

 Virtual screening is a useful tool for disclosing new chemical species able to inhibit mPGES-1.
Virtual screening is a useful tool for disclosing new chemical species able to inhibit mPGES-1.
Abstract
Targeting microsomal prostaglandin E2 synthase-1 (mPGES-1) represents an efficient strategy for the development of novel drugs against inflammation and cancer with potentially reduced side effects. With this aim, a virtual screening was performed on a large library of commercially available molecules using the X-ray structure of mPGES-1 co-complexed with a potent inhibitor. Combining fast ligand-based shape alignment, molecular docking experiments, and qualitative analysis of the binding poses, a small set of molecules was selected for the subsequent steps of validation of the biological activity. Compounds 2 and 3, bearing the 3-hydroxy-3-pyrrolin-2-one nucleus, showed mPGES-1-inhibitory activity in the low micromolar range. These data highlighted the applicability of the reported virtual screening protocol for the selection of new mPGES-1 inhibitors as promising anti-inflammatory/anti-cancer drugs.
Introduction
The functional relationship between inflammation and cancer has been broadly debated in the scientific community.1–4 It is noteworthy that inflammation triggers proliferation, invasion and migration of cancer cells.5 Also, the onset of pre-cancerous lesions at various anatomic sites has been related to inflammatory events.6–8 Prostaglandins (PGs) are bioactive effectors of inflammation, and they represent key mediators involved in physiological functions as well as in further pathologic conditions, such as tumorigenesis.9 PGs are initially formed from released arachidonic acid (AA) by cyclooxygenases (COX), and subsequently they are converted into different PGs species by specific terminal PG synthases.10 Among them, prostaglandin E2 (PGE2) exerts a number of physiological functions, mainly upon activation of downstream signaling cascades through the interaction with transmembrane EP receptors located on the cell surface.11 In this way, PGE2 regulates cell proliferation, apoptosis, angiogenesis as well as inflammation and immune surveillance.12,13 Also, high levels of COX-2 and the elevated production of PGE2 have been detected in human colon adenomas and in adenocarcinomas.14,15
The identification of potent anti-inflammatory agents has become a valuable and synergic pharmacological strategy to develop new anticancer drugs.16 In this context, nonsteroidal drugs (NSAIDs) still represent the main agents used for the treatment of inflammatory symptoms, mainly inhibiting COX enzymes and acting as suppressors of PG biosynthesis.17 Although NSAIDs are widely used for the treatment of inflammation, they are also responsible for important side effects due to their action on COXs, such as cardiovascular, gastrointestinal and renal complications.18 For these reasons, the discovery of new and safer drugs targeting different proteins at the terminal level in the AA cascade is urgently required, especially for long-term therapies. In particular, prostaglandin E2 synthases (PGES, namely mPGES-1, mPGES-2 and cPGES) that are responsible for the biosynthesis of PGE2 from PGH2 (ref. 19 and 20) have been recognized as promising targets for the development of new anti-inflammatory/anticancer agents with potentially lower risk of side effects. Among PGES, mPGES-1 is the inducible membrane-bound isoform, and it is involved in a number of acute and chronic disorders,21 such as pain, fever, rheumatoid arthritis, arthritis, inflammation,22 and cancer.23 Specifically, mPGES-1 inhibition lowers the COX-related side effects and facilitates the development of new promising and safer drugs blocking the chemical conversion of PGH2, enzymatically produced by the COXs, to the terminal PGE2.24 To date, a high number of mPGES-1 inhibitors, showing chemical variability, have been disclosed,25,26 but only two inhibitors entered clinical development phases. Specifically, GRC 27864 (Glenmark Pharmaceuticals Ltd) and LY3023703 (developed by Eli Lilly) are under evaluation in phase II clinical trials.
On the other hand, the rational design and the identification of new inhibitors have been strongly encouraged in the last few years by the release of different crystal structures of mPGES-1, both in the apo form (PDB code: 4AL0)27 and co-complexed with potent mPGES-1 inhibitors (mPGES-1 co-complexed with substrate competitor inhibitors, PDB codes: ; 4BPM, ; 4WAB, ; 4YK5, ; 4YL0, ; 4YL1, ; 4YL3, ; 5BQG, ; 5BQH, ; 5BQI; mPGES-1 co-complexed with substrate/cofactor competitor, ; 4AL1).28–31
Also, the catalysis events required for the conversion of PGH2 to PGE2 have been recently elucidated, disclosing the involvement of the thiolate group on glutathione (GSH) as cofactor, and a mutual interaction between Arg126 and Asp49 (on the adjacent chain).32
In this scenario, we have been interested in the discovery of novel mPGES-1 inhibitors featuring different chemical scaffolds.33–43 A powerful strategy regards the use of fast computational tools for selecting new compounds, and we have shown the efficient use of fragment virtual screening to identify new valuable chemical scaffolds that were subsequently modified, obtaining interesting inhibitory activities and paving the way for the development of novel potent mPGES-1 inhibitors. With the aim of identifying new chemical species able to inhibit mPGES-1, here we report a virtual screening protocol starting from a library of commercially available compounds using the crystal structure of mPGES-1 co-crystallized with the potent inhibitor named LVJ (PDB code: ; 4BPM).28 The use of different filters restricted the initial large library of compounds to even smaller groups of predicted affine hits, with the final identification of two potent mPGES-1 inhibitors. The effects of the selected lead compounds were also tested against 5-lipoxygenase (5-LO), an enzyme that initiates the biosynthesis of leukotrienes (LTs). Indeed, the dual inhibition of PGE2/LTs production is considered a novel strategy for developing new potent and efficient anti-inflammatory drugs, again with a potentially safer profile as compared to classical NSAIDs.44 The identified mPGES-1 inhibitors were then tested against 5-LO, with the final discovery of 3 as new promising mPGES-1 inhibitor also able to partially inhibit 5-LO.
Results and discussion
Structural analysis of mPGES-1
mPGES-1 is organized as homotrimer in the endoplasmic reticulum membrane, featuring three equivalent active site cavities within the membrane-spanning region at each monomer interface.27 Each active site is toward the cytoplasmic part of the protein, between the N-terminal parts of helix II and IV of one monomer and the C-terminal part of helix I and the cytoplasmic domain of the adjacent monomer (Fig. 1). The accurate analysis of the mPGES-1 binding site disclosed several sub-regions featuring hydrophobic/polar residues, shedding light for the structure-based design of opportunely functionalized ligand counterpart (Fig. 1). Moving from the cytoplasmic to the external part of endoplasmic reticulum membrane, the following sites are identifiable:
Fig. 1. mPGES-1 structure: A) on the left, transparent molecular surface representation of the mPGES-1 trimer, with chains A, B and C depicted in red, black, and blue ribbons, respectively; on the right, focused representation of the mPGES-1 binding site with transparent molecular surface colored in yellow, orange, and green (see text for details, glutathione (GSH) cofactor and key residues in the mPGES-1 binding site are represented in sticks, C grey, O red, N, blue, polar H white); B) on the left, mPGES-1 co-complexed with LVJ inhibitor (PDB code: 4BPM, LVJ represented in sticks, C cyan, O red, N, blue, polar H white, Cl green, F light green); on the right, two-dimensional panel representing interactions between LVJ and residues in mPGES-1 binding site.

a) A groove between the GSH binding site and a region close to the cytoplasmic part of the protein, featuring aromatic (C:Phe44, C:His53) and polar (C:Arg52, C:Asp49) residues (colored in green, Fig. 1). We have highlighted that a mPGES-1 binder establishes π–π contacts with these aromatic residues in this region, as occurred for the co-crystallized LVJ,28 4DV, 4DZ, 4U8, 4U9,29 and as emerged from molecular modeling studies performed on various mPGES-1 inhibitors.38,39,43 Also, a recent study that aimed at identifying the catalytic residues through site-directed mutagenesis/activity assays disclosed the interaction between C:Asp49 and A:Arg126 essential for the mPGES-1 catalysis.32
b) The cofactor (GSH) binding region (colored in orange, Fig. 1). Specifically, GSH is characterized by a particular U-shape conformation due to the interactions between its two terminal carboxylic functions and the positively charged residues in the deeper part of the binding site (C:Arg38, A:Arg73). In this region, Ser127 on chain A was firstly supposed to participate to chemical events behind the isomerization of PGH2 to PGE2,27 but recent evidences based on site-directed mutagenesis highlighted that this residue is nonessential for the catalysis;32
c) A binding groove between helix 1 of chain C and helix 4 of chain A (colored in yellow, Fig. 1), with polar (A:Gln134), aliphatic (C:Val24) and aromatic residues (C:Tyr28 and A:Tyr130, the latter interacting with the gamma peptide linkage between the cysteine and the glutamate of GSH).
Virtual screening workflow
The protocol employed for the identification of novel mPGES-1 inhibitors involved ligand-based and structure-based computational methodologies. The workflow is summarized in Fig. 2. With the aim of identifying novel mPGES-1 inhibitors, a virtual screening was performed starting from a large library of commercially available compounds from Otava, Ltd. (∼3.1 × 105 compounds). mPGES-1 crystal structure (PDB code: ; 4BPM) was used for the structure-based molecular docking experiments (3D-protein structure), whereas the co-crystallized 3D structure of the potent inhibitor LVJ was employed for ligand-based computation of the “shape similarity” parameter (see Fig. 2). The preliminary analysis of the starting library highlighted its high promiscuity, since it was composed by small fragments up to “heavy” commercially available compounds (16 Da ≤ molecular weight (MW) ≤ 1307 Da) also showing a large range of hydrogen bond donors (HBD) and acceptors (HBA) (0 ≤ HBD ≤ 8; 0 ≤ HBA ≤ 16). Prior to performing molecular docking experiments, a fast shape alignment was performed between the starting library of compounds and the co-crystallized mPGES-1 inhibitor LVJ as reference compound (Phase software). With the aim of selecting the most promising items, a restricted set of compounds was selected from the original library with a chemical shape comparable to that of LVJ, following the principle that similar molecules could likely show similar binding modes on the protein counterpart, as demonstrated in virtual screening studies.45,46 Specifically, this protocol envisages a first conformational search round, and afterwards the obtained conformers are aligned and compared to the reference compound. Then, the arithmetical parameter termed “shape similarity” is computed for each screened molecule, and it could range from 0 (no one atom matching between screened and reference compounds) to 1 (all atoms matching). Using this filter, the starting library of 310 657 molecules was reduced to 7220 items. In this way, a new library of promising molecules was obtained showing intrinsic conformational and pharmacophoric features, then compatible with the binding mode showed by LVJ reference compound and prone to be submitted to the molecular docking step.
Fig. 2. The virtual screening workflow for the identification of mPGES-1 inhibitors.

Structure-based molecular docking experiments were then performed using the selected library of compounds and the mPGES-1 3D protein structure, using Glide software. Specifically, a first round of docking experiments was performed setting the high-throughput virtual screening (HTVS) precision mode of Glide that allows a first enrichment from the starting library of compounds with a high fastness. The top-ranked poses (selection parameter: docking score, see Materials and methods) were then saved and submitted to a second set of docking experiments, using the standard precision (SP) Glide precision mode in order to obtain more accurate sampling and scoring if compared with the HTVS mode. The output poses were then further filtered computing their “in place” shape similarity against LVJ. In details, differently from the above reported ligand-based shape similarity computation that requires a free conformational sampling for the alignment, here accounted conformers were those arising from the structure-based docking calculations sampling, and then they could be directly compared to the LVJ pose originally co-crystallized with the protein structure (“in place” shape similarity). The selected poses (see Materials and methods) were then submitted to a third docking round, setting in this case the extra-precision (XP) Glide mode that outperforms the SP mode for both sampling and scoring. Again, the docking poses showing a promising “in place” shape similarity referenced to LVJ-co-crystallized structure were filtered and subsequently carefully analyzed for selecting new putative mPGES-1 binders.
The selected docking poses, featuring a promising shape similarity to LVJ, were also visually inspected in order to filter those better interacting with mPGES-1, considering the protein form in the presence of the cofactor. Since the selected docking poses shared a similar shape with that of LVJ-co-crystallized ligand, the binding mode was evaluated ascertaining the respect of the following key interactions as detected for the reference compound:
• edge-to-face π–π interaction with C:Phe44 and/or C:His53;
• contacts with A:Pro124, A:Thr131, C:Asp49;
• contacts with GSH;
• also, the interaction with A:Tyr130, another key residue in the mPGES-1 binding site interacting with the GSH cofactor, was considered evaluating the possible edge to face π–π contacts with ligand counterpart.
Following this procedure, 296 poses were roughly selected, corresponding to 50 unique compounds.
Among them, we selected 8 final compounds as promising mPGES-1 binders after removing compounds showing chemical groups classified as “Pan-Assay Interference Compounds” (PAINS) (SwissADME web tool,47 see Experimental section), and accounting the chemical variability while preserving the most promising XP GlideScore values (Table 1).
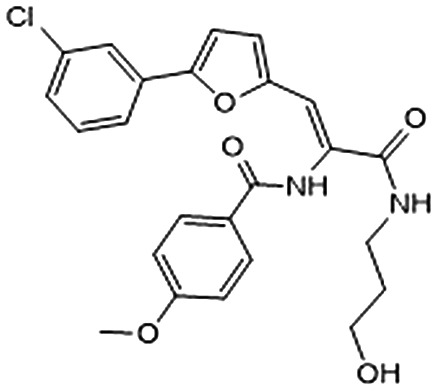
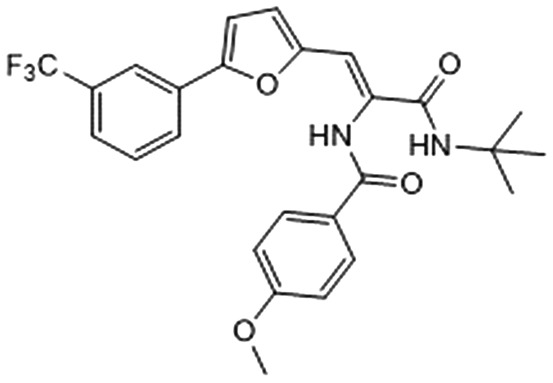
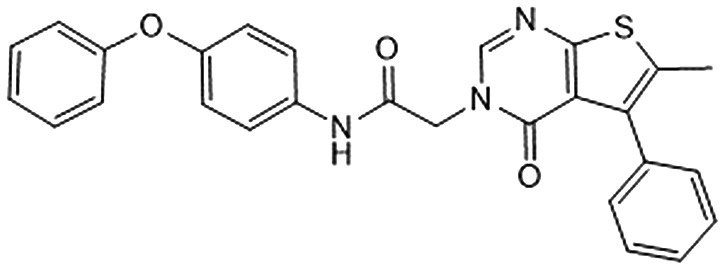
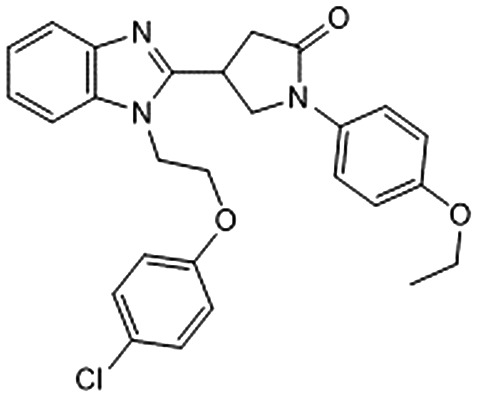
Table 1. Chemical structures of compounds 1–8, with related Glide XP Score, best shape similarity value (related to LVJ reference crystallized conformation), mPGES-1 and 5-LO residual activities tested at 10 μM ligand concentration, and mPGES-1 IC50 values.
| ID | Chemical structure | Glide XP score | LVJ shape similarity | mPGES-1 (residual activity at 10 μM) | mPGES-1 IC50 (μM) | 5-LO (residual activity at 10 μM) |
| 1 |
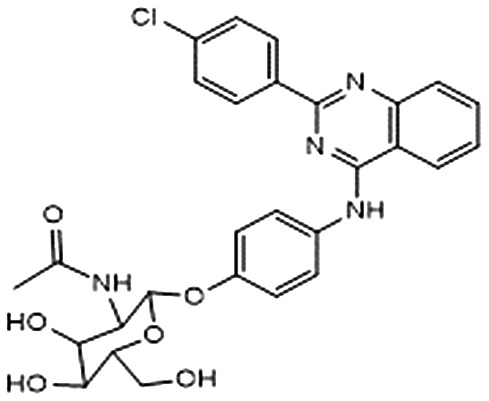
|
–7.462 | 0.506 | 80.1 ± 3.9 | — | — |
| 2 |
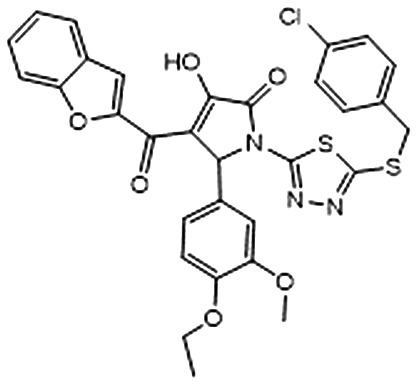
|
–6.494 | 0.585 | 30. 5 ± 3.4 | 3.7 ± 2.7 | 92.5 ± 10.0 |
| 3 |
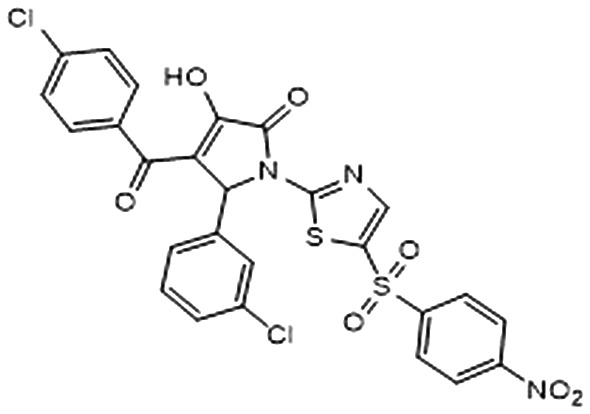
|
–6.406 | 0.538 | 25.0 ± 6.5 | 1.9 ± 1.5 | 56.6 ± 8.5 |
| 4 |
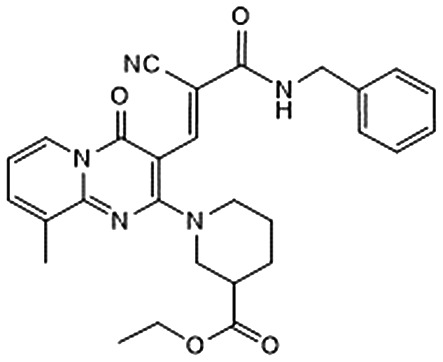
|
–6.323 | 0.657 | 86.8 ± 4.2 | — | — |
| 5 |
|
–6.284 | 0.660 | 72.1 ± 4.1 | — | — |
| 6 |
|
–6.241 | 0.517 | 88.6 ± 3.7 | — | — |
| 7 |
|
–6.193 | 0.515 | 82.6 ± 2.4 | — | — |
| 8 |
|
–6.155 | 0.507 | 65.3 ± 11.1 | — | — |
Biological assessment of mPGES-1 and 5-LO inhibition
Subsequently, in order to assess the ability of compounds 1–8 (Table 1) to interfere with the activity of mPGES-1, a cell-free assay using the microsomal fractions of IL-1β-treated A549 cells (as source for mPGES-1) was applied. In a first screening round, all the compounds were solubilized in DMSO and tested at a final concentration of 10 μM. Biological data (Table 1) disclosed 2 and 3, bearing the 3-hydroxy-3-pyrrolin-2-one nucleus, as the most promising compounds (∼70 up to ∼80% of mPGES-1 inhibition). We were intrigued by this result, since this chemical core has shown a wide spectrum of biological activities, such as anticancer and antiviral, and chemical procedures have been reported for its decoration using multi-component reactions, then paving the way for future studies for the identification of chemical analogues.48–50 On the other hand, 5 and 8 showed a modest activity ranging from ∼30 up to ∼40% of mPGES-1 inhibition, suggesting further investigations to identify the minimal pharmacophoric portions in order to obtain more potent analogues. We then investigated the biological activities of compounds 2 and 3 in more detail. Specifically, the related IC50 values corroborated the activities of the two lead compounds, able to inhibit mPGES-1 in the low micromolar range (IC50 = 3.7 ± 2.7 μM for 2; IC50 = 1.9 ± 1.5 μM for 3). The analysis of three-dimensional poses from molecular docking experiments, showed the good accommodation of the two identified compounds onto the mPGES-1 binding site, establishing a wide set of polar interactions (C:Asp49, A:Ser127, A:Thr131, C:Gln36, C:Arg38, C:His53), H-bonds (A:Ser127, A:Thr131, C:Gln36, C:Arg52), and π–π and π-cation contacts (A:Tyr130, C:Phe44, C:His53, C:Arg52) (docking poses featuring the highest shape similarity with LVJ reported in Fig. 3 and 4). Furthermore, 2 and 3 were also tested at 10 μM ligand concentration against 5-LO, another key enzyme involved in the AA cascade.44 The obtained results highlighted no inhibitory activity for 2 and a modest inhibition exerted by 3 (see Table 1).
Fig. 3. A) Selected 3D pose of 2 (colored by atom types: C, ochre; N, blue; O, red; S, yellow; polar H, light grey; Cl, light green) featuring the highest shape similarity to LVJ, in docking with mPGES-1 (chains A, B and C depicted in red, black, and blue ribbons, transparent molecular surface on the binding site colored in yellow, orange, and green, see text for details, glutathione (GSH) cofactor and key residues in the mPGES-1 binding site are represented in sticks, C grey, O red, N, blue, polar H white); B) related two-dimensional panels representing interactions.

Fig. 4. A) Selected 3D pose of 3 (colored by atom types: C, indigo blue; N, blue; O, red; S, yellow; polar H, light grey; Cl, light green) featuring the highest shape similarity to LVJ, in docking with mPGES-1 (chains A, B and C depicted in red, black, and blue ribbons, transparent molecular surface on the binding site colored in yellow, orange, and green, see text for details, glutathione (GSH) cofactor and key residues in the mPGES-1 binding site are represented in sticks, C grey, O red, N, blue, polar H white); B) related two-dimensional panels representing interactions.

Materials and methods
Input files preparation for molecular docking
The starting library of compounds was retrieved from the Otava Chemicals database (∼3.1 × 105 compounds). The compounds were prepared using LigPrep software (Schrodinger Suite).51 Specifically, all the possible stereoisomers, tautomers, and protonation states at pH = 7.4 ± 1.0 were generated for each compound, and finally the structures were minimized using OPLS 2005 force field. Protein 3D model was prepared using the Schrödinger Protein Preparation Wizard,52 starting from the mPGES-1 X-ray structure in the active form co-complexed with the inhibitor LVJ (2-[[2,6-bis(chloranyl)-3-[(2,2-dimethylpropanoylamino)methyl]phenyl]amino]-1-methyl-6-(2-methyl-2-oxidanyl-propoxy)-N-[2,2,2-tris(fluoranyl)ethyl]benzimidazole-5-carboxamide) (PDB code: ; 4BPM).28 The visual inspection of the protein crystal structure revealed that the binding of the co-crystallized inhibitor LVJ was not assisted by structural water molecules, thus, we proceeded to remove them. All hydrogen atoms were added, and bond orders were assigned. Docking calculations were performed on the protein structure in the presence of the cofactor GSH, whereas LVJ was removed.
Ligand-based shape screening
The starting library of compounds was submitted to a fast “shape screening” alignment, using the 3D structure of the co-complexed mPGES-1 inhibitor LVJ as shape query ligand. With this aim, Phase software (Schrodinger, LLC)53 was employed; in particular, for the screened compounds, the sampling was performed allowing the conformers around the amide bond to vary freely, and finally, 1000 maximum number of conformers were considered for the shape computation. Once associated a shape similarity value for each compound against LVJ, a ranking from the best to worst values was obtained, and we set a threshold = 0.627 (value chosen starting from the best value = 0.727 and reducing it of 0.100) for saving the most promising compounds to be submitted to the subsequent docking calculations. Specifically, a new library of 7220 unique compounds was filtered out.
Molecular docking and structure-based shape screening
The molecular docking virtual screening campaign was performed using the Virtual Screening Workflow using Glide software54 as implemented in Schrodinger Suite, following the scheme:
• High-throughput virtual screening scoring/sampling (HTVS): 7220 unique compounds as input, saved 10 maximum number of poses for each compound, saved first 10 000 ranked poses (selection filter: docking score) as output, corresponding to 4546 unique compounds.
• Standard precision scoring/sampling phase (SP), and structure-based shape screening: 4546 unique compounds from HTVS phase as input, saved 10 maximum number of poses for each compound. Each produced pose was submitted to an “in place” shape screening (see Results and discussion) against LVJ. Choosing a threshold = 0.400, 4120 unique compounds were selected for the subsequent step.
• Extra precision scoring/sampling phase (XP), and structure-based shape screening: 4120 unique compounds from SP/shape screening step as input, saved 100 maximum number of poses for each compound. The produced poses were submitted to an “in place” shape screening (see Results and discussion) against LVJ. Choosing a threshold = 0.500, 914 unique compounds were selected. The selected compounds were then further filtered considering their binding mode with the receptor counterpart (see Results and discussion).
PAINS screening
Selected compounds were screened with SwissADME47 web tool, in order to evaluate the presence of chemical species belonging to “Pan-Assay Interference Compounds” (PAINS) chemical class, implemented from the paper by Baell et al.;55 the final library of compounds selected for the biological assays (1–8) were not PAINS.
Bioactivity assays
Cell-free mPGES-1 activity assay
Microsomes of IL-1β-stimulated A549 cells (source: ATCC) were used as source for mPGES-1. Expression of mPGES-1, preparation of microsomes and determination of mPGES-1 activity was performed as described previously.56 This protocol showed a high robustness and accuracy, since it was applied in previous studies considering mPGES-1 inhibitors as standard drugs, like FLAP/mPGES-1 inhibitor MK-886 for which the related mPGES-1 IC50 value was correctly reproduced.56–58 In brief, A549 cells were treated with IL-1β (1 ng ml–1) for 48 h, cells were harvested, sonicated and the homogenate was subjected to differential centrifugation at a) 10 000×g for 10 min and b) 174 000×g for 1 h at 4 °C. The microsomal fraction (pellet) was resuspended in 1 ml homogenization buffer (0.1 M potassium phosphate buffer, pH 7.4, 1 mM phenylmethanesulfonyl fluoride, 60 μg mL–1 soybean trypsin inhibitor, 1 μg mL–1 leupeptin, 2.5 mM glutathione, and 250 mM sucrose), the total protein concentration was determined, and microsomes were diluted in potassium phosphate buffer (0.1 M, pH 7.4) containing 2.5 mM glutathione.
Test compounds (or DMSO as vehicle) were added, and after 15 min at 4 °C reaction (100 μl total volume) was initiated by addition of 20 μM PGH2. After 1 min at 4 °C, 100 μl of stop solution (40 mM FeCl2, 80 mM citric acid, and 10 μM 11β-PGE2) were added.
PGE2 was separated by solid-phase extraction and analyzed by RP-HPLC as described previously.56
Cell-based 5-LO activity assay
Freshly isolated human neutrophils from peripheral blood were resuspended (5 × 106 cells per ml) in PBS pH 7.4 plus 1 mM CaCl2 containing 1 mg mL–1 glucose, pre-incubated with test compounds (1 μl in DMSO; final DMSO concentration: 0.1%) for 10 min and then treated with 2.5 μM Ca2+ ionophore A23187 plus 20 μM arachidonic acid.
After 10 min at 37 °C the reaction was stopped on ice by addition of 1 mL of methanol. 30 ml 1 N HCl and 500 ml PBS, and 200 ng PGB1 were added and the samples were subjected to solid phase extraction on C18-columns (100 mg, UCT, Bristol, PA, USA). 5-LO products, namely all-trans isomers of LTB4 and 5-hydro(pero)xy-6,8,11,14-eicosatetraenoic acid (5-H(P)ETE), were analyzed by RP-HPLC and quantities calculated on the basis of the internal standard PGB1.
Compounds
Compounds 1–8 were purchased from OTAVA chemicals, Ltd company. For all the compounds the company has undergone quality control to confirm their chemical structures. 1H NMR spectra of 1–8 are provided in ESI.† In addition, we performed analytical reversed-phase HPLC on both the active compounds 2 and 3, in order to verify the purity. An Agilent Technologies 1200 Series high performance liquid chromatography system was used for the analysis. The binary solvent system (A/B) was as follows: 0.1% TFA solution in water (A) and 0.1% TFA solution in CH3CN (B). The adopted conditions were: a Fusion, C18 reversed-phase column (250 × 4.6 mm, 4 μM, 80 Å, flow rate = 1 mL min–1), a gradient from 5% B ending to 100% B over 40 min, and the absorbance was detected at 240 nm (tR compound 2 = 30.209; tR compound 3 = 29.835). Both the samples showed a high grade of purity in the HPLC analysis (>98%).
Conclusions
We have reported the identification of the two novel mPGES-1 inhibitors following a multi-step virtual screening protocol. In particular, compounds 2 and 3 showed interesting inhibitory activities in the low micromolar range, disclosing 3-hydroxy-3-pyrrolin-2-one as a new promising template for developing anti-inflammatory/anticancer agents.
The reported multi-step virtual screening workflow is a useful tool for facilitating the ligand/structure-based identification of novel mPGES-1 inhibitors by means of computational techniques and will be opportunely applied to further libraries of virtual compounds. The discovered 3-hydroxy-3-pyrrolin-2-one scaffold represents an interesting chemical core that could be easily decorated according to convenient multi-component organic reactions. In summary, we provide a suitable strategy based on virtual screening for the identification of new mPGES-1 inhibitors useful for the treatment of cancer and inflammation.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) (grant IG 2015 – IG 17440 to Bifulco Giuseppe). G. L. acknowledges fellowship support from Associazione Italiana per la Ricerca sul Cancro (AIRC) (grant IG 2015 – IG 17440 to Bifulco Giuseppe).
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00497h
References
- Coussens L. M., Werb Z. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E., Nowarski R., Thaiss C. A., Hu B., Jin C., Flavell R. A. Nat. Rev. Cancer. 2013;13:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- Korniluk A., Koper O., Kemona H., Dymicka-Piekarska V. Ir. J. Med. Sci. 2017;186:57–62. doi: 10.1007/s11845-016-1464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crusz S. M., Balkwill F. R. Nat. Rev. Clin. Oncol. 2015;12:584–596. doi: 10.1038/nrclinonc.2015.105. [DOI] [PubMed] [Google Scholar]
- Landen N. X., Li D., Stahle M. Cell. Mol. Life Sci. 2016;73:3861–3885. doi: 10.1007/s00018-016-2268-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman I., Stanford J. L., Kuniyuki A., Berger R. E. Urology. 2004;64:876–879. doi: 10.1016/j.urology.2004.06.071. [DOI] [PubMed] [Google Scholar]
- Loftus, Jr. E. V. Gastroenterol. Clin. North Am. 2006;35:517–531. doi: 10.1016/j.gtc.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Rosenblatt K. A., Wicklund K. G., Stanford J. L. Am. J. Epidemiol. 2001;153:1152–1158. doi: 10.1093/aje/153.12.1152. [DOI] [PubMed] [Google Scholar]
- Wang D., Dubois R. N. Gut. 2006;55:115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk C. D. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y., Narumiya S. J. Biol. Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Ke J., Yang Y., Che Q., Jiang F., Wang H., Chen Z., Zhu M., Tong H., Zhang H., Yan X., Wang X., Wang F., Liu Y., Dai C., Wan X. Tumor Biol. 2016;37:12203–12211. doi: 10.1007/s13277-016-5087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi M., Rosenberg D. W. Semin. Immunopathol. 2013;35:123–137. doi: 10.1007/s00281-012-0342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh S., Thomas G. A. Gut. 1994;35:675–678. doi: 10.1136/gut.35.5.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montrose D. C., Nakanishi M., Murphy R. C., Zarini S., McAleer J. P., Vella A. T., Rosenberg D. W. Prostaglandins Other Lipid Mediators. 2015;116–117:26–36. doi: 10.1016/j.prostaglandins.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayburn E. R., Ezell S. J., Zhang R. Mol. Cell. Pharmacol. 2009;1:29–43. doi: 10.4255/mcpharmacol.09.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadkarni D. H., Wang F., Wang W., Rayburn E. R., Ezell S. J., Murugesan S., Velu S. E., Zhang R. Med. Chem. 2009;5:227–236. doi: 10.2174/157340609788185873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sostres C., Gargallo C. J., Arroyo M. T., Lanas A. Best Pract. Res., Clin. Gastroenterol. 2010;24:121–132. doi: 10.1016/j.bpg.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Koeberle A., Werz O. Biochem. Pharmacol. 2015;98:1–15. doi: 10.1016/j.bcp.2015.06.022. [DOI] [PubMed] [Google Scholar]
- Park J. Y., Pillinger M. H., Abramson S. B. Clin. Immunol. 2006;119:229–240. doi: 10.1016/j.clim.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Samuelsson B., Morgenstern R., Jakobsson P. J. Pharmacol. Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- Trebino C. E., Stock J. L., Gibbons C. P., Naiman B. M., Wachtmann T. S., Umland J. P., Pandher K., Lapointe J. M., Saha S., Roach M. L., Carter D., Thomas N. A., Durtschi B. A., McNeish J. D., Hambor J. E., Jakobsson P. J., Carty T. J., Perez J. R., Audoly L. P. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi M., Gokhale V., Meuillet E. J., Rosenberg D. W. Biochimie. 2010;92:660–664. doi: 10.1016/j.biochi.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberle A., Laufer S. A., Werz O. J. Med. Chem. 2016;56:5970–5986. doi: 10.1021/acs.jmedchem.5b01750. [DOI] [PubMed] [Google Scholar]
- Chang H.-H., Meuillet E. J. Future Med. Chem. 2011;3:1909–1934. doi: 10.4155/fmc.11.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psarra A., Nikolaou A., Kokotou M. G., Limnios D., Kokotos G. Expert Opin. Ther. Pat. 2017;27:1047–1059. doi: 10.1080/13543776.2017.1344218. [DOI] [PubMed] [Google Scholar]
- Sjogren T., Nord J., Ek M., Johansson P., Liu G., Geschwindner S. Proc. Natl. Acad. Sci. U. S. A. 2013;110:3806–3811. doi: 10.1073/pnas.1218504110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Howe N., Dukkipati A., Shah S. T. A., Bax B. D., Edge C., Bridges A., Hardwicke P., Singh O. M. P., Giblin G., Pautsch A., Pfau R., Schnapp G., Wang M., Olieric V., Caffrey M. Cryst. Growth Des. 2014;14:2034–2047. doi: 10.1021/cg500157x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz J. G., Antonysamy S., Kuklish S. L., Condon B., Lee M. R., Allison D., Yu X. P., Chandrasekhar S., Backer R., Zhang A., Russell M., Chang S. S., Harvey A., Sloan A. V., Fisher M. J. J. Med. Chem. 2015;58:4727–4737. doi: 10.1021/acs.jmedchem.5b00330. [DOI] [PubMed] [Google Scholar]
- Weinert T., Olieric V., Waltersperger S., Panepucci E., Chen L., Zhang H., Zhou D., Rose J., Ebihara A., Kuramitsu S., Li D., Howe N., Schnapp G., Pautsch A., Bargsten K., Prota A. E., Surana P., Kottur J., Nair D. T., Basilico F., Cecatiello V., Pasqualato S., Boland A., Weichenrieder O., Wang B. C., Steinmetz M. O., Caffrey M., Wang M. Nat. Methods. 2015;12:131–133. doi: 10.1038/nmeth.3211. [DOI] [PubMed] [Google Scholar]
- Schiffler M. A., Antonysamy S., Bhattachar S. N., Campanale K. M., Chandrasekhar S., Condon B., Desai P. V., Fisher M. J., Groshong C., Harvey A., Hickey M. J., Hughes N. E., Jones S. A., Kim E. J., Kuklish S. L., Luz J. G., Norman B. H., Rathmell R. E., Rizzo J. R., Seng T. W., Thibodeaux S. J., Woods T. A., York J. S., Yu X. P. J. Med. Chem. 2015;59:194–205. doi: 10.1021/acs.jmedchem.5b01249. [DOI] [PubMed] [Google Scholar]
- Brock J. S., Hamberg M., Balagunaseelan N., Goodman M., Morgenstern R., Strandback E., Samuelsson B., Rinaldo-Matthis A., Haeggstrom J. Z. Proc. Natl. Acad. Sci. U. S. A. 2016;113:972–977. doi: 10.1073/pnas.1522891113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone R., Andres R. M., Aquino M., Bruno I., Guerrero M. D., Terencio M. C., Paya M., Riccio R. Chem. Biol. Drug Des. 2010;76:17–24. doi: 10.1111/j.1747-0285.2010.00984.x. [DOI] [PubMed] [Google Scholar]
- De Simone R., Chini M. G., Bruno I., Riccio R., Mueller D., Werz O., Bifulco G. J. Med. Chem. 2011;54:1565–1575. doi: 10.1021/jm101238d. [DOI] [PubMed] [Google Scholar]
- Chini M. G., De Simone R., Bruno I., Riccio R., Dehm F., Weinigel C., Barz D., Werz O., Bifulco G. Eur. J. Med. Chem. 2012;54:311–323. doi: 10.1016/j.ejmech.2012.05.014. [DOI] [PubMed] [Google Scholar]
- Lauro G., Strocchia M., Terracciano S., Bruno I., Fischer K., Pergola C., Werz O., Riccio R., Bifulco G. Eur. J. Med. Chem. 2014;80:407–415. doi: 10.1016/j.ejmech.2014.04.061. [DOI] [PubMed] [Google Scholar]
- Chini M. G., Ferroni C., Cantone V., Dambruoso P., Varchi G., Pepe A., Fischer K., Pergola C., Werz O., Bruno I., Riccio R., Bifulco G. MedChemComm. 2015;6:75–79. [Google Scholar]
- Terracciano S., Lauro G., Strocchia M., Fischer K., Werz O., Riccio R., Bruno I., Bifulco G. ACS Med. Chem. Lett. 2015;6:187–191. doi: 10.1021/ml500433j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauro G., Tortorella P., Bertamino A., Ostacolo C., Koeberle A., Fischer K., Bruno I., Terracciano S., Gomez-Monterrey I. M., Tauro M., Loiodice F., Novellino E., Riccio R., Werz O., Campiglia P., Bifulco G. ChemMedChem. 2016;11:612–619. doi: 10.1002/cmdc.201500598. [DOI] [PubMed] [Google Scholar]
- Di Micco S., Spatafora C., Cardullo N., Riccio R., Fischer K., Pergola C., Koeberle A., Werz O., Chalal M., Vervandier-Fasseur D., Tringali C., Bifulco G. Bioorg. Med. Chem. 2016;24:820–826. doi: 10.1016/j.bmc.2016.01.002. [DOI] [PubMed] [Google Scholar]
- Iranshahi M., Chini M. G., Masullo M., Sahebkar A., Javidnia A., Chitsazian Yazdi M., Pergola C., Koeberle A., Werz O., Pizza C., Terracciano S., Piacente S., Bifulco G. J. Nat. Prod. 2015;78:2867–2879. doi: 10.1021/acs.jnatprod.5b00700. [DOI] [PubMed] [Google Scholar]
- Di Micco S., Terracciano S., Cantone V., Fischer K., Koeberle A., Foglia A., Riccio R., Werz O., Bruno I., Bifulco G. Eur. J. Med. Chem. 2018;143:1419–1427. doi: 10.1016/j.ejmech.2017.10.039. [DOI] [PubMed] [Google Scholar]
- Lauro G., Manfra M., Pedatella S., Fischer K., Cantone V., Terracciano S., Bertamino A., Ostacolo C., Gomez-Monterrey I., De Nisco M., Riccio R., Novellino E., Werz O., Campiglia P., Bifulco G. Eur. J. Med. Chem. 2017;125:278–287. doi: 10.1016/j.ejmech.2016.09.042. [DOI] [PubMed] [Google Scholar]
- Radmark O., Samuelsson B. J. Intern. Med. 2010;268:5–14. doi: 10.1111/j.1365-2796.2010.02246.x. [DOI] [PubMed] [Google Scholar]
- Sastry G. M., Inakollu V. S., Sherman W. J. Chem. Inf. Model. 2013;53:1531–1542. doi: 10.1021/ci300463g. [DOI] [PubMed] [Google Scholar]
- Pala D., Beuming T., Sherman W., Lodola A., Rivara S., Mor M. J. Chem. Inf. Model. 2013;53:821–835. doi: 10.1021/ci4000147. [DOI] [PubMed] [Google Scholar]
- Daina A., Michielin O., Zoete V. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shymanska N. V., Pierce J. G. Org. Lett. 2017;19:2961–2964. doi: 10.1021/acs.orglett.7b01185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M., Tintori C., Christopher R. S., Radi M., Schenone S., Musumeci F., Brullo C., Sanita P., Delle Monache S., Angelucci A., Kissova M., Crespan E., Maga G., Botta M. ChemMedChem. 2013;8:484–496. doi: 10.1002/cmdc.201200480. [DOI] [PubMed] [Google Scholar]
- Pace P., Spieser S. A., Summa V. Bioorg. Med. Chem. Lett. 2008;18:3865–3869. doi: 10.1016/j.bmcl.2008.06.056. [DOI] [PubMed] [Google Scholar]
- LigPrep, Schrödinger, LLC, New York, NY, 2017.
- Maestro, Schrödinger, LLC, New York, NY, 2017.
- Phase, Schrödinger, LLC, New York, NY, 2017.
- Glide, Schrödinger, LLC, New York, NY, 2017.
- Baell J. B., Holloway G. A. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Koeberle A., Siemoneit U., Buehring U., Northoff H., Laufer S., Albrecht W., Werz O. J. Pharmacol. Exp. Ther. 2008;326:975–982. doi: 10.1124/jpet.108.139444. [DOI] [PubMed] [Google Scholar]
- Siemoneit U., Koeberle A., Rossi A., Dehm F., Verhoff M., Reckel S., Maier T. J., Jauch J., Northoff H., Bernhard F., Doetsch V., Sautebin L., Werz O. Br. J. Pharmacol. 2011;162:147–162. doi: 10.1111/j.1476-5381.2010.01020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberle A., Northoff H., Werz O. Biochem. Pharmacol. 2009;77:1513–1521. doi: 10.1016/j.bcp.2009.02.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.