 (m, 6H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 172.5, 156.2, 146.8, 143.6, 138.8, 135.3, 132.5, 124.2, 122.1, 120.1, 113.9, 109.4, 55.6, 51.3, 24.9, 23.5 ppm. HRMS m/z [M+H]+ calcd for C18H20N4OS: 341.1431, found 341.1429.
4-(3-(4,4-Difluoropiperidin-1-yl)isothiazolo[4,3-b]pyridin-6-yl)-2-methoxyaniline (12b)
This compound was prepared from 6-bromo-3-(4,4-difluoropiperidin-1-yl)isothiazolo[4,3-b]pyridine 10b (0.24 mmol, 100 mg). The crude residue was purified by silica gel flash column chromatography (using a mixture of dichloromethane/ethyl acetate in a ratio 90:10 as mobile phase) yielding the title compound (49 mg, 54 %). 1H NMR (300 MHz, DMSO-d6): δ = 8.77 (d, J = 2.1 Hz, 1H), 7.90 (d, J = 2.1 Hz, 1H), 7.27 (d, J = 1.7 Hz, 1H), 7.22 (dd, J = 8.1, 1.8 Hz, 1H), 6.75 (d, J = 8.1 Hz, 1H), 5.09 (s, 2H), 4.20 – 3.98 (m, 4H), 3.89 (s, 3H), 2.42 – 2.10 (m, 4H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 171.6, 156.2, 146.8, 144.6, 138.9, 135.4, 132.7, 124.0, 122.2, 120.2, 113.9, 109.4, 55.6, 47.4, 32.8 ppm. HRMS m/z [M+H]+ calcd for C18H18F2N4OS: 377.1242, found 377.1260.
1-(6-(4-Amino-3-methoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)piperidine-4-carboxamide (12c)
The title compound was prepared from 1-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)piperidine-4-carboxamide 10c (0.29 mmol, 100 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/ethyl acetate in a ratio of 90:10 as mobile phase) yielding the title compound (27 mg, 24%). 1H NMR (300 MHz, DMSO-d6): δ = 8.72 (d, J = 2.0 Hz, 1H), 7.86 (d, J = 2.0 Hz, 1H), 7.39 (bs, 1H), 7.26 (d, J = 1.6 Hz, 1H), 7.21 (dd, J = 8.1, 1.8 Hz, 1H), 6.89 (bs, 1H), 6.74 (d, J = 8.1 Hz, 1H), 5.09 (bs, 2H), 4.65 – 4.53 (m, 2H), 3.88 (s, 3H), 3.38 – 3.22 (m, 3H), 1.95 – 1.67 (m, 4H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 175.9, 172.3, 156.2, 146.8, 143.9, 138.8, 135.3, 132.6, 124.2, 122.1, 120.2, 113.9, 109.4, 55.6, 50.0, 40.9, 27.71ppm. HRMS m/z [M+H]+ calcd for C19H21N5O2S: 384.1489, found 384.1483.
1-(6-(4-Amino-3-methoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)piperidine-4-carbonitrile (12d)
This compound was prepared from 1-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)piperidine-4-carbonitrile 10d (0.31 mmol, 100 mg). The crude product was purified by silica gel flash column chromatography (with a mixture of dichloromethane/acetone in a ratio of 95:5 as mobile phase) yielding the title compound (106 mg, 94%). 1H NMR (300 MHz, DMSO-d6): δ = 8.75 (d, J = 1.8 Hz, 1H), 7.89 (d, J = 1.8 Hz, 1H), 7.29 – 7.18 (m, 2H), 6.74 (d, J = 8.1 Hz, 1H), 5.10 (bs, 2H), 4.34 – 4.08 (m, 2H), 3.89 (s, 3H), 3.84 – 3.70 (m, 2H), 3.32 – 3.14 (m, 1H), 2.05 – 1.88 (m, 2H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 172.1, 156.2, 146.8, 144.3, 138.9, 135.4, 132.6, 124.0, 122.2, 121.9, 120.2, 113.9, 109.4, 55.6, 48.6, 27.4, 25.0 ppm. HRMS m/z [M+H]+ calcd for C19H19N5OS: 366.1383, found 366.1375.
1-(6-(4-Amino-3-methoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)piperidine-3-carboxamide (12e)
This compound was prepared from 1-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)piperidine-3-carboxamide 10e (0.29 mmol, 100 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/ethyl acetate in a ratio of 90:10 as mobile phase), yielding the title compound (63 mg, 57%). 1H NMR (300 MHz, DMSO-d6): δ = 8.74 (d, J = 1.9 Hz, 1H), 7.86 (d, J = 1.9 Hz, 1H), 7.47 (bs, 1H), 7.29 – 7.17 (m, 2H), 7.00 (bs, 1H), 6.74 (d, J = 8.0 Hz, 1H), 5.09 (bs, 2H), 4.81 – 4.64 (m, 1H), 4.47 – 4.35 (m, 1H), 3.89 (s, 3H), 3.35 – 3.21 (m, 2H), 2.67 – 2.54 (m, 1H), 2.10 – 1.59 (m, 4H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 174.67, 172.3, 156.2, 146.8, 143.9, 138.8, 135.3, 132.5, 124.2, 122.1, 120.2, 113.9, 109.4, 55.6, 52.5, 50.9, 41.4, 27.4, 23.8 ppm. HRMS m/z [M+H]+ calcd for C19H21N5O2S: 384.1489, found 384.1483.
4-(6-(4-Amino-3-methoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)thiomorpholine 1,1-dioxide (12f)
This compound was prepared from 4-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)thiomorpholine 1,1-dioxide 10f (0.26 mmol, 90 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio of 95:5 as mobile phase), yielding the title compound (53 mg, 51 %). 1H NMR (300 MHz, DMSO-d6): δ = 8.79 (d, J = 2.1 Hz, 1H), 7.93 (d, J = 2.1 Hz, 1H), 7.28 (d, J = 1.8 Hz, 1H), 7.23 (dd, J = 8.1, 1.9 Hz, 1H), 6.75 (d, J = 8.1 Hz, 1H), 5.11 (bs, 2H), 4.48 – 4.41 (m, 4H), 3.89 (s, 3H), 3.47 – 3.40 (m, 4H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 170.6, 156.3, 146.9, 144.9, 139.0, 135.5, 132.7, 123.9, 122.3, 120.3, 113.9, 109.4, 55.7, 50.1, 49.1 ppm. HRMS m/z [M+H]+ calcd for C17H18N4O3S2: 391.0893, found 391.0883.
6-(4-Amino-3-methoxyphenyl)-N-(tetrahydro-2H-pyran-4-yl)isothiazolo[4,3-b]pyridin-3-amine (12g)
This compound was prepared from 6-bromo-N-(tetrahydro-2H-pyran-4-yl)isothiazolo[4,3-b]pyridin-3-amine 10g (0.25 mmol, 80 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio 90:10 as mobile phase) yielding the title compound (25 mg, 7 %). 1H NMR (300 MHz, DMSO-d6): δ = 8.63 (d, J = 2.0 Hz, 1H), 8.47 (s, 1H), 7.80 (d, J = 2.0 Hz, 1H), 7.23 (d, J = 1.8 Hz, 1H), 7.20 (dd, J = 8.1, 1.9 Hz, 1H), 6.75 (d, J = 8.0 Hz, 1H), 5.06 (bs, 2H), 3.95 (m, 2H), 3.89 (s, 3H), 3.58 (m, 1H), 3.43 (td, J = 10.9 Hz, 3H), 1.99 (dd, J = 12.7 Hz, J = 2.2 Hz, 2H), 1.72 (m, 2H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 170.9, 154.8, 146.9, 143.4, 138.7, 136.3, 132.7, 124.6, 121.8, 120.2, 113.9, 109.5, 66.0, 55.7, 54.4, 31.8 ppm. HRMS m/z [M+H]+ calcd for C18H20N4O2S: 357.1380, found 357.1379.
4-(6-(4-Amino-3-methoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)piperazin-2-one (12h)
This compound was prepared from 6-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)-2-oxa-6-azaspiro[3.3]heptane 10h (0.19 mmol, 60 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio of 90:10 as mobile phase). The residue was further purified by RP-HPLC (eluting isocratically with a mixture of acetonitrile/water in a ratio of 4:6) yielding the title compound (30 mg, 45%). 1H NMR (300 MHz, DMSO-d6): δ = 8.79 – 8.75 (m, 1H), 8.35 (bs, 1H), 7.92 – 7.88 (m, 1H), 7.29 – 7.19 (m, 2H), 6.75 (d, J = 8.1 Hz, 1H), 5.10 (bs, 2H), 4.43 – 4.37 (m, 2H), 4.21 – 4.14 (m, 2H), 3.93 – 3.85 (s, 3H), 3.50 – 3.42 (m, 2H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 170.5, 165.7, 156.0, 146.9, 144.4, 138.9, 135.6, 124.2, 122.1, 120.2, 114.0, 109.5, 55.7, 52.9, 46.6 ppm. HRMS m/z [M+H]+ calcd for C17H17N5O2S: 311.9801, found 311.9815.
4-(3-(1,4-Oxazepan-4-yl)isothiazolo[4,3-b]pyridin-6-yl)-2-methoxyaniline (12i)
This compound was prepared from 4-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)-1,4-oxazepane 10i (0.32 mmol, 100 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio 95:5 as mobile phase) yielding the title compound (60 mg, 53 %). 1H NMR (300 MHz, DMSO-d6): δ = 8.68 (d, J = 2.1 Hz, 1H), 7.83 (d, J = 2.1 Hz, 1H), 7.26 (d, J = 1.8 Hz, 1H), 7.21 (dd, J = 8.1, 1.9 Hz, 1H), 6.74 (d, J = 8.1 Hz, 1H), 5.07 (bs, 2H), 4.24 – 4.18 (m, 2H), 4.08 (t, J = 5.9 Hz, 2H), 3.93 – 3.86 (m, 4H), 3.77 – 3.68 (m, 2H), 2.11 – 1.99 (m, 2H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 171.0, 156.0, 146.8, 143.3, 138.8, 135.3, 131.8, 124.3, 122.0, 120.1, 114.0, 109.4, 69.7, 68.6, 55.6, 54.6, 51.9, 29.0 ppm. HRMS m/z [M+H]+ calcd for C18H20N4O2S: 357.1380, found 357.1379.
4-(3-(2-Oxa-6-azaspiro[3.3]heptan-6-yl)isothiazolo[4,3-b]pyridin-6-yl)-2-methoxyaniline (12j)
This compound was prepared from 6-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)-2-oxa-6-azaspiro[3.3]heptane 10j (0.19 mmol, 60 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio of 9:1 as mobile phase) yielding the title compound (50 mg). This residue was further purified by RP-HPLC (eluting with a mixture of acetonitrile/water in a ratio of 4:6 as mobile phase) yielding the title compound (30 mg, 45%). 1H NMR (300 MHz, CDCl3): δ = 8.6 (d, J = 2.0 Hz, 1H), 7.79 (d, J = 2.0 Hz, 1H), 7.12 (dd, J = 8.0, 1.9 Hz, 1H), 7.07 (d, J = 1.8 Hz, 1H), 6.81 (d, J = 8.0 Hz, 1H), 4.92 (s, 4H), 4.62 (s, 4H), 3.98 (bs, 2H), 3.93 (s, 3H) ppm. 13C NMR (75 MHz, CDCl3): δ = 171.7, 155.9, 147.9, 146.0, 137.2, 136.7, 134.89, 127.9, 123.8, 120.6, 115.3, 109.6, 811, 65.9, 55.9, 42.0 ppm. HRMS m/z [M+H]+ calcd for C18H18N4O2S: 355.1223, found 355.1220.
4-(3-(8-Oxa-3-azabicyclo[3.2.1]octan-3-yl)isothiazolo[4,3-b]pyridin-6-yl)-2-methoxyaniline (12k)
This compound was prepared from 3-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)-8-oxa-3-azabicyclo[3.2.1]octane 10k (0.31 mmol, 100 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/ethyl acetate in a ratio ranging from 99:1 to 90:10 as mobile phase) yielding the title compound (93 mg, 81%). 1H NMR (300 MHz, CDCl3): δ = 8.61 (d, J = 2.1 Hz, 1H), 7.81 (d, J = 2.1 Hz, 1H), 7.13 (dd, J = 8.0, 1.9 Hz, 1H), 7.08 (d, J = 1.9 Hz, 1H), 6.81 (d, J = 8.0 Hz, 1H), 4.55 (s, 2H), 4.32 (d, J = 12.4 Hz, 2H), 3.98 (bs, 2H), 3.93 (s, 3H), 3.52 (dd, J = 12.3, 2.4 Hz, 2H), 2.15 – 2.01 (m, 4H) ppm. 13C NMR (75 MHz, CDCl3): δ = 174.0, 156.9, 147.9, 144.7, 137.2, 136.3, 134.0, 127.9, 124.2, 120.5, 115.3, 109.6, 74.1, 56.0, 55.9, 28.4 ppm. HRMS m/z [M+H]+ calcd for C19H20N4O2S: 369.1380, found 369.1370.
2-Methoxy-4-(3-(2-methylmorpholino)isothiazolo[4,3-b]pyridin-6-yl)aniline (12l)
This compound was prepared from 4-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)-2-methylmorpholine 10l (0.29 mmol, 90 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio of 95:5 as mobile phase) yielding the title compound (53 mg, 51 %). 1H NMR (300 MHz, DMSO-d6): δ = 8.75 (d, J = 2.0 Hz, 1H), 7.89 (d, J = 2.0 Hz, 1H), 7.27 (d, J = 1.6 Hz, 1H), 7.22 (dd, J = 8.1, 1.8 Hz, 1H), 6.75 (d, J = 8.1 Hz, 1H), 5.09 (bs, 2H), 4.45 (d, J = 12.5 Hz, 2H), 4.02 – 3.71 (m, 6H), 3.32 – 3.16 (m, 1H), 2.97 (q, J = 12.3, 10.6 Hz, 1H), 1.19 (d, J = 6.2 Hz, 3H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 172.3, 156.2, 146.8, 144.4, 138.9, 135.5, 132.8, 124.1, 122.2, 120.2, 113.9, 109.4, 70.8, 65.3, 55.6, 49.3, 18.7 ppm. HRMS m/z [M+H]+ calcd C18H20N4O2S: for 357.1380, found 357.1385.
(S)-2-Methoxy-4-(3-(2-methylmorpholino)isothiazolo[4,3-b]pyridin-6-yl)aniline (12m)
This compound was prepared from (S)-4-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)-2-methylmorpholine 10m (0.32 mmol, 100 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio of 95:5 as mobile phase), yielding the title compound (14 mg, 12%). 1H NMR (300 MHz, DMSO-d6): δ = 8.75 (d, J = 2.1 Hz, 1H), 7.89 (d, J = 2.1 Hz, 1H), 7.27 (d, J = 1.8 Hz, 1H), 7.22 (dd, J = 8.1, 1.9 Hz, 1H), 6.74 (d, J = 8.1 Hz, 1H), 5.09 (bs, 2H), 4.46 (d, J = 11.8 Hz, 2H), 3.99 (dd, J = 11.7, 2.7 Hz, 1H), 3.89 (s, 3H), 3.86 – 3.73 (m, 2H), 3.30 – 3.21 (m, 1H), 2.97 (dd, J = 12.3, 10.6 Hz, 1H), 1.19 (d, J = 6.2 Hz, 3H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 172.3, 156.2, 146.9, 144.4, 138.9, 135.5, 132.8, 124.1, 122.2, 120.2, 113.9, 109.4, 70.8, 65.3, 55.7, 55.6, 49.3, 18.7 ppm. HRMS m/z [M+H]+ calcd for C18H20N4O2S: 357.1380, found 357.1376.
(R)-2-Methoxy-4-(3-(2-methylmorpholino)isothiazolo[4,3-b]pyridin-6-yl)aniline (12n)
This compound was prepared from (S)-4-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)-2-methylmorpholine (0.32 mmol, 100 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio of 95:5 as mobile phase) yielding the title compound (23 mg, 20%). 1H NMR (300 MHz, CDCl3): δ = 8.61 (s, 1H), 7.80 (s, 1H), 7.11 (d, J = 8.0 Hz, 1H), 7.06 (s, 1H), 6.78 (d, J = 8.0 Hz, 1H), 4.56 – 4.36 (m, 2H), 4.06 – 3.80 (m, 1H), 3.36 – 3.17 (m, 1H), 3.01 – 2.85 (m, 1H), 1.28 (d, J = 6.2 Hz, 4H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 172.3, 156.2, 146.9, 144.4, 138.9, 135.5, 132.8, 124.1, 122.2, 120.2, 113.9, 109.4, 70.8, 65.3, 55.6, 49.3, 18.7 ppm. HRMS m/z [M+H]+ calcd for C18H20N4O2S: 357.1380, found 357.1378.
4-(3-(3,3-Dimethylpiperidin-1-yl)isothiazolo[4,3-b]pyridin-6-yl)-2-methoxyaniline (12o)
This compound was prepared from 6-bromo-3-(3,3-dimethylpiperidin-1-yl)isothiazolo[4,3-b]pyridine 10o (0.30 mmol, 100 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio of 9:1 as mobile phase). The residue was further purified using RP-HPLC (eluting with a linear gradient of 30% - 95% acetonitrile in water) yielding the title compound (23 mg, 21%). 1H NMR (300 MHz, DMSO-d6): δ = 8.74 (d, J = 1.8 Hz, 1H), 7.87 (d, J = 1.8 Hz, 1H), 7.28 – 7.18 (m, 2H), 6.74 (d, J = 8.1 Hz, 1H), 5.09 (bs, 2H), 3.89 (s, 3H), 3.88 – 3.83 (m, 4H), 3.80 (s, 2H), 1.27 (s, 6H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 172.6, 156.3, 144.1, 138.9, 135.5, 122.2, 120.2, 114.0, 109.5, 71.2, 59.4, 58.6, 55.7, 49.8, 24.4 ppm. HRMS m/z [M+H]+ calcd for C20H24N4OS: 371.1536, found 371.1533.
(S)-2-Methoxy-4-(3-(3-methylmorpholino)isothiazolo[4,3-b]pyridin-6-yl)aniline (12p)
This compound was prepared from (R)-6-bromo-3-(2-methylpiperidin-1-yl)isothiazolo[4,3-b]pyridine 10p (0.32 mmol, 100 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio 9:1 as mobile phase). The residue was further purified by RP-HPLC (eluting with a gradient of acetonitrile/water in a ratio ranging from 40:60 to 95:5) to yield the title compound (40 mg, 35 %). 1H NMR (300 MHz, DMSO-d6): δ = 8.72 (d, J = 2.0 Hz, 1H), 7.88 (d, J = 2.0 Hz, 1H), 7.26 (d, J = 1.8 Hz, 1H), 7.21 (dd, J = 8.1, 1.8 Hz, 1H), 6.74 (d, J = 8.1 Hz, 1H), 5.09 (s, 2H), 5.03 – 4.97 (m, 1H), 4.04 – 3.91 (m, 2H), 3.89 (s, 3H), 3.83 – 3.67 (m, 3H), 3.58 – 3.48 (m, 1H), 1.30 (d, J = 6.7 Hz, 3H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 171.6, 156.2, 146.8, 144.2, 138.9, 135.5, 132.44, 124.1, 122.2, 120.2, 113.9, 109.4, 70.0, 65.8, 55.6, 52.2, 45.1, 13.0 ppm. HRMS m/z [M+H]+ calcd for C18H20N4O2S: 357.1380, found 357.1380.
(R)-2-Methoxy-4-(3-(3-methylmorpholino)isothiazolo[4,3-b]pyridin-6-yl)aniline (12q)
This compound was prepared from (R)-6-bromo-3-(2-methylpiperidin-1-yl)isothiazolo[4,3-b]pyridine 10q (0.32 mmol, 100 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/ethyl acetate in a ratio of 9:1 as mobile phase). The residue was then further purified by RP-HPLC (eluting isocratically with a mixture of acetonitrile/water in a ratio of 6:4 as mobile phase), yielding the title compound (22 mg, 20 %). 1H NMR (300 MHz, DMSO-d6): δ = 8.72 (d, J = 2.1 Hz, 1H), 7.88 (d, J = 2.1 Hz, 1H), 7.26 (d, J = 1.9 Hz, 1H), 7.21 (dd, J = 8.1, 2.0 Hz, 1H), 6.74 (d, J = 8.1 Hz, 1H), 5.09 (bs, 2H), 5.04 – 4.95 (m, 1H), 4.03 – 3.93 (m, 2H), 3.89 (s, 3H), 3.84 – 3.67 (m, 3H), 3.59 – 3.49 (m, 1H), 1.30 (d, J = 6.7 Hz, 3H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 171.6, 156.2, 146.8, 144.2, 138.9, 135.5, 132.5, 124.1, 122.2, 120.2, 113.9, 109.4, 70.0, 65.8, 55.6, 52.2, 45.1, 13.0 ppm. HRMS m/z [M+H]+ calcd for C18H20N4O2S: 357.1380, found 357.1376.
4-(3-(cis-2,6-Dimethylmorpholino)isothiazolo[4,3-b]pyridin-6-yl)-2-methoxyaniline (12r)
This compound was prepared from 6-bromo-3-cis-2,6-dimethylmorpholinoisothiazolo[4,3-b]pyridine 10r (100 mg, 0.30 mmol). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio of 95:5 as mobile phase) yielding the title compound (84 mg, 76%). 1H NMR (300 MHz, DMSO-d6): δ = 8.76 (d, J = 2.1 Hz, 1H), 7.88 (d, J = 2.1 Hz, 1H), 7.27 (d, J = 1.8 Hz, 1H), 7.22 (dd, J = 8.1, 1.9 Hz, 1H), 6.74 (d, J = 8.1 Hz, 1H), 5.08 (bs, 2H), 4.64 – 4.37 (m, 2H), 3.89 (s, 3H), 3.88 – 3.80 (m, 2H), 2.95 – 2.76 (m, 2H), 1.20 (s, 3H), 1.18 (s, 3H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 172.1, 156.2, 146.8, 144.4, 138.9, 135.5, 132.7, 124.1, 122.1, 120.2, 113.9, 109.4, 70.7, 55.6, 54.9, 18.7 ppm. HRMS m/z [M+H]+ calcd for C19H22N4O2S: 371.1536, found 371.1534.
4-(3-(trans-2,6-Dimethylmorpholino)isothiazolo[4,3-b]pyridin-6-yl)-2-methoxyaniline (12s)
This compound was prepared from trans-4-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)-2,6-dimethylmorpholine 10s (0.30 mmol, 100 mg). The crude product was purified by silica gel flash column chromatography (using a mixture of dichloromethane/acetone in a ratio of 97:3 as mobile phase) yielding the title compound (90 mg, 81%). 1H NMR (300 MHz, CDCl3): δ = 8.62 (d, J = 2.0 Hz, 2H), 7.80 (d, J = 2.0 Hz, 1H), 7.17 – 7.05 (m, 2H), 6.80 (d, J = 8.0 Hz, 1H), 4.32 – 4.22 (m, 2H), 4.04 – 3.96 (m, 4H), 3.92 (s, 3H), 3.71 (dd, J = 12.5, 6.2 Hz, 2H), 1.36 (s, 3H), 1.34 (s, 3H) ppm. 13C NMR (75 MHz, CDCl3): δ = 173.6, 157.0, 147.9, 144.6, 137.2, 136.3, 133.7, 127.9, 124.1, 120.5, 115.3, 109.6, 66.3, 55.9, 55.1, 18.1 ppm. HRMS m/z [M+H]+ calcd for C19H22N4O2S: 371.1536, found 384.1483.
1-(6-(4-Amino-3-methoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)piperidin-4-amine (12t)
To a solution of tert-butyl (1-(6-(4-amino-3-methoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)piperidin-4-yl)carbamate 11 (0.22 mmol, 80 mg) in dioxane (5mL) was added a 37% aqueous HCl solution (2.2 mmol, 183 μL). The reaction mixture was stirred at room temperature overnight. The volatiles were evaporated in vacuo. The crude residue was redissolved in water and washed three times with dichloromethane. The organic phases were combined and evaporated in vacuo to yield the title compound (20 mg, 26%). 1H NMR (300 MHz, Pyr-d5): δ = 8.90 (d, J = 2.1 Hz, 1H), 8.15 (d, J = 2.1 Hz, 1H), 7.37 – 7.29 (m, 2H), 7.05 (d, J = 7.9 Hz, 1H), 5.54 (bs, 2H), 4.67 – 4.58 (m, 2H), 4.43 (bs, 2H), 3.79 (s, 3H), 3.38 – 3.19 (m, 2H), 3.01 – 2.87 (m, 1H), 1.95 – 1.81 (m, 2H), 1.70 – 1.45 (m, 2H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 172.3, 156.2, 146.8, 143.7, 138.6, 135.3, 132.5, 124.3, 122.1, 120.2, 114.0, 109.3, 55.6, 49.2, 47.2, 33.7 ppm. HRMS m/z [M+H]+ calcd for C18H21N5OS: 356.1539, found 356.1545.
Plasmids and virus constructs.
DENV2 (New Guinea C strain)32,33 Renilla reporter plasmid used for in vitro assays was a gift from Pei-Yong Shi (The University of Texas Medical Branch). DENV 16681 plasmid (pD2IC-30P-NBX) used for ex vivo experiments was a gift from Claire Huang (CDC).40 Plasmids pOG44 for Flp recombinase expression and pG-LAP were from Thermo Fischer Scientific.
Cells.
Huh7 (Apath LLC), BHK-21 (ATCC), and T-Rex-293 (ThermoFisher Scientific) cells were grown in DMEM (Mediatech) supplemented with 10% FBS (Omega Scientific), nonessential amino acids, 1% L-glutamine, and 1% penicillin-streptomycin (ThermoFisher Scientific) and maintained in a humidified incubator with 5% CO2 at 37 °C. C6/36 cells were grown in Leibovitz’s L-15 media (CellGro) supplemented with 10% FBS and 1% HEPES in a humidified chamber at 28 °C and 0% CO2. Vero 76 and Vero E6 cells were grown in EMEM (ThermoFisher Scientific) supplemented with 5% FBS (HyClone), and 1% penicillin-streptomycin (ThermoFisher Scientific) and maintained in a humidified incubator with 5% CO2 at 37 °C.
Virus Production.
DENV2 RNA was transcribed in vitro using mMessage/mMachine (Ambion) kits. DENV was produced by electroporating RNA into BHK-21 cells, harvesting supernatants on day 10 and titering via standard plaque assays on BHK-21 cells. In parallel, on day 2 post-electroporation, DENV-containing supernatant was used to inoculate C6/36 cells to amplify the virus. CHIKV (strain AF15561) was grown in Vero 76 cells, supernatants were collected and clarified, and stored at −80 °C until further use. Virus titers were determined via standard plaque assay on Vero 76 cells. EBOV (Kikwit isolate) was grown in Vero E6 cells, supernatants were collected and clarified and stored at −80 °C until further use. Virus titers were determined via standard plaque assay on Vero E6 cells.
Infection assays.
Huh7 cells were infected with DENV in replicates (n = 3–10) for 4 hours at MOI of 0.01. Overall infection was measured at 48 hours using a Renilla luciferase substrate. MDDCs were infected with DENV2 (16881) at an MOI of 1. Standard plaque assays were conducted following a 72-hour incubation. Huh7 cells were infected with EBOV at an MOI of 1 under biosafety level 4 conditions. Forty-eight hours after infection, cells were formalin-fixed for twenty-four hours prior to removal from biosafety level 4. Infected cells were detected using an EBOV glycoprotein specific monoclonal antibody (KZ52), and quantitated by automated fluorescence microscopy using an Operetta High Content Imaging System and the Harmony software package (Perkin Elmer). Vero cells were pretreated for 48 prior to infection. Media was removed, and cells were incubated with CHIKV at an MOI of 1 in compound-free media for 1 hour at 37 °C. Cells were then washed and media containing compound 12r or DMSO was applied. Forty eight hours following infection, supernatants were collected, pooled and used to infect naïve cells followed by plaque assays.
Viability assays.
Viability was assessed using AlamarBlue® reagent (Invitrogen) or Cell-Titer-Glo® reagent (Promega) assay according to manufacturer’s protocol. Fluorescence was detected at 560 nm on InfiniteM1000 plate reader and luminescence on InfiniteM1000 plate reader (Tecan) or a Spectramax 340PC.
Generation of MDDCs.
MDDCs were prepared as described with slight modifications.34 Buffy coats were obtained from the Stanford Blood Center. CD14+ cells were purified by EasySep™ Human Monocyte Enrichment Kit without CD16 Depletion (Stemcell Technologies). Cells were seeded in 6-well plates (2 ×106 cells per well), stimulated with 500 U/ml granulocyte-macrophage colony-stimulating and 1,000 U/ml interleukin-4 (Pepro tech), and incubated at 37 °C for 6 days prior to DENV infection (MOI 1).
Gain-of-function assays.
A doxycycline-inducible cell line was established to overexpress GAK using the Flp-In™ recombination system (ThermoFisher)41. T-REx 293 cells with a pFRT/lacZeo site and pcDNA™6/TR were co-transfected with a puromycin-resistant expression vector encoding a Flp-In™ recombination target site and a pOG44 plasmid containing the Flp recombinase followed by selection with puromycin. Eight hours post-induction with doxycycline, cells were treated with compound 12r, infected with DENV (MOI=0.01) and incubated for 72 hours prior to luciferase and viability assays.
Effect of 12r on AP2M1 phosphorylation.
Huh7 cells were kept in serum free medium for 1 hr and then were treated with compound 12r or DMSO in complete medium for 4 hours at 37 °C. To allow capturing of phosphorylated AP2M1, 100 nM of the PP2A inhibitor calyculin A (Cell Signalling) was added 30 min prior to lysis in M-PER lysis buffer (ThermoFisher Scientific) with 1X Halt Protease & phosphatase inhibitor cocktail (ThermoFisher Scientific). Samples were then subjected to SDS-PAGE and blotting with antibodies targeting phospho-AP2M1 (Cell Signaling) and total AP2M1 (Santa Cruz Biotechnology). Band intensity was measured with NIH ImageJ.
GAK Kd assay.
Kd values for GAK were determined as previously described.31 Briefly, the DNA-tagged GAK, an immobilized ligand on streptavidin-coated magnetic beads, and the test compound are combined. When binding occurs between GAK and a test compound, no binding can occur between GAK and the immobilized ligand. Upon washing, the compound-bound, DNA-tagged GAK is washed away. The beads carrying the ligands are then resuspended in elution buffer and the remaining kinase concentration measured by qPCR on the eluate. Kd values are determined using dose-response curves.
Kinase Selectivity assay.
Compound 12r was screened against a diverse panel of 468 kinases (DiscoveRX, KinomeScan) using an in vitro ATP-site competition binding assay at a concentration of 10 μM.31The results are reported as the percentage of kinase/phage remaining bound to the ligands/beads, relative to a control. High affinity compounds have % of control values close to zero, while weaker binders have higher % control values.
In vitro ADME profiling.
The solubility and permeability of compounds 4, 12h and 12r were measured at Chempartner (Shanghai, China). In brief, for solubility assays, 100 μM of the individual compounds were dissolved in 100 mM phosphate buffer (pH 7.4) and incubated at room temperature for one hour followed by measurement of the solubilized compound fraction in the supernatant via LC-MS/MS. For permeability assay, 5 μM of the compounds were incubated with MDCK-MDR1 cells in either donor or receiver chambers for 90 minutes at 37 °C. The transport of the compounds via the MDCK-MDR1 monolayer in the apical to basolateral (A-to-B) or basolateral to apical (B-to-A) directions was then measured via LC-MS/MS.
Statistical analysis.
All data were analyzed with GraphPad Prism software. Fifty percent effective concentration (EC50) values were measured by fitting data to a three-parameter logistic curve. P values were calculated by two-tailed unpaired t-test and one- or two-way ANOVA with either Dunnett’s or Tukey’s multiple comparisons tests.
Supplementary Material
Acknowledgments
This work was supported by award number PR151090 from the Department of Defense (DoD), Congressionally Directed Medical Research Programs (CDMRP) to S.E, J.D., P.H; and seed grant from the Stanford SPARK program. R.W. is the recipient of a doctoral fellowship from the Agency for Innovation by Science and Technology in Flanders (IWT.141103). S.P. was supported by the Child Health Research Institute, Lucile Packard Foundation for Children’s Health, as well as the Stanford CSTA (grant number UL1 TR000093). The opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army or the other funders.
Abbreviations
- AAK1
adaptor-associated kinase 1
- AP
adaptor protein
- CC50
half-maximal cytotoxic concentration
- CCV
clathrin-coated vesicle
- CHIKV
Chikungunya virus
- DENV
Dengue virus
- EBOV
Ebola virus
- EC50
half-maximal effective concentration
- EC90
90% effective concentration
- EGFR
epidermal growth factor receptor
- EVD
Ebola virus disease
- GAK
Cyclin G-associated kinase
- Kd
dissociation constant
- NAK
numb-associated kinase
- TGN
trans-Golgi network
Footnotes
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army.
References
- (1).Bhatt S; Gething PW; Brady OJ; Messina JP; Farlow AW; Moyes CL; Drake JM; Brownstein JS; Hoen AG; Sankoh O; Myers MF; George DB; Jaenisch T; William Wint GR; Simmons CP; Scott TW; Farrar JJ; Hay SI The Global Distribution and Burden of Dengue. Nature 2013, 496, 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).World Health Organization. Dengue: Guidelines for Diagnosis, Treatment, Prevention, and Control In Special Programme for Research and Training in Tropical Diseases; Geneva, Switzerland: WHO, 2009; p 147. [Google Scholar]
- (3).Halstead SB; Russell PK Protective and Immunological Behavior of Chimeric Yellow Fever Dengue Vaccine. Vaccine 2016, 34 (14), 1643–1647. [DOI] [PubMed] [Google Scholar]
- (4).Feldmann H; Geisbert TW Ebola Haemorrhagic Fever. Lancet 2011, 377 (9768), 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Towner JS; Sealy TK; Khristova ML; Albariño CG; Conlan S; Reeder SA; Quan P-L; Lipkin WI; Downing R; Tappero JW; Okware S; Lutwama J; Bakamutumaho B; Kayiwa J; Comer JA; Rollin PE; Ksiazek TG; Nichol ST Newly Discovered Ebola Virus Associated with Hemorrhagic Fever Outbreak in Uganda. PLoS Pathog. 2008, 4 (11), e1000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Negredo A; Palacios G; Vázquez-Morón S; González F; Dopazo H; Molero F; Juste J; Quetglas J; Savji N; de la Cruz Martínez M; Herrera JE; Pizarro M; Hutchison SK; Echevarría JE; Lipkin WI; Tenorio A Discovery of an Ebolavirus-Like Filovirus in Europe. PLoS Pathog. 2011, 7 (10), e1002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wells C; Yamin D; Ndeffo-Mbah ML; Wenzel N; Gaffney SG; Townsend JP; Meyers LA; Fallah M; Nyenswah TG; Altice FL; Atkins KE; Galvani AP Harnessing Case Isolation and Ring Vaccination to Control Ebola. PLoS Negl. Trop. Dis 2015, 9 (5), e0003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Abdelnabi R; Neyts J; Delang L Chikungunya Virus Infections: Time to Act, Time to Treat. Curr. Opin. Virol 2017, 24, 25–30. [DOI] [PubMed] [Google Scholar]
- (9).Henao-Restrepo AM; Longini IM; Egger M; Dean NE; Edmunds WJ; Camacho A; Carroll MW; Doumbia M; Draguez B; Duraffour S; Enwere G; Grais R; Gunther S; Hossmann S; Kondé MK; Kone S; Kuisma E; Levine MM; Mandal S; Norheim G; Riveros X; Soumah A; Trelle S; Vicari AS; Watson CH; Kéïta S; Kieny MP; Røttingen J-A Efficacy and Effectiveness of an RVSV-Vectored Vaccine Expressing Ebola Surface Glycoprotein: Interim Results from the Guinea Ring Vaccination Cluster-Randomised Trial. Lancet 2015, 386 (9996), 857–866. [DOI] [PubMed] [Google Scholar]
- (10).Bekerman E; Einav S Combating Emerging Viral Threats. Science. 2015, 348 (6232), 282–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Edeling MA; Smith C; Owen D Life of a Clathrin Coat: Insights from Clathrin and AP Structures. Nat. Rev. Mol. Cell Biol 2006, 7 (1), 32–44. [DOI] [PubMed] [Google Scholar]
- (12).Zhang CX; Engqvist-Goldstein ÅEY; Carreno S; Owen DJ; Smythe E; Drubin DG Multiple Roles for Cyclin G-Associated Kinase in Clathrin-Mediated Sorting Events. Traffic 2005, 6 (12), 1103–1113. [DOI] [PubMed] [Google Scholar]
- (13).Korolchuk VI; Banting G CK2 and GAK/Auxilin2 Are Major Protein Kinases in Clathrin-Coated Vesicles. Traffic 2002, 3 (6), 428–439. [DOI] [PubMed] [Google Scholar]
- (14).Neveu G; Ziv-Av A; Barouch-Bentov R; Berkerman E; Mulholland J; Einav S AP-2-Associated Protein Kinase 1 and Cyclin G-Associated Kinase Regulate Hepatitis C Virus Entry and Are Potential Drug Targets. J. Virol 2015, 89 (8), 4387–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bekerman E; Neveu G; Shulla A; Brannan J; Pu SY; Wang S; Xiao F; Barouch-Bentov R; Bakken RR; Mateo R; Govero J; Nagamine CM; Diamond MS; De Jonghe S; Herdewijn P; Dye JM; Randall G; Einav S Anticancer Kinase Inhibitors Impair Intracellular Viral Trafficking and Exert Broad-Spectrum Antiviral Effects. J. Clin. Invest 2017, 127 (4), 1338–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Neveu G; Barouch-Bentov R; Ziv-Av A; Gerber D; Jacob Y; Einav S Identification and Targeting of an Interaction between a Tyrosine Motif within Hepatitis C Virus Core Protein and AP2M1 Essential for Viral Assembly. PLoS Pathog. 2012, 8 (8), e1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).McMahon HT; Boucrot E Molecular Mechanism and Physiological Functions of Clathrin-Mediated Endocytosis. Nat. Rev. Mol. Cell Biol 2011, 12 (8), 517–533. [DOI] [PubMed] [Google Scholar]
- (18).Ricotta D; Conner SD; Schmid SL; Von Figura K; Höning S Phosphorylation of the AP2 μ Subunit by AAK1 Mediates High Affinity Binding to Membrane Protein Sorting Signals. J. Cell Biol 2002, 156 (5), 21–9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ghosh P; Kornfeld S AP-1 Binding to Sorting Signals and Release from Clathrin-Coated Vesicles Is Regulated by Phosphorylation. J. Cell Biol 2003, 160 (5), 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Conner SD; Schmid SL Differential Requirements for AP-2 in Clathrin-Mediated Endocytosis. J. Cell Biol 2003, 162 (5), 773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Umeda A; Meyerholz A; Ungewickell E Identification of the Universal Cofactor (Auxilin 2) in Clathrin Coat Dissociation. Eur. J. Cell Biol 2000, 79 (5), 336–342. [DOI] [PubMed] [Google Scholar]
- (22).Xiao F; Wang S; Barouch-Bentov R; Neveu G; Pu S; Beer M; Schor S; Kumar S; Nicolaescu V; Lindenbach BD; Randall G; Einav S Interactions between the Hepatitis C Virus Nonstructural 2 Protein and Host Adaptor Proteins 1 and 4 Orchestrate Virus Release. MBio 2018, 9 (2), e02233-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Fedorov O; Marsden B; Pogacic V; Rellos P; Muller S; Bullock AN; Schwaller J; Sundstrom M; Knapp S A Systematic Interaction Map of Validated Kinase Inhibitors with Ser/Thr Kinases. Proc. Natl. Acad. Sci 2007, 104 (51), 20523–20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Davis MI; Hunt JP; Herrgard S; Ciceri P; Wodicka LM; Pallares G; Hocker M; Treiber DK; Zarrinkar PP Comprehensive Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol 2011, 29 (11), 1046–1051. [DOI] [PubMed] [Google Scholar]
- (25).Karaman MW; Herrgard S; Treiber DK; Gallant P; Atteridge CE; Campbell BT; Chan KW; Ciceri P; Davis MI; Edeen PT; Faraoni R; Floyd M; Hunt JP; Lockhart DJ; Milanov ZV; Morrison MJ; Pallares G; Patel HK; Pritchard S; Wodicka LM; Zarrinkar PP A Quantitative Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol 2008, 26 (1), 127–132. [DOI] [PubMed] [Google Scholar]
- (26).Scagliotti GV; Krzakowski M; Szczesna A; Strausz J; Makhson A; Reck M; Wierzbicki RF; Albert I; Thomas M; Miziara JEA; Papai ZS; Karaseva N; Thongprasert S; Portulas ED; von Pawel J; Zhang K; Selaru P; Tye L; Chao RC; Govindan R Sunitinib plus Erlotinib versus Placebo plus Erlotinib in Patients with Previously Treated Advanced Non-Small-Cell Lung Cancer: A Phase III Trial. J. Clin. Oncol 2012, 30 (17), 2070–2078. [DOI] [PubMed] [Google Scholar]
- (27).Bah EI; Lamah M-C; Fletcher T; Jacob ST; Brett-Major DM; Sall AA; Shindo N; Fischer WA; Lamontagne F; Saliou SM; Bausch DG; Moumié B; Jagatic T; Sprecher A; Lawler JV; Mayet T; Jacquerioz FA; Méndez Baggi MF; Vallenas C; Clement C; Mardel S; Faye O; Faye O; Soropogui B; Magassouba N; Koivogui L; Pinto R; Fowler RA Clinical Presentation of Patients with Ebola Virus Disease in Conakry, Guinea. N. Engl. J. Med 2015, 372 (1), 40–47. [DOI] [PubMed] [Google Scholar]
- (28).Asquith CRM; Laitinen T; Bennett JM; Godoi PH; East MP; Tizzard GJ; Graves LM; Johnson GL; Dornsife RE; Wells CI; Elkins JM; Willson TM; Zuercher WJ Identification and Optimization of 4-Anilinoquinolines as Inhibitors of Cyclin G Associated Kinase. ChemMedChem 2018, 13 (1), 48–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kovackova S; Chang L; Bekerman E; Neveu G; Barouch-Bentov R; Chaikuad A; Heroven C; Šála M; De Jonghe S; Knapp S; Einav S; Herdewijn P Selective Inhibitors of Cyclin G Associated Kinase (GAK) as Anti-Hepatitis C Agents. J. Med. Chem 2015, 58, 3393–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Li J; Kovackova S; Pu S; Rozenski J; De Jonghe S; Einav S; Herdewijn P Isothiazolo[4,3-b]Pyridines as Inhibitors of Cyclin G Associated Kinase: Synthesis, Structure–activity Relationship Studies and Antiviral Activity. Medchemcomm 2015, 6 (9), 1666–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Fabian MA; Biggs WH; Treiber DK; Atteridge CE; Azimioara MD; Benedetti MG; Carter TA; Ciceri P; Edeen PT; Floyd M; Ford JM; Galvin M; Gerlach JL; Grotzfeld RM; Herrgard S; Insko DE; Insko MA; Lai AG; Lélias J-M; Mehta SA; Milanov ZV; Velasco AM; Wodicka LM; Patel HK; Zarrinkar PP; Lockhart DJ A Small Molecule–kinase Interaction Map for Clinical Kinase Inhibitors. Nat. Biotechnol 2005, 23 (3), 329–336. [DOI] [PubMed] [Google Scholar]
- (32).Xie X; Gayen S; Kang C; Yuan Z; Shi P-Y Membrane Topology and Function of Dengue Virus NS2A Protein. J. Virol 2013, 87 (8), 4609–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zou G; Xu HY; Qing M; Wang QY; Shi PY Development and Characterization of a Stable Luciferase Dengue Virus for High-Throughput Screening. Antiviral Res. 2011, 91 (1), 11–19. [DOI] [PubMed] [Google Scholar]
- (34).Rodriguez-Madoz JR; Bernal-Rubio D; Kaminski D; Boyd K; Fernandez-Sesma A Dengue Virus Inhibits the Production of Type I Interferon in Primary Human Dendritic Cells. J. Virol 2010, 84 (9), 4845–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Schmid MA; Diamond MS; Harris E Dendritic Cells in Dengue Virus Infection: Targets of Virus Replication and Mediators of Immunity. Front. Immunol 2014, 5, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).O’Gorman S; Fox DT; Wahl GM Recombinase-Mediated Gene Activation and Site-Specific Integration in Mammalian Cells. Science 1991, 251 (4999), 1351–1355. [DOI] [PubMed] [Google Scholar]
- (37).Clark MJ; Miduturu C; Schmidt AG; Zhu X; Pitts JD; Wang J; Potisopon S; Zhang J; Wojciechowski A; Hann Chu JJ; Gray NS; Yang PL GNF-2 Inhibits Dengue Virus by Targeting Abl Kinases and the Viral E Protein. Cell Chem. Biol 2016, 23 (4), 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).García M; Cooper A; Shi W; Bornmann W; Carrion R; Kalman D; Nabel GJ Productive Replication of Ebola Virus Is Regulated by the C-Abl1 Tyrosine Kinase. Sci. Transl. Med 2012, 4 (123), 123ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Roskoski R Signaling by Kit Protein-Tyrosine Kinase—The Stem Cell Factor Receptor. Biochem. Biophys. Res. Commun 2005, 337 (1), 1–13. [DOI] [PubMed] [Google Scholar]
- (40).Huang CY-H; Butrapet S; Moss KJ; Childers T; Erb SM; Calvert AE; Silengo SJ; Kinney RM; Blair CD; Roehrig JT The Dengue Virus Type 2 Envelope Protein Fusion Peptide Is Essential for Membrane Fusion. Virology 2010, 396 (2), 305–315. [DOI] [PubMed] [Google Scholar]
- (41).Torres JZ; Miller JJ; Jackson PK High-Throughput Generation of Tagged Stable Cell Lines for Proteomic Analysis. Proteomics 2009, 9 (10), 2888–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.