

Abstract
Background
Hepatocellular carcinoma (HCC) is the cancer with the second highest mortality in the world due to its late presentation and limited treatment options. As such, there is an urgent need to identify novel biomarkers for early diagnosis and to develop novel therapies. The availability of next-generation sequencing (NGS) data from tumors of liver cancer patients has provided us with invaluable resources to better understand HCC through the integration of data from different sources to facilitate the identification of promising biomarkers or therapeutic targets.
Findings
Here, we review key insights gleaned from more than 20 NGS studies of HCC tumor samples, comprising approximately 582 whole genomes and 1,211 whole exomes mainly from the East Asian population. Through consolidation of reported somatic mutations from multiple studies, we identified genes with different types of somatic mutations, including single nucleotide variations, insertion/deletions, structural variations, and copy number alterations as well as genes with multiple frequent viral integration. Pathway analysis showed that this curated list of somatic mutations is critically involved in cancer-related pathways, viral carcinogenesis, and signaling pathways. Lastly, we addressed the future directions of HCC research as more NGS datasets become available.
Conclusions
Our review is a comprehensive resource for the current NGS research in HCC, consolidating published articles, potential gene candidates, and their related biological pathways.
Keywords: hepatocellular carcinoma, next-generation sequencing, somatic mutations, viral integration
Introduction
Based on GLOBOCAN 2012, liver cancer is the second most common cause of death from cancer worldwide. Liver cancer is the fifth most common cancer in males (554,000 cases) and the ninth most common cancer in females (228,000 cases) [1]. The incidence rate is higher in males than females at a male-to-female ratio of 2.4 worldwide, and the mortality-to-incidence rate is as high as 0.94 and 0.98 for males and females, respectively. Hepatocellular carcinoma (HCC) is the most dominant form of primary liver cancer. Geographically, there is a high incidence rate in Africa (northern and western) and Asia (eastern and southeastern), particularly in China, which accounts for 50 percent of all HCC cases [2].
HCC is commonly associated with risk factors such as hepatitis B (HBV) and hepatitis C (HCV) infection, alcohol, mycotoxin Aflatoxin, obesity, and non-alcoholic fatty liver disease; the risk varies depending on gender, geographic region, and ethnicity [2–4]. Early evidence shows the association of HBV and HCV infection with the development of liver cirrhosis and HCC [5, 6]. The HBV vaccine has been available since the early 1980s; and implementation of HBV vaccination programs in 177 of 193 World Health Organization member states are successful in decreasing HCC incidence rates in children [7, 8].
While environmental factors play a role in HCC, multiple recurrent genetic aberrations and the disruption of the host genome due to HBV DNA integration in HBV-associated HCC are reported to cause the dysregulation of genes important for the hallmarks of cancer. Initial studies identified HBV integration sites via HBV DNA probes or polymerase chain reaction assay followed by Sanger sequencing [9–13]. Subsequently, somatic alterations such as mutations, gene copy number changes, and chromosomal rearrangements detected in the HCC-derived cell lines were found to affect the expression of oncogenes and tumor suppressor genes [14, 15]. Progress in the mapping of each viral integration site and genetic aberration in HCC patients was ad hoc and slow before the advent of next-generation sequencing (NGS).
NGS technologies, including RNA-sequencing (RNA-seq), whole-exome sequencing (WXS), and whole-genome sequencing (WGS), form the foundation of today's discovery-based genomics research. With the reduced cost of massively parallel sequencing technologies over the last decade [16], there has been an increasing number of genomic liver cancer studies providing new insights about liver cancer. Pioneering NGS studies conducted on patient samples have shown a tremendous increase in our understanding of HBV viral integration patterns [17–19] as well as somatic alterations found in liver cancer [20–22]. The large amount of sequencing data generated has been archived on data servers worldwide, enabling researchers to perform integrative analyses that will lead to new findings. However, maneuvering through literature and data repositories to locate and access this information remains a tedious process.
Here, we introduce and consolidate all existing NGS-based studies on liver cancer (Fig. 1). Only the most relevant studies, conducted using NGS in HCC, have been listed in a recent review [23]. Our NGS-based resource is a complete list of data samples of approximately 582 whole genomes and 1,211 whole exomes. It summarizes the key research and clinical findings from each article with direct links to all publicly available WGS/WXS liver cancer datasets to promote better knowledge and data facilitation. The key findings of somatic mutations, HBV integrations, and mutational signatures reported from recent high-throughput studies and related integrative studies are discussed. We highlight key genes reported across multiple studies found to have recurrence of somatic mutations or HBV integration events. Additionally, we provide a meta-analysis of the pathways that these alterations dysregulate. Finally, we discuss future directions and trends in liver cancer research via the analysis of high-throughput data.
Figure 1:

Summary of NGS databases in liver cancer showing its current and potential research direction.
NGS Resources
Raw sequencing data, read alignment, and annotations from NGS platforms can be accessed via National Center for Biotechnology Information (NCBI)-Sequence Read Archive (SRA) [24], European Molecular Biology Laboratory - European Bioinformatics Institute (EMBL-EBI), European Nucleotide Archive [25], and DNA Data Bank of Japan-SRA [26]. The National Cancer Institute's Genomic Data Commons [27] currently hosts genomic data from the Cancer Genome Atlas (TCGA) project that consists of multiple cancer types. There are currently 377 liver hepatocellular carcinoma samples with data from WXS, single nucleotide polymorphism (SNP)-array, methylation, mRNA, and microRNA profiling. Gigadb [28] is a repository for open-access data associated with the GigaScience journal [29], which currently holds an HCC dataset from 88 individuals [30]. The International Cancer Genome Consortium (ICGC) [31] is a global effort to coordinate large-scale cancer genome studies by providing a comprehensive catalogue of somatic mutations across 50 cancer types, which generates approximately 500 samples each [32]. While primary data files are stored on NCBI and/or EBI, ICGC provides interpreted datasets for somatic mutation calls and incorporates transcriptomic and DNA methylation analyses from the same tumor samples.
We reviewed and consolidated a comprehensive list of liver cancer studies that have analyzed high-throughput genomics data (Table 1). The majority of the studies have their raw and/or processed data available on the above-mentioned public databases (Table 1, Data URL). These studies are mainly focused on liver cancer patients from a single country of the East Asian population (Table 1, Population). Genomics data from the Japanese population constitutes the largest sample size [21, 33–37], including a collection of 300 whole genomes reported in a recent study [33]. NGS studies were also performed with HCC patients from China [38–40], Hong Kong [18, 39, 41, 42], Korea [43–45], Taiwan [46, 47], Singapore [19], and Europe [48–51]. Several studies have a collection of samples from various ethnicities (TCGA) or multiple sources [17, 22, 52–54].
Table 1:
Summary of NGS resources and their key findings from liver cancer studies
| No. | Reference | Data URL | Sample type/total cases | Population | Viral status | Key findings |
|---|---|---|---|---|---|---|
| 1 | TCGA | https://dcc.icgc.org/projects/LIHC-US | 54 WGS (52 HCC, 1 ICC, 1 FC) | 39 White, 9 Asian, 3 African American, 3 Unknown | 7 HCV, 7 HBV, 40 NBNC | TCGA-LIHC-WGS |
| 2 | TCGA | https://portal.gdc.cancer.gov/projects/TCGA-LIHC | 376 WXS (366 HCC, 7 cHCC/ICC, 3 FC) + 371 RNA-seq (361 HCC, 7 cHCC/ICC, 3 FC) | 187/184 White, 160/158 Asian, 17 African American, 2 American Indian/Native, 10 Unknown | 49 HCV, 102 HBV, 8 HBV/HCV, 217 NBNC | TCGA-LIHC-WXS |
| 3 | Letouze et al. (2017) Nature Comm. https://doi.org/10.1038/s41467-017-01358-x | https://www.ebi.ac.uk/ega/studies/EGAS00001002408 | 44 WGS (35 HCC, 5 HCA, 4 FC) | 40 European, 4 African | 4 HCV, 5 HBV, 35 NBNC | 1. Analysis of more than 300 genomes highlighted 10 mutational signatures, including ubiquitous as well as sporadic signatures.2. Reconstruction of the temporal evolution in driver mutations and signatures revealed the clonal architecture in each tumor. |
| 4 | Ng et al. (2017) Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aan6446 | https://www.ebi.ac.uk/ega/studies/EGAS00001002301 | 98 WXS (HCC) | Asian (Taiwan) | 21 HCV, 56 HBV, 3 HBV/HCV, 10 NBNC, 8 N.D. | 1. Distinct mutational signatures were identified in the whole exomes of HCC patients with aristolochic acid exposure.2. The aristolochic acid signature also revealed known cancer driver genes, TP53 and CTNNB1, mutated 54 and 24 percent of the total HCC cases respectively. |
| 5 | Zhang et al., (2017) Gastroenterology. https://doi.org/10.1053/j.gastro.2017.03.024 | Unknown | 49 WGS + 13 WXS (HCC) | Asian (China) | 38 HBV, 9 NB, 2 N.D. | 1. Aflatoxin-associated HCCs were reported to frequently contain C>A transversions, sequence motif GCN, and strand bias.2. Frequent mutations identified in the adhesion G protein-coupled receptor B1 gene (ADGRB1) were found to be associated with increased capillary density of the tumor tissue. |
| 6 | Fujimoto et al. (2016) Nature Genetics. https://doi.org/10.1038/ng.3547 | https://dcc.icgc.org/projects/LIRI-JP https://www.ebi.ac.uk/ega/studies/EGAS00001000671 | 300 WGS (268 HCC, 24 ICC, 8 cHCC/ICC) + 254 RNA-seq | Asian (Japan) | 159 HCV, 82 HBV, 4 HBV/HCV, 55 NBNC | 1. Coding and noncoding regions (including NEAT1 and MALAT1) were identified to have significant mutations.2. Structural variation analysis revealed cancer-related genes (e.g., TERT and NCOR1) that led to altered expression. |
| 7 | Hirotsu et al. (2016) Hepatology Research https://doi.org/10.1111/hepr.12663 | http://trace.ddbj.nig.ac.jp/DRASearch/submission?acc=DRA003210 | 9 WXS (HCC) | Asian (Japan) | 1 HBV, 5 HCV, 3 NBNC | 1. Targeted deep sequencing analysis showed that TP53 (3/9 cases) and CTNNB1 (2/9 cases) were recurrent missense mutations in HCCs.2. Functional analysis of the β-catenin H36P mutant was observed to be resistant to protein degradation and to promote HCC cell proliferation. |
| 8 | Fujimoto et al. (2015) Nature Comm. https://doi.org/10.1038/ncomms7120 | https://dcc.icgc.org/projects/LIRI-JP | 90 WGS (60 HCC, 7 cHCC/ICC, 22 ICC, 1 CoCC) + 69 RNA-seq | Asian (Japan) | 60 HCC: 23 HBV, 29 HCV, 3 HBV/HCV, 5 NBNC | 1. cHCC/ICC and CoCC showing biliary epithelial differentiation (LCB) have recurrent mutations in the TERT promoter and chromatin regulators.2. Hepatitis-positive HCC and cHCC/CC had a larger frequency of TERT promoter mutations and a lower frequency of KRAS and IDH1/2 mutations than hepatitis-negative LCB. |
| 9 | Kang et al. (2015) Genomics https://doi.org/10.1016/j.ygeno.2014.11.005 | http://www.ebi.ac.uk/ena/data/view/ERP001196 http://gigadb.org/dataset/100034 | 9 WGS + RNA-seq (HCC) | Asian (Hong Kong) | HBV | 1. An improved bioinformatics pipeline detects RNA-editing events in HCC tumor and matched adjacent tissues.2. Varying editing degrees were significant in 13 cancer-related genes from 18 editing sites and one gene with editing in the CDS region between normal and tumor tissues. |
| 10 | Schulze et al. (2015) Nature Geneticshttps://doi.org/10.1038/ng.3252 | https://dcc.icgc.org/projects/LICA-FR https://www.ebi.ac.uk/ega/studies/EGAS00001000217 | 236 WXS (HCC) | European (193 France, 9 Spain, 41 Italy) | 57 HCV, 29 HBV, 4 HBV/HCV, 142 NBNC | 1. Mutational signatures were significantly associated with demographic, etiological, molecular features.2. Signature 23 that contained predominantly C>T mutations is consistent with the study by Totoki et al. (2011). |
| 11 | Nault et al. (2015) Nature Geneticshttps://doi.org/10.1038/ng.3389 | https://www.ebi.ac.uk/ega/studies/EGAS00001000217 | 193 WXS (HCC) | European (France) | 36 HCV, 22 HBV, 135 NBNC | 1. Clonal integration of the adeno-associated virus type 2 (AAV2) were identified in 11of 193 HCCs.2. AAV2 integrations occurred in known cancer driver genes including TERT, CCNA2, CCNE1, KMT2B, and TNFSF10. |
| 12 | Dong et al. (2015) PLoS Onehttps://doi.org/10.1371/journal.pone.0123175 | http://www.ncbi.nlm.nih.gov/bioproject/279878 | 55 RNA-seq (HCC) | Asian (China) | 49 HBV, 7 NBNC | 1. MLL4 was identified as the most frequent HBV integration site (8/44 cases).2. Gene expression levels of the 8 MLL4-integration-positive samples were significantly higher than wild-type tumor and adjacent tissues. |
| 13 | Totoki et al. (2014) Nature Geneticshttps://doi.org/10.1038/ng.3126 | http://www.ncbi.nlm.nih.gov/gap/?term=phs000509 https://www.ebi.ac.uk/ega/studies/EGAS00001000389 | 503 WXS (488 HCC, 2 cHCC/ICC, 13 ICC) | 414 Asian (Japan), 50 Caucasian, 14 US-Asian, 11 African American, 14 N.D. | 212 HCV, 117 HBV, 12 HBV/HCV, 150 NBNC, 9 N.D. | 1. Thirty candidate driver genes, including non-recurring mutated genes BRD7, MEN1, TSC2, SCRAP, and NCOR1 were identified.2. Distinct substitution signatures were detected between the various ancestries and gender but not associated with viral status. |
| 14 | Shirashi et al. (2014) PLoS Onehttps://doi.org/10.1371/journal.pone.0114263 | https://www.ebi.ac.uk/ega/datasets/EGAD00001001035 | 22 WGS + RNA-seq (HCC) | Asian (Japan) | HBV | 1. Comparison of genomic and transcriptomic reads identified 292 genomic mutation-related splicing aberrations.2. Twenty-three of 33 HBV-human fusions were reported to affect TERT, FN1, MLL4, as well as concentrated around the HBx genes. |
| 15 | Fernandez-Banet et al. (2014) Genomics https://doi.org/10.1016/j.ygeno.2014.01.003 | http://www.ebi.ac.uk/ena/data/view/ERP001196 http://gigadb.org/dataset/100034 | 88 WGS (HCC) | Asian (Hong Kong) | 81 HBV, 7 NBNC | 1. A total of 4,314 somatic genomic rearrangement (GR) events were detected and annotated at the single-nucleotide resolution.2. Five HCC tumors harbored chromothripsis on chromosomal arms 1q, 8q, and 5p; 13 genes, including CEBPB, MCL1, and AXIN1, were significantly affected by GR. |
| 16 | Jhunjhunwala et al. (2014) Genome Biology https://doi.org/10.1186/s13059-014-0436-9 | https://www.ebi.ac.uk/ega/studies/EGAS00001000824 | 12 WGS + RNA-seq (HCC) | Samples obtained from commercial sources | 11 HBV, 1 NBNC | 1. Recurrent mutations in TP53, AXIN1, and CTNNB1 were detected as well as a rare find in LAMA2 (6/42 cases) and IDH1 (2/42 cases).2. The activation of TERT was either due to viral integrations in its promoter or its translocation to another chromosomal region. |
| 17 | Ahn et al. (2014) Hepatology https://doi.org/10.1002/hep.27198 | Unknown | 231 WXS (HCC) | Asian (Korea) | 167 HBV, 22 HCV, 42 NBNC | 1. Nine significantly mutated genes and cellular pathways such as p53, Wnt, PIK3/Ras, cell cycle, and chromatin remodeling account for ∼80% of the mutations identified in the 231 tumors.2. Genetic aberrations in the cell cycle pathway genes (RB1, MYC, CCND1, RBL2) were associated with cancer-specific and recurrence-free survival. |
| 18 | Woo et al. (2014) PLoS One https://doi.org/10.1371/journal.pone.0115152 | Unknown | 12 WXS (HCC) | Asian (Korea) | HBV | 1. Tumor-specific genes such as CTNNB1, TTN, SETD2, and ALK have been identified.2. The T>A transversions were present significantly and exclusively in tumor-specific variants. |
| 19 | Ouyang et al. (2014) BMC Medical Genomics https://doi.org/10.1186/1755-8794-7-2 | https://trace.ddbj.nig.ac.jp/DRASearch/submission?acc=SRA076160 | 4 WGS (HCC) | Asian (Korea) | HBV | 1. Analysis of the mutational spectrum showed that C>T transition rates within the coding regions were the highest.2. Altered pathways in primary tumor were Wnt, JAK-STAT, cell cycle, and focal adhesion pathways, while tight junction, focal adhesion, and ErbB/MAPK pathways were affected in the metastases. |
| 20 | Kan et al. (2013) Genome Research https://doi.org/10.1101/gr.154492.113 | http://www.ebi.ac.uk/ena/data/view/ERP001196 http://gigadb.org/dataset/100034 | 88 WGS (HCC) | Asian (Hong Kong) | 81 HBV, 7 NBNC | 1. The study reveals recurrent mutations in TP53, CTNNB1 and AXIN1, two genes (JAK1, LRPB1) commonly mutated in other cancers as well as six genes previously not reported.2. Pathways affected include Wnt, cytokine-induced JAK/STAT, G1/S cell cycle, and apoptosis. |
| 21 | Toh et al. (2013) Carcinogenesis https://doi.org/10.1093/carcin/bgs406 | Unknown | 48 FLX-Seq (HCC) | Asian (Singapore) | 48 HBV | 1. Preferential integration of HBV into the TERT promoter (6/97 cases).2. The 3ʹ-end of the HBV X protein is the preferred HBV genomic region detected in the integration events. |
| 22 | Cleary et al. (2013) Hepatology https://doi.org/10.1002/hep.26540 | http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000627.v1.p1 | 87 WXS (HCC) | Samples obtained from Canada, North Carolina, and CHTN | 19 HCV, 38 HBV, 30 NBNC | 1. Thirteen significantly mutated genes identified include CTNNB1, TP53, CPA2, IGSF3, and KEAP1 as well as four significantly mutated gene families.2. Further validation of the MLL gene family revealed MLL4 (6/13 missense mutations) to be a potential driver gene of HCC. |
| 23 | Lin et al. (2013) Oncogene https://doi.org/10.1038/onc.2013.424 | https://trace.ddbj.nig.ac.jp/DRASearch/study?acc=SRP007560 | 55 RNA-seq (HCC) | Asian (Taiwan) | 20 HBV, 18 HCV, 17 NBNC | 1. Putative mRNA sequences filtered via Cufflinks de novo assembly identified, DUNQU1, a 101-amino-acid peptide encoded by 3 exons.2. Analysis of alternative splicing in transcripts revealed three cancer-related events in FGFR2, EXOC7, and ADAM15. |
| 24 | Fujimoto et al. (2012) Nature Genetics https://doi.org/10.1038/ng.2291 | https://dcc.icgc.org/projects/LINC-JP | 27 WGS (HCC) | Asian (Japan) | 11 HBV, 14 HCV, 2 NBNC | 1. TP53 and CTNNB1, as well as ATM, ARID1A, ERRF11, WWP1 mutations were detected in the tumors.2. Gene-set enrichment analysis identified several genes associated with chromatin regulation. |
| 25 | Sung et al. (2012) Nature Genetics https://doi.org/10.1038/ng.2295 | http://www.ebi.ac.uk/ena/data/view/ERP001196 http://gigadb.org/dataset/100034 | 88 WGS (HCC) | Asian (Hong Kong) | 81 HBV, 7 NBNC | 1. A total of 179 of the 399 HBV integration breakpoints were identified in known coding genes.2. HBV integrations led to increased gene expression of TERT, MLL4, and CCNE1. |
| 26 | Guichard et al. (2012) Nature Genetics https://doi.org/10.1038/ng.2256 | https://www.ebi.ac.uk/ega/studies/EGAS00001000217 | 24 WXS (HCC) | European (France) | 4 HCV, 1 HBV, 19 NBNC | 1. A total of 850 mutations corresponded to single-nucleotide variants, particularly C>T changes that occur more frequently in non-cirrhotic liver HCC tumors.2. Major pathways with frequently altered genes identified include Wnt and p53 pathways as well as four recurrent mutations (ARID1A, RPS6KA3, NFE2L2, and IRF2) previously not reported. |
| 27 | Jiang et al. (2012) Genome Research https://doi.org/10.1101/gr.133926.111 | http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000384.v1.p1 | 4 WGS + RNA-seq (HCC) | Samples obtained from commercial sources | 3 HBV, 1 NBNC | 1. RNA-seq expression analysis revealed the impact of HBV integrations on adjacent transcription activation of MLL4 and ANGPT1 in different patients.2. There is a strong bias of viral-fusion transcripts containing HBV genome sequences near its direct repeat 1 (DR1) region. |
| 28 | Huang et al. (2012) Nature Genetics https://doi.org/10.1038/ng.2391 | http://www.ncbi.nlm.nih.gov/bioproject/PRJNA167270 | 10 WXS (HCC) | Asian (China) | 8 HBV, 2 NBNC | 1. The comparison between matched samples of HBV-associated HCC individuals (primary tumor vs. portal vein tumor thromboses) reveals 65 mutations including TP53 and ARID1A.2. ARID1A mutations were also identified in four HCC cell lines with high metastatic potential. |
| 29 | Totoki et al. (2011) Nature Genetics https://doi.org/10.1038/ng.804 | https://dcc.icgc.org/projects/LINC-JP | 1 WGS (HCC) | Asian (Japan) | HCV | 1. The study identified somatic substitutions patterns predominantly from T>C and C>T transitions.2. Somatic alterations include well-known tumor suppressors TP53 and AXIN1 as well as five other genes found commonly mutated in other cancers. |
| 30 | Li et al. (2011) Nature Genetics https://doi.org/10.1038/ng.903 | Unknown | 139 WXS (HCC) | US (44 White, 15 Black, 9 Asian, 1 Hispanic, 1 Arabic, 8 Unknown), China (61 Asian) | 43 HCV, 50 HBV, 2 HBV/HCV, 44 NBNC | 1. Somatic mutations were found in five genes (CTNNB1, TP53, ARID2, DMXL1, and NLRP1).2. Six of nine of the samples containing ARID2 mutations also contained CTNNB1 mutations but none of them contained TP53 mutations. |
| Total | Total | |||||
| 582 WGS; 1211 WXS; 778 RNA-seq; 48 FLX-seq | 43.71% HBV; 21.13% HCV; 34.48% NBNC |
Multiple findings have already been reported on the patient samples from Japan [33–35], Hong Kong [18, 41, 42], and Europe [48–51], as well as integrative studies from multiple sources [54, 55] or commercial sources [17, 53]. Here, we review approximately 582 whole genomes, 1,211 exome, and 778 RNA-seq samples of liver cancer patients (Table 1, Total cases). Of patients with known viral status, 44 percent are infected with HBV, 21 percent with HCV, while 35 percent are not infected by either HBV or HCV (NBNC) (Table 1, Viral status). Several of the groups have also employed NGS to examine HBV integrations in HCC patients [17–19, 56].
Key Findings
Somatic genomic alterations
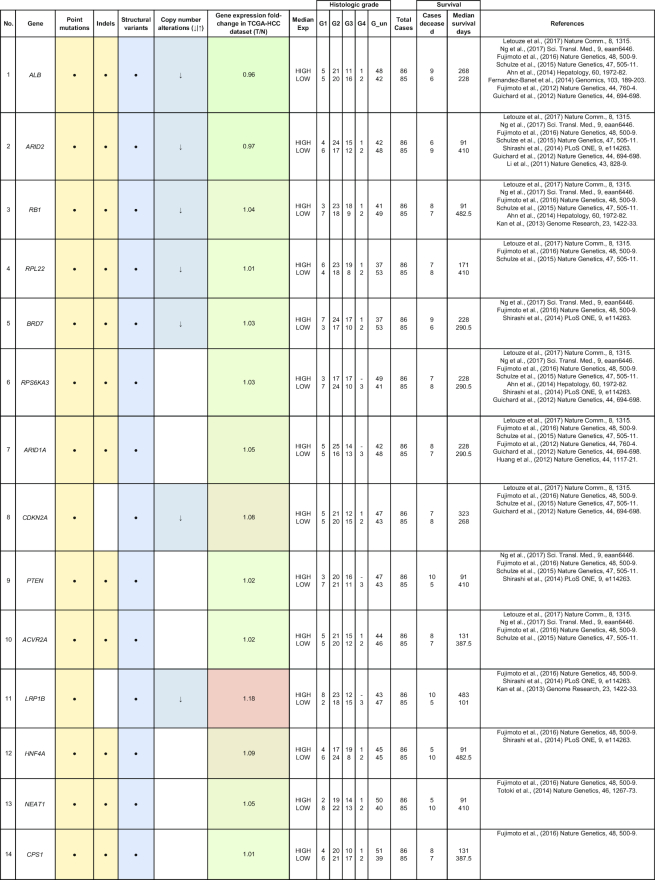
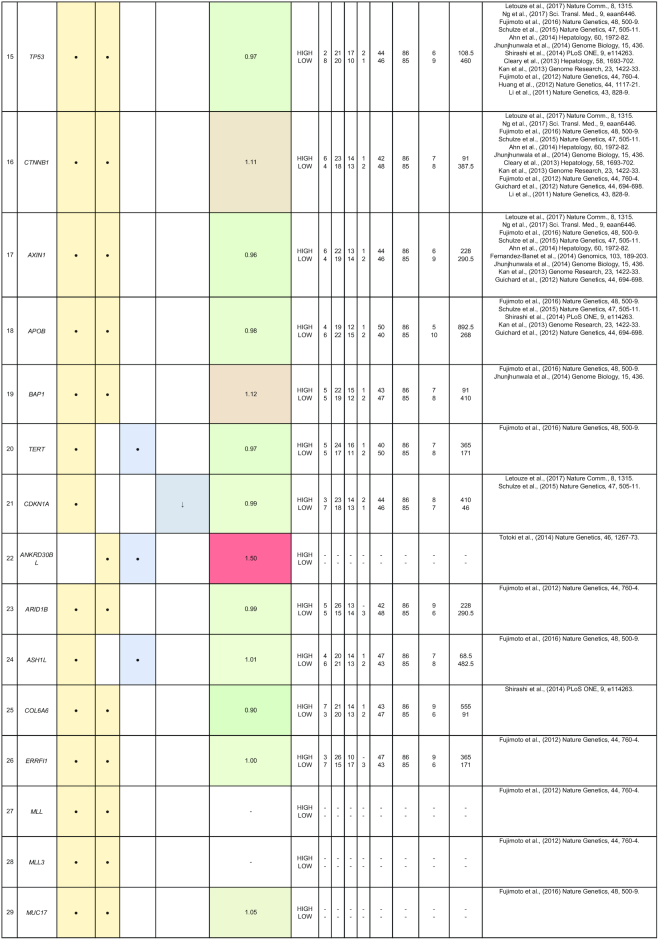
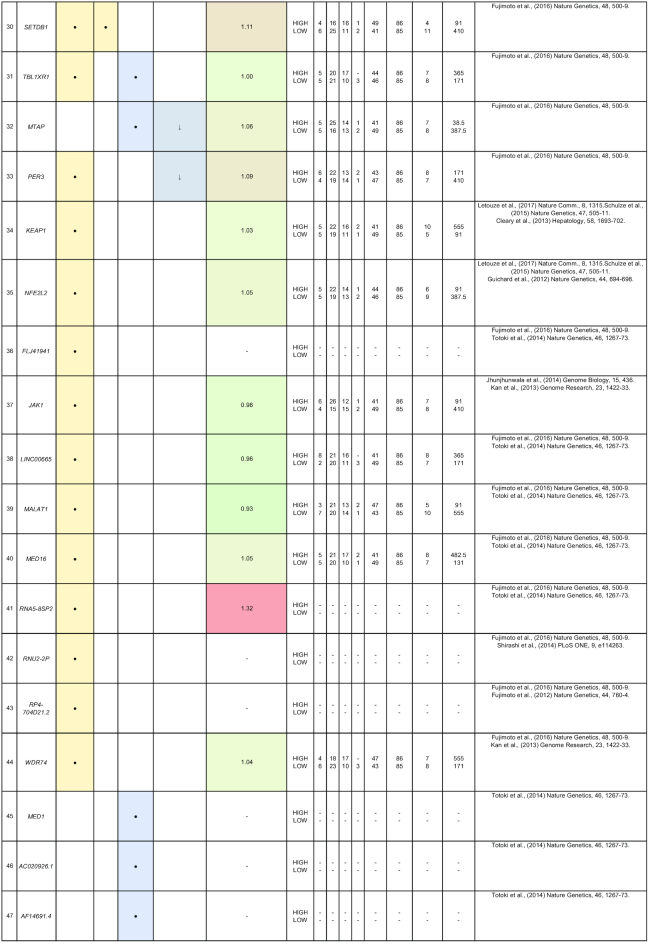
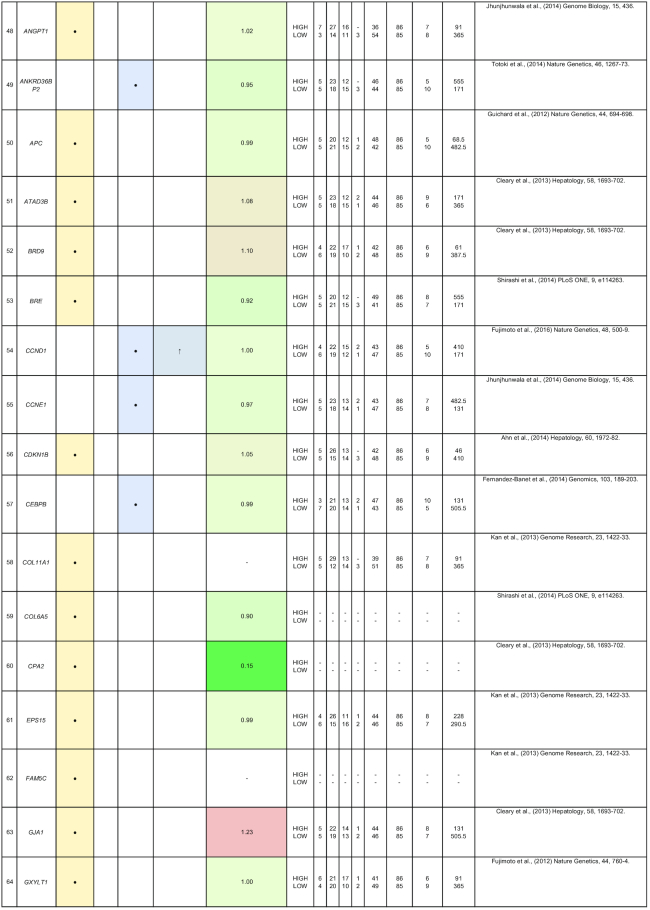
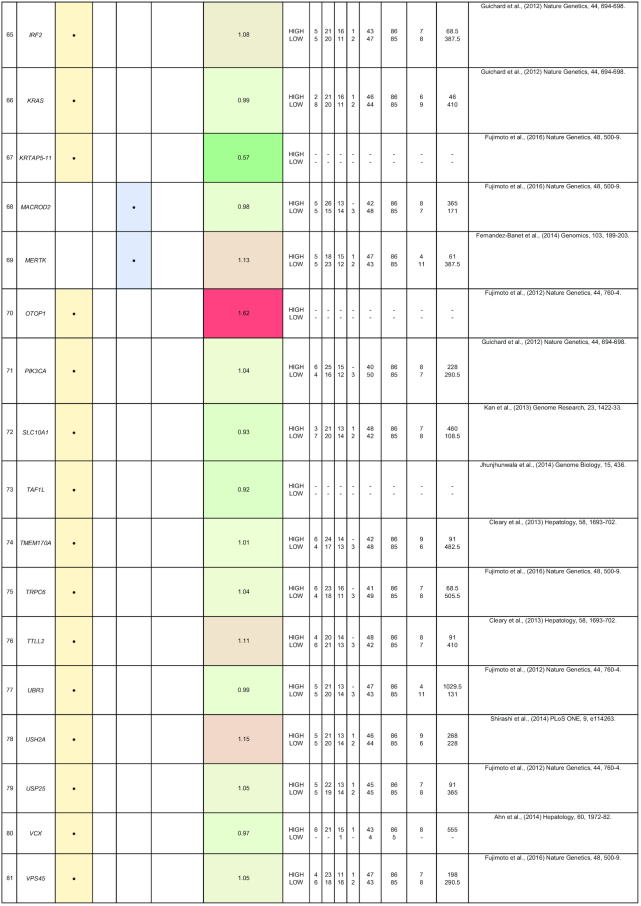
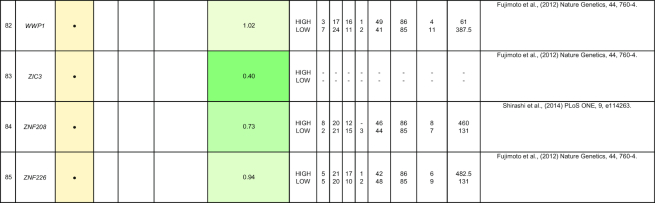
By comparing matched normal and tumor samples, computational algorithms have identified a number of likely cancer-causing point mutations and insertions/deletions (indels). Somatic alterations such as point mutations, indels, structural variants, and copy number alterations have been identified in 1 or more of the 85 genes that we have included in Table 2. Recurrent mutations in 12 genes (TP53, CTNNB1, AXIN1, ALB, ARID2, ARID1A, RPS6KA3, APOB, RB1, CDKN2A, LRP1B, and PTEN) were reported in multiple studies. In this section, we discuss five genes (ALB, ARID2, RB1, BRD7, and RPL22) that were reported to show all four types of somatic alterations. To gain further insights into the genes with reported somatic mutations, their gene expression (tumor/normal fold-change) and clinic-pathological clinical information (histologic grade and survival) from the TCGA HCC cohort are also presented.
Table 2:
Summary of mutations in liver cancer identified through high-throughput genomics data including their association with gene expression and clinical phenotype
|
|
|
|
|
|
The table indicates the nature of the mutation (single-nucleotide variant [SNV], indels, structural variants, or copy number alterations) in the coding regions. The fold-change of the gene is obtained from the TCGA microarray analysis on HCC patient samples. Histologic grade refers to degree of tumor grade: G1 to G4; G_un indicate cases with unidentified histologic grading. The cases are segregated into HIGH or LOW based on their median gene expression (Median Exp). SNVs and indel mutations are indicated by the yellow box (●), structural variants by the blue box (●), and copy number alterations by the gray box (↓|↑).
ARID2 belongs to the SWI/SNF-related chromatin remodeling complexes and is identified as a tumor suppressor that is frequently mutated in HCC patients [22, 42, 48]. In addition, gene expression profiling of ARID2-deficient HCC cell lines reveals negative regulation of UV-response gene sets, suggesting that ARID2 may be involved in DNA repair processes [57]. ARID2 is also involved in HCC via the effects of hepatitis B and C infection. In HBV-related HCC, the HBV X protein is reported to suppress ARID2 expression, leading to increased hepatoma tumorigenesis [58]. ARID2 mutations are also significantly associated (P = 0.046) with HCV-related HCC [22]. These findings suggest that ARID2 is a critical tumor suppressor in hepatitis virus-related HCC progression.
Similar to ARID2, BRD7 is also a component of the SWI/SNF remodeling machinery and a putative tumor suppressor reported with significant truncating mutations in HCC [55]. Loss-of-function mutations at the BRD7 gene locus are frequently observed (7/268) in HBV-associated HCC patients [33]. BRD7 expression is also reported to be associated with the clinical characteristics in HCC (tumor size, tumor stage, and survival) [59]. HCV infections repress BRD7 expression in vitro, resulting in the dysregulation of hepatoma cell proliferation [60]. BRD7 also negatively regulates PI3K signaling by binding to the inter-SH2 (iSH2) domain of p85, leading to the impairment of p88/p110 complex formation [61].
The ALB gene encodes for the most abundant plasma protein, albumin, synthesized exclusively by hepatocytes [41]. Blood albumin tests that deviate from the normal healthy range often indicate dysregulation of protein production in the liver and other liver-associated issues. Somatic mutations at the ALB gene locus were reported in multiple studies, including genomic rearrangements in 10% (9/88) of Chinese HCC patients [41] as well as point mutations clusters and indels in Japanese HCC patients [33]. ALB is touted as a liver cancer driver gene as it is significantly enriched with damaging mutations in the European population [50]. Highly expressed genes such as ALB and APOB have been shown to be strongly enriched with indels, which are characteristic of replication slippage errors resulting from conflicts between the replication and transcription machineries [51]. Hence, low albumin levels may contribute to liver cancer progression.
RB1 is a key inhibitor of cell cycle progression that harbors multiple nonsense mutations and genomic deletions in HCC patients [33, 42, 43, 50]. RB1 is found to be predominantly mutated in Asian Americans (10/53 patients) as compared to European Americans (2/101 patients) [62]. The inactivation of the RB pathway in Rb family triple knockout mice resulted in the development of HCC [63]. A study reveals that in 16/40 HCC patients, DNA methylation abnormalities were observed in CpG island 85 (CpG85) located within intron 2 of the RB1 gene, which can potentially regulate the expression of the RB1-E2B alternative transcript [64]. In addition, RB1 mutations are also significantly associated with reduced cancer-specific and recurrence-free survival after resection in HCC patients [43, 50]. It is thus worthwhile to further characterize RB1 mutations, as they are reported to have a significantly higher mutation rate in HBV-related HCCs [42, 43].
RPL22, another gene that is reported to exhibit all 4 different types of mutations (single-nucleotide variant, indels, structural and copy number variation), encodes for a ribosomal 60S subunit protein. It was reported to be significantly mutated in Japanese (5/268 patients) and European (7/242) HCC patients [33, 50]. RPL22 was identified through pan-genomic characterization as a driver gene with significant somatic alterations in adenocortical carcinoma [65]. A study of microsatellite instability-positive gastric cancers also identified RPL22 as a recurrently mutated gene with single base deletions [66]. Therefore, there is potential for more research to be conducted to fully determine the functional roles of RPL22 in HCC.
HBV integration
The HBV genome often integrates into the chromosomes of liver cells, resulting in alterations of the host genome. Recent findings have confirmed that the viral transcription/replication initiation site, DR1 (located near the 3′ end of the HBx gene and the beginning of the Precore/Core gene), is the preferred region to be integrated into the host chromosome [11, 17, 19]. More HBV integration events were identified in tumor as compared to their matched normal samples [18]. In HCC tumors, studies show that HBV integration was randomly distributed throughout the human genome [17, 18, 33]. In a group of 48 HCC patients from the Singapore cohort, HBV integrations were significantly enriched in the q arm of chromosome 10 and correlated with poorly differentiated tumors [19].
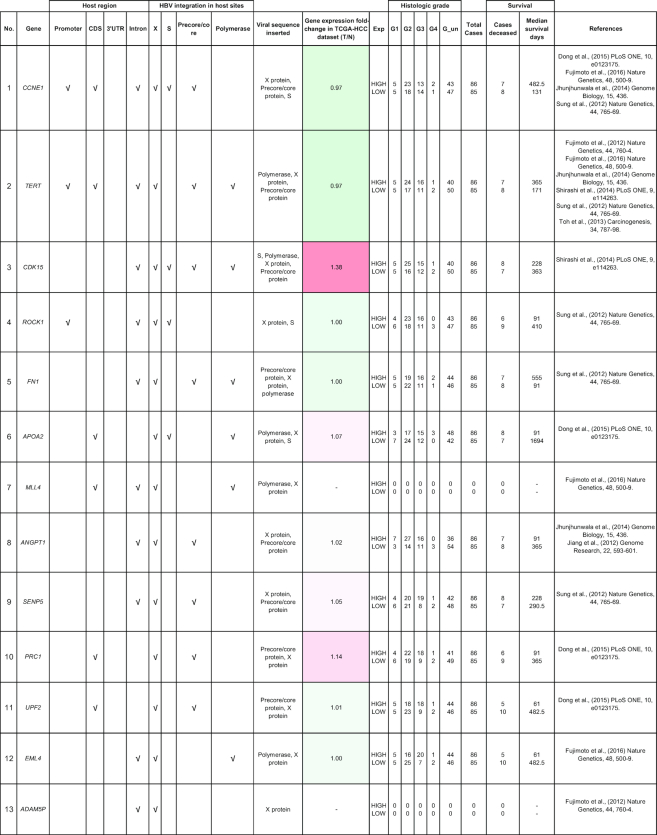
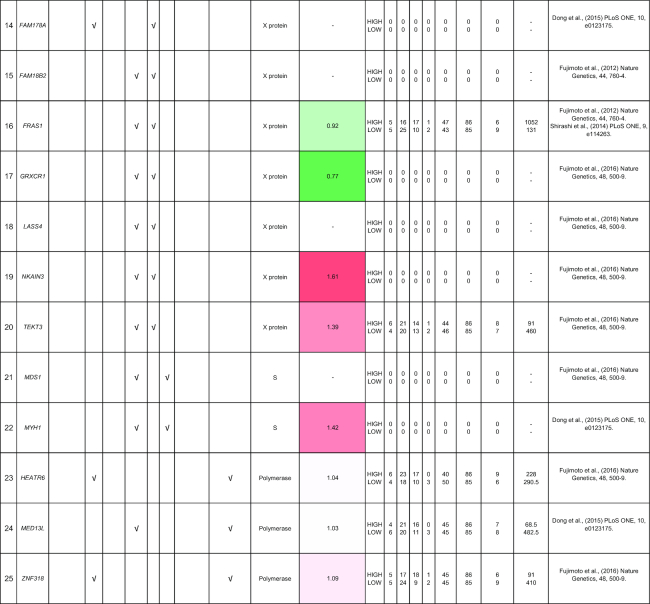
From the NGS studies, we have consolidated a comprehensive table of viral integration events that occurred in HCC patients (Table 3). There are multiple integration events in the promoter, 3′UTR, coding sequence and/or intronic region of the CCNE1 [67], TERT [19, 35, 37], CDK15 [37], ROCK1 [18], FN1 [68], APOA2 [67], and MLL4 [17, 18, 67] genes. HBV was reported in several studies to integrate into the CCNE1 and TERT genes [18, 33, 53]. CDK15, ROCK1, FN1, APOA2, and MLL4 are less frequently reported to be sites of integration for HBV.
Table 3:
Summary of HBV viral integration events occurring in HCC patients identified through high-throughput genomics data
|
|
The table indicates the genes and where the integration events occur. The fold-change of the gene is obtained from the TCGA microarray analysis on HCC patient samples. Histologic grade refers to degree of tumor grade: G1 to G4; G_un indicates cases with unidentified histologic grading. The cases are segregated into HIGH or LOW based on their median gene expression (Median_Exp).
CCNE1 encodes for the cyclin E1 protein that is a regulatory subunit of CDK2 involved in the G1/S phase of the cell cycle. CCNE1 amplification has been reported to be the mechanism of resistance in ER-positive and HER2-positive breast cancers as well as high-grade serous ovarian cancer [69–72]. HBV integrations within the CCNE1 have been reported in 4 of 76 HBV-positive HCC samples and resulted in significantly increased expression of CCNE1 [18]. The molecular mechanism of CCNE1 mutations in HCC patients has yet to be fully elucidated.
The previously reported recurrent integration site at the TERT promoter was found by several high-throughput genomic studies to be the most frequent site for integration [19, 33, 73, 74]. Disruption of the TERT promoter is likely to cause the dysregulation of the telomerase reverse transcriptase (TERT) expression, which plays important roles in cancer development due to its diverse telomere-independent functions in Wnt pathway signaling, cell proliferation, and DNA-damage repair [75]. Viral sequences may act as enhancers where the closer the HBV is integrated to the transcription start site of TERT, the higher the mRNA expression of TERT [19].
Chimeric HBx/MLL4 fusion transcripts containing the HBx promoter and Open Reading Frame (ORF)fused to the exon 4 and 5 of MLL4 were initially detected in 4 of 10 HCC patients [76] and subsequently confirmed in later studies and reported to lead to increased MLL4 expression [17, 18, 67]. In a Chinese cohort, 8 of 44 patients were found to have HBx/MLL4 fusion transcripts, resulting in a higher expression of MLL4 gene [67]. The chimeric transcript lacks the AT-hook DNA-binding domain of MLL4, hence, it may act as a dominant negative allele [17].
CDK15 encodes for the cyclin-dependent kinase 15 and is a serine/threonine protein kinase. In one study, CDK15 contributed to the effects of tumor necrosis factor-related apoptosis-inducing ligand resistance by possibly regulating the phosphorylation of survivin (Thr34) [77]. Interestingly, multiple HBV-CDK15 fusion transcripts were detected in an HCC patient, including one in-frame fusion, which caused CDK15 over-expression [37]. However, like many of the other genes where HBV integrations have been identified, the function of CDK15 in HCC remains unclear. Hence, there is great potential to further investigate HBV integrations in HCC.
It is noteworthy that CCNE1, TERT, and ANGPT1 not only harbor somatic mutations (Table 2), they are also reported to be sites for viral integrations (Table 3). CCNE1 has been reported with structural variant alterations and HBV integrations, while TERT has been reported with point mutations, structural variant alterations, and HBV integrations, suggesting that deregulation of these genes may play important roles in tumorigenesis. ANGPT1 (Angiopoietin-1), a ligand for Tie2 vascular endothelial-specific receptor tyrosine kinase, involved in the induction of HCC neovascularization and disease progression [78–80], was reported to harbor point mutations and HBV integrations in its intronic regions. ANGPT1 and Angiopoietin-2 (ANGPT2) were over-expressed in 68 and 81 percent of poorly differentiated HCC tumors, respectively [81]. However, high ANGPT2 expression, but not ANGPT1, showed correlation in the disease-free survival of 60 HCC patients [82]. The role of ANGPT1 in tumor angiogenesis remains unclear.
Pathways of somatic mutated genes and mutation signatures
Pathway analysis based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID v6.8) to identify pathways that were altered by somatic mutations in the TCGA HCC cohort [83, 84]. Seventy-nine of the 85 genes in our list of somatic mutations have identifiable DAVID IDs, of which 45 genes can be categorized in KEGG pathways. Fifteen significant pathways were identified (FDR <0.05) from the 45 genes, of which 14 genes are found to be involved in more than one of the pathways (Fig. 2). All 14 genes are involved in pathways in cancer, including other significant cancer types: prostate, endometrial, glioma, melanoma, chronic myeloid leukemia, colorectal, pancreatic, bladder, and non-small lung cancer. The association of the genes with the PI3K-Akt signaling pathway and the regulation of pluripotent stem cells also reflect the importance of these somatic mutations. Lastly, the analysis also reported viral-associated pathways such as hepatitis B, viral carcinogenesis, and Human T Lymphotropic Virus Type 1 (HTLV-1) infection, where the interplay between somatic mutations in genes and viral integration events come together to give a bigger picture represented by overall changes in the biological pathways.
Figure 2:

Reported genes with somatic mutations that are significantly involved in KEGG pathways.
Mutational signatures are well-categorized somatic mutations with distinct nucleotide substitutions. These signatures are often identified through principal-component analysis of the trinucleotide mutation context, with 96 possible combinations of the mutated nucleotide including the bases 5′ and 3′ to each site [33]. There are currently 30 mutational signatures listed in the Catalogue of Somatic Mutations in Cancer (COSMIC), where some of these signatures represent exposure to mutagens, errors in the DNA replication machinery, or defective DNA repair [85].
Fujimoto et al. (2016) was able to identify seven distinct mutational signatures (W1-W7) in HCC patients. Three of the seven signatures (W1, W4, and W5) were found in multiple studies [33, 50, 55]. These recurrent signatures correspond well to COSMIC Signature 1, Signature 4, and Signature 16, which are proposed to be caused by the spontaneous deamination of 5-methylcytosine, tobacco mutagens, or unknown factors, respectively [85]. Other COSMIC signatures identified include Signature 9, Signature 12, and Signature 19, which are linked to somatic hypermutation, liver cancer, and unknown factors, respectively [86]. Signature W6 was not associated with any COSMIC signatures and, thus, represents a new mutational signature. A further meta-analysis performed by Letouze et al. (2017) identified 10 mutational signatures including COSMIC Signatures 1, 4, 5, 6, 12, 16, 17, 22, 23, and 24 [51]. A mutational signature characterized with increased C>A transversions was a major contributor to the driver mutations found in HCC patients exposed to aflatoxin B1 [40]. A high proportion of Taiwanese HCC patients marked with aristolochic acid mutagen exposure had T>A mutations that corresponded to COSMIC signature 22 [47]. The AA signature was also found to be higher in HCC patients from China and Southeast Asia and much lower in Japan, America, and Europe. A prominent mutational signature was also identified after cisplatin treatment in human liver cancer cell line HepG2 [87]. Mutational signatures not only allow us to appreciate the mechanisms underlying somatic mutations in HCC tumors but they could relate to mutational processes in other cancer types with related etiology.
Multi-omics analysis combine results from more than one type of data to give us a more comprehensive view of biological profiles. Boyault et al. (2007) conducted an unsupervised transcriptome analysis to identify six subgroups of HCC, G1-G6, where G1-G3 are associated with chromosomal instability, G5-G6 are related to β-catenin mutations, and G4 is a heterogenous group [88]. The association between HCC transcriptome subclasses, G5-G6, involved in Wnt pathway activation and CTNNB1 mutations has been validated using WXS data in a later study [48, 88]. In addition, multi-omics analysis shows that there is a correlation between gene expression profiles from RNA-seq data and allele frequencies of somatic mutations from WGS, highlighting 252 genomic mutations that cause transcriptomic aberrations [37].
With the large number of available NGS-based HCC studies, there is an opportunity to integrate data across studies to provide greater statistical power and elimination of potential biases from a single cohort study. Zhang et al. (2014) collected four datasets containing 99, 88, 10, and 10 HCC samples to identify known and also novel mutated genes and pathways [89]. This study illustrated that larger sample sizes can identify mutations at lower frequencies in HCC than in smaller sample cohorts. As a second example of data integration, using combined liver cancer data from ICGC and TCGA to analyze the association of ancestry to HCC mutational signatures, an increase in T>C substitutions (in the ATA context) in Japanese males and an increase in T>A substitutions (in the CTG context) in US-Asian males and females were also reported [55].
Future
Mutations in the non-coding regulatory regions of the genome
Non-coding DNA makes up more than 98 percent of the human genome and include crucial transcription factor binding sites that regulate the transcription of RNA. Non-coding RNA includes introns, 3′ and 5′ UTR located in pre-mRNAs as well as microRNAs and long non-coding RNAs (lincRNAs) [90, 91]. The functional annotation of non-coding elements from the Encyclopedia of DNA Elements consortium and the US National Institutes of Health Roadmap Epigenomics project have provided support for the study of non-coding regions of human DNA [92, 93]. Cancer whole-genome data from TCGA have been intensively analyzed to identify mutations in the non-coding regions. For example, two pan-cancer studies have shown that TERT promoter mutations are present in at least six cancer types including glioblastoma, bladder, low-grade glioma, melanoma, and lung (and liver which is analyzed in one of the studies) [68, 94].
TERT promoter mutations are detected in 254 of 469 cases of HCC (54%) and more frequently detected in HCV-positive and non-viral cases than HBV-positive cases [55]. A more in-depth study reveals other noncoding mutations in NEAT1, MALAT1, WDR74 promoter, BCL6 promoter, and TFPI2 promoter [33]. Non-coding DNA analysis is challenging because many of the non-coding mutations are reported at lower mutation frequencies and at DNA locus with limited information regarding its function. We may overcome limitations in sample size and statistical power of patient datasets by analyzing an increased number of liver cancer whole genomes. Hence, there is potential to better characterize non-coding regions in the future.
AAV2 viral integration events
In addition to HBV integration, recent reports of the observation of integration of the wild-type adeno-associated virus 2 (AAV2) in 11 of 193 cases of HCC via deep sequencing [49, 95] have sparked a debate regarding the safety issues of using AAV2 as a gene delivery vector in gene therapy [96–99]. Coincidently, the AAV2 integrations were detected in several recurrent mutation sites in HCC including the TERT promoter, MLL4, CCNE1, CCNA2, and TNFSF10 [49, 100].
In an independent study, Fujimoto et al. (2016) detected AAV genome sequences in three liver cancer and three non-cancer liver cases. These three liver cancer cases were also infected with either HBV or HCV, and the AAV2 integration sites were located at MLL4, CCNE1, and an intergenic region of chromosome 5, respectively [33]. HBV integration sites were detected at the CCNA2 locus in one patient in this study as well as an early, well-differentiated HCC patient [12]. With these observations, additional analyses are necessary to evaluate the prevalence and effects of AAV2 integration events in liver cancer and in gene therapy. The extensiveness of WGS data is therefore applicable to the detection of foreign genomic material present in the human genome that may influence the development and the treatment of liver cancer.
RNA editing
RNA editing caused by the deamination of nucleotide bases on an RNA sequence is catalyzed by the nucleotide-specific deaminases. Historically, transgenic mice and rabbits expressing mRNA editing enzyme APOBEC-1 (C-to-U editing) resulted in unexpected liver dysplasia, with a few of the mice developing HCC [101]. The main form of RNA editing is A-to-I editing catalyzed by the adenosine deaminase acting on RNA (ADAR) (A-to-I editing) family [102].
A genome-wide study that used both WGS and RNA-seq data reported normal and tumor-specific RNA editing sites in HCC as well as the positive correlation between editing degree ratio and gene expression ratio [39]. Results show that the increased expression of ADAR1 resulted in the over-editing of the AZIN1 gene in HCC tumors, confirming the findings from a previous study [103]. Another genome-wide study showed that in addition to AZIN1, the BLCAP RNA has been over-edited (A-to-I editing) in HCC, and functional analysis suggests that the over-edited BLCAP resulted in enhanced cell proliferation and the activation of the AKT/mTOR signal pathway [104]. Two pan-cancer studies involving A-to-I RNA editing using data from TCGA reported no significant differences between matched normal and tumor samples, although a high Alu editing index in HCC has been significantly associated with poor survival [105, 106].
Expanding the cancer genome database
With rapidly falling costs and newer technologies, the number of whole genomes sequenced in the next 10 years is projected to increase dramatically [107]. Larger sample sizes will provide better statistical power to detect rare variants and subgroups of liver cancer, particularly in HCC. For example, a large-scale whole-genome study conducted on the Icelandic population identified missense SNP variants in ABCB4 to be associated with gallstone disease, liver cancer, liver cirrhosis, and other liver-specific traits [108, 109]. There are currently several international collaborations to generate more cancer whole genomes. The Pan-Cancer Analysis of Whole Genomes is an international collaboration between ICGC and TCGA to analyze more than 2,800 whole genomes across different cancer types to identify genetic alterations, beginning with 12 tumor types profiled by TCGA, although HCC was not included [110]. Additionally, the 100,000 Genomes Project by Genomics England in the United Kingdom will consist of samples from 25,000 cancer patients [111].
Conclusion
In this review, we have discussed the key findings from WGS information (Fig. 1) and future directions of HCC. WGS is a promising approach that provides genomic information for discovery-based genomic analyses in the future. Hence, it holds great potential for liver cancer research as we seek to understand more about the genetic characteristics of HCC, which is influenced by gender, ethnicity, geolocation, and many risk factors. This review identified genes with somatic mutations (Table 2), many of which are involved in cancer-related pathways (Fig. 2). Many of the mutated genes are yet to be characterized for their molecular function and roles in cancer, presenting great opportunity for future research in this direction. With improved clinical annotation and the automation of data analysis, more genomic sequences can be translated into valuable biological insights.
Abbreviations
AAV2: adeno-associated virus 2; COSMIC: Catalogue of Somatic Mutations in Cancer; DAVID: Database for Annotation, Visualization and Integrated Discovery; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; HCV: hepatitis C virus; ICGC: International Cancer Genome Consortium; indel: insertions and deletions; KEGG: Kyoto Encyclopedia of Genes and Genomes; NGS: next-generation sequencing; RNA-seq: RNA sequencing; SNP: single nucleotide polymorphism; SNV: single-nucleotide variant; SRA: Sequence Read Archive; TCGA: the Cancer Genome Atlas; TERT: telomerase reverse transcriptase; WGS: whole-genome sequencing; WXS: whole-exome sequencing; EMBL-EBI: European Molecular Biology Laboratory - European Bioinformatics Institute;
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by a grant from the Singapore Ministry of Health's National Medical Research Council (NMRC/CBRG/0095/2015) as well as some block funding from the National Cancer Centre Singapore and Duke-NUS Graduate Medical School to C.G.L. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
Conceptualization: C.L., A.S.; data curation: W.H.; formal analysis: W.H.; funding acquisition: C.L.; supervision: C.L.; writing the original draft: .W.H; review and editing: C.L., A.S., W.H.
Supplementary Material
8/10/2018 Reviewed
References
- 1. Ferlay J, Soerjomataram I, Ervik M et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. http://globocan.iarc.fr 2013. Accessed 19 May 2018. [Google Scholar]
- 2. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–76. [DOI] [PubMed] [Google Scholar]
- 3. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–27. [DOI] [PubMed] [Google Scholar]
- 4. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142(6):1264–73 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Di Bisceglie AM, Simpson LH, Lotze MT et al. Development of hepatocellular carcinoma among patients with chronic liver disease due to hepatitis C viral infection. J Clin Gastroenterol. 1994;19(3):222–6. [DOI] [PubMed] [Google Scholar]
- 6. Takano S, Yokosuka O, Imazeki F, et al. Incidence of hepatocellular carcinoma in chronic hepatitis B and C: a prospective study of 251 patients. Hepatology (Baltimore, Md). 1995;21(3):650–5. [PubMed] [Google Scholar]
- 7. Chang MH, Chen CJ, Lai MS, et al. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N Engl J Med. 1997;336(26):1855–9. [DOI] [PubMed] [Google Scholar]
- 8. Aspinall EJ, Hawkins G, Fraser A et al. Hepatitis B prevention, diagnosis, treatment and care: a review. Occup Med (Lond). 2011;61(8):531–40. [DOI] [PubMed] [Google Scholar]
- 9. Paterlini-Brechot P, Saigo K, Murakami Y et al. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22(25):3911–6. [DOI] [PubMed] [Google Scholar]
- 10. Tamori A, Yamanishi Y, Kawashima S, et al. Alteration of gene expression in human hepatocellular carcinoma with integrated hepatitis B virus DNA. Clinical Cancer Research. 2005;11(16):5821–6. [DOI] [PubMed] [Google Scholar]
- 11. Nagaya T, Nakamura T, Tokino T et al. The mode of hepatitis B virus DNA integration in chromosomes of human hepatocellular carcinoma. Genes & Development. 1987;1(8):773–82. [DOI] [PubMed] [Google Scholar]
- 12. Wang J, Chenivesse X, Henglein B et al. Hepatitis B virus integration in a cyclin A gene in a hepatocellular carcinoma. Nature. 1990;343(6258):555–7. [DOI] [PubMed] [Google Scholar]
- 13. Dejean A, Bougueleret L, Grzeschik KH et al. Hepatitis B virus DNA integration in a sequence homologous to v-erb-A and steroid receptor genes in a hepatocellular carcinoma. Nature. 1986;322(6074):70–2. [DOI] [PubMed] [Google Scholar]
- 14. Satoh S, Daigo Y, Furukawa Y, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24(3):245–50. [DOI] [PubMed] [Google Scholar]
- 15. Murakami Y, Hayashi K, Hirohashi S, et al. Aberrations of the tumor suppressor p53 and retinoblastoma genes in human hepatocellular carcinomas. Cancer Res. 1991;51(20):5520–5. [PubMed] [Google Scholar]
- 16. Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang Z, Jhunjhunwala S, Liu J et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012;22(4):593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sung WK, Zheng H, Li S et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44(7):765–9. [DOI] [PubMed] [Google Scholar]
- 19. Toh ST, Jin Y, Liu L et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis. 2013;34(4):787–98. [DOI] [PubMed] [Google Scholar]
- 20. Tao Y, Ruan J, Yeh SH et al. Rapid growth of a hepatocellular carcinoma and the driving mutations revealed by cell-population genetic analysis of whole-genome data. PNAS. 2011;108(29):12042–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Totoki Y, Tatsuno K, Yamamoto S, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43(5):464–9. [DOI] [PubMed] [Google Scholar]
- 22. Li M, Zhao H, Zhang X, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43(9):828–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schulze K, Nault JC, Villanueva A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J Hepatol. 2016;65(5):1031–42. [DOI] [PubMed] [Google Scholar]
- 24. Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Res. 2011;39(Database Issue):D19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. EBML-EBI European Nucleotide Archive (ENA) http://www.ebi.ac.uk/ena 2018. Accessed 31 Oct 2018. [Google Scholar]
- 26. Kaminuma E, Mashima J, Kodama Y, et al. DDBJ launches a new archive database with analytical tools for next-generation sequence data. Nucleic Acids Res. 2010;38(Database Issue):D33–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. National Cancer Institute - Genomic Data Commons. https://gdc.cancer.gov/ 2018. Accessed 31 Oct 2018. [Google Scholar]
- 28. Gigadb. http://gigadb.org/ 2018. Accessed 31 Oct 2018. [Google Scholar]
- 29. Sneddon TP, Li P, Edmunds SC. GigaDB: announcing the GigaScience database. GigaScience. 2012;1(1):1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kan Z, Zheng H, Liu X et al. Hepatocellular carcinoma genomic data from the Asian Cancer Research Group. GigaScience. 2012. [Google Scholar]
- 31. International Cancer Genome Consortium (ICGC). http://icgc.org/ 2018. Accessed 31 Oct 2018. [Google Scholar]
- 32. Hudson TJ, Anderson W, Artez A et al. International network of cancer genome projects. Nature. 2010;464(7291):993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fujimoto A, Furuta M, Totoki Y et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016;48(5):500–9. [DOI] [PubMed] [Google Scholar]
- 34. Fujimoto A, Furuta M, Shiraishi Y et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat Commun. 2015;6:6120 doi:10.1038/ncomms7120. [DOI] [PubMed] [Google Scholar]
- 35. Fujimoto A, Totoki Y, Abe T, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44(7):760–4. [DOI] [PubMed] [Google Scholar]
- 36. Hirotsu Y, Zheng TH, Amemiya K et al. Targeted and exome sequencing identified somatic mutations in hepatocellular carcinoma. Hepatology Research. 2016; doi:10.1111/hepr.12663. [DOI] [PubMed] [Google Scholar]
- 37. Shiraishi Y, Fujimoto A, Furuta M, et al. Integrated analysis of whole genome and transcriptome sequencing reveals diverse transcriptomic aberrations driven by somatic genomic changes in liver cancers. PLoS One. 2014;9(12):e114263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang J, Deng Q, Wang Q et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet. 2012;44(10):1117–21. [DOI] [PubMed] [Google Scholar]
- 39. Kang L, Liu X, Gong Z, et al. Genome-wide identification of RNA editing in hepatocellular carcinoma. Genomics. 2015;105(2):76–82. [DOI] [PubMed] [Google Scholar]
- 40. Zhang W, He H, Zang M et al. Genetic features of aflatoxin-associated hepatocellular carcinoma. Gastroenterology. 2017;153(1):249–62.e2. [DOI] [PubMed] [Google Scholar]
- 41. Fernandez-Banet J, Lee NP, Chan KT, et al. Decoding complex patterns of genomic rearrangement in hepatocellular carcinoma. Genomics. 2014;103(2–3):189–203. [DOI] [PubMed] [Google Scholar]
- 42. Kan Z, Zheng H, Liu X et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23(9):1422–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ahn SM, Jang SJ, Shim JH, et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology (Baltimore, Md). 2014;60(6):1972–82. [DOI] [PubMed] [Google Scholar]
- 44. Ouyang L, Lee J, Park CK et al. Whole-genome sequencing of matched primary and metastatic hepatocellular carcinomas. BMC Med Genomics. 2014;7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Woo HG, Kim SS, Cho H et al. Profiling of exome mutations associated with progression of HBV-related hepatocellular carcinoma. PLoS One. 2014;9(12):e115152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lin KT, Shann YJ, Chau GY et al. Identification of latent biomarkers in hepatocellular carcinoma by ultra-deep whole-transcriptome sequencing. Oncogene. 2014;33(39):4786–94. [DOI] [PubMed] [Google Scholar]
- 47. Ng AWT, Poon SL, Huang MN et al. Aristolochic acids and their derivatives are widely implicated in liver cancers in Taiwan and throughout Asia. Sci Transl Med. 2017;9(412) doi:10.1126/scitranslmed.aan6446. [DOI] [PubMed] [Google Scholar]
- 48. Guichard C, Amaddeo G, Imbeaud S et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44(6):694–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nault JC, Datta S, Imbeaud S et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat Genet. 2015;47(10):1187–93. [DOI] [PubMed] [Google Scholar]
- 50. Schulze K, Imbeaud S, Letouze E, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47(5):505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Letouzé E, Shinde J, Renault V, et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nature Communications. 2017;8(1):1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cleary SP, Jeck WR, Zhao X, et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology (Baltimore, Md). 2013;58(5):1693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jhunjhunwala S, Jiang Z, Stawiski EW, et al. Diverse modes of genomic alteration in hepatocellular carcinoma. Genome Biol. 2014;15(8):436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chaudhary K, Poirion OB, Lu L, et al. Multi-modal meta-analysis of 1494 hepatocellular carcinoma samples reveals significant impact of consensus driver genes on phenotypes. Clinical Cancer Research. 2018; doi:10.1158/1078-0432.ccr-18-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Totoki Y, Tatsuno K, Covington KR, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46(12):1267–73. [DOI] [PubMed] [Google Scholar]
- 56. Ding D, Lou X, Hua D et al. Recurrent targeted genes of hepatitis B virus in the liver cancer genomes identified by a next-generation sequencing-based approach. PLoS Genet. 2012;8(12):e1003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Oba A, Shimada S, Akiyama Y, et al. ARID2 modulates DNA damage response in human hepatocellular carcinoma cells. J Hepatol. 2017;66(5):942–51. [DOI] [PubMed] [Google Scholar]
- 58. Gao Q, Wang K, Chen K et al. HBx protein-mediated ATOH1 downregulation suppresses ARID2 expression and promotes hepatocellular carcinoma. Cancer Sci. 2017;108(7):1328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen CL, Wang Y, Pan QZ et al. Bromodomain-containing protein 7 (BRD7) as a potential tumor suppressor in hepatocellular carcinoma. Oncotarget. 2016;7(13):16248–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Q, Wei L, Yang H et al. Bromodomain containing protein represses the Ras/Raf/MEK/ERK pathway to attenuate human hepatoma cell proliferation during HCV infection. Cancer Lett. 2016;371(1):107–16. [DOI] [PubMed] [Google Scholar]
- 61. Chiu YH, Lee JY, Cantley LC. BRD7, a tumor suppressor, interacts with p85alpha and regulates PI3K activity. Mol Cell. 2014;54(1):193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yao S, Johnson C, Hu Q et al. Differences in somatic mutation landscape of hepatocellular carcinoma in Asian American and European American populations. Oncotarget. 2016;7(26):40491–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Viatour P, Ehmer U, Saddic LA, et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med. 2011;208(10):1963–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Anwar SL, Krech T, Hasemeier B, et al. Deregulation of RB1 expression by loss of imprinting in human hepatocellular carcinoma. J Pathol. 2014;233(4):392–401. [DOI] [PubMed] [Google Scholar]
- 65. Zheng S, Cherniack AD, Dewal N et al. Comprehensive Pan-genomic characterization of adrenocortical carcinoma. Cancer Cell. 2016;29(5):723–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nagarajan N, Bertrand D, Hillmer AM et al. Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol. 2012;13(12):R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dong H, Zhang L, Qian Z et al. Identification of HBV-MLL4 integration and its molecular basis in Chinese hepatocellular carcinoma. PLoS One. 2015;10(4):e0123175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fredriksson NJ, Ny L, Nilsson JA, et al. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat Genet. 2014;46(12):1258–63. [DOI] [PubMed] [Google Scholar]
- 69. Herrera-Abreu MT, Palafox M, Asghar U et al. Early adaptation and acquired resistance to CDK4/6 Inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016;76(8):2301–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Scaltriti M, Eichhorn PJ, Cortes J, et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. PNAS. 2011;108(9):3761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Au-Yeung G, Lang F, Azar WJ, et al. Selective targeting of cyclin E1-amplified high-grade serous ovarian cancer by cyclin-dependent kinase 2 and AKT inhibition. Clinical Cancer Research. 2017;23(7):1862–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Patch AM, Christie EL, Etemadmoghadam D et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521(7553):489–94. [DOI] [PubMed] [Google Scholar]
- 73. Ferber MJ, Montoya DP, Yu C et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene. 2003;22(24):3813–20. [DOI] [PubMed] [Google Scholar]
- 74. Khoury JD, Tannir NM, Williams MD et al. Landscape of DNA virus associations across human malignant cancers: analysis of 3,775 cases using RNA-Seq. J Virol. 2013;87(16):8916–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hanahan D, Weinberg Robert A. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 76. Saigo K, Yoshida K, Ikeda R et al. Integration of hepatitis B virus DNA into the myeloid/lymphoid or mixed-lineage leukemia (MLL4) gene and rearrangements of MLL4 in human hepatocellular carcinoma. Hum Mutat. 2008;29(5):703–8. [DOI] [PubMed] [Google Scholar]
- 77. Park MH, Kim SY, Kim YJ, et al. ALS2CR7 (CDK15) attenuates TRAIL induced apoptosis by inducing phosphorylation of survivin Thr34. Biochem Biophys Res Commun. 2014;450(1):129–34. [DOI] [PubMed] [Google Scholar]
- 78. Tanaka S, Sugimachi K, Yamashita Yi Y, et al. Tie2 vascular endothelial receptor expression and function in hepatocellular carcinoma. Hepatology (Baltimore, Md). 2002;35(4):861–7. [DOI] [PubMed] [Google Scholar]
- 79. Tanaka S, Mori M, Sakamoto Y, et al. Biologic significance of angiopoietin-2 expression in human hepatocellular carcinoma. J Clin Invest. 1999;103(3):341–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mitsuhashi N, Shimizu H, Ohtsuka M et al. Angiopoietins and Tie-2 expression in angiogenesis and proliferation of human hepatocellular carcinoma. Hepatology (Baltimore, Md). 2003;37(5):1105–13. [DOI] [PubMed] [Google Scholar]
- 81. Sugimachi K, Tanaka S, Taguchi K et al. Angiopoietin switching regulates angiogenesis and progression of human hepatocellular carcinoma. J Clin Pathol. 2003;56(11):854–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wada H, Nagano H, Yamamoto H et al. Expression pattern of angiogenic factors and prognosis after hepatic resection in hepatocellular carcinoma: importance of angiopoietin-2 and hypoxia-induced factor-1 alpha. Liver International. 2006;26(4):414–23. [DOI] [PubMed] [Google Scholar]
- 83. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. [DOI] [PubMed] [Google Scholar]
- 84. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Forbes SA, Beare D, Boutselakis H, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777–D83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Alexandrov LB, Nik-Zainal S, Wedge DC et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Boot A, Huang MN, Ng AWT et al. In-depth characterization of the cisplatin mutational signature in human cell lines and in esophageal and liver tumors. Genome Res. 2018;28(5):654–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Boyault S, Rickman DS, de Reynies A et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology (Baltimore, Md). 2007;45(1):42–52. [DOI] [PubMed] [Google Scholar]
- 89. Zhang Y, Qiu Z, Wei L et al. Integrated analysis of mutation data from various sources identifies key genes and signaling pathways in hepatocellular carcinoma. PLoS One. 2014;9(7):e100854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ghidini M, Braconi C. Non-coding RNAs in primary liver cancer. Front Med (Lausanne). 2015;2:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. He Y, Meng XM, Huang C, et al. Long noncoding RNAs: novel insights into hepatocelluar carcinoma. Cancer Lett. 2014;344(1):20–7. [DOI] [PubMed] [Google Scholar]
- 92. The-ENCODE-Project-Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bernstein BE, Stamatoyannopoulos JA, Costello JF et al. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol. 2010;28(10):1045–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Weinhold N, Jacobsen A, Schultz N, et al. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet. 2014;46(11):1160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nault J-C, Datta S, Imbeaud S et al. Adeno-associated virus type 2 as an oncogenic virus in human hepatocellular carcinoma. Molecular & Cellular Oncology. 2016;3(2):e1095271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Berns KI, Byrne BJ, Flotte TR,et al. Adeno-associated virus type 2 and hepatocellular carcinoma?. Hum Gene Ther. 2015;26(12):779–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Buning H, Schmidt M. Adeno-associated vector toxicity-to be or not to be?. Mol Ther. 2015;23(11):1673–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gil-Farina I, Fronza R, Kaeppel C et al. Recombinant AAV integration is not associated with hepatic genotoxicity in nonhuman primates and patients. Mol Ther. 2016; doi:10.1038/mt.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Schmidt M, Gil-Farina I, Buning H. Reply to “Wild-type AAV insertions in hepatocellular carcinoma do not inform debate over genotoxicity risk of vectorized AAV.”. Mol Ther. 2016;24(4):661–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Nault JC, Datta S, Imbeaud S et al. AAV2 and hepatocellular carcinoma. Hum Gene Ther. 2016;27(3):211–3. [DOI] [PubMed] [Google Scholar]
- 101. Yamanaka S, Balestra ME, Ferrell LD et al. Apolipoprotein B mRNA-editing protein induces hepatocellular carcinoma and dysplasia in transgenic animals. PNAS. 1995;92(18):8483–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Brennicke A, Marchfelder A, Binder S. RNA editing. FEMS Microbiol Rev. 1999;23(3):297–316. [DOI] [PubMed] [Google Scholar]
- 103. Chen L, Li Y, Lin CH, et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013;19(2):209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hu X, Wan S, Ou Y, et al. RNA over-editing of BLCAP contributes to hepatocarcinogenesis identified by whole-genome and transcriptome sequencing. Cancer Lett. 2015;357(2):510–9. [DOI] [PubMed] [Google Scholar]
- 105. Ding SL, Yang ZW, Wang J et al. Integrative analysis of aberrant Wnt signaling in hepatitis B virus-related hepatocellular carcinoma. World Journal of Gastroenterology. 2015;21(20):6317–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Paz-Yaacov N, Bazak L, Buchumenski I et al. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Reports. 2015;13(2):267–76. [DOI] [PubMed] [Google Scholar]
- 107. Eisenstein M. Big data: the power of petabytes. Nature. 2015;527(7576):S2–4. [DOI] [PubMed] [Google Scholar]
- 108. Gudbjartsson DF, Helgason H, Gudjonsson SA et al. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet. 2015;47(5):435–44. [DOI] [PubMed] [Google Scholar]
- 109. Lammert F, Hochrath K. A letter on ABCB4 from Iceland: on the highway to liver disease. Clin Res Hepatol Gastroenterol. 2015;39(6):655–8. [DOI] [PubMed] [Google Scholar]
- 110. Weinstein JN, Collisson EA, Mills GB et al. The cancer genome atlas Pan-cancer analysis project. Nat Genet. 2013;45(10):1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Marx V. The DNA of a nation. Nature. 2015;524(7566):503–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
8/10/2018 Reviewed