

Abstract
Preclinical investigations support the concept that donor cells more oriented towards a cardiovascular phenotype favor repair. In light of this philosophy, we previously identified HDAC1 as a mediator of cardiac mesenchymal cell (CMC) cardiomyogenic lineage commitment and paracrine signaling potency in vitro—suggesting HDAC1 as a potential therapeutically exploitable target to enhance CMC cardiac reparative capacity. In the current study we examined the effects of pharmacologic HDAC1 inhibition, using the benzamide class 1 isoform selective HDAC inhibitor entinostat (MS-275), on CMC cardiomyogenic lineage commitment and CMC-mediated myocardial repair in vivo. Human CMCs pretreated with entinostat or DMSO diluent control were delivered intramyocardially in an athymic nude rat model of chronic ischemic cardiomyopathy 30 days after a reperfused myocardial infarction. Indices of cardiac function were assessed by echocardiography and left ventricular (LV) Millar conductance catheterization 35 days after treatment. Compared with naïve CMCs, entinostat-treated CMCs exhibited heightened capacity for myocyte-like differentiation in vitro and superior ability to attenuate LV remodeling and systolic dysfunction in vivo. The improvement in CMC therapeutic efficacy observed with entinostat pretreatment was not associated with enhanced donor cell engraftment, cardiomyogenesis, or vasculogenesis, but instead with more efficient inhibition of myocardial fibrosis and greater increase in myocyte size. These results suggest that HDAC inhibition enhances the reparative capacity of CMCs, likely via a paracrine mechanism that improves ventricular compliance and contraction and augments myocyte growth and function.
Keywords: Cardiac Mesenchymal Cell Therapy, Histone Deacetylase Inhibitors, Myocardial Fibrosis, Myocardial Infarction, Paracrine Signaling, Cardiomyogenic Lineage Commitment
Introduction
As the incidence of heart failure (HF) grows, novel therapeutic interventions that effectively attenuate disease progression remain in extraordinary demand. Though beta-blockers, angiotensin-converting-enzyme (ACE) inhibitors, and aldosterone antagonists provide some clinical benefit to heart failure patients, they essentially comprise only palliative measures that fail to directly address disease etiology—namely the loss of cardiomyocytes, ongoing myocardial attrition, and advancing ventricular fibrosis [31]. To this end, cell therapy has materialized as a promising therapeutic modality that limits ventricular detrimental remodeling and enhances functional recovery in preclinical models of heart failure [4, 9, 18, 30, 48]; nonetheless, even with years of ongoing scientific research, the precise mechanism(s) by which cell therapy promotes post-infarct cardiac recovery largely remains a “black-box,” as are the phenotypic properties that afford donor cells their therapeutic abilities. To date, the cell therapy field is replete with various purported cardiomyocyte progenitors or stem cells; however, there is limited durable evidence that any of these so called cardiac progenitors are sufficiently retained or contribute significantly and directly to cardiomyogenesis after transplantation [4, 9–11, 18]—providing indirect support for a paracrine mechanism of action. Despite such observations, many of these cells with “cardiac potential,” such as c-kit+ sorted [36–38, 40] or unfractionated cardiac-derived mesenchymal cells (CMCs) [6, 14, 28, 41], have been tested and validated as agents that improve cardiac function. It is, therefore, possible that cells with cardiac potential—though never fully realized in the form of becoming adult myocytes—contribute to cardiac repair. Furthermore, it is conceivable that such cardiac potential is necessary for the salutary effects of cell therapy.
The notion that cardiomyogenic lineage commitment is an important element influencing donor cell reparative aptitude is supported by previous studies demonstrating that donor cell cardiogenic transcription factor expression (i.e., Nkx2.5, Tbx5, and Mef2c) can bias cardiac repair [2, 28]. Although the fundamental mechanisms underlying these effects remain unresolved, these observations support the idea that one can effectively enhance donor cell therapeutic efficacy by augmenting their cardiovascular lineage commitment. For this reason, various methodologies have been employed in effort to “forward reprogram” or stimulate mesenchymal progenitor cell cardiomyogenic lineage specification; this has been achieved using various experimental approaches, including recombinant protein overexpression as well as pharmacologic pre-treatment. The former includes ectopic expression of cardiogenic transcription factors (e.g., Gata4, Mef2c, and Tbx5) [12] and the latter, exposure to chromatin modifying agents, such as inhibitors of histone deacetylase (HDACi) [23, 43] or DNA methyltransferase (DNMTi) [17, 24, 27] enzymatic activity. Consistent with these reports, we previously identified HDAC1 as a novel regulator of CMC cardiovascular lineage specification [23]. We found that short-hairpin RNA interference (shRNAi)-targeted depletion of HDAC1 stimulated cardiogenic transcriptional program activation and cardiomyocyte-like differentiation in patient-derived CMCs in vitro [23]. Importantly, relative to naïve CMCs, HDAC1-depleted, cardiomyogenic lineage-committed CMCs exhibited distinct trophic factor secretion profiles and heightened in vitro paracrine signaling potency [22]—highlighting HDAC1 as a novel therapeutic target that may be exploited to augment CMC cardiac reparative capacity. As the next logical step in this work, the current study examined the influence of pharmacologic HDAC1 inhibition (using the benzamide HDAC inhibitor, entinostat) on CMC cardiomyogenic lineage commitment and on CMC-mediated myocardial repair in vivo.
Materials and Methods
Human Cardiac Mesenchymal Stromal Cell Isolation
Human CMCs were isolated from discarded right atrial appendage specimens collected from patients during routine coronary artery bypass surgery at Jewish Hospital (University of Louisville, Louisville, KY) according to a previously established collagenase digestion protocol [22, 23]. Further, all established CMC cell lines were validated via flow cytometry-based verification of the presence and absence of prototypical mesenchymal and hematopoietic cell lineage markers, respectively, as previous [23]. De-identified right atrial appendage specimens were collected via written consent agreement according to the approved protocol by the Institutional Review Board on human subject research (IRB number: 03.052J) at the University of Louisville.
Cells, Cell Culture, and Treatment
Primary patient-derived CMCs were cultured in Ham’s F12 medium (Gibco) supplemented with 10% FBS (Seradigm), 20 ng/ml recombinant human bFGF (PeproTech), 0.2 mM L-Glutamine (Gibco), 0.005 U/ml human erythropoietin (Invitrogen), and 100 U/ml penicillin/streptomycin (Gibco). CMCs were not used beyond passage number 8. All cell lines were maintained under standard incubation conditions at 37°C with 5% atmospheric CO2 and passaged using TrypLE™ (ThermoFisher Scientific) when approaching ≈70% confluence. 100 mM stock solutions of entinostat (S1053; Selleck Chemicals) were prepared by solubilizing lyophilized powder in cell culture grade dimethyl sulfoxide (DMSO).
Western Blotting
Immunoblotting was performed according to previously described protocols [22, 23]. A detailed list of antibodies with corresponding dilutions is available in Supporting Information Table S1.
Quantitative PCR
RNA isolation, reverse transcription, and quantitative real-time PCR was performed as previously described [22, 23]. A detailed list of transcript-specific primers is available in Supporting Information Table S2.
Flow Cytometry
For intracellular myocyte lineage-specific protein detection, CMCs were fixed with 4% formaldehyde for 10 min at room temperature, washed in 1× PBS, and permeabilized with 90% methanol on ice for 30 min. Cells were incubated with primary antibodies or isotype controls diluted in incubation buffer (1% BSA, 1× PBS) for 1 h at room temperature, washed by centrifugation (600g for 5 min at 4°C) in incubation buffer, and resuspended in fluorochrome-conjugated secondary antibodies for 30 min at room temperature. Flow cytometric analyses were performed as previously [23] using a BD LSR II Flow Cytometer (BD Biosciences). Briefly, 50,000 events were collected per sample and analyzed using Flowing Software version 2.5.0 (Perttu Terho, Turku Centre for Biotechnology). The percentage of positive cells was determined by gating against corresponding isotype controls. A complete list of utilized antibodies is detailed in Supporting Information Table S3.
CMC Conditioned Medium Harvest and Cytokine Array Analyses
CMCs were grown in T75 tissue culture flasks with complete medium supplemented with 1 μM entinostat or DMSO diluent control for 5 days. Culture medium was replaced every two days throughout this duration. On day 5, cells were washed with 10 ml of 1× PBS and subsequently incubated with 10 ml of serum free, Ham’s F12 medium (Gibco) supplemented with 0.5% bovine serum albumin for 24 h. Conditioned medium was harvested via a sterile serological pipette and transferred to 15 ml conical tubes. Detached cells were then removed by 600g centrifugation at 4°C for 10 min. Cytokine arrays were generated using the Proteome Profiler Human XL Cytokine Array Kit (R&D Systems, Minneapolis, MN), according to the manufacturers’ protocol. For each assay, a total of 1 ml of unconcentrated cell culture supernatant/conditioned medium was used. Resultant membranes were imaged with a chemiluminescent image analyzer (MyECL; ThermoFisher Scientific) and analyzed by densitometric quantification (ImageJ; NIH, Bethesda, MD). Resultant pixel density values were imported into R version 2.15.1 × 64 bit (The R Project for Statistical Computing) and subject to hierarchical cluster analysis using the heatmap.2 function in the gplots library. Each conditioned medium experiment (corresponding to either entinostat or DMSO diluent treatment) consisted of a 1:1:1 mixture of 3 independent patient-derived CMC lines. Conditioned medium cytokine arrays, sourced from entinostat- or DMSO-treated CMCs, were performed using biological duplicates (n=2 for each).
Ischemia Reperfusion and CMC Injection Procedures
All animal experiments were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of health (Eight Edition, Revised 2010) and with the guidelines of the Animal Care and Use Committee of the University of Louisville, School of Medicine (Louisville, KY, USA); and therefore were carried out in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Similar to our previously performed rat ischemia-reperfusion injury procedure [36], female athymic homozygous nude rats (genotype, Foxn1rnu/rnu; supplier, Charles River Laboratories; age, 2-3 months; weight, 150-180 g) were anesthetized with ketamine (37 mg/kg) and xylazine (5 mg/kg) and intubated with a rodent respirator (Harvard Apparatus). Anesthesia was maintained with isoflurane inhalation and body temperature sustained at 37 ⁰C with a heating pad. Following administration of antibiotics, the chest was opened via left thoracotomy and the heart exposed. All animals subsequently underwent 90-min occlusion of the left anterior descending coronary artery followed by reperfusion, after which the chest wall was sutured closed. Thirty days after reperfusion, rats were re-anesthetized, and the heart exposed via left anterolateral thoracotomy. At this time, rats received intramyocardial injections (peri-infarct) of DMSO-treated CMCs, entinostat-treated CMCs, or vehicle (1× PBS; phosphate-buffered saline). In effort to limit patient-dependent variables (e.g., disease state, age, gender, etc.), each animal received a mixture of CMCs sourced from three randomly selected patient samples. CMC-groups received a total of 2 million CMCs per heart (4 injections; 500,000 cells per injection). Following treatment, all groups received 5-Bromo-2´-deoxyuridine (BrdU) ad libitum in drinking water for 35 d to label newly proliferating cells. All rat groups were euthanized 35 d after injection (65 days after MI).
Power Analyses, Exclusion Criteria, and Randomization
A total of more than 30 rats (2-3 months of age) were required for successful completion of the study. This number was calculated via implementation of a statistical power analysis using SigmaStat Software (SigmaStat, Systat Software, Inc.). Based on our preliminary in vivo studies with human CMCs, we determined a 20% minimum detectable difference in means with an expected standard deviation of residuals of 15%. Further, with these groups, a desired power of 80%, and an alpha of 0.05 (95th percentile), we calculated a requisite number of approximately 10-12 animals per group (vehicle-, DMSO CMC-, and entinostat CMC-treated rats). A priori exclusion criteria were established whereby any animal not exhibiting a ≥ 15% reduction in left ventricular ejection fraction (ΔEF of ≥ 15% 30 days after injury relative to baseline) were removed from the study. Based on such criteria, animals deemed acceptable for study inclusion (a total of 31 rats) were randomly assigned to control (vehicle; n=11) or treatment (DMSO CMC- and entinostat CMC-treated; n=10 for both) groups using the Mersenne Twister algorithm available in Microsoft Excel.
Echocardiographic procedures and analyses
Indices of cardiac function and ventricle dimensions were assessed via echocardiography under light anesthesia (pentobarbital, 25 mg/kg, i.p., as previously described [33, 36]) with serial measurements obtained at baseline (prior to MI), 30 days post MI (prior to treatment), and 65 days post MI (35 days after treatment) (Fig. 4A). The anterior chest was shaved, and rats were placed in the left lateral decubitus position. Body temperature was maintained between 36.9 ⁰C and 37.3 ⁰C. Echocardiographic images were acquired using an HP SONOS 7500 ultrasound system outfitted with a L12-5 linear broadband and an S12 phased array transducer equipped with a 0.3 cm standoff. Hearts were imaged in the para-sternal long axis view to measure LV end-systolic and end-diastolic volumes (LVESV and LVEDV, respectively), as well as ejection fraction (EF). All measurements were averaged in three consecutive cardiac cycles and analyzed off-line by a single blinded observer using COMPACS image analysis software. All calculations were derived using standard formulas and analyzed according to modified American Society for Echocardiography standards [16]. Echocardiograms were collected from all 31 animals, which consisted of control (vehicle; n=11) and CMC treatment (DMSO CMC- and entinostat CMC-treated; n=10 for both) groups.
Fig. 4.

Epigenetically modified CMCs more efficiently attenuate post-MI ventricular remodeling and dysfunction in vivo. a Schematic diagram detailing the timeline of ischemia-reperfusion procedures, serial echocardiogram collection (corresponding to baseline, pre-treatment [pre-Tx], and post-treatment [post-Tx]), and cell injections. At associated time points, ventricular volumes (b end-diastolic volume (μl) and c end-systolic volume (μl)) and functional parameters (d stroke volume (μl), e heart rate (BPM), f cardiac output (ml/min), g ejection fraction (%), and i fractional shorting (%)) were calculated for each group. The change in h ejection fraction (ΔEF) and j fractional shortening (ΔFS) from pre-treatment to post-treatment is depicted in accompanying dot plots. All graphs report mean ± SEM for treatment groups consisting of vehicle- (n=11), DMSO CMC- (n=10), and entinostat CMC- (n=10) injected animals.
Hemodynamic procedures and analyses
Hemodynamic studies were performed 65 days after MI, just prior to euthanasia as previous [36–38]. Rats were anesthetized with ketamine (37 mg/kg) and xylazine (5 mg/kg), intubated, and mechanically ventilated. Anesthesia was maintained with 1% isoflurane and the core temperature sustained at 37 ⁰C with a heating pad for the duration of the procedure. A 2F micro-tip pressure-volume (PV) catheter (SPR-869, Millar Instruments) was inserted into the right carotid artery and advanced into the LV cavity. The right jugular vein was cannulated for fluid administration. After 20 min of stabilization, the PV signals were recorded continuously with an ARIA PV conductance system (Millar Instruments) coupled with a Powerlab/4SP A/D converter (AD instruments), stored, and displayed on a personal computer. PV relations were assessed by transiently compressing the inferior vena cava with a cotton swab. Parallel conductance from surrounding structures was calculated by injecting a small bolus of 15% NaCl through the jugular vein. LV end-diastolic pressure (LVEDP), dP/dtmax and dP/dtmin, end-systolic elastance (Ees), and adjusted maximal power were calculated using the PVAN software program Millar [36–38]. Conductance catheterization was performed on all 31 animals, however 1 vehicle-treated animal died during the procedure, which brought group totals to n=10 for vehicle and n=10 for both DMSO CMC and entinostat CMC treatment groups.
General Histology and Morphometry Procedures
Following hemodynamic measurements, a polyethylene catheter filled with phosphate buffer (0.2 M, pH 7.4) and heparin (100 IU/ml) was advanced to the ascending aorta via the right carotid artery. In rapid succession, the heart was arrested in diastole by injecting 1 ml of a mixture of cadmium chloride (100 mM) and potassium chloride (3 M) through the aortic catheter. Subsequently, the heart was excised and retrogradely perfused with phosphate buffer for 3 min to clear residual blood within the coronary circulation, and perfused with 10% neutral buffered formalin solution for 15 min. Perfusion pressures were maintained between 60 and 80 mmHg while end-diastolic pressures were retained at 8 mmHg. After perfusion-fixation, the atria and right ventricle were dissected from the left ventricle. The LV weight was measured and recorded. Each heart was then cut into 5 transverse slices (each 3 mm thick), processed, paraffin-embedded, sectioned at 4 μm intervals, and stained with Masson’s trichrome, picrosirius red, or antibodies recognizing cardiovascular cell lineage-specific markers. A complete list of utilized antibodies is detailed in Supporting Information Table S4.
Infarct Size Determination by Masson’s Trichrome Staining
Masson’s trichrome staining was performed according to the manufacturer’s instructions (HT15-1KT; Sigma-Aldrich) as previously described [38]. Briefly, formalin-fixed, paraffin-embedded myocardial tissue sections were heated at 80°C for 30 min, deparaffinized in xylene, and stepwise rehydrated via incubation in decreasing concentrations of ethanol (100%, 96%, 90%, and 80%) and deionized water. Tissue sections were subsequently incubated in Bouin’s solution (57211, ThermoFisher Scientific) at 55°C for 15 min and left to cool for 5 min. Sections were then washed with deionized water for 15 min, left to rest in deionized water for 5 min, and then stained with Biebrich Scarlet-Acid Fuchsin solution (HT151, Sigma-Aldrich) for 10 min. Tissue sections were next rinsed in deionized water for 5 min and placed in working phosphotungstic acid (HT152, Sigma-Aldrich)/phosphomolybdic acid (HT153, Sigma-Aldrich)/deionized water solution for 5 min. Sections were successively counter-stained in aniline blue (HT154, Sigma-Aldrich) for 10 min, rinsed in 1% acetic acid for 1 min, and stepwise dehydrated with incubation in increasing concentrations of ethanol (80%, 90%, 96%, and 100%). Tissue sections were finally mounted under glass coverslips using Permount Mounting Media (SP15-100, ThermoFisher Scientific). Images were digitally acquired at 1× magnification using a Nikon Eclipse Ni-E light microscope and analyzed using NIH ImageJ software (1.46r). Morphometric parameters, including total LV area, remote area, risk area, and scar area, were measured in each section. In accordance with our previous work [38], the risk region was defined as the sum of the LV segment containing the infarct scar and the two border zones (the region that encompasses 0.5 mm on either side of the lateral borders of the scar). Total scar size (mass in mg) was enumerated by summation of scar masses corresponding to each transverse myocardial slice (apex to base; 5 slices per heart) .
Quantification of Myocardial Collagen Content by Picrosirius Red Staining
To evaluate cardiac fibrosis, myocardial collagen content was enumerated in Picrosirius Red stained sections via quantitative analysis of polarized light microscopy images, as previously performed [41]. As above, formalin-fixed, paraffin-embedded myocardial tissue sections were heated at 80°C for 30 min, deparaffinized in xylene, and stepwise rehydrated via incubation in decreasing concentrations of ethanol (100%, 96%, 90%, and 80%) and deionized water. Picrosirius Red stain was prepared using 0.1% (w/v) Direct Red 80 (365548-5G, Sigma-Aldrich) in picric acid (P6744-1GA, Sigma-Aldrich). For each heart, mid-papillary muscle level myocardial sections were incubated in Picrosirius Red for 1 h, washed in 0.5% acetic acid (2 times; 1 min each), stepwise dehydrated with ethanol, and mounted under glass coverslips using Permount Mounting Media (SP15-100, ThermoFisher Scientific). Images were digitally acquired using a Nikon Eclipse Ni-E microscope at both 1× magnification under brightfield and at 4× magnification under polarized light. Acquired polarized light images (for each myocardial section) were digitally reconstructed using the Stitching Plug-in for NIH ImageJ (1.46r) [26]. Subsequently, a signal threshold was established and applied to all acquired polarized light images (including stained sections from all groups) prior to digital quantification in ImageJ (1.46r). Results are expressed as the percent area of collagen per myocardial tissue region ± SEM.
General Immunostaining Procedures
Transverse myocardial tissue sections were heated at 80°C for 30 min, deparaffinized with xylene, and stepwise rehydrated by successive rinsing in decreasing concentrations of ethanol (100%, 96%, 90%, and 80%) prior to placing in deionized water. Antigen retrieval was then performed by submerging tissue sections in citrate retrieval buffer [2.4 g/L sodium citrate tribasic dehydrate (S4641, Sigma-Aldrich), 0.35 g/L citric acid (C0759, Sigma-Aldrich), pH 6.0] for 10 min at approximately 100 °C. Sections were allowed to cool for 10 min on ice and washed in deionized water prior to application of diluted antibodies raised against cell lineage-specific markers (detailed in Supporting Information Table S4). Primary and secondary antibodies were diluted in antibody diluent reagent (003218, Invitrogen) prior to use. All sections were counterstained with 1 μg/mL DAPI (D3571, Invitrogen) for 10 min and subsequently incubated with 0.1% Sudan Black solution (199664, Sigma-Aldrich) in 70% ethanol for 15 min to mitigate autofluorescence signal. Sections were then washed with 1× PBS (3 times; 3 min each), rinsed in deionized water, and finally mounted under glass coverslips using PermaFluor Aqueous Mounting Medium (TA-030-FM, ThermoFisher Scientific). Images were acquired digitally using a Nikon Eclipse Ti fluorescence microscope and analyzed using NIH Image J software (1.46r).
Human CMC Myocardial Retention
Human CMC myocardial retention was assessed via antibody-mediated detection of human nucleoli antigen (HNA). Briefly, transverse mid-papillary myocardial tissue sections were deparaffinized and rehydrated as above, and permeabilized in 0.2% Triton X-100 in 1× PBS for 15 min. Sections were then subject to antigen retrieval using citrate retrieval buffer, washed in deionized water, and blocked with 2% horse serum (16050130, Invitrogen) in antibody diluent reagent (003218, Invitrogen) for 2 h at room temperature. Slides were subsequently incubated with anti-HNA antibody (AB190710, Abcam) overnight at 4°C, washed in 1× PBS (3 times), and incubated with Alexa Fluor 488-conjugated secondary antibody (A21202, Invitrogen) at 37°C for 1 h. In effort to identify CMC progeny assuming a mature cardiovascular cell phenotype, tissue sections were also co-stained with cardiovascular cell lineage-specific markers (e.g., sarcomeric and smooth muscle actin) to identify HNA+ cardiomyocytes and/or vessels, respectively. Thus, tissue sections were subsequently counterstained with α-sarcomeric actin antibody (A2172, Sigma-Aldrich) for 1 h at 37°C, washed with 1× PBS (3 times), and incubated in Alexa Fluor 633-conjugated secondary antibody (A21046, Life Tech); lastly, sections were incubated with α-smooth muscle actin-Cy3-conjugated antibody (SMA) (C6198, Sigma-Aldrich) at 37°C for 1 h, and washed with 1× PBS (3 times). Slides were finally counterstained with DAPI, blocked with 0.1% Sudan Black solution, and mounted under glass coverslips. For each heart, an entire mid-papillary level, transverse myocardial tissue section was visually screened for the presence of HNA+ nuclei and enumerated; any of which were identified to be HNA+ by the operator were imaged at 30× magnification.
Myocardial Capillary Density
To detect capillaries, slides were incubated with fluorescein-labeled Isolectin B4 (FL-1201, Vector Labs) for 3 h at 37°C and washed 3 times in 1× PBS. As an added measure to facilitate capillary identification, slides were also counterstained with rhodamine-conjugated wheat germ agglutinin (WGA) (RL-1022, Vector) for 30 min at room temperature and washed in 1× PBS (3 times). Tissue sections were then counterstained with DAPI, autofluourescence quenched with 0.1% Sudan Black solution, and mounted under glass coverslips. Images were randomly collected from regions corresponding to infarct, border, and remote myocardial zones. Myocardial regions exhibiting significant morphologic heterogeneity (i.e., infarct zones) were imaged with near complete coverage—acquiring approximately 60-80 images per infarct zone; compared to regions which were more uniform in nature, namely border and remote regions, wherein 6-8 images and 4-6 images were collected and enumerated for each myocardial tissue section, respectively. Capillary density (for each interrogated region) is expressed as the average number of capillaries per unit area (mm2) ± SEM.
Myocardial Vessel Density and Proliferation
To enumerate vessel density, as well as proliferation, vessel resident smooth muscles cells were assessed for incorporation of BrdU using a 5-Bromo-2′-deoxy-uridine Labeling and Detection Kit (Sigma-Aldrich), according to the manufacturer’s protocol. Briefly, anti-BrdU antibody was diluted in manufacturer supplied diluent buffer (Sigma-Aldrich), placed on mid-papillary level transverse myocardial sections, incubated at 37°C for 1 h, and washed in 1× PBS (3 times). Slides were then incubated with anti-mouse Ig-fluorescein secondary antibody (11296736001, Sigma-Aldrich) (11296736001, Sigma) at 37°C for 1 h and washed in 1× PBS (3 times). Slides were subsequently counterstained with α-sarcomeric actin antibody (α-SA) at 37°C for 1 h, washed 3 times in 1× PBS, and co-incubated in Alexa Fluor 633 secondary antibody and α-smooth muscle actin-Cy3-conjugated antibody (SMA) (C6198, Sigma-Aldrich) at 37°C for 1 h. After a final wash with 1× PBS, slides were counterstained with DAPI, blocked with 0.1% Sudan Black solution, and mounted under glass coverslips. As mentioned above, myocardial regions exhibiting significant morphologic heterogeneity (i.e., infarct zones) were imaged with near complete coverage—acquiring approximately 60-80 images per infarct zone; as opposed to regions that were more uniform in nature, namely border and remote regions, where 6-8 images and 4-6 images were collected and enumerated for each myocardial tissue section, respectively. Vessel density (for each interrogated region) is expressed as the average number of vessels per unit area (mm2) ± SEM. Vessels were stratified into two groups, those of which were greater than or less than 20 μm in diameter. In addition, the number of vessels containing one or more BrdU+ smooth muscle cells were also enumerated and reported as the mean percentage of BrdU+ vessels per unit area ± SEM. Lastly the total number of proliferating vessel resident smooth muscle cells were counted and reported as the mean percentage of BrdU+ nucleated cells in vessels per myocardial region ± SEM
Cardiomyocyte Proliferation and Cross-Sectional Area
To assess cardiomyocyte proliferation, bromodeoxyuridine (BrdU) labeled cardiomyocytes were identified and enumerated using a 5-Bromo-2′-deoxy-uridine Labeling and Detection Kit (Sigma-Aldrich), according to the manufacturer’s instruction. Briefly, anti-BrdU antibody was diluted in manufacturer supplied diluent buffer (Sigma-Aldrich), placed on mid-papillary level transverse myocardial sections, incubated at 37°C for 1 h, and washed in 1× PBS 3 times. Next, kit supplied anti-mouse Ig-fluorescein secondary antibody (11296736001, Sigma-Aldrich) and rhodamine-conjugated WGA (to demarcate cell membranes) were co-diluted in diluent buffer, placed on myocardial tissue sections for 1 h at 37°C, and subsequently washed in 1× PBS. Sections were then incubated with α-sarcomeric actin antibody (α-SA) for 1 h at 37°C, washed 3 times with 1× PBS, and finally incubated with Alexa Fluor 633 secondary antibody for 1 h at 37°C. Finally, slides were counterstained with DAPI, blocked with 0.1% Sudan Black solution, and mounted under glass coverslips. Myocardial regions corresponding to infarct, border, and remote zones were imaged (as above) and the number of BrdU+ nucleated myocytes enumerated. Per region, approximately 1500-2000 nucleated cardiomyocytes were counted in infarct zones, 500-600 in remote zones, and 500-700 in border zones for each myocardial tissue section. Data are presented as the total number of regional BrdU+ cardiomyocytes per 10,000 nucleated cardiomyocytes ± SEM.
In addition to myocyte proliferation, the same staining procedure was used to assess regional cardiomyocyte cross-sectional area. Here, cardiomyocyte areas were determined in transverse-cut cells with centrally located nuclei. In order to only identify cells cut at a right angle to the long axis, Nikon Elements software [64bit, version 4.13.05 (Build 933)] was used for measurements. Specifically, only cells with a “Shape Factor” equal to and/or above 0.895 were considered for area measurements, which is equivalent to a perpendicular radius ratio >1:1.4. For each heart, approximately 50 cardiomyocytes were measured per myocardial region (infarct, border, and remote zones) of a single mid-papillary muscle level, transverse myocardial section. Regional myocyte cross-sectional area (μm2) is reported as the arithmetic mean ± SEM. Further, cardiomyocyte size distribution is reported in accompanying histograms.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism version 7.00 for Windows (GraphPad Software, La Jolla California USA, www.graphpad.com). Flow cytometry data (reporting percentages) were arcsine transformed and subject to 2-way ANOVA with the post hoc Holm-Sidak’s multiple comparison test. Gene expression data were log base 10 (y=log10(y)) transformed prior to performing 2-way ANOVA with post hoc Holm-Sidak’s multiple comparison test. Immunoblot densitometric data were analyzed for statistical significance using the non-parametric Mann-Whitney test. Echocardiographic and hemodynamic values were subject to 1-way or 2-way ANOVA, where appropriate, followed by the post hoc Holm-Sidak’s multiple comparison test. For cardiomyocyte cross-sectional area data (that did not exhibit a Gaussian distribution), statistical significance was assessed using a non-parametric Kruskal-Wallis followed by a post hoc Dunn’s multiple comparison test. For all data sets, the arithmetic mean ± SEM is reported. P-values of less than 0.05 were considered statistically significant.
Results
Pharmacologic HDAC1 inhibition promotes cardiogenic gene transcription in human CMCs
In a previous report we demonstrated that shRNA-mediated knockdown of HDAC1 promotes the activation of a core cardiogenic transcriptional program in human CMCs [23]. Here we sought to determine whether the pharmacologic benzamide HDAC inhibitor entinostat, a class 1 isoform selective HDACi that strongly targets HDAC1 (HDAC1 IC50≈300 nM) and weakly HDAC3 (HDAC3 IC50≈8 μM), would recapitulate the effects of HDAC1 genetic depletion on cardiogenic gene transcription in CMCs. We first performed preliminary dose- (Fig. 1) and time- (Fig. 2) response experiments on patient-derived CMCs to empirically investigate the effect of entinostat treatment on the magnitude of HDAC1 inhibition and gene transcription in vitro. As previous investigations have revealed HDAC inhibition to influence the expression of genetic networks associated with pluripotency [23, 29] and cardiogenic lineage specification [23, 25, 44], RT-PCR was used to assess the expression of stemness (OCT4, NANOG, SOX2, and KLF4)-, cardiomyogenic (GATA4, MEF2C, MYBPC3, NKX2.5, and TBX5)-, endothelial (KDR and CD31)-, and smooth muscle (SM22α, αSMA, and CNN1)-associated mRNA transcripts in response to entinostat exposure. In Figure 1A, the efficiency of entinostat-mediated HDAC inhibition (72 h treatment; concentrations ranging from 0.25 to 2.0 μM) was interrogated by immunoblotting for acetylated-lysine 9 of histone-H3 (AcK9-H3). We observed robust inhibition of HDAC activity at the lowest investigated dose of 0.25 μM, with maximal histone-H3 acetylation observed at 0.50 μM and complete signaling saturation at all subsequent doses (Fig. 1A). In a correlative fashion, quantitative PCR (qPCR)-based gene expression assays at corresponding concentrations (ranging from 0.25 to 5.0 μM) revealed a dose-dependent increase in stemness-, cardiac-, and endothelial-related mRNA transcripts (Fig. 1B-D), many of which achieved statistical significance at or beyond 1.0 μM of entinostat exposure (specifically, OCT4, NANOG, SOX2, KLF4, MYBPC3, NKX2.5, TBX5, and CD31); in contrast, smooth muscle lineage-specific mRNA transcripts (e.g., SM22α and αSMA) exhibited a nominal decrease in expression with exception to CNN1 (Fig. 1E), which showed a statistical increase at 5.0 μM.
Fig. 1.

Dose-dependent effects of entinostat exposure on CMC gene transcription in vitro. a Representative immunoblot utilizing antibodies raised against acetylated lysine 9 of histone H3 (AcK9-H3) and β-actin (loading control). Total protein extracts were harvested from human CMCs incubated with increasing concentrations of entinostat for 72 h (n=2 independent biological replicates). Real-time qPCR-based gene expression assays evaluating the expression of b stemness-, c cardiomyogenic-, d endothelial-, and e smooth muscle-related mRNA transcripts at corresponding entinostat concentrations. Bar graphs denote mean relative mRNA expression ± SEM (n=5 independent biological replicates).
Fig. 2.

Time-dependent effects of entinostat exposure on CMC gene transcription in vitro. a Representative immunoblot using antibodies raised against acetylated lysine 9 of histone H3 (AcK9-H3) and β-actin (loading control). Total protein extracts were harvested from human CMCs incubated with DMSO diluent control or 1 μM entinostat for a duration of 3, 5, and 7 days (n=2 independent biological replicates). Real-time qPCR-based gene expression assays evaluating the expression of b stemness-, c cardiomyogenic-, d endothelial-, and e smooth muscle-related mRNA transcripts at associated time-points. Bar graphs denote mean relative mRNA expression ± SEM (n=5 independent biological replicates).
Next, the temporal effects of entinostat exposure on CMC gene transcription were investigated. Here we chose to use 1 μM concentrations of entinostat as this was determined to be the minimal concentration required to promote cardiogenic gene activation. In Figure 2, CMCs were treated for 3, 5, and 7 days with either 1 μM entinostat or DMSO diluent control. Accompanying immunoblots for acetylated histone-H3 revealed efficient inhibition of HDAC activity by day 3—and maximal signal saturation by day 5 (Fig. 2A). Further, at related endpoints we observed a time-dependent increase in the expression of stemness-, cardiac-, and endothelial-associated mRNA transcripts (Fig. 2B-D), the majority of which began to show statistical increases in relative expression by days 5 and 7 (e.g., OCT4, NANOG, SOX2, GATA4, MYBPC3, NKX2.5, TBX5, KDR, and CD31). Conversely, many smooth muscle lineage-specific transcripts exhibited a time-dependent decrease (i.e., SM22α) or no change (αSMA) in gene expression relative to controls, except for CNN1, which again increased in response to entinostat treatment (Fig. 2E). Together, these results indicate that isoform-selective inhibition of HDAC1 via entinostat exposure, like with shRNA-mediated depletion [23], induce a core cardiogenic transcriptional program in human CMCs and that prolonged exposure (5 days) to a minimal dose of entinostat (1 μM) is sufficient for this transcriptional induction. While the precise mechanism(s) detailing this response are not entirely clear, we previously demonstrated that HDAC1 inhibition-mediated cardiogenic transcriptional program activation in CMCs is partly dependent upon enhanced acetylation and accumulation of the tumor suppressor protein p53 [23]. In fact, p53 stability and its transcriptional activity have been shown to be negatively regulated by HDAC1-mediated deacetylation of lysine residues 373/382 (K373/382) [49]. Moreover, other studies have revealed p53 expression and its transcriptional activity to play a key role in the regulation of myogenic differentiation of embryonic stem cells [7], as well as skeletal muscle precursors [8, 21, 32]. To this end, p53 acetylation and total protein levels were assessed in CMCs treated with 1 μM entinostat or DMSO diluent control for 5 days. While immunoblot analyses revealed only a nominal increase in the total protein levels of p53 (Fig. 3A and 3C), antibody-mediated detection of acetylated p53 revealed significant hyper-acetylation of K382 in entinostat-CMCs compared to DMSO controls (Fig. 3B and 3C)—closely reiterating previously observed effects of genetic HDAC1 depletion on CMC p53 acetylation and cardiogenic gene expression [23].
Fig. 3.

Entinostat-treated CMCs exhibit heightened commitment toward a cardiomyocyte-like phenotype when stimulated to differentiate in vitro. Quantification of a p53 and b acetylated p53 (acetyl-p53; acetylated lysine 382) protein in c immunoblots containing whole cell extracts derived from CMCs treated with DMSO diluent control or 1 μM entinostat for 5 days. Bar graphs denote mean p53 or acetylated p53 protein expression (relative to β-actin loading controls) ± SEM (n=4 independent biological replicates). d In vitro cardiogenic differentiation schematic and timeline. e Flow cytometric detection of myocyte lineage-specific proteins [MEF2C (myocyte-specific enhancer factor 2C), MHC (cardiac myosin heavy chain), and MLC2 (cardiac myosin light chain 2)] in differentiated CMCs pre-treated with DMSO diluent control or 1 μM entinostat for 5 days. Isotype controls (rabbit IgG and mouse IgG2b) were used to evaluate non-specific antibody binding characteristics and establish appropriate gating procedures (blue polygons). f Bar graph depicting flow cytometric-based quantification of the percentage of differentiated CMCs expressing MEF2C, MHC, and MLC2 proteins. Values denote mean percentage of expressing cells ± SEM (n = 6 independent biological replicates).
CMCs Treated with Entinostat Adopt a Cardiomyocyte Cell-Like Fate Following Their Differentiation In Vitro
We next sought to investigate the consequences of entinostat-induced cardiogenic program activation on CMC differentiation in vitro. CMCs were treated with either 1 μM entinostat or DMSO diluent control in Ham’s F12 complete medium for 5 days (Fig. 3D). As previously performed [23], CMCs were then coaxed to differentiate via incubation under reduced serum conditions (DMEM, 5% FBS) for two weeks (Fig. 3D). At the conclusion of differentiation, CMCs were harvested and subject to flow cytometric analyses using antibodies raised against the myocyte lineage-specific transcription factor, MEF2C (myocyte enhancer factor 2C), and myocyte contractile machinery component proteins, MHC (myosin heavy chain) and MLC2 (myosin light chain 2) (Fig. 3F and 3E). Relative to DMSO controls, entinostat-treated CMCs more competently assumed a cardiomyocyte-like phenotype wherein approximately 10% of CMCs expressed cardiomyocyte-specific MHC (9.8±2.3% vs. 1.1±0.2%; entinostat vs. DMSO, p<0.0001), MEF2C (8.8±1.9% vs. 1.3±0.2%; entinostat vs. DMSO, p<0.0005), and MLC2 (11.8±1.8% vs. 4.9±1.7%; entinostat vs. DMSO, p<0.001) (Fig. 3F and 3E). In contrast, DMSO-treated CMCs exhibited no statistical difference in the percentage of cells that express MHC and MEF2C compared to isotype controls, however, as much as 4.9% of the control population was observed to express MLC2 (Fig. 3F and 3E). These results indicate that although entinostat-treated CMCs may be epigenetically/transcriptionally poised for cardiogenic cell-lineage specification, only approximately 10% of the CMC populations are in fact amenable to cardiomyocyte-like differentiation under these conditions—a phenomenon we previously observed with genetic HDAC1 depletion in vitro [23].
Entinostat-Treated CMCs Induce Greater Attenuation of LV Remodeling and Dysfunction
In a previous report we demonstrated that HDAC1-depleted, cardiomyogenic lineage-committed CMCs possess distinct trophic factor secretion profiles and augmented in vitro paracrine signaling activities relative to naïve CMCs [22]. In light of these findings, we postulated that HDAC1 could serve as a therapeutically exploitable target to enhance CMC cardiac reparative capacity. To test this hypothesis, athymic nude rats were subjected to ischemia-reperfusion injury (as detailed in the methods section), randomized into treatment groups, and intramyocardially administered CMCs (entinostat-treated or DMSO diluent control-treated) or PBS vehicle. Indices of cardiac function were assessed via serial echocardiograms on day 0 (baseline), day 30 (immediately before treatment; Pre-Tx), and day 65 (35 days after treatment; Post-Tx) (Fig. 4A). As expected, echocardiographic assessment of chamber dimensions revealed the development of adverse left ventricular (LV) remodeling in vehicle-treated rats, as evidenced by the progressive increase in LV end-diastolic and end-systolic volumes (Fig. 4B and 4C, respectively) relative to baseline. Although vehicle-, DMSO CMC-, and entinostat CMC-treated groups exhibited no difference in LV end-diastolic ventricular volumes after treatment (Fig. 4B), entinostat-treated rats exhibited a statistically significant decrease in end-systolic ventricular volumes compared to that of vehicle-, whereas only a nominal decrease was observed with DMSO CMC-treated animals (vehicle: 192.2±11.7 μL; DMSO CMC: 162.0±18.8 μL; entinostat CMC: 137.9±12.3 μL) (Fig. 4C). Moreover, rats administered entinostat-treated CMCs showed a marked increase in stroke volume (vehicle: 172.3±8.0 μL; DMSO CMC: 186.5±8.4 μL; entinostat CMC: 221.3±9.8 μL) (Fig. 4D) and cardiac output (vehicle: 59.1±3.6 ml/min; DMSO CMC: 60.2±3.4 ml/min; entinostat CMC: 74.1±3.8 ml/min) (Fig. 4F), with no detectable change in heart rate (Fig. 4E), 35 days after treatment compared to both DMSO CMC- and vehicle-treated groups (Fig. 4D). Additional indices of cardiac function, such as ejection fraction (EF) and fractional shortening (FS), were evaluated (Fig. 4G-J). 35 days after treatment, both entinostat- and DMSO-treated CMCs exhibited a statistically significant increase in EF relative to vehicle (vehicle: 47.7±1.0%; DMSO CMC: 56.5±1.9%; entinostat CMC: 60.4±1.5%); however, the magnitude of this increase was significantly greater with entinostat-treated CMCs (Fig. 4G). The change in EF (ΔEF) from pre- to post-treatment was also determined for each animal in each treatment group. Both entinostat- and DMSO-treated CMCs exhibited a statistically significant improvement in ΔEF relative to vehicle (vehicle: −5.4±1.4%; DMSO CMC: +2.9±2.3%; entinostat CMC: +5.6±2.1%), although these improvements were similar among both CMC treatment groups (Fig. 4H). Interestingly, 35 days after treatment, entinostat CMC-treated rats displayed a significant increase in FS relative to vehicle-treated groups whereas DMSO CMCs did not (vehicle: 12.4±1.3%; DMSO CMC: 13.5±1.5%; entinostat CMC: 17.7±1.7%) (Fig. 4I). Such observations were further corroborated after plotting the change in FS (ΔFS) from pre- to post-treatment wherein only entinostat-treated CMCs yielded a significant change in FS relative to vehicle-treated animals (vehicle: −1.1±1.1%; DMSO CMC: +4.0±1.4%; entinostat CMC: +6.3±2.2%) (Fig. 4J). Taken together these results suggest that entinostat-treated CMCs have a greater capacity to improve LV remodeling and global LV function relative to naïve (DMSO-treated) CMCs.
As an additional measure, all experimental groups were also assessed by hemodynamic analyses 35 days after treatment (prior to euthanasia). Here, the consequences of CMC administration on load-dependent (LV ejection fraction, LV dP/dtmax, and LV dP/dtmin) and load-independent (end-systolic elastance and adjusted maximal power) indices of LV systolic function were evaluated. Analogous to our echocardiography-based observations, hemodynamic analyses revealed neither DMSO CMCs nor entinostat-treated CMCs to yield a measurable change in LV end-diastolic volumes relative to vehicle controls (Fig. 5A); however, only entinostat CMC-treated rats exhibited a statistically significant decrease in end-systolic volumes compared to vehicle-treated groups (vehicle: 174.1±13.7 μl; DMSO CMC: 153.2±14.6 μl; entinostat CMC: 126.7±10.5 μl) (Fig. 5B). Further, comparisons of ventricular function revealed both entinostat and DMSO-treated CMCs to yield a significant improvement in EF relative to vehicle-treated rats (vehicle: 48.8±1.1%; DMSO CMC: 54.3±1.9%; entinostat CMC: 60.0±1.3%) (Fig. 5C), although the degree of this improvement was considerably greater with entinostat-treated CMCs over that of control CMCs. Additional physiologic parameters that were measured included heart rate (Fig. 5D), which was notably similar amongst all groups, and LV dP/dtmax and dP/dtmin (Fig. 5E), an indicator of global LV contractility that exhibited only nominal increases in DMSO- and entinostat-treated CMCs relative to vehicle. Bearing this in mind, the observed decreases in LV end-systolic volumes suggest that the beneficial effects imparted by entinostat-treated CMCs may, in part, be attributed to enhancements in cardiac contractility. LV end-systolic elastance (Ees) and preload-adjusted maximal power, both load-insensitive indices of LV contractility, were also measured (Fig. 5F and 5G, respectively). Congruent with the concept of increasing contractility, only entinostat-treated CMCs produced a statistically significant improvement in both end-systolic elastance (vehicle: 0.59±0.05 mmHg/ml; DMSO CMC: 0.74±0.06 mmHg/ml; entinostat CMC: 0.83±0.07 mmHg/ml) (Fig. 5F) and adjusted maximal power (vehicle: 6.8±0.5 mW/μl2; DMSO CMC: 8.1±0.9 mW/μl2; entinostat CMC: 10.6±1.5 mW/μl2) (Fig. 5G) compared to the vehicle-treated animals. Thus, two independent methods of functional assessment (echocardiography and hemodynamic studies with conductance catheters) consistently demonstrate that delivery of entinostat-treated CMCs provides greater improvement in LV systolic performance than naïve CMCs (DMSO control-treated).
Fig. 5.

Hemodynamic analyses indicate a therapeutic advantage with epigenetically modified CMCs compared to naïve CMCs. Conductance catheters were used 35 days after CMC treatment to evaluate ventricular volumes (a end-diastolic volume (μl) and b end-systolic volume (μl)), c ejection fraction (%), d heart rate, e LV dP/dtmax and LV dP/dtmin (mmHg/sec)), and load-independent indices of LV systolic function (f end-systolic elastance (mmHg/ml) and g pre-load adjusted maximal power (mWatts/μl2)). All graphs report mean ± SEM for treatment groups consisting of vehicle- (n=10), DMSO CMC- (n=10), and entinostat CMC- (n=10) injected animals.
Entinostat-CMC Administration Reduces Myocardial Collagen Deposition
Thirty five days after treatment, all groups were subjected to histological analyses to evaluate infarct size, as well as myocardial collagen content. In Figure 6A, resected hearts were transversally cut into 5 blocks (B1-B5), in 3 mm increments, from apex to base. The scar size of each block was determined via planimetry of Masson’s trichrome stained tissue sections (Fig. 6B) and summed to yield total scar size (in mg) for each heart (Fig. 6C). Resultant data revealed that neither entinostat-treated CMCs nor DMSO control-treated CMCs produced a measurable difference in total scar size relative to vehicle (Fig. 6C). Further, there was no detectable difference in scar size calculated as a percentage of the LV, whereby amongst all groups approximately 10% of the LV consisted of scar tissue (Fig. 6D). Intriguingly, assessment of myocardial collagen content displayed dissimilar findings. In Figure 6E, regional collagen content was enumerated via polarized light microscopy of picrosirius red stained mid-papillary level myocardial sections. In the risk region (consisting of scar and border zone), entinostat-treated CMCs exhibited a statistically significant decrease in collagen content relative to vehicle, whereas only a nominal decrease was observed with DMSO-treated CMCs (vehicle: 23.9±2.5%; DMSO CMC: 20.3±2.3%; entinostat CMC: 17.3±1.3%) (Fig. 6F). Such effects were less pronounced in remote zones, wherein entinostat-CMCs yielded an average, yet non-significant, reduction in collagen content within remote regions relative to both vehicle and DMSO CMC-treated groups (vehicle: 3.5±0.7%; DMSO CMC: 3.5±0.6%; entinostat CMC: 2.9±0.3%) (Fig. 6F). These results indicate that although entinostat-treated CMCs do not decrease infarct size, they do, however, impede myocardial collagen deposition—a phenomenon that could facilitate improved ventricular compliance and, importantly, underlie the observed improvements in LV systolic function.
Fig. 6.

Entinostat-CMC administration more efficiently attenuates myocardial fibrosis. a Schematic diagram detailing cardiac dissection for histopathology. Each heart was dissected into 5 × 3 mm blocks (B1-B5) prior to histopathological analysis. Planimetric measurements of b Masson’s trichrome-stained transverse myocardial tissue sections (B1-B5; 4 μm in depth) were used to enumerate scar size, reported in c mass (mg) and d percentage of LV (% scarmass/LVmass; % wt/wt) in vehicle- (n=11), DMSO CMC- (n=10), and entinostat CMC- (n=10) treated rat hearts. Dot plots report the arithmetic mean ± SEM. Scale bars=3 mm. e Myocardial collagen content was digitally enumerated via polarized light microscopy of picrosirius red stained mid-papillary level myocardial sections (top row: brightfield; bottom row: polarized light). Scale bars=1 mm. f Regional collagen content (risk and remote myocardial zones) was quantified in cardiac sections derived from vehicle- (n=11), DMSO CMC- (n=10), and entinostat CMC- (n=10) injected animals. Values are reported as regional collagen content (% area) ± SEM.
Functional Benefits Provided by Epigenetically Modified CMCs are Not Associated with Enhanced Vasculogenesis or Cardiomyogenesis
Having observed significant improvements in cardiac function, as well as measurable reductions in myocardial collagen content with entinostat-treated CMCs, we next sought to determine whether such improvements could be attributed to improved donor cell retention and/or enhanced activation of endogenous repair mechanisms (e.g., angiogenesis and cardiomyocyte proliferation). To address the former, the presence of myocardial retained human CMCs was evaluated via immunohistochemical detection of human nucleoli antigen (HNA) (Fig. 7A). Consistent with previous reports highlighting diminutive retention of donor cells [10], less than approximately 0.5% of the injected 2 million cells per heart were detected in either entinostat- or DMSO-treated CMC hearts—demonstrating that CMC myocardial retention is nearly negligible 35 days after delivery. Of the few HNA+ CMCs identified in either entinostat or DMSO CMC-treated hearts, none expressed mature cardiovascular cell lineage markers or acquired morphological characteristics reminiscent of functional cardiac parenchyma.
Fig. 7.
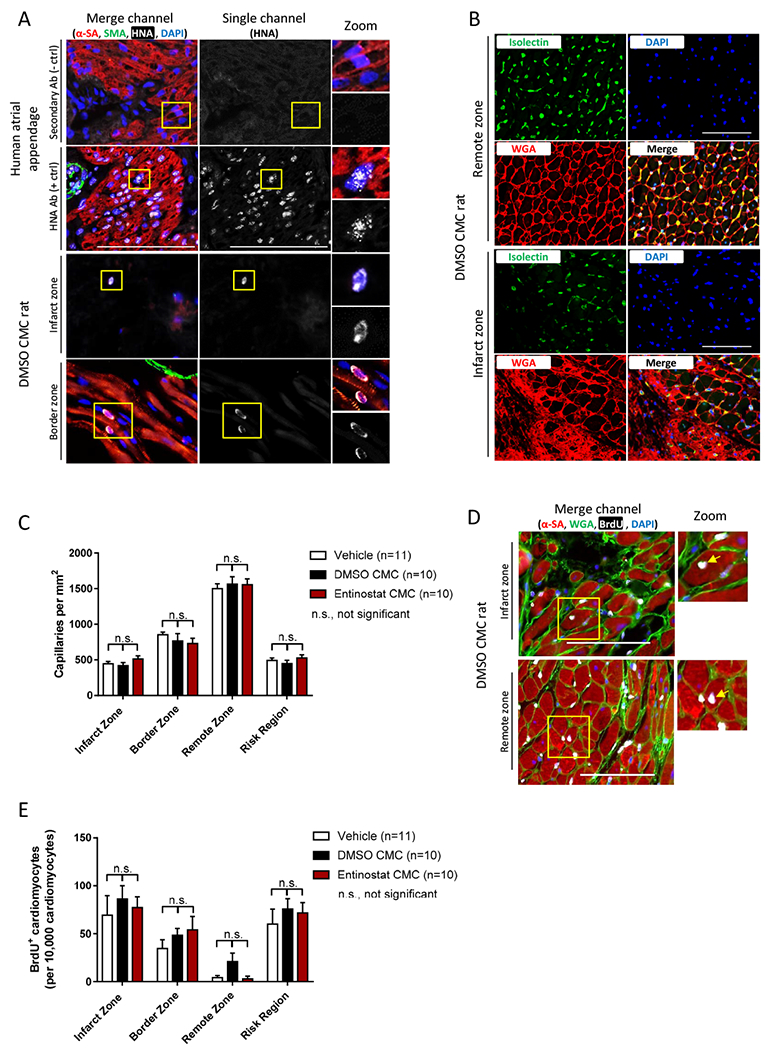
The effects of entinostat on CMC retention, myocardial capillary density, and endogenous myocyte proliferation. a Representative myocardial tissue sections (top four panels: human atrial appendage controls; bottom four panels: DMSO CMC-injected rat) stained with antibodies raised against human nucleolar antigen (HNA; white), α-sarcomeric actin (α-SA; myocytes; red), and smooth muscle actin (SMA; vessels; green). Nuclei are counterstained with DAPI (nuclei; blue). Scale bars=100 μm. b Representative myocardial tissue sections derived from DMSO CMC-injected rat hearts (top four panels: remote zone; bottom four panels: infarct zone) stained with isolectin B4 (capillaries; green), wheat germ agglutinin (WGA; cell membranes; red), and DAPI (blue). Scale bars=100 μm. c Graph depicting regional capillary density in infarct, border, remote, and risk regions of vehicle- (n=11), DMSO CMC- (n=10), and entinostat CMC- (n=10) injected rat hearts. Results are reported as the number of capillaries per mm2 ± SEM. d Representative myocardial tissue sections derived from DMSO CMC-treated rat hearts (top panel: infarct zone; bottom panel: remote zone) stained with antibodies raised against α-sarcomeric actin (α-SA; myocytes; red), wheat germ agglutinin (WGA; cell membranes; green), and 5-Bromo-2′-deoxyuridine (BrdU; proliferating cells; white). Nuclei are counterstained with DAPI (nuclei; blue). Scale bars=100 μm. e Graph illustrating the number of BrdU+ nucleated myocytes within myocardial regions (infarct, border, remote, and risk region) of vehicle (n=11), DMSO CMC (n=10), and entinostat CMC (n=10) treatment groups. Values are reported as the number of BrdU+ myocytes per 10,000 nucleated myocytes ± SEM.
Regional capillary density was next evaluated in all groups via isolectin B4 staining of mid-papillary level, transverse myocardial sections (Fig. 7B); here, entinostat and DMSO-treated CMC groups exhibited no difference in the number of capillaries in any of the investigated myocardial regions, relative to vehicle control rats (Fig. 7C). In corresponding tissue sections, cardiomyocyte proliferation was also interrogated and enumerated via immunohistochemical detection of nuclear incorporated BrdU in mature cardiomyocytes (Fig. 7D). Analyses revealed entinostat and DMSO-treated CMC groups to exhibit no detectable difference in the number of BrdU+ nucleated cardiomyocytes in any of the investigated myocardial regions, compared to vehicle control rats (Fig. 7E). In fact, on average, less than 1% of all nucleated myocytes were BrdU+—suggesting that endogenous cardiomyocyte proliferation is very low and, moreover, unaffected by CMC treatment.
Analogous conclusions were drawn when investigating the effects of CMC administration on regional myocardial vessel density and growth (Fig. 8). Here, α-smooth muscle actin (SMA) antibody was used to detect and quantify myocardial vessels in mid-papillary level transverse myocardial tissue sections (Fig. 8A). For each group, blood vessels were stratified according to luminal diameter, reporting the total number of vessels per unit area that were less than or greater than 20 μm in diameter (Fig. 8B and 8C, respectively). Neither entinostat- nor DMSO-treated CMCs exhibited a change in vessel density in either small (Fig. 8B) or large (Fig. 8C) diameter vessels, in any and all investigated myocardial regions, relative to vehicle. Similar trends were observed when enumerating the total of both small and large luminal diameter vessels together (Fig. 8D)—where again, no measurable change in total vessel density was observed amongst entinostat CMC-, DMSO CMC-, and vehicle-treated groups. Although no change in the absolute number of vessels was evident among these groups, we next sought to investigate the consequences of entinostat CMC administration on endogenous vessel growth. This was achieved via antibody-mediated detection and enumeration of nuclear incorporated BrdU within cells residing in SMA+ vessels (Fig. 8E). The number of small (Fig. 8F) or large (Fig. 8G) vessels per unit area that were BrdU+ (counting vessels that had 1 or more BrdU+ nucleated cells) was no different among entinostat CMC-, DMSO CMC-, and vehicle-treated rats. Moreover, no measurable difference was observed in the percentage of BrdU+ nucleated vessel cells, where in all groups approximately 10-15% of counted nucleated vessel cells were identified to be BrdU+ (Fig. 8H). Taken together, these results indicate that neither naive CMC nor entinostat-treated CMC administration provides any measurable benefits on endogenous myocardial vessel growth or regional vessel density.
Fig. 8.

The consequences of entinostat CMC administration on myocardial vessel density and growth. a Representative myocardial tissues sections stained with α-sarcomeric actin (α-SA; myocytes; red), smooth muscle actin (SMA; vessels; green), 5-Bromo-2′-deoxyuridine (BrdU; proliferating cells; white), and DAPI (nuclei; blue). Sections were sourced from vehicle (n=11), DMSO CMC (n=10), and entinostat CMC (n=10) treatment groups and accompanying images correspond to infarct (top row), border (middle row), and remote (bottom row) myocardial regions. Scale bars=100 μm. Regional vessel density (total number of vessels per mm2 ± SEM) is reported in accompanying graphs and is stratified into vessels with a luminal diameter of b < 20 μm and c > 20 μm; d illustrates the total number of vessels (including both small and large vessels) per unit area. e Illustrative myocardial tissues sections sourced from entinostat CMC-injected rats (infarct zones are shown). Sections were stained with α-sarcomeric actin (α-SA; myocytes; red), smooth muscle actin (SMA; vessels; green), 5-Bromo-2′-deoxyuridine (BrdU; proliferating cells; white), and DAPI (nuclei; blue). Yellow arrows identify BrdU-labeled, SMA+ vessels (proliferating vessels). Scale bars=100 μm. The number of proliferating vessels per unit area (BrdU+ vessels per mm2 ± SEM) is reported in associated graphs and is stratified according to vessel luminal diameters of f < 20 μm and g > 20 μm; h reports the total percentage of nucleated vessel cells that were identified to be BrdU+ (% of BrdU+ nucleated cells in vessels ± SEM).
Entinostat-Treated CMCs Increase Cardiomyocyte Cross-Sectional Area
Previous investigations suggest regression of cardiomyocyte hypertrophy as an additional mechanism of action of donor cells in cardiac cell therapy [35, 39]. To this end, a comprehensive assessment of regional cardiomyocyte cross-sectional area was performed on wheat germ agglutinin (WGA)-stained, transverse mid-papillary level myocardial tissue sections from entinostat CMC-, DMSO CMC-, and vehicle-treated rats (Fig. 9A). Unexpectedly, both DMSO CMC- and entinostat CMC-treated rat hearts exhibited a statistically significant increase in myocyte cross-sectional area in all investigated myocardial regions (e.g., infarct, border, and remote zones (Fig. 9B)), relative to vehicle. The relative increase in myocyte size was similar between entinostat- and DMSO-treated CMCs compared to vehicle within infarct regions (vehicle: 380.8±11.1 μm2; DMSO CMC: 499.9±11.9 μm2; entinostat CMC: 513.0±12.2 μm2) (Fig. 9B); however, entinostat CMCs elicited a significantly greater increase in myocyte cross-sectional area in remote regions compared to DMSO CMCs (vehicle: 269.4±3.8 μm2; DMSO CMC: 290.8±5.7 μm2; entinostat CMC: 352.3±5.8 μm2) (Fig. 9B). All digitally measured myocytes were further graphed in accompanying histograms to ascertain data distribution (Fig. 9C). These histograms illustrate that measured myocytes in CMC-treated hearts, on average, tend to exhibit a larger cross-sectional area compared to vehicle controls in all investigated myocardial regions (Fig. 9C). Moreover, in remote zones, entinostat CMC-treated hearts possessed a greater number of myocytes whose cross-sectional area was greater than 350 μm2, compared to DMSO CMC- and vehicle-treated hearts exhibiting similar distribution patterns (Fig. 9C). These effects on myocyte size were found to be independent of body weight, as all rat groups exhibited similar body mass (Fig. 9D) but appeared to correlate with increasing ventricular weights observed in CMC-treated groups (vehicle: 636.8±62.0 mg; DMSO CMC: 719.7±35.0 mg; entinostat CMC: 848.6±50.2 mg) (Fig. 9E)—although only entinostat CMC-treated rat hearts displayed a statistically significant increase in overall LV mass relative to vehicle. Overall, these results suggest that intramyocardial delivery of epigenetically modified CMCs promotes myocyte growth in recipient hearts, compared to DMSO CMCs, whose effects were considerably milder.
Fig. 9.

The effects of entinostat-treated CMCs on cardiomyocyte cross-sectional area. a Representative myocardial tissue sections derived from vehicle, DMSO CMC, and entinostat CMC treatment groups (infarct regions are shown). Sections were stained with antibodies raised against α-sarcomeric actin (α-SA; myocytes; red) and wheat germ agglutinin (WGA; cell membranes; green). Nuclei are counterstained with DAPI (nuclei; blue). Scale bars=100 μm. b Graphs illustrating mean cardiomyocyte cross-sectional area (μm2) ± SEM in vehicle (n=11), DMSO CMC (n=10), and entinostat CMC (n=10) treatment groups according to myocardial region (infarct, border, and remote). Abscissas indicate the total number of myocytes measured per group. c Accompanying histograms showing myocyte size distribution (number of values vs. cardiomyocyte size [bin center; area in μm2]) according to myocardial region for vehicle (grey), DMSO CMC (black), and entinostat CMC (red) treatment groups. Figure legends indicate the total number of myocytes measured per group. d Bar graph reporting the mean body weight (g) ± SEM of vehicle (n=11), DMSO CMC (n=10), and entinostat CMC (n=10) treatment groups. e Bar graph reporting mean left ventricular mass (mg) ± SEM of vehicle (n=11), DMSO CMC (n=10), and entinostat CMC (n=10) treatment groups.
Discussion
The phenotypic properties underlying donor cell cardiac reparative capacity remain poorly defined, as are their precise mechanism(s) of action. Previous evidence suggests that donor cell cardiomyogenic lineage commitment could be an important element influencing the efficacy of cell therapy [2, 28, 45]; however, whether this is a crucial feature contributing to their therapeutic benefits remains to be resolved. Nonetheless, this view has been the driving force behind numerous studies describing new and efficient methodologies to stimulate mesenchymal cell cardiomyogenic lineage commitment and/or cardiomyocyte differentiation [12, 17, 23, 24, 27, 43]—techniques which could, theoretically, be exploited to increase the therapeutic utility of donor cells in cell therapy-based applications for the treatment of ischemic cardiac injury. Following this line of investigation, we previously identified HDAC1 as an important regulator of CMC cell fate decisions, wherein HDAC1 genetic depletion efficiently promoted activation of a core cardiogenic transcriptional program and facilitated cardiomyocyte-like differentiation of human CMCs in vitro [23]. Further, CMCs depleted of HDAC1 exhibited markedly distinct cytokine secretion profiles and heightened in vitro paracrine signaling potency in vitro [22]—suggesting that HDAC1 inhibition could have pronounced consequences on CMC cardiac reparative capacity in vivo. To this end, the principal goal of the current study was to evaluate the influence of pharmacologic class 1 isoform selective HDAC inhibition on i) CMC cardiomyogenic lineage commitment and ii) its associated effects on CMC therapeutic efficacy in a rat model of ischemic cardiomyopathy.
In the current study we demonstrate that the benzamide HDAC inhibitor entinostat stimulates CMC cardiogenic gene transcription. This was evidenced via both a dose- and time-dependent increase in the expression of early (NKX2.5, TBX5, and GATA4) and late (MEF2C) cardiogenic transcription factors—factors known to cooperatively regulate genetic programs involved in cardiomyogenic differentiation. The precise mechanisms underlying CMC cardiogenic program activation in response to HDAC inhibition are not entirely clear; however, our previous work suggests that this phenomenon occurs via a p53 dependent mechanism, wherein HDAC1 inhibition-mediated cardiogenic transcriptional program activation requires, for the most part, the cellular accumulation of acetylated p53 protein [23]. Congruent with these observations, p53 stability and its transcriptional activity have been shown to be negatively regulated via direct deacetylation of lysine residues 373/382 (K373/382) by HDAC1 [49]. Interestingly, p53 has been identified as an important mediator of myogenic differentiation of embryonic stem cells (via regulation of the mesodermal master genes Brachyury and Mesp1) [7], as well as skeletal muscle precursors [8, 21, 32]. Thus, we postulate that HDAC1 may indirectly regulate cardiomyogenic transcriptional programs in CMCs by directly modifying p53 acetylation status, and consequently, p53 transcriptional activity. In support of this proposed mechanism, entinostat-induced alterations in CMC cardiogenic program activation was associated with marked increases in K382 p53 protein acetylation—notably recapitulating our previously observed effects with genetic HDAC1 knockdown [23]. Resultant transcriptional profiles from entinostat-treated CMCs suggest the acquisition of a developmental state with heightened commitment towards the cardiomyogenic lineage. Consistent with this assessment, entinostat-treated CMCs more efficiently assumed a cardiomyocyte-like phenotype when coaxed to differentiate in vitro under reduced serum conditions. This was indicated by the increased expression of the late cardiogenic transcription factor, MEF2C, and cardiomyocyte contractile machinery component proteins, MHC and MLC2. Despite observing increased expression of these cardiomyocyte proteins in entinostat pre-treated CMCs, at no time were spontaneous beating cardiomyocytes ever detected—suggesting that although entinostat-treated CMCs appear to be transcriptionally poised or rather primed for cardiomyogenic differentiation, additional genetic factors and/or environmental ques are likely necessary for complete maturation.
In addition to its effects on enhancing CMC cardiomyogenic lineage commitment in vitro, entinostat pre-treatment significantly augmented CMC cardiac reparative capacity in vivo. Here, entinostat CMCs more effectively attenuated myocardial remodeling and improved LV systolic performance in a chronic model of heart failure, compared to naïve cells (DMSO-treated CMCs) whose therapeutic benefits were considerably less conspicuous. For instance, echocardiography revealed that only entinostat CMC-treated hearts bore statistically significant improvements in LV end-systolic volume, stroke volume, cardiac output, and fractional shortening relative to vehicle treated hearts; however, both entinostat- and DMSO-treated CMCs were found to produce relatively similar improvements in LV EF using this imaging modality. Comparable conclusions were drawn from hemodynamic studies with conductance catheters; therein, only entinostat CMC-treated hearts exhibited a statistically significant decrease in LV end-systolic volumes compared to vehicle. Although echocardiography showed that both entinostat and DMSO CMCs exhibited similar improvements in LV EF, conductance catheterization demonstrated that entinostat CMCs provided the greatest increase in EF compared to vehicle-treated hearts. Hence, two independent methods of functional assessment indicate that entinostat-treated CMCs are therapeutically more efficacious than naïve CMCs (DMSO control-treated). Clues as to the physiologic basis of these improvements in cardiac function were provided by the evaluation of additional physiologic parameters, namely, LV end-systolic elastance (Ees) and preload adjusted maximal power—both load-insensitive indices of cardiac contractility. Specifically, entinostat-treated CMCs produced a pronounced, statistically significant increase in both Ees and adjusted maximal power relative to vehicle-treated rats, whereas only a minor (non-significant) increase was observed with DMSO CMCs. Taken together, these analyses suggest that the salutary effects provided by entinostat-treated CMCs could largely be ascribed to improvements in myocardial contractility.
Histopathological evaluation of CMC treated hearts indicated that the improvements in ventricular performance were neither related to a decrease in infarct size nor an increase in endogenous cardiomyogenesis and/or angiogenesis. This is in contrast to previous reports which have indicated that infused cardiac-sourced mesenchyme stimulates endogenous myocyte proliferation and induce arteriogenesis/neovascularization [1, 35, 39]. In our study, the only measurable histopathological changes detected were those in myocardial collagen content among entinostat CMC-, DMSO CMC-, and vehicle-treated hearts; wherein entinostat CMC-treated hearts exhibited a more prominent decrease in myocardial collagen deposition compared to that of naïve CMCs. Entinostat CMCs specifically yielded both a 28% and a 17% decrease in myocardial collagen content within risk and remote regions, respectively, compared to naïve CMCs, which produced only a nominal decrease of 15% within risk regions. These findings suggest that entinostat-treated CMCs more efficiently attenuate myocardial fibrosis—findings which suggest diminished myocardial rigidity and improved LV compliance as a potential mechanism contributing to the enhanced therapeutic efficacy of entinostat-treated CMCs. Of course, such a mechanism is consistent with the observed preservation of end-systolic volumes, increased LV end-systolic elastance, and improved preload adjusted maximal power in entinostat CMC-treatment groups 35 days after treatment. In addition to these alterations in cardiac fibrosis, we also observed a significant increase cardiomyocyte cross-sectional area in CMC treatment groups—an effect which, again, was more pronounced with entinostat-treated CMCs. Relative to vehicle-injected groups, entinostat CMC-treated hearts exhibited a 31% increase in cardiomyocyte area within remote myocardial regions compared to naïve CMCs, which showed only an 8% increase. Both entinostat CMC- and naïve CMC-treated hearts exhibited an approximate 31-35% increase in myocyte cross-sectional area relative to vehicle-treated hearts within infarct regions—signifying that CMC treatment may indirectly augment endogenous myocyte biology. Although the physiologic significance of this finding on cardiac function is not known, we postulate that secreted factors from transplanted CMCs may, in addition to limiting myocardial fibrosis, also promote physiologic myocyte growth and/or augment myocyte contractility. In support of this hypothesis, recent studies provide evidence suggesting that mesenchymal cell-derived exosomes contain microRNAs (miRNAs) that function in a paracrine fashion to boost the expression of myocyte calcium handling genes and increase cardiac tissue contractility [19, 20]—a potentially important feature of donor cells that is likely to exhibit cell lineage dependency [3, 13, 22].
In the current study we postulate that cardiomyogenic lineage commitment is an important predictor of CMC therapeutic efficacy and, further, that lineage commitment-dependent adaptation in paracrine signaling likely underlies their enhanced cardiac reparative capacity in vivo. Consistent with this premise, preliminary analyses of conditioned medium (CM) harvested from entinostat- and DMSO-treated CMCs reveal marked alterations in cytokine secretion patterns (Suppl. Fig. 1a and 1b). Specifically, entinostat-treated CMCs show augmented expression of soluble factors implicated to function in pathways that regulate extracellular matrix remodeling, collagen catabolism, and inflammation (Suppl. Fig. 1c). Thus, one may postulate that the enhanced expression of such factors may contribute to the improved antifibrogenic activities of entinostat CMCs in vivo. With this in mind, it is important to note that this particular study has a number of limitations and thus, the data should be interpreted with caution. Although HDAC inhibition does appear to enhance CMC cardiac reparative capacity, whether this is exclusively the result of enhancements in CMC cardiomyogenic lineage commitment cannot be deduced with these experiments alone, as there exists an array of other known and unidentified substrates (both histones and non-histone proteins) that are subject to HDAC activity—making it difficult to decipher the precise mechanisms responsible for this effect. In addition, potential off-target effects elicited by entinostat exposure also preclude the identification of exact underlying mechanisms contributing to the improved therapeutic performance of CMCs in vivo. To this end, current studies are in progress in our lab that employ genetic modification (e.g., ectopic expression of cardiogenic transcription factors) to promote CMC cardiomyogenic lineage commitment—obviating HDACi-induced widespread alterations in chromatin structure and potential off-target drug effects.
In summary, our study demonstrates that entinostat-treated CMCs are epigenetically primed for cardiomyogenic-like differentiation and, further, that these cells are therapeutically more efficacious than naïve CMCs in a model of chronic ischemic cardiomyopathy. Histopathological evaluation suggests that the salutary effects of entinostat CMC treatment cannot be attributed to enhanced cardiomyogenesis or vasculogenesis, but potentially to a combination of factors involving attenuation of ongoing myocardial collagen deposition and alterations in cardiomyocyte biology. In light of the fact that we observed only diminutive rates of CMC retention after infusion with either naïve or entinostat-treated CMCs, paracrine actions must comprise the core substance of the afforded benefits. Inhibitors of HDAC1 activity have proven effective in augmenting CMC cytokine secretion patterns and paracrine signaling activities in vitro [22]; thus, the improvement in reparative capacity of CMCs with entinostat exposure is likely related to elevated paracrine signaling potency, due to enhanced secretion and/or alternative expression of cardiotrophic factors. Taken together, these results implicate HDAC1 as both a modulator of CMC cell fate decisions and paracrine signaling activities and, importantly, support the use of class 1 isoform-selective HDAC inhibitors to enhance the therapeutic efficacy of CMCs in cell therapy-based applications. While our study indicates the pretreatment of CMCs with an isoform selective HDAC inhibitor to afford a reparative advantage to cells transplanted in a model of ischemic cardiomyopathy, it is noteworthy to mention that other studies have shown parenteral administration of pan-HDAC inhibitors alone to mitigate cell death and prevent detrimental ventricular remodeling in rodent [5, 15, 46, 47], as well as lagomorph [42] models of myocardial infarction; however, to our knowledge, comparative studies directly comparing the therapeutic efficacy of systemic HDACi administration to cell therapy HDACi pre-treatment approaches in preclinical models of heart failure have yet to be performed. It is clear, however, that cellular HDACi-pretreatment approaches would likely curtail the many non-specific toxicities [34] associated with systemic HDACi administration in vivo.
Supplementary Material
Acknowledgements:
This work was supported by National Institutes of Health grants R01 HL141081 (JBM), P01 HL078825 (RB), and UM1 HL113530 (RB). Additional funding was provided by the University of Louisville’s School of Medicine Basic Grant Program (JBM).
Footnotes
Disclosure Statement
The authors indicate no potential conflicts of interest.
References
- 1.Avolio E, Meloni M, Spencer HL, Riu F, Katare R, Mangialardi G, Oikawa A, Rodriguez-Arabaolaza I, Dang ZX, Mitchell K, Reni C, Alvino VV, Rowlinson J, Livi U, Cesselli D, Angelini G, Emanueli C, Beltrami AP, Madeddu P (2015) Combined Intramyocardial Delivery of Human Pericytes and Cardiac Stem Cells Additively Improves the Healing of Mouse Infarcted Hearts Through Stimulation of Vascular and Muscular Repair. Circ Res 116:E81–E94 doi: 10.1161/Circresaha.115.306146 [DOI] [PubMed] [Google Scholar]
- 2.Behfar A, Yamada S, Crespo-Diaz R, Nesbitt JJ, Rowe LA, Perez-Terzic C, Gaussin V, Homsy C, Bartunek J, Terzic A (2010) Guided Cardiopoiesis Enhances Therapeutic Benefit of Bone Marrow Human Mesenchymal Stem Cells in Chronic Myocardial Infarction. J Am Coll Cardiol 56:721–734 doi: 10.1016/j.jacc.2010.03.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charoenviriyakul C, Takahashi Y, Morishita M, Matsumoto A, Nishikawa M, Takakura Y (2017) Cell type-specific and common characteristics of exosomes derived from mouse cell lines: Yield, physicochemical properties, and pharmacokinetics. Eur J Pharm Sci 96:316–322 doi: 10.1016/j.ejps.2016.10.009 [DOI] [PubMed] [Google Scholar]
- 4.Gnecchi M, Zhang ZP, Ni AG, Dzau VJ (2008) Paracrine Mechanisms in Adult Stem Cell Signaling and Therapy. Circ Res 103:1204–1219 doi: 10.1161/Circresaha.108.176826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Granger A, Abdullah I, Huebner F, Stout A, Wang T, Huebner T, Epstein JA, Gruber PJ (2008) Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J 22:3549–3560 doi: 10.1096/fj.08-108548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo YR, Wysoczynski M, Nong YB, Tomlin A, Zhu XP, Gumpert AM, Nasr M, Muthusamy S, Li H, Book M, Khan A, Hong KU, Li QH, Bolli R (2017) Repeated doses of cardiac mesenchymal cells are therapeutically superior to a single dose in mice with old myocardial infarction. Basic Res Cardiol 112 doi: 10.1007/s00395-017-0606-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hadjal Y, Hadadeh O, Yazidi CEI, Barruet E, Binetruy B (2013) A p38mapk-p53 cascade regulates mesodermal differentiation and neurogenesis of embryonic stem cells. Cell Death Dis 4 doi:ARTN e737 10.1038/cddis.2013.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halevy O (1993) P53 Gene Is up-Regulated during Skeletal-Muscle Cell-Differentiation. Biochem Bioph Res Co 192:714–719 doi:DOI 10.1006/bbrc.1993.1473 [DOI] [PubMed] [Google Scholar]
- 9.Hodgkinson CP, Bareja A, Gomez JA, Dzau VJ (2016) Emerging Concepts in Paracrine Mechanisms in Regenerative Cardiovascular Medicine and Biology. Circ Res 118:95–107 doi: 10.1161/Circresaha.115.305373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong KU, Guo Y, Li QH, Cao P, Al-Maqtari T, Vajravelu BN, Du J, Book MJ, Zhu X, Nong Y, Bhatnagar A, Bolli R (2014) c-kit+ Cardiac stem cells alleviate post-myocardial infarction left ventricular dysfunction despite poor engraftment and negligible retention in the recipient heart. PloS one 9:e96725 doi: 10.1371/journal.pone.0096725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong KU, Li QH, Guo Y, Patton NS, Moktar A, Bhatnagar A, Bolli R (2013) A highly sensitive and accurate method to quantify absolute numbers of c-kit+ cardiac stem cells following transplantation in mice. Basic Res Cardiol 108:346 doi: 10.1007/s00395-013-0346-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D (2010) Direct Reprogramming of Fibroblasts into Functional Cardiomyocytes by Defined Factors. Cell 142:375–386 doi: 10.1016/j.cell.2010.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.J Pharmacol Exp TherJ Am Heart Assoc Ahn SY, Chang YS, Sung DK, Yoo HS, Sung SI, Choi SJ, Park WS (2015) Cell type-dependent variation in paracrine potency determines therapeutic efficacy against neonatal hyperoxic lung injury. Cytotherapy 17:1025–1035 doi: 10.1016/j.jcyt.2015.03.008 [DOI] [PubMed] [Google Scholar]
- 14.Kanazawa H, Tseliou E, Dawkins JF, De Couto G, Gallet R, Malliaras K, Yee K, Kreke M, Valle I, Smith RR, Middleton RC, Ho CS, Dharmakumar R, Li D, Makkar RR, Fukuda K, Marban L, Marban E (2016) Durable Benefits of Cellular Postconditioning: Long-Term Effects of Allogeneic Cardiosphere-Derived Cells Infused After Reperfusion in Pigs with Acute Myocardial Infarction. J Am Heart Assoc 5 doi: 10.1161/JAHA.115.002796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee TM, Lin MS, Chang NC (2007) Inhibition of histone deacetylase on ventricular remodeling in infarcted rats. Am J Physiol Heart Circ Physiol 293:H968–977 doi: 10.1152/ajpheart.00891.2006 [DOI] [PubMed] [Google Scholar]
- 16.Litwin SE, Katz SE, Morgan JP, Douglas PS (1994) Serial echocardiographic assessment of left ventricular geometry and function after large myocardial infarction in the rat. Circulation 89:345–354 [DOI] [PubMed] [Google Scholar]
- 17.Makino S, Fukuda K, Miyoshi S, Konishi F, Kodama H, Pan J, Sano M, Takahashi T, Hori S, Abe H, Hata J, Umezawa A, Ogawa S (1999) Cardiomyocytes can be generated from marrow stromal cells in vitro. J Clin Invest 103:697–705 doi:Doi 10.1172/Jci5298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martire A, Bedada FB, Uchida S, Poling J, Kruger M, Warnecke H, Richter M, Kubin T, Herold S, Braun T (2016) Mesenchymal stem cells attenuate inflammatory processes in the heart and lung via inhibition of TNF signaling. Basic Res Cardiol 111 doi: 10.1007/s00395-016-0573-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayourian J, Cashman TJ, Ceholski DK, Johnson BV, Sachs D, Kaji DA, Sahoo S, Hare JM, Hajjar RJ, Sobie EA, Costa KD (2017) Experimental and Computational Insight Into Human Mesenchymal Stem Cell Paracrine Signaling and Heterocellular Coupling Effects on Cardiac Contractility and Arrhythmogenicity. Circ Res 121:411–423 doi: 10.1161/Circresaha.117.310796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayourian J, Ceholski DK, Gorski PA, Mathiyalagan P, Murphy JF, Salazar SI, Stillitano F, Hare JM, Sahoo S, Hajjar RJ, Costa KD (2018) Exosomal microRNA-21–5p Mediates Mesenchymal Stem Cell Paracrine Effects on Human Cardiac Tissue Contractility. Circ Res 122:933–944 doi: 10.1161/Circresaha.118.312420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazzaro G, Bossi G, Coen S, Sacchi A, Soddu S (1999) The role of wild-type p53 in the differentiation of primary hemopoietic and muscle cells. Oncogene 18:5831–5835 doi:DOI 10.1038/sj.onc.1202962 [DOI] [PubMed] [Google Scholar]
- 22.Moore JB 4th, Zhao J, Fischer AG, Keith MCL, Hagan D, Wysoczynski M, Bolli R (2017) Histone Deacetylase 1 Depletion Activates Human Cardiac Mesenchymal Stromal Cell Proangiogenic Paracrine Signaling Through a Mechanism Requiring Enhanced Basic Fibroblast Growth Factor Synthesis and Secretion. J Am Heart Assoc 6 doi: 10.1161/JAHA.117.006183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore JB 4th, Zhao J, Keith MC, Amraotkar AR, Wysoczynski M, Hong KU, Bolli R (2016) The Epigenetic Regulator HDAC1 Modulates Transcription of a Core Cardiogenic Program in Human Cardiac Mesenchymal Stromal Cells Through a p53-Dependent Mechanism. Stem cells 34:2916–2929 doi: 10.1002/stem.2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moscoso I, Centeno A, Lopez E, Rodriguez-Barbosa JI, Santamarina I, Filgueira P, Sanchez MJ, Dominguez-Perles R, Penuelas-Rivas GP, Domenech N (2005) Differentiation “in vitro” of primary and immortalized porcine mesenchymal stem cells into cardiomyocytes for cell transplantation. Transpl P 37:481–482 doi: 10.1016/j.transproceed.2004.12.247 [DOI] [PubMed] [Google Scholar]
- 25.Oyama T, Nagai T, Wada H, Naito AT, Matsuura K, Iwanaga K, Takahashi T, Goto M, Mikami Y, Yasuda N, Akazawa H, Uezumi A, Takeda S, Komuro I (2007) Cardiac side population cells have a potential to migrate and differentiate into cardiomyocytes in vitro and in vivo. J Cell Biol 176:329–341 doi: 10.1083/jcb.200603014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Preibisch S, Saalfeld S, Tomancak P (2009) Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25:1463–1465 doi: 10.1093/bioinformatics/btp184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian Q, Qian H, Zhang X, Zhu W, Yan YM, Ye SQ, Peng XJ, Li W, Xu Z, Sun LY, Xu WR (2012) 5-Azacytidine Induces Cardiac Differentiation of Human Umbilical Cord-Derived Mesenchymal Stem Cells by Activating Extracellular Regulated Kinase. Stem Cells Dev 21:67–75 doi: 10.1089/scd.2010.0519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rossini A, Frati C, Lagrasta C, Graiani G, Scopece A, Cavalli S, Musso E, Baccarin M, Di Segni M, Fagnoni F, Germani A, Quaini E, Mayr M, Xu QB, Barbuti A, DiFrancesco D, Pompilio G, Quaini F, Gaetano C, Capogrossi MC (2011) Human cardiac and bone marrow stromal cells exhibit distinctive properties related to their origin. Cardiovasc Res 89:650–660 doi: 10.1093/cvr/cvq290 [DOI] [PubMed] [Google Scholar]
- 29.Ruau D, Ensenat-Waser R, Dinger TC, Vallabhapurapu DS, Rolletschek A, Hacker C, Hieronymus T, Wobus AM, Muller AM, Zenke M (2008) Pluripotency associated genes are reactivated by chromatin-modifying agents in neurosphere cells. Stem cells 26:920–926 doi: 10.1634/stemcells.2007-0649 [DOI] [PubMed] [Google Scholar]
- 30.Sanganalmath SK, Bolli R (2013) Cell Therapy for Heart Failure A Comprehensive Overview of Experimental and Clinical Studies, Current Challenges, and Future Directions. Circ Res 113:810–834 doi: 10.1161/Circresaha.113.300219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Segura AM, Frazier OH, Buja LM (2014) Fibrosis and heart failure. Heart Fail Rev 19:173–185 doi: 10.1007/s10741-012-9365-4 [DOI] [PubMed] [Google Scholar]
- 32.Soddu S, Blandino G, Scardigli R, Coen S, Marchetti A, Rizzo MG, Bossi G, Cimino L, Crescenzi M, Sacchi A (1996) Interference with p53 protein inhibits hematopoietic and muscle differentiation. J Cell Biol 134:193–204 doi:DOI 10.1083/jcb.134.1.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stein AB, Tiwari S, Thomas P, Hunt G, Levent C, Stoddard MF, Tang XL, Bolli R, Dawn B (2007) Effects of anesthesia on echocardiographic assessment of left ventricular structure and function in rats. Basic Res Cardiol 102:28–41 doi: 10.1007/s00395-006-0627-y [DOI] [PubMed] [Google Scholar]
- 34.Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL (2010) Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 3:2751–2767 doi: 10.3390/ph3092751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki G, Weil BR, Leiker MM, Ribbeck AE, Young RF, Cimato TR, Canty JM (2014) Global Intracoronary Infusion of Allogeneic Cardiosphere-Derived Cells Improves Ventricular Function and Stimulates Endogenous Myocyte Regeneration throughout the Heart in Swine with Hibernating Myocardium. PloS one 9 doi:ARTN e113009 10.1371/journal.pone.0113009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang XL, Li Q, Rokosh G, Sanganalmath SK, Chen N, Ou Q, Stowers H, Hunt G, Bolli R (2016) Long-Term Outcome of Administration of c-kit(POS) Cardiac Progenitor Cells After Acute Myocardial Infarction: Transplanted Cells Do not Become Cardiomyocytes, but Structural and Functional Improvement and Proliferation of Endogenous Cells Persist for at Least One Year. Circ Res 118:1091–1105 doi: 10.1161/CIRCRESAHA.115.307647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang XL, Rokosh G, Sanganalmath SK, Tokita Y, Keith MC, Shirk G, Stowers H, Hunt GN, Wu W, Dawn B, Bolli R (2015) Effects of Intracoronary Infusion of Escalating Doses of Cardiac Stem Cells in Rats With Acute Myocardial Infarction. Circ Heart Fail 8:757–765 doi: 10.1161/CIRCHEARTFAILURE.115.002210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang XL, Rokosh G, Sanganalmath SK, Yuan F, Sato H, Mu J, Dai S, Li C, Chen N, Peng Y, Dawn B, Hunt G, Leri A, Kajstura J, Tiwari S, Shirk G, Anversa P, Bolli R (2010) Intracoronary administration of cardiac progenitor cells alleviates left ventricular dysfunction in rats with a 30-day-old infarction. Circulation 121:293–305 doi: 10.1161/CIRCULATIONAHA.109.871905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weil BR, Suzuki G, Leiker MM, Fallavollita JA, Canty JM, Jr. (2015) Comparative Efficacy of Intracoronary Allogeneic Mesenchymal Stem Cells and Cardiosphere-Derived Cells in Swine with Hibernating Myocardium. Circ Res 117:634–644 doi: 10.1161/CIRCRESAHA.115.306850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wysoczynski M, Dassanayaka S, Zafir A, Ghafghazi S, Long BW, Noble C, DeMartino AM, Brittian KR, Bolli R, Jones SP (2016) A New Method to Stabilize C-Kit Expression in Reparative Cardiac Mesenchymal Cells. Front Cell Dev Biol 4:78 doi: 10.3389/fcell.2016.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wysoczynski M, Guo Y, Moore JB 4th, Muthusamy S, Li Q, Nasr M, Li H, Nong Y, Wu W, Tomlin AA, Zhu X, Hunt G, Gumpert AM, Book MJ, Khan A, Tang XL, Bolli R (2017) Myocardial Reparative Properties of Cardiac Mesenchymal Cells Isolated on the Basis of Adherence. J Am Coll Cardiol 69:1824–1838 doi: 10.1016/j.jacc.2017.01.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie M, Kong YL, Tan W, May H, Battiprolu PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, Jiang N, Jessen ME, Warner JJ, Lavandero S, Gillette TG, Turer AT, Hill JA (2014) Histone Deacetylase Inhibition Blunts Ischemia/Reperfusion Injury by Inducing Cardiomyocyte Autophagy. Circulation 129:1139–U1170 doi: 10.1161/Circulationaha.113.002416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang G, Tian J, Feng C, Zhao LL, Liu Z, Zhu J (2012) Trichostatin a promotes cardiomyocyte differentiation of rat mesenchymal stem cells after 5-azacytidine induction or during coculture with neonatal cardiomyocytes via a mechanism independent of histone deacetylase inhibition. Cell transplantation 21:985–996 doi: 10.3727/096368911X593145 [DOI] [PubMed] [Google Scholar]
- 44.Yang G, Tian J, Feng C, Zhao LL, Liu ZG, Zhu J (2012) Trichostatin A Promotes Cardiomyocyte Differentiation of Rat Mesenchymal Stem Cells After 5-Azacytidine Induction or During Coculture With Neonatal Cardiomyocytes Via a Mechanism Independent of Histone Deacetylase Inhibition. Cell Transplant 21:985–996 doi: 10.3727/096368911X593145 [DOI] [PubMed] [Google Scholar]
- 45.Yoon CH, Koyanagi M, Iekushi K, Seeger F, Urbich C, Zeiher AM, Dimmeler S (2010) Mechanism of improved cardiac function after bone marrow mononuclear cell therapy: role of cardiovascular lineage commitment. Circulation 121:2001–2011 doi: 10.1161/CIRCULATIONAHA.109.909291 [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Qin X, Zhao Y, Fast L, Zhuang S, Liu P, Cheng G, Zhao TC (2012) Inhibition of histone deacetylases preserves myocardial performance and prevents cardiac remodeling through stimulation of endogenous angiomyogenesis. J Pharmacol Exp Ther 341:285–293 doi: 10.1124/jpet.111.189910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang LX, Zhao Y, Cheng G, Guo TL, Chin YE, Liu PY, Zhao TC (2010) Targeted deletion of NF-kappaB p50 diminishes the cardioprotection of histone deacetylase inhibition. Am J Physiol Heart Circ Physiol 298:H2154–2163 doi: 10.1152/ajpheart.01015.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao J, Ghafghazi S, Khan AR, Farid TA, Moore 4th JB (2016) Recent Developments in Stem and Progenitor Cell Therapy for Cardiac Repair. Circ Res 119:e152–e159 doi: 10.1161/CIRCRESAHA.116.310257 [DOI] [PubMed] [Google Scholar]
- 49.Zhao Y, Lu SL, Wu LP, Chai GL, Wang HY, Chen YQ, Sun J, Yu Y, Zhou W, Zheng QH, Wu M, Otterson GA, Zhu WG (2006) Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol Cell Biol 26:2782–2790 doi: 10.1128/Mcb.26.7.2782-2790.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.