

SUMMARY
Tumor-derived extracellular vesicles (TEV) “educate” healthy cells to promote metastases. We found that melanoma TEV downregulated type I interferon (IFN) receptor and expression of IFN-inducible cholesterol 25-hydroxylase (CH25H). CH25H produces 25-hydroxycholesterol, which inhibited TEV uptake. Low CH25H levels in leukocytes from melanoma patients correlated with poor prognosis. Mice incapable of downregulating the IFN receptor and Ch25h were resistant to TEV uptake, TEV-induced pre-metastatic niche and melanoma lung metastases; however, ablation of Ch25h reversed these phenotypes. An antihypertensive drug reserpine suppressed TEV uptake and disrupted TEV-induced formation of the pre-metastatic niche and melanoma lung metastases. These results suggest the importance of CH25H in defense against education of normal cells by TEV and argue for use of reserpine in adjuvant melanoma therapy.
Keywords: metastasis, adjuvant therapy, extracellular vesicles, melanoma, 25-hydroxycholesterol, reserpine, interferon
Graphical Abstract
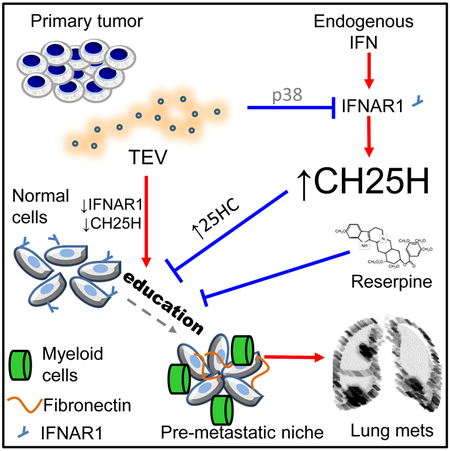
In Brief
Ortiz et al. reveal melanoma-derived extracellular vesicles (EV) downregulate type I interferon receptor (IFNAR1) and cholesterol 25-hydroxylase (CH25H) in normal cells to facilitate EV uptake and pre-metastatic niche development. IFNAR1-CH25H upregulation or reserpine treatment inhibits EV uptake and metastasis.
INTRODUCTION
Whereas metastases are the most important determinants of cancer lethality (Chaffer and Weinberg, 2011), there is a relative dearth of clinically validated specific targets and agents that either eliminate the existing metastatic lesions, or, at the very least prevent initial metastases and suppress their colonization (Steeg, 2016). Normal cells in target organs can form a hospitable “soil” (Fuchs, 1882; Paget, 1889) enabling malignant cells to home and colonize. Given genomic instability and extreme plasticity of malignant cells, targeting these normal cells might represent a promising therapeutic strategy against metastatic disease.
The metastatic process in many cancer types is stimulated by tumor-derived extracellular vesicles (TEV), including exosomes (Costa-Silva et al., 2015; Peinado et al., 2012). These vesicles can dramatically affect normal cells by delivering biomolecules that alter the function of these cells (Aleckovic and Kang, 2015; Becker et al., 2016; Kalluri, 2016; Lobb et al., 2017; Peinado et al., 2012). Such “education” by TEV can co-opt normal cells to contribute to tumor progression and metastasis via multiple mechanisms including formation of the pre-metastatic niches in distant healthy organs such as the lungs (Liu et al., 2016; Lobb et al., 2017).
However, the majority of normal cells in a tumor-bearing organism exhibits limited uptake of TEV (Mendt et al., 2018) and appears to remain unaffected suggesting the existence of yet to be identified defensive mechanisms that restrict the extent of education by TEV. Here we sought to identify the nature of such defenses in normal cells as well as potential mechanisms underlying their breach and failure during tumor progression and metastasis. In addition, we aimed to identify and characterize the agents that can disrupt the education of normal cells by TEV and thus may have therapeutic activities in the adjuvant/neoadjuvant setting.
RESULTS
TEV inactivate the IFN pathway to facilitate TEV uptake and education of normal cells
TEV-driven education of normal cells is attributed to transfer of diverse biomolecules including signaling receptors (reviewed in (Aleckovic and Kang, 2015; Becker et al., 2016; Kalluri, 2016; Lobb et al., 2017; Peinado et al., 2012)). We used a Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)-based Plasma Membrane Profiling proteomic screen (Matheson et al., 2015; Weekes et al., 2013) (Figure S1a) to determine changes in the repertoire of cell surface signaling receptors in human cells exposed to TEV (characterized in Figure S1b). Comparative mass spectrometry revealed that TEV treatment decreased the levels of several plasma membrane proteins (Table S1) including the IFNAR1 chain of the type I interferon (IFN) receptor (Figure 1a). We further focused on IFNAR1 because of the important role of IFN in defense against tumor metastases (Parker et al., 2016) and clinical use of these cytokines in adjuvant therapy of melanoma (Eggermont et al., 2014).
Figure 1: TEV downregulate IFNAR1 in normal cells.

A. Volcano plot summarizing SILAC-based plasma membrane profiling results from THP-1 cells treated with 1205Lu human melanoma TEV. Mean log2(fold change) and -log10(p value) are shown for 366 proteins identified by >1 unique peptide and quantitated in every replicate. Downregulated proteins with a fold change >2 and p<0.05 are outlined in red.
B. Representative histogram of IFNAR1 cell surface levels measured by flow cytometry on THP-1 cells treated with 1205Lu human melanoma TEV (10 μg/106 cells for 2 hr) or PBS.
C. Effects of TEV (10 μg/106 cells for 30 min) on IFNAR1 phosphorylation and ubiquitination in THP-1 cells. Immunoblot (IB) analyses of immunoprecipitated (IP) IFNAR1 using indicated antibodies are shown. Immunoblot analysis of βTRCP and GADPH levels in the whole cell lysate are also shown.
D. Flow cytometry analysis of IFNAR1 cell surface levels on THP-1 cells treated with extracellular vesicles (5 μg for 2 hr) isolated from plasma of healthy donors (HDEV, n=8) or melanoma patients (MPEV, n=13) or PBS (n=7). A representative histogram (top) and quantification (bottom) are shown.
E. IFNAR1 cell surface levels on peripheral blood leukocytes from healthy donors (HD; n=10) or melanoma patients (MP) from two independent cohorts: MP1 (40 patients of stages I-IV) and MP2 20 patients in each of two groups that were positive (SLN+) or negative (SLN−) for sentinel lymph node metastases. A representative histogram (top) and ΔMFI quantification (bottom) are shown.
F. Effect of extracellular vesicles from B16F10 (TEV, 10 μg/106 cells, 2 hr) or from primary mouse lung fibroblasts (FEV, 20 μg/106 cells, 2 hr) on IFNAR1 surface level (analyzed by flow cytometry, top) or p38 activation (analyzed by immunoblotting, bottom) in primary mouse splenocytes.
G. Effect of B16F10 TEV treatment (10 μg/106 cells for 2 hr) on IFNAR1 surface level on splenocytes of indicated genotypes.
H. Analysis of IFNAR1 cell surface levels on peripheral blood leukocytes from WT and SA mice (naive or bearing B16F10 tumors of equal size ~200 mm2, each n=4)
Data are represented as mean ± SEM; p values: * p<0.05; ** p<0.01; *** p<0.001; NS, not significant from unpaired Student’s t test (panels A, D, E, H).
TEV derived from melanoma cells did not alter the levels of IFNAR1 mRNA (Figure S1c) but notably decreased IFNAR1 protein (Figure 1b and S1d) without inducing activation of STAT1 (Figure S1d) suggesting that TEV downregulate IFNAR1 in an IFN-independent manner. Previous reports demonstrated that non-ligand stimuli activate p38α protein kinase-dependent phosphorylation of IFNAR1 on Ser residues (Bhattacharya et al., 2011). This phosphorylation enables the βTrcp E3 ubiquitin ligase to facilitate IFNAR1 ubiquitination and degradation (Bhattacharya et al., 2013; Fuchs, 2013; Huangfu et al., 2012; Liu et al., 2009; Qian et al., 2011). Given that TEV encompass diverse types of RNA that can trigger a number of signaling cascades including p38 activation (Liu et al., 2016), we examined the impact of TEV on this pathway. TEV treatment had a modest effect on βTrcp but activated p38 kinase (Figure S1d) and stimulated IFNAR1 Ser phosphorylation and ubiquitination (Figure 1c). Furthermore, a p38 inhibitor prevented IFNAR1 loss (Figure S1e) suggesting that TEV act as inducers of p38-dependent IFNAR1 downregulation.
To determine the relevance of these observations to naturally occurring melanoma, we examined whether extracellular vesicles isolated from melanoma patients can downregulate IFNAR1 and whether IFNAR1 is indeed downregulated in normal cells from melanoma patients. Plasma-derived vesicles from melanoma patients (but not from healthy donors) indeed decreased the levels of IFNAR1 protein but not mRNA in human cells in vitro (Figure 1d and S1f). Furthermore, lower levels of the cell surface IFNAR1 protein (but not mRNA) were found in leukocytes from melanoma patients compared to those from healthy donors (Figure 1e and S1g). Importantly, the extent of IFNAR1 downregulation was greater in patients with documented metastatic lesions (Figure 1e). Given that an increase in TEV was associated with metastatic melanoma (Peinado et al., 2012), these data are consistent with downregulation of IFNAR1 on normal cells as a hallmark of their education by TEV in patients.
Understanding of importance of IFNAR1 downregulation as a part of education of normal cells by TEV requires modeling the effects of TEV in vitro and in vivo. Extracellular vesicles isolated from cultured B16F10 mouse melanoma cells (but not from normal primary mouse lung fibroblasts) robustly induced p38 kinase activation and downregulated IFNAR1 in primary mouse splenocytes (Figure 1f). Furthermore, ablation of Mapk14 (gene that encodes p38α) in these splenocytes attenuated the loss of IFNAR1 upon exposure to TEV (Figure 1g) suggesting that normal mouse cells can recapitulate the p38-driven loss of IFNAR1.
In order to evaluate the importance of TEV-induced downregulation of IFNAR1, we used Ifnar1S526A knock-in mice, which are deficient in IFNAR1 ubiquitination and degradation (characterized in (Bhattacharya et al., 2014; Katlinski et al., 2017) and henceforth termed “SA”). SA splenocytes were resistant to TEV-induced downregulation of IFNAR1 in vitro (Figure 1g). Furthermore, intravenous injection of TEV in vivo downregulated IFNAR1 levels on the surface of wild-type (WT) (but not SA) leukocytes (Figure S1h). Finally, lower levels of IFNAR1 on peripheral blood leukocytes were detected in tumor-bearing WT (but not SA) mice compared to naive animals (Figure 1h) indicating that SA cells and animals can be used as controls for assessing the importance of IFNAR1 loss in TEV-induced education.
IFNAR1 is required for all effects of Type I IFN but not Type II IFNγ (Platanias, 2005). Accordingly, pre-treatment of mouse cells with TEV attenuated phosphorylation of STAT1 induced specifically by IFNβ but not by IFNγ (Figure 2a and S2a) suggesting that TEV can inactivate IFNAR1 signaling. Whereas extracellular vesicles from normal fibroblasts induced expression of some of IFN-stimulated genes (ISG) consistent with a previous report (Boelens et al., 2014), TEV notably suppressed this expression (Figure 2b). An even greater suppression of ISG was elicited by multiple treatments imitating continuous education by TEV (Figure S2b). These observations in mouse cells were faithfully recapitulated in human cells where expression of ISG was inhibited by extracellular vesicles from melanoma patients (but not from healthy donors, Figure 2c) indicating that TEV can inactivate the IFN-IFNAR1 pathway.
Figure 2: TEV and IFN counteract effects of each other.

A. Effect of pre-treatment with B16F10 TEV (10 μg/106 cells for 90 min) on activation of STAT1 by IFNβ (250 IU/mL for 30 min) or IFNγ (50 IU/mL for 30 min) in indicated mouse splenocytes analyzed by immunoblotting.
B. qPCR analysis of mRNA for indicated genes in indicated mouse splenocytes treated with PBS, or extracellular vesicles from primary lung fibroblasts (FEV, n=3), or B16F10 (TEV, n=8) at 10 μg for 4 hr.
C. qPCR analysis of mRNA for indicated genes in THP-1 cells treated with PBS (n=7) or with vesicles (5 μg for 4 hr) from healthy donors (HDEV) (n=8) or melanoma patients (MPEV). (n=13)
D. Uptake of DiD-labeled B16F10 TEV (10 μg for 4 hr) by WT or SA splenocytes pre-treated with PBS or mouse IFNβ (1000 IU/mL for 8 hr); n=6 for each group.
E. Uptake of DiD-labeled B16F10 TEV (10 μg for 4 hr) by indicated splenocytes (106/mL) pre-treated with IgG control or anti-IFNAR1 (0.33 μg/mL for 1 hr); n=6 for each group.
F. In vivo uptake of i.v. administered DiD-labeled B16F10 TEV (30 μg for 24 hr) by splenocytes and bone marrow cells of indicated mice; n=6 for each group.
Data are represented as mean ± SEM; p values: * p<0.05; ** p<0.01; *** p<0.001; NS, not significant from Student’s t test.
See also Figure S2
The ISG were not suppressed if cells were exposed to IFN before TEV (Figure S2c) indicating that IFN may cancel the effect of TEV. Importantly, control experiments revealed that pre-treatment of cells with IFN impeded TEV uptake in splenocytes (Figure 2d and S2d). These results may suggest that IFN and TEV can mutually counteract each other’s effects: whereas IFNAR1 will be inactivated in cells first exposed to TEV, signaling through active IFNAR1 should limit the education by TEV.
Indeed, SA cells resistant to TEV-induced IFNAR1 downregulation (Figure 1g-h, S1h) also did not respond to TEV by decreasing IFN signaling (Figure 2a and S2a) or the ISG expression (Figure 2b). Importantly, SA cells were relatively refractory to TEV uptake in vitro compared to WT or Ifnar1-null cells (Figures 2d, e and S2d, e). TEV uptake in SA cells was restored by neutralizing antibody against IFNAR1 (Figures 2e and S2e) demonstrating a critical role of IFNAR1 signaling in this phenotype. The SA mice were also resistant to in vivo uptake of TEV by splenocytes and bone marrow cells (Figure 2f and S2f). These results suggest the importance of IFNAR1 in limiting TEV uptake and TEV-elicited education of normal cells.
TEV administration in vivo was shown to educate normal cells and induce the pre-metastatic niche in the lungs characterized by pulmonary myeloid infiltration and fibronectin deposition (Kaplan et al., 2005; Peinado et al., 2012). Intriguingly, treatment of WT (but not SA) mice with TEV led to a notable loss of IFNAR1 protein (Figure 3a) and decreased ISG mRNAs (Figure 3b) in lung tissues. These results suggest that education of normal cells by TEV in vivo includes inactivation of IFNAR1 signaling and further justifies using SA animals as a negative control in experiments assessing the importance of this education.
Figure 3: Inactivation of IFNAR1 promotes education of normal cells by TEV and generation of the pre-metastatic niche.

A. Immunofluorescence and quantification of IFNAR1 levels in lung tissues collected from indicated mice treated with indicated TEV (8 μg, 3× per week, 3 weeks). Red, IFNAR1; Blue, DAPI; Scale bar, 100 μm; Quantification representing averages from 10 random fields in sections from each of 3 different animals is shown in the right panel
B. qPCR analysis of mRNA for indicated genes from lung tissues from panel A
C. Immunofluorescence analysis of CD11b+ (green) clusters (>2 cells) in the lungs of WT and SA animals treated with PBS or B16F10 TEV (i.v., 8 μg 3× per week, 3 weeks). Scale bar - 100 μm; quantification representing averages from 7 random fields in sections from each of 3 different animals is shown in the bottom panel
D. Immunofluorescence analysis of fibronectin (green) deposits in the lungs of WT and SA animals treated with PBS or B16F10 TEV (i.v., 8 μg 3× per week, 3 weeks). Scale bar, 100 μm; quantification representing averages from 7 random fields in sections from each of 3 different animals is shown in the bottom panel.
E. Schematic of the experiments to evaluate the education of WT and SA bone marrow-derived cells by TEV and effect of this education on B16F10 tumor growth and metastasis
F. Analysis and quantification of lungs from the td-Tomato-B16F10 tumors-bearing WT hosts that received GFP+ bone marrow derived cells (BMDCs) from WT or SA mice educated or not by TEV. Scale bar, 100 μm. Quantification represents averages from 7 random fields in sections from each of four different animals is shown in the right panel.
Data are represented as mean ± SEM; p values: * p<0.05; *** p<0.001; NS, not significant from Student’s t test.
See also Figure S3
Consistent with published results (Peinado et al., 2012) we found that administration of TEV promoted formation of the CD11b+ myeloid clusters (Figure 3c and S3a), and fibronectin deposits (Figure 3d and S3b) in the lungs of WT mice. Importantly, lung tissues from TEV-treated SA mice exhibited fewer CD11b+ cell clusters or fibronectin deposits and lower overall lung myeloid infiltration compared to WT mice (Figures 3c-d and S3b-c). Given that poor myeloid infiltration in SA lungs was reversed by administration of the neutralizing antibody against IFNAR1 (Figure S3d), these data suggest that formation of the TEV-induced pre-metastatic niche requires inactivation of IFNAR1 and ensuing disruption of the IFNAR1-mediated defenses against education of normal cells by TEV.
Lyden and co-authors demonstrated a notable stimulation of primary tumor growth and metastases in irradiated host mice that received transfer of the educated bone marrow from TEV-treated donor mice (Peinado et al., 2012) (Figure 3e). Consistent with this report, we found that transplantation of bone marrow from TEV-treated WT mice indeed stimulated lung metastases in WT recipients inoculated with B16F10 tumors (Figures 3f). Importantly, we did not observe such stimulation when bone marrow was taken from TEV-treated SA mice (Figure 3f). These results suggest that IFNAR1 in normal cells is important to interrupt their education by TEV and, consequently, to provide defense against TEV-stimulated metastases.
IFN-induced CH25H limits uptake of TEV and interrupts the education of normal cells
We next sought to determine how IFNAR1 signaling can suppress TEV uptake and disrupt TEV-induced education of normal cells. Uptake of extracellular vesicles can occur through a number of mechanisms including phagocytosis, endocytosis, micropinocytosis, direct membrane fusion, etc (French et al., 2017). Bone marrow macrophages from SA mice were not deficient in phagocytosis compared to WT macrophages (Figure S4a). Fortuitously, we also observed that IFN suppressed the uptake of empty protein-less liposomes (Figure S4b) suggesting an inhibitory effect of IFN on lipid membrane fusion, which is involved in many types of TEV uptake (French et al., 2017).
Indeed effects of IFN on lipid membrane fluidity have been previously reported (Chatterjee et al., 1982). Recent findings suggest that cholesterol 25-hydroxylase (CH25H), an enzyme generating 25-hydroxycholesterol (25HC) that alters membrane characteristics (Luu and Moog, 1991) can be induced by IFN (Anggakusuma et al., 2015; Blanc et al., 2013; Liu et al., 2013). Pre-treatment of cells with 25HC suppressed uptake of liposomes in WT cells as well as in cells lacking Ifnar1 or Ch25h (Figure S4b) suggesting that this oxysterol acts downstream of these genes. Cells exposed to 25HC also exhibited a decreased uptake of TEV (Figure 4a and S4c). Importantly, IFN did not inhibit uptake of liposomes or TEV in cells lacking Ch25h (Figure 4a and S4b-c). Furthermore, ablation of Ch25h in SA mice restored the ability of tissues from these animals to uptake TEV in vivo (Figures 4b and S4d) suggesting that Ch25h acts downstream of IFNAR1 to restrict the education of normal cells by TEV.
Figure 4: CH25H functions downstream of IFNAR1 to limit education of normal cells by TEV.

A. Uptake of DiD-labeled B16F10 TEV (10 μg for 4 hr) by splenocytes from indicated mice pre-treated with IFNβ (1000 IU/mL for 8 hr) or 25HC (4 μM for 2 hr) or vehicles as indicated; n=6 for each group.
B. Analysis of DiD+ splenocytes and bone marrow cells isolated from mice of indicated genotypes 24 hr after i.v. administration of DiD-labeled B16F10 (30 μg); n=6 for each group.
C. qPCR analysis of relative Ch25h mRNA levels in B16-F10 TEV-treated (10 μg, 4 hr) splenocytes or bone marrow cells of indicated genotypes (value of 1.0 ascribed to untreated WT cells) ; n=4 for each group.
D. qPCR analysis of relative mRNA levels of Ch25h and other genes encoding cholesterol monooxygenases (Cyp27a and Cyp46a) in the lungs of WT or SA mice administered with PBS or B16F10 TEV (i.v., 8 μg, 3× per week, 3 weeks); n=4 for each group.
E. Analysis of CD11b+ cell clusters and fibronectin deposits in the lungs of indicated mice treated with B16-F10 TEV (i.v., 8 μg, 3× per week, 3 weeks). Quantifications averaging from 7 random fields in sections from each of three different animals are shown. Scale bar, 100 μm.
Data are represented as mean ± SEM; p values: * p<0.05; ** p<0.01; *** p<0.001; NS, not significant from Student’s t test.
See also Figure S4
Of note, TEV downregulated Ch25h expression whereas vesicles from primary fibroblasts did not (Figure S4e). Opposite effects on Ch25h were elicited by IFN and TEV, but these stimuli did not affect other monooxygenases involved in cholesterol oxidation such as Cyp46a and Cyp27a (Figure S4e). Tissues from naive SA mice expressed higher levels of Ch25h compared to those from WT mice (Figure S4f). Furthermore, WT cells exposed to TEV in vitro exhibited lower levels of Ch25h compared to those from SA mice (Figure 4c). Importantly, administration of TEV in vivo decreased Ch25h in lung tissues from WT but not SA mice (Figure 4d) indicating the importance of IFNAR1 loss for downregulation of Ch25h. Remarkably, ablation of Ch25h in SA mice restored TEV-induced formation of pre-metastatic niche as seen from accumulation of CD11b+ clusters and fibronectin deposits in the lungs (Figure 4e). This result indicates a defensive role of the IFNAR1-CH25H pathway against TEV-induced education of normal cells and the ensuing development of a pre-metastatic niche.
Suboptimal bioavailability of 25HC (Li et al., 2017) prompted us to search for small molecules that can mimic the suppressive effects of this oxysterol on TEV uptake. A limited hypothesis-driven screen tested the agents previously reported to inhibit biological processes that involve lipid membrane fusion such as viral membrane fusion (Boriskin et al., 2006; Shoemaker et al., 2013), neuromediator re-uptake (Erickson et al., 1992), maturation of myocytes and osteoclasts (Bar-Sagi and Prives, 1983; Okazaki et al., 1999; Yamashita et al., 2010), etc (Table S2). Among six tested compounds, the anti-hypertensive drug reserpine was found to interfere with B16F10 TEV uptake by mouse splenocytes or bone marrow cells (Figure S5a). These results were further validated in vitro in human cells (Figure 5a and S5b).
Figure 5: Reserpine suppresses education of normal cells by TEV.

A. Uptake of DiD-labeled 1205Lu TEV (10 μg for 4 hr) by THP-1 cells pre-treated with vehicle or reserpine (10 μM for 2 hr); n=3 for each group.
B. Uptake of DiD-labeled B16F10 TEV (30 μg) by splenocytes and bone marrow cells isolated from WT mice 24 hr after i.p. injection of vehicle or reserpine (1 μg/kg i.p., given 30 min before TEV injection) followed by i.v. administration of TEV; n=6 for each group.
C. Flow cytometry analysis of IFNAR1 levels on CD45+ peripheral blood leukocytes isolated from WT mice administered i.v. treatment with PBS or B16F10 TEV (8 μg, 3× per week, 3 weeks) and receiving i.p. vehicle or reserpine treatment (as in panel B) as indicated; n=3 for each group.
D. Immunofluorescence analysis of CD11b+ cell clusters and fibronectin deposits in the lungs of WT mice receiving i.p. vehicle or reserpine treatment (as in panel B) followed by i.v. treatment with B16F10 TEV (8 μg, 3× per week, 3 weeks).
Data are represented as mean ± SEM; p values: * p<0.05; ** p<0.01; *** p<0.001; NS – not significant from Student’s t test.
Furthermore, administration of reserpine in vivo also suppressed TEV uptake (Figure 5b and S5c), prevented TEV-induced downregulation of IFNAR1 (Figure 5c) and stimulated ISG including Ch25h (Figure S5d) in normal cells. Importantly, treatment with reserpine abolished TEV-induced formation of the pre-metastatic niche as evident from analysis of lung myeloid infiltrates and fibronectin deposits (Figure 5d and S5e). These results suggest that reserpine can be used to disrupt TEV-induced education of normal cells and the generation of the pre-metastatic niche.
Anti-metastatic role of the IFNAR1-CH25H pathway and potential for adjuvant therapy utilizing inhibitors of TEV uptake
Given that the IFNAR1-dependent expression of Ch25h limits the education of normal cells by TEV, which, in turn, can stimulate tumor metastases (Chin and Wang, 2016a; Chin and Wang, 2016b; Costa-Silva et al., 2015; Peinado et al., 2012; Peinado et al., 2011), we sought to examine the putative anti-metastatic role of this pathway. First, we evaluated its status in human melanoma. Extracellular vesicles isolated from melanoma patients (but not healthy donors) decreased CH25H expression in human cells in vitro (Figure 6a). In addition, leukocytes from melanoma patients with metastatic disease expressed less CH25H than healthy donors or melanoma patients without metastases (Figure 6b). Furthermore, melanoma patients expressing lower CH25H than the average level in healthy donors had increased occurrence of distant metastases (Figure 6c) and displayed a poor survival (Figure 6d). These results suggest that downregulation of CH25H in normal cells may breach the CH25H-mediated defenses against melanoma progression and metastases.
Figure 6: The IFNAR1-CH25H pathway protects against melanoma progression and metastasis.

A. qPCR analysis of CH25H mRNA levels in THP-1 cells treated with extracellular vesicles from healthy donors (HDEV, n=8) or melanoma patients (MPEV, n=13).
B. qPCR analysis of CH25H mRNA levels in blood leukocytes from healthy donors (HD, n=10) or melanoma patients with (MET, n=17) or without (Non-MET, n=20) documented metastatic disease.
C. Documented distant metastases in MP whose lymphocytes expressed greater (CH25Hhigh) or lower (CH25Hlow) levels of CH25H mRNA compared to healthy donors (HD); Fisher’s exact test: p value = 0.000661
D. Kaplan-Meier analysis of survival of melanoma patients classified as CH25Hhigh or CH25Hlow
E. Representative H&E staining of lungs from indicated mice harvested 30 days after resection of s.c. B16F10 tumors of ~200 mm2. Examples of metastatic lesions are pointed by arrowheads.
F. Quantification of total area of metastatic load in lungs from mice of indicated genotypes from panel
G. Kaplan-Meier analysis of cancer-related survival of indicated mice that underwent resection of B16F10 tumors as described in panel E.
Data are represented as mean ± SEM; p values: * p<0.05; ** p<0.01; *** p<0.001; NS, not significant from Student’s t test (panels A, B, F), Fisher test (panel C) or log-rank test (panels D, G).
See also Figure S6
To further examine this possibility we used a syngeneic metastatic tumor model in mice harboring WT or SA IFNAR1. In these experiments, we did not administer additional TEV but rather relied on TEV production by subcutaneous B16F10 tumors, which were shown to generate abundant endogenous TEV (Liu et al., 2016; Peinado et al., 2012). Many more of disseminated B16F10-TdTomato cells were found in the lungs of WT mice compared to SA animals (Figure S6a-b) indicating the importance of IFNAR1 inactivation for efficient metastatic process.
A delayed growth of primary B16F10 tumors observed in SA mice (Figure S6c) was similar to other tumor models (Katlinskaya et al., 2016; Katlinski et al., 2017). This delay was partially alleviated by knockout of Ch25h in SA mice. Resection of tumors growing in SA mice was postponed until they reached a volume ~200 mm2 similar to that in other genotypes (Figure S6c). At that point, we excised primary tumors and examined post-surgical survival and appearance of lung metastases. Under these conditions, WT and Ch25h−/− mice developed massive lung metastatic disease (Figure 6e-f) that led to 100% lethality within a month (Figure 6g). When primary tumors of similar size were resected from SA mice, they displayed virtually no detectable lung metastases and a dramatic improvement in post-surgical survival (Figure 6e-f-g). These data further support the importance of IFNAR1 signaling in normal cells in protection against metastatic disease.
Remarkably, the ablation of Ch25h in SA mice completely reversed the suppression of lung metastases (Figure 6e-f) and abolished the extended survival (Figure 6g). This result suggest that CH25H acts epistatically downstream of IFNAR1 to elicit the defenses against tumor metastasis in normal cells. Given that the IFNAR1-CH25H pathway also protects these normal cells against TEV uptake (Figure 4b) and pre-metastatic niche-stimulating education by TEV (Figure 4e), it is plausible that preventing education by TEV may restore the defenses against metastases, for example, in the context of adjuvant or neoadjuvant therapies.
Thus, we examined the anti-metastatic activities of reserpine that can inhibit TEV uptake (Figure 5a-b), remediate the loss of IFNAR1 and Ch25h (Figures 5c and S5d) and disrupt formation of TEV-induced pre-metastatic niche (Figure 5d). Reserpine treatment of B16F10 cells in vitro did not dramatically affect their proliferation (Figure S7a) or characteristics of produced TEV (Figure S7b) or ability of these TEV to induce the myeloid infiltration in the lungs in vivo (Figure S7c).
We next assessed the effects of reserpine in the B16F10 tumor-bearing mice. Early studies reported that administration of reserpine inhibited tumor growth but attributed this effect to a severely tranquilized state and dramatic loss of weight due to a failure of sleeping animals to eat or drink for protracted periods of time (Belkin and Hardy, 1957; Burton et al., 1957). Here we used moderate reserpine doses, which were well tolerated and did not decrease animal weight throughout the experiments (Figure S7d). Importantly, reserpine treatment restored IFNAR1 levels in peripheral blood leukocytes (Figure 7a) and upregulated Ch25h and other ISGs in the lungs of tumor-bearing animals (Figure S7e) suggesting a successful disruption of education of normal cells by endogenously produced TEV.
Figure 7: Activity of reserpine in the context of adjuvant/neoadjuvant therapy.

A. Flow cytometry analysis of IFNAR1 cell surface levels on peripheral blood leukocytes from naive WT mice (n=4) or WT mice bearing similar sized B16F10 tumors treated with vehicle or reserpine (i.p. 1 mg/kg, 3x per week for 1 week; n=6 each).
B. Kaplan-Meier survival analysis of B16F10 tumor bearing mice treated with vehicle or reserpine (i.p. 1 mg/kg every 48 hr for 1 week)
C. Schematic description of experiments assessing activity of reserpine as neoadjuvant/adjuvant therapeutic agent
D. Kaplan-Meier analysis of cancer-related survival of mice described in panel C.
E. Quantification of total area of metastatic load in lungs from mice described in panel C (Vehicle n=12, Reserpine n=16).
F. H&E staining of lungs from mice treated with vehicle or reserpine as in panel C and harvested 30 days after surgery. Examples of metastatic lesions are pointed by arrowheads.
Data are represented as mean ± SEM; p values: * p<0.05; ** p<0.01; *** p<0.001 from Student’s t test (panels A, E) or log-rank test (panels B, D).
See also Figure S7
When administered alone, reserpine had rather modest effects on primary tumor growth (Figure S7f) and cancer-related survival (Figure 7b) indicating its limited utility as a single agent. We next examined the efficacy of reserpine in the context of adjuvant/neo-adjuvant therapy (Figure 7c). Primary tumors were grown to ~200 mm2 (Figure S7g) followed by their surgical removal and reserpine was administered before and after surgery (Figure 7c). Although recurrence of primary tumors and tumors growing in the lymph nodes (that were not removed during the surgery) forced the euthanasia of some of animals, the overall survival was significantly improved by reserpine (Figure 7d). Subsequent analysis showed that reserpine virtually eliminated lung metastases (Figure 7e-f) indicating a great potency of reserpine as an adjuvant therapeutic agent.
DISCUSSION
Data presented here suggest that IFNAR1-mediated expression of CH25H in healthy tissues limits the extent of education of these tissues by TEV and, accordingly, restricts the overall area of the pre-metastatic niche in the lungs. Accordingly, these results suggest a defensive role of the IFNAR1-CH25H pathway against development of distant lung metastases. Whereas limited analysis of leukocytes from melanoma patients is suggestive of association between the loss of CH25H and poor disease outcome, future extensive studies should determine the value of CH25H expression as potential prognostic biomarker in patients with melanoma and other highly metastatic cancers.
Among other signaling pathways, activation of p38 kinase was observed during the education of normal lung cells by TEV that encompass RNA and other damage-associated molecular patterns (Liu et al., 2016). Whereas activation of p38 is important for TEV-driven loss of IFNAR1, additional studies should characterize the nature of these diverse (and likely redundant) TEV-carried substances that elicit IFNAR1 inactivation. Given that CH25H is an IFN-inducible gene (Anggakusuma et al., 2015; Liu et al., 2013) and knockout of this gene abolishes the suppression of metastases in mice that maintain the expression of downregulation-resistant IFNAR1 mutant, it is likely that CH25H functions downstream of IFNAR1.
Accordingly, continuous exposure to TEV and TEV-induced loss of IFNAR1 may contribute to suppression of this defensive pathway in normal cells. Despite re-synthesis of IFNAR1, its rapid ubiquitination and degradation in cells, which encounter TEV before IFN, would further stimulate uptake of TEV and, thereby, perpetuate the positive feedback loop of IFNAR1/CH25H inactivation. In turn, this positive feedback would ensure continuous education of normal cells leading to generation of the metastatic niches and promoting cancer metastasis.
Conversely, enabling normal cells to restore IFNAR1 and respond to IFN by induction of CH25H is expected to interrupt the education by TEV. Indeed, IFN counteracts the effects of TEV on normal cells through maintaining CH25H expression and limiting TEV uptake. It remains to be determined, which of many types of TEV uptake (e.g. endocytosis, macropinocytosis, direct fusion, etc) are regulated by CH25H. Although CH25H product 25HC can act as a ligand for activating the nuclear Liver X Receptor (that can play anti-tumorigenic roles) a number of oxysterol-elicited effects were shown to be independent of the Liver X Receptor (reviewed in (Traversari et al., 2014)). Importantly, another ligand of this receptor, 27-hydroxycholesterol, was recently shown to promote the metastatic process through acting on immune cells (Baek et al., 2017). Notably, IFN did not induce the expression of Cyp27a, which catalyzes production of 27-hydroxycholesterol. Regardless of these considerations and, given that anti-tumorigenic agonists of the Liver X Receptor are being developed (Flaveny et al., 2015), future studies should determine its role in 25HC-dependent protection of normal cells against TEV education.
Furthermore, 25HC and reserpine may possess additional anti-tumorigenic activities unrelated to their ability to restrict TEV uptake. Although we did not observe that reserpine affects TEV production or ability of these TEV to educate normal cells, additional mechanisms of action for reserpine cannot be entirely ruled out. Independent of these considerations, our data suggest that reserpine (and perhaps other agents capable of disrupting TEV-induced education) can elicit potent adjuvant therapeutic activities.
Limited benefits of IFN in patients who already have developed distant metastases restricts application of IFN to the sphere of adjuvant/neoadjuvant therapy (Eggermont et al., 2014; Tarhini et al., 2012) aimed to disrupt the pre-metastatic niche and limit metastatic colonization. Our findings that protection of normal cells can be compromised by TEV-induced IFNAR1/CH25H downregulation argues for shifting the strategy towards IFNAR1- and CH25H-independent modalities such as a low-dose reserpine regimen. Given that metastasis is a progressive process involving continuous education of normal cells, reserpine might be relevant not only for preventing this process but also for restricting the ongoing metastatic expansion. Being approved by the FDA since 1955 and deemed tolerable for patients with conditions far less grave than metastatic cancer, the clinical efficacy of reserpine in the context of adjuvant melanoma therapy should be tested posthaste.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Serge Y. Fuchs (syfuchs@vet.upenn.edu)
EXPERIMENTAL MODEL and SUBJECT DETAILS
Animal Studies
All experiments with animals were carried out under the protocol 803995 approved by the Institutional Animal Care and Use Committee (IACUC) of The University of Pennsylvania. All mice were of C57Bl/6 background and had water and chow ad libitum. Mice were maintained in a specific pathogen-free facility in accordance with American Association for Laboratory Animal Science guidelines. Littermate animals from different cages were randomly assigned into the experimental groups. These randomized experimental cohorts were either co-housed or systematically exposed to other groups’ bedding to ensure equal exposure to all group’s microbiota. Ch25h−/− and Ifnar1−/− mice were obtained from the Jackson Laboratory. Littermate Ifnar1+/+ (“WT”) and Ifnar1S526A mice (“SA”) were described previously (Bhattacharya et al., 2014). Other strains were generated by intercrossing and the littermates of 6-8 weeks of age were used in the experiments.
Cell Culture
Human and mouse cell lines such as human monocytic leukemia THP-1 cells (ATCC®TIB-202 ™), human 1205Lu melanoma (ATCC® CRL-2812™), and mouse B16F10 melano ma (ATCC® CRL-6475™) were purchased from ATCC and cultured according to the ATCC recommendations. Mouse mammary adenocarcinoma EO7771 cell line was purchased from CH3 Biosystems and cultured according to this provider’s recommendations.
Human Samples
Informed consent was obtained from all study participants. Study approval was given by the Institutional Review Board of the University of Pittsburgh. Use of previously collected lymphocyte samples that could not be directly or indirectly linked to individual human subjects was conducted under the strict guidance of the institutional review at the University of Pittsburgh Medical Center.
METHOD DETAILS
Analysis of human samples.
The cell surface IFNAR1 levels on lymphocytes from healthy donors or patients from two independent cohorts of melanoma patients were analyzed by flow cytometry. The MP1 cohort consisted of 40 patients with melanoma (stage I, II, III or IV). The MP2 cohort was described in details elsewhere (Tarhini et al., 2017)). These patients had a sentinel lymph node involved (SLN+, n=20) or not (SLN−, n=20) by melanoma metastasis as assessed by H&E and immunohistochemistry. Combined samples from patients from both cohorts (minus two patient samples lacking sufficient amount of material to extract RNA) were analyzed for CH25H. Peripheral blood was aseptically collected into (green top) Na Heparin vacutainer tubes from healthy donors and melanoma patients. The blood was then stored at room temperature (16-30°C) and processed as follows. Whole blood was diluted (1:2) in PBS and gently mixed before adding 10 mL of Lymphocyte Separation media. The sample was centrifuged for 5 min at 1000 rpm and without stopping the speed was increased to 1800-2000 rpm for 20 min with the brake off. The peripheral blood leukocyte layer between the clear lymphocyte separation media and upper PBS layer was gently pipetted off.
The cells were washed with PBS three times and suspended in media (Iscov’s Modified Dulbecco’s Medium, 10% Human Serum, 1% Pen-Strep solution, 1% L-Glutamine, 1% MEM non-essential amino acids 10mM, 1% HEPES Buffer solution 1M) with 10% DMSO for incubation at −80°C overnight before long term storage at −140°C. For recovery, frozen cells were thawed rapidly (< 1 minute) in a 37°C water bath by gently swishing the tube in the water without submerging. The thawed cells were slowly, but immediately, transferred into 10 ml of PBS. The cells were twice washed with PBS before flow cytometry analyses or RNA extraction.
Blood samples were collected from patients with melanoma (n = 13) or healthy volunteers (n = 8) to isolate extracellular vesicles from plasma. Blood specimens were immediately processed by centrifugation at 1,000 × g for 10 min at room temperature and plasma was separated and frozen in aliquots at −80°C. Frozen plasma samples were thawed and centrifuged at 2,000 × g for 10 min at room temperature and at 14,000 × g for 30 min at 4°C for thrombocyte removal. Centrifuged plasma was applied to a 0.22 μm-pore Millipore filter. Extracellular vesicle isolation was performed on mini-size exclusion chromatography columns as previously described (Hong et al., 2016). Briefly, plastic columns (Bio-Rad Econo-Pac columns, 1.5 cm x 12 cm) were packed with 10ml Sepharose 2B including a frit on top (Sigma-Aldrich, St. Louis, MO, USA) and washed with phosphate buffered saline (PBS). Clarified plasma (1mL) was loaded on top of the column and then eluted with 1mL PBS per fraction. Fraction 4 was used for further investigations. Detailed characterization of these extracellular vesicles is described elsewhere (Sharma et al., 2018).
Extracellular vesicles isolation from cell culture and labeling.
The media from 80% confluent cells grown in Extracellular Vesicle-free (EV-free) was collected. The cancer cell extracellular vesicles were separated via ultracentrifugation as previously described (Thery et al., 2006). Extracellular vesicles were characterized in size and number by the Characterization Core Facility in the University of North Carolina at Chapel Hill while corresponding images were taken by Analytical and Nanofabrication Laboratory in the University of North Carolina at Chapel Hill using TEM JEOL 100CX II scope. Protein concentration was assessed using the Pierce BCA Protein Assay Kit. Extracellular vesicles were stored at −80°C for long term storage and thawed on ice before use. For labelling, extracellular vesicles were suspended in sterile PBS solution containing 5 μg/mL of the lipophilic dye DiD (Biotium Cat# 60014) dissolved in DMSO, incubated at 37°C for 10 min, transferred into ultracentrifugation tubes and washed twice with 35 mL of sterile PBS and re-suspended in PBS.
Plasma Membrane Profiling Proteomic Screen.
Plasma Membrane Profiling uses selective aminooxy-biotinylation and quantitative proteomics to compare the expression of cell surface receptors in experimental and control cells, and was performed as previously described (Matheson et al., 2015; Weekes et al., 2012; Weekes et al., 2013; Weekes et al., 2014). Briefly, for Stable Isotope Labelling with Amino Acids in Cell Culture (SILAC)-based quantitation, THP-1 cells were pre-labeled for 7 days with light, medium or heavy isotopes of lysine and arginine (Cambridge Isotope Laboratories). Cells were then treated with either PBS or 1205Lu TEV (50 μg per 2×107 cells for 2 hr), washed in ice-cold PBS and mixed in a 1:1 ratio. For aminooxy-biotinylation of plasma membrane proteins, cells were resuspended in a “one pot” ami nooxy-biotinylation mix comprising 1mM sodium meta-periodate (Thermo Scientific), 100 μM aminooxy-biot in (Biotium) and 10mM aniline (Sigma) in ice-cold PBS pH 6.7 and incubated for 30 min in the dark. After lysis in 1% Triton X-100 (Thermo Scientific), biotinylated proteins were captured on High Capacity Streptavidin Agarose Resin (Thermo Scientific), stringently washed, reduced and alkylated prior to on-bead digestion with trypsin (Promega). Eluted peptides were subjected to LC–MS/MS using an RSLCnano 3000 (Thermo Scientific) coupled to a Q Exactive (Thermo Scientific). MS data were acquired from m/z 400 to 1650 with MS/MS spectra acquired in a top 10 DDA fashion. Raw MS files were processed using MaxQuant 1.5.0.0 and searched against a UniProt Homo sapiens database (downloaded 03/03/2014). Oxidation (M), deamidation (N/Q) and acetylation (Protein N-terminus) were set as variable modifications and carbamidomethyl (C) as a fixed modification. Peptide re-quantify was enabled and quantitation utilized razor and unique peptides. Parallel experiments were conducted in triplicate, including a three-way SILAC label swap (TEV/PBS, replicate 1: heavy/light; replicate 2: medium/heavy; replicate 3: light/medium). For each protein, TEV/PBS abundance ratios were compared using a one-sample t-test (XLSTAT) with theoretical mean log2(ratio)=0.
Isolation of primary mouse splenocytes, fibroblasts, bone marrow cells and peripheral blood leukocytes.
Mice (3-5 week old) were euthanized and their spleens were immediately removed before forcibly traversed through a 70 μm cell strainer and washed with PBS. The samples were incubated with red blood cell lysis buffer and washed three times with PBS. The splenocytes were counted and seeded in suspension plates in complete media [RPMI with 10% EV-free FBS and 1:2000 dilution of cell culture grade β-mercaptoethanol] and allowed to rest for at least 4 hr and no more than 8 hr before experimental set up.
Lungs from these mice were aseptically removed, washed in ice-cold PBS, minced with a scalpel blade and incubated in dissociation solution (8 mL of 100 mg Collagenase II and 5 mg DnaseI in HBSS) at 37°C for 30–45 min. The collagenase reaction was quenched by diluting with equal volume of 100% EV-Free FBS. The dissociated tissue traversed a 100 μm cell strainer and the flow through was applied to a 40 μm cell strainer. The cells were washed twice with PBS before plating on a 15 cm tissue culture plate in antibiotic-free media (DMEM with 10% EV-free FBS). After 20-40 min, the non-adherent cells were removed; the remaining cells were washed thoroughly with PBS and left to rest in complete media (DMEM with 10% EV-Free FBS).
Bone marrow was flushed from femurs and tibia of these mice with PBS using 26 gauge needles. The cells were traversed a 70 μm cell strainer and then treated with red blood cell lysis buffer before washed and suspended in complete media (RPMI with 10% EV-Free FBS and 1:2000 dilution of cell culture grade β-mercaptoethanol). The bone marrow cells were seeded in suspension plates for immediate experimental use. Peripheral blood was aseptically collected from the mouse tail vein into (green top) Na Heparin capiject tubes. The blood samples were suspended in red blood cell lysis buffer and washed thoroughly with PBS prior to experimental use.
Cell treatment in vitro.
25-Hydroxycholesterol was dissolved in 100% Ethanol to make a 4 mM working stock solution. Cells were washed with PBS to remove residual media and suspended in serum-free media containing 4 μM of 25-Hydroxycholesterol. Reserpine and other drugs were dissolved in DMSO or water as indicated. For reserpine, a 100 μM stock solution was used. Cultured human and mouse cells were washed with PBS and suspended in complete media with 10 μM of Reserpine or DMSO alone for 2 hr.
For in vitro treatment with tumor derived extracellular vesicles, the cells were washed and counted to 1×106 cells per mL and suspended in appropriate media with EV-free FBS. For each mL, 10 μg of B16F10 TEV added for mouse cells and 20 μg of 1205Lu TEV to human cells. A 2 hr incubation time point was used to measure changes in IFNAR1 cell surface level via flow cytometry. The 4 hr incubation time point was used to analyze TEV uptake and prepare samples for qPCR.
For IFN pretreatment, human cells were treated with 1000 U/mL of human IFNα or IFNβ for 12 hr and were then given 20 μg of labeled 1205Lu TEV for 4 hr. Cells were then thoroughly washed with PBS and suspended in PBS with DAPI for flow cytometry analysis. Freshly isolated mouse splenocytes and bone marrow cells were seeded in suspension plates at 1×106 cells/mL and treated with 1000 U/mL with mouse IFNβ for 8 hr. The mouse cells were then treated with 10 μg of B16F10 TEV for 2 hr (with gentle mixing every 30 min) to examine changes in IFNAR1 level, or 4 hr to examine TEV uptake via flow cytometry.
Lung Tissue Dissociation for Flow Cytometry.
Lung tissues were collected from euthanized mice and washed with ice cold PBS. The lung tissues are then cut into small pieces using sharp dissection scissors. The tissue homogenate is then incubated in dissociation solution (3 ml/lung) (dissociation solution: 2 mg/ml Collagenase II, 1 mg/ml Collagenase D plus 100 μg/ml Dnase I solution) for around 1 hr with continuous agitation at 37 degree. The digested tissue was passed through 70 μm cell strainer and the digestion quenched with 10 ml 10% FBS RPMI medium. The cells were centrifuged prior to incubation with RBC lysis buffer for 5 min to remove red blood cells. The cells are washed three times with PBS before prepped for flow cytometry experiments.
EV and liposomes uptake.
For in vitro assays, cells were washed and counted to 1×106 cells per mL and re-suspended in appropriate media with EV-free FBS. For each mL, 10 μg of DiD-labeled B16F10 TEV for 4 h r or 1 μL of DiD-labeled liposomes were added for 2 hr. The cells were then washed thoroughly and analyzed by flow cytometry.
Short or long term experiments were used to analyze in vivo TEV Uptake. For short term experiments, 30 μg of DiD-labeled TEV were injected into the tail vein of 5 to 7 week old mice. Mice that received vehicle or Reserpine were injected 30 min prior to TEV tail vein injection. 24 hr after injection, the spleens and bone marrow were isolated from the euthanized mice for immediate analysis. For long term experiments, 8 μg of B16F10 TEV were injected into the tail veins of 5 to 7 week old mice three times a week for 3 weeks. Mice that received vehicle or reserpine were injected 30 min prior to each TEV tail vein injection. The tissues were collected 3 days after the final injection and splenocytes or bone marrow cells were analyzed by flow cytometry.
Cells that uptake EV and liposomes incorporate the DiD-dye into their plasma membranes, which allowed us to gate for DiD positive, DAPI negative cells using BD FACS Canto II (660/20 Red laser) or BD LSR Fortessa (670/30 Red laser) flow cytometers with BD FACS DIVA software. Comparison of DiD positivity was analyzed using FlowJo 10.4.1 (FlowJo, LLC, Ashland, OR).
For fluorescence, lungs collected from the bone marrow transfer recipients following survival surgery were cut in 8.0 μm sections onto glass slides. The sections were incubated in 1% paraformaldehyde in PBS for 3 min to fix tissue without quenching GFP and RFP signals. The tissues were washed four times with PBS before excess liquid removed and sections sealed with ProLong Gold Antifade Reagent with DAPI (Life Technologies) and glass coverslip.
For immunofluorescence, OCT stored tissues were cut into 8.0 μm sections onto glass slides and fixed in 100% acetone at −20°C for 10 min before allowed to dry at room temperature. The tissues were re-hydrated and washed with PBS. The samples were incubated in 0.2% Triton in PBS for 10 min at room temperature and washed 3 times in PBS before blocking in 5% Goat Serum in PBS. Tissue samples were incubated with primary antibodies overnight at 4°C and washed three times with PBS before incubated with respective secondary antibody at room temperature for 1 hr. ProLong Gold Antifade Reagent with DAPI was added. Non-confocal images were taken using the Olympus BX51 microscope. Confocal images were taken using Leica SP5-II FLIM.
For Flow Cytometry Analysis, cells were washed with PBS and incubated with human or mouse Fc block diluted in 0.5% BSA-PBS on ice for 10 min. Cells were then incubated with APC-conjugated anti-human CD45 antibody and anti-human AA3 IFNAR1 antibody (Goldman et al., 1999) 30 min on ice. FITC-anti-Rabbit Alexa Fluor antibody was added to bind AA3 for 30 min on ice. Cells were washed to remove unbound primary and secondary antibodies before suspended in PBS with DAPI for flow cytometry analysis on a LSR Fortessa (BD Biosciences) equipped with FACS DIVA software.
Mouse cells were washed with PBS and incubated with mouse Fc block diluted in 0.5% BSA-PBS. Afterward, cells were incubated with APC-conjugated anti-mouse CD45 and PE-conjugated anti-mouse IFNAR1. Cells were washed to remove unbound antibodies then suspended in PBS with DAPI for flow cytometry analysis on FACS Canto II and LSR Fortessa (BD Biosciences) equipped with FACS DIVA. In all cases, the changes in mean fluorescence intensity (ΔMFI) was calculated as follows:
Quantitative Real-Time PCR Analysis.
Total RNA was isolated from samples using Trizol Reagent and chloroform. RNA concentration and purity was measured using a NanoDrop spectrophotometer. Applied Biosystems High-Capacity RNA-to-cDNA Kit was used to make cDNA. The mRNA expression levels were measured by quantitative real-time PCR using SYBR Green Master Mix.
For immunoprecipitation and immunoblot analysis, THP-1 cells were grown to no more than 2×106 cells per mL with media changed every two days. Six hr before experiment, cells were seeded in suspension plates with fresh media. Cells treated with 1000 IU/mL of human IFNα-a2 or 20 μg/mL of 1205Lu TEV for 30 min or 2 hr with some gentle agitation every 30 min. The cells were transferred to pre-chilled conical tubes to remove media and wash cells with ice-cold PBS twice to removed media. The cell pellets were lysed using lysis buffer with protease and phosphatase inhibitors as previously described. Protein concentration detected using the Pierce BCA Protein Assay Kit. For immunoprecipitation, 1mg of lysate was pre-cleared with Protein A/G beads for one hour prior to overnight incubation with anti-IFNAR1 (EA12) antibody (Goldman et al., 1999) with fresh Protein A/G beads. The supernatant removed and the beads were washed four times with 1xTBS with 0.001% Tween-20. The beads were then boiled in 4xSDS loading dye with β-mercaptoethanol for 10 min at 98°C. The whole cell lysates and immunoprecipitation samples were separated on SDS PAGE, transferred onto PVDF membranes and analyzed as previously described (Bhattacharya et al., 2013; Bhattacharya et al., 2014).
Generating Bone Marrow Chimeric Animals.
5 week old mice were injected with 10 μg of B16F10 TEV or PBS into the tail vein three times a week for four weeks. The mice were euthanized and the bone marrow cells were flushed with PBS from the long bones. Recipient mice were irradiated with 900 rads and allowed to rest for 3 hr before injected with 500,000 bone marrow cells through the tail vein. The mice were given 4-6 weeks to recover post transplantation before being inoculated with B16F10 tumors followed by analysis of primary tumor growth and metastases.
In Vivo Subcutaneous Tumor Growth.
B16F10 cells at 60-70% confluence were lifted off the plate using 2mM EDTA in PBS. The lifted cells were washed with PBS and suspended in fresh PBS to 1×106 cells per 100 μL. The cells were kept on ice and 100 μL were injected subcutaneously into the flanks of mice. Tumors were measured every other day and resected when tumor volume reached approximately 200 mm2.
Reserpine Adjuvant Treatment.
Mice with s.c. B16F10 tumors that were approximately 30-50 mm2, began pre-surgical treatment with reserpine (1 mg/kg dissolved in ascorbic acid and diluted in ddH2O) or vehicle (0.1% ascorbic acid diluted in ddH2O) three times (every other day) for one week. Once the tumor reaches approximately 200 mm2, the tumors were resected. One week after surgery or upon healed sutures, the animals began weekly treatment with reserpine (or vehicle). When animals displayed signs of respiratory stress or became moribund, they were sacrificed and their lungs were analyzed for metastatic lesions.
Collection of Lungs for frozen sections or H&E.
Mice were euthanized and the lungs were gently perfused with ice cold PBS to wash out blood and prevent tissue collapse. For H&E analysis, the lungs were then perfused with 4% paraformaldehyde solution before processed for paraffin embedding. For fluorescence and immunofluorescence analysis, PBS perfused lungs were then perfused with 50% OCT solution before freezing in 100% OCT for cryosections.
Analysis of Tumor Number and Size in Lung Tissue.
The Leica DM6000 widefield microscope was used to take low magnification images to generate composite images of whole, H&E stained lungs. The composite images were opened in ImageJ to accentuate tumor-rich areas, remove background noise, and define size of neoplasm. Composite images of H&E stained primary tumor were used as controls to set ImageJ parameters. ImageJ then creates a mask that was superimposed to the original H&E pictures to verify areas that contained cancer cells. The ImageJ mask was then analyzed to count number of nodules and also to calculate area of lung with tumor burden.
QUANTIFICATION AND STATISTICAL ANALYSIS
All described results are representative of at least three independent experiments (n≥5 mice per group in each experiment unless specified otherwise). All in vitro analyses using cells or tissues from each of these animals were done at least in biological triplicates (e.g. samples from 3 tumors, 3 spleens, etc). Data are presented as average ± S.E.M. Statistical analysis was performed using Microsoft Excel (Microsoft) or GraphPad Prism 7 software (GraphPad Prism Software Inc). For analysis of proteomics data, For each protein, TEV/PBS abundance ratios were compared using a one-sample t-test (XLSTAT) with theoretical mean log2(ratio)=0. Unpaired Student t test was used for the comparison between two groups. One-way ANOVA or two-way ANOVA analysis followed by the Bonferroni post-hoc test were used for the multiple comparisons. Repeated-measure two-way ANOVA (mixed-model) followed by the Bonferroni post-hoc test was used for the analysis of tumor growth curve. The Kaplan-Meier curves were used to depict the survival function from lifetime data for mice and human patients; the Peto’s log-rank test was used to estimate the differences between the groups. The Student t-test or Fisher test were used for other comparisons. A value of p <0.05 was considered significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-phospho p38(T180/Y182) (28B10) | Cell Signaling | Cat#9216S |
| Rabbit monoclonal anti-phospho p38(T180/Y182) (28B10) | Cell Signaling | Cat#4631S |
| Rabbit monoclonal anti-phospho STAT1(Y701) (58D6) | Cell Signaling | Cat# 9167S |
| Rabbit monoclonal anti-STAT1 (9H2) | Cell Signaling | Cat#9176S |
| Rabbit monoclonal anti-βTRCP (D13F10) | Cell Signaling | Cat#4394S |
| Rabbit monoclonal anti-GAPDH (D16H11) | Cell Signaling | Cat#5174S |
| Rabbit monoclonal anti-β-Tubulin | Cell Signaling | Cat#2146S |
| Rabbit monoclonal anti- p38 (N-20) | Santa Cruz | Cat#SC-728 |
| Rat monoclonal anti-CD11b unconjugated (clone M1/70) | BioLegend | Cat#101202 |
| Mouse monoclonal anti-IFNAR1-PE (clone MAR1-5A3) | BioLegend | Cat#127311 |
| Mouse monoclonal anti-CD11b-FITC (clone M1/70) | BioLegend | Cat#101205 |
| Mouse monoclonal anti-CD45-APC (clone 30-F11) | BioLegend | Cat#103112 |
| Mouse monoclonal anti-Ubiquitin (FK2) | Millipore Sigma | Cat#ST1200 |
| Rabbit monoclonal anti-human IFNAR1 | Abcam | Cat#ab45172 |
| Rabbit polyclonal anti-Fibronectin | Abcam | Cat#23750 |
| Rabbit polyclonal anti-IFNAR1 | Sinobiological | Cat#50469-RP02 |
| mouse IgG | Leinco Technologies | cat# I-536 |
| Mouse monoclonal anti-mouse IFNAR1 (clone MAR1-5A3) | Leinco Technologies | cat# I-401 |
| Mouse monoclonal anti-human IFNAR1 (clone AA3) | (Goldman et al., 1999) | N/A |
| Mouse monoclonal anti-human IFNAR1 (clone EA12) | (Goldman et al., 1999) | N/A |
| Rabbit polyclonal anti-phosphoSer-IFNAR1 | (Bhattacharya et al., 2010; Bhattacharya et al., 2011) | N/A |
| Biological Samples | ||
| Human extracellular vesicles | This paper and (Sharma et al., 2018) | N/A |
| Human leukocytes | This paper and (Tarhini et al., 2017) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 25-hydroxycholesterol | Sigma-Aldrich | Cat# H1015 CAS#2140-46-7 |
| Reserpine | Sigma-Aldrich | Cat#83580 CAS#50-55-5 |
| Simvastatin | Sigma-Aldrich | Cat#S6196 CAS#79902-63-9 |
| Roglitone | Sigma-Aldrich | Cat#R2408 CAS#122320-73-4 |
| Toremifene | Sigma-Aldrich | Cat#T7204 CAS#89778-27-8 |
| Enclomiphene | Sigma-Aldrich | Cat#SML0719 CAS#14158-65-7 |
| Trifluoperazine | Sigma-Aldrich | Cat#T8516 CAS#79902-63-9 |
| Simvastatin | Sigma-Aldrich | Cat#S6196 CAS#440-17-5 |
| Latex beads, fluorescent yellow-green | Sigma-Aldrich | Cat#L5155 |
| LY294002 (p38 inhibitor) | Selleckchem | Cat# S1494 CAS# 862507-23-1 |
| Recombinant mouse IFNβ | PBL Assay | Cat#12401-1 |
| Human IFNβ (1a) | PBL Assay | Cat# 11415-1 |
| human IFNα (2a) | PBL Assay | Cat#11100-1 |
| Recombinant mouse IFNγ | BioLegend | Cat#575306 |
| DiD lipophilic dye | Biotium | Cat# 60014 CAS#127274-91-3 |
| DiD-labeled Liposomes (100 nanometers) | FormuMax Liposome Reagents | Cat#F60103F |
| Collagenase II | MP Biomedicals | Cat#1005002 CAS#9001-12-1 |
| Collagenase D | Roche Diagnostics | Cat#11088882001 |
| DNaseI, grade II | Roche Diagnostics | Cat#10104159001 |
| Critical Commercial Assays | ||
| Power Sybr Green PCR Master Mix | Applied Biosystems | Cat#4367659 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
| Pierce BCA Protein Assay Kit | ThermoFisher | Cat#23225 |
| Experimental Models: Cell Lines | ||
| Human: THP-1 monocytic leukemia | ATCC | Cat# TIB-202 |
| Human: 1205Lu melanoma | ATCC | Cat# CRL-2812 |
| Mouse: B16F10 melanoma | ATCC | Cat# CRL-6475 |
| Mouse: EO771 mammary adenocarcinoma | CH3 Biosystems | Cat#940001 |
| Experimental Models: Organisms/Strains | ||
| Mouse WT C57BL/6 | The Jackson Laboratory | Cat#000664 |
| Mouse: C57BL/6 Ch25h−/− | The Jackson Laboratory | Cat#016263 |
| Mouse: C57BL/6 Ifnar1S526A | (Bhattacharya et al., 2014; Yu et al., 2015) | N/A |
| Mouse: C57BL/6 Ifnar1S526A;Ch25h−/− | This paper | N/A |
| Mouse: C57BL/6 Mapk14f/f; Ubc9-CreERT | (Katlinski et al., 2017) | N/A |
| Oligonucleotides | ||
| Primers for qPCR and genotyping, see Table S3 | This paper | N/A |
| Software and Algorithms | ||
| FlowJo software version 10 | FlowJo Software | https://www.flowjo.com/ |
| GraphPad Prism software | GraphPad Software | http://www.graphpad.com |
| Microsoft Excel 2010 | Microsoft Office | N/A |
Highlights.
Tumor-derived extracellular vesicles (TEV) downregulate IFNAR1 and CH25H
CH25H acts to restrict TEV uptake and limit the education of healthy cells
Downregulation of CH25H in normal cells promotes melanoma metastasis
Disruption of TEV uptake and education by reserpine elicits anti-metastatic effects
SIGNIFICANCE.
Education of normal cells by factors delivered via the tumor-derived vesicles promotes tumor growth, progression and metastasis. Results of this study uncover the key role of CH25H in defense against normal cells education by tumor-derived vesicles and, accordingly, against melanoma progression and metastatic disease. In addition, these data provide a proof-of-principle for interfering with tumor-derived extracellular vesicles uptake as a strategy for adjuvant treatment of metastatic melanoma and suggest the repurpose of reserpine for such application.
ACKNOWLEDGEMENTS
This work was supported by the NIH/NCI PO1 CA165997 grant (to C.K., J.A.D., and S.Y.F.), R01 grants CA092900, CA216936 and CA188575 (to S.Y.F. and H.R.), The Wellcome Trust (PRF 101835/Z/13/Z to P.J.L.), MRC (CSF MR/P008801/1 to N.J.M.) and NHSBT (WPA15-02 to N.J.M.)as well as by additional support from F32 CA206431 (to A.O.), T32 CA115299 (to S.B.), T32 CA009140 (to K.V.K.), the Pittsburgh-Essen-Partnership Program (to S.L.), Undergraduate Student Scholars Program in the Center for Molecular Studies in Digestive and Liver Disease, University of Pennsylvania (to S.H.) and the Merial Veterinary Scholars Program Fellowship (to V.H.). We thank Ze’ev Ronai and the members of Gabrilovich, Fuchs, Diehl, Koumenis, and Minn labs for critical suggestions
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and two tables and can be found with this article online.
DECLARATION OF INTERESTS: All authors declare no competing financial interests
REFERENCES
- Aleckovic M, and Kang Y (2015). Regulation of cancer metastasis by cell-free miRNAs. Biochim Biophys Acta 1855, 24–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggakusuma, Romero-Brey I, Berger C, Colpitts CC, Boldanova T, Engelmann M, Todt D, Perin PM, Behrendt P, Vondran FW, et al. (2015). Interferon-inducible cholesterol-25-hydroxylase restricts hepatitis C virus replication through blockage of membranous web formation. Hepatology 62, 702–714. [DOI] [PubMed] [Google Scholar]
- Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, Pillai RV, McDowell HB, Thompson JW, Dubois LG, et al. (2017). The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun 8, 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Sagi D, and Prives J (1983). Trifluoperazine, a calmodulin antagonist, inhibits muscle cell fusion. The Journal of cell biology 97, 1375–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, and Lyden D (2016). Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 30, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkin M, and Hardy WG (1957). Effect of reserpine and chlorpromazine on sarcoma 37. Science 125, 233–234. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, HuangFu WC, Dong G, Qian J, Baker DP, Karar J, Koumenis C, Diehl JA, and Fuchs SY (2013). Anti-tumorigenic effects of Type 1 interferon are subdued by integrated stress responses. Oncogene 32, 4214–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, HuangFu WC, Liu J, Veeranki S, Baker DP, Koumenis C, Diehl JA, and Fuchs SY (2010). Inducible priming phosphorylation promotes ligand-independent degradation of the IFNAR1 chain of type I interferon receptor. The Journal of biological chemistry 285, 2318–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Katlinski KV, Reichert M, Takano S, Brice A, Zhao B, Yu Q, Zheng H, Carbone CJ, Katlinskaya YV, et al. (2014). Triggering ubiquitination of IFNAR1 protects tissues from inflammatory injury. EMBO Mol Med 6, 384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Qian J, Tzimas C, Baker DP, Koumenis C, Diehl JA, and Fuchs SY (2011). Role of p38 protein kinase in the ligand-independent ubiquitination and down-regulation of the IFNAR1 chain of type I interferon receptor. The Journal of biological chemistry 286, 22069–22076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ, et al. (2013). The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman-Saint Victor C, Wiemann BZ, Ishwaran H, et al. (2014). Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 159, 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boriskin YS, Pecheur EI, and Polyak SJ (2006). Arbidol: a broad-spectrum antiviral that inhibits acute and chronic HCV infection. Virology journal 3, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton RM, Goldin A, Humphreys SR, and Venditti JM (1957). Antileukemic action of reserpine. Science 125, 156–157. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, and Weinberg RA (2011). A perspective on cancer cell metastasis. Science 331, 1559–1564. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Cheung HC, and Hunter E (1982). Interferon inhibits Sendai virus-induced cell fusion: an effect on cell membrane fluidity. Proc Natl Acad Sci U S A 79, 835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin AR, and Wang SE (2016a). Cancer-derived extracellular vesicles: the 'soil conditioner' in breast cancer metastasis? Cancer Metastasis Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin AR, and Wang SE (2016b). Cancer Tills the Premetastatic Field: Mechanistic Basis and Clinical Implications. Clin Cancer Res 22, 3725–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H, et al. (2015). Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 17, 816–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont AM, Spatz A, and Robert C (2014). Cutaneous melanoma. Lancet 383, 816–827. [DOI] [PubMed] [Google Scholar]
- Erickson JD, Eiden LE, and Hoffman BJ (1992). Expression cloning of a reserpine-sensitive vesicular monoamine transporter. Proc Natl Acad Sci U S A 89, 10993–10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaveny CA, Griffett K, El-Gendy Bel D, Kazantzis M, Sengupta M, Amelio AL, Chatterjee A, Walker J, Solt LA, Kamenecka TM, and Burris TP (2015). Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell 28, 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French KC, Antonyak MA, and Cerione RA (2017). Extracellular vesicle docking at the cellular port: Extracellular vesicle binding and uptake. Semin Cell Dev Biol 67, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E (1882). Das Sarkom des Uvealtractus. Graefe's Arch Ophthalmol, Wien XII. [Google Scholar]
- Fuchs SY (2013). Hope and fear for interferon: the receptor-centric outlook on the future of interferon therapy. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research 33, 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman LA, Zafari M, Cutrone EC, Dang A, Brickelmeier M, Runkel L, Benjamin CD, Ling LE, and Langer JA (1999). Characterization of antihuman IFNAR-1 monoclonal antibodies: epitope localization and functional analysis. J Interferon Cytokine Res 19, 15–26. [DOI] [PubMed] [Google Scholar]
- Huangfu WC, Qian J, Liu C, Liu J, Lokshin AE, Baker DP, Rui H, and Fuchs SY (2012). Inflammatory signaling compromises cell responses to interferon alpha. Oncogene 31, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R (2016). The biology and function of exosomes in cancer. J Clin Invest 126, 1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al. (2005). VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katlinskaya YV, Katlinski KV, Yu Q, Ortiz A, Beiting DP, Brice A, Davar D, Sanders C, Kirkwood JM, Rui H, et al. (2016). Suppression of Type I Interferon Signaling Overcomes Oncogene-Induced Senescence and Mediates Melanoma Development and Progression. Cell Rep 15, 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, Carbone CJ, Beiting DP, Girondo MA, Peck AR, et al. (2017). Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 31, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Deng YQ, Wang S, Ma F, Aliyari R, Huang XY, Zhang NN, Watanabe M, Dong HL, Liu P, et al. (2017). 25-Hydroxycholesterol Protects Host against Zika Virus Infection and Its Associated Microcephaly in a Mouse Model. Immunity 46, 446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Carvalho LP, Bhattacharya S, Carbone CJ, Kumar KG, Leu NA, Yau PM, Donald RG, Weiss MJ, Baker DP, et al. (2009). Mammalian casein kinase 1alpha and its leishmanial ortholog regulate stability of IFNAR1 and type I interferon signaling. Mol Cell Biol 29, 6401–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA, et al. (2013). Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, Huang B, Xu X, Zheng J, and Cao X (2016). Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell 30, 243–256. [DOI] [PubMed] [Google Scholar]
- Lobb RJ, Lima LG, and Moller A (2017). Exosomes: Key mediators of metastasis and pre-metastatic niche formation. Semin Cell Dev Biol 67, 3–10. [DOI] [PubMed] [Google Scholar]
- Luu B, and Moog C (1991). Oxysterols: biological activities and physicochemical studies. Biochimie 73, 1317–1320. [DOI] [PubMed] [Google Scholar]
- Matheson NJ, Sumner J, Wals K, Rapiteanu R, Weekes MP, Vigan R, Weinelt J, Schindler M, Antrobus R, Costa AS, et al. (2015). Cell Surface Proteomic Map of HIV Infection Reveals Antagonism of Amino Acid Metabolism by Vpu and Nef. Cell Host Microbe 18, 409–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendt M, Kamerkar S, Sugimoto H, McAndrews KM, Wu CC, Gagea M, Yang S, Blanko EVR, Peng Q, Ma X, et al. (2018). Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki R, Toriumi M, Fukumoto S, Miyamoto M, Fujita T, Tanaka K, and Takeuchi Y (1999). Thiazolidinediones inhibit osteoclast-like cell formation and bone resorption in vitro. Endocrinology 140, 5060–5065. [DOI] [PubMed] [Google Scholar]
- Paget S (1889). The distribution of secondary growths in cancer of the breast. Lancet 133, 571–573. [PubMed] [Google Scholar]
- Parker BS, Rautela J, and Hertzog PJ (2016). Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 16, 131–144. [DOI] [PubMed] [Google Scholar]
- Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, et al. (2012). Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 18, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H, Lavotshkin S, and Lyden D (2011). The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol 21, 139–146. [DOI] [PubMed] [Google Scholar]
- Platanias LC (2005). Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5, 375–386. [DOI] [PubMed] [Google Scholar]
- Qian J, Zheng H, Huangfu WC, Liu J, Carbone CJ, Leu NA, Baker DP, and Fuchs SY (2011). Pathogen recognition receptor signaling accelerates phosphorylation-dependent degradation of IFNAR1. PLoS Pathog 7, e1002065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Ludwig S, Muller L, Hong CS, Kirkwood JM, Ferrone S, and Whiteside TL (2018). Immunoaffinity-based isolation of melanoma cell-derived exosomes from plasma of patients with melanoma. J Extracell Vesicles 7, 1435138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Schornberg KL, Delos SE, Scully C, Pajouhesh H, Olinger GG, Johansen LM, and White JM (2013). Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection. PloS one 8, e56265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeg PS (2016). Targeting metastasis. Nat Rev Cancer 16, 201–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarhini AA, Floros T, Lin HM, Lin Y, Rahman Z, Ashraf M, Vallabhaneni P, Sander C, Rao UNM, Panelli M, et al. (2017). A unique gene expression signature is significantly differentially expressed in tumor-positive or tumor-negative sentinel lymph nodes in patients with melanoma. Melanoma Res 27, 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarhini AA, Gogas H, and Kirkwood JM (2012). IFN-alpha in the treatment of melanoma. J Immunol 189, 3789–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traversari C, Sozzani S, Steffensen KR, and Russo V (2014). LXR-dependent and -independent effects of oxysterols on immunity and tumor growth. Eur J Immunol 44, 1896–1903. [DOI] [PubMed] [Google Scholar]
- Weekes MP, Antrobus R, Talbot S, Hor S, Simecek N, Smith DL, Bloor S, Randow F, and Lehner PJ (2012). Proteomic plasma membrane profiling reveals an essential role for gp96 in the cell surface expression of LDLR family members, including the LDL receptor and LRP6. J Proteome Res 11, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekes MP, Tan SY, Poole E, Talbot S, Antrobus R, Smith DL, Montag C, Gygi SP, Sinclair JH, and Lehner PJ (2013). Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science 340, 199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang EC, Aicheler R, Murrell I, Wilkinson GW, et al. (2014). Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157, 1460–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Otsuka F, Mukai T, Yamanaka R, Otani H, Matsumoto Y, Nakamura E, Takano M, Sada KE, and Makino H (2010). Simvastatin inhibits osteoclast differentiation induced by bone morphogenetic protein-2 and RANKL through regulating MAPK, AKT and Src signaling. Regulatory peptides 162, 99–108. [DOI] [PubMed] [Google Scholar]
- Yu Q, Zhao B, Gui J, Katlinski KV, Brice A, Gao Y, Li C, Kushner JA, Koumenis C, Diehl JA, and Fuchs SY (2015). Type I interferons mediate pancreatic toxicities of PERK inhibition. Proc Natl Acad Sci U S A 112, 15420–15425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.