

Abstract
Sex hormones contribute to sex differences in blood pressure. Inappropriate activation of the renin-angiotensin system is involved in vascular dysfunction and hypertension. This study evaluated the role of androgens (testosterone) in angiotensin II (Ang II)-induced increase in blood pressure, vascular reactivity, and cardiac hypertrophy. Eight-week-old male Wistar rats underwent sham operation, castration, or castration with testosterone replacement. After 12 weeks of chronic changes in androgen status, Ang II (120 ng/kg per minute) or saline was infused for 28 days via subcutaneous miniosmotic pump, and changes in blood pressure was measured. Vascular reactivity and Ang II receptor levels were examined in mesenteric arteries. Heart weight, cardiac ANP mRNA levels, and fibrosis were also assessed. Ang II infusion increased arterial pressure in intact males. The Ang II-induced increase in hypertensive response was prevented in castrated males. Testosterone replacement in castrated males restored Ang II-induced hypertensive responses. Castration reduced vascular AT1R/AT2R ratio, an effect that was reversed by testosterone replacement. Ang II-induced hypertension was associated with increased contractile response of mesenteric arteries to Ang II and phenylephrine in intact and testosterone-replaced castrated males; these increases were prevented in castrated males. Ang II infusion induced increased left ventricle-to-body weight ratio and ANP mRNA expression, indicators of left ventricular hypertrophy, and fibrosis in intact and testosterone-replaced castrated males, and castration prevented the increase in these parameters caused by Ang II. This study demonstrates that testosterone plays a permissive role in development and maintenance of Ang II-induced vascular dysfunction, hypertension, and cardiac hypertrophy.
Keywords: gonadal steroids, hormone action, blood pressure, angiotensin II, hypertrophy, angiotensin receptors, mesenteric arteries
Testosterone, at physiologically relevant concentrations, plays a permissive role in development and maintenance of Ang II-induced hypertension, cardiac hypertrophy, and fibrosis via heightened angiotensin II receptor-mediated signaling.
Introduction
Cardiovascular diseases (CVD) are the leading cause of mortality and morbidity in the United States. Among CVD, hypertension ranks first, affecting more than 75 million people—nearly 1 in 3 adults (Center for Disease Control statistics, 2017). Epidemiological and clinical studies have demonstrated a role for sex-dependent mechanisms in the pathophysiology of hypertension and cardiac hypertrophy. For example, the incidence and severity of hypertension have been shown to be greater in men than age matched premenopausal women [1, 2]. Also, sex differences in hypertension and cardiac hypertrophy have been demonstrated in both genetic and induced models, including Dahl salt-sensitive rats [3], deoxycorticosterone acetate hypertension [4], spontaneously hypertensive rats [5], one-kidney renal wrap hypertension [6], and in utero programmed hypertension [7–10]. In these animals, hypertension developed more rapidly and severely in male compared to female rats. Also, orchiectomy prevented blood pressure elevation and testosterone replacement restored hypertension in males. In the females, ovariectomy increased blood pressure and estrogen replacement attenuated this increase. These studies suggest that sex hormones participate in hypertension development, with testosterone exacerbating blood pressure in males and estrogen protecting against blood pressure increase in females. Although numerous studies have examined the protective mechanisms of estrogens on cardiovascular function, few studies have examined how androgens contribute for cardiovascular pathology.
Among the various regulatory systems that impact blood pressure and cardiac function, the renin-angiotensin system (RAS) has a key role. Inappropriate activation of RAS leads to profound hypertension and cardiovascular morbidity [11, 12]. Angiotensin II (Ang II), the main effector of RAS, is increased in patients and animal models of hypertension and cardiac hypertrophy [13, 14]. At the cellular level, responsiveness to Ang II is conferred by the expression of the two classes of angiotensin receptors (AT1R and AT2R). These receptors are expressed in a variety of tissues, including the heart and vasculature [15], and RAS blockade, especially AT1R, effectively reduces blood pressure and ameliorate cardiovascular complications [16, 17], suggesting that dysregulation of the RAS contributes to their pathophysiology.
Studies in humans and hypertensive animal models have demonstrated the importance of interaction between testosterone and RAS in regulating cardiovascular function and blood pressure [18, 19]. Much evidence supports that males have greater expression of classical RAS components Ang II and AT1R and lower levels of AT2R [20, 21]. Androgens augment renal vascular responses to Ang II in New Zealand genetically hypertensive rats [22]. However, the functional impact of androgens in Ang II-induced hypertension and cardiac hypertrophy has given seemingly contradictory information. Some studies have shown that androgens protect against Ang II-induced cardiovascular dysfunction [23–25], while others demonstrated that testosterone enhance the cardiovascular detrimental effects of Ang II [22, 26–28]. Our and other recent studies show that Ang II-induced vascular contraction depends on androgen status, with elevated androgen levels contributing for exaggerated vasoconstriction in in utero programmed hypertension [29–31]. This led us to hypothesize that androgens contribute to Ang II-induced hypertension and cardiac hypertrophy. Studies in men show that testosterone supplements improve libido, protect against osteoporosis, and improve overall feelings of well-being [32, 33]; however, no long-term studies have carefully documented the cardiovascular consequences of chronic androgen changes in men. Hence, we determined whether chronic changes in the androgen environment could induce alterations in Ang II-induced hypertension, vascular reactivity, and cardiac hypertrophy in intact, castrated and testosterone-replaced castrated male rats. The results show that Ang II-induced increase in blood pressure and associated cardiac pathophysiological changes, including hypertrophy and fibrosis were minimized by castration, and reversed by testosterone replacement. These findings indicate that testosterone plays a permissive role in Ang II-induced hypertension and associated cardiac effects.
Methods
Animals
All experiments were carried out according to the protocols approved by our Institutional Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Eight-week-old male Wistar rats were purchased from Charles River Laboratories (Houston, TX). Since male rats reach sexually maturity and have stable testosterone levels after 6–7 weeks of age, we used 8-week-old post pubertal rats for this study. After 1-week acclimatization, rats were divided into three groups: (a) intact (sham operated), (b) castrated, and (c) castrated with testosterone replacement using subcutaneous implanted pellets (150 mg, 150-day release, Innovative Research of America, Sarasota, FL) [20]. Castration was done by standard procedure as described previously [8]. Rats were housed in a temperature-controlled room (23°C) with a 12:12-hour light/dark cycle with food and water available ad libitum.
Ang II infusion and blood pressure measurement
After 12 weeks of chronic changes in androgen status, rats in the three groups were implanted with subcutaneous osmotic pumps (Alzet, Cupertino, CA) to infuse Ang II (120 ng/kg/min, n = 6 in each group) or saline (vehicle, n = 6 in each group) for 28 days. Blood pressure was measured every fourth day using a noninvasive tail cuff method (Kent Scientific, Torrington, CT) as described previously [20]. Prior to implanting the osmotic pumps, rats were acclimated to the blood pressure measuring device for 1 week. After completing infusion of Ang II or its vehicle from above treatment groups, rats were weighed and then sacrificed by CO2 inhalation. Blood was collected to determine plasma testosterone levels, mesenteric arteries were isolated and a portion of it was processed for western blotting and the reminder was used for wire myography. The hearts were removed for hypertrophic measurements.
Plasma testosterone levels
Plasma was separated by centrifugation and stored at –80°C until the time of measurement. Testosterone levels in the samples were measured by using ELISA kit (Enzo Life Sciences, Farmingdale, NY) as in our previous publication [8]. The minimum detectable concentration is 6 pg/mL. The intra- and interassay coefficients of variation were lower than 5%.
Western blotting for measurement of mesenteric arterial AT1R, AT2R, and androgen receptor protein levels
Mesenteric arteries were hgenized in ice-cold radioimmunoprecipitation assay buffer (Cell Signaling Technology, Danvers, MA) containing a protease inhibitor tablet and phosphatase inhibitor cocktail-2 and -3 (Sigma-Aldrich, St Louis, MO). Lysates were centrifuged (14,000× g for 10 min at 4°C), and the protein content was measured using the bicinchoninic acid protein assay kit (Pierce; Thermo Scientific, Grand Island, NY). The supernatant was resuspended in neutral pH polyacrylamide gel electrophoresis (NuPAGE) lithium dodecyl sulfate sample buffer and reducing agent (Invitrogen; Thermo Scientific). Proteins (30 μg) alongside Precision Plus Standard (Kaleidoscope; Bio-Rad, Hercules, CA) were resolved on 4–12% gradient NuPAGE Bis-Tris gels (Invitrogen) at 100 V for 2 h at room temperature and then transferred onto Immobilon-P membranes (Millipore, Billerica, MA) at 100 V for 1 h. The membranes were blocked with 5% nonfat dry milk for 1 h and then incubated overnight at 4°C with primary antibodies. The primary antibodies were mouse monoclonal AT1R (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit monoclonal AT2R (1:1000 dilution; Abcam, Cambridge, MA), rabbit polyclonal androgen receptor (1:1000 dilution; Abcam) and β-actin (1:2500 dilution; Cell Signaling Technology). After being washed, the membranes were incubated with secondary antibodies (anti-rabbit or -mouse conjugated with horseradish peroxidase) at 1:5000 dilutions and detected with the enhanced chemiluminescence detection kits (Pierce; Thermo Scientific). Densitometric measurement was done using ImageJ software. Results were expressed as ratios of the intensity of a specific band to that of β-actin.
Ex vivo vascular reactivity studies
Mesenteric arteries were dissected free of adherent connective tissues. Arterial rings of 2 mm length were suspended in a Halpern-Mulvany myograph (Model 610 M, Danish Myo Technology A/S, Denmark) using 25 μm stainless steel wires for recording of isometric contractile force (PowerLab, ADInstruments, Colorado Springs, CO). The preparations were immersed in Krebs-Ringer bicarbonate solution (37°C, aerated with a 95% O2 and 5% CO2 gas mixture, pH 7.4) of the following composition (in mM): NaCl 118, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, and glucose 11.1. Studies were done in endothelium-denuded mesenteric arterial rings. Endothelium was removed by gently rubbing the intimal surface of rings with a tungsten wire. Endothelium removal was verified by the absence of relaxation to acetylcholine (10 μM) in rings precontracted with phenylephrine (3 μM). The rings were allowed to equilibrate in Krebs solution for 1 h at a resting tension. After stabilization, the rings were normalized to an internal diameter of 0.9 of L13.3kPa using a normalization software package (Myodata; Danish Myotechnologies). Dose-response curves were constructed using cumulative doses of Ang II (3 × 10−11 to 3 × 10−8M; Sigma) and phenylephrine (10−9 to 10−5M; Sigma).
Measurement of left ventricular hypertrophy
Left ventricle:body weight ratio
The hearts were removed and left ventricle was separated and weighted. The ratio of left ventricle (mg) to body weight (g) was calculated and used as a measure of left ventriclar hypertrophy.
Quantification of Left Ventricular ANP mRNA Expression
Left ventricular tissues were processed for the total RNA extraction (TRIZOL, Invitrogen, Carlsbad, CA). All RNA isolates were made DNA-free by treatment with DNAse and further purified with RNeasy clean up kit (QIAGEN Inc, Valencia, CA). Total RNA concentration and integrity were determined using an ND-1000 Nanodrop spectrophotometer (Thermo Fisher Scientific, Newark, DE). One microgram of total RNA was reverse transcribed using a modified Maloney murine leukemia virus-derived RT (New England Biolabs Inc, Ipswich, MA) and a blend of oligo (dT) and random hexamer primers (Invitrogen). The reaction was carried out at 28°C for 15 min and 42°C for 50 min, then stopped by heating at 94°C for 5 min followed by 4°C before storage at −20°C until further analysis. One microliter of the diluted cDNA corresponding to 100 ng RNA was amplified by real-time PCR using FAM (Invitrogen) as the fluorophore in a CFX96 real-time thermal cycler (Bio-Rad, Hercules, CA). PCR conditions used were 2 min at 50°C for 1 cycle; 10 min at 95°C, 15 s at 95°C, and 1 min at 60°C for 40 cycles; and a final dissociation step (0.05 s at 65°C and 0.5 s at 95°C). Results were calculated using the 2−ΔΔCT method and expressed in fold increase/decrease of the gene of interest in intact, castrated and testosterone-replaced with and without Ang II infusion. All reactions were performed in duplicate, and GAPDH was used as an internal control. TaqMan assays were done in 10 μL volume for real-time PCR at a final concentration of 250 nM TaqMan probe and 900 nM of each primer; Assays-on-Demand for atrial natriuretic peptide (ANP) (Rn00664637_g1) were obtained from Applied Biosystems.
Histopathologic Analysis
Left ventricular tissues were washed in phosphate-buffered saline, fixed overnight in 4% paraformaldehyde, and embedded in paraffin, cut into 5-μm sections, and stained with Masson trichrome as described previously [34]. Fibrosis areas within sections were measured by investigators blinded to treatment, by visualizing blue-stained areas, exclusive of staining that colocalized with perivascular or intramural vascular structures, the endocardium, or LV trabeculae. Using ImageJ software, blue-stained areas and nonstained myocyte areas from each section were determined using color-based thresholding [35]. The percentage of total fibrosis area was calculated as the summed blue-stained areas divided by total ventricular area, as described previously [36].
Statistical analysis
For the comparison, analysis was performed using 2 × 3 ANOVA model in which the factors were treatment (Ang II and vehicle exposure) and gonadal status (intact, castration, and castration with testosterone replacement) and used contrasts to make pairwise comparisons. Cumulative concentration-response curves were analyzed by computer fitting to a 4-parameter sigmoid curve using the Prism 6 program (GraphPad, San Diego, CA) to evaluate pD2 (negative log molar concentration for half-maximal effect) and Emax (maximum asymptote of the curve). All values are expressed as means ± SE. A P < 0 .05 was considered significant.
Results
Plasma testosterone levels in rats
Testosterone levels were markedly reduced following castration (n = 6; P < 0.05) compared with their intact counterparts (n = 6; Figure 1). Testosterone replacement reinstated plasma testosterone levels in castrated rats (n = 6; P < 0.05) similar to levels in intact males (Figure 1).
Figure 1.
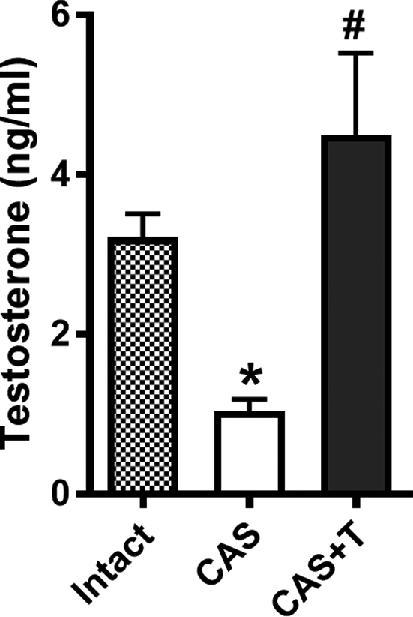
Plasma testosterone levels. Plasma testosterone levels were measured using ELISA in intact (sham-operated), castrated, and testosterone-replaced castrated males. Blood was collected following CO2 inhalation. *P < 0.05 compared with intact gonad. #P < 0.05 compared to castrated group. All data are expressed as means ± SEM. CAS—castrated, CAS+T—castrated with testosterone replacement.
Castration counteracts the hypertensive effect of Ang II in rats
Ang II infusion increased blood pressure in males with intact testes (152 ± 3 mm Hg; mean increase of 29 mm Hg; n = 6; P < 0.05; Figure 2). Castration alone did not alter blood pressure but prevented Ang II-induced increase in blood pressure (113 ± 2 mmHg; n = 6; mean decrease of 4 mmHg) (Figure 2). Testosterone replacement in castrated males restored the ability of Ang II to increase arterial pressure to levels not different from those observed in intact males (156 ± 3 mmHg; mean increase of 28 mmHg; n = 6; P < 0.05; Figure 2).
Figure 2.

Castration counteracts the hypertensive effect of Ang II, which was restored by testosterone. Intact, castrated and castrated with testosterone replaced rats were infused with Ang II or vehicle for 28 days, as described in Methods. Blood pressures were measured in the experimental groups before and during 28 days of Ang II infusion. Data are expressed as mean ± SEM. *P < 0.05 Ang II vs vehicle, n = 6 for all experiments.
Castration prevents increased AT1R/AT2R ratio in the mesenteric arteries of Ang II-treated rats
Since Ang II mediates effect through its receptors, we determined whether mesenteric arterial AT1R and AT2R expression were altered in response to Ang II infusion and castration. Ang II infusion significantly increased mesenteric arterial AT1R levels in intact males (35% increase; n = 6; P < 0.05; Figure 3). Castration significantly decreased AT1R (40% decrease; n = 6; P < 0.05) compared to intact males (Figure 3). Ang II infusion to these castrated rats minimized the increase in AT1R (20% increase, n = 6; P < 0.05; Figure 3). Testosterone replacement in castrated males restored AT1R, and Ang II infusion increased AT1R to levels similar to that in intact males (35% increase, n = 6; P < 0.05; Figure 3).
Figure 3.

Castration decreases AT1R/AT2R ratio in mesenteric arteries, which is restored by testosterone. Intact, castrated and castrated with testosterone-replaced rats were infused with vehicle or Ang II for 28 days as described in the Methods section. After the end of Ang II infusion, mesenteric arteries were removed and processed for western blotting. Representative western blots for AT1R and AT2R are shown at top; blot density obtained from densitometric scanning of AT1R and AT2R normalized to β-actin is shown at bottom. The ratio of AT1R/AT2R is presented. Values are given as means ± SEM of six rats in each group. *P < 0.05 compared with respective vehicle-infused group. #P < 0.05 compared with intact vehicle-infused group. §P < 0.05 compared with intact Ang II-infused group. CAS—castrated, CAS+T—castrated with testosterone replacement.
Ang II infusion increased AT2R levels in intact males (100% increase; n = 6 each; P < 0.05; Figure 3). While castration by itself significantly increased AT2R (160% increase; n = 6; P < 0.05), Ang II further increased AT2R levels (29% increase; n = 6; P < 0.05; Figure 3). Testosterone replacement restored AT2R levels, and Ang II infusion increased AT2R levels (53% increase; n = 6; P < 0.05) comparable to that in intact males (Figure 3).
The AT1R/AT2R ratio was higher in the intact males, decreased by castration (75% decrease, n = 6; P < 0.05), and restored by testosterone replacement (Figure 3). Ang II infusion did not significantly affect this ratio in these groups (Figure 3).
Castration did not alter androgen receptor levels in the mesenteric arteries of Ang II-treated rats
Androgen receptor levels were not significantly altered in intact, intact + Ang II, castration, castration + Ang II, testosterone replacement, or testosterone replacement + Ang II groups (Figure 4).
Figure 4.
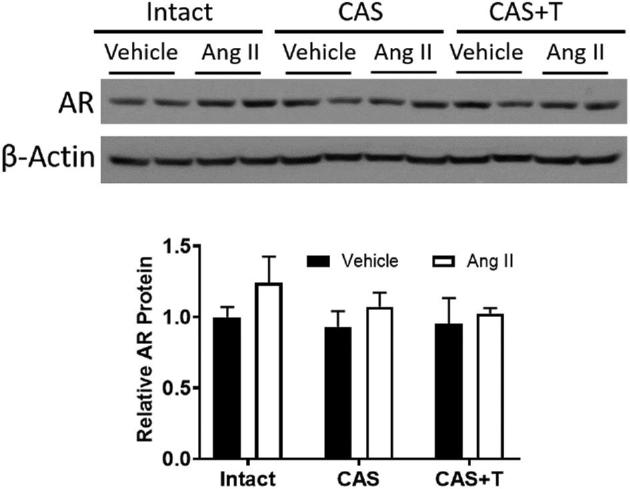
Castration or testosterone replacement does not alter androgen receptor (AR). Intact, castrated and castrated with testosterone-replaced rats were infused with vehicle or Ang II for 28 days as described in the Methods section. After the end of Ang II infusion, mesenteric arteries were removed and processed for western blotting. Representative western blots for AR are shown at top; blot density obtained from densitometric scanning of AR normalized to β-actin is shown at bottom. Values are given as means ± SEM of six rats in each group. CAS—castrated, CAS+T—castrated with testosterone replacement.
Castration prevents increased vascular reactivity in Ang II-treated rats
We reasoned that activation of Ang II in the vasculature might cause hypertension by influencing peripheral vascular resistance. Therefore, we compared vascular reactivity of resistance-sized mesenteric arteries between the experimental groups. Mesenteric arteries isolated from Ang II infused intact males exhibited exaggerated vasoconstriction to ex vivo Ang II (Emax: 93 ± 3.5 vs 81 ± 3.5 %; pD2: 8.9 ± 0.08 vs 9.1 ± 0.08 in vehicle infused intact males, n = 6; P < 0.05; Figure 5A). In contrast, arteries from castration rats showed no evidence of exaggerated Ang II contractile response in castrated males (Emax: 56 ± 6.1 vs 61 ± 1.5 %; pD2: 8.6 ± 0.06 vs 8.5 ± 0.09 in vehicle infused castrated males, n = 6; Figure 5A). Castration alone attenuated contractile response to ex vivo Ang II when compared to intact males. In testosterone-replaced castrated males, Ang II infusion restored the ability of ex vivo Ang II to induce exaggerated vasoconstriction (Emax: 98 ± 3.3 vs 84 ± 5.9 %; pD2: 9.0 ± 0.06 vs 8.7 ± 0.13 in vehicle infused testosterone-replaced castrated males, n = 6; P < 0.05) identical to that in the intact group (Figure 5A).
Figure 5.

Castration attenuates exaggerated mesenteric arterial contractile responses, which is restored by testosterone. Intact, castrated, and castrated with testosterone-replaced rats were infused with vehicle or Ang II for 4 weeks as described in the Methods section. After the end of Ang II infusion, mesenteric arteries were removed and processed for wire myography. Contractile responses were taken to cumulative additions of (A) Ang II, and (B) phenylephrine. Ang II and phenylephrine contractions are presented as percentage of 80 mM KCl contractions. Values are given as means ± SEM. n = 6 for all experiments.
Ang II infusion in intact males significantly increased phenylephrine-induced contractile response (pD2: 6.7 ± 0.08 vs 6.3 ± 0.10; n = 6; P < 0.05; Figure 5B). In contrast, in the castrated group, there was no increase in phenylephrine-induced contractile response after Ang II infusion (pD2: 6.1 ± 0.09 vs 6.3 ± 0.04, n = 6; Figure 5B). In testosterone-replaced castrated males, Ang II infusion reinstated the ability of phenylephrine to induce increased vasoconstriction (pD2: 6.9 ± 0.06 vs 6.49 ± 0.03, n = 6; P < 0.05; Figure 5B). There was no difference in Emax for phenylephrine between respective vehicle and Ang II infusion in all groups. These results indicate that Ang II infusion leads to exaggerated vasoconstriction to Ang II and phenylephrine in mesenteric arteries of intact males, which was prevented by castration and restored by testosterone replacement.
Castration protects against cardiac hypertrophy and fibrosis in Ang II-treated rats
In patients with hypertension, one of the early and most common consequences of chronic hypertension is left ventricular hypertrophy [37, 38]. Clinical evidence suggests that the RAS contributes to the development of left ventricular hypertrophy in hypertension [39, 40]. Accordingly, we compared left ventricular weights in all the experimental groups. With Ang II infusion, the intact males developed ventricular hypertrophy as indicated by increased left ventricle-body weight ratio (n = 6; P < 0.05; Figure 6A). In contrast, in the castrated group, which was without hypertension, there was no evidence of ventricular hypertrophy after Ang II infusion (n = 6; Figure 6A). In testosterone-replaced castrated males, Ang II induced increased ventricular weights identical to those of the intact group (n = 6; P < 0.05; Figure 6A). These results suggest that Ang II infusion leads to cardiac hypertrophy in vivo in presence of testosterone.
Figure 6.

Castration prevents Ang II-induced cardiac hypertrophy, which is restored by testosterone. Intact, castrated, and castrated with testosterone-replaced rats were infused with vehicle or Ang II for 28 days as described in the Methods section. After the end of Ang II infusion, heart was removed and weighed. (A) Mean left ventricle-to-body weight ratios, and (B) expression of ANP mRNA in left ventricle. Real-time PCR was used to assess ANP mRNA expression. Quantitation of ANP expression was normalized relative to GAPDH. Data are expressed as mean ± SEM of n = 6 for all experiments. *P < 0.05 compared with respective vehicle-infused group. §P < 0.05 compared with intact Ang II-infused group. CAS—castrated, CAS+T—castrated with testosterone replacement.
During hypertrophic cardiac remodeling, gene expression in myocardium undergoes characteristic alterations, including enhanced expression of ANP and fibrosis [41]. Previous studies have suggested that Ang II may stimulate these processes [42]. We therefore examined left ventricular mRNA expression of ANP and fibrosis. As shown in Figure 6B, Ang II infusion increased levels of ANP mRNA in hearts from in intact males (n = 6; P < 0.05). In contrast, Ang II-induced upregulation of ANP mRNAs was not detected in castrated males (n = 6; Figure 6B). A similar pattern of Ang II induced upregulation was observed in testosterone replaced group as in the intact males (n = 6; P < 0.05; Figure 6B).
Hearts from Ang II-infused intact males also displayed increased fibrosis, as indicated by Masson's trichrome staining (n = 3; P < 0.05; Figure 7). Castration alone had no effect but minimized Ang II-induced cardiac fibrosis (n = 3; Figure 7). Testosterone replacement in castrated males restored the ability of Ang II to induce fibrosis (n = 3; P < 0.05; Figure 7).
Figure 7.

Castration minimizes Ang II-induced cardiac fibrosis, which is restored by testosterone. Intact, castrated and castrated with testosterone-replaced rats were infused with vehicle or Ang II for 4 weeks as described in the Methods section. After the end of Ang II infusion, heart was removed and processed for immunohistochemistry. Masson trichrome staining revealed intracardiac collagen deposition (intense blue staining). Photomicrographs are representative images of at least three animals from each experimental group. Presented below is the percentage of total fibrosis area as calculated as the summed blue-stained areas divided by total ventricular area. *P < 0.05 compared with respective vehicle-infused group. §P < 0.05 compared with intact Ang II-infused group. CAS—castrated, CAS+T—castrated with testosterone replacement.
Discussion
The major findings of this study are testosterone, at physiologically relevant concentrations, leads to the development of Ang II-induced hypertension with associated increase in (1) the AT1R/AT2R ratio, and contractile response of mesenteric arteries to Ang II and phenylephrine; (2) left ventricle:body weight ratio and the expression of ANP mRNA in the left ventricle; and (3) fibrosis in the myocardium. A major finding in our study is that presence of testosterone is primarily responsible for the actions of Ang II to cause hypertension, vascular dysfunction, and cardiac hypertrophy since castration decreased plasma testosterone levels and also prevented Ang II-induced cardiovascular defects. Moreover, testosterone replacement in castrated rats restored the ability of Ang II to increase blood pressure and cardiac dysfunction to the same levels as in testes intact males. We suggest that the maintenance of higher AT1R/AT2R ratio in the vasculature by testosterone may contribute to androgenic potentiation of the hypertensive and vascular responses to Ang II in this model.
The development of hypertension is complex and is associated with the alteration of a wide range of systems. Sex steroid hormones, androgens, and estrogens are known to play an important role in blood pressure regulation. Men are generally at greater risk for CVD than are age-matched, premenopausal women. Experimental studies support the importance of sex hormones in blood pressure regulation by showing that either treatment with or absence of sex steroids alters the course of hypertension development [9, 10]. In the present study, chronic depletion of testosterone levels by castration abolished Ang II-induced blood pressure increase (mean decrease of 4 mmHg); however, the Ang II-induced hypertensive effect is greater in the males with intact testes (mean increase of 29 mmHg) and testosterone-replaced castrated males (mean increase of 28 mmHg). These findings show that testosterone is important in the hypertensive effect induced by Ang II as withdrawal (castration) prevented Ang II effects and replacement (testosterone) reversed the castration effects on Ang II-induced changes in blood pressure. This suggests that the development of Ang II-induced hypertension is androgen dependent, which appears to be similar to that observed in other in utero programming of increases in blood pressure [20, 43–45].
The protective role of castration may not be directly related to the level of testosterone per se, but due to the testosterone withdrawal effect on other systems that control blood pressure. One potential mechanism for the protective status mediated by castration against Ang II-induced increase in blood pressure may involve modulation of Ang II receptor expression. Ang II acts through AT1R and AT2R. Increasing evidence has shown that AT1R is a key player for blood pressure increase [16, 46]. AT2R acts as a negative regulator of the vasoconstrictor effects of Ang II, and has an important blood pressure lowering effect [47]. It is suggested that when AT1R activity is greater than AT2R, vasoconstriction to Ang II will predominate and hypertension will be induced [7, 29, 48]. Since testosterone is shown to regulate AT2R protein levels in the aorta of rat [20], we determined whether mesenteric arterial AT1R and AT2R expression were altered in response to castration. Castration induced a significant decrease in AT1R and increase in AT2R protein expression, and this effect was restored by testosterone replacement. Thus, in intact and testosterone-replaced castrated males, the constitutive high levels of AT1R and lower AT2R with higher ratio of AT1R/AT2R may be important in maintaining the normal physiological and biological effects of Ang II. Loss of testosterone by castration decreased the vasoconstrictor AT1R and increased the vasodilator AT2R (i.e., decreased AT1R/AT2R ratio), yet there were no difference in blood pressure in the castrated rats compared to intact rats at basal state. The reason for this is in unclear but possibly compensatory systems are activated to maintain blood pressure at physiological levels. The important finding is that decreased AT1R/AT2R ratio in castrated rats negated the Ang II effects to increase blood pressure, which is consistent with the studies that indicate AT1R/AT2R ratio plays a crucial role in the development of the hypertensive phenotype, and the vascular AT1R/AT2R ratio relates to the magnitude of blood pressure elevation observed in hypertensive rats [7, 29, 48]. Therefore, the protection against Ang II-induced increase in blood pressure in castrated rats may reflect alterations in the Ang II receptors systems, which are known to play an important role in long-term control of arterial pressure. Indeed, studies show that chronic blockade of AT1R or activation of AT2R prevents development of hypertension [49]. Although, we have shown that testosterone can directly downregulate vascular AT2R through an androgen-receptor-mediated ERK1/2 MAP kinase pathway [20], how testosterone modulates to increase the expression of AT1R need to be evaluated in future investigations. It is also important to note that Ang II, by itself also regulates its own receptors [50, 51]; however, this study shows that presence of testosterone may potentiate the ability of Ang II to regulate its receptor.
Increased peripheral vascular reactivity has been linked to the development and maintenance of hypertension [52]. In particular, enhanced peripheral vascular reactivity to Ang II has been identified in the spontaneously hypertensive rats [53] and prehypertensive humans [54]. To dissect out the functional effect of alterations in AT1R/AT2R ratio and the contribution of the peripheral vasculature to contractile responses and to blood pressure increase, we examined vascular reactivity to Ang II in mesenteric arteries. In the present study, Ang II-induced contractile responses were significantly increased in intact and testosterone-replaced castrated rats. The Emax were greater in the Ang II-infused intact and testosterone-replaced rats, but was prevented by castration in proportion with the decreased AT1R/AT2R ratio and blood pressure decreases. Such data are consistent with classical receptor theory which predicts that increasing the contractile receptor number will result in an increase of the maximal responsiveness to agonists [55]. Other studies also reported testosterone-dependent exaggerated vasomotor responses to Ang II in growth-restricted offspring [29, 56], genetically hypertensive rats [57] and testosterone-treated pregnant rats [31]. Interestingly, the vasomotor response to another vasoconstrictor, phenylephrine, was also enhanced in Ang II-infused intact and testosterone-replaced rats, but was prevented in castrated rats. Thus, it is likely that androgen potentiation of contractile effect may occur for multiple vasoconstrictors reflecting alterations not only occur at the receptor level, but at a common intracellular signaling level. Consistently, testosterone is shown to upregulate RhoA [58], Rho kinase [59], and PKC [57, 60], which are important second messenger systems that amplifies the vascular smooth muscle contractile sensitivity.
Although we focused on the peripheral vascular reactivity, it is important to acknowledge that protection against Ang II-induced blood pressure increases in castrated rats may have a contribution from activation of brain [61], kidney [62] and immune cells [63]. Similarly, testosterone metabolites, 6β-hydroxytestosterone and dihydrotestosterone are known players in mediating vascular and cardiac dysfunction [26], hence it is possible that a component of Ang II-induced increase in arterial pressure be due to formation of these metabolites. It is also possible that the effect of androgens to facilitate Ang II actions is mediated via genomic or nongenomic androgen receptor [64]. However, restoration by testosterone of Ang II actions in castrated males was not due to alterations in expression of androgen receptor because expression of androgen receptor was not altered in these rats.
A major goal of hypertension treatment is to prevent or ameliorate injury to key target organs, including the heart. One of the most prevalent manifestations of end-organ damage in hypertension is the development of left ventricular hypertrophy and fibrosis [37], and its presence confers substantial cardiovascular risk [37, 38]. Although pressure load from elevated blood pressure clearly contributes to left ventricular hypertrophy and fibrosis, several lines of evidence suggest that activation of the RAS also plays a role. For example, Ang II, acting through AT1R, stimulates hypertrophy of cardiac myocytes and interstitial fibrosis [65, 66]. In addition, clinical studies have demonstrated RAS blockage cause regression of cardiac hypertrophy and fibrosis more effectively than other classes of antihypertensive agents with similar levels of blood pressure control [39, 40, 67]. Thus, in this study it is difficult to distinguish whether the restoration of Ang II-induced cardiac remodeling in castrated males is due to direct cardiac Ang II receptor activation or secondary to restoration of blood pressure. However, the finding that the effects of Ang II were restored by testosterone replacement in castrated rats suggests that androgens play a permissive role in the development of Ang II-induced hypertension and associated cardiac effects.
In conclusion, this study demonstrates that testosterone play a permissive role in development and maintenance of Ang II-induced hypertension, increased vascular reactivity to vasoconstrictor agents, cardiac hypertrophy and fibrosis. Modulation of ratio of AT1R/AT2R in the vasculature may be one mechanism critical to normalization of blood pressure in castrated males. Recently, the effect of sex chromosome complement has been implicated in Ang II-induced hypertension [68]. Therefore, it appears that, in addition to sex chromosomes, testosterone contributes to Ang II-induced hypertension and associated pathogenesis in males. Identifying agents that prevent androgen-mediated upregulation of Ang II-induced hypertension could be useful for treatment of hypertension and associated CVD.
Acknowledgements
Financial Support from the National Institute of Health (NIH) through grants, HL119869 and HL134779 to SK is greatly appreciated. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interest: The authors have declared that no conflict of interest exists.
Notes
Edited by Dr. T. Rajendra Kumar, PhD, University of Colorado Anschutz Medical Campus
Footnotes
Grant Support: National Institute of Health (NIH) grants, HL119869 and HL134779.
References
- 1. Kotchen JM, McKean HE, Kotchen TA. Blood pressure trends with aging. Hypertension 1982; 4(5_Pt_2):III128–III128. [DOI] [PubMed] [Google Scholar]
- 2. Regitz-Zagrosek V, Oertelt-Prigione S, Seeland U, Hetzer R. Sex and gender differences in myocardial hypertrophy and heart failure. Circ J 2010; 74(7):1265–1273. [DOI] [PubMed] [Google Scholar]
- 3. Rowland NE, Fregly MJ. Role of gonadal hormones in hypertension in the Dahl salt-sensitive rat. Clin Exp Hypertens A 1992; 14:367–375. [DOI] [PubMed] [Google Scholar]
- 4. Crofton JT, Share L. Gonadal hormones modulate deoxycorticosterone-salt hypertension in male and female rats. Hypertension 1997; 29(1):494–499. [DOI] [PubMed] [Google Scholar]
- 5. Reckelhoff JF. Gender differences in the regulation of blood pressure. Hypertension 2001; 37(5):1199–1208. [DOI] [PubMed] [Google Scholar]
- 6. Haywood JR, Hinojosa-Laborde C. Sexual dimorphism of sodium-sensitive renal-wrap hypertension. Hypertension 1997; 30(3):667–671. [DOI] [PubMed] [Google Scholar]
- 7. Sathishkumar K, Balakrishnan M, Chinnathambi V, Gao H, Yallampalli C. Temporal alterations in vascular angiotensin receptors and vasomotor responses in offspring of protein-restricted rat dams. Am J Obstet Gynecol 2012; 206(6):507.e1–507.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chinnathambi V, Balakrishnan M, Yallampalli C, Sathishkumar K. Prenatal testosterone exposure leads to hypertension that is gonadal Hormone-Dependent in adult rat male and female offspring. Biol Reprod 2012; 86(5):137, 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sathishkumar K, Elkins R, Yallampalli U, Yallampalli C. Protein restriction during pregnancy induces hypertension in adult female rat offspring – influence of oestradiol. Br J Nutr 2012; 107(5):665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grigore D, Ojeda NB, Alexander BT. Sex differences in the fetal programming of hypertension. Gender Med 2008; 5(Suppl A):S121–S132. [Google Scholar]
- 11. Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 2007; 59:251–287. [DOI] [PubMed] [Google Scholar]
- 12. Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci 2006; 103:17985–17990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mazzolai L, Pedrazzini T, Nicoud F, Gabbiani G, Brunner HR, Nussberger J. Increased cardiac angiotensin II levels induce right and left ventricular hypertrophy in normotensive mice. Hypertension 2000; 35:985–991. [DOI] [PubMed] [Google Scholar]
- 14. Schelling P, Fischer H, Ganten D. Angiotensin and cell growth: a link to cardiovascular hypertrophy? J Hypertens 1991; 9:3–15. [PubMed] [Google Scholar]
- 15. Bader M, Peters J, Baltatu O, Muller DN, Luft FC, Ganten D. Tissue renin-angiotensin systems: new insights from experimental animal models in hypertension research. J Mol Med 2001; 79:76–102. [DOI] [PubMed] [Google Scholar]
- 16. Billet S, Aguilar F, Baudry C, Clauser E. Role of angiotensin II AT1 receptor activation in cardiovascular diseases. Kidney Int 2008; 74:1379–1384. [DOI] [PubMed] [Google Scholar]
- 17. Hansson L, Lindholm LH, Niskanen L, Lanke J, Hedner T, Niklason A, Luomanmaki K, Dahlof B, de Faire U, Morlin C, Karlberg BE, Wester PO et al. . Effect of angiotensin-converting-enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPPP) randomised trial. Lancet North Am Ed 1999; 353:611–616. [DOI] [PubMed] [Google Scholar]
- 18. Komukai K, Mochizuki S, Yoshimura M. Gender and the renin-angiotensin-aldosterone system. Fundam Clin Pharmacol 2010; 24:687–698. [DOI] [PubMed] [Google Scholar]
- 19. Reckelhoff JF, Zhang H, Srivastava K. Gender differences in development of hypertension in spontaneously hypertensive rats: role of the renin-angiotensin system. Hypertension 2000; 35:480–483. [DOI] [PubMed] [Google Scholar]
- 20. Mishra JS, Hankins GD, Kumar S. Testosterone downregulates angiotensin II type-2 receptor via androgen receptor-mediated ERK1/2 MAP kinase pathway in rat aorta. J Renin Angiotensin Aldosterone Syst 2016; 17(4):1470320316674875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sullivan JC, Bhatia K, Yamamoto T, Elmarakby AA. Angiotensin (1–7) receptor antagonism equalizes angiotensin II-induced hypertension in male and female spontaneously hypertensive rats. Hypertension 2010; 56:658–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song J, Kost CK Jr, Martin DS. Androgens augment renal vascular responses to ANG II in New Zealand genetically hypertensive rats. Am J Physiol Regul Integr Comp Physiol 2006; 290(6):R1608–R1615. [DOI] [PubMed] [Google Scholar]
- 23. Chung CC, Hsu RC, Kao YH, Liou JP, Lu YY, Chen YJ. Androgen attenuates cardiac fibroblasts activations through modulations of transforming growth factor-beta and angiotensin II signaling. Int J Cardiol 2014; 176:386–393. [DOI] [PubMed] [Google Scholar]
- 24. Kang NN, Fu L, Xu J, Han Y, Cao JX, Sun JF, Zheng M. Testosterone improves cardiac function and alters angiotensin II receptors in isoproterenol-induced heart failure. Arch Cardiovas Dis 2012; 105:68–76. [DOI] [PubMed] [Google Scholar]
- 25. Ikeda Y, Aihara K, Yoshida S, Sato T, Yagi S, Iwase T, Sumitomo Y, Ise T, Ishikawa K, Azuma H, Akaike M, Kato S et al. . Androgen-androgen receptor system protects against angiotensin II-induced vascular remodeling. Endocrinology 2009; 150:2857–2864. [DOI] [PubMed] [Google Scholar]
- 26. Pingili AK, Kara M, Khan NS, Estes AM, Lin Z, Li W, Gonzalez FJ, Malik KU. 6beta-hydroxytestosterone, a cytochrome P450 1B1 metabolite of testosterone, contributes to angiotensin II-induced hypertension and its pathogenesis in male mice. Hypertension 2015; 65:1279–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rocha FL, Carmo EC, Roque FR, Hashimoto NY, Rossoni LV, Frimm C, Aneas I, Negrao CE, Krieger JE, Oliveira EM. Anabolic steroids induce cardiac renin-angiotensin system and impair the beneficial effects of aerobic training in rats. Am J Physiol Heart Circ Physiol 2007; 293:H3575–H3583. [DOI] [PubMed] [Google Scholar]
- 28. Xue B, Pamidimukkala J, Hay M. Sex differences in the development of angiotensin II-induced hypertension in conscious mice. Am J Physiol Heart Circ Physiol 2005; 288:H2177–H2184. [DOI] [PubMed] [Google Scholar]
- 29. Sathishkumar K, Balakrishnan MP, Yallampalli C. enhanced mesenteric arterial responsiveness to angiotensin II is androgen Receptor-Dependent in prenatally Protein-Restricted adult female rat offspring. Biol Reprod 2015; 92:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chinnathambi V, More AS, Hankins GD, Yallampalli C, Sathishkumar K. Gestational exposure to elevated testosterone levels induces hypertension via heightened vascular angiotensin II type 1 receptor signaling in rats. Biol Reprod 2014; 91:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chinnathambi V, Blesson CS, Vincent KL, Saade GR, Hankins GD, Yallampalli C, Sathishkumar K. Elevated testosterone levels during rat pregnancy cause hypersensitivity to angiotensin II and attenuation of endothelium-dependent vasodilation in uterine arteries. Hypertension 2014; 64:405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Malkin CJ, Pugh PJ, Jones RD, Kapoor D, Channer KS, Jones TH. The effect of testosterone replacement on endogenous inflammatory cytokines and lipid profiles in hypogonadal men. J Clin Endocrinol Metab 2004; 89:3313–3318. [DOI] [PubMed] [Google Scholar]
- 33. Liu PY, Death AK, Handelsman DJ. Androgens and cardiovascular disease. Endocr Rev 2003; 24:313–340. [DOI] [PubMed] [Google Scholar]
- 34. Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, Kim JB, Schmitt JP et al. . Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest 2010; 120:3520–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen Y, Yu Q, Xu CB. A convenient method for quantifying collagen fibers in atherosclerotic lesions by ImageJ software. Int J Clin Exp Med 2017; 10:14904–14910. [Google Scholar]
- 36. Caglayan E, Stauber B, Collins AR, Lyon CJ, Yin F, Liu J, Rosenkranz S, Erdmann E, Peterson LE, Ross RS, Tangirala RK, Hsueh WA. Differential roles of cardiomyocyte and macrophage peroxisome proliferator-activated receptor gamma in cardiac fibrosis. Diabetes 2008; 57:2470–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med 1991; 114:345–352. [DOI] [PubMed] [Google Scholar]
- 38. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990; 322:1561–1566. [DOI] [PubMed] [Google Scholar]
- 39. Devereux RB, Dahlof B, Gerdts E, Boman K, Nieminen MS, Papademetriou V, Rokkedal J, Harris KE, Edelman JM, Wachtell K. Regression of hypertensive left ventricular hypertrophy by losartan compared with atenolol: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) trial. Circulation 2004; 110:1456–1462. [DOI] [PubMed] [Google Scholar]
- 40. Schmieder RE, Martus P, Klingbeil A. Reversal of left ventricular hypertrophy in essential hypertension. A meta-analysis of randomized double-blind studies. JAMA 1996; 275:1507–1513. [PubMed] [Google Scholar]
- 41. Lee RT, Bloch KD, Pfeffer JM, Pfeffer MA, Neer EJ, Seidman CE. Atrial natriuretic factor gene expression in ventricles of rats with spontaneous biventricular hypertrophy. J Clin Invest 1988; 81:431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sadoshima J, Izumo S. Molecular characterization of angiotensin II–induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res 1993; 73:413–423. [DOI] [PubMed] [Google Scholar]
- 43. Dalmasso C, Patil CN, Yanes Cardozo LL, Romero DG, Maranon RO. Cardiovascular and metabolic consequences of testosterone supplements in young and old male spontaneously hypertensive Rats: Implications for testosterone supplements in men. J Am Heart Assoc 2017; 6:e007074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dasinger JH, Intapad S, Rudsenske BR, Davis GK, Newsome AD, Alexander BT. Chronic blockade of the androgen receptor abolishes Age-Dependent increases in blood pressure in female Growth-Restricted Rats: Novelty and significance. Hypertension 2016; 67:1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. More AS, Mishra JS, Hankins GD, Yallampalli C, Sathishkumar K. Enalapril normalizes endothelium-derived hyperpolarizing factor-mediated relaxation in mesenteric artery of adult hypertensive rats prenatally exposed to testosterone. Biol Reprod 2015; 92:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wright JW, Mizutani S, Harding JW. Pathways involved in the transition from hypertension to hypertrophy to heart failure. Treatment strategies. Heart Fail Rev 2008; 13:367–375. [DOI] [PubMed] [Google Scholar]
- 47. Carey RM. Blood pressure and the renal actions of AT2 receptors. Curr Hypertens Rep 2017; 19:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Silva-Antonialli MM, Tostes RC, Fernandes L, Fior-Chadi DR, Akamine EH, Carvalho MH, Fortes ZB, Nigro D. A lower ratio of AT1/AT2 receptors of angiotensin II is found in female than in male spontaneously hypertensive rats. Cardiovasc Res 2004; 62:587–593. [DOI] [PubMed] [Google Scholar]
- 49. Moroni C, Tolone S, Lopreiato F, Scrofani AR, Bossini A, Affricano C, Cassone R, Gaudio C. Effects of losartan on left ventricular mass: a three-year follow-up in elderly hypertensives with myocardial hypertrophy despite successful conventional antihypertensive treatment. Eur Rev Med Pharmacol Sci 2017; 21:1323–1328. [PubMed] [Google Scholar]
- 50. Wei SG, Yu Y, Zhang ZH, Felder RB. Angiotensin II upregulates hypothalamic AT1 receptor expression in rats via the mitogen-activated protein kinase pathway. Am J Physiol Heart Circ Physiol 2009; 296:H1425–H1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li JY, Avallet O, Berthelon MC, Langlois D, Saez JM. Transcriptional and translational regulation of angiotensin II type 2 receptor by angiotensin II and growth factors. Endocrinology 1999; 140:4988–4994. [DOI] [PubMed] [Google Scholar]
- 52. Mayet J, Hughes A. Cardiac and vascular pathophysiology in hypertension. Heart 2003; 89:1104–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Endemann D, Touyz RM, Li JS, Deng LY, Schiffrin EL. Altered angiotensin II-induced small artery contraction during the development of hypertension in spontaneously hypertensive rats. Am J Hypertens 1999; 12:716–723. [DOI] [PubMed] [Google Scholar]
- 54. Palmgren E, Widgren B, Aurell M, Herlitz H. Increased renal vascular sensitivity to angiotensin II in hypertension is due to decreased response to prostaglandins. J Hypertens 2003; 21:969–976. [DOI] [PubMed] [Google Scholar]
- 55. Kenakin TP. Pharmacologic Analysis of Drug-Receptor Interaction, 3rd ed Philadelphia: Lippincott-Raven Publishers; 1997:289–330. [Google Scholar]
- 56. Ojeda NB, Royals TP, Black JT, Dasinger JH, Johnson JM, Alexander BT. Enhanced sensitivity to acute angiotensin II is testosterone dependent in adult male growth-restricted offspring. Am J Physiol Regul Integr Comp Physiol 2010; 298:R1421–R1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Song J, Eyster KM, Kost CK Jr., Kjellsen B, Martin DS. Involvement of protein kinase C-CPI-17 in androgen modulation of angiotensin II-renal vasoconstriction. Cardiovasc Res 2010; 85:614–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guilluy C, Bregeon J, Toumaniantz G, Rolli-Derkinderen M, Retailleau K, Loufrani L, Henrion D, Scalbert E, Bril A, Torres RM, Offermanns S, Pacaud P et al. . The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med 2010; 16:183–190. [DOI] [PubMed] [Google Scholar]
- 59. Song J, Kost CK Jr, Martin DS. Androgens potentiate renal vascular responses to angiotensin II via amplification of the Rho kinase signaling pathway. Cardiovasc Res 2006; 72:456–463. [DOI] [PubMed] [Google Scholar]
- 60. Blesson CS, Chinnathambi V, Hankins GD, Yallampalli C, Sathishkumar K. Prenatal testosterone exposure induces hypertension in adult females via androgen receptor-dependent protein kinase Cdelta-mediated mechanism. Hypertension 2015; 65:683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR, Davisson RL. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res 2002; 91:1038–1045. [DOI] [PubMed] [Google Scholar]
- 62. Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, Flannery PJ, Spurney RF, Kim HS, Smithies O, Le TH, Coffman TM. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest 2005; 115:1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II–induced hypertension and vascular dysfunction. J Exp Med 2007; 204:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science 2005; 308:1583–1587. [DOI] [PubMed] [Google Scholar]
- 65. Schunkert H, Sadoshima J, Cornelius T, Kagaya Y, Weinberg EO, Izumo S, Riegger G, Lorell BH. Angiotensin II-induced growth responses in isolated adult rat hearts. Evidence for load-independent induction of cardiac protein synthesis by angiotensin II. Circ Res 1995; 76:489–497. [DOI] [PubMed] [Google Scholar]
- 66. Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell 1993; 75:977–984. [DOI] [PubMed] [Google Scholar]
- 67. Mathew J, Sleight P, Lonn E, Johnstone D, Pogue J, Yi Q, Bosch J, Sussex B, Probstfield J, Yusuf S Heart Outcomes Prevention Evaluation I . Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ramipril. Circulation 2001; 104:1615–1621. [DOI] [PubMed] [Google Scholar]
- 68. Ji H, Zheng W, Wu X, Liu J, Ecelbarger CM, Watkins R, Arnold AP, Sandberg K. Sex chromosome effects unmasked in angiotensin II-induced hypertension. Hypertension 2010; 55:1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]