

Abstract
A rapid, non-invasive urine test for early stage alcohol-induced liver disease (ALD) would permit risk stratification and treatment of high-risk individuals before ALD leads to irreversible liver damage and death. Urinary metabolomic studies were carried out to identify ALD-associated metabolic biomarkers using Ppara-null mouse model that is susceptible to ALD development on chronic alcohol consumption. Two successive studies were conducted to evaluate the applicability of mass spectrometry-based metabolomics in identification of ALD-specific signatures and to examine the robustness of these biomarkers against genetic background. Principal components analysis of ultraperformance liquid chromatography coupled with electrospray ionization quadrupole time-of-flight mass spectrometry (UPLC- ESI-QTOFMS)-generated urinary metabolic fingerprints showed that alcohol- treated wild-type and Ppara-null mice could be distinguished from control animals. It also showed that a combined endogenous biomarker panel helps to identify sub- jects with ALD as well as those at risk of developing ALD even without any information on alcohol intake or genetics. Quantitative analysis showed that increased excretion of indole-3-lactic acid and phenyllactic acid was a genetic background- independent signature exclusively associated with ALD pathogenesis in Ppara-null mice that showed liver pathologies similar to those observed in early stages of human ALD. These findings demonstrated that mass spectrometry-based metabolomic analysis could help in the identification of ALD-specific signatures, and that metabolites such as indole-3-lactic acid and phenyllactic acid, may serve as robust noninvasive biomarkers for early stages of ALD.
Keywords: Alcohol-induced liver disease, PPARα, Ppara-null mouse, Steatosis, Metabolomics, UPLC-ESI-QTOFMS, Multivariate data analysis, Biomarker, Genetic background, Indole-3-lactic acid, Phenyllactic acid
1. Introduction
1.1. Alcohol and Alcohol-Induced Liver Disease
Alcohol consumption is the third most common cause of lifestyle-associated mortality in the United States 2003 [1]. Alcohol consumption is also an emerging problem in developing countries [2]. The 2011 World Health Organization (WHO) status report [3] stated that “almost 4 % of all deaths worldwide are attributed to alcohol, greater than deaths caused by HIV/AIDS, violence or tuberculosis.” Additionally, epidemiological studies have shown significant variation exists in susceptibility to alcohol use and alcohol-dependent health conditions depending on an individual’s genetic background [2, 4–11]. Genetic polymorphisms related to alcohol metabolism affect incidence of alcoholism and physiological response [8, 10, 12], as well the development alcohol-induced liver disease (ALD) and associated outcomes [4, 5, 9, 13, 14]. Even in developed countries such as United States, more than half of alcoholism-related deaths are attributable to ALD [1]. Thus ALD poses a significant challenge to public health all over the world.
ALD pathogenesis is characterized by three stages; steatosis, alcoholic hepatitis, and fibrosis/cirrhosis [2]. Approximately 90 % of alcoholics develop fatty liver (steatosis) that resolves when alcohol consumption is discontinued [2]. However, continued excessive drinking with concomitant steatosis increases the risk of developing cirrhosis by 37 % [15], an irreversible stage of ALD [16]. Overall 5-year survival rates for patients with cirrhosis are as low as 35 %. Liver cirrhosis is also associated with increased risk of development of liver cancer [17, 18]. Although, at earlier stages of ALD (steatosis), liver damage is reversible and patients can recover completely [2, 11, 19, 20], it is largely asymptomatic and, thus, evades diagnosis to proceed to irreversible liver damage. Detection of ALD at this stage is, therefore, key to improve quality of life, maximize therapeutic benefit, and reduce mortality and healthcare burden.
1.2. The Role of PPARα in ALD
Since the first observable change in ALD pathogenesis is the deposition of free fatty acids in the liver [21], many scientific studies have focused on understanding path-ways involved in fatty acid metabolism. The nuclear receptor peroxisome proliferator-activated receptor alpha (PPARα) [22] is a key regulator of the genes involved in lipid metabolism [23, 24], particularly catabolism of fatty acids in the liver. Expression of PPARα and its target genes are attenuated on chronic alcohol consumption [25]. Consistent with these observations, chronic alcohol treatment of the peroxisome proliferator-activated receptor alpha knockout (Ppara-null) mice was shown to result in the development of liver pathologies very similar to the early stages of the human ALD whereas wild-type animals remained protected [26].
1.3. Diagnosis of ALD
Currently, ALD diagnosis is based on biochemical assays including enzymatic activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and gamma-glutamyl transpeptidase (GGT), along with patient history and other clinical symptoms [20, 27]. Serum-based enzymatic activity assays are non-specific with respect to etiology [11, 19]. This leaves liver biopsy as the only confirmatory tool for diagnosis [11, 19, 28]. However, in the absence of detailed life-style associated information, especially acknowledgement of alcohol consumption, often biopsy alone cannot be used to readily distinguish ALD from other liver disorders [29, 30]. The invasiveness of biopsies, and its associated complications [31] also precludes it as a routine screening and diagnostic tool, particularly, given the fact that ALD is largely asymptomatic initial stages [20]. Therefore, an early, noninvasive, high-throughput, ALD-specific biomarker is highly warranted.
1.4. Scope of Metabolomics
Metabolomics is an emerging field in chemical biology that seeks to identify and quantify changes in distribution of all endogenous and exogenous biochemicals (metabolites) in the biological matrix of interest. Since the production of a metabolite is dependent on interaction among of biological molecules (i.e. DNA, RNA, and proteins), the collection of metabolites (e.g. the metabolome), is essentially a reflection of physiological state of an organism at systems level. Therefore, in principle, every physiological state is expected to have a characteristic biochemical fingerprint represented by the metabolome and differences between these signatures can be used to predict a pathology. The latent signatures can also be used to elucidate the changes in biochemical landscape during pathogenesis. Metabolomics has yielded promising findings in recent studies of complex systems including pharmacometabolomics, radiation biodosimetry, and cancer biology [32–35]. As such, the application of metabolomics to elucidate biochemical changes associated with ALD represents a powerful approach to identify early biomarkers of the disease that could reveal novel aspects of underlying biology.
2. Methodological Overview for Urinary Metabolomics
2.1. Animal Model
Since, wild-type mice remain protected whereas Ppara-null mice develop ALD on chronic alcohol consumption; they together represent an excellent model for delineating ALD-specific changes. The studies discussed herein combine the power of metabolomics with the well-characterized Ppara-null mouse model to search for ALD-specific changes in urinary metabolome. Age-matched male wild-type and Ppara-null mice were fed control or an alcohol-containing liquid diet. Urine samples collected from these mice were analyzed using ultraperformance liquid chromatography coupled with electrospray ionization quadrupole time-of-flight mass spectrometry (UPLC-ESI-QTOFMS) to identify metabolomic changes associated with the development of ALD and to differentiate them from those related to the metabolomic changes due to of alcohol consumption [36–38]. A summary of the workflow is shown in Fig. 1.
Fig. 1.
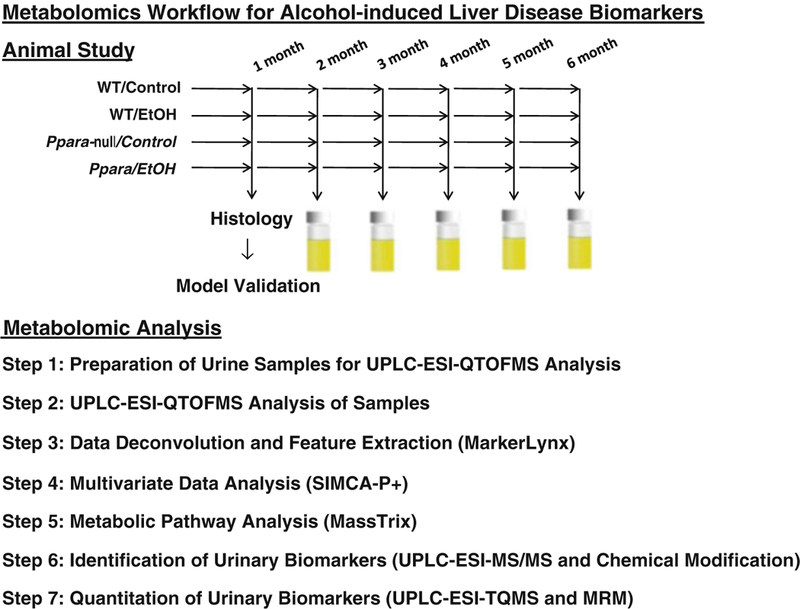
Step-wise workflow for urinary metabolomic analysis to identify biomarkers of alcohol-induced liver disease (ALD). Wild-type and Ppara-null B6 and 129S mice were placed in the 4 % EtOH or control group. After 1 month of alcohol treatment, histological and biochemical analysis was performed to confirm ALD onset in Ppara-null mice. From 2 to 6 months, 24 h urine samples were collected monthly and subjected to UPLC-ESI-QTOFMS analysis, multivariate and pathway analyses, and identification and quantitation of urinary metabolites
2.2. Step 1: Preparation of Urine Samples for UPLC-ESI-QTOFMS Analysis
Urine was diluted 1:2 (v/v) with 50 % aqueous acetonitrile containing internal standards (50 μM 4-nitrobenzoic acid and 1 μM debrisoquine) in a Sirroco™ protein precipitation plate (Waters Corp.) and briefly vortexed. The deproteinated extracts were collected into 96-well collection plates under vacuum, and a 5 μL aliquot was injected into a Waters UPLC-ESI-QTOFMS system.
2.3. Step 2: UPLC-ESI-QTOFMS Analysis of Urine Samples
An Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm, Waters Corp.) was used for chromatographic separation of metabolites before introduction into electrospray. The mobile phase comprised of a mixture of 0.1 % aqueous formic acid (A) and acetonitrile containing 0.1 % formic acid (B). A 0.5 mL/min flow rate was maintained over a 10-min run with a gradient elution: 2 % B for 0.5 min, 2–20 % B in 4 min, 20–95 % B in 8 min, 95–99 % B in 8.1 min, holding at 99% B up to 9.0 min, bringing back to 2 % at 9.1 min and holding at 2 % till end. Column temperature was maintained at 40 °C throughout sample runs. The QTOF Premier mass spectrometer was operated in electrospray ionization positive (ESI+) and negative (ESI−) mode. Capillary voltage and cone voltage were maintained at 3 kV and 20 V, respectively. Source and desolvation temperatures were set at 120 °C and 350 °C, respectively. Nitrogen was used as both cone gas (50 L/h) and desolvation gas (600 L/h), and argon was used as collision gas. Sulfadimethoxine was used as the lock mass (m/z 311.0814+) for accurate mass calibration in real time. Collision energy ranging from 10 to 40 eV was applied for MS/MS fragmentation of target ions. All urine samples were analyzed in a randomized fashion to avoid complications due to artifacts related to injection order and changes in instrument efficiency.
2.4. Step 3: Data Deconvolution and Feature Extraction
Ion chromatogram and mass spectral data were acquired using MassLynx software (Waters Corp.) in centroid format. Chromatograms were inspected for consistency of sample injection, reproducibility of retention time, and mass accuracy using internal standards and quality control samples. Data was binned, features extracted and area under the peak was calculated through integration using MarkerLynx soft- ware (Waters Corp.)
2.5. Step 4: Multivariate Data Analysis
Individual ion intensities were normalized with respect to the total ion count (TIC) in order to generate a data matrix consisting of the retention time, m/z value, and the normalized peak area. The data matrix was analyzed by SIMCA-P+12 software (Umetrics, Kennelon, NJ). Unsupervised segregation of control and alcohol-treated metabolomes was checked by principal components analysis (PCA) using Pareto- scaled data [39]. The supervised orthogonal projection to latent structures (OPLS) model was used to identify ions that contributed significantly to group discrimination. OPLS analysis concentrated group discrimination into the first component with remaining unrelated variation contained in subsequent components. The magnitude of the parameter p(corr)[1] obtained from the loadings S-plot generated by OPLS analysis correlates with the group discriminating power of a variable. A list of ions was then generated from the loadings S-plot showing considerable group discriminating power (−0.8 > p(corr)[1] or p(corr)[1] > 0.8) (statistically significant (P < 0.05) difference in relative abundance between control and alcohol-treated animals). The p(corr)[1] values represent the interclass difference and w(1) values indicate the relative abundance of the ions. Ions that contribute highly to the interclass differences were selected for further identification and quantitation as candidate biomarkers.
2.6. Step 5: Metabolic Pathway Analysis
MassTRIX (http://metabolomics.helmholtz-muenchen.de/masstrix/) is a web-based tool designed to assign ions of interest from a metabolomics experiment to annotated pathways [40]. It can be used to find metabolic pathways even without any systematic identification [41]. This was used to identify metabolic pathways affected by alcohol treatment. The masses of the ions that are significantly elevated (p(corr) [1] > 0.8) or depleted (p(corr)[1] < −0.8) upon alcohol treatment were used to identify metabolic pathways of interest using the KEGG (http://www.genome.jp/kegg/) database (including HMDB, Lipidmaps, and updated KEGG). A mass error of <5 ppm in the respective ionization modes and the possibility of formation of Na+- adducts in the electrosprayer (ESI+ mode) was also taken into account.
2.7. Step 6: Identification of Urinary Biomarkers
Elemental compositions were derived using the Seven Golden Rules [42] considering a mass error <5 ppm. Possible candidates were also searched using metabolomic databases [43, 44]. Finally, authentic standards were used to confirm the identities of these ions by comparison of retention time (UPLC) and fragmentation pattern (ESI-MS/MS). Sulfatase (Sigma-Aldrich) treatment followed by retention time and fragmentation comparison of deconjugated metabolites with authentic standards, were used to confirm sulfate conjugates. Urine samples and standards were incubated with 40 U/mL of the enzyme solution in 200 mM sodium acetate buffer (pH 5.0) overnight at 37 °C. The enzyme and other particulates were precipitated with 50 % aqueous acetonitrile, and the supernatant was analyzed by UPLC-ESI- QTOFMS/MS. 4-Nitrocatechol sulfate was used as a positive control for the sulfatase activity. Deconjugation was also carried out using acid hydrolysis by heating the urine samples with 6 M HCl at 100 °C for 1 h under refluxing conditions.
2.8. Step 7: Quantitation of Urinary Metabolites
An Acquity® UPLC system coupled with a XEVO™ triple-quadrupole tandem mass spectrometer (Waters Corp.) was used to quantitate urinary metabolites by multiple reaction monitoring (MRM). Standard compounds were mixed together to optimize the condition for separation and detection of metabolites from a complex mixture such as urine. Standard calibration plots for quantitation were generated using authentic standards. Deproteinated urine samples containing 0.5 μM debrisoquine (internal standard) were analyzed in the same fashion as that of authentic compounds. The mobile phase was comprised of 0.1 % aqueous formic acid (A) and acetonitrile containing 0.1 % formic acid (B). The gradient elution was performed over 6 min at a flow rate of 0.3 mL using: 1–99 % B in 4 min, holding at 99%B up to 5.0 min, bringing back to 1 % at 5.5 min and holding at 1 % till end. The area under the peak for each metabolite was divided by that for the internal standard to calculate response and a serial dilution was performed to generate a standard calibration plot of response vs. concentration. Serially diluted urine samples containing 0.5 μM debrisoquine were analyzed in the same way as the authentic standards. The quantitative abundances were calculated from the response using the linear range of detection of the calibration plot. All analyses were performed using TargetLynx software (Waters Corp.). One-way ANOVA with Bonferroni’s correction for multiple comparisons was performed using GraphPad Prism 4 software (San Diego, CA) with a two-sided P < 0.05 considered statistically significant.
According to their fragmentation pattern, the following MRM transitions were monitored for the respective compounds: indole-3-lactic acid (206→118; ESI+), indole-3-pyruvic acid (204→130; ESI+), tryptophan (205→118; ESI+), 2-hydroxyphenylacetic acid (151→107; ESI−), 4-hydroxyphenylacetic acid (151→107; ESI−), adipic acid (147→101; ESI+), pimelic acid (159→97; ESI−), debrisoquine (176→134; ESI+), phenylalanine (166→120; ESI+), phenyllactic acid (165→103; ESI−), suberic acid (173→111; ESI−), N-hexanoylglycine (174→76; ESI+), xanthurenic acid (206→160, ESI−), N-acetylglycine (116→74, ESI−), taurine (124→80, ESI−), and creatinine (114→86; ESI+). All concentrations were normalized with respect to creatinine to account for any change in glomerular filtration rates.
2.9. Effect of Genetic Background on Metabolomic Signatures
Genetic background is well-known to influence outcome of alcoholism including alcohol-induced liver disease [4, 5, 13, 14]. Since, metabolome reflects the phenotype; robustness of metabolomic biomarkers against genetic background needs to be investigated. C57BL/6 (B6) and 129/Sv has earlier shown to differ considerabl with respect to physiological functions [45] as well as the biochemical response and outcome of xenobiotic insults [46, 47]. Thus these two strains of mice were used to characterize the influence of genetic background on overall metabolome and ALD biomarkers.
3. Animal Study Design
-
Study 1: Identification of ALD-associated metabolic signatures in Ppara-null Mice.
Male (6- to-8-week-old, N = 4/group) wild-type and Ppara-null on 129/Sv back- ground were fed a 4 % ethanol-containing liquid diet ad libitum (Lieber-DeCarli Diet, Dyets, Inc.). Control animals (N = 4/group) were fed an isocaloric diet supplemented with maltose dextran ad libitum (Dyets, Inc.).
-
Study 2: Identification of genetic background-independent ALD biomarkers.
The design in Study 1 was replicated but with the addition of two genetic back- grounds: wild-type and Ppara-null mice (6- to 8-week-old male, N = 4/group) on B6 (C57BL/6 N-Ppara<tm1Gonz>/N) as well as their counterparts on a 129/Sv (129S4/SvJae-Ppara<tm1Gonz>/N) background.
In both studies, a subset of mice were euthanized after 1 month on the alcohol diet, serum was collected, and portions of the liver were harvested for histology to confirm that Ppara-null mice were developing steatosis. Livers were formalin-fixed, paraffin-embedded, sectioned, and stained with hematoxylin and eosin. Serum AST and ALT activities were measured using VetSpec™ kits (Catachem, Inc.). Liver and serum triglycerides were estimated using a colorimetric assay kit from Wako. At 2 months, after mice were accustomed well to the liquid diets, they were transferred to a urinary metabolomics protocol where urine samples were collected monthly using Nalgene metabolic cages (Tecniplast USA, Inc.). Urines were collected over 24 h and stored at −80 °C in glass vials until analyzed. All mice were acclimated to the metabolic cages by placing them in the metabolic cages before the actual sample collection.
4. Results and Discussion
4.1. PCA Analysis of Metabolomic Data
In agreement with an earlier report [26], only Ppara-null mice on alcohol treatment showed lipid accumulation after 1 month (Fig. 2a) indicating ALD onset [48, 49]. Mass spectrometry-based metabolomic analysis revealed that alcohol exposed Ppara-null mice had a distinct urinary metabolic profile compared to those on control diet even at the earliest time point, i.e., after 2 months of alcohol treatment (Fig. 2b). After 6 months of alcohol treatment, when Ppara-null mice exclusively developed alcoholic steatosis, the urinary metabolomic data showed distinct segregation of control and alcohol-treated mice as well as wild-type and Ppara-null animals on the scores-scatter plot for unsupervised principal components analysis (Fig. 2c). These data indicated that each of these four groups represents a distinct metabolic signature. The separation of these mice along first principal component was according to their alcohol exposure. Interestingly, the separation along second principal component, which was influenced by the genotype, was more prominent in Ppara-null mice compared to wild-type. This indicated that in agreement with the liver pathology, the Ppara-null metabolome is also more susceptible to chronic alcohol consumption. Subsequently, supervised orthogonal projection to latent structures (OPLS) analysis was performed. As the loadings S-plots showed (Fig. 2d, e) there were a number of ions that showed similar trends of elevation (such as P1, P1a, and P8) or depletion (such as P4, P5, and P5a) on alcohol treatment in both wild-type and Ppara-null mice. However, few ions were found to be exclusively elevated (such as P2) in the urine of alcohol-treated Ppara-null mice (Fig. 2e) that developed ALD. These ions might represent ALD-specific metabolic derangements.
Fig. 2.
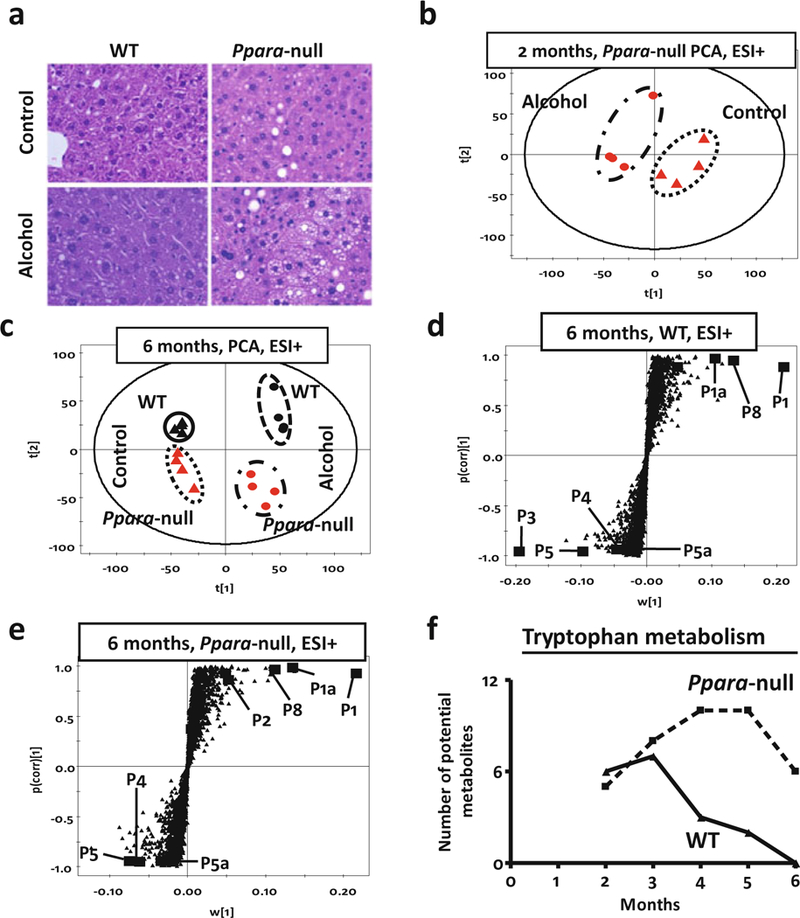
(a) Liver histology (HE stain) of wild-type (WT, left panel) and peroxisome proliferator- activated receptor alpha knockout (Ppara-null, right panel) mice after a duration of 1 month on control (upper panel) or 4 % alcohol-containing liquid diet (lower panel). Histology shows increased fat deposition in Ppara-null animals on the 4 % alcohol containing liquid diet. (b) Scores scatter plot from principal components analysis (PCA) showing unsupervised segregation of the urinary metabolome (ESI+ mode) from control and alcohol-treated Ppara-null mice at 2 months. (c) PCA scores scatter plot showing a larger difference between wild-type and Ppara-null metabolomic data as a result of chronic alcohol treatment (over 6 months). The triangles and dots indicate mice on control and alcoholic diet, respectively, with black and red color representing wild-type and Ppara-null mice, respectively. (d) Loading S-plots from the supervised orthogonal projection to latent structures (OPLS) analysis of ESI+ mode metabolic signatures (at 6 months) for candidate markers of chronic alcohol exposure in wild-type and (e) Ppara-null mice. Each triangle represents an ion characterized by unique mass and retention time. Representative candidates have been high- lighted (solid box) in the plots. A differential response was characterized by biomarkers that were exclusive to wild-type (P3) or Ppara-null (P2) mice. (f) MassTRIX analysis of putative metabolites related to tryptophan metabolism detected in ESI+ mode show variation over time during alcohol treatment. The solid and dotted lines represent wild-type and Ppara-null animals, respectively
4.2. Metabolic Pathway Analysis
To further identify possible metabolic pathways affected by alcohol treatment and ALD pathogenesis, ions that significantly contributed to the separation of alcohol-treated and control animals were analyzed using MassTRIX. Following alcohol exposure, metabolites potentially originating from tryptophan metabolism were found to be significantly significantly elevated (Fig. 2f). However, in wild-type animals, the number of such metabolites gradually decreased over time, while in Ppara-null animals, the corresponding number of metabolites increased. Thus, the MassTRIX analysis indicated that alcohol consumption impacted tryptophan metabolism more in the Ppara-null mice as compared to wild-type mice.
4.3. Identification and Quantitation of Metabolites
Identities of a number of these ions were subsequently confirmed using authentic standard and their concentrations were measured. Tables 1 and 2 show metabolites deranged in the urine of wild-type and Ppara-null mice on alcohol treatment. Both wild-type and Ppara-null animals showed an elevation of ethanol metabolites such as ethyl sulfate and ethyl-β-d-glucuronide, albeit to a different extent. In addition, metabolites such as 2-hydroxyphenylacetic acid, 4-hydroxyphenylacetic acid, 4-hydroxyphenylacetic acid sulfate and xanthurenic acid were elevated, whereas adipic acid and pimelic acid were depleted in the urine of alcohol-treated mice. Similar to alcohol metabolites, many of these endogenous metabolites also showed significant difference in their excretion in wild-type and Ppara-null animals. However, it was interesting to note that indole-3-lactic acid was exclusively elevated in the urine of alcohol-treated Ppara-null mice (Fig. 2a, b).
Table 1.
Metabolic signature of chronic alcohol exposure in the wild-type mice
| Identity | Putative origin | Trend in B6 | Trend in 129S |
|---|---|---|---|
| Ethyl sulfate | Alcohol metabolism | − | ↑ |
| Ethyl-β-d-glucuronide | Alcohol metabolism | ↑ | ↑ |
| N-Acetylglycine | Alcohol metabolism | ↑ | ↑ |
| 4-Hydroxyphenylacetic acid | Phenylalanine metabolism and gut flora | − | ↑ |
| 4-Hydroxyphenylacetic acid sulfate | Phenylalanine metabolism and gut flora | − | ↑ |
| 2-Hydroxyphenylacetic acid | Phenylalanine metabolism and gut flora | − | ↓ |
| Xanthurenic acid | Tryptophan metabolism and gut flora | ↑ | − |
| Adipic acid | Fatty acid ω-oxidation | − | ↓ |
| Pimelic acid | Fatty acid ω-oxidation | − | ↓ |
| Taurine | Cysteine metabolism | ↓ | ↑ |
| N-hexanoylglycine | Fatty acid β-oxidation and gut flora | ↑ | − |
Table 2.
Metabolic signature of chronic alcohol exposure in the Ppara-null mice
| Identity | Putative origin | Trend in B6 | Trend in 129S |
|---|---|---|---|
| Ethyl sulfate | Alcohol metabolism | − | ↑ |
| Ethyl-β-d-glucuronide | Alcohol metabolism | ↑ | ↑ |
| N-Acetylglycine | Alcohol metabolism | ↑ | ↑ |
| 4-Hydroxyphenylacetic acid | Phenylalanine metabolism and gut flora | − | ↑ |
| 4-Hydroxyphenylacetic acid sulfate | Phenylalanine metabolism and gut flora | − | ↑ |
| 2-Hydroxyphenylacetic acid | Phenylalanine metabolism and gut flora | − | ↓ |
| Xanthurenic acid | Tryptophan metabolism and gut flora | ↑ | − |
| Adipic acid | Fatty acid ω-oxidation | − | ↓ |
| Pimelic acid | Fatty acid ω-oxidation | − | ↓ |
| Taurine | Cysteine metabolism | ↓ | ↑ |
| Indole-3-lactic acid | Tryptophan metabolism | ↑ | ↑ |
| Phenyllactic acid | Phenylalanine metabolism | ↑ | ↑ |
For biomarker discovery, reproducibility of measurements is a very important issue. Nuclear magnetic resonance (NMR)-based metabolic profiling has an advantage of being very reproducible as well as for giving direct structural information about the metabolite. However, it is interesting to note that concentrations of indole- 3-lactic acid in these urine samples were in the low micromolar range. The sensitivity of the analytical method used for measuring changes in the metabolic profile becomes crucial to detect changes in the excretion of such metabolites. NMR typically fails to capture changes in abundance of metabolites at these concentration levels whereas mass spectrometry, as evident from these results, is sensitive enough to measure concentrations down to nanomolar and even picomolar ranges. Thus in spite of inferior reproducibility compared to NMR, mass spectrometry has a distinct advantage in increasing sensitivity and capturing miniscule changes in excretion of larger number of metabolites present in such low concentrations. Mass spectrometry can also increase the chance of identification novel metabolites that may be low in abundance but specific to the pathology. On the other hand, NMR typically measures only few hundreds of known and relatively abundant metabolites. Thus, mass spectrometry is often superior as a platform, particularly, for discovery of metabolic biomarkers.
4.4. Potential Use of Metabolic Signature in Detection of Alcohol Intake and ALD Susceptibility
Diagnosis of ALD is often complicated due to lack or fidelity of information on alcohol intake. The principal components analysis including all endogenous and alcohol metabolites showed clear clustering of mice according to genotype and alcohol exposure as early as after 2 months of alcohol treatment (Fig. 3c). At 3 months, these clusters separated into different quadrants with first principal component reflecting genotype and the second representing alcohol exposure (Fig. 3d). Alcohol metabolites such as ethyl sulfate and ethyl-β-d-glucuronide are used in forensic analysis for alcohol consumption [50, 51]. Thus, a combined metabolic panel could be used for detection of recent alcohol consumption as well as liver damage. However, none of these metabolites are detectable beyond 3 days [50, 51]. This could present a challenge in ALD diagnosis if the patient stops drinking just 3 days prior to examination and denies alcoholism. Interestingly, it was found that endogenous metabolites also showed similar discriminatory power between these groups of animals (Fig. 3e, f). At 3 months, mice clustered in four different quadrants with horizontal separation indicating ALD susceptibility (genotype), vertical separation indicating alcohol consumption and diagonal separation indicating interaction between them resulting in ALD pathogenesis. This indicated that metabolomic signature alone may not only help to diagnose ALD but also detect alcohol intake and predict ALD susceptibility prospectively.
Fig. 3.
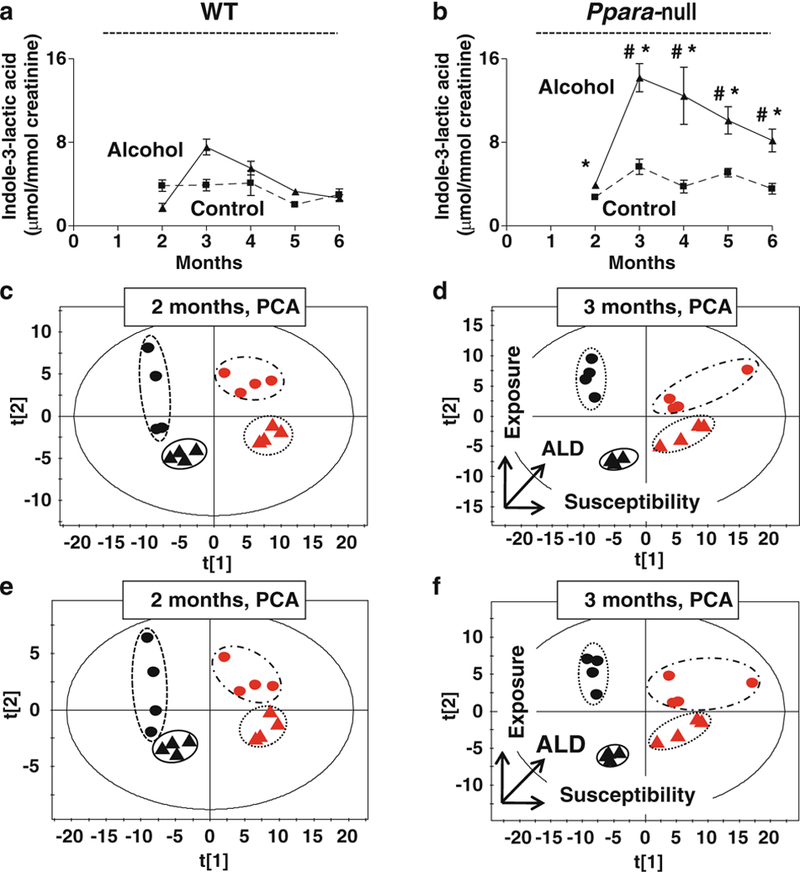
Discriminatory power of non-invasive ALD urinary biomarkers. Variation of the urinary excretion of indole-3-lactic acid in wild-type mice (a) and Ppara-null mice (b) that develop alcohol- induced liver disease (ALD). The dashed and solid lines represent the variation in the concentration of urinary indole-3-lactic acid from control and alcohol-treated mice, respectively (One-way ANOVA with Bonferroni’s correction for multiple comparisons, significance at P < 0.05; #, significantly different from control Ppara-null mice; *, significantly different from the alcohol-treated wild-type mice). PCA scores scatter plot for the variation in the urinary excretion of the endogenous metabolites (indole-3-lactic acid, 4-hydroxyphenylacetic acid, 4-hydroxyphenylacetic acid sulfate, 2-hydroxyphenylacetic acid, adipic acid, and pimelic acid) as well as alcohol metabolites (ethyl sulfate, ethyl-β-d-glucuronide) at 2 months (c) and at 3 months (d) after beginning of alcohol treatment. The triangles and dots indicate mice on control and alcoholic diet, respectively, with black and red color representing wild-type and Ppara-null mice, respectively. Horizontal separation in the scatter plots correlates with Ppara-null expression that determines the susceptibility towards ALD with the horizontal arrow indicating a decrease in PPARα expression and an increase in ALD susceptibility. Vertical separation in the scatter plots correlates with alcohol exposure (in the direction of the vertical arrow). (e) The scores scatter plot for the PCA of endogenous urinary metabolites shows their collective discriminatory power to identify phenotypes at 2 months and (f) at 3 months of alcohol treatment. The triangles and dots indicate mice on control and alcoholic diet, respectively, with black and red color representing wild-type and Ppara-null mice, respectively
4.5. Effect of Genetic Background on Metabolic Signatures
Similar to 129/Sv mice, Ppara-null mice on B6 background also showed an increase in steatosis compared to their wild-type counterparts on alcohol treatment. However, the overall metabolic fingerprint of B6 mice was distinctly different from the 129S mice irrespective of Ppara expression and ethanol treatment throughout the course of the study (Fig. 4a). This represents intrinsic difference between biochemical landscapes of these two strains due to difference in genetic background. In fact, these mice were also found to be different in terms of alcohol metabolism. Ethyl sulfate, which showed a huge increase in the urine of alcohol-treated 129/Sv mice, was not detected in the urine of B6 mice (Tables 1 and 2). Such differences in alcohol metabolism is common in people with different genetic backgrounds and contributes to difference in the outcome of alcohol-induced liver injury [4, 5, 13, 14]. However, MassTRIX analysis showed (Fig. 4a) that similar to that observed in case of 129S mice, a number of metabolites potentially belonging to tryptophan metabolism were elevated on alcohol treatment in B6 mice and the number of elevated metabolites in Ppara-null was also higher than that in the wild-type mice. In addition, Ppara-null mice also showed a progressive increase in number of potential metabolites belonging to phenylalanine metabolism on alcohol treatment whereas wild-type mice showed a decrease. All metabolites were identified and quantitated using authentic standards. The results showed an elevation in urinary excretion of indole-3-lactic acid exclusively in alcohol-treated Ppara-null mice (Fig. 5a, b) on B6 background similar to that observed in 129/Sv mice. This was accompanied with an elevation in the urinary excretion of phenyllactic acid exclusively in alcohol- treated Ppara-null mice (Fig. 5c, d). Phenyllactic acid was also measured and found to be elevated in the urine of alcohol-treated Ppara-null mice on 129/Sv background (Fig. 5e, f).
Fig. 4.
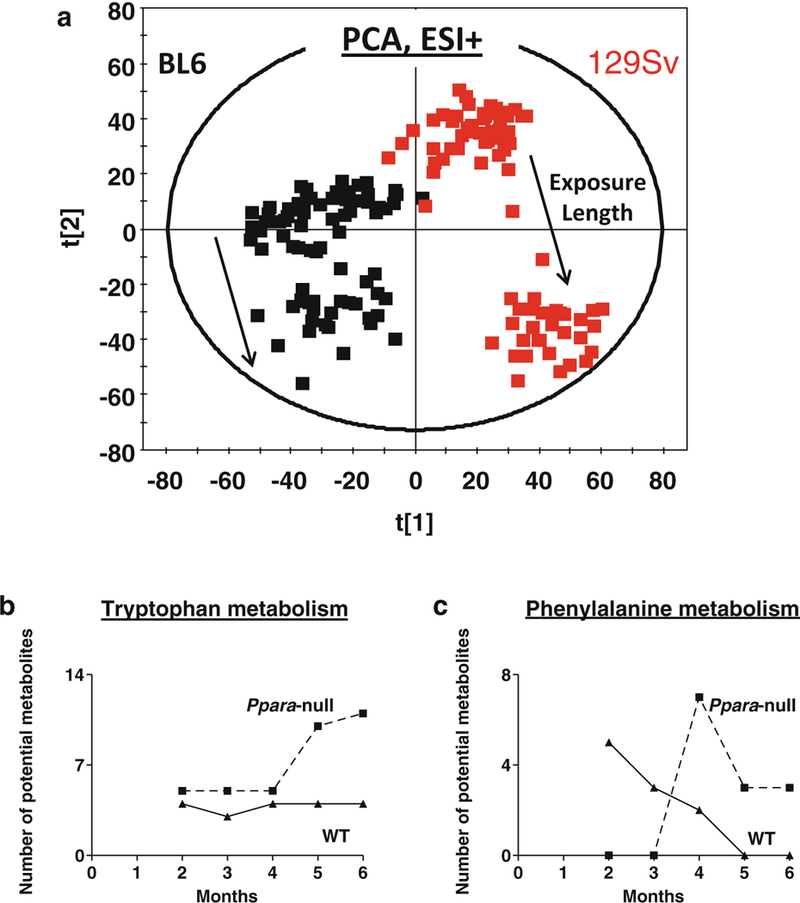
(a) PCA scores scatter plot showing mice on B6 (black) and 129/Sc (red) backgrounds possess distinct metabotypes associated throughout the study duration, irrespective of genotype and alcohol exposure. The solid arrows indicate the shift along second principal component over time due to alcohol exposure. MassTRIX analysis of putative metabolites related to (b) tryptophan and (c) phenylalanine metabolism detected in ESI+ mode show variation over time during alcohol treatment. The solid and dotted lines represent wild-type and Ppara-null animals, respectively
Fig. 5.
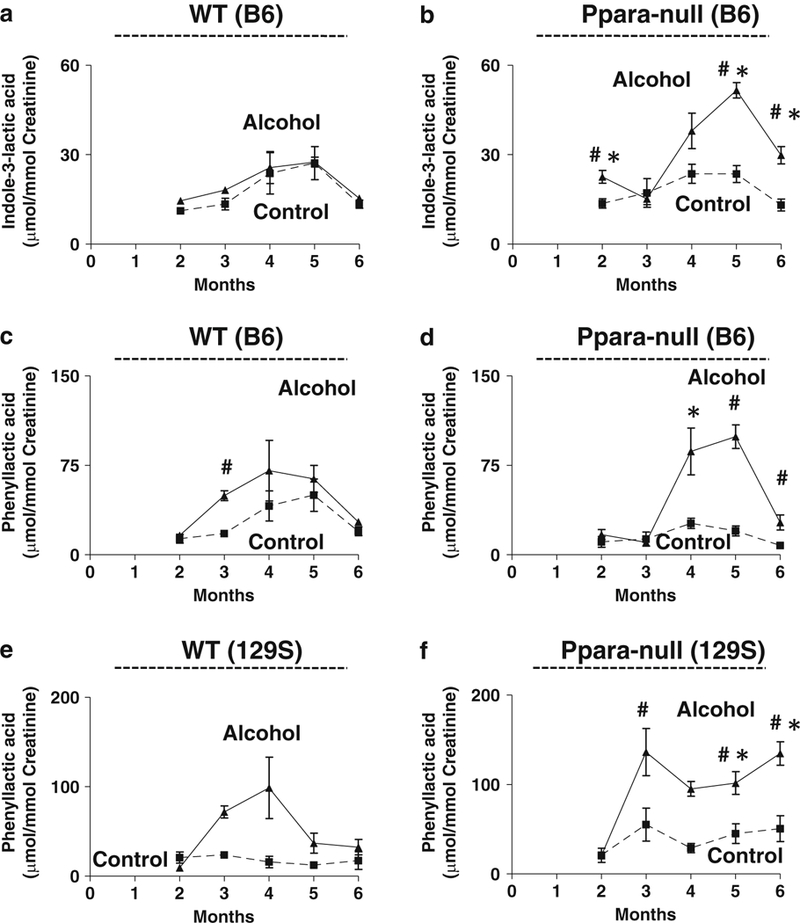
Genetic background-independent increases in phenylalanine and tryptophan metabolites in an ALD mouse model. Urinary excretion of indole-3-lactic acid in (a) wild-type and (b) Ppara-null mice on the B6 background. Urinary excretion of phenyllactic acid in (c) wild-type and (d) Ppara-null mice on the B6 background. Urinary excretion of phenyllactic acid in (e) wild-type and (f) Ppara-null mice on the 129S background. Dashed and solid lines represent control and alcoholic diet-treated mice, respectively. (One-way ANOVA with Bonferroni’s correction for multiple comparisons, significance at P < 0.05 with ‡, significantly different from Ppara-null mice of same treatment group; #, significantly different from control mice of same genotype; *, significantly different from alcohol-treated wild-type mice)
4.6. The Biochemical Origin of ALD Biomarkers
These results showed a significant difference between B6 and 129/Sv animals with respect to alcohol metabolism, fatty acid metabolism, amino acid metabolism and gut flora metabolism as shown in Fig. 6. In spite such widespread difference due to genetic background, two α-hydroxy acid metabolites, namely, indole-3-lactic acid and phenyllactic acid were exclusively elevated in the urine of alcohol-treated Ppara-null mice of both backgrounds. This indicates plausible mechanistic association of these metabolites with molecular events associated with ALD pathogenesis in Ppara-null mice. PPARα, which is a master regulator of genes involved in fatty acid β-oxidation, activates the tryptophan-quinolinic acid-NAD+ pathway by down-regulating α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase [52]. This results in the attenuation of NAD+ production in Ppara-null. Since NAD+ is a cofactor for fatty acid oxidation by both β- and ω-oxidation path- ways, the reduced NAD+ biosynthesis makes Ppara-null mice more susceptible to fat deposition in the liver compared to their wild-type counterparts.
Fig. 6.
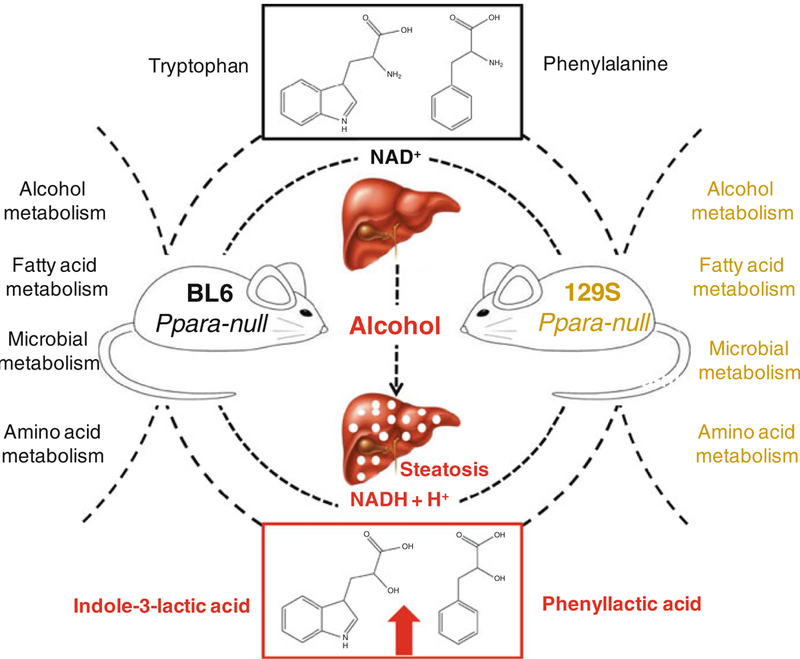
Effect of genetic background on urinary metabolic signatures associated with chronic alcohol treatment of Ppara-null mice. These mice differed with respect to derangement of urinary excretion of metabolites related to alcohol metabolism, fatty acid metabolism, amino acid metabolism and gut flora were affected. However, elevation of indole-3-lactic acid and phenyllactic acid was exclusively associated with ALD pathogenesis irrespective of their genetic background
Alcohol is oxidized stepwise by alcohol dehydrogenase (EC 1.1.1.2) into acetaldehyde in the liver and acetaldehyde to acetic acid by aldehyde dehydrogenase (EC 1.2.1.3) (Fig. 7). Acetic acid can enter the TCA cycle or be a substrate for fatty acid synthesis [53]. However, both reactions consume NAD+ and produce NADH. Therefore, chronic alcohol consumption further decreases the ratio of NAD+/NADH [54] in Ppara-null mice, shifting cellular redox balance more towards reduced state to impair fatty acid catabolism and results in fat deposition in the liver (Fig. 7).
Fig. 7.
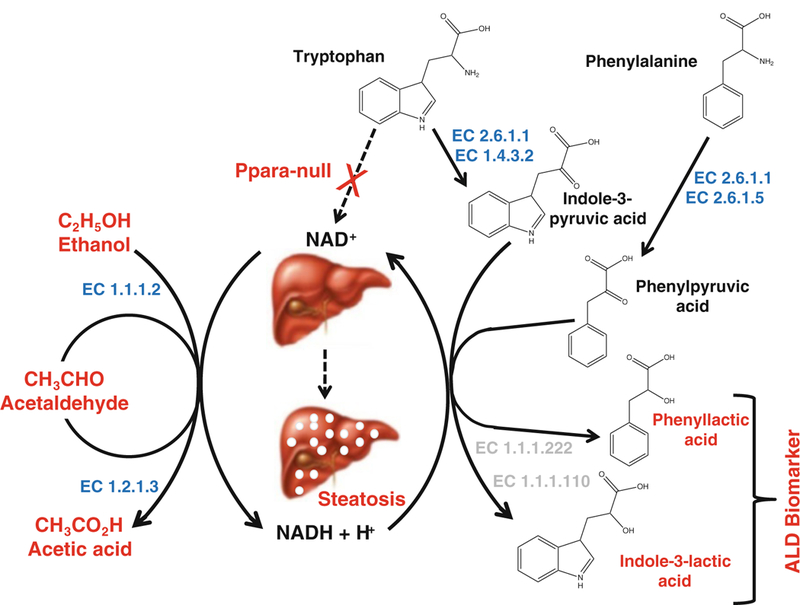
Proposed biochemical mechanism explaining origin of genetic background-independent noninvasive biomarkers of alcohol-induced liver disease in Ppara-null mice. Together with impairment of NAD+ biosynthesis in these mice, oxidation of alcohol leads to a marked shift in redox balance occurs with an increased NADH/NAD+ ratio resulting in impairment of fatty acid oxidation and steatosis in alcohol-treated Ppara-null mice. Concurrent increase in aspartate aminotransferase activity (EC 2.6.1.1) due to liver injury might enhance the deamination of phenylalanine and tryptophan to produce α-keto acids: phenylpyruvic acid and indole-3-pyruvic acid. The elevated NADH/NAD+ ratio could drive the reduction α-keto acid intermediates to the corresponding α-hydroxy acids: phenyllactic acid and indole-3-lactic acid. Enzyme numbers mentioned in blue and light gray indicate enzymes annotated and unannotated in mammals, respectively
Tryptophan is an essential amino acid that is metabolized in the liver. Tryptophan is typically deaminated by l-amino acid oxidase (EC 1.4.3.2) to indole-3-pyruvic acid, an α-keto acid intermediate. It was shown that some microbial AST (EC 2.6.1.1) can also catalyze this reaction, albeit with lower efficiency [55, 56]. Interestingly, aspartate aminotransferase (AST) level also increases during liver injury. Thus it may also contribute to increase in production of to indole-3-pyruvic acid. In the presence of elevated NADH due to alcohol consumption, indole-3-pyruvic acid may be readily reduced to corresponding α-hydroxy acid, i.e., indole-3-lactic acid (Fig. 7). Enzymes responsible for this interconversion have been reported in microbes (EC 1.1.1.120, indolelactate dehydrogenase; and EC 1.1.1.222 (R)-4-hydroxyphenyllactate dehydrogenase) [57]. Tryptophan conversion to indole-3-lactate has been shown in protozoa [58].
Another α-hydroxy acid, phenyllactic acid, was also found to be elevated in the urine of Ppara-null mice following alcohol-treatment. Phenyllactic acid is a product of reduction of the deaminated phenylalanine. Apart from tyrosine aminotransferase (EC 2.6.1.5), aspartate aminotransferase can also catalyze the deamination of phenylalanine to phenylpyruvic acid [56, 59]. However, the shift in the redox balance in the alcohol-treated Ppara-null mice may drive the reduction of this intermediate to phenyllactate, possibly by the action of (R)-4-hydroxyphenyllactate dehydrogenase (EC 1.1.1.222) [60] as depicted in Fig. 7. This enzyme is also not known in mammals.
Taken together, the elevation of these α-hydroxy acids in the alcohol-treated Ppara-null mice strongly suggests common enzymatic pathways linking alcohol- induced liver injury and shift in redox balance. It shows that the increase in metabolic biomarkers is essentially driven by same biochemical events that are associated with alcohol-induced liver damage and steatosis. However, the proposed biochemical pathways highlight lacunae in our understanding of metabolic pathways, particularly, under pathological conditions.
5. Summary and Future Directions
This study showed that mass spectrometry-based metabolic fingerprinting can be a powerful tool in identification of noninvasive signature for detection of ALD at early stage. The simultaneous use of wild-type and Ppara-null animals helped to distinguish signatures of alcohol exposure from those associated with ALD pathogenesis. It showed that an endogenous metabolomic signature may be helpful in prediction of ALD susceptibility as well as ALD diagnosis even in absence of reliable information on alcohol consumption. Validation of metabolomic signatures in different genetic backgrounds helped to identify robust biomarkers for ALD. These noninvasive biomarkers appeared to simultaneously reflect change in redox balance and ongoing liver injury due to chronic alcohol consumption. In conclusion, this study demonstrated that metabolic signatures can be helpful in early noninvasive screening and diagnosis of ALD. However, these results are yet to be validated in human samples. In addition, to the effect of human genetic background, careful analysis on the effect of food habit, life-style as well as orthogonality of metabolic biomarkers to other liver disease/disorders remains to be undertaken.
Acknowledgements
This work was supported by the National Cancer Institute Intramural Research Program, the National Institute of Environmental Health Sciences grant (U01ES016013).
Abbreviations
- ALD
Alcohol-induced liver disease
- ALT
Alanine aminotransferase
- ANOVA
Analysis of variance
- AST
Aspartate aminotransferase
- ESI+
Electrospray ionization in positive mode
- ESI−
Electrospray ionization in negative mode
- MRM
Multiple reaction monitoring
- NAD+
Oxidized nicotinamide adenine dinucleotide
- NADH
Reduced nicotinamide adenine dinucleotide
- OPLS
Orthogonal projection to latent structures
- PCA
Principal components analysis
- Ppara-null
Peroxisome proliferator-activated receptor alpha knock-out mouse model
- PPARα
Peroxisome proliferator-activated receptor alpha
- UPLC-ESI-QTOF-MS
Ultraperformance liquid chromatography coupled with electrospray ionization quadrupole time-of-flight mass spectrometry
References
- 1.Hoyert DL, Heron MP, Murphy SL, Kung HC (2006) Deaths: final data for 2003. Natl Vital Stat Rep 54(13):1–120 [PubMed] [Google Scholar]
- 2.Mandayam S, Jamal MM, Morgan TR (2004) Epidemiology of alcoholic liver disease. Semin Liver Dis 24(3):217–232 [DOI] [PubMed] [Google Scholar]
- 3.WHO (2011) Global status report on alcohol and health World Health Organization, Geneva [Google Scholar]
- 4.Shibuya A, Yoshida A (1988) Genotypes of alcohol-metabolizing enzymes in Japanese with alcohol liver diseases: a strong association of the usual Caucasian-type aldehyde dehydrogenase gene (ALDH1(2)) with the disease. Am J Hum Genet 43(5):744–748 [PMC free article] [PubMed] [Google Scholar]
- 5.Pirmohamed M, Kitteringham NR, Quest LJ et al. (1995) Genetic polymorphism of cytochrome P4502E1 and risk of alcoholic liver disease in Caucasians. Pharmacogenetics 5(6):351–357 [DOI] [PubMed] [Google Scholar]
- 6.Tanaka F, Shiratori Y, Yokosuka O, Imazeki F, Tsukada Y, Omata M (1996) High incidence of ADH2*1/ALDH2*1 genes among Japanese alcohol dependents and patients with alcoholic liver disease. Hepatology 23(2):234–239 [DOI] [PubMed] [Google Scholar]
- 7.Zintzaras E, Stefanidis I, Santos M, Vidal F (2006) Do alcohol-metabolizing enzyme gene polymorphisms increase the risk of alcoholism and alcoholic liver disease? Hepatology 43(2):352–361 [DOI] [PubMed] [Google Scholar]
- 8.Sherva R, Rice JP, Neuman RJ, Rochberg N, Saccone NL, Bierut LJ (2009) Associations and interactions between SNPs in the alcohol metabolizing genes and alcoholism phenotypes in European Americans. Alcohol Clin Exp Res 33(5):848–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Auguet T, Vidal F, Broch M et al. (2010) Polymorphisms in the interleukin-10 gene promoter and the risk of alcoholism and alcoholic liver disease in Caucasian Spaniard men. Alcohol 44(3):211–216 [DOI] [PubMed] [Google Scholar]
- 10.Linneberg A, Gonzalez-Quintela A, Vidal C et al. (2010) Genetic determinants of both ethanol and acetaldehyde metabolism influence alcohol hypersensitivity and drinking behaviour among Scandinavians. Clin Exp Allergy 40(1):123–130 [DOI] [PubMed] [Google Scholar]
- 11.O’Shea RS, Dasarathy S, McCullough AJ (2010) Alcoholic liver disease. Am J Gastroenterol 105(1):14–32; quiz 33 [DOI] [PubMed] [Google Scholar]
- 12.Chen AC, Manz N, Tang Y et al. (2010) Single-nucleotide polymorphisms in corticotropin releasing hormone receptor 1 gene (CRHR1) are associated with quantitative trait of event-related potential and alcohol dependence. Alcohol Clin Exp Res 34(6):988–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grove J, Brown AS, Daly AK, Bassendine MF, James OF, Day CP (1998) The RsaI polymorphism of CYP2E1 and susceptibility to alcoholic liver disease in Caucasians: effect on age of presentation and dependence on alcohol dehydrogenase genotype. Pharmacogenetics 8(4):335–342 [DOI] [PubMed] [Google Scholar]
- 14.Wong NA, Rae F, Bathgate A, Smith CA, Harrison DJ (2000) Polymorphisms of the gene for microsomal epoxide hydrolase and susceptibility to alcoholic liver disease and hepatocellular carcinoma in a Caucasian population. Toxicol Lett 115(1):17–22 [DOI] [PubMed] [Google Scholar]
- 15.Teli MR, Day CP, Burt AD, Bennett MK, James OF (1995) Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 346(8981):987–990 [DOI] [PubMed] [Google Scholar]
- 16.MacSween RN, Burt AD (1986) Histologic spectrum of alcoholic liver disease. Semin Liver Dis 6(3):221–232 [DOI] [PubMed] [Google Scholar]
- 17.Schutte K, Bornschein J, Malfertheiner P (2009) Hepatocellular carcinoma—epidemiological trends and risk factors. Dig Dis 27(2):80–92 [DOI] [PubMed] [Google Scholar]
- 18.Sherman M (2010) Hepatocellular carcinoma: New and emerging risks. Dig Liver Dis 42(Suppl 3):S215–S222 [DOI] [PubMed] [Google Scholar]
- 19.Levitsky J, Mailliard ME (2004) Diagnosis and therapy of alcoholic liver disease. Semin Liver Dis 24(3):233–247 [DOI] [PubMed] [Google Scholar]
- 20.Menon KV, Gores GJ, Shah VH (2001) Pathogenesis, diagnosis, and treatment of alcoholic liver disease. Mayo Clin Proc 76(10):1021–1029 [DOI] [PubMed] [Google Scholar]
- 21.Crabb DW, Liangpunsakul S (2006) Alcohol and lipid metabolism. J Gastroenterol Hepatol 21(Suppl 3):S56–S60 [DOI] [PubMed] [Google Scholar]
- 22.Lee SS, Pineau T, Drago J et al. (1995) Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol 15(6):3012–3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin PG, Guillou H, Lasserre F et al. (2007) Novel aspects of PPARalpha-mediated regulation of lipid and xenobiotic metabolism revealed through a nutrigenomic study. Hepatology 45(3):767–777 [DOI] [PubMed] [Google Scholar]
- 24.Rakhshandehroo M, Sanderson LM, Matilainen M et al. (2007) Comprehensive analysis of PPARalpha-dependent regulation of hepatic lipid metabolism by expression profiling. PPAR Res 2007:26839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sozio M, Crabb DW (2008) Alcohol and lipid metabolism. Am J Physiol Endocrinol Metab 295(1):E10–E16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakajima T, Kamijo Y, Tanaka N et al. (2004) Peroxisome proliferator-activated receptor alpha protects against alcohol-induced liver damage. Hepatology 40(4):972–980 [DOI] [PubMed] [Google Scholar]
- 27.Mancinelli R, Ceccanti M (2009) Biomarkers in alcohol misuse: their role in the prevention and detection of thiamine deficiency. Alcohol Alcohol 44(2):177–182 [DOI] [PubMed] [Google Scholar]
- 28.Sharpe PC (2001) Biochemical detection and monitoring of alcohol abuse and abstinence. Ann Clin Biochem 38(Pt 6):652–664 [DOI] [PubMed] [Google Scholar]
- 29.Saadeh S (2007) Nonalcoholic Fatty liver disease and obesity. Nutr Clin Pract 22(1):1–10 [DOI] [PubMed] [Google Scholar]
- 30.Calvaruso V, Craxi A (2009) Implication of normal liver enzymes in liver disease. J Viral Hepat 16(8):529–536 [DOI] [PubMed] [Google Scholar]
- 31.Cadranel JF, Rufat P, Degos F (2000) Practices of liver biopsy in France: results of a prospective nationwide survey. For the Group of Epidemiology of the French Association for the Study of the Liver (AFEF). Hepatology 32(3):477–481 [DOI] [PubMed] [Google Scholar]
- 32.Tyburski JB, Patterson AD, Krausz KW et al. (2008) Radiation metabolomics. 1. Identification of minimally invasive urine biomarkers for gamma-radiation exposure in mice. Radiat Res 170(1):1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patterson AD, Lanz C, Gonzalez FJ, Idle JR (2009) The role of mass spectrometry-based metabolomics in medical countermeasures against radiation. Mass Spectrom Rev 29(3): 503–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sreekumar A, Poisson LM, Rajendiran TM et al. (2009) Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 457(7231):910–914 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 35.MacIntyre DA, Jimenez B, Lewintre EJ et al. (2010) Serum metabolome analysis by 1H-NMR reveals differences between chronic lymphocytic leukaemia molecular subgroups. Leukemia 24(4):788–797 [DOI] [PubMed] [Google Scholar]
- 36.Loftus N, Barnes A, Ashton S et al. (2011) Metabonomic investigation of liver profiles of non- polar metabolites obtained from alcohol-dosed rats and mice using high mass accuracy MSn analysis. J Proteome Res 10(2):705–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bradford BU, O’Connell TM, Han J et al. (2008) Metabolomic profiling of a modified alcohol liquid diet model for liver injury in the mouse uncovers new markers of disease. Toxicol Appl Pharmacol 232(2):236–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernando H, Kondraganti S, Bhopale KK et al. (2010) (1)H and (3)(1)P NMR lipidome of ethanol-induced fatty liver. Alcohol Clin Exp Res 34(11):1937–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pearson K (1901) On lines and planes of closest fit to systems of points in space. Philos Mag 2(6):559–572 [Google Scholar]
- 40.Suhre K, Schmitt-Kopplin P (2008) MassTRIX: mass translator into pathways. Nucleic Acids Res 36(Web Server Issue):W481–W484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jansson J, Willing B, Lucio M et al. (2009) Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS One 4(7):e6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kind T, Fiehn O (2007) Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics 8:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cui Q, Lewis IA, Hegeman AD et al. (2008) Metabolite identification via the Madison Metabolomics Consortium Database. Nat Biotechnol 26(2):162–164 [DOI] [PubMed] [Google Scholar]
- 44.Smith CA, O’Maille G, Want EJ et al. (2005) METLIN: a metabolite mass spectral database. Ther Drug Monit 27(6):747–751 [DOI] [PubMed] [Google Scholar]
- 45.Nguyen PV, Abel T, Kandel ER, Bourtchouladze R (2000) Strain-dependent differences in LTP and hippocampus-dependent memory in inbred mice. Learn Mem 7(3):170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Syn WK, Yang L, Chiang DJ et al. (2009) Genetic differences in oxidative stress and inflammatory responses to diet-induced obesity do not alter liver fibrosis in mice. Liver Int 29(8): 1262–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu J, Corton C, Dix DJ, Liu Y, Waalkes MP, Klaassen CD (2001) Genetic background but not metallothionein phenotype dictates sensitivity to cadmium-induced testicular injury in mice. Toxicol Appl Pharmacol 176(1):1–9 [DOI] [PubMed] [Google Scholar]
- 48.Manna SK, Patterson AD, Yang Q et al. (2010) Identification of noninvasive biomarkers for alcohol-induced liver disease using urinary metabolomics and the Ppara-null mouse. J Proteome Res 9(8):4176–4188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manna SK, Patterson AD, Yang Q et al. (2011) UPLC–MS-based urine metabolomics reveals indole-3-lactic acid and phenyllactic acid as conserved biomarkers for alcohol-induced liver disease in the Ppara-null mouse model. J Proteome Res 10(9):4120–4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Helander A, Bottcher M, Fehr C, Dahmen N, Beck O (2009) Detection times for urinary ethyl glucuronide and ethyl sulfate in heavy drinkers during alcohol detoxification. Alcohol Alcohol 44(1):55–61 [DOI] [PubMed] [Google Scholar]
- 51.Hoiseth G, Bernard JP, Stephanson N et al. (2008) Comparison between the urinary alcohol markers EtG, EtS, and GTOL/5-HIAA in a controlled drinking experiment. Alcohol Alcohol 43(2):187–191 [DOI] [PubMed] [Google Scholar]
- 52.Shin M, Kim I, Inoue Y, Kimura S, Gonzalez FJ (2006) Regulation of mouse hepatic alpha- amino-beta-carboxymuconate-epsilon-semialdehyde decarboxylase, a key enzyme in the tryptophan-nicotinamide adenine dinucleotide pathway, by hepatocyte nuclear factor 4alpha and peroxisome proliferator-activated receptor alpha. Mol Pharmacol 70(4):1281–1290 [DOI] [PubMed] [Google Scholar]
- 53.Lumeng L, Crabb DW (2001) Alcoholic liver disease. Curr Opin Gastroenterol 17(3): 211–220 [DOI] [PubMed] [Google Scholar]
- 54.Kalant H, Khanna JM, Loth J (1970) Effect of chronic intake of ethanol on pyridine nucleotide levels in rat liver and kidney. Can J Physiol Pharmacol 48(8):542–549 [DOI] [PubMed] [Google Scholar]
- 55.Recasens M, Benezra R, Basset P, Mandel P (1980) Cysteine sulfinate aminotransferase and aspartate aminotransferase isoenzymes of rat brain. Purification, characterization, and further evidence for identity. Biochemistry 19(20):4583–4589 [DOI] [PubMed] [Google Scholar]
- 56.Yagi T, Kagamiyama H, Motosugi K, Nozaki M, Soda K (1979) Crystallization and properties of aspartate aminotransferase from Escherichia coli B. FEBS Lett 100(1):81–84 [DOI] [PubMed] [Google Scholar]
- 57.Jean M, DeMoss RD (1968) Indolelactate dehydrogenase from Clostridium sporogenes. Can J Microbiol 14(4):429–435 [DOI] [PubMed] [Google Scholar]
- 58.Leelayoova S, Marbury D, Rainey PM, Mackenzie NE, Hall JE (1992) In vitro tryptophan catabolism by Leishmania donovani donovani promastigotes. J Protozool 39(2):350–358 [DOI] [PubMed] [Google Scholar]
- 59.Owen TG, Hochachka PW (1974) Purification and properties of dolphin muscle aspartate and alanine transaminases and their possible roles in the energy metabolism of diving mammals. Biochem J 143(3):541–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bode R, Lippoldt A, Birnbaum D (1986) Purification and properties of D-aromatic lactate dehydrogenase, an enzyme involved in the catabolism of the aromatic amino acids of Candida maltosa. Biochem Physiol Pflanz 181:189–198 [Google Scholar]