

Summary
Endocytosis is implicated in the maintenance of embryonic stem cell (ESC) pluripotency, although its exact role and the identity of molecular players remain poorly understood. Here, we show that the clathrin heavy chain (CLTC), involved in clathrin-mediated endocytosis (CME), is vital for maintaining mouse ESC (mESC) pluripotency. Knockdown of Cltc resulted in a loss of pluripotency accompanied by reduced E-cadherin (E-CAD) levels and increased levels of transforming growth factor β (TGF-β) and extracellular signal-regulated kinase (ERK) signaling. We demonstrate that both E-CAD and TGF-β receptor type 1 (TGF-βR1) are internalized through CME in mESCs. While E-CAD is recycled, TGF-βR1 is targeted for lysosomal degradation thus maintaining inverse levels of these molecules. Finally, we show that E-CAD interacts with ERK, and that the decreased pluripotency upon CME loss can be rescued by inhibiting TGF-βR, MEK, and GSK3β, or overexpressing E-CAD. Our results demonstrate that CME is critical for balancing signaling outputs to regulate ESC pluripotency, and possibly cell fate choices in early development.
Keywords: embryonic stem cells, pluripotency, trafficking, E-cadherin, clathrin, recycling, TGF-β, epithelial
Graphical Abstract
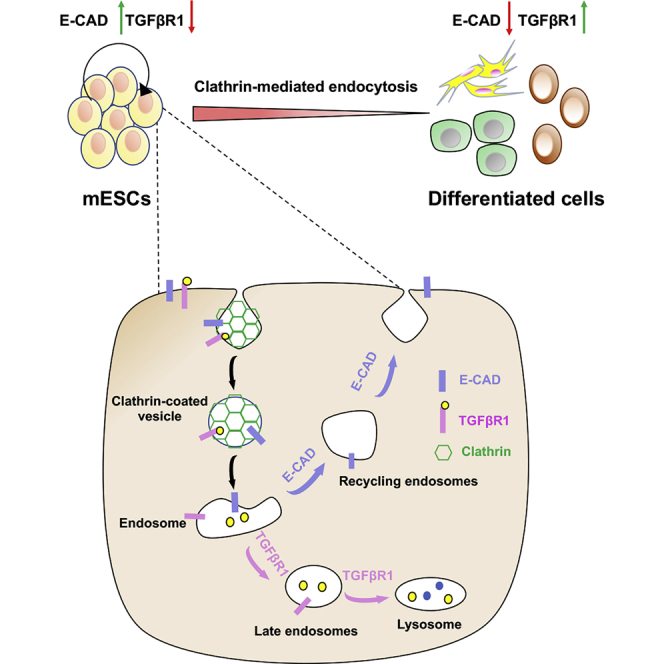
Highlights
-
•
Knockdown of Cltc results in loss of mESC pluripotency
-
•
CME regulates E-CAD and TGF-βR1 trafficking in mESCs
-
•
ESCs lacking CME can be rescued by TGF-βR1/MEK inhibition or E-CAD overexpression
-
•
CME balances opposing signaling outputs to maintain ESC pluripotency
Subramanyam and colleagues demonstrate that clathrin-mediated endocytosis (CME) is necessary to maintain ESC pluripotency by regulating the trafficking of E-CAD and TGF-βR1. Loss of CME results in a loss of pluripotency characterized by decreased E-CAD and enhanced TGF-βR1 and MEK signaling. Expression of pluripotency markers in ESCs lacking Cltc can be restored by inhibiting TGF-βR1 or MEK or by overexpressing E-CAD.
Introduction
Mammalian development is an exquisitely controlled process involving the concerted action of numerous signaling pathways and cellular processes. Embryonic stem cells (ESCs) isolated from the inner cell mass of the blastocyst represent a wonderful model system to follow developmental decisions and cell fate transitions in vitro (Evans, 2011, Evans and Kaufman, 1981, Martin, 1981). Recent reports have demonstrated that endocytosis plays a role in regulating the acquisition, as well as the maintenance of the pluripotent state (Dambournet et al., 2018, Li et al., 2010, Mote et al., 2017, Qin et al., 2014, Subramanyam et al., 2011). Endocytosis is a fundamental eukaryotic process, wherein membrane-bound proteins are trafficked to different compartments of the cell depending on their function, with the help of small GTPases called RABs (Doherty and McMahon, 2009, Traub, 2009).
Endocytosis is known to play a role in cell fate specification during early metazoan development (De Renzis et al., 2006, Fabrowski et al., 2013, Kawamura et al., 2012, Song et al., 2013). However, the exact mechanistic role played by endocytosis in the regulation of early mammalian development remains to be elucidated.
To this end, we undertook a small interfering RNA (siRNA) screen in mouse ESCs (mESCs), wherein transcripts encoding for endocytic components were depleted, and the pluripotency of ESCs was assessed. We found that knocking down clathrin heavy chain (Cltc), a major coat protein required for clathrin-mediated endocytosis (CME), resulted in a decrease in mESC pluripotency. Further, we show that CME regulated mESC pluripotency by trafficking two opposing molecular players, namely E-cadherin (E-CAD or CDH1) and transforming growth factor β receptor type 1 (TGF-βR1). In mESCs, E-CAD is recycled back to the membrane after internalization, while TGF-βR1 is targeted to the lysosome for degradation. We further discovered a novel interaction between E-CAD and extracellular signal-regulated kinase (ERK) in mESCs, and found that loss of CME resulted in elevated ERK signaling. β-CATENIN levels were also reduced upon loss of CME. A decrease in CME thus caused an imbalance between key signaling pathways, resulting in a loss of pluripotency. Our results demonstrate that trafficking of molecules via endocytosis is required to maintain the pluripotent state and identity of mESCs, and that a change in endocytic thresholds may facilitate cell fate transitions.
Results
Clathrin Heavy Chain Is an Essential Molecule for Maintaining the Pluripotency of mESCs
While the contribution of endocytosis to the state of pluripotency has been minimally described over the last few years (Dambournet et al., 2018, Li et al., 2010, Mote et al., 2017, Qin et al., 2014, Subramanyam et al., 2011), a systematic study looking at specific endocytic elements that are intimately involved in the maintenance of ESC pluripotency is lacking. We undertook an unbiased, small-scale siRNA-based screen, targeting 112 different components of the endocytic machinery. Efficiency of individual pools of siRNA was not determined. Levels of alkaline phosphatase (AP) activity, a marker of ESC pluripotency (Wang et al., 2013), was used as a readout based on the analysis described in the Experimental Procedures. High-throughput screening based on detection of AP was performed 4 days after transfection of siRNAs into mESCs. Knockdown of five genes resulted in a significant increase in AP staining (shown as red bars), while knockdown of two genes resulted in a significant decrease in AP staining (shown in blue) (Figure 1A). We were specifically interested in understanding the role of endocytic genes in the maintenance of pluripotency, and hence focused on genes whose knockdown resulted in a loss of AP staining. One such gene was the clathrin heavy chain (Cltc) (Figure 1A).
Figure 1.

CME Is Required for the Maintenance of mESC Pluripotency and Self-Renewal
(A) Dot plot showing the results of the endocytic siRNA screen in mESCs, based on AP staining. Knockdowns resulting in a significant decrease in AP staining are marked in blue, while those resulting in an increase in AP staining are shown in red.
(B) Bright-field images show AP staining and morphology of mESCs 2 days post Cltc knockdown, or expression of K44A Dnm (K44A). Scale bar, 50 μm. NTi, non-targeting siRNA control; Cltci, Cltc siRNA; control, vector control.
(C) Bar graph showing mESC colony number, 3 days post indicated conditions.
(D and E) Line graph showing the proliferation rate of (D) mESCs and (E) MEFs over 3 days post indicated conditions.
(F) Cell-cycle analysis of mESCs and MEFs 3 days post indicated conditions. Bar graph shows the percent of cells in G1, S, and G2 phases of the cell cycle for both mESCs and MEFs.
(G) Bar graph showing the expression of pluripotency markers in mESCs under indicated conditions relative to control (n = 3). Control is shown as a dotted line at 1.
(H) Bar graph showing the expression of differentiation markers in embryoid bodies generated from Cltc knockdown mESCs by qRT-PCR analysis (n = 3). SCi, scrambled shRNA control; Cltci, Cltc shRNA.
Error bars represent mean ± SD from three independent experiments (n = 3). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 by two-tailed Student’s t test.
Cltc encodes for the heavy chain of clathrin, which is an integral component of the clathrin triskelion, which forms the clathrin coat during CME (Kirchhausen et al., 2014, McMahon and Boucrot, 2011). The clathrin coat stabilizes endocytic structures through polymerization at the sites of endocytosis (Saffarian et al., 2009, Tebar et al., 1996), followed by scission of the clathrin-coated pit with the help of the small GTPase, Dynamin (Ferguson and De Camilli, 2012).
We validated the screen results by generating independent endoribonuclease-prepared siRNAs (esiRNAs) (Kittler et al., 2007), targeting Cltc, which showed a significant reduction of CLTC protein in mESCs (Figure S1A). Knockdown of Cltc resulted in a decrease in the uptake of labeled transferrin (a well-studied cargo for CME) (Kirchhausen et al., 2014) into mESCs, indicative of a block of CME (Figures S1B and S1C). As previously reported, transferrin uptake was also significantly reduced upon introduction of the dominant negative mutant of Dynamin, K44A Dnm (Bitoun et al., 2009, Herskovits et al., 1993), in mESCs (Figures S1D and S1E). We found that blocking CME in mESCs through either the knockdown of Cltc, or upon overexpression of K44A Dnm, resulted in a decrease in AP staining (Figure 1B), colony-forming ability (Figure 1C), a reduction in the proliferation rate (Figure 1D), and an altered cell-cycle profile with increased number of cells in G1 (Figure 1F). However, changes in the proliferation, or cell-cycle profile were not observed in terminally differentiated mouse embryonic fibroblasts (MEFs) upon blocking CME (Figures 1E and 1F). Reduced CME resulted in a significant decrease in the expression of pluripotency markers (Figures 1G, S1F, and S1H), and an increase in the expression of differentiation markers corresponding to all three germ layers (Figures 1H, S1G, and S1I). However, no induction of apoptosis was observed upon blocking CME (Figure S1J). Teratomas generated from Cltc knockdown cells were smaller compared with those generated from wild-type cells (Figure S1K), while containing structures corresponding to all three germ layers (Figure S1L). However, teratomas generated from Cltc knockdown mESCs displayed excessive hemorrhage, and appeared to have a greater bias toward mesodermal differentiation (Figure S1L). Our results thus demonstrate that CME plays an important role in maintaining the pluripotent state of mESCs.
Clathrin-Dependent Endocytosis of E-CAD Maintains mESC Pluripotency
Interestingly, the knockdown of Cltc, or the introduction of K44A Dnm in mESCs, resulted in a striking phenotype, with cells showing a dispersed and flattened morphology and loss of cell-cell contacts, reminiscent of the phenotype of ESCs lacking E-cad (Redmer et al., 2011) (Figures 1B and S2). These morphological features are different from normal mESCs, which grow in tight, three-dimensional, domed colonies that are AP positive (Figure 1B). Indeed, we found that E-CAD levels were decreased upon blocking CME in mESCs (Figures 2A, S3A, S3B, S4A, and S4B). In addition, E-CAD levels were also depleted from the plasma membrane under Cltc knockdown conditions (Figure S3B).
Figure 2.

E-CAD Trafficking Is Required to Maintain mESC Pluripotency
(A) Western blot showing E-CAD levels upon knockdown of Cltc in mESCs.
(B and C) Representative confocal micrographs showing the co-localization of E-CAD with CLTC (B) and RAB11 (C). Scale bars, 7.5 μm (B) and 10 μm (C).
(D) Representative confocal micrographs showing the increased localization of E-CAD with RAB11-positive recycling endosomes in the presence of the E-CAD neutralizing antibody, DECMA1 (1 hr treatment). Scale bars, 10 μm (top panel) and 25 μm (lower panel). Mouse IgG was used as a control.
(E) Scatterplot showing the quantitation of E-CAD levels, membrane versus cytoplasmic, in the presence of the E-CAD neutralizing antibody, DECMA1. ∗∗p < 0.01 by two-tailed Student’s t test.
(F) Graph showing levels of pluripotency marker genes by qRT-PCR analysis in mESCs treated with the E-CAD neutralizing antibody, DECMA1 or mouse IgG (24 hr treatment). ∗p < 0.05; ∗∗p < 0.01 by two-tailed Student’s t test.
(G) Representative confocal micrographs showing E-CAD accumulation in RAB11-positive endosomes in mESCs treated with BAF-A1. DMSO was used as a vehicle control. Scale bars, 25 μm (top panel) and 10 μm (lower panel).
(H) Scatterplot showing the quantitation of E-CAD levels, membrane versus cytoplasmic; in mESCs treated with BAF-A1. ∗∗∗p < 0.001 by two-tailed Student’s t test.
(I) Graph showing levels of pluripotency marker genes by qRT-PCR analysis in mESCs treated with BAF-A1 or DMSO.
(J) Graph showing levels of pluripotency marker genes by qRT-PCR analysis upon Rab11 knockdown in mESCs. SCi, scrambled shRNA control; Rab11i, Rab11 shRNA.
Error bars represent mean ± SD from three independent experiments (n = 3). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 by two-tailed Student’s t test.
The transmembrane, Ca2+-dependent protein, E-CAD, is known to act by maintaining cell contacts through the formation of adherens junctions (Leckband and de Rooij, 2014). It is involved in maintenance of the pluripotent state of mESCs (Redmer et al., 2011), and is required for normal mammalian development (Larue et al., 1994, Stephenson et al., 2010). Trafficking of E-CAD has been shown to be essential for the generation and turnover of cell-cell contacts in the context of vertebrate development (Nanes and Kowalczyk, 2012, Song et al., 2013). However, it is unclear whether E-CAD undergoes endocytosis in mESCs and, if so, which pathways of intracellular trafficking are utilized, and whether this impacts pluripotency.
We observed that E-CAD co-localized with CLTC (Figures 2B and S3D), and co-immunoprecipitated with the clathrin adaptor AP2 complex subunit, AP2α (Figure S3C), confirming that E-CAD indeed associates with the CME machinery in mESCs. E-CAD localization was also abundantly detected in RAB11-positive recycling endosomes (Figures 2C and S3E). RAB7-positive late endosomes and LAMP2-positive lysosomes, which target cargo for lysosomal degradation, were largely devoid of E-CAD (Figures S3F and S3G).
E-CAD was predominantly present on the membrane in mESCs (Figure 2B). We disrupted its normal localization through two approaches: (1) treatment with an E-CAD neutralizing antibody (DECMA1), which prevents the dimerization of E-CAD, resulting in its internalization; and (2) treatment with EDTA, a metal ion chelating agent that chelates the extracellular Ca2+, preventing E-CAD dimerization. Treatment of mESCs with either DECMA1 or EDTA resulted in a loss of cell-cell contacts and decreased E-CAD on the cell surface (Figures 2D, 2E, S3H, and S3I). This was accompanied by a decrease in the expression of pluripotency marker genes (Figures 2F and S3J), similar to what was previously observed in E-cad null mESCs (Redmer et al., 2011).
Treatment of mESCs with DECMA1, or EDTA, resulted in increased accumulation of E-CAD in RAB11-positive recycling endosomes (Figures 2D, 2E, S3H, and S3I). To determine whether recycling of E-CAD was essential for the pluripotency of mESCs, we knocked down Rab11 (Figure S3K), or blocked endosomal recycling by treating cells with the pharmacological inhibitor, Bafilomycin A1 (vesicular ATPase inhibitor that blocks endocytic recycling) (Presley et al., 1997). This resulted in an increased accumulation of E-CAD in RAB11-positive endosomes (Figures 2G and 2H) and a decrease in the expression of pluripotency marker genes (Figures 2I and 2J). Taken together, our results imply that the normal membrane localization, internalization through CME, and the recycling of E-CAD are essential for the pluripotency of mESCs. Thus, the normal trafficking of E-CAD is imperative to maintain the pluripotency of mESCs.
CME Suppresses TGF-β/SMAD Signaling to Maintain E-CAD Expression and mESC Pluripotency
The loss of CME resulted in a dramatic phenotype, with loss of cell-cell contacts and a decrease in levels of E-cad/E-CAD at both the transcript and protein levels (Figures 2A, 3A, S2, S3A, S4A, and S4B), highly reminiscent of the action of a core epithelial-mesenchymal transition driver, TGF-β signaling (Lamouille et al., 2014, Nieto et al., 2016, Xu et al., 2009). Increased TGF-β signaling is associated with the differentiation of ESCs (Fei et al., 2010), and is also known to play a negative role during somatic cell reprogramming (Ichida et al., 2009, Li et al., 2011, Subramanyam et al., 2011). Activation of the TGF-β/SMAD signaling pathway results in the expression of transcription factors Snai1 (Snail), Snai2 (Slug), and Zeb1, which transcriptionally repress E-cad (Xu et al., 2009). Blocking CME resulted in the stabilization of TGF-βR1 levels accompanied by elevated levels of active TGF-β/SMAD signaling, determined by phospho-SMAD2/3 levels (Figures 3B, 3C, S4C, S4E, and S4F). Blocking CME resulted in increased expression of Snai1, Snai2, Zeb1, and Cdh2 in mESCs (Figures 3D and S4D).
Figure 3.

CME Promotes Degradation of TGF-βR1 to Maintain E-CAD Levels in mESCs
(A) Graph showing levels of the E-cad transcript by qRT-PCR analysis upon knockdown of Cltc in mESCs. NTi, non-targeting siRNA control; Cltci, Cltc siRNA.
(B and C) Western blots showing TGF-βR1 (B), pSMAD2/3, and total SMAD2/3 (C) levels in mESCs, 72 hr post Cltc knockdown.
(D) Graph showing levels of indicated genes by qRT-PCR analysis after 3 days of Cltc knockdown in mESCs.
(E and F) Representative micrographs showing the co-localization between CLTC and TGF-βR1 (E) and between TGF-βR1 and RAB7 in mESCs (F). Scale bar, 10 μm.
(G and H) Western blots showing TGF-βR1, pSMAD2/3, and total SMAD2/3 levels 72 hr post Rab7 knockdown in mESCs. Rab7i, Rab7 siRNA.
(I) Graph showing levels of indicated genes by qRT-PCR analysis upon knockdown of Rab7 in mESCs.
(J) Graph showing levels of E-cad by qRT-PCR analysis upon knockdown of Rab7 in mESCs.
(K) Construction of a model to show the inter-relationships between TGF-βR, E-CAD, and CLTC in mESCs. E, E-CAD; T, TGF-βR; m, membrane; c, cytoplasm; e, endosome; r, recycling endosome; l, lysosome.
(L) Graph showing levels of E-CAD and TGF-βR based on simulation of the constructed model. WT (wild-type) represents the pre-treatment concentrations of membrane-bound TGF-βR and E-CAD. The “reduced clathrin” set shows the steady-state values following clathrin depletion. The “reduced clathrin and TGF-βR dependence” set is obtained by simulating a decrease in TGF-β signaling following CLTC depletion, by changing the parameter Kitmec, which changes the sensitivity of the E-CAD formation rate to TGF-βR levels by 5-fold, keeping other parameters constant.
Error bars represent mean ± SD from three independent experiments (n = 3). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 by two-tailed Student’s t test.
TGF-βR1 co-localized with CLTC and the late endosomal marker, RAB7 (Figures 3E, 3F, S4G, and S4H), suggesting that TGF-βR1 is internalized through CME and targeted for lysosomal degradation. Minimal to no co-localization was observed with RAB11, suggesting minimal or no recycling of TGF-βR1 in mESCs (Figure S4I). To determine whether TGF-βR1 was indeed targeted for lysosomal degradation, we used siRNAs to knock down Rab7 (Figure S4J). We observed an accumulation of LC3-2, a protein that is degraded in the lysosome (Figure S4K), indicating a block in lysosomal degradation on loss of Rab7. This was accompanied by an increase in the levels of TGF-βR1 (Figure 3G), pSMAD2/3 (Figure 3H), and mesenchymal markers (Figure 3I). The mesenchymal genes Snai1, Snai2, and Zeb1 are known to transcriptionally repress E-cad expression. Indeed, knockdown of Rab7 resulted in a decrease in E-cad/E-CAD expression, presumably through a feedback loop involving downstream targets of TGF-βR1 signaling (Figures 3J and S4K), similar to what was observed upon Cltc knockdown. These observations were further validated by treatment of mESCs with the lysosomal inhibitor, chloroquine, resulting in increased levels of LC3-2 (Figure S4L), TGF-βR1 (Figure S4M), and pSMAD2/3 (Figure S4N). Blocking lysosomal degradation also resulted in a decrease in E-CAD levels (Figure S4O), similar to what we observed upon knockdown of Rab7 (Figure S4K). Together these results show that CME negatively regulates TGF-β/SMAD signaling to maintain E-CAD expression and the pluripotency of mESCs.
Based on these observations, we constructed a mathematical model for the inter-relationships between TGF-β signaling, E-CAD, and CLTC (Figure 3K; Table S1). Keeping all other parameters constant, clathrin depletion was modeled by decreasing the parameter for clathrin levels by 10-fold. The depletion of clathrin resulted in a decrease in E-CAD levels, due to an increase in TGF-β signaling, consistent with the experimental observations. In addition, the model predicted an initial transient increase in membrane-bound E-CAD (not shown here) upon clathrin depletion, which is explained as the effect of decreased internalization resulting from the depletion of CLTC. We further used the model to ask whether decreasing TGF-β signaling upon clathrin depletion could restore the cell to its original state. We simulated a decrease in TGF-β signaling following Cltc depletion, by changing the parameter Kitmec (Table S1), which changes the sensitivity of the E-CAD formation rate to TGF-βR levels by 5-fold, keeping other parameters constant. From the simulation results (“reduced clathrin and TGF-βR dependence” set) we predicted that this leads to a restoration of E-CAD (membrane bound), and possibly even a higher level due to the reduced internalization even though there is no effect on the membrane-bound TGF-βR levels (Figure 3L).
Based on the constructed model, we predicted that perhaps the normal balance of signaling could be restored in mESCs in the absence of CME by blocking the aberrant increase in TGF-β/Smad signaling. To test this, we treated Cltc-depleted mESCs with TGF-βR1 kinase inhibitors, RepSox (Ichida et al., 2009), or SB431542. Inhibition of TGF-β/Smad signaling resulted in a partial rescue of the phenotype with restoration of cell-cell contacts of mESCs, along with increased AP activity (Figure 4A), OCT3/4 and E-CAD expression (Figures 4B and 4C). The overexpression of E-cad alone also (Figure 5A) resulted in a rescue of pluripotency in the background of decreased CME (Figures 5B and 5C).
Figure 4.

Effect of Cltc Knockdown in mESCs Can Be Partially Rescued by Inhibiting TGF-β Signaling
(A) Bright-field micrographs showing AP staining and morphology of Cltc knockdown mESCs in the presence of TGF-β inhibitors RepSox or SB431542 (SB). Scale bar, 50 μm.
(B) Immunofluorescence micrographs showing OCT3/4 and E-CAD staining upon RepSox or SB431542 (SB) treatment in mESCs with Cltc knockdown. Scale bar, 50 μm.
(C) Bar graph showing the quantitation of OCT3/4 and E-CAD-positive mESC colonies upon RepSox or SB431542 (SB) treatment in Cltc knockdown mESCs. Error bars represent mean ± SD from three independent experiments (n = 3). ∗∗p < 0.01 by two-tailed Student’s t test. One-way ANOVA with post-hoc Tukey-Kramer test was used.
Figure 5.

Effect of Cltc Knockdown in mESCs Can Be Partially Rescued by Overexpressing E-cad
(A) Western blot showing the overexpression of E-CAD in mESCs post transfection with the pEF1-E-cad vector. The EF1 empty vector is used as a control.
(B) Representative micrographs showing the morphology of mESCs in the presence of empty vector (EF1), and vector-expressing E-cad (pEF1-E-cad) in combination with either NTi or Cltci. Scale bar 50 μm.
(C) Bar graph showing the levels of pluripotency marker genes in the presence of the empty vector or upon E-cad overexpression. EF1, empty vector; E-cad, EF1-E-cad vector; NTi, non-targeting siRNA control; Cltci, Cltc siRNA. Error bars represent mean ± SD from three independent experiments (n = 3). ∗p < 0.05; ∗∗p < 0.01 by two-tailed Student’s t test. One-way ANOVA with post-hoc Tukey-Kramer test was used.
These results indicate that CME plays a vital role in regulating the balance between opposing signals such as E-CAD (pro-pluripotency) and TGF-β (pro-differentiation) in ESCs.
E-CAD is also known to associate with other proteins such as β-CATENIN (Nanes and Kowalczyk, 2012). β-CATENIN signaling is required for the maintenance of mESC pluripotency and self-renewal (Anton et al., 2007, Miyabayashi et al., 2007). Indeed we found that as reported earlier (Nanes and Kowalczyk, 2012), β-CATENIN associated with E-CAD in mESCs (Figure S5A). Knockdown of Cltc resulted in decreased β-CATENIN levels (Figure S5B) and enhanced phospho-β-CATENIN levels (Figure S5B), suggestive of it being targeted for degradation (Perez-Moreno and Fuchs, 2006). In addition, we also observed enhanced levels of active phospho-ERK1/2 levels (Figure S5E), which is associated with the differentiation of mESCs toward the neural and mesodermal lineages (Kunath et al., 2007, Stavridis et al., 2007). Interestingly, we also observed for the first time an interaction between E-CAD and ERK in mESCs (Figures S5C and S5D), indicating that E-CAD may function as a hub to regulate the activity of other proteins involved in the pluripotency of mESCs.
The naïve state of ESCs is maintained by culturing them in a media containing mitogen-activated protein kinase (MEK) and glycogen synthase kinase 3β (GSK3β) inhibitors (Ying et al., 2008). We therefore sought to determine whether the loss of pluripotency upon Cltc knockdown could be rescued by treatment with either the MEK inhibitor, or the GSK3β inhibitor (1i), or both (2i). Indeed, we found that the knockdown of Cltc in mESCs cultured in MEK inhibitor or 2i medium did not result in a significant decrease in pluripotency (Figures S5F and S5G), suggesting that the elevation in MEK signaling alone, or in combination with decreased β-CATENIN levels in the absence of CME, plays a significant role in exiting the pluripotent state.
In summary, our data indicate that CME plays a crucial role in maintaining a fine balance between antagonistic signaling pathways, such as TGF-β/SMAD, ERK, β-CATENIN, and E-CAD. CME is required for the internalization and recycling of E-CAD, and for the lysosomal degradation of TGF-βR1 in mESCs. Any disruption in this trafficking mechanism leads to dominance of the differentiation signal, TGF-β, along with elevated levels of phospho-ERK, and reduced levels of β-CATENIN, resulting in a cell fate change. We find that repression of the differentiation signal is sufficient to some extent to restore the pluripotent state. Our results thus demonstrate that CME is required for the normal pluripotent state of mESCs, and that altering CME levels may permit ESCs to exit the pluripotent state.
Discussion
While endocytosis plays a major role in metazoan development, it remains largely unknown as to whether and how it can function as a regulator of cell fate decisions. Here we demonstrate that CME functions as a novel regulator of ESC pluripotency. Decrease in the level of CME in ESCs results in an exit from the pluripotent state. We further demonstrate that, in ESCs, CME facilitates the internalization and recycling of E-CAD, while simultaneously being required for the internalization and lysosomal targeting of TGF-βR. It is well appreciated that cell-cell adhesion mediated by E-CAD is crucial for maintaining mESC self-renewal (Murray et al., 2013, Redmer et al., 2011, Soncin et al., 2011). Our findings show that the internalization and recycling of E-CAD via CME is crucial to maintain the stem cell state of mESCs. Although the mechanistic underpinnings of this regulation are unclear, based on our data and previous studies with the NOTCH receptor and GPCRs (Windler and Bilder, 2010, Eichel et al., 2016) we speculate that E-CAD trafficking via CME may facilitate its transition through different intracellular compartments to either undergo conformational changes or association with other proteins, ultimately resulting in an altered signaling output. While RAP1 has been previously described to play a role in the recycling of E-CAD, it is unclear as to whether this is dependent on CME (Li et al., 2010). Our data are also strengthened by our observation that E-CAD is enriched in ESC-derived endosomes (Narayana et al. unpublished data). A similar mechanism has also been described with pSTAT3 in mESCs, where its association with the endosomal protein ASRIJ regulates STAT3 signaling, as well as the expression of pluripotency genes (Sinha et al., 2013). In addition to the recycling of E-CAD, CME also plays a major role in maintaining the expression of E-cad by lysosomal degradation of the TGF-β receptor. Earlier studies in non-stem cells show that TGF-β receptors were recycled via CME (Chen, 2009, Hayes et al., 2002, Mitchell et al., 2004). Previous studies have also demonstrated that CME is involved in enhancing signals downstream of the TGF-β receptor, while the caveolin-mediated endocytic route targets these receptors for degradation (Di Guglielmo et al., 2003, Scita and Di Fiore, 2010), indicating a requirement for specific compartmentalization of the same receptor to achieve opposing outcomes. We have previously demonstrated that caveolin-mediated endocytosis is largely absent in mESCs (Mote et al., 2017), and here we demonstrate that CME promotes the lysosomal degradation of the TGF-β receptor. Such varying utilizations of trafficking pathways in the context of the same receptor may be a reflection of the specific state of a cell, and an attempt to maintain a balance in signaling outcomes to support a particular cell fate. While it remains elusive as to how CME is involved in sorting a specific cargo to different compartments in stem versus non-stem cells, our findings show that CME balances opposing signals to maintain ESC pluripotency by sorting molecules to different compartments.
In addition, we also see that both β-CATENIN and ERK associate with E-CAD, implicating yet another layer of regulation in ESCs, linked to E-CAD. It remains to be seen whether the activity of ERK and β-CATENIN are dependent on the internalization of E-CAD. Our results demonstrate that we could rescue the decrease in pluripotency caused by the loss of CME by either inhibiting the TGF-β or MEK signaling pathway, or by overexpressing E-cad, indicating that CME does indeed regulate the activity of multiple signaling pathways and molecules in mESCs. Together, our data demonstrate that an intact trafficking machinery is essential for regulating the activity of key signaling molecules, all of which are intimately linked to the pluripotent state of a stem cell.
It is well established that CME is one of the major routes of endocytosis in differentiated cells (Doherty and McMahon, 2009, Traub, 2009). Recent results comparing the rate of CME between human ESCs and differentiated cells show that the rate of endocytosis decreases as stem cells differentiate (Dambournet et al., 2018). Further, this was also accompanied by changes in types of clathrin coats that were observed as ESCs underwent differentiation. This is in line with our observation that ESCs differentiate upon reduction of CME. In addition, our data suggest that a temporary reduction of CME, or a fine-tuning of levels of endocytosis in a cell that possesses the potential to switch fate, such as a stem cell, can provide a novel handle to regulate cell fate changes. Further studies in understanding the mechanistic role of endocytosis will expand our understanding about the role of cellular trafficking in stemness, cell fate decisions and development.
Experimental Procedures
Cell Culture, Chemical Treatments, Plasmids, and Antibodies
V6.5 and R1 mESCs were cultured on 0.2% gelatin-coated plastic tissue culture dishes (Corning), and cultured in media containing knockout DMEM (Invitrogen), 15% fetal bovine serum (Invitrogen), 2 mM L-glutamate (Invitrogen), 1× penicillin/streptomycin (Invitrogen), 1 mM non-essential amino acids (Invitrogen), 100 μM β-mercaptoethanol (Invitrogen), and 5,000 U leukemia inhibitory factor (LIF) (ESC medium). MEFs were isolated from embryonic day 13.5 stage mouse embryos from the CF1 strain of mice. Inhibitors and chemicals were used as follows: chloroquine (Sigma) was used at 100 μM final concentration; RepSox (Sigma) 25 μM final concentration; SB431542 (Sigma) 5 μM final concentration; PD0325901 (Sigma) was used at 0.1 μM final concentration; CHIR99021 (Sigma) was used at 0.3 μM final concentration; Bafilomycin A1 (Sigma) 1 μM final concentration; EDTA (pH 8.0), 2.5 μM final concentration. K44A Dynamin2-GFP mutant was obtained from Addgene (no. 34687). Empty pEGFP N1 vector was used as a control. The following antibodies were used at the appropriate concentrations: CLTC (sc-6579, Santa Cruz, goat), TGF-βR1 (sc-398, Santa Cruz, rabbit), NANOG (sc-30328, Santa Cruz, goat), SMAD2/3 (sc-7960, Santa Cruz, mouse), RAB7 (sc-81922, Santa Cruz, rabbit), OCT3/4 (no. 2849, Cell Signaling Technology, rabbit), E-CAD (no. 3195, Cell Signaling Technology, rabbit), pSMAD2/3 (no. 8828, Cell Signaling Technology, rabbit), SOX2 (ab97951, Abcam, rabbit), E-CAD (no. 610182, BD Transduction Laboratories, mouse), RAB11 (no. 610657, BD Transduction Laboratories, mouse), CLTC (no. 610500, BD Transduction Laboratories, mouse), TUBULIN (T5168, Sigma, mouse), RAB5 (no. 3547, Cell Signaling Technology, rabbit), RAB7 (no. 9367, Cell Signaling Technology, rabbit), GAPDH (no. SC25778, Santa Cruz, rabbit), Na,K+ ATPASE (no. 21713, Santa Cruz, goat), LC3 1/2 (no. 12741, Cell Signaling Technology, rabbit), LAMP2 (ab13542, Abcam, rat). E-CAD neutralizing antibody (DECMA-1, Abcam ab11512) was used at 5 μg/mL final concentration. Rat immunoglobulin G (IgG) was used as an antibody control. For imaging, cells were treated with the antibody for 1 hr, and for pluripotency assays, 24 hr in ESC media. Mouse E-cad open reading frame was amplified from cDNA and cloned into Ecor1 and Not1 sites of pEF1/myc-His vector (a gift from Dr. Ramkumar Sambasivan). Transfections were performed using Lipofectamine 2000 (no. 11668019, Invitrogen) according to the manufacturer's instructions. All experiments were performed using V6.5 ESCs. Experiments where R1 ESCs were used are clearly indicated.
Lentivirus Production and Infection
Plko.1 scrambled, Cltc, and Rab11 small hairpin RNA (shRNA) vectors (obtained from shRNA Resource Center, Indian Institute of Science, Bangalore) were co-transfected with psPAX2 (Addgene, no. 12260), pMD2.G (Addgene, no. 12259) in HEK293T cells using FuGENE HD (no. E2311, Promega). Viral supernatants were harvested and used for infection 48 hr post transfection.
qRT-PCR
Total RNA was isolated using TRIzol (Invitrogen) and quantified using a NanoDrop Spectrophotometer (Thermo Fisher Scientific). For mRNA amplification, 1 μg RNA was treated with DNaseI (Invitrogen), and reverse transcribed using the Superscript III kit (Invitrogen) using random hexamers. Total cDNA thus obtained was diluted 1:10 and 1 μL was used for qRT-PCR using gene-specific primers and Power SYBR Green PCR Master Mix (Applied Biosystems). Primers sets are listed in Table S2. For qRT-PCR analysis, mRNA expression is normalized to GAPDH and represented relative to control or non-targeting siRNA.
Colony Counting and Cell Doubling Time
A total of 50,000 mESCs were plated in a 6-well plate and transfected the next day with Cltc esiRNA. Colonies were counted on day 4. For determination of cell doubling time, 20,000 cells were plated in a 24-well dish and transfected with Cltc esiRNA next day. Post transfection, cells were counted every 24 hr.
Cell-Cycle Analysis
Cells (20,000) were grown in a 24-well dish and treated as indicated. After 72 hr, cells were trypsinized and washed twice with PBS, fixed in 95% ethanol at 4°C overnight, stained with propidium iodide (10 μg/mL) and RNase A (10 μg/mL), and 2 mM MgCl2. Stained cells were analyzed on a BD FACS Calibur.
Alkaline Phosphatase Staining
ESCs were transfected with siRNAs or the mentioned plasmid, and stained for AP activity using a kit (Vector Laboratories), as per manufacturer's instructions.
esiRNA Preparation and Transfection
Template for esiRNA production against specific genes was prepared by PCR amplification from mESC or MEF cDNA. In vitro transcription was performed using T7 RNA polymerase, followed by digestion of the double-stranded RNA using RNase III (Fazzio et al., 2008, Kittler et al., 2007). esiRNAs were transfected into ESCs using DharmaFECT 1 transfection reagent (Dharmacon). After 72 hr, cells were lysed in TRIzol for RNA, or in RIPA lysis buffer to prepare whole-cell protein extract. Non-targeting esiRNA was prepared using GFP as a template in all experiments.
Western Blotting
Protein lysates were separated on SDS-PAGE, blotted onto a polyvinylidene fluoride membrane, and blocked with 5% BSA or skimmed milk. Primary antibodies were used at specific concentrations overnight, followed by incubation with the appropriate secondary antibody.
Immunocytochemistry and Imaging
mESCs were cultured on glass coverslips coated with 0.2% gelatin. Cells were fixed with 4% paraformaldehyde (PFA) for 10 min at 37°C, followed by blocking and permeabilization using 5% BSA containing 0.1% Triton X-100. Incubation with primary antibody was done overnight at 4°C, followed by washing and incubation with secondary antibody for 1 hr at room temperature, and washing and mounting using VECTASHIELD (Vector Laboratories). All confocal imaging was done using the Leica SP5 confocal microscope, and imaged using a 63× 1.4 NA objective. Bright-field images were taken using Nikon Eclipse Ti2 microscope with either the 20× or 10× objective.
Teratoma Formation Assay
mESCs (25 × 105) stably expressing scrambled or Cltc shRNA were suspended in 50 μL knockout DMEM and 10 μL of Matrigel. Nude mice were anesthetized with diethyl ether and 60 μL of cell suspension was injected subcutaneously into the dorsal flank of mice. Tumors were surgically dissected and measured and fixed in 4% PFA 3 weeks post injection, and embedded in paraffin. Sections were stained with hematoxylin and eosin. All protocols involving animal use were approved by the Institutional Animal Ethics Committee.
Embryoid Body Formation
Embryoid bodies were formed by hanging drop method (500 cells/20 μL drop). Embryoid bodies were formed in ES media without LIF for 3 days. After 3 days these embryoid bodies were cultured on Matrigel-coated dishes for 6 days. Post 6 days, cells were collected for RNA isolation.
Co-immunoprecipitation
Co-immunoprecipitation was performed by incubating the cell lysate (lysed using 1% NP40 cell lysis buffer) with specific antibody and Dynabeads (Invitrogen). The supernatant was separated using Dynamag and analyzed by western blotting. Normal IgG was used as antibody control.
Membrane-Cytoplasmic Fractionation
For fractionation, cells were osmotically lysed in buffer (20 mM HEPES [pH 8], 0.1 mM KCl, 0.1 mM MgCl2, and 20% glycerol), followed by passing of the lysate through an insulin syringe. The supernatant (cytoplasmic fraction) was collected by spinning at 14,000 rpm for 60 min. The membrane fraction was collected by lysing the pellet in buffer (20 mM HEPES [pH 8], 0.1 mM KCl, 0.1 mM MgCl2, 20% glycerol, and 0.5% Triton-X 100), followed by spinning at 14,000 rpm, for 30 min.
Mathematical Modeling
TGF-βR and E-CAD are assumed to be present in membrane-bound, cytoplasmic, endosomal, and lysosomal forms. In addition, E-CAD is assumed to also exist in a recycling endosome. The presence of additional compartments, such as recycling endosome, or lumping/merging compartments, coupled with a compensatory change in the rate constants does not qualitatively change these results. The rates of interactions are assumed to follow mass-action kinetics. TGF-β ligand is assumed to be constantly available, and therefore the inhibition of E-cad expression by TGF-β signaling is assumed to be mediated by the level of the receptors present at the membrane, and consequently the downstream signaling. Inhibition is incorporated by including a Hill-like inhibitory term (with a coefficient of 2) to the zero-order formation kinetics. Inhibition of TGF-β signaling is modeled as an increase in the inhibition constant, which is equivalent to a reduction in the amount of membrane-bound TGF-βR available for inhibition. Based on these kinetics, a mass balance for each of the nine components of the system shown, leads to a system of 9 coupled ordinary differential equations, which include the effect of the 22 reactions and 28 parameters (equations and parameters are listed in Table S1).
The equations were integrated with the ode15s function of MATLAB version 9.2 and SimBiology version 5.6 with relative and absolute tolerance 10−8 until steady state was achieved, and this state was assumed to be the pre-treatment state of the system. The values were scaled to the maximum value following all the simulations (clathrin depletion, clathrin depletion with decreased TGF-β signaling).
Transferrin Uptake Assay
mESCs (20,000) were plated followed by transfection with Cltc esiRNAs or the mentioned plasmids after 24 hr. Next day, the cells were plated on 0.2% gelatin-coated glass coverslips. After 48 hr, cells were serum starved in plain knockout DMEM at 37°C. After 1 hr, cells were incubated with Transferrin-Alexa Fluor 488 (5 μg/100 μL) (no. 015-540-050, Jackson ImmunoResearch) for 10 min at 37°C, washed with acid wash buffer (0.2 M glycine, 0.15 M NaCl [pH 3.0]) for 30 s to remove surface-bound and non-specific transferrin. Cells were washed three times in PBS, fixed in 4% PFA, and imaged. All images were analyzed using ImageJ software (NIH). The total amount of internalized transferrin was quantified from cells as mean gray value, and background-subtracted values are represented from three independent experiments.
Statistical Analysis
Error bars represent mean ± SD for three independent experiments (n = 3). Statistical significance, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001, was determined by two-tailed Student’s t test. mRNA expression is normalized to GAPDH and represented relative to non-targeting siRNA control. For comparing multiple groups of data, one-way ANOVA was used. If the p value for the comparison was <0.05, then individual groups were compared with each other using a post-hoc Tukey-Kramer test.
Apoptosis Analysis
Cells were fixed with 4% PFA for 10 min, followed by staining with Hoechst 33342 for 10 min. Cells were scored as apoptotic based on the presence of fragmented and/or condensed DNA, as described previously (Chakrabarti et al., 2004). As a positive control for apoptosis, cells were treated with camptothecin (6 μM) for 12 hr.
siRNA Screening
mESCs (2,000) were plated per well in a 96-well plate on day 0. On day 1, cells were transfected with siRNAs from the Dharmacon Membrane Trafficking siGENOME Library (GU-015500-05). None of the siRNAs were validated for their efficiency at the time of screening. Each well received a pool of four siRNAs against a single gene. At the time of transfection, cells were shifted to ESC medium minus LIF. Cells were fixed on day 4 and stained for AP (Vector Laboratories) and co-stained with DAPI. Each plate included ten control wells that were mock-transfected. Five were maintained in ESC medium and the other five in ESC minus LIF.
Image Analysis
All micrographs were analyzed using ImageJ software (NIH). Images were background-subtracted and quantified as mean gray values. For transferrin uptake assay, total cell fluorescence was quantified from three independent experiments. For E-CAD membrane/cytoplasmic quantification, total and cytoplasmic protein expressions were quantified from the middle section of the z stack. The membrane E-CAD expression was calculated by subtracting cytoplasmic from total expression, and represented as membrane versus cytoplasmic ratio from three independent experiments.
Screen Analysis
Every individual well of screen plates was imaged using the BD pathway 855, 4× objective, 5 × 5 matrix. All analysis was done using custom programs written in MATLAB (MathWorks). All images were read into MATLAB as two-dimensional matrices of pixel intensities. Images for the control wells were visually screened for the presence of imaging artifacts and wells with obvious artifacts like reflections were excluded. All pixels with intensity greater than a specified threshold value in the DAPI image were considered to be cells. In a subset of wells, the choice of this threshold value was visually checked using two criteria: (1) all chosen pixels did indeed correspond to cells and (2) no cells were omitted. A threshold value of 950 was found to satisfy the above criteria and was used for all wells in all plates. Once pixels corresponding to cells were identified, the locations of these pixels were transferred to the Texas Red image and a cumulative distribution function (cdf) of the intensities of these pixels was calculated using ecdf (MATLAB function) for each well. The pixel values for all of the control wells (ESCs–LIF) in a given plate were combined and an average cdf was generated to represent the Control cdf in a given plate. The largest difference between the cdf for each well and the Control cdf was then calculated for each well (kstest2). These differences for each of the individual control wells gave us a measure of differences that could occur purely by chance. The differences for all wells were normalized into a Z score as follows:
Wells with a Z score >2.5 or ≤2.5 were considered as wells that had significant changes in their cdf and the direction of change was analyzed to see if they had more or less Texas Red staining compared with the Control cdf.
Author Contributions
D.S. and Y.V.N. conceived and designed the study. D.S. carried out the screening in Figure 1A. R.R. carried out the analysis for Figure 1A. C.G. did the modeling for Figures 3K and 3L. R.D.M. did experiments for Figure S1J and helped with Figure S2. All other experiments were performed by Y.V.N.. D.S., and Y.V.N. wrote the manuscript with help from R.R. and C.G. All authors read and approved the manuscript.
Acknowledgments
The authors thank Richa Rikhy and Ayantika (IISER Pune) for help with image analysis; IISER Pune for access to imaging facilities; Amitabha Majumdar and Jomon Joseph for discussions; NCCS Experimental Animal Facility and Subramanyam lab for critical reading of the manuscript and feedback. This work was supported by an Intermediate Fellowship from The Wellcome Trust/DBT India Alliance (IA/I/12/1/500507) to D.S.; Y.V.N. is a recipient of a CSIR-SRF Fellowship; R.R. is supported by a DBT Ramalingaswami Fellowship (BT/HRD/35/02/2006).
Published: December 13, 2018
Footnotes
Supplemental Information includes five figures and two tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.11.018.
Supplemental Information
References
- Anton R., Kestler H.A., Kühl M. Beta-catenin signaling contributes to stemness and regulates early differentiation in murine embryonic stem cells. FEBS Lett. 2007;581:5247–5254. doi: 10.1016/j.febslet.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Bitoun M., Durieux A.-C., Prudhon B., Bevilacqua J.A., Herledan A., Sakanyan V., Urtizberea A., Cartier L., Romero N.B., Guicheney P. Dynamin 2 mutations associated with human diseases impair clathrin-mediated receptor endocytosis. Hum. Mutat. 2009;30:1419–1427. doi: 10.1002/humu.21086. [DOI] [PubMed] [Google Scholar]
- Chakrabarti O., Veeraraghavalu K., Tergaonkar V., Liu Y., Androphy E.J., Stanley M.A., Krishna S. Human papillomavirus type 16 E6 amino acid 83 variants enhance E6-mediated MAPK signaling and differentially regulate tumorigenesis by notch signaling and oncogenic Ras. J. Virol. 2004;78:5934–5945. doi: 10.1128/JVI.78.11.5934-5945.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-G. Endocytic regulation of TGF-β signaling. Cell Res. 2009;19:58–70. doi: 10.1038/cr.2008.315. [DOI] [PubMed] [Google Scholar]
- Dambournet D., Sochacki K.A., Cheng A.T., Akamatsu M., Taraska J.W., Hockemeyer D., Drubin D.G. Genome-edited human stem cells expressing fluorescently labeled endocytic markers allow quantitative analysis of clathrin-mediated endocytosis during differentiation. J. Cell Biol. 2018;217:3301–3311. doi: 10.1083/jcb.201710084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Renzis S., Yu J., Zinzen R., Wieschaus E. Dorsal-ventral pattern of delta trafficking is established by a snail-tom-neuralized pathway. Dev. Cell. 2006;10:257–264. doi: 10.1016/j.devcel.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Di Guglielmo G.M., Le Roy C., Goodfellow A.F., Wrana J.L. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat. Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- Doherty G.J., McMahon H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- Eichel K., Jullié D., von Zastrow M. β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat. Cell Biol. 2016;18:303–310. doi: 10.1038/ncb3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M. Discovering pluripotency: 30 years of mouse embryonic stem cells. Nat. Rev. Mol. Cell Biol. 2011;12:680–686. doi: 10.1038/nrm3190. [DOI] [PubMed] [Google Scholar]
- Evans M.J., Kaufman M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- Fabrowski P., Necakov A.S., Mumbauer S., Loeser E., Reversi A., Streichan S., Briggs J.A.G., Renzis S.D. Tubular endocytosis drives remodelling of the apical surface during epithelial morphogenesis in Drosophila. Nat. Commun. 2013;4:2244. doi: 10.1038/ncomms3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzio T.G., Huff J.T., Panning B. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell. 2008;134:162–174. doi: 10.1016/j.cell.2008.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei T., Zhu S., Xia K., Zhang J., Li Z., Han J.-D.J., Chen Y.-G. Smad2 mediates Activin/Nodal signaling in mesendoderm differentiation of mouse embryonic stem cells. Cell Res. 2010;20:1306–1318. doi: 10.1038/cr.2010.158. [DOI] [PubMed] [Google Scholar]
- Ferguson S.M., De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 2012;13:75–88. doi: 10.1038/nrm3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes S., Chawla A., Corvera S. TGFβ receptor internalization into EEA1-enriched early endosomes: role in signaling to Smad2. J. Cell Biol. 2002;158:1239–1249. doi: 10.1083/jcb.200204088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskovits J.S., Burgess C.C., Obar R.A., Vallee R.B. Effects of mutant rat dynamin on endocytosis. J. Cell Biol. 1993;122:565–578. doi: 10.1083/jcb.122.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichida J.K., Blanchard J., Lam K., Son E.Y., Chung J.E., Egli D., Loh K.M., Carter A.C., Di Giorgio F.P., Koszka K. A small molecule inhibitor of Tgf-β signaling replaces Sox2 in reprogramming by inducing nanog. Cell Stem Cell. 2009;5:491–503. doi: 10.1016/j.stem.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura N., Sun-Wada G.-H., Aoyama M., Harada A., Takasuga S., Sasaki T., Wada Y. Delivery of endosomes to lysosomes via microautophagy in the visceral endoderm of mouse embryos. Nat. Commun. 2012;3:1071. doi: 10.1038/ncomms2069. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T., Owen D., Harrison S.C. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb. Perspect. Biol. 2014;6:a016725. doi: 10.1101/cshperspect.a016725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler R., Surendranath V., Heninger A.-K., Slabicki M., Theis M., Putz G., Franke K., Caldarelli A., Grabner H., Kozak K. Genome-wide resources of endoribonuclease-prepared short interfering RNAs for specific loss-of-function studies. Nat. Methods. 2007;4:337–344. doi: 10.1038/nmeth1025. [DOI] [PubMed] [Google Scholar]
- Kunath T., Saba-El-Leil M.K., Almousailleakh M., Wray J., Meloche S., Smith A. FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development. 2007;134:2895–2902. doi: 10.1242/dev.02880. [DOI] [PubMed] [Google Scholar]
- Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue L., Ohsugi M., Hirchenhain J., Kemler R. E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc. Natl. Acad. Sci. U S A. 1994;91:8263–8267. doi: 10.1073/pnas.91.17.8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leckband D.E., de Rooij J. Cadherin adhesion and mechanotransduction. Annu. Rev. Cell Dev. Biol. 2014;30:291–315. doi: 10.1146/annurev-cellbio-100913-013212. [DOI] [PubMed] [Google Scholar]
- Li L., Wang S., Jezierski A., Moalim-Nour L., Mohib K., Parks R.J., Retta S.F., Wang L. A unique interplay between Rap1 and E-cadherin in the endocytic pathway regulates self-renewal of human embryonic stem cells. Stem Cells. 2010;28:247–257. doi: 10.1002/stem.289. [DOI] [PubMed] [Google Scholar]
- Li Z., Yang C.-S., Nakashima K., Rana T.M. Small RNA-mediated regulation of iPS cell generation. EMBO J. 2011;30:823–834. doi: 10.1038/emboj.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. U S A. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon H.T., Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- Mitchell H., Choudhury A., Pagano R.E., Leof E.B. Ligand-dependent and -independent transforming growth factor-β receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol. Biol. Cell. 2004;15:4166–4178. doi: 10.1091/mbc.E04-03-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabayashi T., Teo J.-L., Yamamoto M., McMillan M., Nguyen C., Kahn M. Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. U S A. 2007;104:5668–5673. doi: 10.1073/pnas.0701331104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mote R.D., Mahajan G., Padmanabhan A., Ambati R., Subramanyam D. Dual repression of endocytic players by ESCC microRNAs and the Polycomb complex regulates mouse embryonic stem cell pluripotency. Sci. Rep. 2017;7:17572. doi: 10.1038/s41598-017-17828-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P., Prewitz M., Hopp I., Wells N., Zhang H., Cooper A., Parry K.L., Short R., Antoine D.J., Edgar D. The self-renewal of mouse embryonic stem cells is regulated by cell-substratum adhesion and cell spreading. Int. J. Biochem. Cell Biol. 2013;45:2698–2705. doi: 10.1016/j.biocel.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanes B.A., Kowalczyk A.P. Adherens junction turnover: regulating adhesion through cadherin endocytosis, degradation, and recycling. Subcell. Biochem. 2012;60:197–222. doi: 10.1007/978-94-007-4186-7_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto M.A., Huang R.Y.-J., Jackson R.A., Thiery J.P. EMT: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Perez-Moreno M., Fuchs E. Catenins: keeping cells from getting their signals crossed. Dev. Cell. 2006;11:601–612. doi: 10.1016/j.devcel.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley J.F., Mayor S., McGraw T.E., Dunn K.W., Maxfield F.R. Bafilomycin A1 treatment retards transferrin receptor recycling more than bulk membrane recycling. J. Biol. Chem. 1997;272:13929–13936. doi: 10.1074/jbc.272.21.13929. [DOI] [PubMed] [Google Scholar]
- Qin H., Diaz A., Blouin L., Lebbink R.J., Patena W., Tanbun P., LeProust E.M., McManus M.T., Song J.S., Ramalho-Santos M. Systematic identification of barriers to human iPSC generation. Cell. 2014;158:449–461. doi: 10.1016/j.cell.2014.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmer T., Diecke S., Grigoryan T., Quiroga-Negreira A., Birchmeier W., Besser D. E-cadherin is crucial for embryonic stem cell pluripotency and can replace OCT4 during somatic cell reprogramming. EMBO Rep. 2011;12:720–726. doi: 10.1038/embor.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffarian S., Cocucci E., Kirchhausen T. Distinct dynamics of endocytic clathrin-coated pits and coated plaques. PLoS Biol. 2009;7:e1000191. doi: 10.1371/journal.pbio.1000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scita G., Di Fiore P.P. The endocytic matrix. Nature. 2010;463:464–473. doi: 10.1038/nature08910. [DOI] [PubMed] [Google Scholar]
- Sinha A., Khadilkar R.J., S V.K., Roychowdhury Sinha A., Inamdar M.S. Conserved regulation of the Jak/STAT pathway by the endosomal protein asrij maintains stem cell potency. Cell Rep. 2013;4:649–658. doi: 10.1016/j.celrep.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soncin F., Ward C.M., Soncin F., Ward C.M. The function of E-cadherin in stem cell pluripotency and self-renewal. Genes. 2011;2:229–259. doi: 10.3390/genes2010229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S., Eckerle S., Onichtchouk D., Marrs J.A., Nitschke R., Driever W. Pou5f1-dependent EGF expression controls E-cadherin endocytosis, cell adhesion, and zebrafish epiboly movements. Dev. Cell. 2013;24:486–501. doi: 10.1016/j.devcel.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavridis M.P., Lunn J.S., Collins B.J., Storey K.G. A discrete period of FGF-induced Erk1/2 signalling is required for vertebrate neural specification. Development. 2007;134:2889–2894. doi: 10.1242/dev.02858. [DOI] [PubMed] [Google Scholar]
- Stephenson R.O., Yamanaka Y., Rossant J. Disorganized epithelial polarity and excess trophectoderm cell fate in preimplantation embryos lacking E-cadherin. Development. 2010;137:3383–3391. doi: 10.1242/dev.050195. [DOI] [PubMed] [Google Scholar]
- Subramanyam D., Lamouille S., Judson R.L., Liu J.Y., Bucay N., Derynck R., Blelloch R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 2011;29:443–448. doi: 10.1038/nbt.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebar F., Sorkina T., Sorkin A., Ericsson M., Kirchhausen T. Eps15 is a component of clathrin-coated pits and vesicles and is located at the rim of coated pits. J. Biol. Chem. 1996;271:28727–28730. doi: 10.1074/jbc.271.46.28727. [DOI] [PubMed] [Google Scholar]
- Traub L.M. Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat. Rev. Mol. Cell Biol. 2009;10:583–596. doi: 10.1038/nrm2751. [DOI] [PubMed] [Google Scholar]
- Wang Y., Melton C., Li Y.-P., Shenoy A., Zhang X.-X., Subramanyam D., Blelloch R. miR-294/miR-302 promotes proliferation, suppresses G1-S restriction point, and inhibits ESC differentiation through separable mechanisms. Cell Rep. 2013;4:99–109. doi: 10.1016/j.celrep.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windler S.L., Bilder D. Endocytic internalization routes required for Delta/Notch signaling. Curr. Biol. 2010;20:538–543. doi: 10.1016/j.cub.2010.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Lamouille S., Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q.-L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.