

 Novel benzoyl thioureido benzene sulfonamides as CA IX inhibitors such as 6a displayed inhibitory activity against CA IX with an IC50 value of 0.48 nM and about 1500-fold selectivity over CA II were reported.
Novel benzoyl thioureido benzene sulfonamides as CA IX inhibitors such as 6a displayed inhibitory activity against CA IX with an IC50 value of 0.48 nM and about 1500-fold selectivity over CA II were reported.
Abstract
CA IX has attracted much attention as a promising target for the development of new anticancer agents. In this study, a series of sulfonamide derivatives were designed and synthesized as potential CA IX inhibitors from a lead compound (benzoyl thioureido benzene sulfonamide) discovered by virtual screening. The bioassay demonstrated that some of the synthesized compounds exhibited potent inhibitory activity against CA IX in the subnanomolar range and high selectivity over isozymes CA I and CA II. Among them, compound 6a displayed inhibitory activity against CA IX with an IC50 value of 0.48 nM and about 1500-fold selectivity over CA II. The structure–activity relationship and CA IX selectivity of the new sulfonamide derivatives were also analyzed by molecular docking.
1. Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are abundant zinc metalloenzymes found in a diversity of organisms including higher vertebrates, green plants, algae, bacteria, and archaea.1,2 These CAs catalyze a fundamental physiological reaction, hydration of carbon dioxide to bicarbonate and a proton.3 So far, sixteen different CA isoforms (designated as h CA) belonging to the α-CA class are presently known in humans.4 They are involved in many biological processes, such as pH homeostasis,5 ion transport,6 respiration, gluconeogenesis,7 glaucoma,8 bone resorption,9 renal acidification,10 and formation of cerebrospinal fluid and gastric acid.11
In addition, many reports have been published on the role of CA IX in tumor physiology as well,12 such as the control of tumor pH and its influence on other processes in the cell microenvironment that promotes cell proliferation, invasion, and metastasis.13,14 Hypoxia is an important characteristic of the tumor microenvironment.13 Under hypoxia, hypoxia-inducible factor 1α (HIF-1α) translocates to the nucleus and dimerizes with the HIF-1β constitutive subunit to form an active transcription factor.15 Then it binds to a hypoxia response element (HRE) and induces the expression of CA IX.16 Thus, the level of CA IX is dramatically increased in a variety of human tumors, while it has low expression in normal tissues.17,18 Considering the abnormally high expression of CA IX in many hypoxic tumors and its demonstrated role in tumor acidification processes and oncogenesis, CA IX has become a potential drug target for anticancer therapy.19–21
The sulfonamide group (R-SO2NH2) is one of the most important and largely used zinc-binding groups for the design of CA inhibitors. Some classical sulfonamides/sulfamates and their derivatives such as acetazolamide (AZA), methazolamide (MZA), ethoxzolamide (EZA), dichlorphenamide (DCP), etc. (Fig. 1) have been reported to be effective inhibitors of CAs.22–25
Fig. 1. Structures of clinically used CA inhibitors.

There are so many CA isoforms in humans, so it is difficult to find the specific inhibitors of one isoform. In fact, until now, there haven't been any clinically used CAIs that show selectivity for a specific CA isoform.2,26 Therefore, it's of great importance to develop novel isozyme-specific CAI drugs for avoiding various undesired side effects. Through complex X-ray crystallography studies, the selectivity to small molecules of classical CAIs is low, which means that the interaction of small molecules with the CA isoforms at active sites is indiscriminate.27 To improve the CAIs' selectivity, a tail approach of designing effective sulfonamides is utilized, in which some groups are introduced by special links and substituents to enhance the activity and selectivity efficiently due to the favorable interactions with some specific amino acid residues of CA IX catalytic sites.28,29 In this way, benzene sulfonamides containing ureido, thioureido and acyl selenoureido moieties show selective CA inhibitory properties against different CA isozymes30 because the flexible linker makes the inhibitors adopt suitable conformations and bind to the most energetically favored hydrophobic region.
We previously discovered the lead compound 1a as a CA IX inhibitor with a carbonyl thioureido linker by virtual screening.31 It exhibits inhibition against CA IX with an IC50 value of 2.86 nM and some selectivity over CA I and CA II. Based on previously reported CA inhibitors,30 tail approaches and molecular docking, two new series of structurally optimized derivatives from the lead compound 1a were designed and synthesized to improve the bioactivity and selectivity to CA IX, as shown in Fig. 2.
Fig. 2. Structures of the lead compound 1a and the target compounds.

2. Results and discussion
2.1. Docking analysis and the target compound design
The discovery of the lead compound was conducted by virtual screening in our previous work.31 To achieve structural optimization efficiency, we first analyzed the potential binding mode of the lead compound 1a shown in Fig. 3. According to the binding mode, we found that the compound bound in slightly twisted conformations with CA IX allowed for the formation of stable intermolecular hydrogen bonds. The deprotonated sulfonamide nitrogen coordinates to Zn2+ in the same way as other benzene sulfonamide inhibitors. Besides, the coordinated sulfonamide nitrogen atom also forms a hydrogen bond with the hydroxyl group of Thr199, and one of the oxygen atoms of the sulfonamide moiety forms a hydrogen bond with the backbone amide of Thr199 as well. These directional hydrogen bond interactions are beneficial for stabilizing the ligand in the active site. At the hydrophilic part of the pocket (including residues Asn62, His64, Ser65, Gln67, Thr69, Gln92 and His94), the polar sulfur atom and oxygen atom of the carbonyl thioureido moiety can form hydrogen bonds with the side chains of Asn62 and Gln67. Compared with benzenesulfonamides containing an ureido or thioureido moiety,30 compound 1a may form stronger hydrogen bond interactions in the hydrophilic region since the carbonyl thioureido moiety provides one more H-bond acceptor from the carbonyl group which can form an additional hydrogen bond with Gln67. Meanwhile, in the hydrophobic region, the benzene moiety can interact with Leu91, Val131, Leu135 and Leu141, and there is still some space around the benzene ring for further substitution. These common factors contribute to the high affinities with CA IX. Based on the docking analysis, some substituents such as chloride and methoxy were introduced to the benzene ring of the lead compound 1a to improve its inhibitory potency against CA IX and the selectivity over isozyme CA II. The structures of the target compounds are shown in Fig. 2.
Fig. 3. Docking result of compound 1a against CA IX (PDB code: ; 3IAI). The protein and the key residues are colored light pink and green, respectively, and the docked molecule is colored cyan. The gray sphere represents the zinc ion. The hydrogen bonds and coordinate interactions are shown as red dashed lines.

2.2. Chemistry
The compounds were obtained from the coupling of the corresponding substituted benzoyl isothiocyanate and 3- and 4-aminobenzenesulfonamide (Scheme 1). The preparation route of the target compounds was as follows.
Scheme 1. The preparation route of compounds 1a–7a and 1b–7b.

2.3. Bioactivity and inhibition analysis
Based on the lead compound 1a, thirteen novel compounds were synthesized (Scheme 1) and their bioactivities were evaluated (Table 1).
Table 1. The inhibition assay against CA IX, CA I and CA II in vitro.
| Compounds | IC50 (nM) |
Selectivity ratios (I/IX) | Selectivity ratios (II/IX) | ||
| CA IX | CA I | CA II | |||
| AZA | 7.72 | 509.60 | 37.59 | 66.01 | 4.87 |
| EZA | 0.25 | 67.64 | 4.83 | 270.56 | 19.32 |
| 1a | 2.86 | 197.64 | 60.93 | 69.10 | 21.30 |
| 1b | 25.75 | 866.74 | 166.58 | 33.66 | 6.47 |
| 2a | 4.14 | 423.42 | 12.94 | 102.28 | 3.13 |
| 2b | 42.23 | 1541.18 | 239.58 | 36.50 | 5.67 |
| 3a | 3.42 | 660.65 | 22.86 | 193.17 | 6.68 |
| 3b | 18.98 | 772.88 | 276.83 | 40.72 | 14.58 |
| 4a | 0.47 | 693.64 | 25.58 | 1475.83 | 54.42 |
| 4b | 25.50 | 664.71 | 908.13 | 26.07 | 35.61 |
| 5a | 0.30 | 266.56 | 2.09 | 888.53 | 6.97 |
| 5b | 54.35 | 44.38 | 20.75 | 0.82 | 0.38 |
| 6a | 0.48 | 503.74 | 749.28 | 1049.46 | 1561 |
| 6b | >104 | 60.46 | 142.48 | — | — |
| 7a | 0.12 | 631.92 | 11.82 | 5266 | 98.50 |
| 7b | 4.42 | 1341.59 | 949.64 | 303.53 | 214.85 |
According to the bioassay results, the sulfonamide group at the para-position (compounds 1a–7a) showed better inhibitory activity than that at the meta-position (compounds 1b–7b) on the benzene ring. Based on docking analysis, as shown in Fig. 4, we deduced that the main reason was that the carbonyl thioureido moiety in compounds 1a–7a can form more stable hydrogen bonds with residues Asn62 and Gln67 than those formed by the same functional group in 1b–7b in the hydrophilic region, since the carbonyl thioureido moiety of the latter deviated from these hydrophilic residues. The linker moiety was kept, because its hydrophilicity can be beneficial to the binding affinities. In the hydrophobic region, the bis-substituents at the 2,4-position on the benzene ring could improve the activity compared to the mono-substituted benzene at the 2-position (7avs.3a) or 3-position (7avs.2a) alone. It is presumed that the substituted phenyl can strengthen the van der Waals interactions with the hydrophobic residues around.
Fig. 4. Docking result of compounds 4a (A) and 4b (B) against CA IX (PDB code: ; 3IAI). The protein and the key residues are colored light pink and green respectively, and the docked molecules are colored cyan. The gray sphere represents the zinc ion. The hydrogen bonds and coordinate interactions are shown as red dashed lines.

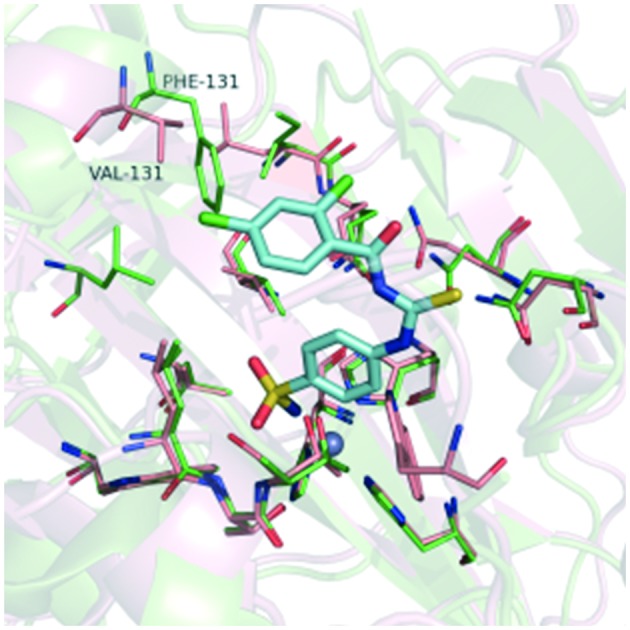
Meanwhile, the data in Table 1 clearly show that all the compounds had relatively high selectivity ratios for CA IX over CA I and CA II. Research has shown that CA II is the most susceptible to be inhibited by sulfonamides,32 whereas CA I generally has a lower affinity for this type of inhibitor and much lower for inorganic anions, such as cyanide, cyanate, and thiocyanate.33 Therefore, we focused on the selectivity for CA IX over CA II. An important difference between these two isozymes is the amino acid in position 131 (Fig. 5). The phenylalanine with its bulky side chain locates in CA II, while the valine with its less sterically demanding side chain locates in CA IX. As a consequence, CA IX has a larger room at the active site than CA II. Therefore, the phenyl group of compound 7a may form strong clashing interactions with Phe131 of CA II, while compound 7a can interact well with Val131 by hydrophobic interaction.
Fig. 5. Superposition of the crystal structures of CA II and CA IX. The binding poses of compound 7a (cyan stick) were predicted by molecular docking. The pink structure is CA IX (PDB code: ; 3IAI); the green structure is CA II (PDB code: ; 3R16); the gray spheres represents the zinc ion.

3. Conclusion
In this paper, some potent CA IX inhibitors with high selectivity over other CA isozymes (CA I and CA II) were designed and synthesized based on the lead compound 1a. Compound 7a was the most potent CA IX inhibitor with an IC50 value of 0.12 nM and exhibited moderate selectivity over CA I and CA II. To our satisfaction, compound 6a displayed inhibitory activity against CA IX with an IC50 value of 0.48 nM and about 1500-fold selectivity over CA II. The structure–activity relationship and CA IX selectivity of the newly synthesized compounds were analyzed by docking analysis. The compounds with the novel scaffold discovered in this work may serve as new lead compounds for further development of therapeutics to treat cancer.
4. Experimental
4.1. Chemistry: synthetic methods and experimental
All the starting materials and reagents were of analytical grade and used without further purification. 1H NMR was measured on a Bruker AV-500 spectrometer with chemical shifts reported in ppm (in DMSO-d6/CDCl3, TMS as an internal standard). The mass spectra were measured on an HP 1100 LC-MS spectrometer. The melting points were determined using an X-6 micro-melting point apparatus and uncorrected.
All the reactions were monitored by thin-layer chromatography (TLC) using UV light (254 nm). Silica gel (300–400 mesh) was used for column chromatography.
General procedure for target compounds 1a–7a and 1b–7b
Substituted benzoyl isothiocyanate (5.0 mmol) was dissolved in 20 mL acetone in a 50 mL round-bottom flask. Then 3- and 4-aminobenzenesulfonamide (4.0 mmol) were added. After reflux overnight, the solution was evaporated under reduced pressure. The product was purified by recrystallization or silica gel column chromatography, and the structures were identified by 1H-NMR, 13C-NMR and MS.
4.1.1. N-((4-Sulfamoylphenyl)carbamothioyl)benzamide (1a)
White crystal; yield: 35%; m.p: 204.7–205.4 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.72 (s, 1H), 11.69 (s, 1H), 8.01 (d, J = 7.6 Hz, 2H), 7.92 (d, J = 8.8 Hz, 2H), 7.87–7.85 (m, 2H), 7.68 (t, J = 7.6 Hz, 1H), 7.55 (t, J = 7.6 Hz, 2H), 7.40 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 180.00, 168.69, 141.80, 141.59, 134.00, 132.54, 129.20, 128.95, 128.67, 127.91, 126.76, 124.84; HRMS (ESI) calcd. for C14H13N3O3S2 [M – H+]: 334.0320, found: 334.0320.
4.1.2. N-((3-Sulfamoylphenyl)carbamothioyl)benzamide (1b)
White crystal; yield: 30%; m.p: 189.1–190.0 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.68 (s, 1H), 11.68 (s, 1H), 8.21 (s, 1H), 8.01 (d, J = 8.0 Hz, 2H), 7.91 (d, J = 7.6 Hz, 1H), 7.74–7.72 (m, 1H), 7.70–7.61 (m, 2H), 7.58 (t, J = 7.6 Hz, 2H), 7.46 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 180.04, 168.66, 145.00, 139.00, 133.68, 132.50, 129.85, 129.20, 128.95, 128.17, 123.79, 122.00; HRMS (ESI) calcd. for C14H13N3O3S2 [M – H+]: 334.0320, found: 334.0321.
4.1.3. 4-Chloro-N-((4-sulfamoylphenyl)carbamothioyl)benzamide (2a)
White crystal; yield: 32%; m.p: 204.9–206.7 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.62 (s, 1H), 11.79 (s, 1H), 8.00 (d, J = 8.4 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H), 7.86 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 8.4 Hz, 2H), 7.40 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 179.78, 167.62, 141.84, 141.35, 138.55, 131.42, 131.18, 129.02, 126.76, 124.83; HRMS (ESI) calcd. for C14H12ClN3O3S2 [M – H+]: 367.9930, found: 367.9929.
4.1.4. 4-Chloro-N-((3-sulfamoylphenyl)carbamothioyl)benzamide (2b)
White crystal; yield: 28%; m.p: 192.7–194.2 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.57 (s, 1H), 11.78 (s, 1H), 8.20 (s, 1H), 8.01 (d, J = 8.0 Hz, 2H), 7.90 (d, J = 7.6 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.63 (d, J = 8.0 Hz, 3H), 7.46 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 179.93, 167.58, 145.00, 138.94, 138.53, 131.45, 131.19, 129.86, 129.03, 128.18, 123.82, 122.01; HRMS (ESI) calcd. for C14H12ClN3O3S2 [M – H+]: 367.9930, found: 367.9931.
4.1.5. 2-Chloro-N-((4-sulfamoylphenyl)carbamothioyl)benzamide (3a)
White crystal; yield: 40%; m.p: 206.3–206.9 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.46 (s, 1H), 12.12 (s, 1H), 7.92 (d, J = 8.8 Hz, 2H), 7.86 (d, J = 8.8 Hz, 2H), 7.64 (d, J = 5.6 Hz, 1H), 7.59–7.54 (m, 2H), 7.50–7.46 (m, 1H), 7.40 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 179.40, 172.34, 168.10, 141.90, 141.21, 134.66, 132.70, 130.46, 130.18, 130.09, 129.82, 127.65, 126.76, 125.03; HRMS (ESI) calcd. for C14H12ClN3O3S2 [M – H+]: 367.9930, found: 367.9930.
4.1.6. 2-Chloro-N-((3-sulfamoylphenyl)carbamothioyl)benzamide (3b)
White crystal; yield: 34%; m.p: 162.0–162.5 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.41 (s, 1H), 12.10 (s, 1H), 8.19 (s, 1H), 7.91 (d, J = 7.2 Hz, 1H), 7.73 (d, J = 7.2 Hz, 1H), 7.64 (dd, J1 = 6.8 Hz, J2 = 2.4 Hz, 2H), 7.56 (d, J = 5.6 Hz, 2H), 7.46 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 179.58, 168.08, 145.00, 138.81, 134.73, 132.66, 130.47, 130.09, 129.87, 129.81, 128.39, 127.63, 123.91, 122.22; HRMS (ESI) calcd. for C14H12ClN3O3S2 [M + Na+]: 391.9906, found: 391.9896.
4.1.7. 4-Methoxy-N-((4-sulfamoylphenyl)carbamothioyl)benzamide (4a)
White crystal; yield: 38%; m.p: 206.0–206.9 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.85 (s, 1H), 11.51 (s, 1H), 8.04 (d, J = 9.2 Hz, 2H), 7.92 (d, J = 8.0 Hz, 2H), 7.87–7.85 (m, 2H), 7.40 (s, 2H), 7.08 (d, J = 8.0 Hz, 2H), 3.87 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 180.00, 167.78, 163.81, 141.71, 141.42, 131.55, 126.75, 124.78, 124.20, 114.31, 56.09; HRMS (ESI) calcd. for C15H15N3O4S2 [M – H+]: 364.0426, found: 364.0420.
4.1.8. 4-Methoxy-N-((3-sulfamoylphenyl)carbamothioyl)benzamide (4b)
White crystal; yield: 35%; m.p: 223.0–223.8 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.65 (s, 1H), 11.31 (s, 1H), 8.23 (s, 1H), 7.93 (s, 2H), 7.74 (d, J = 6.4 Hz, 1H), 7.65 (d, J = 7.2 Hz, 2H), 7.47 (s, 2H), 7.30 (d, J = 6.4 Hz, 1H), 7.18 (s, 1H), 4.02 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 178.94, 165.84, 158.02, 145.03, 138.76, 135.59, 131.60, 129.91, 128.10, 123.93, 121.91, 121.79, 120.03, 113.31, 57.17; HRMS (ESI) calcd. for C15H15N3O4S2 [M – H+]: 364.0426, found: 364.0424.
4.1.9. 2-Methoxy-N-((4-sulfamoylphenyl)carbamothioyl)benzamide (5a)
White crystal; yield: 42%; m.p: 198.0–198.5 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.70 (s, 1H), 11.32 (s, 1H), 7.94 (d, J = 7.6 Hz, 3H), 7.88 (d, J = 7.6 Hz, 2H), 7.67 (t, J = 7.2 Hz, 1H), 7.42 (s, 2H), 7.30 (d, J = 8.0 Hz, 1H), 7.18 (t, J = 7.2 Hz, 1H), 4.02 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 178.79, 165.88, 158.02, 141.94, 141.15, 135.59, 131.57, 126.78, 124.77, 121.78, 120.04, 113.31, 57.16; HRMS (ESI) calcd. for C15H15N3O4S2 [M – H+]: 364.0426, found: 364.0423.
4.1.10. 2-Methoxy-N-((3-sulfamoylphenyl)carbamothioyl)benzamide (5b)
White crystal; yield: 28%; m.p: 222.2–223.2 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.65 (s, 1H), 11.32 (s, 1H), 8.23 (s, 1H), 7.93 (d, J = 6.8 Hz, 2H), 7.74 (d, J = 7.6 Hz, 1H), 7.69–7.61 (m, 2H), 7.48 (s, 2H), 7.30 (d, J = 8.4 Hz, 1H), 7.18 (t, J = 7.2 Hz, 1H), 4.02 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 178.94, 165.85, 158.02, 145.04, 138.76, 135.57, 131.58, 129.92, 128.10, 123.92, 121.91, 121.78, 120.07, 131.32, 57.17; HRMS (ESI) calcd. for C15H15N3O4S2 [M – H+]: 364.0426, found: 364.0425.
4.1.11. 4-Nitro-N-((4-sulfamoylphenyl)carbamothioyl)benzamide (6a)
Faint yellow; yield: 32%; m.p: 202.0–202.3 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.51 (s, 1H), 12.07 (s, 1H), 8.36 (d, J = 9.2 Hz, 2H), 8.18 (d, J = 8.8 Hz, 2H), 7.92 (d, J = 8.8 Hz, 2H), 7.87 (d, J = 8.8 Hz, 2H), 7.42 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 179.59, 167.09, 150.34, 141.92, 141.32, 138.49, 130.76, 126.79, 124.88, 123.85; HRMS (ESI) calcd. for C14H12N4O5S2 [M – H+]: 379.0717, found: 379.0710.
4.1.12. 4-Nitro-N-((3-sulfamoylphenyl)carbamothioyl)benzamide (6b)
Faint yellow; yield: 30%; m.p: 207.0–207.8 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.47 (s, 1H), 12.07 (s, 1H), 8.36 (d, J = 8.8 Hz, 2H), 8.19 (d, J = 8.8 Hz, 3H), 7.91 (d, J = 7.2 Hz, 1H), 7.75 (d, J = 7.6 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.49 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 179.73, 167.05, 150.32, 145.03, 138.91, 138.52, 130.77, 129.90, 128.22, 123.92, 123.87, 122.05; HRMS (ESI) calcd. for C14H12N4O5S2 [M + Na+]: 403.0147, found: 403.0149.
4.1.13. 2,4-Dichloro-N-((4-sulfamoylphenyl)carbamothioyl)benzamide (7a)
White crystal; yield: 32%; m.p: 206.0–206.6 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.38 (s, 1H), 12.14 (s, 1H), 7.91 (d, J = 8.8 Hz, 2H), 7.87 (d, J = 8.8 Hz, 2H), 7.79 (d, J = 2.0 Hz, 1H), 7.70 (d, J = 8.4 Hz, 1H), 7.59–7.57 (m, 1H), 7.41 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 179.28, 167.16, 141.97, 141.17, 136.51, 133.85, 131.26, 129.69, 127.88, 125.04; HRMS (ESI) calcd. for C14H10Cl2N3O3S2 [M – H+]: 401.9541, found: 401.9539.
4.1.14. 2,4-Dichloro-N-((3-sulfamoylphenyl)carbamothioyl)benzamide (7b)
White to pale yellow crystal; yield: 28%; m.p: 186.8–187.7 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.35 (s, 1H), 12.13 (s, 1H), 7.92 (t, J = 8.0 Hz, 1H), 7.47 (d, J = 7.6 Hz, 1H), 7.69 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 9.6 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.47 (s, 3H); HRMS (ESI) calcd. for C14H10Cl2N3O3S2 [M + Na+]: 425.9517, found: 425.9511.
4.2. Biological assay
The purification of recombinant protein h CA IX was performed as described previously,28 and CA I and CA II were both purchased from Sigma. The stock solutions of the inhibitors (50 mM) were prepared and diluted to different concentrations with DMSO. An Applied Photophysics SX 20 stopped-flow instrument was used to test the CA-catalyzed CO2 hydration activity, as described by I. Nishimori et al.34 The initial rates of 4-nitrophenyl acetate hydrolysis catalyzed were monitored using a Synergy 2 Biotek Microplate reader, as described by E. Truppo et al.35 The inhibitor and enzyme solutions were pre-incubated together for 15 min prior to the assay study. The enzyme concentrations were 1 ng μL–1 for CA II, 50 ng μL–1 for CA I and 3 ng μL–1 for CA IX. Triplicate experiments were done for each inhibitor concentration, and the values reported throughout the paper are the mean of such results.
4.3. Molecular modeling
For better understanding, compounds 1a, 4a, 4b and 7a were studied by molecular docking. First, the four compounds were prepared by using the Builder tool and then optimized with the Ligprep module in Maestro 9.0 (Schrödinger Inc.). The protein structures of CA II (PDB ID: ; 3R16) and CA IX (PDB ID: ; 3IAI) were derived from the Protein Data Bank. Then we utilized the Glide module in Maestro to optimize these structures and all the water molecules were removed. When defining the active site, the grid-enclosing box was generated based on the ligand in the complex structure, and other parameters were set up by default. All of the compounds were docked into the protein using Glide with the standard precision (SP) approach, and a scaling factor of 1.0 was set to the van der Waals (VDW) radii of protein atoms with partial atomic charges less than 0.25. The best 20 docking poses of each compound ranked by GlideScore were kept for further analysis.
Conflicts of interest
The authors declare no competing interests, no conflicts to declare.
Acknowledgments
This research is supported in part by the National Key Research and Development Program (Grant 2016YFA0502304) and the Shanghai Committee of Science and Technology (Grant 14431902100).
References
- Supuran C. T. Curr. Pharm. Des. 2008;14:603. doi: 10.2174/138161208783877884. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Nat. Rev. Drug Discovery. 2008;7:168. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Pastorekova S., Parkkila S., Pastorek J., Supuran C. T. J. Enzyme Inhib. Med. Chem. 2004;19:199. doi: 10.1080/14756360410001689540. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Bioorg. Med. Chem. Lett. 2010;20:3467. doi: 10.1016/j.bmcl.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Ivanov S., Liao S.-Y., Ivanova A., Danilkovitch-Miagkova A., Tarasova N., Weirich G., Merrill M. J., Proescholdt M. A., Oldfield E. H., Lee J. Am. J. Pathol. 2001;158:905. doi: 10.1016/S0002-9440(10)64038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry R. P. Annu. Rev. Physiol. 1996;58:523. doi: 10.1146/annurev.ph.58.030196.002515. [DOI] [PubMed] [Google Scholar]
- Breton S. J. Oncol. Pract. 2001;2:159. [Google Scholar]
- Maren T. H. Drug Dev. Res. 1987;10:255. [Google Scholar]
- Teitelbaum S. L. Science. 2000;289:1504. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- Kaunisto K., Parkkila S., Rajaniemi H., Waheed A., Grubb J., Sly W. S. Kidney Int. 2002;61:2111. doi: 10.1046/j.1523-1755.2002.00371.x. [DOI] [PubMed] [Google Scholar]
- Oppelt W., Maren T., Owens E., Rall D. Exp. Biol. Med. 1963;114:86. doi: 10.3181/00379727-114-28593. [DOI] [PubMed] [Google Scholar]
- Pouysségur J., Dayan F., Mazure N. M. Nature. 2006;441:437. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- Brahimi-Horn M. C., Chiche J., Pouysségur J. J. Mol. Med. 2007;85:1301. doi: 10.1007/s00109-007-0281-3. [DOI] [PubMed] [Google Scholar]
- Harris A. L. Nat. Rev. Cancer. 2002;2:38. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Ke Q., Costa M. Mol. Pharmacol. 2006;70:1469. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- Jubb A., Pham T., Hanby A., Frantz G., Peale F., Wu T., Koeppen H., Hillan K. J. Clin. Pathol. 2004;57:504. doi: 10.1136/jcp.2003.012963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Švastová E., Hulíková A., Rafajová M., Zat'ovičová M., Gibadulinová A., Casini A., Cecchi A., Scozzafava A., Supuran C. T., Pastorek J. FEBS Lett. 2004;577:439. doi: 10.1016/j.febslet.2004.10.043. [DOI] [PubMed] [Google Scholar]
- Chiche J., Ilc K., Laferrière J., Trottier E., Dayan F., Mazure N. M., Brahimi-Horn M. C., Pouysségur J. Cancer Res. 2009;69:358. doi: 10.1158/0008-5472.CAN-08-2470. [DOI] [PubMed] [Google Scholar]
- Supuran C. T., Scozzafava A. Expert Opin. Ther. Pat. 2000;10:575. [Google Scholar]
- Potter C., Harris A. L. Cell Cycle. 2004;3:159. [PubMed] [Google Scholar]
- Tennant D. A., Durán R. V., Gottlieb E. Nat. Rev. Cancer. 2010;10:267. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- Ahlskog J. K., Dumelin C. E., Trüssel S., Mårlind J., Neri D. Bioorg. Med. Chem. Lett. 2009;19:4851. doi: 10.1016/j.bmcl.2009.06.022. [DOI] [PubMed] [Google Scholar]
- Jaiswal M., Khadikar P. V., Scozzafava A., Supuran C. T. Bioorg. Med. Chem. Lett. 2004;14:3283. doi: 10.1016/j.bmcl.2004.03.099. [DOI] [PubMed] [Google Scholar]
- Garaj V., Puccetti L., Fasolis G., Winum J.-Y., Montero J.-L., Scozzafava A., Vullo D., Innocenti A., Supuran C. T. Bioorg. Med. Chem. Lett. 2004;14:5427. doi: 10.1016/j.bmcl.2004.07.087. [DOI] [PubMed] [Google Scholar]
- Purichia N., Erway L. C. Dev. Biol. 1972;27:395. doi: 10.1016/0012-1606(72)90178-9. [DOI] [PubMed] [Google Scholar]
- Pacchiano F., Carta F., McDonald P. C., Lou Y., Vullo D., Scozzafava A., Dedhar S., Supuran C. T. J. Med. Chem. 2011;54:1896. doi: 10.1021/jm101541x. [DOI] [PubMed] [Google Scholar]
- Alterio V., Di Fiore A., D'Ambrosio K., Supuran C. T., De Simone G. Chem. Rev. 2012;112:4421. doi: 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]
- Bozdag M., Ferraroni M., Nuti E., Vullo D., Rossello A., Carta F., Scozzafava A., Supuran C. T. Bioorg. Med. Chem. 2014;22:334. doi: 10.1016/j.bmc.2013.11.016. [DOI] [PubMed] [Google Scholar]
- De Simone G., Alterio V., Supuran C. T. Expert Opin. Drug Discovery. 2013;8:793. doi: 10.1517/17460441.2013.795145. [DOI] [PubMed] [Google Scholar]
- Mincione F., Starnotti M., Masini E., Bacciottini L., Scrivanti C., Casini A., Vullo D., Scozzafava A., Supuran C. T., Mincione F., Starnotti M., Masini E., Bacciottini L., Scrivanti C., Casini A., Vullo D., Scozzafava A., Supuran C. T. Bioorg. Med. Chem. Lett. Bioorg. Med. Chem. 2005;2017;1525:3821. 3567. doi: 10.1016/j.bmcl.2005.06.054. [DOI] [PubMed] [Google Scholar]
- Wang L., Yang C., Lu W., Liu L., Gao R., Liao S., Zhao Z., Zhu L., Xu Y., Li H., Huang J., Zhu W. Bioorg. Med. Chem. Lett. 2013;23:3496–3499. doi: 10.1016/j.bmcl.2013.04.048. [DOI] [PubMed] [Google Scholar]
- Vullo D., Franchi M., Gallori E., Pastorek J., Scozzafava A., Pastorekova S., Supuran C. T. Bioorg. Med. Chem. Lett. 2003;13:1005. doi: 10.1016/s0960-894x(03)00091-x. [DOI] [PubMed] [Google Scholar]
- Supuran C. T., Scozzafava A., Casini A. Med. Res. Rev. 2003;23:146. doi: 10.1002/med.10025. [DOI] [PubMed] [Google Scholar]
- Nishimori I., Vullo D., Innocenti A., Scozzafava A., Mastrolorenzo A., Supuran C. T. J. Med. Chem. 2005;48:7860. doi: 10.1021/jm050483n. [DOI] [PubMed] [Google Scholar]
- Truppo E., Supuran C. T., Sandomenico A., Vullo D., Innocenti A., Di Fiore A., Alterio V., De Simone G., Monti S. M. Bioorg. Med. Chem. Lett. 2012;22:1560. doi: 10.1016/j.bmcl.2011.12.134. [DOI] [PubMed] [Google Scholar]