

Abstract
Autism spectrum disorders are often associated with atypical sensory processing and sensory hypersensitivity, which can lead to maladaptive behaviors, such as tactile defensiveness. Such altered sensory perception in autism spectrum disorders could arise from disruptions in experience-dependent maturation of circuits during early brain development. Here, we tested the hypothesis that synaptic structures of primary somatosensory cortex (S1) neurons in Fragile X syndrome (FXS), which is a common inherited cause of autism, are not modulated by novel sensory information during development. We used chronic in vivo two-photon microscopy to image dendritic spines and axon “en passant” boutons of layer 2/3 pyramidal neurons in S1 of male and female WT and Fmr1 KO mice, a model of FXS. We found that a brief (overnight) exposure to dramatically enhance sensory inputs in the second postnatal week led to a significant increase in spine density in WT mice, but not in Fmr1 KO mice. In contrast, axon “en passant” boutons dynamics were impervious to this novel sensory experience in mice of both genotypes. We surmise that the inability of Fmr1 KO mice to modulate postsynaptic dynamics in response to increased sensory input, at a time when sensory information processing first comes online in S1 cortex, could play a role in altered sensory processing in FXS.
SIGNIFICANCE STATEMENT Very few longitudinal in vivo imaging studies have investigated synaptic structure and dynamics in early postnatal mice. Moreover, those studies tend to focus on the effects of sensory input deprivation, a process that rarely occurs during normal brain development. Early postnatal imaging experiments are critical because a variety of neurodevelopmental disorders, including those characterized by autism, could result from alterations in how circuits are shaped by incoming sensory inputs during critical periods of development. In this study, we focused on a mouse model of Fragile X syndrome and demonstrate how dendritic spines are insensitive to a brief period of novel sensory experience.
Keywords: cortex, dendritic spines, development, en passant boutons, Fragile X syndrome, two-photon microscopy
Introduction
Fragile X syndrome (FXS) is a leading hereditary cause of autism and intellectual disability (Bailey et al., 2008; Hernandez et al., 2009). Although phenotypically heterogeneous, FXS is generally characterized by learning impairments, anxiety, hyperarousal, and atypical sensory processing (Cascio, 2010; Smith et al., 2012). The latter is associated with sensory hypersensitivity and avoidance responses, such as tactile defensiveness (Butler et al., 1991; Baranek et al., 1997). This is likely due to circuit dysfunction across brain regions, and of particular importance for the integration and processing of tactile inputs, in the primary somatosensory (S1) cortex. The onset of sensory-related phenotypes occurs in infancy and might therefore arise from alterations in sensory experience-dependent circuit assembly, which in mouse S1 cortex takes place during a critical period in the second postnatal week (Wen and Barth, 2011; Itami and Kimura, 2012; van der Bourg et al., 2017).
Neuropathological studies of FXS patients have revealed a subtle synaptic defect, namely, an overabundance of dendritic spines with immature morphologies in cortical pyramidal neurons (Rudelli et al., 1985; Hinton et al., 1991; Irwin et al., 2001). The same synaptic abnormality has been consistently described in the Fmr1 KO mouse model of FXS (for review, see Portera-Cailliau, 2012). Immature spines are long, thin, and highly dynamic; as they mature, spines become more stable and adopt a lollipop or mushroom-like morphology. Therefore, the morphology and turnover of spines are proxies for their maturity. In vivo imaging studies have shown that spines of cortical pyramidal neurons in Fmr1 KO mice are both morphologically immature and abnormally unstable during early postnatal development and in the adult (Cruz-Martín et al., 2010; Pan et al., 2010; Padmashri et al., 2013; Nagaoka et al., 2016). Based on these results, we have argued that the most consistent synaptic abnormality in FXS is a defect in experience-dependent maturation and plasticity of spines (He and Portera-Cailliau, 2013).
One way to understand how sensory experience modulates synapses is to image dendritic spines in vivo before and after a period of sensory deprivation (Trachtenberg et al., 2002; Holtmaat et al., 2006; Hofer et al., 2009; Pan et al., 2010; Miquelajauregui et al., 2015). Interestingly, whereas adult WT mice have reduced rates of spine elimination following sensory deprivation, spines of Fmr1 KO mice fail to demonstrate changes in spine dynamics in the absence of sensory input (Pan et al., 2010). But whether dendritic spines (or axonal boutons) in FXS are also insensitive to new sensory experiences, which is more relevant to normal brain maturation, has never been tested in vivo during development. To address this, we used a brief period of dramatic novel sensory experience in the form of environmental enrichment (EE) in postnatal day (P)14 mice. We found that, at baseline, layer (L)2/3 pyramidal neuron spines in S1 cortex of 2-week-old Fmr1 KO mice appear structurally immature. Importantly, whereas EE leads to a transient increase in spine density in WT animals, Fmr1 KO neurons were insensitive to such novel sensory inputs. On the other hand, we did not detect any differences in “en passant” bouton (EPB) density, regardless of genotype or environment.
Materials and Methods
Reagents.
The plasmid vector pCAG-GFP was obtained through Addgene (plasmid #11150). All other reagents were from Sigma-Aldrich unless otherwise noted.
Experimental animals.
All experiments followed the U.S. National Institutes of Health guidelines for animal research, under an animal use protocol (ARC #2006-016) approved by the Chancellor's Animal Research Committee and Office for Animal Research Oversight at the University of California, Los Angeles. We used male and female C57BL/6J mice (HSD C57Bl/6NHsd) housed in a vivarium with a 12 h light-dark cycle and food and water ad libitum. Imaging was performed during the light cycle, and enrichment experiments were performed during the dark cycle.
In utero electroporation (IUE) for GFP expression in L2/3 pyramidal cells.
IUE was performed as previously reported (Cruz-Martín et al., 2010, 2012). Briefly, pregnant female mice at gestation day 16 were anesthetized with isoflurane (5% induction, 1.5%–2% maintenance v/v). Under sterile conditions, a medial incision was made along the abdomen to expose the uterine horns. Using ringed forceps to gently hold embryos, we pressure injected a plasmid encoding pCAG-GFP (500 ng/μl) through a glass needle into the left lateral ventricle with a Picospritzer (Parker; 40 psi, 10 ms pulses). A set of 3 square pulses (50 ms duration, 35 V, with 500 ms between each pulse) was administered to each embryo via a custom-built electroporator with the positive electrode paddle placed over the left somatosensory cortex. Throughout the procedure, the embryos were frequently irrigated with warm saline (37°C). The embryos were placed back inside the mother, and the abdominal wall was sutured with absorbable sutures (muscle: Surgical specialties LOOK, nylon 0.7 metric, 1279B) and nylon sutures (skin: Surgical specialties LOOK, polysyn 0.7 metric, 492B). The dam was observed for 2 h following the surgery to monitor full recovery.
Cranial window surgery for chronic in vivo two-photon microscopy.
We followed a previously described protocol (Mostany and Portera-Cailliau, 2008; Cruz-Martín, Portera-Cailliau et al., 2010; Cruz-Martín et al., 2010). In short, pups (P10–P12) were anesthetized with isoflurane and placed in a stereotaxic frame. A 2.5-mm-diameter circular craniotomy was performed over the electroporated S1 cortex in the left hemisphere and covered with a 3 mm glass coverslip (Electron Microscopy Sciences). The coverslip was secured to the skull with Krazy Glue and dental cement. At P14, a small titanium headbar was attached to the skull with dental cement to secure the animal to the microscope stage for imaging.
Histology.
Two-month-old mice were perfused intracardially first with 0.1 m phosphate buffer until blood clearance, then with ice-cold 4% PFA in 0.1 m phosphate buffer. The brains were harvested, postfixed overnight at 4°C, and then sliced in 100-μm-thick sections on a vibratome. Sections were mounted onto coverslips coated with Vectashield containing DAPI (Vector Laboratories). Sections were then imaged with a structure illumination Apotome microscope (Carl Zeiss) and 10× objective (0.3 NA, Carl Zeiss). Images were acquired and stitched via ZEN (Carl Zeiss).
In vivo two-photon imaging in head-restrained mice.
Structural synaptic imaging was performed on a custom-built two-photon microscope with a Chameleon Ultra II Ti:Sapphire laser (Coherent), a 40× objective (0.8 NA, Olympus), and ScanImage software (Pologruto et al., 2003). Mice were anesthetized for imaging sessions with isoflurane (1%–2% maintenance v/v) and kept warm with an electric heating blanket (Harvard Apparatus). Throughout the imaging sessions (typically <1 h), mice were monitored for taxed breathing and proper coloration of paws/ears. Z stacks with 1 μm steps were collected at 512 × 512 pixels, and a ScanImage zoom of 1× for low-magnification images (0.71 μm/pixel) (see Fig. 1D1,D2) and 5× for dendrite and axon imaging (0.153 μm/pixel) (see Figs. 1, 2, 4, 5). Imaging sessions were performed in the evening at P14 and in the morning at P15 after being housed with their dam and littermates in either standard cages or enriched cages (see EE). A typical imaging experiment was performed as follows: t0: in the evening, P14 mice were imaged (<1 h), allowed to recover (∼1 h), and returned to their home cage with their dam and littermates; t1: 4 h after t0, the mice were imaged again (same FOVs), allowed to recover, and placed overnight with their dam and littermates in either a standard cage or an enriched cage (see EE); t2: the next morning (∼8–10 h after t1; at P15), mice were imaged a third time, allowed to recover, and returned to their home cage (standard or enriched); and t3: 4 h after t2, mice were imaged one final time.
Figure 1.

Plasmid transduction, in vivo imaging of L2/3 neurons at P14. A, IUE scheme. pCAG-GFP plasmid is injected into the lateral ventricle of embryos and electroporated at embryonic day 16. B, GFP expression localized to L2/3 pyramidal cells in S1 cortex. Coronal slice of 2-month-old WT brain following IUE with pCAG-GFP (green) and counterstained with DAPI (blue). C, Representative cranial window implanted over the S1 cortex at P12 and photographed at P14. D1, Representative L2/3 neuron imaged in vivo (maximum intensity xyz projection of 42 slices, 5 μm apart). D2, Side view of the same L2/3 neuron. D3, Apical dendritic segment from the same L2/3 neuron (see box in D1; maximum intensity xyz projection of 38 slices, 1 μm apart). E, Representative axon from a L2/3 neuron (sum xyz projection of 30 slices, 1 μm apart).
Figure 2.

Spine density and EPB density/volume of L2/3 neurons are similar in WT and Fmr1 KO mice at P14, but Fmr1 KO spines are longer. A, In vivo two-photon images (best projections xyt) of representative apical dendrites and axons in WT and Fmr1 KO mice acquired at P14. Scale bar, 10 μm. B, Density of spines in L2/3 neurons in WT mice (gray) and Fmr1 KO mice (red) is not significantly different. C, Dendritic spines in Fmr1 KO mice (red) are significantly longer than those in WT mice (gray) in L2/3 dendrites. D, No difference in the density of EPBs was detected between WT (gray) and Fmr1 KO (red) mice. E, Example heat map generated by MATLAB code illustrating how EPBs are detected. Intensity profiles (solid blue line) above threshold (gray dashed line) are considered EPBs (white arrows) for further analysis and estimation of volume. Representative axon shown is the same as in A (far right). F, No difference in the volume of EPBs was detected between WT (gray) and Fmr1KO (red) mice.
Figure 4.

EPB density and turnover are similar between WT and Fmr1 KO mice and do not change following EE regardless of genotype. A, In vivo two-photon images of axonal EPBs in WT mice were acquired at P14 (t1). Animals were then housed overnight (for 8–10 h) either in SE or in an EE and imaged again at P15 (t2). Subsets of representative EPBs were tracked with arrows: yellow indicates persistent EPBs; magenta indicates lost EPBs; and green indicates newly formed EPBs (with respect to overnight period); empty arrows indicate the position along the axon that corresponds to the location of an eliminated EPB (magenta) or newly formed EPB (green). B, Changes in the density of EPBs were not detected from P14 to P15 regardless of genotype and or housing condition. Top, Density of EPBs in L2/3 neurons of WT mice housed overnight in SE or EE. Each line indicates the EPB density for an individual axonal segment. Green, magenta, and yellow lines indicate an increase, a reduction, or no change in EPB density, respectively. Thick black lines indicate the average for all the axonal segments. Bottom, Density of EPBs in L2/3 neurons of Fmr1 KO mice housed overnight in SE or EE at P14. There was no significant change detected in EPB density after EE or SE. C, Differences in the turnover of EPBs of L2/3 neurons were not detected between WT mice (top) and Fmr1 KO mice (bottom), nor was turnover of EPB affected by EE in either genotype. There was a slight trend for EPB turnover to be lower after EE compared with after SE in WT mice, but this was not significant. D, After SE (brown) or EE (blue) housing overnight (P15, t2), all EPB volumes were pooled per axonal segment. Differences of EPB volumes were not detected between any groups. E, The volumes of only persistent EPBs were tracked from t1 to t2 and averaged for each axonal segment (represented by individual lines). Significant changes in EPB volumes were not detected. Green and magenta lines indicate an increase or reduction in EPB averages, respectively. Thick black lines indicate the average for all the axonal segments.
Figure 5.

Dendritic spines of L2/3 neurons in Fmr1 KO mice are insensitive to EE at P14. A, Best projection in vivo two-photon images of dendritic spines of representative L2/3 neurons from WT mice. Images for EE experiments were acquired as follows: at P14, 4 h apart (t0 and t1); animals were then housed overnight (for 8–10 h) either in SE or in EE and imaged again twice at P15, 4 h apart (t2 and t3). Subsets of representative spines are pseudo-colored: yellow represents persistent spines; magenta represents lost spines; and green represents newly formed spines (with respect to overnight period). B, ToR of spines in L2/3 neurons of WT (top) and Fmr1 KO (bottom) mice housed overnight in SE or EE. C, Density of spines in L2/3 neurons of WT (top) and Fmr1 KO (bottom) mice housed overnight in SE or EE. There is significant increase in spine density after EE in WT mice. Each line indicates the average of all ROIs for an individual neuron. Green, magenta, and gray lines indicate an increase, a reduction, or no change in spine density, respectively, for individual cells. Thick black lines indicate the average for all the cells. D, Rates of spine elimination for L2/3 neurons in WT mice (left) and Fmr1 KO mice (right). E, Rates of formation of new dendritic spines of L2/3 neurons in WT mice (left) and Fmr1 KO mice (right). The rate of spine formation was significantly higher in WT mice immediately after EE, but not in Fmr1 KO mice or WT mice housed in SE.
EE.
Animals were housed in their home cages for standard enrichment (SE) or in EE cages overnight (8–10 h) between P14 and P15 during the dark cycle when rodents are most active and awake. Standard cage dimensions were 13–1/4 inch × 7–1/8 inch × 5–7/16 inch with two pads for nest-building and a layer of bedding. EE cage dimensions were 13–1/4 inch × 16–19/32 inch × 7–3/8 inch (1800 Mouse Cage, Lab Products) and contained brightly colored balls and plastic tunnels, a running wheel, beaded necklaces (Mardi Gras beads) strewn from the top of the cage, and a variety of toys, multiple nest pads, and a layer of bedding (see Fig. 3B).
Figure 3.
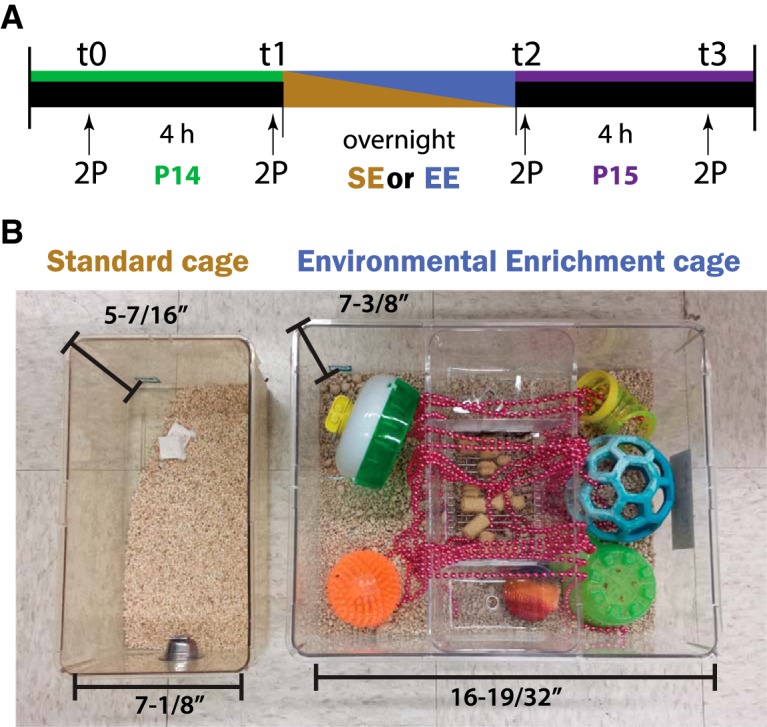
EE cages. A, Chronic in vivo 2-photon (2P) imaging scheme for mice housed overnight in either SE (gold) or EE (blue) cages at P14 (green bar) and P15 (purple bar). B, SE cages (left) are small and only contain bedding and 1 or 2 nesting pads, whereas EE cages (right) contain various objects chosen to engage different sensory modalities, including toys of bright colors and various shapes, sizes, and textures, as well as a running wheel.
Statistical analysis.
Spine imaging data were analyzed using the semiautomated ScanImage (Pologruto et al., 2003) spine analysis software in MATLAB (The MathWorks). Raw, unprocessed images were used for the analysis. If a time point was missing or one or more imaging sessions was of poor quality, the FOV was excluded from the analysis for time-series calculations. Spines >5 μm were excluded from analysis (<1% of all spines). Dendritic and axonal segments were analyzed only in regions >10 μm from branch points and distal tips, and only if the segment was at least 15 μm in length, and the segment was isolated (i.e., it had only minimal overlap with other processes).
Custom-written code in MATLAB was used to determine EPB volume. Z stacks with 1 μm steps were collected at 512 × 512 pixels and a ScanImage zoom 5× for imaging (0.153 μm/pixel) (see Figs. 1, 2, 4, 5). To minimize background signal interference, slices above and below ROIs were deleted from stacks. Raw images were first aligned using the StackReg plugin in ImageJ and then sum-projected. Images were manually binarized before skeletonization. Then, the signal baseline along the skeleton was estimated on the projected images using an asymmetric least-squares smoothing algorithm (Eilers, 2003). Peaks along the skeleton of the axon were detected using the Fast 2D Peak Finder algorithm version 1.7 written in MATLAB (Natan, 2013). This algorithm is based on the smoothing of the noisy point spread function associated to each peak. This was achieved through median filtering and convolution-based smoothing of the input image, so that each peak corresponds to the local maximum of the point spread function. The threshold for EPB detection was empirically set to detect what visibly appeared as a plausible EPB, rather than an irregularity of the axon shaft (see Fig. 2E). The volume of EPBs was defined as the sum of the pixel intensities of the subregions containing the peaks detected by the Fast 2D Peak Finder algorithm, divided by the baseline local value for normalization. Statistical analysis was performed using MATLAB for each peak along the axonal shaft.
In Results, nc and nas indicate the number of cells or axonal segments analyzed, respectively, and N indicates the number of animals used. In total, we tracked and analyzed 7969 individual spines in 165 cells over a total dendritic length of 6.90 mm (average dendritic segment was 42 μm in length) and 703 individual EPBs in 124 axonal segments measuring on average 52 μm (total axonal length analyzed was 6.56 mm). For statistical comparisons, we used nc or nas as the sample size.
For statistical analysis where ANOVAs were used, data were distributed normally as determined by the Shapiro–Wilk test for normality (with the p value set at < 0.05) and met the criteria for Levene's test of homogeneity of variances in each group. Outliers were defined as data that were >2 or <2 SDs from the mean. For baseline length, density, and turnover of WT and Fmr1 KO spines, one-way ANOVAs were used. Turnover was calculated as follows: . All pairwise comparisons were corrected for multiple comparisons with the Bonferroni method. For baseline density of WT and Fmr1 KO EPB, data were not normally distributed, so comparisons were made using the Mann–Whitney U test.
For EE versus SE comparisons in spine imaging experiments, mixed-design or split plot repeated-measures ANOVAs were used with Bonferroni-corrected pairwise comparisons across all groups, with the exception of spine formation rate at P14 versus P15 (see Fig. 5C). Because the Fmr1 KO SE group was not normally distributed, comparisons for all groups were determined using the nonparametric Wilcoxon signed ranks test (with significance set at p < 0.05, two-tailed). For EE EPB analysis, data were not normally distributed, so we used a Kruskal–Wallis H test for turnover rate (ToR) and P15 volume comparisons between groups. For within-group comparisons of EPB density and volume, Wilcoxon signed-rank tests were used (with significance set at p < 0.05, two-tailed). All statistics were performed with SPSS 24 (IBM). Graphs were generated using Excel 2017 (Microsoft) and SigmaPlot 13 (Systat). In box-and-whisker plots, error bars indicate the SEM and horizontal bars indicate medians. Figures were generated with SPSS 24 (IBM), Excel 2017 (Microsoft), FIJI (FIJI is just ImageJ), Photoshop 2018 (Adobe), and Illustrator 2018 (Adobe).
Code accessibility.
Access to the EPB volumetric analysis MATLAB code will be made available upon request.
Results
For chronic two-photon imaging of synaptic structures, IUE was performed at embryonic day 16 to express GFP in L2/3 pyramidal neurons of S1 cortex (Fig. 1A). To ensure sparse labeling of GFP-expressing neurons and minimal overlap of their apical dendrites and axons in L1 (Fig. 1B), a low concentration of pCAG-GFP (500 ng/μl) was injected into the lateral ventricle during the IUE procedure. At P10–P12, cranial windows were implanted over the S1 cortex. Only mice with windows that were optically transparent with no signs of bleeding or dural damage were used for subsequent imaging (Fig. 1C). This allowed us to repeatedly image the same dendrites and axons of L2/3 neurons (Fig. 1D,E).
High-resolution in vivo imaging of L2/3 dendrites was first performed at P14 (Fig. 2A, left). We did not find a difference of spine density in WT and Fmr1 KO mice (0.68 ± 0.01 spines/μm vs 0.65 ± 0.02 spines/μm, respectively; nc/N = 81/18 for WT and nc/N = 73/15 for Fmr1 KO; one-way ANOVA, F(1,152) = 1.61, p = 0.20, partial η2 = 0.01; Fig. 2B), which is in agreement with several prior in vivo imaging studies during development and in adulthood (Cruz-Martín et al., 2010; Pan et al., 2010; Padmashri et al., 2013). Because immature dendritic protrusions are often longer and thinner (filopodia-like) than their mature counterparts (Portera-Cailliau, 2012), we also compared the length of spines in both genotypes. We found that spines in Fmr1 KO mice at P14 were significantly longer than those of WT mice, although the difference was small (1.50 ± 0.03 μm vs 1.38 ± 0.03 μm; one-way ANOVA, F(1,165) = 8.069, p = 0.005, partial η2 = 0.047; Fig. 2C).
Because FMRP is also expressed in axon tips, where it plays a role in growth cone navigation, and along the axon shaft near synapses (Antar et al., 2006; Christie et al., 2009), we imaged axons of L2/3 neurons coursing through L1 at P14 and tracked their EPBs (Fig. 2A, right). We did not find any significant differences in EPB density between WT and Fmr1 KO mice (Fig. 2D; 0.12 ± 0.01 EPBs/μm vs 0.11 ± 0.01 EPBs/μm, respectively; nas/N = 65/18 for WT, and nas/N = 59/14 for Fmr1 KO; Mann–Whitney U test, Z = −0.82, p = 0.41, partial η2 = 0.01), nor any differences in EPB volume (Fig. 2F; 34.2 ± 3.8 A.U. and 33.1 ± 5.0 A.U. in nas/N = 55/15 in WT and nas/N = 38/11 Fmr1 KO, respectively, Mann–Whitney U test, Z = −0.15, p = 0.88, partial η2 = 2 E-4).
Multiple independent in vivo imaging studies have revealed a significant increase in spine turnover in juvenile or adult Fmr1 KO mice (Cruz-Martín et al., 2010; Pan et al., 2010; Padmashri et al., 2013; Nagaoka et al., 2016). We have interpreted this synaptic instability to mean that neurons in Fmr1 KO mice are unable to respond to new sensory inputs related to novel sensory experiences or to learning (He and Portera-Cailliau, 2013). To test this hypothesis, WT and Fmr1 KO mice were placed in either EE overnight at P14–P15 or kept in their SE cage (Fig. 3). The EE cages provided a dramatic novel sensory experience across visual, auditory and, especially, tactile modalities. To determine dynamic changes before and after EE/SE, mice were imaged twice at P14, 4 h apart, then placed overnight in either EE or SE cages, and then imaged again at P15 twice, 4 h apart (Fig. 3A).
We first imaged axons before and after either EE or SE (Fig. 4A) and found that neither the ToR nor the density of EPBs was significantly different in WT mice or Fmr1 KO mice, regardless of whether they were housed overnight in EE or in SE (Fig. 4B; nas/N = 32/10 for WT EE, nas/N = 32/8 for Fmr1 KO EE, nas/N = 33/8 for WT SE, nas/N = 27/6 for Fmr1 KO SE; Wilcoxon signed ranks tests for density; Fig. 4C; WT SE, Z = −1.55, p = 0.12, r = −0.27; WT EE, Z = −1.06, p = 0.29, r = 0.19; Fmr1 KO SE, Z = −1.66, p = 0.096, r = −0.32; Fmr1 KO EE, Z = −0.096, p = 0.92, r = −0.017; Kruskal–Wallis H test for ToR, F(3,124) = 1.97, p = 0.58, partial η2 = 0.016). We also did not detect any global differences in EPB volumes at P15 across all groups (Fig. 4D; nas/N = 29/7 for WT SE, nas/N = 29/9 for WT EE, nas/N = 18/5 for Fmr1 KO SE, and nas/N = 17/6 for Fmr1 KO EE; Kruskal–Wallis H test, F(3,89) = 3.088, p = 0.38, partial η2 = 0.034). To account for varied EPB sizes at baseline, we then tracked EPBs present at t1 and t2 to determine whether or not EPB volumes change following EE, but again, we did not detect any significant differences (Fig. 4E; nas/N = 21/7 and 71 EPBs for WT SE, nas/N = 27/9 and 97 EPBs for WT EE, nas/N = 17/5 and 64 EPBs for KO SE, nas/N = 17/6 and 60 EPBs for KO EE; Wilcoxon signed-ranks tests for EPB volumes; WT SE, Z = −1.87, p = 0.061, r = −0.36; WT EE, Z = −1.13, p = 0.26, r = −0.22; KO SE, Z = −0.64, p = 0.53, r = −0.16; KO EE, Z = −1.11, p = 0.27, r = −0.27).
We then imaged the apical dendrites of L2/3 neurons before and after either EE or SE (Fig. 5A). Because at young ages (P10-P12) and in adult animals, spine ToR is known to be higher in Fmr1 KO mice, we examined ToR over 4 h in mice kept overnight in SE or EE. Exposure to EE did not result in any significant change in spine ToR for mice of either genotype (Fig. 5B; nc/N = 40/9 for WT SE, nc/N = 40/9 for WT EE, nc/N = 32/7 for Fmr1 KO SE, and nc/N = 41/8 for Fmr1 KO; Kruskal–Wallis H test, F(3,153) = 2.32, p = 0.51, partial η2 = 0.015). When we compared spine density in WT mice housed in SE versus EE, we found a significant increase in spine density in WT mice following the novel sensory experience in EE (from 0.66 ± 0.02 to 0.70 ± 0.02 spines/μm; nc/N = 40/9; mixed-model repeated-measures ANOVA was used to compare across all groups, p values reported were corrected via Bonferroni, F(3,147) = 2.96, p = 0.023, partial η2 = 0.34; Fig. 5C, top). WT mice kept overnight in SE showed no difference in spine density (from 0.66 ± 0.02 to 0.64 ± 0.02 spines/μm; nc/N = 40/9; F(3,147) = 2.46, p = 0.84, partial η2 = 0.029). Although the increase in spine density with EE in WT mice was subtle (∼6%), the vast majority (67.5%, 27 of 40 cells) of the neurons analyzed added spines following this relatively brief period of novel sensory experience, whereas the majority of neurons in WT mice that remained in the more impoverished standard environment (62.5%, 25 of 40 cells) showed a decrease in spine density (Fig. 5C, top). Approximately 65% of spines that were newly added during EE in WT mice persisted until t3 (4 h). In contrast to WT mice, the density of spines from P14 Fmr1 KO mice kept overnight in EE or SE did not change significantly (0.64 ± 0.02 spines/μm at P14 to 0.61 ± 0.02 spines/μm at P15 for Fmr1 KO SE, nc/N = 32/7, F(3,147) = 2.78, p = 0.45, partial η2 = 0.14; and 0.64 ± 0.02 spines/μm to 0.62 ± 0.02 spines/μm for Fmr1 KO mice in EE, nc/N = 41/8, F(3,147) = 2.46, p = 0.67, partial η2 = 0.061; Figure 5C, bottom).
Changes in spine density are due to changes in the rates at which spines are added or eliminated, and/or to changes in their lifetime. Thus, the higher spine density in WT mice after a novel sensory experience (EE) could arise from a gain in new spines or from a reduction in the rate of elimination of preexisting spines. First, we examined how the rates of spine elimination were affected by EE and did not find changes in either WT mice (repeated-measures ANOVA, F(1,40) = 1.42, p = 0.24, partial η2 = 0.035; Fig. 5D, left), or Fmr1 KO mice (repeated-measures ANOVA, F(1,43) = 3.85, p = 0.056, partial η2 = 0.082; Fig. 5D, right). However, when examining spine formation, we discovered that the rate at which new spines were added in WT mice was significantly increased by EE (from 0.14 ± 0.01 new spines/μm before vs 0.16 ± 0.01 new spines/μm after EE; Wilcoxon signed-rank test, Z = −2.11, p = 0.035, r = −0.33; Fig. 5E, left). This effect was specific to the enriched environment because neither spine elimination nor new spine formation changed in WT mice that remained in SE overnight (formation: 0.14 ± 0.01 spines/μm at P14 and 0.15 ± 0.01 spines/μm at P15, Wilcoxon signed rank test, Z = −0.16, p = 0.87, r = −0.025; Fig. 5E, left; elimination: Wilcoxon signed-rank test, Z = −0.038, p = 0.97, r = −0.0061; Fig. 5D, left). In contrast to WT mice, Fmr1 KO mice housed overnight in EE showed no change in the rates of spine formation (0.16 ± 0.01 spines/μm before vs 0.14 ± 0.01 spines/μm, signed rank test; Z = −1.11, p = 0.27, r = −0.17; Fig. 5E, right).
Discussion
We set out to investigate whether or not presynaptic and postsynaptic elements of S1 cortical L2/3 neurons in WT and Fmr1 KO mice are affected in similar ways by novel sensory experience at a time during development when sensory information guides behaviors, such as navigation, object recognition, social interaction, and sensorimotor function, which are relevant to autism spectrum disorders. This was an important question because, previously, in vivo imaging studies of dendritic spines had focused only on sensory deprivation, which is not something that happens during typical brain development. Our main findings are as follows: (1) spine density is normal at P14 in Fmr1 KO mice; (2) the density and size of axonal EPBs are not different between genotypes and are unaltered by EE; (3) a very brief period (8–10 h) of EE at P14 leads to a significant increase in spine density in WT mice, due to an increase in the rate of spine formation; and (4) dendritic spines of Fmr1 KO mice are insensitive to EE.
The fact that spine density was normal in the S1 cortex of Fmr1 KO mice is in line with several other in vivo imaging studies (Cruz-Martín et al., 2010; Pan et al., 2010; Padmashri et al., 2013) and helps support the notion that prior reports of higher spine density in the somatosensory cortex of mutant mice from fixed tissue studies may have been due to sampling bias (He and Portera-Cailliau, 2013). It is also possible that differences in spine density in Fmr1 KO mice may occur in only certain neuronal cell types or be specific to certain brain regions (Levenga et al., 2011; Scotto-Lomassese et al., 2011; Suresh and Dunaevsky, 2017).
Previous work from our laboratory shows that L2/3 spine density at P10–P12 is ∼0.35 spines/μm but increases to ∼0.99 spines/μm by P21–P24 (Cruz-Martín et al., 2010). Here, we show that, by P14, spine density nearly doubles from that seen at P10–P12, reaching 0.68 spines/μm in WT and 0.65 spines/μm in Fmr1 KO mice, suggesting that the bulk of the spine gain between P10–P12 and P21-P24 happens in the first few days. The data also emphasize the notion that the second postnatal week is an important developmental period during which cortical L2/3 neurons rapidly increase the number of dendritic spines, presumably in response to incoming sensory information. The Cruz-Martín et al. (2010) study also showed that, at P10–P12, ToR is higher in Fmr1 KO but that this high ToR stabilizes by P21. In the present study, we did not find that ToR was significantly different between genotypes at P14. There are several possible explanations for this finding: First, in Cruz-Martín (2010), we measured ToR over a 60 min interval, whereas for this study we imaged mice 4 h apart. Second, the ages in the two studies do not match (P14–P15 here vs P10–P12 and P21–P24 in Cruz-Martín et al., 2010), which is important because we previously reported an elevated ToR of spines for L2/3 neurons in S1 at P10–P12, but no differences in dynamics by P21–P24. Thus, it is possible that any genotypic differences in ToR (over short periods of time, 1–4 h) could disappear by P14–P15. In general, however, our present data are in good agreement with the data presented in Cruz-Martín (2010), as well as with data from several other in vivo imaging studies of dendritic spines (Pan et al., 2010; Padmashri et al., 2013; Nagaoka et al., 2016; Suresh and Dunaevsky, 2017).
Despite the fact that FMRP is expressed presynaptically, we did not observe any alteration in axon EPB density and volume in L2/3 neurons of Fmr1 KO mice. This could mean that, because of the relative abundance of FMRP in dendrites (compared with axons), dendritic spines are selectively affected by its absence in Fmr1 KO mice. However, due to the much lower density of EPBs compared with spines, it is conceivable that, with a larger sample size (i.e., more mice), we might have detected differences in EPBs in Fmr1 KO mice. Still, even during normal aging, we detected significant changes in EPB density and dynamics with a similar sample size (Mostany et al., 2013). A perplexing result about axon EPB data is the discrepancy between the lack of plasticity in EPBs in light of changes in spine density and dynamics. We previously identified a similar mismatch in the context of normal aging in mice, which led to significant changes in spine density and dynamics, while changes in in axons were underwhelming (Mostany et al., 2013). One interesting possibility is that newly formed spines after EE will primarily contact preexisting EPBs (rather than inducing de novo EPB formation). In this scenario, new spines would either replace preexisting spines (competition) or eventually disappear themselves (unless the animals are maintained in an enriched environment); this would leave total synapse numbers more or less stable. Alternatively, new spines might form contacts on the same EPBs as preexisting spines, in essence forming multiple-synapse EPBs. However, in our previous study, we did not find evidence of such multiple-synapse EPBs when examining serial section electron microscopic reconstructions (Mostany et al., 2013). Clearly, the role of axonal EPBs in experience-dependent synaptic plasticity is less well understood because we do not yet know the relationship between the appearance or disappearance of an EPB and synapse remodeling. On the other hand, changes in EPB volume clearly reflect changes in synaptic strength (for review, see Kasai et al., 2010). However, we did not find any significant changes in EPB volume after overnight period of EE. Reasons for this may have to do with our lower sample size for EPBs than for spines, the fact that the period of enrichment was relatively brief, or perhaps that EPBs are less plastic in young mice than in adults.
The most significant finding of our study is that a novel sensory experience leads to new spines being added onto dendrites of L2/3 neurons in WT mice, but not in Fmr1 KO mice. A previous in vivo imaging study found that a period of EE for 2 d in 1-month-old WT mice led to a significant increase in the rate of spine formation (Yang et al., 2009), which is in line with our findings. However, our study is the first to demonstrate that this effect of EE is already apparent during the critical period of S1 cortex development (P14) and requires only 8–10 h of novel experience. The Yang et al. (2009) study showed that experience-induced spines persisted longer than spines formed during SE, indicating that they were permanently incorporated into the circuit. Similarly, we found that, although the modest increase in spine density was only transient, spines formed during EE persisted, suggesting that newly added spines are retained. The fact that new spines are added in the neocortex in response to new sensory inputs in early postnatal development is a critical step in circuit wiring in the cortex.
Our finding that spines in 2-week-old Fmr1 KO mice are insensitive to novel sensory experience is reminiscent of results in motor cortex of adult Fmr1 KO mice, where spines do not respond to motor learning (Padmashri et al., 2013). This fits our theory that a central defect of dendritic spines in FXS is their lack of plasticity in response to learning or incoming sensory information (He and Portera-Cailliau, 2013). However, we do not rule out the possibility that the defect is simply a delay in their responsivity to EE, and that, if we had extended the period of EE, Fmr1 KO mice would have eventually shown an increase in spine density. Indeed, there is some evidence that the increased abundance of morphologically immature spines in L2/3 neurons is rescued when adult Fmr1 KO mice are reared in EE since birth (Restivo et al., 2005; Nagaoka et al., 2016). In an extension of that study, our data support the notion that the end of the second postnatal week is a critical time when external sensory information shapes and refines circuits in mice (Wen and Barth, 2011; Itami and Kimura, 2012; van der Bourg et al., 2017).
In the future, it will be important to determine the functional impact of spine gains after sensory experience. For example, using calcium imaging, one could investigate whether newly added spines in barrel cortex have larger whisker-evoked signals in WT mice. It is also fair to assume that our EE paradigm would yield similar synaptic gains in other brain regions, including visual or auditory cortex, as well as motor cortex, but that Fmr1 KO mice would not show those changes. Because EE-induced improvements in synaptic and behavioral abnormalities have been shown in other models of autism (Lonetti et al., 2010; Yamaguchi et al., 2017), we believe our results have broad implications regarding the effects of EE in autism.
Footnotes
This work was supported by a University of California Los Angeles dissertation year fellowship, University of California Los Angeles Neurobehavioral Genetics Training Grant T32 5T32MH073526-08, and University of California Los Angeles Eugene V. Cota-Robles Fellowship to E.D.A.; W81XWH-14-1-0433 (United States Army Medical Research and Materiel Command, Department of Defense), Developmental Disabilities Translational Research Program Grant 20160969 (John Merck Fund), SFARI Grant 295438 (Simons Foundation), and National Institutes of Health, National Institute of Child Health and Human Development Grant R01 HD054453 to C.P.-C. D.F. was supported by Direction Générale de l'Armement (France) postdoctoral fellowship. We thank Annasha Vyas for helping troubleshoot the custom MATLAB code and performing some of the early EPB volume analysis; Amaya Miquelajauregui for guidance in surgical and imaging techniques; Michael Condro (University of California Los Angeles IDDRC core facility) for assistance with the apotome microscopy training; and Aditi Newadkar and Nazim Kourdougli for mouse perfusions and histology in Figure 1.
The authors declare no competing financial interests.
References
- Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ (2006) Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci 32:37–48. 10.1016/j.mcn.2006.02.001 [DOI] [PubMed] [Google Scholar]
- Bailey DB Jr, Raspa M, Olmsted M, Holiday DB (2008) Co-occurring conditions associated with FMR1 gene variations: findings from a national parent survey. Am J Med Genet A 146A:2060–2069. 10.1002/ajmg.a.32439 [DOI] [PubMed] [Google Scholar]
- Baranek GT, Foster LG, Berkson G (1997) Tactile defensiveness and stereotyped behaviors. Am J Occup Ther 51:91–95. 10.5014/ajot.51.2.91 [DOI] [PubMed] [Google Scholar]
- Butler MG, Mangrum T, Gupta R, Singh DN (1991) A 15-item checklist for screening mentally retarded males for the fragile X syndrome. Clin Genet 39:347–354. 10.1111/j.1399-0004.1991.tb03041.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascio CJ. (2010) Somatosensory processing in neurodevelopmental disorders. J Neurodev Disord 2:62–69. 10.1007/s11689-010-9046-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie SB, Akins MR, Schwob JE, Fallon JR (2009) The FXG: a presynaptic fragile X granule expressed in a subset of developing brain circuits. J Neurosci 29:1514–1524. 10.1523/JNEUROSCI.3937-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Martín A, Crespo M, Portera-Cailliau C (2010) Delayed stabilization of dendritic spines in fragile X mice. J Neurosci 30:7793–7803. 10.1523/JNEUROSCI.0577-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Martín A, Portera-Cailliau C, Yuste R (2010) In Vivo Imaging of Axonal and Dendritic Structures in Developing Cortex. In: Imaging in Developmental Biology: a Laboratory Manual, Ed 1 (Sharpe J, Wong RO, eds), pp 513–522. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Martín A, Crespo M, Portera-Cailliau C (2012) Glutamate induces the elongation of early dendritic protrusions via mGluRs in wild type mice, but not in fragile X mice. PLoS One 7:e32446. 10.1371/journal.pone.0032446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers PH. (2003) A perfect smoother. Anal Chem 75:3631–3636. 10.1021/ac034173t [DOI] [PubMed] [Google Scholar]
- He CX, Portera-Cailliau C (2013) The trouble with spines in fragile X syndrome: density, maturity and plasticity. Neuroscience 251:120–128. 10.1016/j.neuroscience.2012.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez RN, Feinberg RL, Vaurio R, Passanante NM, Thompson RE, Kaufmann WE (2009) Autism spectrum disorder in fragile X syndrome: a longitudinal evaluation. Am J Med Genet A 149A:1125–1137. 10.1002/ajmg.a.32848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton VJ, Brown WT, Wisniewski K, Rudelli RD (1991) Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet 41:289–294. 10.1002/ajmg.1320410306 [DOI] [PubMed] [Google Scholar]
- Hofer SB, Mrsic-Flogel TD, Bonhoeffer T, Hübener M (2009) Experience leaves a lasting structural trace in cortical circuits. Nature 457:313–317. 10.1038/nature07487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K (2006) Experience-dependent and cell-type-specific spine growth in the neocortex. Nature 441:979–983. 10.1038/nature04783 [DOI] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, Greenough WT (2001) Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet 98:161–167. [DOI] [PubMed] [Google Scholar]
- Itami C, Kimura F (2012) Developmental switch in spike timing-dependent plasticity at layers 4–2/3 in the rodent barrel cortex. J Neurosci 32:15000–15011. 10.1523/JNEUROSCI.2506-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J (2010) Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci 33:121–129. 10.1016/j.tins.2010.01.001 [DOI] [PubMed] [Google Scholar]
- Levenga J, de Vrij FM, Buijsen RA, Li T, Nieuwenhuizen IM, Pop A, Oostra BA, Willemsen R (2011) Subregion-specific dendritic spine abnormalities in the hippocampus of Fmr1 KO mice. Neurobiol Learn Mem 95:467–472. 10.1016/j.nlm.2011.02.009 [DOI] [PubMed] [Google Scholar]
- Lonetti G, Angelucci A, Morando L, Boggio EM, Giustetto M, Pizzorusso T (2010) Early environmental enrichment moderates the behavioral and synaptic phenotype of MeCP2 null mice. Biol Psychiatry 67:657–665. 10.1016/j.biopsych.2009.12.022 [DOI] [PubMed] [Google Scholar]
- Miquelajauregui A, Kribakaran S, Mostany R, Badaloni A, Consalez GG, Portera-Cailliau C (2015) Layer 4 pyramidal neurons exhibit robust dendritic spine plasticity in vivo after input deprivation. J Neurosci 35:7287–7294. 10.1523/JNEUROSCI.5215-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostany R, Portera-Cailliau C (2008) A method for 2-photon imaging of blood flow in the neocortex through a cranial window. J Vis Exp 12:678. 10.3791/678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostany R, Anstey JE, Crump KL, Maco B, Knott G, Portera-Cailliau C (2013) Altered synaptic dynamics during normal brain aging. J Neurosci Methods 33:4094–4104. 10.1523/JNEUROSCI.4825-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka A, Takehara H, Hayashi-Takagi A, Noguchi J, Ishii K, Shirai F, Yagishita S, Akagi T, Ichiki T, Kasai H (2016) Abnormal intrinsic dynamics of dendritic spines in a fragile X syndrome mouse model in vivo. Sci Rep 6:26651. 10.1038/srep26651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natan A. (2013) Fast 2D peak finder File Exchange: MATLAB Central. Available at https://www.mathworks.com/matlabcentral/fileexchange/37388-fast-2d-peak-finder. Accessed August 16, 2018.
- Padmashri R, Reiner BC, Suresh A, Spartz E, Dunaevsky A (2013) Altered structural and functional synaptic plasticity with motor skill learning in a mouse model of fragile X syndrome. J Neurosci 33:19715–19723. 10.1523/JNEUROSCI.2514-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan F, Aldridge GM, Greenough WT, Gan WB (2010) Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A 107:17768–17773. 10.1073/pnas.1012496107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K (2003) ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online 2:13. 10.1186/1475-925X-2-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portera-Cailliau C. (2012) Which comes first in fragile X syndrome, dendritic spine dysgenesis or defects in circuit plasticity? Neuroscientist 18:28–44. 10.1177/1073858410395322 [DOI] [PubMed] [Google Scholar]
- Restivo L, Ferrari F, Passino E, Sgobio C, Bock J, Oostra BA, Bagni C, Ammassari-Teule M (2005) Enriched environment promotes behavioral and morphological recovery in a mouse model for the fragile X syndrome. Proc Natl Acad Sci U S A 102:11557–11562. 10.1073/pnas.0504984102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, Wisniewski HM (1985) Adult fragile X syndrome: clinico-neuropathologic findings. Acta Neuropathol 67:289–295. 10.1007/BF00687814 [DOI] [PubMed] [Google Scholar]
- Scotto-Lomassese S, Nissant A, Mota T, Néant-Fery M, Oostra BA, Greer CA, Lledo PM, Trembleau A, Caillé I (2011) Fragile X mental retardation protein regulates new neuron differentiation in the adult olfactory bulb. J Neurosci 31:2205–2215. 10.1523/JNEUROSCI.5514-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LE, Barker ET, Seltzer MM, Abbeduto L, Greenberg JS (2012) Behavioral phenotype of fragile X syndrome in adolescence and adulthood. Am J Intellect Dev Disabil 117:1–17. 10.1352/1944-7558-117.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh A, Dunaevsky A (2017) Relationship between synaptic AMPAR and spine dynamics: impairments in the FXS mouse. Cereb Cortex 27:4244–4256. 10.1093/cercor/bhx128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K (2002) Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420:788–794. 10.1038/nature01273 [DOI] [PubMed] [Google Scholar]
- van der Bourg A, Yang JW, Reyes-Puerta V, Laurenczy B, Wieckhorst M, Stüttgen MC, Luhmann HJ, Helmchen F (2017) Layer-specific refinement of sensory coding in developing mouse barrel cortex. Cereb Cortex 27:4835–4850. 10.1093/cercor/bhw280 [DOI] [PubMed] [Google Scholar]
- Wen JA, Barth AL (2011) Input-specific critical periods for experience-dependent plasticity in layer 2/3 pyramidal neurons. J Neurosci 31:4456–4465. 10.1523/JNEUROSCI.6042-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Hara Y, Ago Y, Takano E, Hasebe S, Nakazawa T, Hashimoto H, Matsuda T, Takuma K (2017) Environmental enrichment attenuates behavioral abnormalities in valproic acid-exposed autism model mice. Behav Brain Res 333:67–73. 10.1016/j.bbr.2017.06.035 [DOI] [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB (2009) Stably maintained dendritic spines are associated with lifelong memories. Nature 462:920–924. 10.1038/nature08577 [DOI] [PMC free article] [PubMed] [Google Scholar]