

Abstract
Preterm birth (PTB) affects approximately 1 in 10 pregnancies and contributes to approximately 50% of neonatal mortality. However, despite decades of research, little is understood about the etiology of PTB, likely due to the multifactorial nature of the disease. In this study, we examined preterm and term placentas, from unassisted conceptions and those conceived using in vitro fertilization (IVF). IVF increases the risk of PTB and causes epigenetic change in the placenta and fetus; therefore, we utilized these patients as a unique population with a potential common etiology. We investigated genome-wide DNA methylation in placentas from term IVF, preterm IVF, term control (unassisted conception) and preterm control pregnancies and discovered epigenetic dysregulation of multiple genes involved in cell migration, including members of the ADAMTS family, ADAMTS12 and ADAMTS16. These genes function in extracellular matrix regulation and tumor cell invasion, processes replicated by invasive trophoblasts (extravillous trophoblasts (EVTs)) during early placentation. Though expression was similar between term and preterm placentas, we found that both genes demonstrate high expression in first- and second-trimester placenta, specifically in EVTs and syncytiotrophoblasts. When we knocked down ADAMTS12 or ADAMTS16in vitro, there was poor EVT invasion and reduced matrix metalloproteinase activity, reinforcing their critical role in placentation. In conclusion, utilizing a population at high risk for PTB, we have identified a role for ADAMTS gene methylation in regulating early placentation and susceptibility to PTB.
Introduction
Preterm birth (PTB), defined by the World Health Organization as birth before 37 weeks of gestation, is a global epidemic and the leading cause of infant morbidity and mortality worldwide (1–3). Infants born premature can experience multiple complications including neurodevelopmental, cardiovascular, pulmonary and renal abnormalities (4). These adverse outcomes are not limited to early childhood and adolescence. PTB also has a significant impact on adult chronic cardiovascular, metabolic and renal diseases (5).
Despite significant research efforts, the rate of PTB has not changed significantly over the past three decades (6). The ability to identify interventions that prevent and reduce the risk of PTB rests upon a clear understanding of the pathophysiology of PTB. Identification of causal pathways contributing to PTB is complicated by the multifactorial nature of this complex disease. Infection, inflammation, activation of the maternal/fetal Hypothalamic-Pituitary-Adrenal (HPA) axis through stress, uterine distension and placental abnormalities have all been implicated in the etiology of PTB (7,8). While PTB was traditionally thought to originate from events occurring late in gestation, recent work suggests that abnormalities in embryo implantation and early placentation may contribute to PTB (8). Early in gestation, extravillous trophoblast (EVT) cells must migrate into the maternal decidua and a subset of these cells remodel the spiral arteries and replace the maternal endothelial cells. In a study examining the histology of the placental bed, patients with PTB had a higher frequency of inadequately remodeled spiral arteries than patients who had a spontaneous delivery at term (9). Additional studies support the hypothesis that, at least in a subset of patients, abnormalities in the placenta are present long before the onset of preterm contractions: infants delivered preterm have been found to have altered placental blood flow, with higher uterine artery resistance indices as early as 16 weeks gestation, when compared to infants that delivered full term (10). Studies have also demonstrated changes in serum corticotropin-releasing hormone levels, produced abundantly by the placenta and known to affect fetal–placental blood flow, during midgestation, prior to contractions, in pregnancies complicated by preterm delivery (11,12). These data support the hypothesis that defects early on in gestation, potentially during implantation and early placentation, may contribute to at least a subset of PTB. However, of all the potential contributors to PTB, the early events of implantation and placentation are the most difficult to study in vivo and a mechanistic link between implantation abnormalities and PTB has been challenging to establish.
One population with known increased risk of PTB is patients undergoing assisted reproductive technologies (ARTs) such as in vitro fertilization (IVF). While this was initially thought to be a consequence of the increased incidence of multiple gestations (13), we now know that even singleton pregnancies conceived by IVF are at increased risk for PTB (14–16). The etiology of this association is unknown but is thought to stem, at least in part, from procedures involved in IVF. While subfertility alone could be responsible for PTB (17), studies controlling for infertility continue to demonstrate higher rates of adverse pregnancy outcomes in pregnancies conceived by IVF (18,19). In addition, specific procedures used during IVF, such as extended embryo culture to the blastocyst stage, have been found to increase the risk of PTB, suggesting that early, pre-implantation exposures to the embryo can increase the risk of PTB (20).
The mechanism by which early embryonic exposures affect pregnancy and neonatal outcomes has been studied extensively over the past several decades, and epigenetic changes have been implicated in many of these phenotypes. Our group and others have demonstrated epigenetic changes in pregnancies resulting from ART. Specifically, we have found alterations in DNA methylation both in the placenta and in infants conceived by IVF (21–23). These observations lead to our hypothesis that early exposures, such as IVF, lead to epigenetic changes in the placenta that affect implantation and placentation resulting in an increased risk for PTB. In this study, we utilize well-characterized and controlled pregnancies following IVF and natural conception and examine placental DNA methylation in patients who deliver either term or preterm. Using this population of patients with a potential common etiology for their PTB, we identify novel candidate genes and pathways that may play a role in PTB. Additionally, we utilize in vitro studies to test the function of these genes, furthering our understanding of how early pregnancy exposures can affect early gestational events and lead to PTB.
Results
Sample characteristics
Placental samples were collected from 71 singleton pregnancies conceived via IVF or natural conception (control) following term and PTB through the Penn Center for the Study of Epigenetics in Reproduction as well as the University of Pennsylvania March of Dimes Prematurity Research Center. Strict inclusion criteria were applied to the preterm samples, utilizing only spontaneous PTB (including preterm premature rupture of membranes) in the absence of any infection, pregnancy complications (such as hypertension or gestational diabetes) or preexisting maternal condition. Control placentas were age and sex matched to the IVF pregnancies to account for parental age, ethnicity and fetal sex. The demographic profile and relevant clinical characteristics of all samples are shown in Table 1. The average gestational age of the preterm samples was 34.1 weeks in the IVF patients compared to 39 weeks in term IVF samples and 34.6 weeks in the preterm control samples compared to 38.9 weeks in term control samples. Term versus preterm gestational age differences were statistically significant in both control and IVF groups (P = 0.0001).
Table 1.
Demographic data and clinical characteristics of study subjects
| Term IVF(n = 24) | Preterm IVF(n = 11) | Term control(n = 24) | Preterm control(n = 12) | P-value | |
|---|---|---|---|---|---|
| Maternal age (years) | 34.9[33.0–36.7] | 34.6[33.0–39.0] | 34.3[31.4–35.9] | 30.2[25.1–34.2] | 0.04 |
| Paternal age (years)a | 36.5[35.0–40.5] | 36.6[32.9–43.0] | 35.4[32.7–39.8] | 30.2[26.6–41.1] | 0.53 |
| Maternal BMI | 23.4[21.7–35.8] | 24.1[21–26.3] | 22.8[21.1–28.5] | 25.2[21.5–26.4] | 0.91 |
| Maternal race | |||||
| White | 79.2% | 54.6% | 58.3% | 58.3% | |
| Black | 22.7% | 36.4% | 33.3% | 41.7% | 0.54 |
| Other | 0% | 9% | 8.3% | 0% | |
| Gestational age (weeks) | 39.0[38.4–39.9] | 34.1[32.9–35.7] | 38.9[38.4–40] | 34.6[33.6–35.7] | 0.0001 |
| Male:female offspring | 12:12 | 7:4 | 12:12 | 6:6 | 0.869 |
| Infertility diagnosis | N/A | N/A | 0.90 | ||
| Male factor | 20.8% | 27.3% | |||
| Uterine factor | 0% | 0% | |||
| Diminished ovarian reserve | 4.2% | 9.1% | |||
| Unexplained | 20.8% | 36.4% | |||
| PCOS | 12.5% | 9.1% | |||
| Tubal factor | 12.5% | 9.1% | |||
| Endometriosis | 20.8% | 9.1% | |||
| Other | 8.3% | 0% | |||
| Protocol | N/A | N/A | 0.02 | ||
| LPL | 8.3% | 36.4% | |||
| Antagonist | 91.7% | 54.6% | |||
| Flare | 0% | 9.1% | |||
| Fertilization | N/A | N/A | 0.06 | ||
| Conventional | 8.3% | 36.4% | |||
| ICSI (some or all) | 91.7% | 63.6% | |||
| Type of transfer | N/A | N/A | 0.55 | ||
| Fresh | 50% | 54.6% | |||
| Frozen | 50% | 45.6% |
BMI, body mass index; ICSI, intra-cytoplasmic sperm injection; IVF, in vitro fertilization; LPL, luteal phase Lupron; PCOS, polycystic ovarian syndrome.
n = number of placentas collected in each group.
a Paternal age has incomplete data (7 in term controls, 23 in term IVF, 3 in PTB controls and 11 in PTB IVF).
Preterm placentas show differential methylation in a large number of genes
We used the Illumina MethylationEPIC BeadChip array to interrogate over 850 000 CpG sites throughout the genome, representing proximal promoters, 5′/3′ untranslated regions, gene bodies, intergenic regions and regulatory elements such as enhancers. Within these regions, CpG islands, CpG island shores and shelves as well as ‘open sea’ CpGs were represented (24,25). Results from the EPIC array highly correlate with the current ‘gold standard’ for fine mapping of methylation, whole genome bisulfite sequencing, suggesting that it is a cost-effective and reliable method to obtain genome-wide DNA methylation data (25).
Four independent groups were analyzed: term IVF (n = 24), preterm IVF (n = 11), term controls (n = 24) and preterm controls (n = 12). Analysis was limited to CpG sites with a mean methylation difference (βvalue) of at least 5%, and genes with at least two differentially methylated CpG sites. As the goal of this study was to utilize the IVF population to enrich for a population with a common etiology for their PTB, the initial analysis compared DNA methylation in the placenta of IVF pregnancies delivering at term to those delivering preterm. We found 7321 CpG sites within 1980 genes differentially methylated between term IVF and preterm IVF placenta. When comparing term versus preterm control placentas we found 4379 differentially methylated CpGs within 1316 genes. Of the genes evaluated, 613 genes were commonly dysregulated in IVF and control preterm samples (Fig. 1, Supplementary Material, Table 2). We examined CpG methylation in the common genes and found that in both preterm IVF and preterm control samples, the majority of CpG sites was hypomethylated when compared to term placenta (70.4% and 68.8%, respectively). In addition, there was concordance between groups—77% of CpGs that were differentially methylated in IVF preterm placentas were similarly changed in preterm controls. Both raw and processed DNA methylation data have been deposited to the NCBI GEO database.
Figure 1.
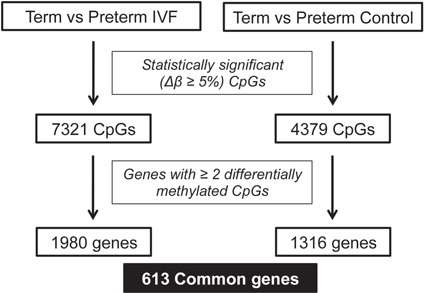
850 K MethylationEPIC BeadChip array results showing number of differentially methylated CpGs and genes when comparing term versus preterm IVF and term versus preterm control placentas.
Ingenuity Pathway Analysis suggests epigenetic dysregulation of gene families involved in early pregnancy
To gain insight into the underlying biology of differentially methylated genes, we carried out Ingenuity Pathway Analysis (IPA) on genes that were differentially methylated in both IVF and control groups (common genes). IPA identified cancer and reproductive system disease as the disease/biological function most likely to be influenced by differential expression of the genes in our dataset. (Supplementary Material, Fig. 1A). In addition, we examined upstream regulators that could potentially affect the expression of genes in our dataset and found epigenetic factors involved in chromatin remodeling, including SETDB1, HDAC4 and Histone H4, suggesting that epigenetic dysregulation may play a role in PTB. We also identified TGFβ1, a factor known to regulate trophoblast invasion, as a master regulator that could potentially affect the expression of multiple genes within our dataset (26) (27,28) (Supplementary Material, Fig. 1B). Therefore, IPA supported the hypothesis that PTB could occur due to epigenetic dysregulation of genes involved in processes important to the establishment of pregnancy, specifically trophoblast invasion.
Methylation array identified ADAMTS as potential candidate gene family involved in PTB development
As the goal of this study was to identify and functionally evaluate genes that could play a role in the etiology of PTB, we focused on gene families highly represented in our datasets and with biological significance for pyrosequencing validation and functional analysis. We found 15 families that had 4 or more members represented in genes commonly affected by PTB in control and IVF groups (Supplementary Material, Table 3). Several of these families, in keeping with our IPA results, have been reported to be involved in cell invasion and adhesion (ADAM/ADAMTS, CADHERIN, GALNT, PROTOCADHERIN, COLLAGEN). In this study, we have focused on the ADAMTS (A disintegrin and metalloproteinase with thrombospondin motifs) family, as these genes encode proteases that have been identified in early placental cells and have been suggested to be involved in several aspects of female fertility including placentation (29). Work in animal models and in vitro human systems indicate a specific involvement of members of this family in the regulation of trophoblast function (29,30). We found multiple members of the ADAMTS gene family differentially methylated within our datasets: nine ADAMTS genes were differentially methylated in IVF–PTB placenta and five in control–PTB placenta, and five were common to both the IVF–PTB and control–PTB groups. Each of these genes contained several CpG sites that were hypo- or hypermethylated consistently within each gene. Two genes with multiple significantly affected CpG sites were ADAMTS12 and ADAMTS16: ADAMTS12 had 6 differentially methylated CpGs in the IVF group and 5 in the control group while ADAMTS16 had 9 differentially methylated CpGs in the IVF groups and 16 in the control group (Supplementary Material, Table 2, Supplementary Material, Fig. 2). We validated changes by pyrosequencing in ADAMTS12 and ADAMTS16 and found statistically significant hypomethylation in ADAMTS12 cg05950015 (P = 0.009) and significant hypermethylation in ADAMTS16 cg25536014 (P = 0.019) in the preterm versus term IVF group (Fig. 2A and B).
Figure 2.
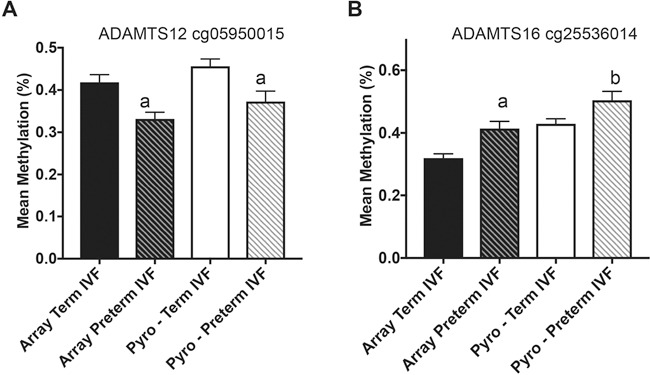
Pyrosequencing validation of candidate ADAMTS genes differentially methylated in both IVF and control groups after PTB. Graph shows mean methylation values (%) in term IVF placenta from the methylation array (black bars), preterm IVF placenta from the methylation array (black-striped bars), term IVF placenta after pyrosequencing (white bars) and preterm IVF placenta after pyrosequencing (white-striped bars). (A) ADAMTS12 cg05950015 showed significant hypomethylation in preterm IVF placenta from the array and after pyrosequencing. (B) ADAMTS16 cg25536014 showed significant hypermethylation in preterm IVF placenta from the array and after pyrosequencing. aP ≤ 0.01 versus term placenta, bP ≤ 0.05 versus term placenta. Data are expressed as mean ± SEM.
ADAMTS genes are expressed during early gestation and are present in all trophoblast cells
Once differences in DNA methylation in ADAMTS12 and ADAMTS16 were validated, we next examined whether these epigenetic changes translated to changes in gene expression. We carried out quantitative real-time polymerase chain reaction (qRT-PCR) on mRNA extracted from term and preterm placenta and found no difference in mRNA levels of ADAMTS12 between term and preterm placentas (data not shown). ADAMTS16 mRNA was undetectable in third-trimester placenta. However, previously published data suggested that members of the ADAM/ADAMTS gene family are expressed at high levels early in gestation (30,31). We therefore examined ADAMTS12 and ADAMT16 mRNA expression in first- and second-trimester placental tissue and found that both genes are expressed at high levels early in gestation (Fig. 3). ADAMTS12 mRNA expression peaks at the end of the first trimester between 7 and 11 weeks gestation age (P = 0.0209) and then decreases during the second trimester (Fig. 3A). ADAMTS16 mRNA appears to gradually increase during the first and second trimester, though differences were not statistically significant (Fig. 3B). We used representative samples from each gestational range to examine protein expression and found that a similar expression pattern can be seen at the protein level. ADAMTS12 protein appeared to be the highest at the end of the first trimester (Fig. 3C), and ADAMT16 protein levels trended towards an increase through the first and second trimester (Fig. 3D). As no differences in ADAMTS12 and ADAMTS16 expression were noted at the time of delivery, the expression of these genes early in gestation may indicate that the epigenetic perturbations found in the third trimester represent changes that impacted expression during implantation and establishment of the placenta.
Figure 3.
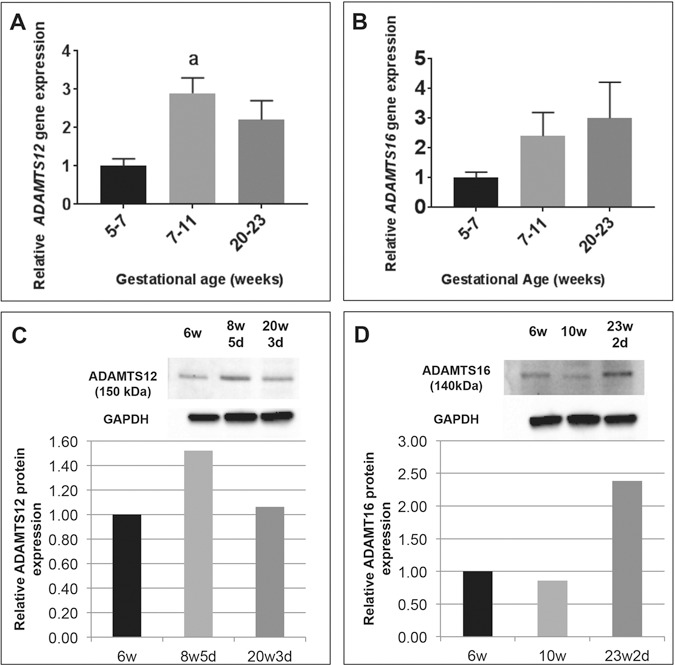
Relative expression of ADAMTS12 and ADAMTS16 in first and second trimester of pregnancy compared to 5–7 weeks samples (black bars). (A) ADAMTS12 mRNA levels peak towards the end of the first trimester (7–11 weeks, light grey bars) and then decrease by the end of the second trimester (20–23 weeks, dark grey bars). (B) ADAMTS16 mRNA levels appear to increase through the first and second trimester of pregnancy. n = 5 in each group. aP ≤ 0.05. Data are expressed as mean ± SEM. (C) Western blot for ADAMTS12 on representative samples from each gestational age range (6 weeks, black bar; 8w5d, light grey bar; 20w3d, dark grey bar) shows similar expression patterns, quantified relative to 6-week sample. (D) Western blot for ADAMTS16 on representative samples from each gestational age range (6 weeks, black bar; 10 weeks, light grey bar; 23w2d, dark grey bar) shows similar expression patterns, quantified relative to 6-week sample.
To localize the source of ADAMTS12 and 16 proteins, we carried out immunofluorescence using validated commercial antibodies. We found that both ADAMTS12 and ADAMTS16 localize to all trophoblast cells using a cytokeratin 7 (CK7) antibody (which stains both cytotrophoblasts and syncytiotrophoblasts) (Fig. 4A and C) including the invasive EVTs (using the EVT specific marker HLA-G) (Fig. 4B and D). Additionally, we found ADAMTS12 and ADAMTS16 protein expression in cytokeratin-negative cells within the stroma of floating villi, potentially representing fibroblast cells and/or macrophages (Fig. 4A–D). No non-specific staining was seen using an IgG as well as a secondary antibody only control (Supplementary Material, Fig. 3A–D).
Figure 4.
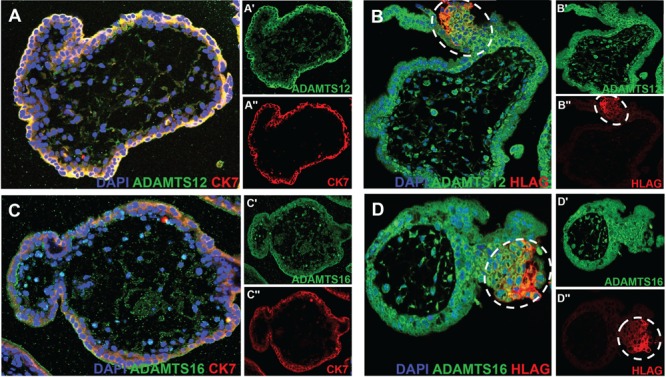
Localization of ADAMTS12 and ADAMTS16 using immunofluorescence on slides from formalin-fixed paraffin embedded 10.5-week placenta. Images show DAPI in blue, ADAMTS12 or ADAMTS16 in green as indicated and CK7 (staining all trophoblasts) or HLA-G (staining EVTs) in red as indicated. Single-channel images of ADAMTS12 or ADAMTS16 and CK7 or HLA-G as indicated are seen next to each merged image and indicated with a ‘ or “. (A) ADAMTS12 is present in all trophoblasts as seen by co-localization with CK7. (B) ADAMTS12 is also present in invasive EVTs as seen by co-localization with HLA-G (white dotted circles). (C) ADAMTS16 is similarly present in all trophoblasts. (D) ADAMTS16 is also present in invasive EVTs (white dotted circles).
ADAMTS genes that are epigenetically altered by PTB play a role in trophoblast invasion
Since ADAMTS genes are expressed during early pregnancy and members of these gene families have been implicated in normal implantation, we next examined the role of ADAMTS12 and ADAMTS16 in trophoblast invasion. The direction of expected expression changes with the observed epigenetic changes is difficult to predict, though ADAMTS12 is hypomethylated at CpGs located in the gene body, which should result in decreased expression and decreased invasion. ADAMTS16 is hypermethylated at CpGs in the promoter, which should reduce expression. Therefore, knockdowns (KDs) were performed for both based on these assumptions.
To do this, we carried out siRNA KD in a human first-trimester EVT cell line, HTR8/SVneo, and evaluated an effect on invasion using multiple assays. EVTs are invasive trophoblasts that migrate deep into the superficial myometrium during implantation and attach the placenta to the uterus. We used Matrigel invasion assays to examine involvement of these genes in trophoblast invasion. After 48 h of siRNA incubation, we confirmed efficiency of KD using qRT-PCR and found that ADAMTS12 and ADAMTS16 mRNA levels were decreased by ∼90% (P = 0.0058 and P = 0.002, respectively) (Fig. 5A and B). We next evaluated EVT invasion and found that compared to control cells (Fig. 5C), EVTs with reduced ADAMTS12 (Fig. 5D) and ADAMTS16 (Fig. 5E) mRNA had significantly less invasion 24 h post-seeding on a Matrigel transwell insert (P ≤ 0.0001) (Fig. 5F).
Figure 5.
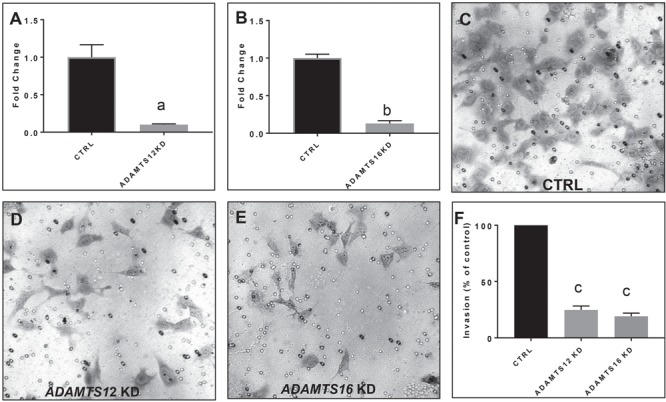
Matrigel invasion assays show decreased invasion of an EVT cell line, HTR8-SVneo after ADAMTS12 or ADAMTS16 KD. (A) qRT-PCR of ADAMTS12 demonstrating KD efficiency of approximately 90% of ADAMTS12. (B) qRT-PCR of ADAMTS16 demonstrating KD efficiency of approximately 90% of ADAMTS16 mRNA. (C) Representative image of HTR8 cells treated with negative control siRNA construct after 24 h of invasion through a transwell insert coated with Matrigel. (D) Representative image of HTR8-SVneo cells treated with ADATMS12 siRNA shows a reduction in the number of cells that invade through a transwell insert coated with Matrigel after 24 h. (E) Representative image of HTR8-SVneo cells treated with ADAMTS16 siRNA shows a reduction in the number of cells that invade through a transwell insert coated with Matrigel after 24 h. (F) siRNA KD of ADAMTS12 (light grey bar) or ADAMTS16 (dark grey bar) in HTR8-SVneo invasion shows a significant decrease in invasion. Three independent experiments were carried out, and five representative images from each experiment were quantified. Fold change values for ADAMTS12 and ADAMTS16 were obtained through normalization to HPRT, a placental housekeeping gene, and then comparison to CTRL cells treated with a negative control siRNA construct. aP ≤ 0.01, bP ≤ 0.001, cP ≤ 0.0001. Data are expressed as mean ± SEM.
A key component of successful EVT invasion is degradation of the extracellular matrix (ECM) (28). The matrix metalloproteinases (MMPs) 2 and 9 (also known as Gelatinase A and B) are known to play an essential role in degradation of the ECM during trophoblast invasion and can be utilized as markers of trophoblast invasion (32). ADAMTS12 KD significantly reduced MMP2 mRNA levels (P = 0.0093) while ADAMTS16 KD did not change MMP2 mRNA levels (Fig. 6A). When examining MMP9 mRNA levels, ADAMTS12 and ADAMTS16 KD significantly affected expression (P = 0.0009 and P = 0.0406, respectively) (Fig. 6B). Since qRT-PCR only examines mRNA levels of inactive MMPs, we carried out gel zymography to assess gelatinolytic activity of MMP2 and 9 present in conditioned media (CM) 24 h after siRNA KD of our candidate genes. Compared to treatment with control siRNA, HTR-8 cells with KD of either ADAMTS12 or 16 significantly reduced secretion of pro-MMP-9 (Fig. 6C and E) but not pro-MMP-2 (Fig. 6D and E) as seen by clear bands indicating cleaved gelatin at that location. These results suggest that epigenetic dysregulation of ADAMTS12 and 16 genes in placentas after PTB could affect trophoblast invasion through regulation of MMP activity.
Figure 6.
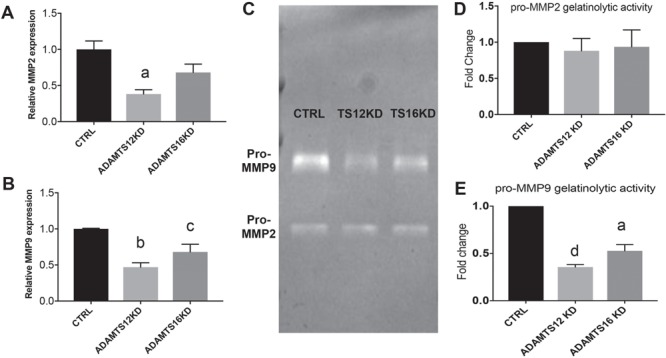
MMP 2 and 9 expression and activity is affected by ADAMTS12 and ADAMTS16 siRNA KD. (A)ADAMTS12 siRNA KD significantly reduces MMP2 mRNA levels (P = 0.009) but reducing ADAMTS16 expression does not affect MMP2 mRNA levels. (B) MMP9 mRNA levels are significantly reduced by both ADAMTS12 and ADAMTS16 siRNA KD. (C) Gel zymography indicates reduction of pro-MMP9 levels as seen by less gelatinolytic activity after ADAMTS12 and ADAMTS16 reduction while no effect on pro-MMP2 levels is seen. (D) Quantification of pro-MMP2 gelatinolytic activity. (E) Quantification of pro-MMP9 gelatinolytic activity. aP ≤ 0.01, bP ≤ 0.001, cP ≤ 0.05, dP ≤ 0.0001.
Discussion
In this study, we examined epigenetic changes in placentas from term and PTB to identify genes playing a role in the etiology of this complex disease. In order to identify a population with a potential common etiology for PTB, we examined genome-wide DNA methylation from term and preterm pregnancies following IVF, a known risk factor for PTB, as well as term and preterm natural conceptions. In both IVF and control datasets, we identified methylation differences in multiple members of the ADAMTS family including ADAMTS12 and ADAMTS16. Both our IPA analysis and prior publications implicate the ADAMTS family in tumor invasion and reproductive processes. Early pregnancy establishment is often thought to mimic malignant tumor cell invasion, as there are multiple parallels between the processes involved in both trophoblast invasion and cancer cell invasion (33). We demonstrate that not only are ADAMTS12 and ADAMTS16 mRNAs expressed during early gestation during the critical window of early trophoblast invasion, but that they are expressed at high levels in the EVTs, cells responsible for anchoring the placenta to the maternal decidua and establishing the maternal–fetal circulation. In order to further understand the function of these two genes, we utilized in vitro assays and demonstrated that KD of either gene leads to changes in EVT invasion. These findings suggest that at least a subset of PTB, both in patients following IVF and natural conception, may be due to epigenetic alteration of genes critical to implantation and early placentation.
Current hypotheses on the etiology of PTB suggest that an early insult affecting establishment of the early implantation site could lead to development of this condition (8). However, there are few molecular or mechanistic studies that support these hypotheses. One important aspect of early establishment of the placenta is appropriate invasion of EVTs through the maternal decidua, a process that requires controlled synthesis and regulation of the ECM. Inappropriate ECM modulation that affects ECM structure has been implicated in several genetic conditions associated with PTB and is a well-studied biological pathway in the context of PTB development (8,34). For example, in heritable disorders such as Ehlers–Danlos syndrome, a disease affecting collagen synthesis, there is a 40–60% risk of PTB (34,35). Similarly, mutations in certain collagen genes, such as those associated with type II osteogenesis imperfecta, have been associated with PTB and PPROM (35). However, examination of ECM modulation defects as a contributor to PTB has primarily been examined in the context of fetal membrane tensile strength and premature rupture of membranes, events that affect later stages of gestation (34,36).
Multiple members of the ADAMTS gene family have been implicated in cell invasion and migration through ECM modulation (37). Similar to the related ADAM protein family, ADAMTS genes have metalloproteinase activity that affects ECM composition. ADAMTS family members, however, lack the transmembrane domain and are therefore secreted proteins that can affect multiple ECM targets (38). ADAMTS genes, and specifically ADAMTS12 and ADAMTS16, have been implicated in ECM modulation responsible for tumor progression (39). In MCF-7 breast cancer cells, ADATMS12 potentiates the migration of the cancer cells (40), while high levels of ADAMTS16 have been demonstrated in samples from esophageal squamous cell carcinomas (40,41).
Our study indicates that the mRNA expression of certain ECM modulators, specifically ADAMTS12 and ADAMTS16, is not changed in the placenta at the time of delivery. These genes are however expressed at high levels in the first and second trimesters of pregnancy in trophoblast cells. We believe the observed epigenetic disruptions are a ‘fossil record’ of events that took place early in gestation and affected processes critical to placentation such as EVT migration. This is challenging to prove since it would require comparison of candidate gene expression during the first trimester between term and preterm pregnancies, and currently there is no way to predict PTB during early gestation.
However, multiple studies from the oncology literature have demonstrated epigenetic regulation of ADAMTS genes and some of these studies have able to correlate these changes with altered expression and cell invasion. For example, both ADAMTS12 and ADAMTS16 expressions have been shown to be controlled by DNA methylation in primary colorectal carcinomas and pancreatic cancer cells, respectively (42,43). In addition, epigenetic changes in other members of the ADAMTS family, such as ADAMTS18, affect both expression and cell invasion in breast cancer cells (44).
A major limitation of these studies is the inability to account for the effect of gestational age on the methylation status. As there is no ‘Control’ PTB, gestational age-matched healthy control placentas cannot be obtained. Therefore, it is possible that the alterations in methylation we see are a result of gestational age or differences in cellular distribution between samples. When examining DNA methylation of ADAMTS12 and ADAMTS16 within the preterm placentas (spanning 31 to 36 weeks gestation), there was no statistically significant correlation between gestational age and methylation, though we are limited by sample number (Supplementary Material, Fig. 3A and B). Moving forward, we hope to collect additional preterm samples with iatrogenic preterm delivery (e.g. preeclampsia, fetal growth restriction) as well as samples from amniocentesis to further examine DNA methylation changes through gestation.
Though this is the first study to associate ADAMTS genes with PTB, recent data have implicated these genes in other disorders of placentation. Two recent studies have found lower ADAMTS12 levels in the serum of patients with preeclampsia (45,46). In addition, Beristain et al. demonstrate that ADATMS12 plays a role in trophoblast invasion through regulating both ECM binding and cell invasion. Other ADAMTS genes have also been implicated in placentation, with increased levels of ADAMTS5 in gestational trophoblastic disease (47). It is likely that multiple insults may be necessary for the early onset of parturition. While one function of ADAMTS12 and ADAMTS16 could be appropriate ECM modulation during early placentation, these genes have also been implicated in the inflammatory response. ADAMTS12 deficient mice display an increase in inflammatory markers and a delay in recovery from inflammatory challenge (48). Inflammation is associated with a large proportion of PTB (8,49). Though our patients had no evidence of clinical infection at the time of delivery, potentially, the combination of disordered implantation, along with later effects on the innate immune system, may be necessary for pathogenesis of this complex disease.
As discussed, several other gene families of interest are highly represented in our dataset. Included in these are other genes critical to ECM establishment. For example, multiple collagen genes were epigenetically dysregulated within the PTB cohort. Collagen is essential during implantation and in maintaining the structural integrity of fetal membranes, suggesting that alterations in these genes could similarly impact PTB development (50). Our dataset also found numerous genes that have been identified in prior epigenetic studies on PTB (Supplementary Material, Table 4). Further validation and investigation into several of these genes are currently ongoing.
This study identifies a possible role for novel ECM modulators active during early pregnancy in the development of spontaneous PTB through examination of an at-risk IVF population, whose underlying PTB etiology may be more homogenous than PTB among natural conceptions. Our findings additionally highlight a potential epigenetic mechanism involving IVF-associated alterations in DNA methylation, which could underlie dysregulation of these genes. This work identifies novel candidates involved in the development of PTB that could be potential targets for identification of at-risk pregnancies as well as preventative treatment options. Further investigations into the mechanisms responsible for these epigenetic changes are currently underway while additionally exploring other genes identified through this unique approach.
Materials and Methods
Sample collection
Placentas were acquired from live-born term and preterm deliveries resulting from IVF pregnancies and naturally conceived pregnancies (controls). For IVF pregnancies, all patients underwent IVF at Penn Fertility Care between 2012 and 2016 using standard protocols. Superovulation was performed using recombinant or purified-urinary follicle stimulating hormone and/or human menopausal gonadotropin. Gonadotropin dose and protocol was chosen based on patient characteristics and was adjusted during stimulation as clinically indicated based on patient response. Oocyte maturation was induced with human chorionic gonadotropin and/or leuprolide acetate followed by transvaginal egg retrieval 36 h later. Fertilization was either conventional or by intracytoplasmic sperm injection (ICSI) (performed for either male factor or unexplained infertility as clinically indicated). Embryo culture was performed utilizing appropriate media (VitroLife; Gothenburg, Sweden) in microdroplets under oil at 5% CO2 in air (5% O2, 5% CO2 and 90% N2), and on day 5 blastocysts were either transferred to the uterus or frozen by vitrification. Luteal support was provided by intramuscular progesterone (50 mg). Frozen embryo transfer cycles were done either using a hormonally programmed cycle with increasing doses of oral micronized estradiol (2–6 mg) followed by intramuscular progesterone to induce a receptive endometrium or timed to a natural cycle. Placentas from natural conceptions, which delivered either at term (greater than 37 weeks) or preterm (<37 weeks), were collected from the Labor and Delivery Unit of the Hospital of the University of Pennsylvania, from patients with no comorbidities, as a control group.
Placentas were collected at delivery and processed for DNA analysis as previously described (22,51,52). Briefly, placental tissue (1.5–2.5 cm3) was excised from the fetal surface of the placenta, directly behind the cord insertion site. The sample was rinsed extensively with sterile saline solution to minimize maternal blood contamination. The tissue was transferred to a 15-ml Falcon tube for DNA and RNA extraction and initially stored at 4°C, and then stored at −80°C until nucleic acid extractions were performed.
First- and second-trimester tissue was collected from terminations performed at the Penn Family and Pregnancy Loss Center. Only intrauterine pregnancies confirmed and dates by last menstrual period and confirmed by ultrasound were collected. Patients with preexisting medical conditions were excluded from the study. Placental villi were isolated and frozen at −80°C until nucleic acid extractions were performed or fixed in formalin for immunofluorescence.
DNA and RNA extraction
Placental genomic DNA was extracted using Invitrogen PureLinkTM Genomic DNA kit (Thermo Fischer Scientific) according to the manufacturer’s guidelines. The isolated DNA was dissolved in 50-ul elution buffer and stored at −20°C until further use. RNA extraction and isolation were performed using the RNAeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA concentration and quality was determined with a NanoDrop spectrophotometer (Thermo Fischer Scientific).
Illumina 850 K Methylation Array
Sample preparation
DNA samples were processed for Infinium Methylation EPIC BeadChip (Illumina) at Center for Applied Genomics, The Children’s Hospital of Philadelphia. The genomic DNA was converted to bisulfite DNA using Qiagen EpiTect Bisulfite conversion kit using the manufacturer’s protocol for sodium bisulfite conversion of unmethylated cytosines in DNA from low-concentration solutions.
Array
The automated protocol for the Infinium HD methylation assay was used for the array run. Briefly, the bisulfite-converted DNA was amplified and then enzymatically fragmented. The fragmented DNA was then precipitated and resuspended followed by hybridization to BeadChips. The unhybridized and non-specifically hybridized DNA sample was washed. The primers hybridized to the DNA was extended using labeled nucleotides and stained. The BeadChip was imaged using the iScan System. The project file was generated from the raw IDAT files using the GenomeStudio version 2011.1. MethylationEPIC_v-1-0_B4.bpm manifest file was used for the downstream data processing.
Data analysis
The resulting methylation data (_v-1-0_B4.bpm) manifest file quantile normalization. The normalized data was analyzed for differential methylation between the preterm and the term groups in both IVF and groups. Differentially methylated CpGs with a P-value less than 0.05 in two-tailed unpaired t-test and a mean difference in methylation of greater than 0.05 were considered to be differentially methylated between the preterm and the term groups. GraphPad Prism version 6 software was used for all the graphical representations. Genes common to both datasets were analyzed by IPA to identify the gene families and processes that may play a role in the etiology of PTB.
Pyrosequencing
Unmethylated cytosines in genomic DNA (0.5–1 μg) were converted to uracil by treatment with sodium bisulfite using the EZ DNA MethylationTM Kit (Zymo Research Corp., USA), following the manufacturer’s guidelines. The bisulfite-converted DNA was dissolved in 20-μl elution buffer and stored at −20°C until further use.
CpG sites differentially methylated in the 850 K array in genes identified as candidate genes through the IPA were considered for validation. Among these, the CpGs for validation were selected based on the quality of pyrosequencing assay designs. Custom assays were designed using Pyromark Assay Design 2.0 software as shown in Supplementary Material, Table 1. Pyrosequencing was done using manufacturer’s guidelines on PyroMark Q96 MD platform.
Statistical analysis
Two-tailed t-tests were used for differential methylation analysis of the 850 K array data and for validation studies. A P-value of less than 0.05 was considered to be significant.
Gene expression analysis
Synthesis of cDNA was performed using the iScript Reverse Transcription Supermix (BioRad) using 1 μg of RNA. qRT-PCR was performed with the QuantStudio 7 qRT-PCR system (Applied Biosystems). Each reaction was performed in 10 μ with 50 ng of cDNA template. Each sample was run in triplicate, and the reactions were carried out for 40 cycles. The following TaqMan probes were used (ThermoFisher): ADAMTS12 (Hs00917098_m1), ADAMTS16 (Hs00373626_m1), MMP2 (Hs01548727_m1) and MMP9 (Hs00234579_m1). Gene expression was normalized to reference gene HPRT (Hs99999909_m1), and relative gene expression compared to the reference group was calculated using the tary Table 1.
Immunofluorescence
Fresh first-trimester placental tissue was formalin fixed at 4°C overnight. Tissue was then placed in 70% ethanol (EtOH) and paraffin embedded before blocks were thinly cut and placed on slides with the aid of the Penn Molecular Pathology and Imaging Core. To antibody stain sections, slides were deparaffinized by immersion in xylene 2X for 5 mins and then rehydrated in 100% EtOH, 95% EtOH, 70% EtOH and 50% EtOH (2 mins in each dilution). Slides were rinsed in dI water for 5 mins, and antigen retrieval was carried out by immersion in citrate buffer (Sigma, C999) at 95°C for 10 mins. After cooling down to room temperature, slides were washed in 0.1% Tris-buffered saline, Tween-20 (TBST) for 5 mins and blocked in 2% Fetal Bovine Serum (FBS) (in TBST) for 30 mins. Slides were then incubated in primary antibody overnight at 4°C. Secondary antibody was added to slides after 3X 5 min washes in 0.1% TBST and incubated overnight at 4°C. After three 0.1% TBST washes, slides were incubated with DAPI for 5 mins before coverslips were mounted on Vectashield. Imaging was carried out using a Leica TCS SP8 Multiphoton Confocal.
The following primary and secondary antibodies and dilutions were used: Rabbit ADAMTS12 (Proteintech, 24934–1-AP, 1:100), Rabbit ADAMTS16 (Abcam, ab45048, 1:100), Mouse CK7 (Novus, NBP2–44814-0.1 mg, 1:100), Mouse HLA-G 4H84 (BD Biosciences, 557577, 1:100), Goat Anti-Rabbit Alexa 488 (Abcam, ab150077, 1:400), Goat Anti-Mouse Alexa 594 (Abcam, ab150116, 1:400) and Rabbit IgG control (Abcam, ab172730, 1:100).
Western blotting
First- and second-trimester placenta were homogenized in 1X RIPA buffer (Cell Signaling, #9806) with protease inhibitors for two rounds of 2 mins each using a Tissue Lyser. Homogenates were spun down at full speed for 5 mins and lysate was removed. A Pierce BSA protein assay (ThermoFisher, 23225) was used to quantify total protein, and 20 ug of each sample was run on a BioRad Mini-Protean Precast Tris 4–15% Gel. Wet transfer was carried out for 1 h at 60 V onto a Polyvinylidene Difluoride (PVDF) membrane. Samples were probed with ADAMTS12 (1:250 dilution), ADAMTS16 (1:2500 dilution) (specified above) and GAPDH (rabbit polyclonal, ProteinTech Group, 10494–1-AP, 1:15000 dilution) primary antibodies and HRP anti-rabbit secondary antibody (Jackson labs 111–035-003). ECL Select (GE Life Sciences, RPN2235) chemiluminescence was used to develop bands, which were quantified using ImageJ (NIH, public domain software).
siRNA KD
KD experiments using siRNA were carried out in HTR8-SVneo cells (ATCC CRL-3271) seeded in 12-well plates at 1 × 105 cells/ml, 24 h after passage. Transfection was performed using Lipofectamine RNAi max (ThermoFisher, Cat. No.: 13778030). To prepare siRNA-Lipofectamine complexes, 2 ul of siRNA constructs at 10 uM was diluted in 100-ul Opti-MEM Reduced Serum medium (Cat. No. 31985–062) and 6 ul of Lipofectamine RNAi max was separately diluted in 100 ul of Opti-MEM medium. Diluted siRNA constructs and Lipofectamine were combined and incubated for 5 mins. 100 ul of siRNA-Lipofectamine complex was then added to each well. The final concentration of siRNA used per well was 10 pmol. KD was confirmed by qRT-PCR. Preliminary time-course experiments (data not shown) suggested that siRNA KD began after 24 h and persisted for at least 120 h. The following SilencerSelect siRNA constructs were used: ADAMTS12 (n290721) and ADAMTS16 (s223419). SilencerSelect Negative Control 1 siRNA (Cat.No. 4390843) was used as a control in all experiments.
Matrigel Invasion Assays
HTR8-SVneo cell invasion through Matrigel was measured using 24-well Corning BioCoat Invasion Chambers pre-coated with Matrigel (Catalog number: 354880). To prepare the invasion chambers, Transwell inserts were incubated at room temperature for 30 mins before inserts were coated with 350 ul of serum-free (SF) medium (RPMI, 1% P/S) and wells were coated with 500 ul of SF medium. Inserts were then placed at 37°C for 2 h to rehydrate the Matrigel. Forty-eight hours after treatment with the siRNA, as described above, confluent HTR8 cells grown in 12-well plates were moved to SF medium for 24 h. Cells from control and KD conditions were harvested using 0.25% trypsin; cells were pelleted, washed with PBS and resuspended in SF medium at a concentration of 1 × 105 cells/ml. 500 ul of the cell suspension was placed in each well and 750 ul of medium containing 20% FBS was placed in each well. After 24 h, non-invading cells were removed from the upper surface of inserts using a dry and then medium-soaked cotton swab. Invaded cells on the lower surface of each insert were fixed and stained using the Diff-Quick staining kit (EMS Catalog Number: 26096–25). Inserts were air-dried for 24 h and imaged using a bright field microscope. Stained cells from 5 microscope fields at 10X magnification were photographed, counted and analyzed. Data presented is expressed as a percentage of invaded cells from control (non-targeting siRNA control) cells. Each invasion assay was repeated three times, and statistical analysis was calculated based on the three independent replicates using a two-tailed t-test.
Gel zymography
Gel zymography was performed on CM obtained from HTR-8 cells treated with siRNAs towards ADAMTS12 and ADAMTS16 to examine the biological activity of MMPs 2 and 9. Twenty-four hours after siRNA KD, cells were serum-starved for 24 h before CM was collected and centrifuged at 1400 rpm to obtain supernatant. Protein content was estimated using the BCA Protein Assay Kit (Pierce, Catalog number: 23227), and equal amounts of protein were loaded on a Novex 10% Zymogram Plus (Gelatin) Gel (ThermoFisher, Catalog number: ZY00105BOX) to determine MMP 2 and 9 activity under non-reducing conditions. After electrophoresis, gels were rinsed in 100 ml of Novex Zymogram renaturing buffer for 30 mins (ThermoFisher, LC2670). Gels were next gently agitated in 100 ml of Novex Developing buffer (ThermoFisher, LC2671) for 30 mins before being replaced with fresh developing buffer and left at 37°C overnight in a humidified chamber. Gels were rinsed with dI water 3X, 5 mins each, and stained under gentle agitation with 40 ml of SimplyBlue Safe Stain (ThermoFisher, LC6060). Gelatinolytic activity was seen as bands of clarity and was imaged using a ChemiDoc MP Imaging system and quantified via ImageJ. Three separate KD experiments were performed and gelatinase activity compared to supernatant from control cells treated with negative control siRNA. Two-tailed t-tests were used to assess statistical significance.
Study Approval
The present study was approved by the University of Pennsylvania Institutional Review Board (approval numbers 804530, 821376 and 827072). Written informed consent was received from all participants prior to inclusion in the study.
Supplementary Material
Acknowledgements
March of Dimes Prematurity Research Center at the University of Pennsylvania and the National Institutes of Health (grant number P50-HD-068157).
Conflict of Interest statement. None declared.
References
- 1. Blencowe H., Cousens S., Chou D., Oestergaard M., Say L., Moller A.B., Kinney M., Lawn J. and Born Too Soon Preterm Birth Action, G (2013) Born too soon: the global epidemiology of 15 million preterm births. Reprod. Health, 10(suppl. 1), S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Frey H.A. and Klebanoff M.A. (2016) The epidemiology,etiology, and costs of preterm birth. Semin. Fetal Neonatal Med., 21, 68–73. [DOI] [PubMed] [Google Scholar]
- 3. Simmons L.E., Rubens C.E., Darmstadt G.L. and Gravett M.G. (2010) Preventing preterm birth and neonatal mortality: exploring the epidemiology, causes, and interventions. Semin. Perinatol., 34, 408–415. [DOI] [PubMed] [Google Scholar]
- 4. Luu T.M., Rehman Mian M.O. and Nuyt A.M. (2017) Long-term impact of preterm birth: neurodevelopmental and physical health outcomes. Clin. Perinatol., 44, 305–314. [DOI] [PubMed] [Google Scholar]
- 5. Nuyt A.M., Lavoie J.C., Mohamed I., Paquette K. and Luu T.M. (2017) Adult consequences of extremely preterm birth: cardiovascular and metabolic diseases risk factors, mechanisms, and prevention avenues. Clin. Perinatol., 44, 315–332. [DOI] [PubMed] [Google Scholar]
- 6. Martin, J., Osterman M., Sutton, P (2010), In NCHS Data Brief, no. 39, pp. 1--8. [PubMed] [Google Scholar]
- 7. Goldenberg R.L., Culhane J.F., Iams J.D. and Romero R. (2008) Epidemiology and causes of preterm birth. Lancet, 371, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Behrman R.E. and Butler A.S. (eds) (2007) Preterm Birth: Causes, Consequences, and Prevention. National Academies Press, Washington, DC. [PubMed] [Google Scholar]
- 9. Kim Y.M., Bujold E., Chaiworapongsa T., Gomez R., Yoon B.H., Thaler H.T., Rotmensch S. and Romero R. (2003) Failure of physiologic transformation of the spiral arteries in patients with preterm labor and intact membranes. Am. J. Obstet. Gynecol., 189, 1063–1069. [DOI] [PubMed] [Google Scholar]
- 10. Romero R., Kusanovic J.P., Chaiworapongsa T. and Hassan S.S. (2011) Placental bed disorders in preterm labor, preterm PROM, spontaneous abortion and abruptio placentae. Best Pract. Res. Clin. Obstet. Gynaecol., 25, 313–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leung T.N., Chung T.K., Madsen G., McLean M., Chang A.M. and Smith R. (1999) Elevated mid-trimester maternal corticotrophin-releasing hormone levels in pregnancies that delivered before 34 weeks. Brit. J. Obstet. Gynaecol., 106, 1041–1046. [DOI] [PubMed] [Google Scholar]
- 12. Smith R. and Nicholson R.C. (2007) Corticotrophin releasing hormone and the timing of birth. Front. Biosci., 12, 912–918. [DOI] [PubMed] [Google Scholar]
- 13. Shapiro-Mendoza C.K. and Lackritz E.M. (2012) Epidemiology of late and moderate preterm birth. Semin. Fetal Neonatal. Med., 17, 120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cavoretto P., Candiani M., Giorgione V., Inversetti A., Abu-Saba M.M., Tiberio F., Sigismondi C. and Farina A. (2017) IVF/ICSI treatment and the risk of spontaneous preterm birth in singleton pregnancies: a meta-analysis of cohort studies. Ultrasound Obstet. Gynecol., 51, 43–53. [DOI] [PubMed] [Google Scholar]
- 15. McDonald S.D., Han Z., Mulla S., Murphy K.E., Beyene J., Ohlsson A. and Knowledge Synthesis G. (2009) Preterm birth and low birth weight among in vitro fertilization singletons: a systematic review and meta-analyses. Eur. J. Obstet. Gynecol. Reprod. Biol., 146, 138–148. [DOI] [PubMed] [Google Scholar]
- 16. Jackson R.A., Gibson K.A., Wu Y.W. and Croughan M.S. (2004) Perinatal outcomes in singletons following in vitro fertilization: a meta-analysis. Obstet. Gynecol., 103, 551–563. [DOI] [PubMed] [Google Scholar]
- 17. Hayashi M., Nakai A., Satoh S. and Matsuda Y. (2012) Adverse obstetric and perinatal outcomes of singleton pregnancies may be related to maternal factors associated with infertility rather than the type of assisted reproductive technology procedure used. Fertil. Steril., 98, 922–928. [DOI] [PubMed] [Google Scholar]
- 18. Bergh T., Ericson A., Hillensjo T., Nygren K.G. andWennerholm U.B. (1999) Deliveries and children born after in-vitro fertilisation in Sweden 1982-95: a retrospective cohort study. Lancet, 354, 1579–1585. [DOI] [PubMed] [Google Scholar]
- 19. Schieve L.A., Meikle S.F., Ferre C., Peterson H.B., Jeng G. and Wilcox L.S. (2002) Low and very low birth weight in infants conceived with use of assisted reproductive technology. N. Engl. J. Med., 346, 731–737. [DOI] [PubMed] [Google Scholar]
- 20. Kalra S.K., Ratcliffe S.J., Barnhart K.T. and Coutifaris C. (2012) Extended embryo culture and an increased risk of preterm delivery. Obstet. Gynecol., 120, 69–75. [DOI] [PubMed] [Google Scholar]
- 21. Katagiri Y., Aoki C., Tamaki-Ishihara Y., Fukuda Y.,Kitamura M., Matsue Y., So A. and Morita M. (2010) Effects of assisted reproduction technology on placental imprinted gene expression. Obstet. Gynecol. Int., 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katari S., Turan N., Bibikova M., Erinle O., Chalian R., Foster M., Gaughan J.P., Coutifaris C. and Sapienza C. (2009) DNA methylation and gene expression differences in children conceived in vitro or in vivo. Hum. Mol. Genet., 18, 3769–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Santos F., Hyslop L., Stojkovic P., Leary C., Murdoch A., Reik W., Stojkovic M., Herbert M. and Dean W. (2010) Evaluation of epigenetic marks in human embryos derived from IVF and ICSI. Hum. Reprod., 25, 2387–2395. [DOI] [PubMed] [Google Scholar]
- 24. Moran S., Arribas C. and Esteller M. (2016) Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics, 8, 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pidsley R., Zotenko E., Peters T.J., Lawrence M.G., Risbridger G.P., Molloy P., Van Djik S., Muhlhausler B., Stirzaker C. and Clark S.J. (2016) Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol., 17, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jones R.L., Stoikos C., Findlay J.K. and Salamonsen L.A. (2006) TGF-beta superfamily expression and actions in the endometrium and placenta. Reproduction, 132, 217–232. [DOI] [PubMed] [Google Scholar]
- 27. Cheng J.C., Chang H.M. and Leung P.C. (2013) Transforming growth factor-beta1 inhibits trophoblast cell invasion by inducing Snail-mediated down-regulation of vascular endothelial-cadherin protein. J. Biol. Chem., 288, 33181–33192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Knofler M. and Pollheimer J. (2012) IFPA Award in Placentology lecture: molecular regulation of human trophoblast invasion. Placenta, 33(suppl), S55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Russell D.L., Brown H.M. and Dunning K.R. (2015) ADAMTS proteases in fertility. Matrix Biol., 44–46, 54–63. [DOI] [PubMed] [Google Scholar]
- 30. Beristain A.G., Zhu H. and Leung P.C. (2011) Regulated expression of ADAMTS-12 in human trophoblastic cells: a role for ADAMTS-12 in epithelial cell invasion? PLoS One, 6, e18473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takahashi H., Yuge K., Matsubara S., Ohkuchi A., Kuwata T., Usui R., Suzuki M. and Takizawa T. (2014) Differential expression of ADAM (a disintegrin and metalloproteinase) genes between human first trimester villous and extravillous trophoblast cells. J. Nippon Med. Sch., 81, 122–129. [DOI] [PubMed] [Google Scholar]
- 32. Zhu J.Y., Pang Z.J. and Yu Y.H. (2012) Regulation of trophoblast invasion: the role of matrix metalloproteinases. Rev. Obstet. Gynecol., 5, e137–e143. [PMC free article] [PubMed] [Google Scholar]
- 33. Holtan S.G., Creedon D.J., Haluska P. and Markovic S.N. (2009) Cancer and pregnancy: parallels in growth, invasion, and immune modulation and implications for cancer therapeutic agents. Mayo Clin. Proc., 84, 985–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Strauss J.F., 3rd. (2013) Extracellular matrix dynamics and fetal membrane rupture. Reprod. Sci., 20, 140–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Anum E.A., Hill L.D., Pandya A. and Strauss J.F. 3rd. (2009) Connective tissue and related disorders and preterm birth: clues to genes contributing to prematurity. Placenta, 30, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parry S. and Strauss J.F. 3rd. (1998) Premature rupture of the fetal membranes. N. Engl. J. Med., 338, 663–670. [DOI] [PubMed] [Google Scholar]
- 37. Porter S., Clark I.M., Kevorkian L. and Edwards D.R. (2005) The ADAMTS metalloproteinases. Biochem. J., 386, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tang B.L. (2001) ADAMTS: a novel family of extracellular matrix proteases. Int. J. Biochem. Cell Biol., 33, 33–44. [DOI] [PubMed] [Google Scholar]
- 39. Cal S. and Lopez-Otin C. (2015) ADAMTS proteases and cancer. Matrix Biol., 44–46, 77–85. [DOI] [PubMed] [Google Scholar]
- 40. Fontanil T., Rua S., Llamazares M., Moncada-Pazos A., Quiros P.M., Garcia-Suarez O., Vega J.A., Sasaki T., Mohamedi Y., Esteban M.M. et al. (2014) Interaction between the ADAMTS-12 metalloprotease and fibulin-2 induces tumor-suppressive effects in breast cancer cells. Oncotarget, 5, 1253–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sakamoto N., Oue N., Noguchi T., Sentani K., Anami K., Sanada Y., Yoshida K. and Yasui W. (2010) Serial analysis of gene expression of esophageal squamous cell carcinoma: ADAMTS16 is upregulated in esophageal squamous cell carcinoma. Cancer Sci., 101, 1038–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moncada-Pazos A., Obaya A.J., Fraga M.F., Viloria C.G., Capella G., Gausachs M., Esteller M., Lopez-Otin C. and Cal S. (2009) The ADAMTS12 metalloprotease gene is epigenetically silenced in tumor cells and transcriptionally activated in the stroma during progression of colon cancer. J. Cell. Sci., 122, 2906–2913. [DOI] [PubMed] [Google Scholar]
- 43. Mishra N.K. and Guda C. (2017) Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer. Oncotarget, 8, 28990–29012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu H., Xiao Q., Fan Y., Xiang T., Li C., Li C., Li S., Hui T., Zhang L., Li H. et al. (2017) Epigenetic silencing of ADAMTS18 promotes cell migration and invasion of breast cancer through AKT and NF-kappaB signaling. Cancer Med., 6, 1399–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eda Gokdemir I., Ozdegirmenci O., Elmas B., Sarikaya E., Tokmak A., Kazanci F.H., Gok S., Erkaya S. and Demircan K. (2016) Evaluation of ADAMTS12, ADAMTS16, ADAMTS18 and IL-33 serum levels in pre-eclampsia. J. Matern. Fetal Neonatal Med., 29, 2451–2456. [DOI] [PubMed] [Google Scholar]
- 46. Namli Kalem M., Kalem Z., Yuce T. and Soylemez F. (2018) ADAMTS 1, 4, 12, and 13 levels in maternal blood, cord blood, and placenta in preeclampsia. Hypertens. Pregnancy, 37, 9–17. [DOI] [PubMed] [Google Scholar]
- 47. Lee S.Y., Lee H.S., Gil M., Kim C.J., Lee Y.H., Kim K.R. and Park C.S. (2014) Differential expression patterns of a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) −1, −4, −5, and −14 in human placenta and gestational trophoblastic diseases. Arch. Pathol. Lab. Med., 138, 643–650. [DOI] [PubMed] [Google Scholar]
- 48. Moncada-Pazos A., Obaya A.J., Llamazares M., Heljasvaara R., Suarez M.F., Colado E., Noel A., Cal S. and Lopez-Otin C. (2012) ADAMTS-12 metalloprotease is necessary for normal inflammatory response. J. Biol. Chem., 287, 39554–39563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lannon S.M., Vanderhoeven J.P., Eschenbach D.A., Gravett M.G. and Adams Waldorf K.M. (2014) Synergy and interactions among biological pathways leading to preterm premature rupture of membranes. Reprod. Sci., 21, 1215–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Damsky C.H., Librach C., Lim K.H., Fitzgerald M.L.,McMaster M.T., Janatpour M., Zhou Y., Logan S.K. and Fisher S.J. (1994) Integrin switching regulates normal trophoblast invasion. Development, 120, 3657–3666. [DOI] [PubMed] [Google Scholar]
- 51. Song S., Ghosh J., Mainigi M., Turan N., Weinerman R., Truongcao M., Coutifaris C. and Sapienza C. (2015) DNA methylation differences between in vitro- and in vivo-conceived children are associated with ART procedures rather than infertility. Clin. Epigenetics, 7, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Turan N., Katari S., Gerson L.F., Chalian R., Foster M.W., Gaughan J.P., Coutifaris C. and Sapienza C. (2010) Inter- and intra-individual variation in allele-specific DNA methylation and gene expression in children conceived using assisted reproductive technology. PLoS Genet., 6, e1001033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.