

Abstract
Pain is a complex process that involves both detection in the peripheral nervous system and perception in the CNS. Individual-to-individual differences in pain are well documented, but not well understood. Here we capitalized on inherited erythromelalgia (IEM), a well characterized human genetic model of chronic pain, and studied a unique family containing related IEM subjects with the same disease-causing NaV1.7 mutation, which is known to make dorsal root ganglion (DRG) neurons hyperexcitable, but different pain profiles (affected son with severe pain, affected mother with moderate pain, and an unaffected father). We show, first, that, at least in some cases, relative sensitivity to pain can be modeled in subject-specific induced pluripotent stem cell (iPSC)-derived sensory neurons in vitro; second, that, in some cases, mechanisms operating in peripheral sensory neurons contribute to interindividual differences in pain; and third, using whole exome sequencing (WES) and dynamic clamp, we show that it is possible to pinpoint a specific variant of another gene, KCNQ in this particular kindred, that modulates the excitability of iPSC-derived sensory neurons in this family. While different gene variants may modulate DRG neuron excitability and thereby contribute to interindividual differences in pain in other families, this study shows that subject-specific iPSCs can be used to model interindividual differences in pain. We further provide proof-of-principle that iPSCs, WES, and dynamic clamp can be used to investigate peripheral mechanisms and pinpoint specific gene variants that modulate pain signaling and contribute to interindividual differences in pain.
SIGNIFICANCE STATEMENT Individual-to-individual differences in pain are well documented, but not well understood. In this study, we show, first, that, at least in some cases, relative sensitivity to pain can be modeled in subject-specific induced pluripotent stem cell-derived sensory neurons in vitro; second, that, in some cases, mechanisms operating in peripheral sensory neurons contribute to interindividual differences in pain; and third, using whole exome sequencing and dynamic clamp, we show that it is possible to pinpoint a specific gene variant that modulates pain signaling and contributes to interindividual differences in pain.
Keywords: dynamic clamp, induced pluripotent stem cells, pain, potassium channel, voltage-gated sodium channel, whole exome sequencing
Introduction
Chronic pain affects >250 million individuals worldwide, and the lack of effective pain treatment has contributed to the opioid crisis. Interindividual differences in pain are well documented, with some individuals reporting more severe pain, and others reporting less severe pain in response to similar noxious insults. However, individual-to-individual variation in pain has not been accurately modeled in the laboratory and its mechanistic basis remains incompletely understood, partially because pain involves both detection in the peripheral nervous system and perception in the CNS, and involves processes that operate at multiple levels, including genetic, epigenetic, environmental, and social.
Inherited erythromelalgia (IEM) is an autosomal-dominant disorder characterized by episodes of intense burning pain in the distal extremities in response to mild warmth that provides a human genetic model of chronic pain with a well defined causative molecular substrate (Drenth and Waxman, 2007). IEM is caused by gain-of-function mutations in the voltage-gated sodium channel NaV1.7, which is expressed mainly in the peripheral nervous system, that produce hyperexcitability in peripheral sensory [dorsal root ganglion (DRG)] neurons. More than a dozen NaV1.7 channel mutations have been reported to cause IEM via this mechanism (Dib-Hajj et al., 2013). Interestingly, even for patients carrying the same NaV1.7 mutation, differences in pain have been documented (Geha et al., 2016; McDonnell et al., 2016). Little is known about the cellular or molecular basis for differences in pain in patients with the same NaV1.7 mutation, and, thus far, the difference in pain has not been modeled at the bench.
We have capitalized on IEM as a well characterized genetic model of chronic pain, and studied a unique kindred containing two IEM subjects from the same family (mother and son), both carrying the NaV1.7-S241T mutation, which is known to enhance channel activation (Lampert et al., 2006) and produce hyperexcitability of DRG neurons (Yang et al., 2012). The son displayed a much more severe pain profile (higher number and longer duration of attacks, and higher number of nightly awakenings) compared with his mother (Geha et al., 2016). We used subject-specific induced pluripotent stem cells (iPSCs) to ask whether the individual-to-individual difference in pain profiles might be modeled in a “disease-in-a-dish” model (McNeish et al., 2015) in the laboratory and studied this phenomenon at the cellular and molecular levels. We asked whether the difference in pain between these two individuals might, at least in part, be a result of different firing properties of their peripheral sensory neurons, and further, asked whether we could identify molecular contributors to the differences in these pain profiles.
Cells derived from iPSCs retain the genetic background and native transcriptional machinery of affected patients (Inoue et al., 2014; Zeltner and Studer, 2015; Soliman et al., 2017). We prepared iPSCs from blood samples of the affected son (P300; severe pain) and mother (P301; mild pain) carrying the NaV1.7-S241T mutation, and from an unaffected family member (P303, P300's father) and differentiated these iPSCs into peripheral sensory neurons (iPSC-SNs) for disease modeling. We demonstrate that iPSC-SNs derived from these subjects display significant differences in firing frequency and spontaneous activity that parallel their different pain profiles. Using whole exome sequencing (WES), we discovered multiple gene variants that might contribute to neuronal excitability and that might serve as modifiers of sensory neuron firing. We then identified a variant of one particular gene (KCNQ in this kindred) as a contributor to differences in pain between these two individuals. While different gene variants may affect DRG neuron excitability and thereby contribute to interindividual differences in pain in other families, this study shows that it is possible to model interindividual differences in pain using subject-specific iPSCs. We further provide proof-of-concept that WES and dynamic clamp can be used to investigate peripheral mechanisms and pinpoint specific gene variants that modulate pain signaling and contribute to interindividual differences in pain.
Materials and Methods
Generation of induced pluripotent stem cells
iPSCs were generated from the blood samples of two IEM subjects [mother (P301) and son (P300) carrying the NaV1.7-S241T mutation] and an unaffected individual [father (P303)] using CytoTune-iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher Scientific) according to the manufacturer protocol. Cells were screened for pluripotent stem cell markers and tested for normal karyotype. iPSCs were cultured for at least 10 generations before the start of differentiation into sensory neurons (i.e., iPSC-SNs). The study was approved by the Yale Human Investigation Committee.
Differentiation of iPSC into sensory neurons
Differentiation was initiated using a modified Chambers protocol using LSB and 3i inhibitors (Chambers et al., 2012; Young et al., 2014; Cao et al., 2016). Differentiated neurons were maintained in Neurobasal Medium supplemented with N2/B27 GlutaMAX (Thermo Fisher Scientific) and nerve growth factors [recombinant human β nerve growth factor, brain-derived neurotrophic factor, glial cell line-derived neurotrophic factor, and neurotrophin-3 (NT-3; 25 μg/ml; PeproTech)] for 8 weeks before functional assessment.
Immunocytochemistry
iPSC-SNs were immunostained with markers for sensory neurons. Primary antibodies were incubated overnight at 4°C in PBS-T (0.1% Triton X-100, 2% BSA, 4% donkey serum in PBS; Pan Neuronal Marker-Alexa Fluor 488 conjugate, 1:100, MAB2300X - Millipore; peripherin 1:200, SC-7604, Santa Cruz Biotechnology; BRN3A, 1:200, AB5945, Millipore; NaV1.7, 1:250, Y083; Islet 1, 1:200, ab86501, Abcam). Secondary antibodies were incubated for 2 h at room temperature in PBS-T. Images were acquired using a Nikon C1 confocal microscope.
Multielectrode array recordings
Multielectrode array (MEA) experiments were performed with a multiwell MEA system (Maestro, Axion Biosystems) according to our recently developed protocol (Yang et al., 2016). Briefly, iPSC-SNs were dissociated and cultured on MEA plates, maintained at 37°C in a 5% CO2 incubator. A 12-well recording plate was used, embedded with a total of 768 electrodes. For each experiment, three wells (with ∼192 available electrodes for recording) were used to assess iPSC-SNs derived from P301, P300, and P303.
Whole-cell current-clamp electrophysiology
Whole-cell current-clamp recordings were obtained for head-to-head comparisons from iPSC-SNs from paired differentiations prepared contemporaneously and processed in parallel by the same technician and studied by the same electrophysiologist. Recordings were amplified using an Axon MultiClamp 700B amplifier. Data were digitized via an analog-to-digital converter (Digidata 1440a, Molecular Devices) and stored on a personal computer using pClamp 10.6 software, which was also used to define and execute protocols. The data were filtered at 5 kHz and acquired at 50 kHz. Electrodes used for the recordings had resistance of <1.5 MΩ when filled with the internal solution, which consisted of the following (in mm): KCl 140, HEPES 5, EGTA 0.5, Mg-ATP 3, and dextrose 20, at pH 7.3 and 295–300 mOsm. iPSC-SNs were continuously perfused with external recording solution containing the following (in mm): NaCl 140, KCl 3, HEPES-NaOH 10, MgCl2 2, CaCl2 2, and dextrose 15, at pH 7.3 and ∼320 mOsm.
Whole exome sequencing and analysis
WES was performed at the Yale Center for Genome Analysis following a previously published protocol (Zaidi et al., 2013; Jin et al., 2017). The following three subjects were included for sequencing analysis: proband carrying the NaV1.7-S241T mutation, proband's mother carrying the NaV1.7-S241T mutation, and proband's unaffected father. The obtained reads were filtered and trimmed for quality and aligned to the hg19 version of the human genome (GRCh37) using an aligner (BWA-MEM algorithm). From the aligned reads, we used variant caller (GATK) to call the variants from each sample. We extracted the significant variants based on genotyping quality score and coverage of the reference and alternative base (the criteria are at least three reads with alternative base, and at least 20% of coverage is alternative base). All the variants that passed the filter were then collected across all the samples using custom-built python scripts.
The Ensembl variant effect predictor was used to determine the effect of the resulting variants, and ingenuity pathway analysis (IPA; build 470319M, version 43605602, Qiagen) was used to carry out functional annotation analyses for gene ontology functions analyses (http://www.ingenuity.com/).
RT-PCR and sequencing
RNA was isolated from iPSC-SNs from P300, P301, and P303 using the RNeasy Plus Kit (catalog #74134, Qiagen) according to the manufacturer protocol. RNA concentration was measured on a Nanodrop, and total RNA (100 ng) was used to generate cDNA using the iScript Reverse Transcription Supermix (catalog #170–8841, Bio-Rad). One milliliter of cDNA was used as a template for PCR amplification in a final volume of 50 ml. High-fidelity AccuPrime TaqDNA Polymerase (catalog #12346-086, Thermo Fisher Scientific) was used for amplification. At least one of the primers crosses an exon–intron boundary to distinguish cDNA products from potential genomic DNA contamination.
Thermal cycling was initiated at 94°C for 2 min followed by 35 cycles of 30 s at 94°C, annealing for 30 s at 55°C for NaV1.7 (60°C for KV7.2), and an extension for 60 s at 68°C. Because of high GC content, PCR was performed with 6% DMSO for KV7.2. The following primers were used: 5′-ATCACGGACAAGGACCGCACC-3′ and 5′-TCCTGCCGCAGGAACTCCATG-3′ generating a 512 bp fragment for KV7.2; and 5′-TGCAAGAGGCTTCTGTGTAGG-3′ and 5′- GCTCGTGTAGCCATAATCAGG-3′ generating a 514 bp fragment for NaV1.7. The identity of the amplicon was verified by Sanger sequencing using the purified PCR product (PCR clean-up, Gel extraction kit, catalog #740609.50, Macherey-Nagel), and the same forward and reverse primers that were used for PCR amplification. Sequencing was performed at the Keck DNA Sequencing facility at Yale University.
Perforated-patch M-current recordings in iPSC-SNs
Recordings were obtained using an EPC-10 amplifier and the PatchMaster program (HEKA Elecktronik). Data were sampled at 4 kHz and filtered at 2.9 kHz with a low-pass Bessel filter. Patch pipette resistance was 2–3 MΩ, and series resistance was compensated for (60–90%).
Extracellular bath solution contained the following (in mm): 144 NaCl, 2.5 KCl, 2 CaCl2, 0.5 MgCl, 5 HEPES, and 10 glucose, with pH adjusted to 7.4 with NaOH. The bath was supplemented with 5 mm 4-aminopyridine (4-AP) to block the fast-activating KV1 channels, 1 μm tetrodotoxin (TTX) to inhibit sodium currents and 20 μm 4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride (ZD-7288) to block hyperpolarization-activated cyclic nucleotide (HCN)-gated currents. Pipettes were filled with an intracellular solution containing the following (in mm): 80 K-acetate, 30 KCl, 40 HEPES, 3 MgCl, 3 EGTA, and 1 CaCl2, with pH adjusted to 7.4 with NaOH. Liquid junction potential (LJP) was corrected (+8.2 mV). All the recordings were performed at room temperature. Data were analyzed using Fitmaster (HEKA Elektronik) and Origin (MicroCal Software).
iPSC-SNs were recorded in the perforated patch configuration using Amphotericin B to reduce rundown, and a stock solution of 1 mg/20 μl DMSO was prepared and stored in the dark. For recordings, 2 μl of the stock were dissolved in 1 ml of intracellular solution using an ultrasonicator. A fresh batch of solution was remade every 2 h.
M-current (IM) was identified by using a standard deactivation voltage protocol (Adams and Brown, 1982), in which cells are held at −20 mV to activate the current and then deactivated by intermittent hyperpolarizing steps.
The current–voltage (I–V) curves were calculated according to Adams et al. (1982). We measured the instantaneous current and the steady-state current, the total current measured after the slow relaxation is complete; the intersection of these two currents give the reversal potential and under our conditions the reversal potential was −78.2 mV, close to the calculated potassium reversal potential (EK). The leak obtained by extrapolation of the linear portion of the I–V curve between −100 and −70 was then subtracted according to the findings of Passmore et al. (2003). The IM (leak-subtracted-steady-state) currents were normalized and plotted versus membrane voltages.
The conductance of IM was assessed according to Adams et al. (1982) The current values (IM) were divided by the driving force and normalized to the maximal value to obtain the conductance (gKV7/M); ΔGM = ΔIM(V)/(V − Vm) (Adams and Brown, 1982; Adams et al., 1982). Data were fitted with a Boltzmann curve: g/gmax = A1 − A2 (1 + exp (V − V1/2)/ Kl) − 1, where V1/2 is the half-maximal activation voltage, A1 and A2 are the minimum and maximum values, and Kl is the curve slope. LJP was corrected (+8.2 mV).
Dynamic clamp recordings
iPSC-SNs were dynamically clamped in whole-cell configuration (Petrovic et al., 2012; Battefeld et al., 2014; Vasylyev et al., 2014) to introduce model IM conductance based on the kinetic model of IM. The extracellular and pipette solutions had the same composition as those used for current-clamp recordings. Electrode resistance was ∼1 MΩ when filled with the intracellular solution. Membrane voltages and currents were recorded in dynamic clamp with a MultiClamp 700B Amplifier (Molecular Devices) interfaced with CED Power 1401 mkII DAI and Signal 6 software (CED), digitized by a Digidata 1440A digital-to-analog converter, and stored on hard disk for off-line analysis using pClamp 10.6 software (Molecular Devices). Recordings were performed at room temperature.
Kinetic model of IM
The gating variable for IM is described using a Hodgkin–Huxley differential equation dn/dt = αn(1 − n) − βnn, where n is the channel activation variable and α(β) is forward (reverse) rate constants, respectively. IM steady-state parameters and kinetics obtained from electrophysiological recordings were converted into rate constants at respective voltages using the equations α = n/τ and β = (1 − n)/τ. Liquid junction potentials (+8.2 mV) were adjusted for all parameters. Reaction rate constants were fitted with a Boltzmann equation and converted into a steady-state activation variable and a time constant according to n = α/(α+ β) and τ = 1/(α+ β). Wild-type (WT) and T730A IM models were calculated in a 28 pF equipotential sphere of 1 μF/cm2 capacitance with a conductance density of 0.00014 S/cm2.
The following rate constants were used for the P300 (homozygous KCNQ2-WT) KV7.2 channel model:
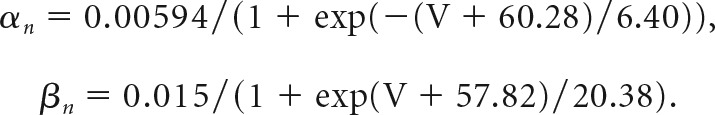 |
P301 KV7.2 channel (heterozygous WT/T730A) was described by the following rate constant:
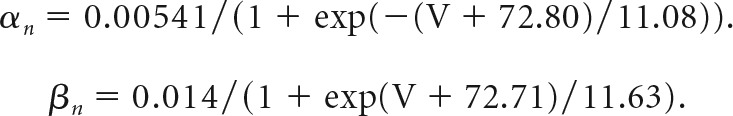 |
IM conductance was modeled using Hodgkin–Huxley formalism as a noninactivating current described by IM = gM * n (V − EK), where gM is the maximal conductance, n represents an activation gate, and V is the membrane potential. Currents evoked by different voltage protocols were calculated in 10 μs precision with a custom program written in OriginPro 8.5 LabTalk.
Experimental design and statistical analysis
MEA.
To minimize potential variations during the recordings, iPSC-SNs from all three subjects were differentiated on the same day with same reagents. iPSC-SNs were always plated on MEA plates by the same investigator. A spike detection criterion of >6 SDs above background signals was used to separate monophasic and biphasic action potential (AP) spikes from noise. We defined active electrodes as registering more than one recorded spike over a 200 s period (Yang et al., 2016). MEA data were analyzed using Axion Integrated Studio AxIS2.1 (Axion Biosystems) and NeuroExplorer (Nex Technologies), as previously described (Yang et al., 2018).
To assess the firing properties under different temperatures, the precise temperature control of the MEA system was used, which enables continuous monitoring of neuronal firing during temperature ramps. iPSC-SNs from P301, P300, and P303 were plated on the same MEA plate for the temperature ramp study, and assessed by an investigator blinded to the genotype. Three different temperatures (33°C, 37°C, and 40°C) were used during the study, and each temperature was maintained for 7–10 min to allow analysis of steady-state neuronal firing at each condition.
Whole-cell current-clamp.
Only iPSC-SNs with stable membrane potential were chosen for analysis. Resting membrane potential was determined immediately after switching into current-clamp mode as the mean membrane voltage in the absence of current stimulation. Set stimulus membrane potentials were established by manual injection of bias currents of appropriate amplitudes for the experiments. Current threshold was defined as the minimum amount of current necessary to trigger an AP and was determined by injecting depolarizing 200 ms current steps in 5 pA increments until an AP was triggered. To assess the firing properties, incremental depolarizing 500 ms current steps up to 500 pA were applied. The elicited APs were counted and plotted against the current injection intensity. Recorded data were processed offline using pClamp version 10.6, Origin 2017, and Excel.
Unless otherwise stated, data are expressed as mean ± SEM. Analyses were performed with SPSS 24 (SPSS) and Origin 2017. Statistical tests used for each individual dataset and exact p values are stated in the Results section.
Results
Differences in pain in individuals carrying the same NaV1.7-S241T mutation are paralleled by differences in excitability of iPSC-SNs
The clinical features of P300 and P301 were evaluated in two previous studies (Geha et al., 2016; McDonnell et al., 2016). Despite carrying the same NaV1.7-S241T mutation (Fig. 1A), subjects P300 and P301 reported very different pain profiles (different number and duration of attacks, and number of awakenings from pain). In one study (McDonnell et al., 2016), P300 reported an average of 11.8 pain attacks per week, while P301 reported 2.8 pain attacks per week. The mean duration of each pain attack for P300 was 378.3 min, while for P301 it was 56.1 min. In the second study (Geha et al., 2016), P300 reported time in pain of 424 min per day, while P301 contemporaneously reported 61 min; P300 reported average pain attack duration of 615 min, while P300 reported 91.5 min; and P300 reported 101 awakenings from pain over a 15 d period, while P301 reported 1 awakening. Although variations in pain profiles between individuals may reflect differences in processing at multiple levels including higher CNS levels, we reasoned that differences in peripheral neurons might also play a role, particularly in individuals with pain of peripheral origin, such as those (e.g., P300 and P301) who carry a gain-of-function mutation in NaV1.7, a channel that is mainly expressed in peripheral sensory neurons where it confers hyperexcitability on them. To assess whether differences in pain profiles might be modeled in an in vitro system containing only their peripheral neurons, we derived iPSC-SNs from the affected son (P300) and mother (P301), as well as the unaffected father (P303) using a differentiation protocol that produces pain-sensing sensory-like neurons (Chambers et al., 2012, 2016; Cao et al., 2016). The S241T mutation was verified by Sanger sequencing in the iPSCs from P300 and P301 and shown to be absent in P303. The iPSC-SNs stained positively for peripheral neuronal marker (peripherin), sensory neuronal marker (Brn3a), as well as NaV1.7 channel (Fig. 1B), and displayed neuronal morphology and electrophysiological properties characteristic of mature neurons (Fig. 1C). The expression of NaV1.7 was verified by RT-PCR and shows that both P300 and P301 iPSC-SNs produce both wild-type and S241T mutant transcripts, while samples from P303 produced only WT transcripts (Fig. 1-1).
Figure 1.

MEA recordings reveal differences in excitability between iPSC-SNs from subjects carrying the NaV1.7-S241T mutation and an unaffected control. A, Affected subjects P301 (mother) and P300 (son) carry the NaV1.7-S241T mutation, whereas P303 (unaffected father) carries only wild-type alleles. B, iPSC-SNs from P300, P301, and P303 express canonical sensory neuron markers and NaV1.7. Left, Peripherin (red), Brn3a (green), and Islet1 (gray). Figure 1-1, shows that the NaV1.7-S241T mutation is present in iPSC-SNs from P300 and P301, but not P303. Right, Peripherin (red), NaV1.7 (green), and Pan Neuronal Marker (gray). C, iPSC-SNs from all three subjects showed a neuronal morphology (microphotograph; scale bar, 20 μm), produced large sodium and potassium currents, and fired APs [examples from P301 (middle) and P300 (right)]. D, MEA recordings of AP firing of iPSC-SNs from P300, P301, and 303. Heatmaps show representative MEA recordings. The firing frequency of each active electrode is color coded: white/red, high firing frequency; blue/black, low firing frequency. Each circle represents an active electrode within an 8 × 8 electrode array. Top panels, Recordings from iPSC-SNs from P300 at 33°C, 37°C, and 40°C. Middle panels, Recordings from iPSC-SNs from P301 at 33°C, 37°C, and 40°C. Bottom panels, Recordings from iPSC-SNs from P303 at 33°C, 37°C, and 40°C. E, Representative MEA recordings showing neuronal firing at 33°C. F, Top, Average firing frequencies of neurons from P300, P301, and P303. Bottom, Average numbers of active electrodes for P300, P301, and P303 (average of three wells). *p < 0.05.
Nav1.7-S241T mutation is expressed in iPSC-SNs from the affected son (P300) and mother (P301), but not the unaffected father (P303). Agarose gel electrophoresis showing the amplification of the expected product (514 bp) from cDNA samples of iPSC-SNs (the M lane shows the 100 bp molecular weight marker) and chromatogram of the sequence of the obtained products confirms the expression of both WT (TCA) and mutant (ACA) alleles in the samples from P300 and P301, and only the WT allele in the sample from P303. Download Figure 1-1, TIF file (1.4MB, tif)
We first studied the excitability of these iPSC-SNs using MEA, a noninvasive, high-throughput, extracellular recording approach, that can assess the excitability of intact neurons (Spira and Hai, 2013). MEA is capable of accurately recording AP firing of neurons as temperature is altered. Because pain in individuals with IEM (including subjects P300 and P301) is triggered by warmth, we assessed the firing of these intact iPSC-SNs at the following three different temperatures: skin temperature (33°C), core body temperature (37°C), and non-noxious warmth (40°C). Neurons from both P300 and P301 displayed temperature-induced increases in firing, as reflected by heat maps (Fig. 1D). Elevating the temperature increased both the mean firing frequency and number of neurons firing APs without electrical stimulation, with neurons from P300 and P301, which carry the S241T mutation, more excitable than these from P303, the unaffected father who does not carry the mutation (Fig. 1D–F). Indeed, we did not observe any firing from iPSC-SNs derived from P303 at frequencies >0.2 Hz, even at 40°C. Notably, while the significant effect of the mutation on mean firing rate (F = 24.7, p = 0.00002, one-way repeated-measures ANOVA; six independent differentiations from two independent clones for each line) and on the number of active electrodes (F = 192, p < 0.0001, one-way repeated-measures ANOVA) was expected, we also observed significant differences in the excitability of iPSC-SNs between P300 and P301 (Fig. 1F), who both carry the same NaV1.7-S241T mutation but reported differences in their pain. Compared with iPSC-SNs from P301 (less pain), iPSC-SNs from P300 (more pain) displayed a significantly higher firing frequency (33°C: P300 = 0.99 ± 0.16 Hz, P301 = 0.32 ± 0.07 Hz, p = 0.01; 37°C: P300 = 1.56 ± 0.27 Hz, P301 = 0.51 ± 0.08 Hz; p = 0.001; 40°C: P300 = 2.1 ± 0.35 Hz, P301 = 0.66 ± 0.09 Hz; p = 0.001; Bonferroni corrections) and a significantly higher number of active electrodes (33°C: P300 = 99 ± 5, P301 = 72 ± 5, p = 0.01, 37°C: P300 = 106 ± 5, P301 = 84 ± 3, p = 0.001, 40°C: P300 = 110 ± 4, P301 = 88 ± 3, p = 0.0004; Bonferroni corrections), suggesting that for these individuals it might be possible to model differences in pain profiles in an in vitro system of only subject-specific iPSC-SNs.
Subject-specific iPSC-SNs from P300 and P301 display differences in membrane excitability that parallel differences in pain profiles
A spectrum of differences in membrane properties might contribute to interindividual differences in the activity of DRG neurons in different kindreds. To extend the findings from the MEA assay to the level of membrane excitability in the family under study, we used whole-cell patch clamp for a head-to-head comparison of iPSC-SNs from P300, P301, and P303, from differentiations prepared contemporaneously and processed in parallel. These experiments revealed pronounced excitability differences among iPSC-SNs from the three subjects. We assessed the following four major parameters that reflect the levels of neuronal excitability: induced firing, percentage of neurons firing repetitively, current threshold, and resting membrane potential (RMP). For all of these four parameters, we observed very clear and consistent differences in excitability among iPSC-SNs from P300, P301, and P303 that again paralleled differences in pain profiles. In response to graded suprathreshold 500 ms depolarizing stimuli, across the entire current injection range, neurons derived from P300 fired the highest number of APs, followed by neurons derived from P301, with P303 neurons firing at the lowest rate (F = 12, p = 0.00006, one-way repeated measures ANOVA; Bonferroni corrections: P300 vs P301, p = 0.037; P300 vs P303, p = 0.00003; P301 vs P303, p = 0.044; Fig. 2A,B). When we considered the percentage of multiple-spiking neurons, we observed a parallel set of differences in excitability, with iPSC-SNs from P300 having the highest and P303 the lowest proportion of multiple-spiking (>1 AP) iPSC-SNs (Fig. 2C).
Figure 2.

Passive and firing properties of iPSC-SNs at RMP show differences in excitability among P300, P301, and P303. A, Input–output relationships for iPSC-SNs from P300 (orange), P301 (blue), and P303 (gray) subjects. Data are reported as the mean ± SEM. P300, N = 18; P301, N = 19; P303, N = 15; F = 12, p = 0.00006, one-way repeated measures ANOVA; Bonferroni corrections: P300–P301 = 0.037; P300–P303 = 0.00003; P301–P303 = 0.044. B, Example traces showing AP firing in iPSC-SNs from P300, P301, and P303 in response to 500 ms 300 pA steps. C, Charts showing the percentage of single-spiking iPSC-SNs (P300, 12%; P301, 32%; P303, 87%). D, Current threshold values. Each symbol refers to an individual neuron; to the right are mean (diamond symbol), median (line), SE (box), and confidence intervals (whiskers). P300, N = 16; P301, N = 17; P303, N = 12; H = 19.7, p < 0.0005, nonparametric ANOVA; Bonferroni corrections: P300–P301 = 0.02; P300–P303 < 0.0005; P301–P303 = 0.2. E, Example traces showing the difference in current threshold between iPSC-SNs from P300, P301, and P303. F, RMP values for the three subjects. Each symbol refers to an individual neuron. To the right are mean (diamond symbol), median (line), SE (box), and confidence intervals (whiskers). P300, N = 18; P301, N = 19; P303, N = 15; F = 7, p = 0.002, one-way ANOVA; Bonferroni corrections: P300–P301 = 0.04; P300–P303 = 0.002; P301–P303 = 0.6. *p < 0.05, **p < 0.01, ***p < 0.001.
iPSC-SNs from P300 were also found to have the lowest current threshold (H = 19.7, p < 0.0005, nonparametric ANOVA; Bonferroni corrections: P300 vs P301, p = 0.02; P300 vs P303, p ≤ 0.0005; P301 vs P303, p = 0.2). The data in Figure 2, D and E, show that it is much harder to trigger an AP in neurons derived from P303 (unaffected control), easier for P301 (less pain), and easiest for P300 (more pain), also consistent with the pain reported by these individuals.
Interestingly, the RMP from these iPSC-SNs showed a similar pattern, with the membrane potential of P303 neurons most hyperpolarized, P300 neurons most depolarized, and P301 neurons in between (RMP: P300 = −54 ± 1 mV; P301 = −58 ± 1 mV and P303 = −61 ± 1 mV; F = 7, p = 0.002, one-way ANOVA; Bonferroni corrections: P300 vs P301, p = 0.04; P300 vs P303, p = 0.002; P301 vs P303, p = 0.6; Fig. 2F).
Membrane potential contributes to the excitability difference observed between iPSC-SNs
It has been shown that in DRG neurons, neuronal excitability is dependent on RMP (Harty et al., 2006; Huang et al., 2017). Following up on our observations of a significant difference of 4 mV in RMP between iPSC-SNs from P301 and P300, in a separate set of experiments we assessed the impact of membrane potential on the excitability of these neurons, again via a head-to-head comparison of iPSC-SNs from paired differentiations prepared contemporaneously and processed in parallel. In these experiments, we studied the current threshold and firing rate of individual iPSC-SNs at RMP before either depolarizing (P301) or hyperpolarizing (P300) the membrane potential of each given neuron by 4 mV. We found that depolarizing neurons derived from P301 by 4 mV resulted in a 25 ± 4% decrease in the current threshold, whereas hyperpolarizing neurons derived from P300 caused a 24 ± 5% increase in current threshold (Fig. 3A,B). Consequently, there was not a significant difference in current threshold between P300 and P301 when the difference in RMP was removed by depolarizing neurons from P301 by 4 mV (Fig. 3C; P300 = 35 ± 4 pA, n = 12; P301 = 36 ± 4 pA, n = 14; t = 0.1, p = 0.9, two-tailed unpaired t test) or by hyperpolarizing neurons from P300 by 4 mV (Fig. 3D; P300 = 51 ± 6 pA, n = 12; P301 = 45 ± 5 pA, n = 14; t = 0.1, p = 0.9, two-tailed unpaired t test). Similarly, using the same paradigm to study the firing rate of both groups of iPSC-SNs, we found that there was no significant difference in firing rate between iPSC-SNs derived from P300 and P301 when studied at matched membrane potentials by hyperpolarizing iPSC-SNs from P300 by 4 mV (Fig. 3E; F = 0.9, p = 0.4, repeated-measures one-way ANOVA; P300, n = 11; P301, n = 10) or by depolarizing neurons from P301 by 4 mV (Fig. 3F–G; F = 1.3, p = 0.3; repeated-measures one-way ANOVA; P300, n = 11; P301, n = 10). These results indicate that the difference in RMP between iPSC-SNs from P300 and P301 contributes to the difference in excitability of these neurons.
Figure 3.

Firing properties of iPSC-SNs from subjects P300 and P301 are not significantly different at matched membrane potentials. A, Graph showing the percentage change in current threshold after depolarizing (P301, n = 10; blue) or hyperpolarizing (P300, n = 12; orange) the RMP of iPSC-SNs by 4 mV (the average difference in RMP between P300 and P301). B, Example recordings from neurons derived from P300 (orange) and P301 (blue) showing the change in current threshold after either depolarizing (P301) or hyperpolarizing (P300) the RMP of iPSC-SNs by 4 mV. C, Scatter plot showing individual current threshold values for iPSC-SNs from P300 (orange) at RMP and P301 (blue) depolarized by 4 mV. D, Scatter plot showing individual current threshold values for iPSC-SNs from P301 (blue) at RMP and P300 (orange) hyperpolarized by 4 mV. E, Input–output relationships for iPSC-SNs from P300 and P301 at matched membrane potentials (obtained by hyperpolarizing P300 neurons by 4 mV). F, Input–output relationships for iPSC-SNs at matched membrane potentials (obtained by depolarizing P301 neurons by 4 mV). G, Example recordings from a neuron derived from P300 (orange) and P301 (blue) showing the change in firing frequency after either depolarizing (P301) or hyperpolarizing (P300) the RMP by 4 mV.
Whole exome sequencing reveals potential modifiers of sensory neuron excitability
Because subjects P300 (severe pain) and P301 (moderate pain) share the same NaV1.7-S241T mutation, we hypothesized that additional genetic variations might contribute to the difference in excitability between iPSC-SNs from P300 and P301. To identify these potential modifiers in the family under study, we performed WES on samples from this family and filtered the resulting variants according to their expression in DRG neurons using IPA (Build 470319M version 43605602), a manually curated knowledge database created from the peer-reviewed biomedical literature. WES confirmed the S241T mutation in both P300 and P301, but not in P303.
The WES analysis identified 90 variants in P300 and P301 in genes known to be expressed in DRG neurons (9 in P300 and 81 in P301; Fig. 4A and Fig. 4-1). Since we found significant differences in excitability between iPSC-SNs from P300 and P301 (Fig. 2A–E), we interrogated specific gene ontology processes and functions related to neuronal excitability (“excitation of neuron” and “neuronal action potential”). These terms identified a variant in KCNQ2, the gene that encodes potassium channel KV7.2, in subject P301 (mother), but not in P300 (son; Fig. 4B). Notably, KV7.2 contributes to the noninactivating IM (produced by KV7.2, KV7.3, and KV7.5 channels in DRG neurons), which has been shown to be a major determinant of the RMP of small-diameter rat DRG neurons, where it regulates excitability (Passmore et al., 2003; Du et al., 2014). KV7.2 has also been found to be the main KV7 channel isoform expressed in rat DRG neurons (Rose et al., 2011). The identified heterozygous missense variant c.2188A>G in exon 17 of the KCNQ2 gene results in the substitution of polar and hydrophilic threonine to nonpolar and hydrophobic alanine at p.730 in the C terminus of the KV7.2 channel (p.Thr730Ala; Fig. 4D, top), has not been previously reported, and is found exclusively in P301 (Fig. 4C). After confirming the expression of KV7.2-WT in iPSC-SNs from P300, and the KV7.2-T730A variant in P301 (Fig. 4D, bottom), we proceeded to validating the impact of this variant on the excitability of iPSC-SNs from P301.
Figure 4.

Whole exome sequencing reveals a variant in KCNQ2 gene as a potential modulator of neuronal excitability in iPSC-SNs from P301. A, Venn diagram showing the numbers of detected variants in samples from P300, P301, and P303. A full list of gene variants is included in Figure 4-1. B, Targeted gene ontology analysis directed toward neuronal excitability indicated KCNQ2 as a potential excitability modulator in P301. C, The KCNQ2 variant is present in the genome of P301 (mother), but not of P300 (son) or P303 (father). D, Top, Location of the T730A mutation in the KV7.2 channel. Boxes in the C terminus indicate the four α-helical regions (A–D) and the ankyrin-G binding domain. Bottom, KV7.2 is expressed in P300 and P301; agarose gel electrophoresis showing the amplification of the expected product (512 bp) from cDNA samples of iPSC-SNs (the M lane shows the100 bp molecular weight marker); and a chromatogram of the sequence of the obtained products showing only the KV7.2-WT allele (ACC) in P300, and both KV7.2-WT and KV7.2-T730A (GCC) alleles in P301. E, Representative perforated patch-clamp recordings of IM from P300 (orange) and P301 (blue) neurons; currents evoked by a series of 1 s, 10 mV hyperpolarizing voltage steps from a holding potential of −20 mV. F, Left, Normalized I–V curves for IM recorded from P300 and P301. Right, G-V curves for IM recorded from P300 and P301 iPSCs-SNs. Data were corrected for LJP (+8.2 mV).
The Whole Exome Sequencing analysis identified variants in P300, P301, and P303 in genes known to be expressed in DRG neurons. P300 – gene variants identified in subject P300 (145). P301 – gene variants identified in subject P301 (184), P303 – gene variants identified in subject P303 (175). P300_unique – gene variants specific to subject P300 (9), P301_unique – gene variants specific to subject P301 (81), P303_unique – gene variants specific to subject P303 (105). Download Figure 4-1, TIF file (5.9MB, tif)
To establish whether the current produced by KV7.2-T730A contributes to the excitability of iPSC-SNs derived from P301 by modulating IM in these neurons, we determined the effect of the mutation on IM via perforated patch-clamp analysis. We then used these data to investigate the influence of this variant on the excitability of iPSC-SNs via dynamic clamp, an approach that combines the strategy of patch-clamp and computer simulation methods (Prinz et al., 2004), permitting the current produced by a mutant ion channel to be replaced with a precisely titrated amount of WT current so that, in each cell studied, the effect of the mutant channel on excitability can be assessed (Vasylyev et al., 2014).
The T730A substitution in KV7.2 causes a gain of function of IM
To characterize the IM in iPSC-SNs from P300 and P301 in voltage-clamp, we used perforated-patch recordings. The current was activated by holding the membrane at a steady depolarized potential (−20 mV) and then deactivated by hyperpolarizing steps. Since IM does not inactivate, this protocol minimizes potential contamination by other voltage-gated currents (Adams and Brown, 1982). We inhibited sodium currents, fast-activating Kv1- and Kv3-type potassium current and HCN currents by including in the recording solution their respective blockers: TTX, 4-AP, and ZD-7288. Representative traces of IM recorded from iPSC-SNs from P300 and P301 are shown in Figure 4E. The I–V curves and conductance, obtained using an established protocol (Adams and Brown, 1982; Wang et al., 1998; Passmore et al., 2003), are shown in Figure 4F. There was a 6 mV hyperpolarized shift in the V1/2 of IM conductance in neurons derived from P301 (P300: V1/2 = −60.9 ± 1.8 mV, n = 11; P301: V1/2 = −67.3 ± 1.8 mV, n = 10; t = 2.6, p = 0.02, two-tailed unpaired t test). The hyperpolarizing shift is a gain-of-function attribute, suggesting an enhancement in IM around RMP.
The KV7.2-T730A variant hyperpolarizes resting membrane potential and reduces excitability of iPSC-SNs
To further establish the role of KV7.2-T730A expression in modulating the excitability of sensory neurons, we used dynamic clamp to subtract the current produced by these mutant channels in iPSC-SNs from P301 and replace it with precisely titrated injections of WT KV7.2 current. To simulate IM conductance at physiologically relevant levels in iPSC-SNs, we constructed a model built from the experimentally determined values of WT IM current (obtained from P300 iPSC-SNs) and mutant IM current (obtained from P301 iPSC-SNs expressing the KV7.2-T730A variant). Figure 5A shows computer simulation of current traces and n activation gate from WT IM and KV7.2-T730A IM models. Steady-state current values from the IM models show a similar shift in normalized I–V relationship and voltage dependence of activation (Fig. 5B), as in the perforated patch recordings (Fig. 4F). We obtained a value of 4 nS as the maximum conductance, equivalent to the maximal current measured in the voltage-clamp recordings (WT IM: 220 ± 36 pA, n = 11; T730A IM: 195 ± 25 pA, n = 10; t = 0.003, p = 0.99, two-tailed unpaired t test). This is a very conservative estimate of the maximal conductance, as the voltage dependence and kinetics of IM in iPSC-SNs were examined using the classical deactivation protocol with the final voltage step of −20 mV to prevent contamination by other voltage-dependent potassium currents (Shah et al., 2008). The maximal opening of KV7 channels is expected to occur at potentials more positive than −20 mV. Indeed, extrapolation of our experimental data suggests a maximum current close to 400 pA (Fig. 5-1), corresponding to ∼8 nS maximum conductance. Hence, we examined the contribution of KV7.2-T730A IM to changes in RMP and current threshold in P301 iPSC-SNs, by substituting KV7.2-T730A IM, which we expect to be 50% of the total current, with 50% WT IM conductance, using dynamic clamp and implementing 4, 6, 8, and 10 nS maximum conductance levels. The effect of KV7.2-T730A IM on RMP was measured, as shown in Figure 5C. Substituting the KV7.2-T730A IM with an equivalent amount of WT conductance caused RMP depolarization in an incremental fashion with increasing amounts of overall conductance. The average values are presented in Figure 5D and reveal depolarization of 2.2 ± 0.3 mV with 4 nS conductance (t = 7, p = 0.001, two-tailed paired t test; n = 9), 3.6 ± 0.6 mV with 6 nS conductance (t = 6, p = 0.005, two-tailed paired t test; n = 5), 4.6 ± 0.6 mV with 8 nS conductance (t = 7, p = 0.002, two-tailed paired t test; n = 5), and 6.4 ± 1 mV with 10 nS conductance (t = 6, p = 0.004, two-tailed paired t test, n = 5). Spontaneous firing was observed in two additional iPSC-SNs at conductances >4 nS (Fig. 5E, example trace) and 6 nS (trace not shown).
Figure 5.

Dynamic clamp recordings confirm that KV7.2-T730A IM reduces the excitability of iPSC-SNs derived from P301. A, Current traces obtained from KV7.2-WT IM and KV7.2-T730A IM model. Currents were evoked from −110 to −20 mV from a holding potential of −20 mV. Time sequence of n variable obtained in the model (bottom panels) in response to a series of voltage steps ranging from −110 to −20 mV. B, Left, Comparison of steady-state activation of the kinetic model of KV7.2-WT IM (orange) and KV7.2-T730A IM (blue). Right, Normalized I–V relationship from P300 (KV7.2-WT IM; orange) and P301 (KV7.2-T730A IM; blue) models. C, An example membrane potential response of a P301 neuron to substituting the T730A IM with WT IM (−50% T730A IM, +50% WT IM) at increasing amounts of conductance, based on the values from extrapolated data of the maximum IM value in iPSC-SNs in Figure 5-1. D, Average response of P301 iPSC-SNs to the protocol from C (n = 5–8). E, Example trace showing spontaneous firing of a P301 iPSC-SN in response to the protocol from C. F, Substituting the T730A IM value with WT IM (−50% T730A IM, +50% WT IM) at increasing amounts of conductance in P301 neurons results in significant reduction in current threshold. Insets show the responses of a representative neuron at each conductance. **p < 0.01.
Extrapolation of the maximum IM from iPSC-SNs derived from P300 and P301. The activation curve of IM obtained from the experimental data was fit to Boltzmann equation to find the peak current of 390 pA and V½ of -30 mV for KCNQ2-T730A IM (P301) and -25mV for WT IM (P300). Download Figure 5-1, XLSX file (35.2KB, xlsx)
We also assessed the effect of the KV7.2-T730A variant on current threshold of iPSC-SNs from P301. The reduction in current threshold from baseline (dynamic clamp off - DC off) is presented in Figure 5F for individual iPSC-SNs (Fig. 5F, gray symbols) and the average (Fig. 5F, blue symbols) for the same range of conductances of 4–10 nS. Substitution of 50% KV7.2-T730A IM with 50% WT IM with 4 nS maximal IM conductance produced a significant reduction in current threshold of 26 ± 3% (p = 0.004, two-tailed paired t test; n = 8). Conductances of 6, 8, and 10 nS resulted in average threshold reductions of 39 ± 4% (p = 0.005, two-tailed paired t test; n = 6), 60 ± 9% (p = 0.003, two-tailed paired t test; n = 6), and 63 ± 13% (p = 0.01, two-tailed paired t test; n = 5), respectively. One cell became spontaneously active in response to substitution at 8 nS, and one at 10 nS (represented by a reduction of 100% in threshold in response to switching on the dynamic-clamp model). Together with the data from Figure 5C–E, these results confirm that, in the family we studied, the T730A variant in KV7.2 significantly reduces the excitability of iPSC-SNs derived from subject P301 (less pain), even at the most conservative estimate of the maximal conductance of 4 nS.
Discussion
Pain is universal but individual-to-individual differences are well documented. Here we show, first, that, at least in some cases, interindividual differences in pain can be modeled in a “disease-in-a-dish” model using subject-specific iPSC-SNs; second, that in some cases mechanisms operating in peripheral sensory neurons can contribute to interindividual differences in pain; and third, we provide proof of concept that subject-specific iPSCs and WES can be used to investigate peripheral mechanisms and pinpoint specific gene variants that modulate pain signaling and contribute to interindividual differences in pain within a single family.
In this study, we demonstrate that a “pain-in-a-dish” in vitro disease model using subject-specific iPSC-SNs parallels differences in pain, as reported by human subjects included in this study. This iPSC-derived model revealed differences in current threshold, firing frequency, responses to elevated temperature, and number of spontaneously active sensory neurons. Our results suggest that depolarized membrane potential is a major factor responsible for the difference in excitability seen between subjects P300 (more pain) and P301 (less pain) in the family under study. These data demonstrate, for the first time, that in some cases interindividual differences in chronic pain can be modeled and studied in vitro. Using WES, we identified multiple candidate genes (Fig. 4-1) that may serve as modifiers of sensory neuron excitability in these individuals. Building upon these observations, we used gene ontology analysis to focus on a variant in one gene, KCNQ2, as a candidate pain-modifier gene that might contribute to interindividual differences in pain in the family we studied, and demonstrated by dynamic clamp that a variant in KCNQ2 reduces sensory neuron excitability and thus is a contributor to pain resilience in the subject with less pain.
Inherited IEM, a severe pain syndrome characterized by episodes of intense burning pain triggered by mild warmth, is caused by mutations in the sodium channel NaV1.7, which is preferentially expressed in peripheral sensory neurons. Microneurographic recordings from IEM patients point to the firing of C-fibers as a cause of pain (Ørstavik et al., 2003; Namer et al., 2015). Our previous studies indicate that rodent DRG neurons expressing pathogenic human NaV1.7 mutant channels from patients with IEM are more excitable than DRG neurons expressing WT NaV1.7 channels (Dib-Hajj et al., 2013), consistent with the notion that increased firing of DRG neurons is associated with neuropathic pain (Ochoa and Torebjörk, 1989; Kleggetveit et al., 2012; Devor, 2013; Zhang et al., 2013; Haroutounian et al., 2014; Vaso et al., 2014). Individual-to-individual variations in pain profiles have been well documented in the clinical domain, even within relatively homogenous patient groups, such as family members with IEM due to the same NaV1.7 mutation (McDonnell et al., 2016). However, these earlier studies have not provided any mechanistic insights regarding differences in pain experience in different individuals.
In the present study, we capitalized on differences in pain profiles in a unique family containing two related IEM individuals (mother and son) carrying the same NaV1.7-S241T mutation (Geha et al., 2016; McDonnell et al., 2016). This mother/son pair differed markedly in terms of overall time in pain, the duration of pain attacks, and the number of awakenings due to pain. The mother and son pair carried the same mutation, which is known to make the DRG neurons hyperexcitable (Yang et al., 2012), but their distinct pain profiles presented an opportunity to study interindividual differences in pain within a single family, in an iPSC model. Given that NaV1.7 channels are mainly expressed in the peripheral nervous system (Toledo-Aral et al., 1997; Dib-Hajj et al., 2013), a fundamental question was whether neurons of the peripheral nervous system could contribute to differences in pain without the inclusion of a CNS component. In our “pain-in-a-dish” disease model, we confirmed that subject-specific iPSC-SNs generated from family members carrying a NaV1.7 mutation are more excitable than neurons derived from an unaffected family member. Using this “pain-in-a-dish” disease model, we observed, for the first time, that an in vitro model with peripheral sensory neurons alone, without a central component, can recapitulate the difference in pain reported by different individuals. Although we acknowledge that CNS components may still play an important role in the overall “pain experience,” and our study cannot rule out the involvement of central processes in modulating the pain experienced by our subjects, our data indicate that the difference in pain between these individuals is at least partially due to the difference in the excitability of their peripheral neurons.
In the current study, we used WES to search for putative genetic modifiers that might contribute to the difference in pain in these two clinically well studied individuals. Since the present study focused on the excitability of sensory neurons, we filtered the obtained variants according to the expression in DRG neurons. Our data revealed 90 genetic variations between mother and son, with 81 variants specific to the mother and 9 to the son. Further interrogation of those variants focused on their suggested role in neuronal excitability and identified the KV7.2-T730A variant in iPSC-SNs derived from the mother as a potential modifier of sensory neuron excitability. The KV7.2 channel is known to regulate the excitability of nociceptive DRG neurons (Passmore et al., 2003; Young et al., 2014). Using dynamic clamp, we established that heterozygous expression of this variant plays a significant role in downregulating the excitability of iPSC-SNs derived from the mother, via a hyperpolarization of RMP and an increase in current threshold, providing a genomic and mechanistic basis for the difference in pain in these two individuals.
It is possible that additional gene variants, including variants with small effects, may contribute to the differences we observed in excitability of iPSC-SNs derived from the two subjects. We cannot rule out variations in genes not currently known to be involved in neuronal excitability, which might indirectly influence peripheral neuron firing. Further studies will be needed to assess the contributions of any variants of this type and to address whether epigenetic factors that influence sensory neuron firing or differences in pain processing at higher levels in the CNS contribute to intrafamilial variability. Importantly, we note that other gene variants might contribute to interindividual differences in pain in other families. Nevertheless, our study provides “proof-of-concept” that subject-specific iPSCs and WES can be used to investigate peripheral mechanisms and pinpoint specific gene variants that modulate pain signaling and contribute to interindividual differences in pain.
In summary, this study shows that an in vitro model of subject-specific iPSC-SNs from two related subjects can recapitulate aspects of individual-to-individual differences in pain, highlighting the value of studying iPSCs from individual subjects to create “disease-in-a-dish” models. Our results indicate that interindividual differences in peripheral sensory neurons can, at least in some cases, contribute to differences in pain and provide proof-of-principle that it is possible to pinpoint, within a single family, a specific gene that contributes to interindividual differences in pain.
Footnotes
This work was supported by Center Grant B9253-C from the U.S. Department of Veterans Affairs Rehabilitation Research and Development Service, by a grant from The Erythromelalgia Association, and by the Regenerative Medicine Research Fund of CT Innovations. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University. Whole exome sequencing data are available through the database of Genotypes and Phenotypes (accession #phs001724.v1.p1). We thank Palak Shah, Dr. Mark Estacion, Dr. Sameet Mehta, and Christopher Castaldi for technical support.
The authors declare no competing financial interests.
References
- Adams PR, Brown DA (1982) Synaptic inhibition of the M-current: slow excitatory post-synaptic potential mechanism in bullfrog sympathetic neurones. J Physiol 332:263–272. 10.1113/jphysiol.1982.sp014412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PR, Brown DA, Constanti A (1982) M-currents and other potassium currents in bullfrog sympathetic neurones. J Physiol 330:537–572. 10.1113/jphysiol.1982.sp014357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battefeld A, Tran BT, Gavrilis J, Cooper EC, Kole MH (2014) Heteromeric KV7.2/7.3 channels differentially regulate action potential initiation and conduction in neocortical myelinated axons. J Neurosci 34:3719–3732. 10.1523/JNEUROSCI.4206-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, McDonnell A, Nitzsche A, Alexandrou A, Saintot PP, Loucif AJ, Brown AR, Young G, Mis M, Randall A, Waxman SG, Stanley P, Kirby S, Tarabar S, Gutteridge A, Butt R, McKernan RM, Whiting P, Ali Z, Bilsland J, et al. (2016) Pharmacological reversal of a pain phenotype in iPSC-derived sensory neurons and patients with inherited erythromelalgia. Sci Transl Med 8:335ra356. 10.1126/scitranslmed.aad7653 [DOI] [PubMed] [Google Scholar]
- Chambers SM, Qi Y, Mica Y, Lee G, Zhang XJ, Niu L, Bilsland J, Cao L, Stevens E, Whiting P, Shi SH, Studer L (2012) Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol 30:715–720. 10.1038/nbt.2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Mica Y, Lee G, Studer L, Tomishima MJ (2016) Dual-SMAD Inhibition/WNT activation-based methods to induce neural crest and derivatives from human pluripotent stem cells. Methods Mol Biol 1307:329–343. 10.1007/7651_2013_59 [DOI] [PubMed] [Google Scholar]
- Devor M. (2013) Neuropathic pain: pathophysiological response of nerves to injury. In: Wall and Melzack's textbook of pain, Ed 6 (McMahon SL, Koltzenburg M, Tracey I, Turk DC, eds), pp 861–888. London: Churchill Livingstone. [Google Scholar]
- Dib-Hajj SD, Yang Y, Black JA, Waxman SG (2013) The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci 14:49–62. 10.1038/nrn3404 [DOI] [PubMed] [Google Scholar]
- Drenth JP, Waxman SG (2007) Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest 117:3603–3609. 10.1172/JCI33297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Hao H, Gigout S, Huang D, Yang Y, Li L, Wang C, Sundt D, Jaffe DB, Zhang H, Gamper N (2014) Control of somatic membrane potential in nociceptive neurons and its implications for peripheral nociceptive transmission. Pain 155:2306–2322. 10.1016/j.pain.2014.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geha P, Yang Y, Estacion M, Schulman BR, Tokuno H, Apkarian AV, Dib-Hajj SD, Waxman SG (2016) Pharmacotherapy for pain in a family with inherited erythromelalgia guided by genomic analysis and functional profiling. JAMA Neurol 73:659–667. 10.1001/jamaneurol.2016.0389 [DOI] [PubMed] [Google Scholar]
- Haroutounian S, Nikolajsen L, Bendtsen TF, Finnerup NB, Kristensen AD, Hasselstrøm JB, Jensen TS (2014) Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 155:1272–1279. 10.1016/j.pain.2014.03.022 [DOI] [PubMed] [Google Scholar]
- Harty TP, Dib-Hajj SD, Tyrrell L, Blackman R, Hisama FM, Rose JB, Waxman SG (2006) Na(V)1.7 mutant A863P in erythromelalgia: effects of altered activation and steady-state inactivation on excitability of nociceptive dorsal root ganglion neurons. J Neurosci 26:12566–12575. 10.1523/JNEUROSCI.3424-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Vanoye CG, Cutts A, Goldberg YP, Dib-Hajj SD, Cohen CJ, Waxman SG, George AL Jr (2017) Sodium channel NaV1.9 mutations associated with insensitivity to pain dampen neuronal excitability. J Clin Invest 127:2805–2814. 10.1172/JCI92373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Nagata N, Kurokawa H, Yamanaka S (2014) iPS cells: a game changer for future medicine. EMBO J 33:409–417. 10.1002/embj.201387098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, Hung WC, Haider S, Zhang J, Knight J, Bjornson RD, Castaldi C, Tikhonoa IR, Bilguvar K, Mane SM, Sanders SJ, et al. (2017) Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet 49:1593–1601. 10.1038/ng.3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleggetveit IP, Namer B, Schmidt R, Helås T, Rückel M, Ørstavik K, Schmelz M, Jørum E (2012) High spontaneous activity of C-nociceptors in painful polyneuropathy. Pain 153:2040–2047. 10.1016/j.pain.2012.05.017 [DOI] [PubMed] [Google Scholar]
- Lampert A, Dib-Hajj SD, Tyrrell L, Waxman SG (2006) Size matters: erythromelalgia mutation S241T in Nav1.7 alters channel gating. J Biol Chem 281:36029–36035. 10.1074/jbc.M607637200 [DOI] [PubMed] [Google Scholar]
- McDonnell A, Schulman B, Ali Z, Dib-Hajj SD, Brock F, Cobain S, Mainka T, Vollert J, Tarabar S, Waxman SG (2016) Inherited erythromelalgia due to mutations in SCN9A: natural history, clinical phenotype and somatosensory profile. Brain 139:1052–1065. 10.1093/brain/aww007 [DOI] [PubMed] [Google Scholar]
- McNeish J, Gardner JP, Wainger BJ, Woolf CJ, Eggan K (2015) From dish to bedside: lessons learned while translating findings from a stem cell model of disease to a clinical trial. Cell Stem Cell 17:8–10. 10.1016/j.stem.2015.06.013 [DOI] [PubMed] [Google Scholar]
- Namer B, Ørstavik K, Schmidt R, Kleggetveit IP, Weidner C, Mørk C, Kvernebo MS, Kvernebo K, Salter H, Carr TH, Segerdahl M, Quiding H, Waxman SG, Handwerker HO, Torebjörk HE, Jørum E, Schmelz M (2015) Specific changes in conduction velocity recovery cycles of single nociceptors in a patient with erythromelalgia with the I848T gain-of-function mutation of Nav1.7. Pain 156:1637–1646. 10.1097/j.pain.0000000000000229 [DOI] [PubMed] [Google Scholar]
- Ochoa J, Torebjörk E (1989) Sensations evoked by intraneural microstimulation of C nociceptor fibres in human skin nerves. J Physiol 415:583–599. 10.1113/jphysiol.1989.sp017737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ørstavik K, Weidner C, Schmidt R, Schmelz M, Hilliges M, Jørum E, Handwerker H, Torebjörk E (2003) Pathological C-fibres in patients with a chronic painful condition. Brain 126:567–578. 10.1093/brain/awg060 [DOI] [PubMed] [Google Scholar]
- Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, Dickenson AH, Brown TA, Burbidge SA, Main M, Brown DA (2003) KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci 23:7227–7236. 10.1523/JNEUROSCI.23-18-07227.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic MM, Nowacki J, Olivo V, Tsaneva-Atanasova K, Randall AD, Mellor JR (2012) Inhibition of post-synaptic Kv7/KCNQ/M channels facilitates long-term potentiation in the hippocampus. PLoS One 7:e30402. 10.1371/journal.pone.0030402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz AA, Abbott LF, Marder E (2004) The dynamic clamp comes of age. Trends Neurosci 27:218–224. 10.1016/j.tins.2004.02.004 [DOI] [PubMed] [Google Scholar]
- Rose K, Ooi L, Dalle C, Robertson B, Wood IC, Gamper N (2011) Transcriptional repression of the M channel subunit Kv7.2 in chronic nerve injury. Pain 152:742–754. 10.1016/j.pain.2010.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA (2008) Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A 105:7869–7874. 10.1073/pnas.0802805105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman MA, Aboharb F, Zeltner N, Studer L (2017) Pluripotent stem cells in neuropsychiatric disorders. Mol Psychiatry 22:1241–1249. 10.1038/mp.2017.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira ME, Hai A (2013) Multi-electrode array technologies for neuroscience and cardiology. Nat Nanotechnol 8:83–94. 10.1038/nnano.2012.265 [DOI] [PubMed] [Google Scholar]
- Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, Wolf JJ, Silos-Santiago I, Halegoua S, Mandel G (1997) Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci U S A 94:1527–1532. 10.1073/pnas.94.4.1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaso A, Adahan HM, Gjika A, Zahaj S, Zhurda T, Vyshka G, Devor M (2014) Peripheral nervous system origin of phantom limb pain. Pain 155:1384–1391. 10.1016/j.pain.2014.04.018 [DOI] [PubMed] [Google Scholar]
- Vasylyev DV, Han C, Zhao P, Dib-Hajj S, Waxman SG (2014) Dynamic-clamp analysis of wild-type human Nav1.7 and erythromelalgia mutant channel L858H. J Neurophysiol 111:1429–1443. 10.1152/jn.00763.2013 [DOI] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282:1890–1893. 10.1126/science.282.5395.1890 [DOI] [PubMed] [Google Scholar]
- Yang Y, Dib-Hajj SD, Zhang J, Zhang Y, Tyrrell L, Estacion M, Waxman SG (2012) Structural modelling and mutant cycle analysis predict pharmacoresponsiveness of a Na(V)1.7 mutant channel. Nat Commun 3:1186. 10.1038/ncomms2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Huang J, Mis MA, Estacion M, Macala L, Shah P, Schulman BR, Horton DB, Dib-Hajj SD, Waxman SG (2016) NaV1.7-A1632G mutation from a family with inherited erythromelalgia: enhanced firing of dorsal root ganglia neurons evoked by thermal stimuli. J Neurosci 36:7511–7522. 10.1523/JNEUROSCI.0462-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Adi T, Effraim PR, Chen L, Dib-Hajj SD, Waxman SG (2018) Reverse pharmacogenomics: carbamazepine normalizes activation and attenuates thermal hyperexcitability of sensory neurons due to Nav 1.7 mutation I234T. Br J Pharmacol 175:2261–2271. 10.1111/bph.13935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young GT, Gutteridge A, Fox H, Wilbrey AL, Cao L, Cho LT, Brown AR, Benn CL, Kammonen LR, Friedman JH, Bictash M, Whiting P, Bilsland JG, Stevens EB (2014) Characterizing human stem cell-derived sensory neurons at the single-cell level reveals their ion channel expression and utility in pain research. Mol Ther 22:1530–1543. 10.1038/mt.2014.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, Carriero NJ, Cheung YH, Deanfield J, DePalma S, Fakhro KA, Glessner J, Hakonarson H, Italia MJ, Kaltman JR, Kaski J, et al. (2013) De novo mutations in histone-modifying genes in congenital heart disease. Nature 498:220–223. 10.1038/nature12141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeltner N, Studer L (2015) Pluripotent stem cell-based disease modeling: current hurdles and future promise. Curr Opin Cell Biol 37:102–110. 10.1016/j.ceb.2015.10.008 [DOI] [PubMed] [Google Scholar]
- Zhang XY, Wen J, Yang W, Wang C, Gao L, Zheng LH, Wang T, Ran K, Li Y, Li X, Xu M, Luo J, Feng S, Ma X, Ma H, Chai Z, Zhou Z, Yao J, Zhang X, Liu JY (2013) Gain-of-function mutations in SCN11A cause familial episodic pain. Am J Hum Genet 93:957–966. 10.1016/j.ajhg.2013.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nav1.7-S241T mutation is expressed in iPSC-SNs from the affected son (P300) and mother (P301), but not the unaffected father (P303). Agarose gel electrophoresis showing the amplification of the expected product (514 bp) from cDNA samples of iPSC-SNs (the M lane shows the 100 bp molecular weight marker) and chromatogram of the sequence of the obtained products confirms the expression of both WT (TCA) and mutant (ACA) alleles in the samples from P300 and P301, and only the WT allele in the sample from P303. Download Figure 1-1, TIF file (1.4MB, tif)
The Whole Exome Sequencing analysis identified variants in P300, P301, and P303 in genes known to be expressed in DRG neurons. P300 – gene variants identified in subject P300 (145). P301 – gene variants identified in subject P301 (184), P303 – gene variants identified in subject P303 (175). P300_unique – gene variants specific to subject P300 (9), P301_unique – gene variants specific to subject P301 (81), P303_unique – gene variants specific to subject P303 (105). Download Figure 4-1, TIF file (5.9MB, tif)
Extrapolation of the maximum IM from iPSC-SNs derived from P300 and P301. The activation curve of IM obtained from the experimental data was fit to Boltzmann equation to find the peak current of 390 pA and V½ of -30 mV for KCNQ2-T730A IM (P301) and -25mV for WT IM (P300). Download Figure 5-1, XLSX file (35.2KB, xlsx)