

Conspectus

Nature’s catalytic machinery has provided endless inspiration for chemists. While the enzymatic ideal has yet to be fully realized, the field has made tremendous strides toward synthetic, small-molecule catalysts for a wide array of transformations, often drawing upon biological concepts in their design. One strategy that has been particularly influenced by enzymology is peptide catalysis, wherein oligopeptides are implemented as chiral catalysts in synthetically relevant reactions. The fundamental goal has been to mimic enzymatic active sites by taking advantage of secondary structures that allow for multifunctional activation of substrates within a framework of significantly reduced molecular complexity.
Our group has now been studying peptide-based catalysis for over two decades. At the outset, there were many reasons to be concerned that general contributions might not be possible. Precedents existed, including the Juliá–Colonna epoxidations mediated by helical oligopeptides, among others. However, we sought to explore whether peptide catalysts could find broad applications in organic synthesis despite what was expected to be their principal liability: conformational flexibility. Over time, we have been able to identify peptidic catalysts for a variety of site- and enantioselective transformations ranging from hydroxyl group and arene functionalizations to redox and C–C bond forming reactions. The peptides often exhibited excellent catalytic activities, in many cases enabling never-before-seen patterns of selectivity. Recent studies even suggest that, in certain situations, the conformational flexibility of these catalysts may be advantageous for asymmetric induction.
In the course of our studies, opportunities to employ peptide-based catalysis to solve long-standing and stereochemically intriguing problems in asymmetric synthesis presented themselves. For example, we have found that peptides provide exceptional enantiotopic group differentiation in catalytic desymmetrization reactions. Early results with symmetrical polyol substrates, such as myo-inositols and glycerols, eventually spurred the development of remote desymmetrizations of diarylmethanes, in which the enantiotopic groups are separated from the prochiral center by ∼6 Å and from one another by nearly 1 nm. Various hydroxyl group functionalizations and electrophilic brominations, as well as C–C, C–O, and C–N cross-coupling reactions using peptidic ligands on copper(I) have now been developed within this reaction archetype. Additionally, the preponderance of axially chiral, atropisomeric compounds as ligands, organocatalysts, and pharmacophores encouraged us to employ peptides as atroposelective catalysts. We have developed peptide-catalyzed brominations of pharmaceutically relevant biaryl, non-biaryl, and hetero-biaryl atropisomers that take advantage of dynamic kinetic resolution schemes. These projects have vastly expanded the reach of our original hypotheses and raised new questions about peptide-based catalysts and the extent to which they might mimic enzymes.
Herein, we recount the development and optimization of these stereochemically complex reactions, with a particular focus on structural and mechanistic aspects of the peptide-based catalysts that make them well-suited for their respective functions. The ability of these peptides to address important yet fundamentally challenging issues in asymmetric catalysis, combined with their modularity and ease-of-synthesis, make them primed for future use in organic synthesis.
1. Introduction
The ability to access enantiopure compounds is a fundamental and critical challenge in modern organic synthesis. The widespread use of single-enantiomer substances in chemical industries, materials science, and chemical biology provides a steady demand for asymmetric methodologies. While resolution continues to be a mainstay of industrial synthesis, asymmetric catalysis is now widely accepted as a reliable way to access enantioenriched compounds. Since its inception, the field of asymmetric catalysis has drawn inspiration from nature’s catalysts. Enzymes have evolved to catalyze biochemical reactions with exceptional rate acceleration and selectivity by taking advantage of their well-defined folded structures and their ability to undergo dynamic conformational changes.1 Within active sites, substrate activation is achieved through the precise orientation of catalytic residues, allowing for significant stabilization of transition states.2 The kinetic advantages provided by active site organization are typically bolstered by protein dynamics, as conformational changes often occur upon binding to provide additional stabilization of intermediates and transition states along a continuous reaction coordinate.3,4 Together, these characteristics contribute to the remarkable catalytic activities exhibited by enzymes and provide an ideal for the conception of synthetic catalysts.
Inspired by the possibility of developing small-molecule enzyme mimics with broad synthetic applicability, our group5,6 and others7,8 have sought to design short-sequence peptides that capture essential features of enzymatic active sites within greatly simplified molecular frameworks (Figure 1a). Peptides are well suited as asymmetric organocatalysts. They are easily synthesized from readily available amino acid residues, and their oligomeric nature renders them modular and tunable. These are helpful assets for deriving structure–selectivity relationships during catalyst development. Moreover, their conformations can be biased toward folded secondary structures by incorporating known sequence elements.9,10 Peptides are dense with stereochemical information: the chirality of the constituent residues often translates into 3D-folded structures, providing a robust platform for asymmetric induction.
Figure 1.

(a) Peptide catalysis summarized. (b) Catalytic residues targeting specific transformations.
The scope of peptide catalysis has expanded dramatically over the past 20 years. Peptides have now been implemented in myriad synthetically relevant transformations, including hydroxyl and amine group transfers,5 oxidations,11 reductions,12 and C–C bond forming reactions.13 In addition to asymmetric operations on small molecules, peptides have also proven useful in the site-selective modification of complex, polyfunctional structures.14 Our own pursuits have been assisted by the design and incorporation of unique catalytic residues, including proteinogenic, non-proteinogenic, and synthetic amino acids, within peptide sequences (Figure 1b). Although these catalytic residues can directly mediate the targeted reaction, the peptide backbone often contributes to rate acceleration and selectivity via stabilizing noncovalent interactions, such as H-bonds.15 Such multifunctionality is a hallmark of enzymatic catalysis.
In the course of our studies, opportunities arose that allowed us to employ oligopeptides to address unsolved problems in asymmetric catalysis, including the desymmetrization of meso and prochiral compounds and DKR reactions of prochiral atropisomers. Regarding the former, the ability to differentiate enantiotopic groups in symmetrical molecules has long been considered a great challenge, especially in cases where the pro-stereogenic element is remote from the reacting groups.16 Despite the practical advantages of catalytic remote desymmetrizations, highly selective examples remain uncommon. General methods to catalytically discriminate between rapidly interconverting atropisomers have also been elusive. The prevalence of axially chiral, atropisomeric scaffolds in pharmaceuticals,17 ligands,18 and organocatalysts19 has spurred the need for facile access to atropo-enantioenriched compounds. However, while resolutions and diastereoselective transformations of atropisomers have been known for decades,20,21 atroposelective catalysis represents a new frontier in asymmetric synthesis.22,23
This Account describes our efforts to develop peptide-based catalysts for remote desymmetrizations of diarylmethanes and atroposelective brominations of biaryl, non-biaryl, and hetero-biaryl atropisomers (Figure 2). While not obviously related, several unique aspects of peptide catalysis contributed to the eventual synergy of these projects. The story highlights the interplay of academic curiosity and industrial collaboration, which, in turn, spawned new questions at the heart of stereoselective catalysis. In each case, peptide catalysis was found to be an enabling strategy, providing highly enantioenriched compounds of demonstrated relevance to medicinal and pharmaceutical chemistry. The structural and mechanistic details leading to the development of selective catalysts are also discussed, as these themes prove central to ongoing work in our group and in the community.
Figure 2.

(a) Remote desymmetrization and (b) atroposelective bromination reactions.
2. Early Studies on the Desymmetrization of Polyols
Our entry into peptide-catalyzed desymmetrization was inspired by kinases, which are known to phosphorylate polyfunctional substrates with site selectivity in vivo. Histidine-dependent kinases mediate phosphorylation events using Lewis base catalysis, wherein an active site His residue transfers phosphate to the substrate through a reactive phosphorylimidazolium intermediate. Encouraged by our early work on peptide-catalyzed acylations,5 which capitalized on embedded π-methylhistidine (Pmh) residues to deliver acyl groups via an acylimidazolium ion, we sought to extrapolate to the site and enantioselective phosphorylation of myo-inositol derivatives (1, Figure 3).24 A biomimetic transformation of this type would streamline access to phosphorylated myo-inositol natural products.
Figure 3.

Enantiodivergent phosphorylation of a myo-inositol derivative.
We discovered pentapeptide 3 from a small catalyst library synthesized using a random-number generator. Remarkably, 3 provided d-myo-inositol-1-phosphate derivative 2 in 65% yield and >98% ee, presumably via group transfer from the phosphorylated catalyst (Figure 3).24,25 Expansion of the library led to the identification of peptide 4 that delivered ent-2 in 56% yield and >98% ee under identical conditions.26 The absolute configuration of the catalytically active Pmh residue is the same in both 3 and 4, suggesting that the observed enantiodivergence is a function of their different secondary structures. While catalyst 3 possesses no readily identifiable structural motif, 4 is biased toward a β-turn conformation by virtue of its central Pro-Cle sequence and its internal H-bonding network (Cle = cycloleucine).9,10 Additionally, both peptides accelerated the reaction greatly over simple achiral catalysts, consistent with rate enhancement through H-bonding interactions. The discovery of these enantiodivergent catalysts enabled direct and efficient syntheses of d-myo-inositol-1-phosphate and its enantiomer following global deprotection of 2 and ent-2, respectively, facilitating biological study of these agents.27
Building upon this strategy, we later developed a peptide-based catalyst for the desymmetrization of flexible glycerol-type substrates (Figure 4). High throughput screening revealed Pmh-containing peptide 8 that catalyzes the conversion of glycerol 5 to mono(acetate) 6 with excellent enantioselectivity, albeit with significant secondary KR in forming bis(acetate) 7.28 The catalyst was proposed to selectively transfer an acetyl group from the acylimidazolium intermediate to the gauche–gauche conformer of 5 through multipoint association. These results are comparable to state-of-the-art biocatalytic methods for related transformations.29
Figure 4.
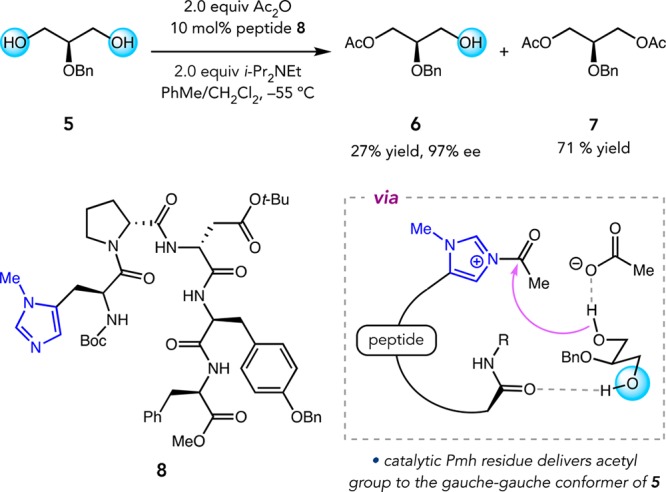
Desymmetrization of glycerols.
3. Remote Desymmetrization of Diarylmethane Bis(phenol)s
The inositol and glycerol desymmetrizations demonstrated that peptide-based catalysts can differentiate enantiotopic sites in close proximity. Possibly as a result, a collaboration with Merck Research Laboratories surfaced that introduced remote desymmetrizations, wherein the reactive functional groups are many bonds removed from one another and from the pro-stereogenic element. A long-standing challenge, remote asymmetric induction is often viewed as the purview of enzymes, given their macromolecular dimensions.16 However, oligopeptides seemed well suited for this task, given their tunable lengths and structures.
We thus undertook the remote desymmetrization of bis(phenol) 9, wherein the enantiotopic hydroxyl groups are separated from the prochiral center by 5.7 Å and from one another by ∼1 nm (Figure 5a). Researchers at Merck had explored lipases to catalyze the desymmetrizing hydrolysis of bis(acetate) 11. In a screen of >450 enzymes, synthetically useful enantioselectivities were only achieved with substantial contribution from the secondary KR of mono(acetate) 10, resulting in low yields. We therefore set out to develop a Pmh-containing peptide that might do better via acylation of 9.30,31
Figure 5.

(a) Peptide-catalyzed remote desymmetrization of a bis(phenol). (b) Methine substituent effects.
Our catalyst library was designed around two structural elements. First, we estimated that hexapeptides would be sufficiently long to engage both phenols of 9 if a bifunctional mechanism were operable. Second, we sought to mirror the structural domains of the substrate: the terminal residues would have aromatic side chains, while the central ones would be aliphatic. After five rounds of screening, we arrived at hexapeptide 12, which delivered 10 in 68% yield and 72% ee (Figure 5a). Sequence truncation studies indicated that the two C-terminal residues were unnecessary for enantioselectivity, so 12 was shortened to a tetramer retaining the critical N-terminal residues. The C-terminal cap also had an effect on selectivity, leading to the (S,S)-N-tosyl-diamine moiety in the optimal scaffold. Tetrapeptide 13 provided mono(acetate) 10 in 80% isolated yield and 95% ee. Although 20% of bis(acetate) 11 was also formed, secondary KR of 10 only minimally contributed to the observed ee (krel = 1.4). NMR studies showed that the degeneracy in the aryl resonances of 9 is lost upon catalyst–substrate association, suggesting that 13 breaks the bis(phenol) symmetry via noncovalent interactions.
While investigating the scope of this reaction, we found that yield and enantioselectivity diminished steadily with decreasing steric demand of the prochiral substituent (Figure 5b). In collaboration with the Sigman Group, we identified LFERs correlating the enantioselectivity with various steric parameters, including Sternhell interference energies.32 Steep negative slopes suggested sensitivity to the size of the substituent. One interpretation is that the steric profile of the substituent influences the propeller-like twisting of 9, impacting attendant interactions with 13. The Sigman Group was later able to correlate the enantioselectivity trends with computed arene vibrational frequencies, which are sensitive and directional readouts of both steric and electronic substituent effects.33
We surmised that the catalytic Pmh residue of peptide 13 could mediate the desymmetrization of 9 via two limiting mechanisms: (1) Lewis base activation of acetic anhydride or (2) Brønsted base activation of the bis(phenol) (Figure 6). While acylations of aliphatic alcohols (e.g., 1 and 5) are more likely to proceed through the former mechanism, both possibilities are accessible to 9 due to the increased acidity of phenols. Although the precise activation mode remains undetermined, these mechanistic questions motivated us to consider other reactivity manifolds amenable to peptide-mediated Brønsted base catalysis.
Figure 6.

Mechanistic questions in the bis(phenol) desymmetrization.
We ultimately arrived at electrophilic aromatic halogenation, which we hypothesized could be accelerated by Brønsted base activation of a phenol, rendering it more phenolate-like. Furthermore, Lewis basic functional groups were known to catalyze halenium ion transfer reactions with substantial rate acceleration.34 Taken together, we thought peptides might catalyze SEAr reactions through a mechanism involving Brønsted basic phenol activation and backbone amide-mediated halenium transfer. We initially tested this hypothesis in the bromination of 9 (Figure 7a).35 Upon exposure to NBS, peptide 13 furnished monobromide 14 in 35% yield and 28% ee; the remainder of the mass balance consisted of polybromides. Because the Pmh residue was also brominated under these conditions, we pursued a new catalytic residue, tertiary amine-containing β-dimethylaminoalanine (Dmaa), which was expected to be more Brønsted basic, yet less prone to irreversible side reactivity. Fortunately, peptide 15 also provided 14 with appreciable selectivity (Figure 7b). Encouraged by these preliminary results, we turned our attention toward new applications of this catalytic activation scheme.
Figure 7.

Desymmetrizing bis(phenol) bromination catalyzed by (a) Pmh-containing and (b) Dmaa-containing peptides.
4. Atroposelective Bromination Reactions
Following our studies of bis(phenol) desymmetrizations, we sought a mode of enantioselective SEAr that might be of broad synthetic utility. The growing interest in atropisomeric biaryls in pharmaceuticals and natural products,17,20 as well as in chiral ligands and organocatalysts,18,19 encouraged us to develop peptide-catalyzed atroposelective brominations. We hypothesized that if a biaryl phenol (e.g., 16) rapidly racemizes in situ, preferential bromination of one enantiomer by a peptide-based catalyst could establish a Curtin–Hammett scenario (Figure 8). This type of DKR would require a low substrate enantiomerization barrier, such that racemization is rapid relative to bromination, and that the product is configurationally stable about the chiral axis. Rotational barriers of mono-ortho-substituted biaryls are known to be quite low, ensuring rapid racemization in substrates like 16. We initially proposed a tribromination of 16 to avoid complicated product mixtures. This strategy also proved fruitful regarding the latter point, as DFT calculations suggested that two ortho-bromides would be required to configurationally lock the products. Interestingly, the asymmetry of tribromide 17 stems from a meta-hydroxyl group distal from the chiral axis, reminiscent of the central challenge in our Merck collaboration.
Figure 8.

Biaryl DKR via atroposelective bromination.
At the outset of this study, the field of atroposelective catalysis was quite young, and most reported examples involved asymmetric cross-coupling strategies.20 However, Bringmann and co-workers pioneered a different DKR-based approach—the atroposelective, nucleophilic ring-opening of strained biaryl lactones (Figure 9).20 These groundbreaking studies, in addition to a report by Murai involving C–H functionalization,36 informed many aspects of our proposed atroposelective bromination.
Figure 9.

DKR of Bringmann lactones.
As our catalyst design centered on Dmaa, we chose to incorporate a carboxylic acid directing group within the biaryl substrate to enforce intermolecular association between 16a and the peptide through salt-bridging. Catalyst optimization led to 18, which furnished tribromide 17a in 80% isolated yield and 97:3 er (Figure 10a).37 Tripeptide 18 was effective over a broad scope that included sterically and electronically diverse biaryls, and even hetero-biaryls. Control experiments showed that the reaction is sluggish and low-yielding both in the absence of catalyst and using substoichiometric Hünig’s base to mimic the Dmaa residue. However, catalytic (±)-Boc-Val-NMe2, the isolated C-terminal residue of 18, boosted the yield to 91%. These mechanistic queries suggest that backbone amides of 18 are essential for catalysis (Figure 10b). In pursuit of further insights, we partnered with the Johnson Group to investigate the noncovalent association of 18 with 16a using cryogenic gas-phase infrared spectroscopy.38 The results were consistent with multidentate docking model 18•16a, wherein the Dmaa side-chain engages the carboxylic acid while upstream backbone amides interact with the phenol, holding 16a in the (aR)-configuration (Figure 10c). Though difficult to study directly, it is possible that bromenium ion is delivered to 16a by nearby amide, perhaps that of the C-terminal Val residue.
Figure 10.

(a) Peptide-catalyzed, atroposelective biaryl bromination. (b) Mechanism-driven experiments. (c) Proposed model for stereoinduction.
One of the advantages of selective electrophilic aromatic bromination is the possibility for derivatization via cross-coupling. Since products 17 contain three sterically and electronically distinct bromides, we wondered if it might be possible to access highly functionalized scaffolds through regioselective cross-coupling sequences. Following optimization, we developed an “A–B−C coupling” method that enables the synthesis of pentaphenyls and other derivatives 20 via sequential, Pd-catalyzed cross-coupling of methyl esters 19 (Figure 11).39
Figure 11.
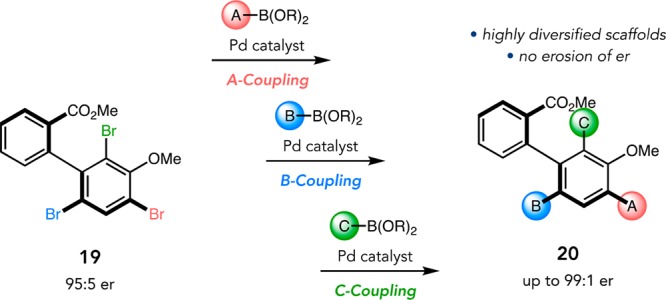
A–B–C cross-coupling of biaryl tribromides.
Having established a peptide-catalyzed atroposelective methodology, we sought to expand its scope to include non-biaryl atropisomers.21 We were particularly attracted to the tertiary benzamide-type scaffolds that had been studied extensively by Clayden and others and which were frequently identified in biologically active compounds.17 We expected substrates like 21 (Figure 12) to present new challenges for the peptide-catalyzed bromination protocol. Since benzamides might catalyze their own bromination, we opted to employ the bulky N,N-diisopropyl amide motif to both disfavor the autocatalytic reaction and achieve higher enantiomerization barriers. Recognition between 21 and the Dmaa-containing peptide was proposed to be nucleated by H-bonding between the phenol moiety and the Dmaa side-chain, which we hypothesized could also activate 21 toward SEAr.
Figure 12.

(a) Peptide-catalyzed, atroposelective benzamide bromination. (b) Low-conversion LC/MS studies provide mechanistic insight. (c) Proposed model for stereoinduction. (d) Regioselective tribromide functionalizations.
We examined a peptide library based on the type II′ β-hairpin architecture that we had previously identified in highly selective catalysts for various transformations.5,6 Tetrapeptides can be biased toward this secondary structure by strategic incorporation of a central d-Pro-Xaa sequence, where Xaa is either an l-amino acid or an α,α-disubstituted residue.10,40 Sequence optimization led to 23, which delivered (aS)-tribromide 22 in 89% yield and 94:6 er (Figure 12a).41 Catalyst 23 also accommodated substitution at the 4- and 5-positions of 21, but pre-installation of ortho-substituents elicited more interesting effects. The 6-methyl substrate was selectively brominated by 23, while the 6-bromo substrate provided nearly racemic product. These results are consistent with a barrier effect, wherein the more electronically demanding bromide may restrict rotation, preventing effective DKR. At the 2-position, both methyl- and bromide-substitution furnished low er values. Using LC/MS to analyze low-conversion reaction mixtures, we determined that 23 overturns the inherent regioselectivity of the reaction by favoring monobromination at the 2-position (Figure 12b). Therefore, 2-substituted benzamides may be poor substrates because they block the enantiodetermining monobromination event. NMR studies of the peptide–substrate complex were consistent with multidentate association between 23 and 21, likely involving H-bonding (e.g., 23·21, Figure 12c). Many other transformations catalyzed by this class of peptides have revealed similar evidence of multifunctional catalysis, prompting us to consider whether these well-defined β-turns might be considered “privileged” chiral scaffolds.42
The products of benzamide bromination also proved amenable to functionalization with no loss of configurational integrity (Figure 12d).43 Regioselective Suzuki–Miyaura cross-coupling of (aS)-22 delivers 4-arylated benzamides 24 in high yields and ers. Other Pd-catalyzed cross-coupling methods, including aminations and carboxymethylations, also selectively target this position. Diversification of 24 via ortho-lithiation/electrophile trapping provides an array of derivatives 25 with no erosion of er.
We next targeted benzamides such as 26 that possess two stereogenic axes, one defined by the chiral benzamide axis and the other implicit in differentially N,N-disubstituted amides (Figure 13). We initially considered the possibility of optimizing four peptide catalysts that each selectively furnishes one of the four possible product diastereomers 27. This would be predicated on the assumption that both axes are configurationally locked following bromination, which did not turn out to be the case. Instead, the 23-catalyzed bromination of 26 led to a kinetically controlled mixture of diastereomers, the ratios of which fluctuated over time due to slow amide isomerization.44 During equilibration, the kinetically favored cis-27 redistributes its enantiomeric composition to the thermodynamically more stable trans-27, resulting in a spontaneous increase in the er of the latter, albeit with compensatory enantioerosion of the cis-isomer. This intriguing phenomenon represents a rare example of spontaneous enantioenrichment in homogeneous solution, and its observation was only possible due to the ability of peptide 23 to differentially process the amide isomers of 26.
Figure 13.

Spontaneous chirality transfer in a two-axis system.
Motivated by the continued identification of hetero-biaryl motifs in pharmaceutically relevant compounds, we became interested in further expanding the atroposelective bromination methodology to include hetero-biaryl substrates.21 Many such compounds are known to exhibit atropisomerism, though few catalytic methods had been reported for their asymmetric preparation. We were drawn to the 3-arylquinazolin-4(3H)-one scaffold (e.g., 28), which occurs in countless drug compounds spanning a wide range of biochemical functions (Figure 14a).45 Quinazolinones 28 also presented an interesting challenge, as enantiomerization barriers were expected to be higher than similar biaryls due to the shorter C–N bond defining the chiral axis. Indeed, DFT computations predicted a 19 kcal/mol barrier for 28a, corresponding to a half-life of 7 s.46 Thus, racemization could be slow on the rapid bromination time scale, limiting the dynamic aspect of the transformation.
Figure 14.

(a) Peptide-catalyzed, atroposelective 3-arylquinazolinone bromination. (b) Tribromide functionalization.
We initially examined a library of 21 Dmaa-containing peptides biased toward type II′ β-hairpin secondary structures. Peptide 30, which possesses a critical 1-aminocyclopropane carboxamide (Acpc) residue at the (i + 2)-position, was found to be a highly selective catalyst, delivering tribromide 29a in 86% yield and 97:3 er (Figure 14a).46 Slow addition of NBS was crucial to achieving this level of selectivity, as it possibly allows more complete reracemization of 28a and thereby enables a more efficient DKR. Under these conditions, 30 addressed a broad scope of quinazolinones. Notable exceptions include 2-CF3-containing 28b and ortho′-fluoride 28c. While the former was likely unselective due to ineffective catalyst–substrate interactions, the latter had too high of an enantiomerization barrier (ΔG⧧ = 26.7 kcal/mol) to undergo DKR. Accordingly, 30 delivered tribromide 29c in 93:7 er at low conversion, demonstrating that 28c is an effective substrate for classical KR. Analogous to the benzamide bromination, low conversion LC/MS studies showed that 30 overturns the inherent para-selectivity by promoting stereodetermining monobromination at the ortho-position of 28a. Additionally, NMR studies of equimolar 28a and 30 revealed chemical shift changes, intramolecular NOEs in 30, and even intermolecular NOEs consistent with catalyst–substrate complex 30·28a. This model predicts that the observed (aS)-configuration of 29a derives from a multifunctional binding and activation mechanism, wherein the site of enantiodetermining bromination is oriented nearby the peptide backbone. It remains a possibility that 30 delivers bromenium ion to 28a via backbone amides,34 though evidence of this mechanism remains elusive.
Tribromides 29 were amenable to regioselective derivatization, providing access to a variety of functionalized, drug-like compounds with no erosion of optical purity (Figure 14b).46 Buchwald–Hartwig amination of (aS)-29a selectively targets the para-bromide, furnishing amines 31 in good yields. Additionally, regioselective hydrogenolysis of 29a delivers configurationally stable ortho-monobromide 32, which was subsequently subjected to Suzuki–Miyaura cross-coupling to biarylquinazolinones 33.
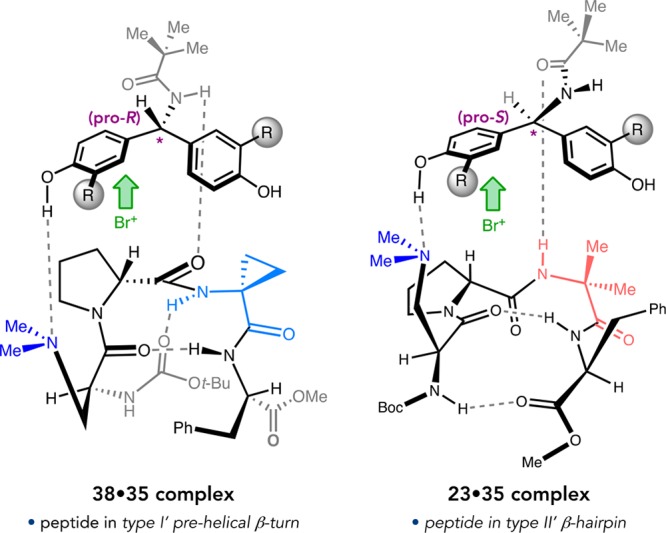
The quinazolinone bromination project also provided a platform from which we launched a thorough structural investigation of β-turn-biased, peptidic catalysts. These studies were initially inspired by a curious finding: three distinct conformations of peptide 30 were observed by X-ray crystallography (Figure 15a).47 The first two, 30a and 30b, were observed within the same asymmetric unit and exhibit the expected type II′ β-hairpin structure based on the ϕ and ψ dihedrals of the loop residues and the intramolecular H-bonding network.9 A polymorphic crystal structure revealed 30c, which adopts an overall prehelical conformation defined by a central type I′ β-turn. The realization that these turn-biased tetrapeptides can nucleate such globally different secondary structures raised new questions about this class of catalysts.
Figure 15.

(a) Crystallographic conformers of peptide 30. (b) Homologous i + 2 series of catalysts for the bromination of 28a. (c) Correlation between crystallographic τ(i + 2) and enantioselectivity.
In a structural analysis of 35 peptides by X-ray crystallography and solution NMR, we observed more structural heterogeneity than was expected from the catalyst design principles we often employ.48 In a particularly striking example, we obtained X-ray structures for seven (i + 2)-homologues of peptide 34. Only Aib-containing 34g was found to adopt the expected type II′ β-hairpin conformation in the solid state, while 34a–f exhibited prehelical type I′ turns analogous to 30c (Figure 15b). In solution, all of the peptides showed NOE signatures satisfying multiple secondary structures, suggesting that these peptides are conformationally dynamic under catalytically relevant conditions.49
We also identified a correlation between the crystallographic main-chain angle (τ) of the i + 2 residue and the enantioselectivity observed in the bromination of quinazolinone 28a (Figure 15c), with larger values of τ(i + 2) delivering higher ers.48 Wide τ-angles are often associated with structural flexibility, suggesting that catalyst dynamics could play an important role in the reaction. Indeed, NMR titration studies show that 30 undergoes a conformational change from a prehelical type I′ turn to a type II′ β-hairpin in the presence of 28a, reminiscent of conformational selection.50 While the same overall change occurs for poorly selective substrate 28b, the degree of structural homogeneity in the catalyst–substrate complex is notably less. These data are consistent with a model wherein effective substrates trigger more significant changes toward a catalytically active conformation, as in 30·28a. MD simulations performed with the Jorgensen Group were consistent with these observations, providing atomic-level resolution on the associated conformational changes.51 Moreover, in another collaborative study with the Sigman Group, peptides of types II′ and I′ were subjected to multidimensional parametrization.52 Features of both limiting conformations correlated well with ΔΔG⧧ (Figure 16), suggesting that the observed enantioselectivity might emerge from an ensemble of transition states in which the catalyst adopts multiple conformations.
Figure 16.

Multidimensional parameterization of the limiting catalyst conformations in the bromination of quinazolinone 28a.
5. Remote Desymmetrization of Diarylmethylamido-Bis(phenol)s
We eventually returned to the original impetus for our study of enantioselective bromination, the desymmetrization of bis(phenol)s. As diarylmethane scaffolds continued to accumulate in the medicinal chemistry literature,53 a variety of enantioselective approaches had been reported,54 including two remarkable examples of desymmetrizing C–H functionalizations reported by Shi/Hartwig and Wang/Yu.55,56 In a complementary way, we chose to revisit desymmetrizing bromination in the context of diarylmethylamido-bis(phenol)s 35 utilizing a Dmaa-containing peptide to remotely differentiate enantiotopic phenols (Figure 17). We proposed that enantio-discrimination could either occur analogously to the mechanism proposed for bis(phenol) 9 (Figure 6) or by taking advantage of a different intermolecular H-bond between the catalyst and the prochiral amido group. Polybromination, which could erode yields with complicated product mixtures, was a concern for this system, so we blocked one of the phenol ortho-positions with an electronically deactivating carbonyl group, which might also slow bromination to permit more robust enantioinduction.
Figure 17.

Desymmetrizing bromination of diarylmethylamido-bis(phenol)s.
Following optimization, we identified Acpc-containing peptide 38 that effectively differentiates the enantiotopic arenes of pivalamides 35, providing monobromides 36 in 55–69% yield and 83:17–97:3 er (Figure 18a).57 Substrates bearing different ortho-functionality were also effectively addressed by 38. In each case, overbromination to the symmetrical dibromide 37 (31–44% yield) also occurs with minimal contribution from secondary KR (krel = 3.2), indicating that the primary mode of asymmetric induction is indeed enantiotopic arene differentiation.
Figure 18.

(a) Peptide-catalyzed, desymmetrizing bromination of diarylmethylamido-bis(phenol)s. (b) Correlation between crystallographic τ(i + 2) and enantioselectivity.
During catalyst optimization, we noticed that 2-aminoisobutyramide (Aib)-containing peptide 23 provided similar yields of ent-36, the opposite enantiomer favored by 38, with modest enantioselectivity (Figure 18a).57 The degree of enantiodivergence was found to be substrate-dependent, with ers ranging from 49:51 to 26:74 as a result of a single point mutation at an achiral residue within an otherwise homologous sequence. Peptides 38 and 23 only differ in mass by an equivalent of H2, and yet they deliver opposite enantiomers of 36. This fascinating phenomenon inspired us to consider possible conformational effects on enantioselectivity. We eventually uncovered an i + 2 τ-angle correlation even more pronounced than that of the quinazolinone example (Figure 18b).48 Again, higher selectivities were observed for wider τ(i + 2) values. This possibly suggests that peptides 38 and 23 interact with 35 from different conformations, as prehelical type I′ β-turns are significantly more accessible when τ(i + 2) is wide. The observed enantiodivergence might be rationalized by a model wherein 35 preferentially interacts with prehelical 38 and hairpin 23 using either the N–H or C=O of the prochiral pivalamide group, respectively (Figure 19).
Figure 19.
Possible enantiodivergent binding modes in the bromination of 35.
Thus, our adventures in atroposelective bromination and peptide structure ultimately enabled us to address the unmet challenge that initially inspired those studies, the desymmetrizing bromination of a bis(phenol). Conceptually related, Lewis and co-workers recently reported a biocatalytic system, wherein an engineered halogenase mediates the desymmetrizing chlorination of a bis(aniline) with excellent enantioselectivity.58 Our studies demonstrate that oligopeptides can be similarly effective for remote desymmetrizations via designed noncovalent interactions.
6. Asymmetric Cross-Coupling with Peptide-Based Ligands
The catalytic bromination of pharmacologically interesting compounds begged the question of whether selective cross-coupling strategies might be feasible. This challenge first presented to us during our studies on regioselective functionalizations of atropisomeric tribromides.39,43,46 It was further brought into focus when we developed site-selective halogenations of glycopeptide antibiotics, such as vancomycin and teicoplanin, which provided templates to pursue the effectively unknown area of site-selective cross-coupling.59 We thus wondered if peptide-ligated transition metals could provide a handle for site-selective cross-couplings. As a prelude to these efforts, we pursued enantioselective cross-coupling as a testing-ground for a new peptide-based ligand architecture.
Given the ongoing interest in diarylmethane-type scaffolds,53 we devised symmetrical dibromide 39 to investigate enantioselective cross-couplings via distal stereocontrol (Figure 20). The incorporation of trifluoroacetamido groups adjacent to the bromides was inspired by an asymmetric Hurtley reaction reported by Ma,60 wherein ortho-trifluoroacetamides served to direct oxidative addition of a Cu(I)-complex. Moreover, the effectiveness of the simple l-trans-Hyp ligand in Ma’s system suggested that a peptidic ligand might also work well with Cu(I) to achieve asymmetric cross-couplings. We proposed that a tetraalkylguanidine-containing residue could be a synthetically tractable ligand motif to install within a peptide that both engages in N,O-chelation of Cu(I) and is sufficiently electron-rich to promote oxidative addition. By incorporating β-linked, tetramethylguanidinylated aspartic acid (TMG-Asp) as the N-terminal residue, we were able to design peptide-based ligands for asymmetric C–C,61 C–O,62 and C–N63 cross-couplings of 39, delivering malonates 40, ethers 41, and benzimidazoles 42 with excellent enantioselectivities (Figure 21).
Figure 20.

Asymmetric cross-coupling of diarylmethanes.
Figure 21.

Desymmetrizing (a) C–C, (b) C–O, and (c) C–N cross-couplings with peptidic ligands.
The initial desymmetrizing malonate coupling was developed in collaboration with Pfizer and Boehringer–Ingelheim (Figure 21a).61 Tripeptide 43 was identified as the lead ligand following structure–function optimization. Notably, a C-terminal carboxylate was found to play an important role in asymmetric induction. Under the optimal conditions, ligand 43 provides up to 76% yield and 93:7 er of mono(malonate)s 40, with t-butyl substitution on the prochiral C atom delivering the best results.32 Control and mechanistic experiments, including KR studies on racemic analogs of 39, such as (±)-46, led us to propose that remote stereocontrol is achieved via a distal cation−π interaction (i.e., 43·39, Figure 22).
Figure 22.

KRs provide mechanistic detail in the malonate cross-coupling.
Tetrapeptide 44 was optimized for the C–O cross-coupling of 39, furnishing mono(ether)s 41 in up to 71% yield and 99:1 er (Figure 21b).62 The reaction worked exceptionally well for a wide scope of alcohol nucleophiles. Good yields and very high selectivities were observed for hindered phenols, which are difficult substrates in standard cross-couplings. Compounds possessing both phenol and amine functionality selectively couple at the phenol with ∼8:1 selectivity and 98:2 er.
Similarly, ligand 45 enabled the highly enantioselective C–N cross-coupling of 39 with various amine nucleophiles (Figure 21c).63In situ acid-mediated cyclodehydration converted the amine-coupled products into benzimidazoles 42 to avoid complicated product mixtures in cases where small quantities of benzimidazole formed during cross-coupling. Simple aniline and alkylamine nucleophiles delivered mono(benzimidazole)s 42 in up to 71% yield and 98:2 er. Hindered amines, such as naphthylamines and 2,6-disubstituted anilines, did not cyclodehydrate spontaneously and gave similar yields of mono(amine)s with up to >99:1 er. Our experience with atropisomerism led us to hypothesize that cyclodehydration of these amines (e.g., 48a) would give rise to diastereomers upon cyclodehydration due to the formation of a configurationally stable chiral axis within a point-chiral framework (Figure 23). Indeed, treatment of 48a with a CPA catalyst cleanly delivered benzimidazole 42a in 90% yield, 17:1 dr, and >99:1 er. We were also able to synthesize all four diastereomers of 42a with complete catalyst control.
Figure 23.

Diastereo- and enantioselective cyclodehydration.
7. Conclusions
The chemistry detailed herein has expanded the purview of peptide-based catalysts in an intriguing way. Initially stimulated by fundamental questions about enzymes but challenged further by practical problems posed by industrial colleagues, our research program has shifted in focus to what might be called “outer sphere” issues. Our forays into remote asymmetric induction and atroposelective halogenation have unveiled a golden opportunity for oligopeptide catalysis. These processes share a multivalency at their mechanistic core with respect to catalyst–substrate interactions, which appear to operate decisively over great distances from the loci of bond formation. Outer sphere interactions are also central to the selective functionalization of complex systems, including natural products and proteins. Catalyst-controlled site-selectivity seems a field poised to break out of its infancy. Perhaps peptide-based catalysts will have a special role to play in this pursuit. If so, this might prove to be an additional aspect of their biomimetic capacity, as outer sphere interactions are often central to the selectivity of enzymes.64 The crafting of the proper outer sphere catalyst environment is currently a major focus of our research.
Acknowledgments
We are grateful to the past and present members of our research group for their contributions to this work. We thank the NIGMS of the NIH (GM-068649 and GM-096403) for financial support. We are also grateful to all of our collaborators, including Prof. Matthew Sigman (University of Utah), and to the NIH for support of recent collaborative efforts (GM-121383). We also thank Dr. Karl Hansen (Boston Pharmaceuticals), who initiated our stimulating collaboration with Merck around the desymmetrization of diarylmethane bis(phenol)s.
Glossary
Abbreviations
- Acbc
1-aminocyclobutane-1-carboxylic acid
- Achc
1-aminocyclohexane-1-carboxylic acid
- Acpc
1-aminocyclopropane-1-carboxylic acid
- Aib
2-aminoisobutyric acid
- Aic
2-aminoindane-2-carboxylic acid
- Cle
cycloleucine
- CPA
chiral phosphoric acid
- DBDMH
1,3-dibromo-5,5-dimethylhydantoin
- DFT
density functional theory
- DKR
dynamic kinetic resolution
- Dmaa
β-dimethylaminoalanine
- Gly
glycine
- H-bond
hydrogen bond
- His
histidine
- Hyp
hydroxyproline
- KR
kinetic resolution
- LFER
linear free energy relationship
- MD
molecular dynamics
- NBP
N-bromopthalimide
- NBS
N-bromosuccinimide
- Pmh
π-methylhistidine
- Pro
proline
- TMG-Asp
tetramethylguanidinylated aspartic acid
- Val
valine
Biographies
Anthony J. Metrano was raised in North Attleborough, MA. He received an A.B. in chemistry from the College of the Holy Cross (2011) and a Ph.D. in organic chemistry from Yale University (2017), where he was an NSF Graduate Research Fellow under Professor Scott J. Miller. He is currently an NIH Postdoctoral Fellow in the Knowles Group at Princeton University.
Scott J. Miller is from Buffalo, NY. He received his B.A. (1989), M.A. (1989), and Ph.D. (1994) from Harvard University, working with Professor David Evans as an NSF Predoctoral Fellow. He was an NSF Postdoctoral Fellow at the California Institute of Technology with Robert Grubbs before joining the faculty at Boston College in 1996. Since 2006, he has been on the faculty at Yale University.
Author Present Address
A.J.M.: Department of Chemistry, Princeton University, Princeton, NJ 08544, United States.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Chemical Biology of Peptides”.
References
- Davidi D.; Longo L. M.; Jabłońska J.; Milo R.; Tawfik D. S. A Bird’s-Eye View of Enzyme Evolution: Chemical, Physicochemical, and Physiological Considerations. Chem. Rev. 2018, 118, 8786–8797. 10.1021/acs.chemrev.8b00039. [DOI] [PubMed] [Google Scholar]
- Wolfenden R.; Snider M. J. The Depth of Chemical Time and the Power of Enzymes as Catalysts. Acc. Chem. Res. 2001, 34, 938–945. 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- Kohen A. Role of Dynamics in Enzyme Catalysis: Substantial Versus Semantic Controversies. Acc. Chem. Res. 2015, 48, 466–473. 10.1021/ar500322s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausen R. S.; Kennedy C. R.; Hyde A. M.; Jacobsen E. N. Chiral Thioureas Promote Enantioselective Pictet-Spengler Cyclization by Stabilizing Every Intermediate and Transition State in the Carboxylic Acid-Catalyzed Reaction. J. Am. Chem. Soc. 2017, 139, 12299–12309. 10.1021/jacs.7b06811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S. J. Search of Peptide-Based Catalysts for Asymmetric Organic Synthesis. Acc. Chem. Res. 2004, 37, 601–610. 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]
- Davie E. A. C.; Mennen S. M.; Xu Y.; Miller S. J. Asymmetric Catalysis Mediated by Synthetic Peptides. Chem. Rev. 2007, 107, 5759–5812. 10.1021/cr068377w. [DOI] [PubMed] [Google Scholar]
- Wennemers H. Asymmetric Catalysis with Peptides. Chem. Commun. 2011, 47, 12036–12041. 10.1039/c1cc15237h. [DOI] [PubMed] [Google Scholar]
- Akagawa K.; Kudo K. Development of Selective Peptide Catalysts with Secondary Structural Frameworks. Acc. Chem. Res. 2017, 50, 2429–2439. 10.1021/acs.accounts.7b00211. [DOI] [PubMed] [Google Scholar]
- Wilmot C. M.; Thornton J. M. Analysis and Prediction of the Different Types of β-Turn in Proteins. J. Mol. Biol. 1988, 203, 221–232. 10.1016/0022-2836(88)90103-9. [DOI] [PubMed] [Google Scholar]
- Haque T. S.; Little J. C.; Gellman S. H. Stereochemical Requirements for β–Hairpin Formation: Model Studies with Four-Residue Peptides and Depsipeptides. J. Am. Chem. Soc. 1996, 118, 6975–6985. 10.1021/ja960429j. [DOI] [Google Scholar]
- Alford J. S.; Abascal N. C.; Shugrue C. R.; Colvin S. M.; Romney D. K.; Miller S. J. Aspartyl Oxidation Catalysts That Dial in Functional Group Selectivity, Along with Regio- and Stereoselectivity. ACS Cent. Sci. 2016, 2, 733–739. 10.1021/acscentsci.6b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugrue C. R.; Miller S. J. Phosphothreonine as a Catalytic Residue in Peptide-Mediated Asymmetric Transfer Hydrogenations of 8-Aminoquinolines. Angew. Chem. 2015, 127, 11325–11328. 10.1002/ange.201505898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbofana C. T.; Miller S. J. Diastereo- and Enantioselective Addition of Anilide-Functionalized Allenoates to N-Acylimines Catalyzed by a Pyridylalanine-Based Peptide. J. Am. Chem. Soc. 2014, 136, 3285–3292. 10.1021/ja412996f. [DOI] [PubMed] [Google Scholar]
- Shugrue C. R.; Miller S. J. Applications of Nonenzymatic Catalysts to the Alteration of Natural Products. Chem. Rev. 2017, 117, 11894–11951. 10.1021/acs.chemrev.7b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle A. G.; Jacobsen E. N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743. 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]
- García-Urdiales E.; Alfonso I.; Gotor V. Enantioselective Enzymatic Desymmetrizations in Organic Synthesis. Chem. Rev. 2005, 105, 313–354. 10.1021/cr040640a. [DOI] [PubMed] [Google Scholar]
- Clayden J.; Moran W. J.; Edwards P. J.; LaPlante S. R. The Challenge of Atropisomerism in Drug Discovery. Angew. Chem., Int. Ed. 2009, 48, 6398–6401. 10.1002/anie.200901719. [DOI] [PubMed] [Google Scholar]
- Tang W.; Zhang X. New Chiral Phosphorus Ligands for Enantioselective Hydrogenation. Chem. Rev. 2003, 103, 3029–3070. 10.1021/cr020049i. [DOI] [PubMed] [Google Scholar]
- Terada M. Chiral Phosphoric Acids as Versatile Catalysts for Enantioselective Transformations. Synthesis 2010, 2010, 1929–1982. 10.1055/s-0029-1218801. [DOI] [Google Scholar]
- Bringmann G.; Price Mortimer A. J.; Keller P. A.; Gresser M. J.; Garner J.; Breuning M. Atroposelective Synthesis of Axially Chiral Biaryl Compounds. Angew. Chem., Int. Ed. 2005, 44, 5384–5427. 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]
- Kumarasamy E.; Raghunathan R.; Sibi M. P.; Sivaguru J. Nonbiaryl and Heterobiaryl Atropisomers: Molecular Templates with Promise for Atropselective Chemical Transformations. Chem. Rev. 2015, 115, 11239–11300. 10.1021/acs.chemrev.5b00136. [DOI] [PubMed] [Google Scholar]
- Zilate B.; Castrogiovanni A.; Sparr C. Catalyst-Controlled Stereoselective Synthesis of Atropisomers. ACS Catal. 2018, 8, 2981–2988. 10.1021/acscatal.7b04337. [DOI] [Google Scholar]
- Wang Y.-B.; Tan B. Construction of Axially Chiral Compounds via Asymmetric Organocatalysis. Acc. Chem. Res. 2018, 51, 534–547. 10.1021/acs.accounts.7b00602. [DOI] [PubMed] [Google Scholar]
- Sculimbrene B. R.; Morgan A. J.; Miller S. J. Nonenzymatic Peptide-Based Catalytic Asymmetric Phosphorylation of Inositol Derivatives. Chem. Commun. 2003, 1781–1785. 10.1039/b304015c. [DOI] [PubMed] [Google Scholar]
- Sculimbrene B. R.; Miller S. J. Discovery of a Catalytic Asymmetric Phosphorylation Through Selection of a Minimal Kinase Mimic: A Concise Total Synthesis of d-myo-Inositol-1-Phosphate. J. Am. Chem. Soc. 2001, 123, 10125–10126. 10.1021/ja016779+. [DOI] [PubMed] [Google Scholar]
- Sculimbrene B. R.; Morgan A. J.; Miller S. J. Enantiodivergence in Small-Molecule Catalysis of Asymmetric Phosphorylation: Concise Total Syntheses of the Enantiomeric d-myo-Inositol-1-Phosphate and d-myo-Inositol-3-Phosphate. J. Am. Chem. Soc. 2002, 124, 11653–11656. 10.1021/ja027402m. [DOI] [PubMed] [Google Scholar]
- Wang Y. K.; Morgan A.; Stieglitz K.; Stec B.; Thompson B.; Miller S. J.; Roberts M. F. The Temperature Dependence of the Inositol Monophosphatase Km Correlates with Accumulation of d-myo-Inositol 1,1’-Phosphate in Archaeoglobus Fulgidus. Biochemistry 2006, 45, 3307–3314. 10.1021/bi052467y. [DOI] [PubMed] [Google Scholar]
- Lewis C. A.; Sculimbrene B. R.; Xu Y.; Miller S. J. Desymmetrization of Glycerol Derivatives with Peptide-Based Acylation Catalysts. Org. Lett. 2005, 7, 3021–3023. 10.1021/ol051061a. [DOI] [PubMed] [Google Scholar]
- Wang Y. F.; Wong C. H. Lipase-Catalyzed Irreversible Transesterification for Preparative Synthesis of Chiral Glycerol Derivatives. J. Org. Chem. 1988, 53, 3127–3129. 10.1021/jo00248a046. [DOI] [Google Scholar]
- Lewis C. A.; Chiu A.; Kubryk M.; Balsells J.; Pollard D.; Esser C. K.; Murry J.; Reamer R. A.; Hansen K. B.; Miller S. J. Remote Desymmetrization at Near-Nanometer Group Separation Catalyzed by a Miniaturized Enzyme Mimic. J. Am. Chem. Soc. 2006, 128, 16454–16455. 10.1021/ja067840j. [DOI] [PubMed] [Google Scholar]
- Lewis C. A.; Gustafson J. L.; Chiu A.; Balsells J.; Pollard D.; Murry J.; Reamer R. A.; Hansen K. B.; Miller S. J. A Case of Remote Asymmetric Induction in the Peptide-Catalyzed Desymmetrization of a Bis(Phenol). J. Am. Chem. Soc. 2008, 130, 16358–16365. 10.1021/ja807120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson J. L.; Sigman M. S.; Miller S. J. Linear Free-Energy Relationship Analysis of a Catalytic Desymmetrization Reaction of a Diarylmethane-Bis(Phenol). Org. Lett. 2010, 12, 2794–2797. 10.1021/ol100927m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo A.; Bess E. N.; Sigman M. S. Interrogating Selectivity in Catalysis Using Molecular Vibrations. Nature 2014, 507, 210–214. 10.1038/nature13019. [DOI] [PubMed] [Google Scholar]
- Denmark S. E.; Burk M. T. Lewis Base Catalysis of Bromo- and Iodolactonization, and Cycloetherification. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20655–20660. 10.1073/pnas.1005296107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson J. L.Application of Low Molecular Weight Peptides to Effect Enantioselective and Regioselective Functionalizations of Phenols. Ph.D. Dissertation, Yale University, New Haven, CT, 2011. [Google Scholar]
- Kakiuchi F.; Le Gendre P.; Yamada A.; Ohtaki H.; Murai S. Atropselective Alkylation of Biaryl Compounds by Means of Transition Metal-Catalyzed C–H/Olefin Coupling. Tetrahedron: Asymmetry 2000, 11, 2647–2651. 10.1016/S0957-4166(00)00244-5. [DOI] [Google Scholar]
- Gustafson J. L.; Lim D.; Miller S. J. Dynamic Kinetic Resolution of Biaryl Atropisomers via Peptide-Catalyzed Asymmetric Bromination. Science 2010, 328, 1251–1255. 10.1126/science.1188403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garand E.; Kamrath M. Z.; Jordan P. A.; Wolk A. B.; Leavitt C. M.; McCoy A. B.; Miller S. J.; Johnson M. A. Determination of Noncovalent Docking by Infrared Spectroscopy of Cold Gas-Phase Complexes. Science 2012, 335, 694–698. 10.1126/science.1214948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson J. L.; Lim D.; Barrett K. T.; Miller S. J. Synthesis of Atropisomerically Defined, Highly Substituted Biaryl Scaffolds Through Catalytic Enantioselective Bromination and Regioselective Cross-Coupling. Angew. Chem., Int. Ed. 2011, 50, 5125–5129. 10.1002/anie.201101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman J.; Shankaramma S. C.; Balaram P. Design of Folded Peptides. Chem. Rev. 2001, 101, 3131–3152. 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]
- Barrett K. T.; Miller S. J. Enantioselective Synthesis of Atropisomeric Benzamides Through Peptide-Catalyzed Bromination. J. Am. Chem. Soc. 2013, 135, 2963–2966. 10.1021/ja400082x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon T. P.; Jacobsen E. N. Privileged Chiral Catalysts. Science 2003, 299, 1691–1693. 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
- Barrett K. T.; Miller S. J. Regioselective Derivatizations of a Tribrominated Atropisomeric Benzamide Scaffold. Org. Lett. 2015, 17, 580–583. 10.1021/ol503593y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett K. T.; Metrano A. J.; Rablen P. R.; Miller S. J. Spontaneous Transfer of Chirality in an Atropisomerically Enriched Two-Axis System. Nature 2014, 509, 71–75. 10.1038/nature13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I.; Zaib S.; Batool S.; Abbas N.; Ashraf Z.; Iqbal J.; Saeed A. Quinazolines and Quinazolinones as Ubiquitous Structural Fragments in Medicinal Chemistry: An Update on the Development of Synthetic Methods and Pharmacological Diversification. Bioorg. Med. Chem. 2016, 24, 2361–2381. 10.1016/j.bmc.2016.03.031. [DOI] [PubMed] [Google Scholar]
- Diener M. E.; Metrano A. J.; Kusano S.; Miller S. J. Enantioselective Synthesis of 3-Arylquinazolin-4(3H)-Ones via Peptide-Catalyzed Atroposelective Bromination. J. Am. Chem. Soc. 2015, 137, 12369–12377. 10.1021/jacs.5b07726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metrano A. J.; Abascal N. C.; Mercado B. Q.; Paulson E. K.; Miller S. J. Structural Studies of Beta-Turn-Containing Peptide Catalysts for Atroposelective Quinazolinone Bromination. Chem. Commun. 2016, 52, 4816–4819. 10.1039/C6CC01428C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metrano A. J.; Abascal N. C.; Mercado B. Q.; Paulson E. K.; Hurtley A. E.; Miller S. J. Diversity of Secondary Structure in Catalytic Peptides with β-Turn-Biased Sequences. J. Am. Chem. Soc. 2017, 139, 492–516. 10.1021/jacs.6b11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigling C.; Kisunzu J. K.; Duschmalé J.; Häussinger D.; Wiesner M.; Ebert M.-O.; Wennemers H. Conformational Properties of a Peptidic Catalyst: Insights from NMR Spectroscopic Studies. J. Am. Chem. Soc. 2018, 140, 10829–10838. 10.1021/jacs.8b05459. [DOI] [PubMed] [Google Scholar]
- Bhabha G.; Biel J. T.; Fraser J. S. Keep on Moving: Discovering and Perturbing the Conformational Dynamics of Enzymes. Acc. Chem. Res. 2015, 48, 423–430. 10.1021/ar5003158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X. C.; Metrano A. J.; Robertson M. J.; Abascal N. C.; Tirado-Rives J.; Miller S. J.; Jorgensen W. L. Molecular Dynamics Simulations of a Conformationally Mobile Peptide-Based Catalyst for Atroposelective Bromination. ACS Catal. 2018, 8, 9968–9979. 10.1021/acscatal.8b03563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford J. M.; Stone E. A.; Metrano A. J.; Miller S. J.; Sigman M. S. Parameterization and Analysis of Peptide-Based Catalysts for the Atroposelective Bromination of 3-Arylquinazolin-4(3H)-Ones. J. Am. Chem. Soc. 2018, 140, 868–871. 10.1021/jacs.7b11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameen D.; Snape T. J. Chiral 1,1-Diaryl Compounds as Important Pharmacophores. MedChemComm 2013, 4, 893–907. 10.1039/c3md00088e. [DOI] [Google Scholar]
- Schmidt F.; Stemmler R. T.; Rudolph J.; Bolm C. Catalytic Asymmetric Approaches Towards Enantiomerically Enriched Diarylmethanols and Diarylmethylamines. Chem. Soc. Rev. 2006, 35, 454–470. 10.1039/b600091f. [DOI] [PubMed] [Google Scholar]
- Laforteza B. N.; Chan K. S. L.; Yu J.-Q. Enantioselective ortho-C–H Cross-Coupling of Diarylmethylamines with Organoborons. Angew. Chem. 2015, 127, 11295–11298. 10.1002/ange.201505204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B.; Zhou T.-G.; Li X.-W.; Shao X.-R.; Xu P.-L.; Wu W.-L.; Hartwig J. F.; Shi Z.-J. A Chiral Nitrogen Ligand for Enantioselective, Iridium-Catalyzed Silylation of Aromatic C–H Bonds. Angew. Chem. 2017, 129, 1112–1116. 10.1002/ange.201609939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtley A. E.; Stone E. A.; Metrano A. J.; Miller S. J. Desymmetrization of Diarylmethylamido Bis(Phenols) Through Peptide-Catalyzed Bromination: Enantiodivergence as a Consequence of a 2 Amu Alteration at an Achiral Residue Within the Catalyst. J. Org. Chem. 2017, 82, 11326–11336. 10.1021/acs.joc.7b02339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne J. T.; Butkovich P. H.; Gu Y.; Kunze K. N.; Park H. J.; Wang D.-S.; Lewis J. C. Enantioselective Desymmetrization of Methylenedianilines via Enzyme-Catalyzed Remote Halogenation. J. Am. Chem. Soc. 2018, 140, 546–549. 10.1021/jacs.7b09573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak T. P.; Miller S. J. Chemical Tailoring of Teicoplanin with Site-Selective Reactions. J. Am. Chem. Soc. 2013, 135, 8415–8422. 10.1021/ja4038998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X.; Chen Y.; Ma D. Enantioselective Arylation of 2-Methylacetoacetates Catalyzed by CuI/trans-4-Hydroxy-l-Proline at Low Reaction Temperatures. J. Am. Chem. Soc. 2006, 128, 16050–16051. 10.1021/ja066991j. [DOI] [PubMed] [Google Scholar]
- Kim B.; Chinn A. J.; Fandrick D. R.; Senanayake C. H.; Singer R. A.; Miller S. J. Distal Stereocontrol Using Guanidinylated Peptides as Multifunctional Ligands: Desymmetrization of Diarylmethanes via Ullman Cross-Coupling. J. Am. Chem. Soc. 2016, 138, 7939–7945. 10.1021/jacs.6b03444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinn A. J.; Kim B.; Kwon Y.; Miller S. J. Enantioselective Intermolecular C–O Bond Formation in the Desymmetrization of Diarylmethines Employing a Guanidinylated Peptide-Based Catalyst. J. Am. Chem. Soc. 2017, 139, 18107–18114. 10.1021/jacs.7b11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y.; Chinn A. J.; Kim B.; Miller S. J. Divergent Control of Point and Axial Stereogenicity: Catalytic Enantioselective C–N Bond-Forming Cross-Coupling and Catalyst-Controlled Atroposelective Cyclodehydration. Angew. Chem., Int. Ed. 2018, 57, 6251–6255. 10.1002/anie.201802963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeymer C.; Hilvert D. Directed Evolution of Protein Catalysts. Annu. Rev. Biochem. 2018, 87, 131–157. 10.1146/annurev-biochem-062917-012034. [DOI] [PubMed] [Google Scholar]