

The nuclear factor kappa B (NF-κB) family of transcription factors plays a central role in coordinating the expression of genes that control inflammation, immune responses, cell proliferation, and a variety of other biological processes. In an attempt to identify novel regulators of this pathway, we performed whole-genome RNA interference (RNAi) screens in physiologically relevant human macrophages in response to lipopolysaccharide and tumor necrosis factor alpha (TNF-α).
KEYWORDS: NF-κB, RNAi screen, transcriptional regulation
ABSTRACT
The nuclear factor kappa B (NF-κB) family of transcription factors plays a central role in coordinating the expression of genes that control inflammation, immune responses, cell proliferation, and a variety of other biological processes. In an attempt to identify novel regulators of this pathway, we performed whole-genome RNA interference (RNAi) screens in physiologically relevant human macrophages in response to lipopolysaccharide and tumor necrosis factor alpha (TNF-α). The top hit was SNW1, a splicing factor and transcriptional coactivator. SNW1 does not regulate the cytoplasmic components of the NF-κB pathway but complexes with the NF-κB heterodimer in the nucleus for transcriptional activation. We show that SNW1 detaches from its splicing complex (formed with SNRNP200 and SNRNP220) upon NF-κB activation and binds to NF-κB’s transcriptional elongation partner p-TEFb. We also show that SNW1 is indispensable for the transcriptional elongation of NF-κB target genes such as the interleukin 8 (IL-8) and TNF genes. SNW1 is a unique protein previously shown to be involved in both splicing and transcription, and in this case, its role involves binding to the NF-κB–p-TEFb complex to facilitate transcriptional elongation of some NF-κB target genes.
INTRODUCTION
Ever since its discovery in 1986 in David Baltimore’s laboratory (1), the NF-κB pathway has been a prime model of inducible transcription in various cell types and in response to multiple stimuli. NF-κB family members in mammals contain five Rel proteins, p50, p52, p65 (RelA), RelB, and c-Rel, which form homo- and heterodimeric complexes (2). In the classical NF-κB signaling pathway, p65 and p50 subunits heterodimerize and are sequestered in an inactive complex in the cytoplasm bound to IκBα (3). Upon activation by proinflammatory stimuli, such as tumor necrosis factor alpha (TNF-α) or lipopolysaccharide (LPS), the IκB kinase (IKK) complex, which is composed of two functionally nonredundant kinases, IKK1 and IKK2 (4); a regulatory subunit, NEMO; and ELKS (5, 6), phosphorylates IκBα at serines 32 and 36 (7), targeting it for ubiquitination and proteasomal degradation (8). This allows the p65/p50 complex to translocate into the nucleus, where p65 binds to the promoter regions of target genes, recruits general transcription machinery, and induces the expression of the corresponding mRNA. One of NF-κB’s target genes is the IκBα gene itself, which acts in an autoregulatory feedback loop to repress NF-κB activity (9).
The NF-κB pathway plays an important role in human physiology. In innate immunity, the pathway is downstream of multiple Toll-like receptors (TLRs) and is activated in response to various pathogenic stimuli, including bacterial lipids (TLR1, -2, -4, and -6) and viral or bacterial nucleic acids (TLR3, -7, -8, and -9). The activation of the pathway leads to the upregulation of proinflammatory proteins, for example, proteins that help in leukocyte migration (VCAM1 and ICAM1), cytokines (TNF-α, interleukin-1 [IL-1], and IL-6), and defensins, etc. In adaptive immunity, NF-κB has been shown to play a role in B- and T-cell proliferation (10) along with dendritic cell maturation. Finally, NF-κB has also been implicated in the development of the liver, skin, and limbs (11, 12). Hence, it is not a surprise that the misregulation of this pathway (via mutations or aberrant activation) plays an important and driving role in a number of diseases, such as rheumatoid arthritis and atherosclerosis (13).
In cancers of hematopoietic origins, members of the NF-κB pathway are found to be mutated. For example, some non-Hodgkin’s B-cell lymphomas have amplification and rearrangement of c-Rel. Also, NF-κB2/p100 is frequently activated through chromosomal translocations in lymphomas and leukemias (14). The NF-κB pathway is known to be active in multiple solid tumors, including glioblastoma (15) and lung cancer (16). This leads to a chronic inflammatory phenotype, which contributes to genomic instability that drives tumor development (14).
Despite rigorous study of NF-κB due to its ubiquity and importance, several unanswered questions remain about this pathway. The canonical NF-κB heterodimer (p65-p50) responds to multiple stimuli and is activated by the same signal transduction steps (phosphorylation and degradation of IκBα, translocation of the heterodimer into the nucleus, binding to CBP/p300 [17], and recruitment of the general transcription machinery). However, the genes transcribed by the pathway differ significantly based on the stimulus, timing, and cell type. Observations like these provide a hint that there are many layers (and, hence, permutations and combinations) in the nuclear regulation of NF-κB target gene transcription. What are the cofactors that bind to NF-κB in the nucleus? What is the order of events of NF-κB-dependent transcription for different genes? How are stimulus-specific signals relayed to the nucleus to cause differences in the genes transcribed? With all these questions in mind, we used an RNA interference (RNAi)-based screening approach in a human monocytic cell line (THP-1 cells differentiated into macrophages) to identify novel genes that regulate the NF-κB pathway in response to two different stimuli (LPS and TNF-α). In follow-up experiments, we validated the splicing factor and transcription coactivator SNW1 as a novel regulator of NF-κB target gene transcription.
RESULTS
SNW1 is required for NF-κB activation in response to TNF-α and LPS.
We developed and optimized three levels of screening (Fig. 1) to stringently filter genes required for the activation of the NF-κB pathway in response to both LPS and TNF-α. The primary screen (Fig. 1A) was a high-throughput luciferase-based assay using a whole-genome small interfering RNA (siRNA) library (4 pooled siRNAs/gene). Differentiated THP-1 human myeloid cells (18) stably transduced with a 5×-NF-κB–luciferase reporter were reverse transfected with siRNAs for 72 h and then treated with TNF-α or LPS at 10 ng/ml for 6 h. The efficacy of the screen was validated by luciferase analysis of siRNA against p65 (sip65), siRNA against luciferase (siLuciferase), and mock controls added to each plate. The LPS and TNF-α screens were carried out in duplicates, and we calculated the Z-score for each gene (twice each for LPS and TNF-α) based on the medians and median absolute deviations of the luciferase values. A stringent Z-score cutoff of less than −2.0 was used for each replicate for both the LPS and TNF-α screens. A fraction of known NF-κB pathway regulators controlling the response to LPS or TNF-α were identified from this primary screen, establishing its validity (Fig. 2). The TNF-α and LPS screens gave 232 and 104 hits, respectively, with 43 of them being common, including known regulators such as RELA, NEMO, and UBC (19). Stimulus-specific mediators, such as TNF receptor (TNFR) for TNF-α and TLR4 for LPS, were also correctly identified.
FIG 1.

Three levels of screening for novel regulators of NF-κB regulation. (A) Primary screen where siRNA oligonucleotides (4 for every gene and 1 gene per well) were prearrayed into 384-well plates. THP-1 macrophages with the NF-κB reporter were reverse transfected for 72 h before being treated with TNF-α or LPS for 6 h and assayed for luciferase activity. Each whole-genome library was assayed in duplicate for each of the two stimuli. (B) Secondary screen where the top hits from panel A were screened again using TNF-α and LPS, with a test for cell viability to rule out essential genes from the final hit list (n = 3 replicates). (C) Tertiary screen where hits from panel B were validated using an endogenous NF-κB reporting system in response to TNF-α (COX2 protein readout, measured via Western blotting and subsequent ImageJ analysis) in U87 cells (n = 2 replicates).
FIG 2.

TNF-α and LPS primary screens. (A) In the TNF-α primary screen, most genes have a Z-score of between −2 and +2, as evident from the smooth line in that interval. Only 232 genes have a Z-score lower than the cutoff of −2 (marked by the dashed line). They include known genes for NF-κB regulators, such as NEMO and p65, and the TNF-α-specific regulator TNF receptor 1 (TNFR1). (B) Correspondingly, in the LPS primary screen, most genes also have a Z-score of between −2 and +2, as evident from the smooth line in that interval. Only 104 genes have a Z-score lower than the cutoff of −2 (marked by the dashed line). They include genes for known NF-κB regulators, such as NEMO and p65, and the LPS-specific regulator Toll-like receptor 4 (TLR4).
Next, we conducted a secondary screen on these hits (Fig. 1B), with an additional measure of cell viability to cull false-positive results from the primary screen by excluding genes that reduced cell viability as opposed to NF-κB activity. We used redundant siRNA analysis (20), which is an accepted statistical method to interpret data from large-scale RNAi screens while minimizing off-target effects. In particular, we were looking for genes whose siRNAs (at least 2 out of 4) showed significant reductions in luciferase activity upon both TNF-α and LPS treatments and did not cause a significant loss of cell viability. We report the results of the screens in Tables SA1 and SA2 in the supplemental material.
Finally, we validated our hits using an endogenous readout of NF-κB activity in a different cell line (Fig. 1C). Here, we used the U87 glioblastoma-like cell line that has an inducible NF-κB pathway (15) and can be transfected with siRNAs at very high efficiency. We used the two most active siRNAs for every gene (identified from the secondary screen) and tested the induction of the NF-κB-inducible molecule COX2 in response to TNF-α treatment by immunoblotting for the COX2 protein. siRNAs against p65, TAK1, and NEMO were used as positive controls. The protein readouts were normalized to actin (loading control). The final list of 14 genes validated by three levels of screening (Table SA3) was headed by the splicing factor and transcription coactivator SNW1.
To confirm the initial screening results, we replicated the primary screening process for SNW1 by transfecting differentiated THP-1 cells (NF-κB luciferase reporter) with sip65, siLuciferase, siSNW1, and a nontargeting siRNA. After stimulation with TNF-α or LPS (Fig. 3A and B), we saw a significant knockdown of relative luciferase activity with siSNW1 along with the positive controls sip65 and siLuciferase. Next, we replicated the secondary screening conditions for SNW1 by transfecting differentiated THP-1 cells (NF-κB luciferase reporter) with sip65, siLuciferase, siDeath (a positive control for cell toxicity that targets essential protein translation genes), and the four siRNAs against SNW1. Upon stimulation with either LPS or TNF-α, we observed that three out of four siRNAs against SNW1 significantly knocked down NF-κB-dependent luciferase activity, comparable to the levels for the positive controls (Fig. 3C to E). Also, none of the siRNAs against SNW1, p65, and luciferase showed significant cell toxicity, but siDeath killed almost 90% of the transfected cells. Next, to replicate the tertiary screen for SNW1, we transfected U87 cells with siSNW1 or a nontargeting siRNA and activated the NF-κB pathway by treating them with TNF-α. NF-κB activation induced the production of COX2 up to 12-fold, while knockdown of SNW1 almost completely attenuated COX2 protein expression (Fig. 3F). This confirmed SNW1’s involvement in NF-κB activation in another cell line using an endogenous target gene as a reporter. We also tested SNW1’s involvement in the transcription of other NF-κB target genes by using quantitative PCR (qPCR). Knockdown of SNW1 in differentiated THP-1 cells reduces the TNF-α-induced expression of the mRNAs for the NF-κB target genes IκBα gene, IL-1 gene, CCL2 gene, and IL-6 gene after 60 min of TNF-α stimulation (Fig. 3G). The knockdown of SNW1 mRNA upon siRNA transfection was also verified (Fig. 3H).
FIG 3.

SNW1 depletion attenuates NF-κB pathway activation in multiple cell lines, with minimal loss of cell viability. (A and B) Reduction of activated NF-κB-dependent luciferase activity (via TNF-α or LPS) under SNW1 RNAi (pool of 4 siRNAs) conditions in THP-1 cells (P < 0.05). The reduction is comparable to those with positive controls (RNAi against p65 or luciferase) (n = 2 replicates). RLuc, Renilla luciferase. (C and D) Three out of four individual siRNAs against SNW1 used in our secondary screen attenuated the NF-κB-dependent luciferase response upon stimulation with TNF-α or LPS (P < 0.05) (n = 3 replicates). (E) The reduction in NF-κB-dependent luciferase activity by siRNAs against SNW1 is not due to a loss of cell viability. siRNA against an essential ribosomal protein (siDeath) was used as a positive control (n = 3 replicates). (F) TNF-α-stimulated NF-κB-dependent expression of COX2 protein is attenuated in U87 cells transfected with siRNA against SNW1 (n = 2 replicates [1 replicate is shown]). (G) Knockdown of SNW1 downregulates the expression of NF-κB target genes upon TNF-α treatment in THP-1 cells, as determined by qPCR (P < 0.05 for negative-control [–ve] siRNA versus other conditions for all genes). The degree of downregulation is similar to those under sip65 and siNEMO conditions (positive controls) (n = 3 replicates). Note that the y axis shows fold changes of NF-κB target gene expression with respect to the non-TNF-α-stimulated control. (H) siSNW1 transfection leads to a reduction of SNW1 mRNA levels (P < 0.05) (n = 3 replicates).
SNW1 is not a general transcriptional coactivator and specifically modulates the NF-κB pathway in response to multiple stimuli.
SNW1 (also termed NCoA62, SKIP in vertebrates, Prp45 in Saccharomyces cerevisiae, and BX42 in Drosophila melanogaster) is a highly conserved protein associated with splicing and transcription (21). Well-established roles of SNW1 in transcriptional regulation include its coregulatory effect on nuclear hormone receptors, such as the vitamin D receptor (22), androgen receptor (23), and retinoid X receptor, which it antagonistically regulates in association with the SIRT1 histone deacetylase (24). SNW1 also positively regulates HIV Tat transcription in vitro (25). To firmly establish SNW1 as a specific regulator of NF-κB, we needed to rule out its involvement in general, constitutive transcription. To test this, we transfected THP-1 cells with both an NF-κB-driven luciferase reporter and a pTK (weak constitutive promoter)-driven Renilla luciferase reporter (Fig. 4A). We knocked down SNW1 in these cells (along with a negative control) and read luminescence values for both luciferases. SNW1 knockdown decreased NF-κB-dependent luciferase expression but had no significant effect on Renilla luciferase expression. We repeated this experiment in U87 cells and obtained similar results (data not shown). These observations support the hypothesis that SNW1 is not involved in general transcription, as knockdown of SNW1 had no effect on constitutive transcription in multiple cell lines.
FIG 4.

SNW1 is not a general transcription coactivator and modulates the NF-κB pathway in response to multiple stimuli. (A) THP-1 cells (with negative-control siRNA or SNW1 siRNA) transfected with constitutively active pTK Renilla luciferase show no significant change in constitutively active Renilla luciferase activity under siSNW1 conditions. (B) Knockdown of the NF-κB pathway (via sip65) increases TNF-α-mediated apoptosis in THP-1 cells. A similar result is seen when SNW1 is knocked down, providing evidence for SNW1’s role in the NF-κB pathway upon TNF-α treatment (P < 0.05 for both sip65 and siSNW1). (C) THP-1 cells (transduced with NF-κB luciferase) were transfected with a negative-control siRNA (si–ve), siSNW1, or sip65 and stimulated for 6 h with the above-mentioned stimuli. siRNA against SNW1 attenuates NF-κB-driven luciferase activity, while siRNA against p65 shows a similar effect with all stimuli (P < 0.05 for negative-control siRNA versus any condition for all stimulants tested).
Furthermore, we tested SNW1’s specificity in regulating TNF-α-dependent NF-κB by testing its ability to mediate the suppression of TNF-α-induced apoptosis. TNF-α stimulation of cells is known to activate two opposing programs: the NF-κB pathway and programmed cell death (apoptosis). We previously established (26) that the TNF-α-induced prosurvival NF-κB pathway suppresses programmed cell death. Therefore, when the NF-κB pathway is attenuated, cells undergo apoptosis at a much higher rate due to the lack of inhibition of the programmed cell death pathway. Hence, our hypothesis was that SNW1 depletion would lead to NF-κB repression and, therefore, increased apoptosis in TNF-α-treated cells. This would provide evidence supporting that SNW1 directly and specifically affects the NF-κB pathway. To test this, we transfected THP-1 cells with siRNAs against p65 and SNW1 and a nontargeting control, treated the cells with TNF-α, and checked caspase 3/7 activity (measured by a luciferase assay) as a readout of apoptosis (Fig. 4B). As expected, apoptotic activity was increased in sip65-treated cells after TNF-α treatment but not in negative-control siRNA (scrambled siRNA)-treated cells. SNW1 depletion also increased apoptosis activity upon TNF-α treatment, supporting our hypothesis that SNW1 specifically regulates the NF-κB pathway.
Finally, we wanted to determine SNW1’s involvement in the NF-κB pathway in response to multiple stimuli. We therefore knocked down SNW1 in THP-1 cells (transduced with the NF-κB luciferase reporter) and treated the cells with a variety of inflammatory stimuli, including PAM3CSK4 (lipoprotein mimic) (TLR1 and TLR2 agonist), PGN (peptidoglycan) (TLR2 agonist), P2C (lipoprotein) (TLR2 agonist), FLG (flagellin) (TLR5 agonist), and IL-1 (cytokine) (IL-1 receptor [IL-1R] agonist). We found that knockdown of SNW1 reduced NF-κB activation from all stimuli (Fig. 4C). Since knockdown of SNW1 attenuated the NF-κB pathway induced by multiple stimuli, we hypothesized that it is not involved upstream of the IKK complex.
SNW1 affects the NF-κB pathway in the nucleus by forming a complex with the NF-κB transcription factor and p-TEFb.
Next, we wanted to explore the mechanism of SNW1’s involvement in the NF-κB pathway. We first checked its role in the cytoplasmic part of the pathway by knocking down SNW1 and probing for changes in the cytoplasmic arc of the NF-κB pathway, i.e., phosphorylation and degradation of IκBα along with nuclear translocation of NF-κB. SNW1 depletion did not affect any of the cytoplasmic processes (data not shown), and hence, to develop our hypothesis, we searched for SNW1’s potential interactions with proteins involved in NF-κB regulation in the nucleus. SNW1/SKIP is known to promote HIV-1 Tat transactivation by interacting with the elongation factor p-TEFb, which is composed of cyclin-dependent kinase 9 (CDK9) and cyclin T1 (27). p-TEFb was previously shown to be recruited to the promoters of NF-κB target genes (IL-8 gene, etc.) in response to TNF-α treatment (28). In the same study, p-TEFb was shown to bind to p65 (upon TNF-α treatment) to initiate transcriptional elongation and to help activate the preinitiation complex (PIC), which includes RNA polymerase II (Pol II). p-TEFb has also been shown to be bound to the promoters of rapidly transcribed NF-κB target genes upon pathway activation.
Hence, we tested the binding of SNW1 with p-TEFb upon TNF-α treatment in THP-1 cells (Fig. 5A). The results show that SNW1 is in the same complex as p-TEFb, even in untreated cells, and it maintains this interaction through stimulation with TNF-α. We also checked SNW1’s interaction with RNA Pol II and observed a considerable increase in the SNW1-Pol II interaction upon TNF-α treatment. Next, we wanted to check p65’s interaction with this complex, so we treated THP-1 cells with TNF-α, immunoprecipitated p65, and probed for proteins bound to it (Fig. 5B). As expected, p65 showed increased interactions with p-TEFb upon TNF-α treatment, and SNW1 and p65 were part of the same complex upon TNF-α treatment. We used IκBα and p50 as positive controls for p65 binding, while β-actin served as a negative control. Taken together, these results suggest SNW1’s involvement as part of the p65-pTEFb complex that cobinds to Pol II upon treatment with TNF-α.
FIG 5.

Both SNW1 and p65 complex with p-TEFb upon TNF-α treatment. THP-1 cells were treated with TNF-α for 30 or 60 min and immunoprecipitated for either SNW1 (A) or p65 (B). (A) Constitutive binding of p-TEFb (CDK9) to SNW1 inside THP-1 cells, with significantly increased binding to RNA Pol II, along with p65, upon TNF-α treatment. (B) Increased binding of p65 to p-TEFb and SNW1 upon treatment with TNF-α, as expected. The binding of IκBα and p50 to p65 is used as a positive control, while the lack of binding of actin (a ubiquitous highly expressed protein) to either SNW1 or p65 is used as a negative control. Note that for panel A, SNW1, Pol II, and p-TEFb were blotted on the same gel, while p65 and actin were blotted using the same samples but on a different gel (due to similar molecular weights of p65, SNW1, p-TEFb, and actin). Similarly, for panel B, p65, p-TEFb, and IκBα were blotted on the same gel, while SNW1, p50, and actin were blotted using the same samples but on a different gel.
SNW1 is necessary for Pol II-dependent elongation of NF-κB target genes IL-8 gene and TNF gene.
To investigate the role of SNW1 in the transcription of NF-κB target genes, we knocked down SNW1 in THP-1 cells, treated the cells with TNF-α, and immunoprecipitated DNA bound to p65, CDK9, and Pol II (Fig. 6). The antibody against Pol II was raised against the N terminus of its RPB1 subunit (to determine the recruitment of total Pol II). The results show that there was increased binding of p65, CDK9, and Pol II (as a percentage of the input) to the promoters of IL-8 and TNF genes upon treatment with TNF-α and that binding was not significantly affected by the depletion of SNW1 (Fig. 6A to F). In control experiments, we did not observe any change in the chromatin immunoprecipitation (ChIP) signal for the Pol III-driven U6 snRNA promoter (negative control), indicating that the TNF-α response is sequence specific. The binding of CDK9, p65, and Pol II to the U6 snRNA promoter was minimal (Fig. 7). These results show that SNW1 has no effect on the assembly of the preinitiation complex comprised of p65, p-TEFb, and Pol II on the promoter of NF-κB target genes, the IL-8 gene and TNF gene, upon treatment with TNF-α.
FIG 6.

SNW1 is necessary for Pol II-dependent elongation of NF-κB target genes but not for the formation of the preinitiation complex. THP-1 cells under normal or siSNW1 conditions were treated with TNF-α for 60 min and immunoprecipitated for chromatin that complexed with either CDK9, p65, or Pol II. (A to F) Recruitment of CDK9 (A), p65 (B), and Pol II (C) on the IL-8 promoter is increased significantly upon TNF-α treatment, and the effect remains unchanged in cells that have SNW1 depleted. (A to C) The corresponding effects are similar for the TNF promoter. (D to H) The recruitment of Pol II on the coding sequence region of the IL-8 gene is increased upon TNF-α treatment in normal cells, but the effect is significantly reduced in SNW1-depleted cells (G), which is also observed for the coding sequence region of the TNF gene (H). Note that panels A to F have statistically insignificant differences (at an alpha value of 0.05) between the negative-control siRNA (TNF) and siSNW1 (TNF) columns, while in panels G and H, the corresponding differences are statistically significant.
FIG 7.

Validation of CDK9, p65, and Pol II antibodies for ChIP using a negative-control U6 snRNA sequence. In THP-1 cells, CDK9 (A), p65 (B), and Pol II (C) antibodies bind minimally to the Pol III-driven U6 snRNA sequence (as evidenced by the low percentage of the input compared to that in Fig. 6), and the binding is unaffected by TNF-α treatment or under SNW1 depletion conditions (statistically insignificant at an alpha value of 0.05) (n = 3 replicates).
Since p-TEFb has been shown to be necessary for transcriptional elongation of NF-κB target genes (28), we investigated if knockdown of SNW1 had any effect on this activity by checking for Pol II recruitment in the coding regions of the NF-κB target genes IL-8 gene and TNF gene (Fig. 6G and H). The results showed an increased occupancy of Pol II on the coding regions of IL-8 and TNF genes upon TNF-α treatment, as expected, but importantly, this occupancy was significantly reduced upon SNW1 depletion. Cumulatively, these observations suggest that SNW1 depletion affects the ability of NF-κB and p-TEFb to stimulate the elongation of some NF-κB target genes while maintaining the preinitiation complex at these NF-κB target gene promoters. SNW1’s role in the transcriptional elongation of these NF-κB target genes is novel and showcases another level of control in NF-κB gene regulation.
SNW1’s role in NF-κB transcription may be independent of splicing.
We have provided evidence for SNW1’s role as an adapter protein between p-TEFb, p65, and Pol II that is necessary for NF-κB target gene elongation (IL-8 and TNF) upon TNF-α treatment. SNW1 is also a known splicing protein that binds with other splicing factors (SNRNP200, SNRNP220, and EFTUD2) to facilitate the splicing of certain genes (29). Interestingly, it has been known to act as a transcriptional coactivator for a target gene of the transcription factor p53 while acting as a splicing factor for another p53 target gene (30). Hence, we wanted to determine its possible role as a splicing factor in the context of NF-κB and TNF-α treatment.
Using THP-1 cells, we immunoprecipitated SNW1 with and without TNF-α treatment and probed for known splicing binding partners (Fig. 8). We observed binding of SNW1 to SNRNP200 and SNRNP220 under basal conditions; however, this binding was lost upon treatment with TNF-α. Interestingly, SNW1-SNRNP200 and SNW1-SNRNP220 binding was partially recovered after 60 min of TNF-α treatment. This shows that SNW1’s involvement in the NF-κB pathway is potentially independent of its splicing role. The fact that the NF-κB-driven luciferase construct used to conduct the primary and secondary screens did not have an intron in it further supports this claim.
FIG 8.
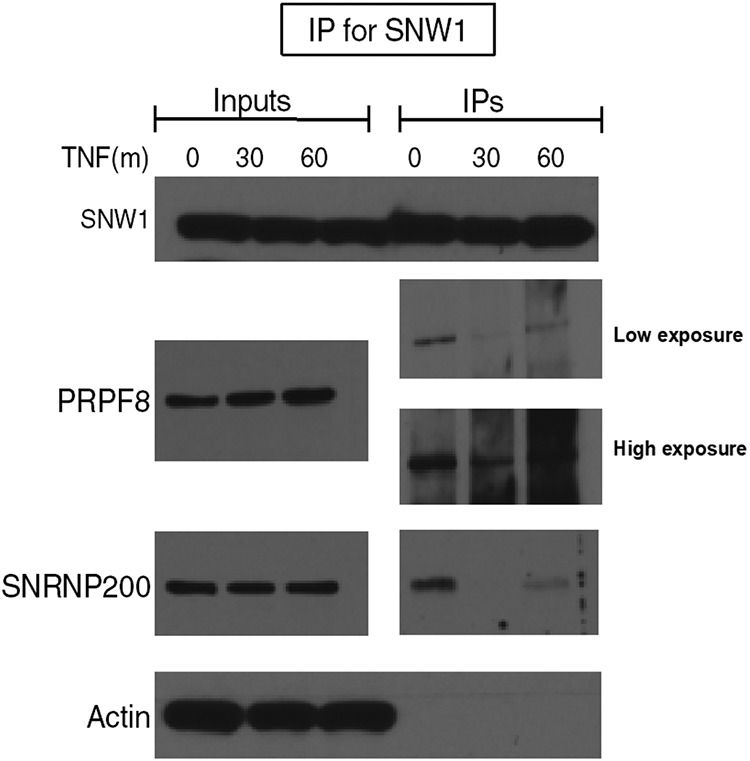
SNW1 binds to splicing factors in the basal (but not TNF-α-treated) state. THP-1 cells were treated with TNF-α for 30 or 60 min, immunoprecipitated for SNW1, and probed for its binding partners. The splicing factors PRPF8 (SNRNP220) and SNRNP200 appear to bind to SNW1 under basal conditions (absence of TNF-α) but lose that binding 30 min after TNF-α treatment, only to partially regain it 60 min after TNF-α treatment.
In total, we have provided evidence toward SNW1 being an adapter protein that is bound to splicing factors and p-TEFb under basal conditions, but upon treatment with TNF-α, it is bound to the NF-κB–p-TEFb–Pol II complex on the promoters of some NF-κB target genes. Our hypothesis is that SNW1 is involved in cotranscriptional splicing in the basal state (as it is bound to both the splicing factors SNRNP200 and SNRNP220 and the transcriptional elongator p-TEFb), but upon activation of the NF-κB pathway, it becomes indispensable for the gene elongation of some NF-κB target genes.
DISCUSSION
The NF-κB family of transcription factors responds to a plethora of diverse agents to transcriptionally activate a large number of target genes. Our RNAi screens identified SNW1 as a novel regulator of NF-κB pathway activation in human macrophages. SNW1 depletion led to a reduced expression of NF-κB target genes such as the IL-1, CCL2, and COX2 genes (Fig. 3G). SNW1 appears to regulate the NF-κB pathway in response to various stimuli (Fig. 4C). SNW1 depletion also led to increased apoptosis of THP-1 cells upon TNF-α treatment (Fig. 4B). This highlights the specificity of SNW1 as an NF-κB regulator in the context of TNF-α activation, since knocking down SNW1 affects only the NF-κB pathway and not the TNF-α-mediated activation of caspase, which leads to apoptosis (31). In a previous genome-wide pooled CRISPR-Cas9 library screen in dendritic cells to identify genes that control the induction of TNF by bacterial lipopolysaccharide, the SNW1 gene was among the top 10% of hits but was not further analyzed (32).
The next and most important question that we tried to answer was the following: what is SNW1’s role in the NF-κB pathway? The literature has shown that SNW1 functions as a splicing cofactor, transcription coactivator, and transcription repressor. In our cell lines, SNW1 (tagged with green fluorescent protein [GFP]) was always localized to the nucleus (data not shown), and hence, it was not a surprise when we found that it did not affect the cytoplasmic part of the NF-κB pathway (data not shown), including NF-κB’s translocation into the nucleus. The nature and role of NF-κB’s binding partners and transcriptional coactivators are still not completely characterized (with the exception of CBP/p300) (17). The p-TEFb protein complex was previously shown to interact with both SNW1 and p65, albeit independently, in the context of transcription. SNW1 was reported to bind p-TEFb in the context of transcriptional activation via HIV-1 Tat, while p65 was shown to use p-TEFb as a transcription elongator. Using this information as our basis, we tested for SNW1’s interaction with p-TEFb in the context of NF-κB activation.
We report that SNW1 constitutively binds to p-TEFb inside the nucleus with or without NF-κB activation (Fig. 5A). We also show that once NF-κB translocates into the nucleus upon TNF-α treatment, it interacts with the SNW1–p-TEFb complex (Fig. 5A).
SNW1 did not have any effect on the TNF-α-dependent assembly of Pol II, p-TEFb, and p65 on the promoters of NF-κB target genes, the IL-8 and TNF genes. SNW1 depletion, however, led to reduced NF-κB target gene elongation, shown by the reduced abundance of Pol II in the coding sequence regions of the IL-8 and TNF genes (Fig. 6G and H). Since SNW1 was known to be involved in splicing, we investigated its potential role in this activity in the absence and presence of TNF-α. We found that SNW1 binds to its splicing cofactors SNRNP200 and SNRNP220 in the basal state (no TNF-α treatment) but dissociates from them when the NF-κB pathway is activated (Fig. 8). Given that SNW1 was always found bound to p-TEFb (Fig. 5A), we hypothesize that in the basal state, SNW1 could be involved in cotranscriptional splicing, connecting nascent RNAs that are being transcribed, with the help of p-TEFb, to various splicing factors. Similar observations regarding SNW1 have been reported in multiple papers (27, 33), in the contexts of vitamin D- and HIV-1 Tat-driven transcription. Interestingly, upon activation of the NF-κB pathway, it dissociated from splicing partners and facilitated the formation of the NF-κB–p-TEFb complex for efficient transcriptional elongation of NF-κB target genes. This suggests that the impact of SNW1 on NF-κB activation is likely mediated through the regulation of transcription but not splicing. This hypothesis garners more support when coupled with the fact that the NF-κB luciferase construct used for the screening did not have an intron in it, and yet SNW1 affected its transcription.
Our study has some limitations: we do not know if the SNW1-p65 interaction upon TNF-α treatment (Fig. 5B) is direct or indirect. Presently, SNW1 knockout mice are not available, and our attempts to knock out SNW1 in 293T and THP-1 cells using CRIPSR-Cas9 failed due to lethal cell cycle defects in the knockout lines (data not shown). We also have limited knowledge about SNW1’s interactions with other binding partners of the NF-κB activation pathway. For example, we were unable to observe cobinding of SNW1 with either CBP/p300, SRC-1, or histone deacetylase 1 (HDAC1) (three known binding partners of p65) upon TNF-α treatment. We cannot discount the possibility that SNW1’s role in NF-κB transcription is independent of these proteins or that these interacting complexes are transient and unstable.
Going forward, we believe that there are several important questions that are still to be answered about SNW1’s role in the NF-κB pathway. First, it is important to determine if SNW1’s regulation of the NF-κB pathway is specific to certain target genes. We hypothesize that SNW1 depletion will lead to a significant decrease in p65 binding to the promoters of the p-TEFb-dependent NF-κB target genes (primary target genes that are not dependent on de novo protein synthesis for transcription) (34). It would be important to see how many and which genes are regulated in an SNW1-dependent manner following NF-κB activation using an RNA Pol II-specific ChIP sequencing method. Second, it will be important to understand the process by which SNW1 loses its binding with splicing factors and forms a complex with NF-κB and p-TEFb upon TNF-α treatment. One likely possibility is that SNW1 undergoes posttranslational modification (e.g., phosphorylation or acetylation) upon treatment with TNF-α, which drives its transition from being a splicing factor to being a transcriptional coactivator. Recently, it was shown that SNW1 undergoes c-Abl-mediated tyrosine phosphorylation to act as a transcription coactivator for the transforming growth factor β (TGF-β) pathway (35). It would also be interesting to know the status of splicing in cells treated with TNF-α, since we have observed that SNW1 dissociates from its splicing partners upon TNF-α treatment. Taken together, the results of this study reveal that SNW1 is a critical regulator of NF-κB-dependent transcriptional responses and is likely to impact a number of physiological responses governed by this pathway.
MATERIALS AND METHODS
Primary and secondary screens.
On day 0, THP-1 monocytes were treated with phorbol myristate acetate (PMA) (final concentration of 10 ng/ml). The cells were at a concentration of ∼250,000 cells per ml at the time of treatment. On day 1, all siRNAs were prearrayed in 384-well plates with 2 μl of a 1.25 μM stock (the primary screen had 4 siRNAs per gene per well, while the secondary screen had 1 siRNA per gene per well [hence 4 wells in total per gene per replicate]). A master stock containing 10 μl of prewarmed Opti-MEM and 0.1 μl Lipofectamine RNAiMAX for each well was prepared, incubated for 5 min at room temperature, and then added to each well. The plates were shaken for 1 min to generate a homogenous siRNA-lipid solution and incubated for 20 min at room temperature. A 50-μl suspension of 7,500 differentiated THP-1 cells in growth medium was added, and cells were incubated at 37°C with 5% CO2 for 72 h. On day 4, cells were stimulated with LPS or TNF-α diluted in growth medium to a final concentration of 10 ng/ml for 6 h each. Cells were then read using a plate reader and the Promega Bright-Glo and Cell Titer-Glo (secondary screen only) assay systems according to the manufacturer’s protocols.
Tertiary screen.
On day 0, U87 cells were seeded in 6-well plates at ∼50% confluence. On day 1, the seeded cells were transfected with individual siRNAs (2 siRNAs per gene and, hence, 2 wells per gene) using Lipofectamine RNAiMAX according to the manufacturer’s default protocol. On day 4, at 72 h posttransfection, the cells were stimulated with TNF-α at a final concentration of 10 ng/ml for 6 h. After 6 h of stimulation, cells were washed once with ice-cold phosphate-buffered saline (PBS) and collected in radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich) mixed with protease and phosphatase inhibitors (Cell Signaling). The cells were lysed, and protein was quantified and run on an SDS-PAGE system according to standard Western blotting protocols (http://www.abcam.com/protocols/general-western-blot-protocol) using the NuPage (ThermoFisher) reagents and apparatus. The gels used were 4 to 12% Bis-Tris NuPage gels, and the proteins were transferred to a polyvinylidene difluoride (PVDF) membrane. Primary antibodies were incubated overnight in 5% milk at a final concentration of 1:2,000, followed by secondary antibody incubation at a 1:10,000 dilution for 2 h at room temperature. The blots were developed using the Amersham ECL Western blotting detection reagent and X-ray films according to the protocol mentioned above.
qPCRs.
Primer sequences for IκBα, IL-1, CCL2, and IL-6 were sourced from PrimerBank (https://pga.mgh.harvard.edu/primerbank/). Total mRNA was extracted using TRIzol reagent (Invitrogen), qPCR was performed using SYBR green PCR master mix (Applied Biosystems), and mRNA expression levels were normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) level.
Caspase 3/7 activity.
The experiment to determine caspase 3/7 activity was performed using Promega’s Caspase-Glo 3/7 assay system (catalog number G8090) according to the manufacturer’s protocol.
Testing SNW1 as an NF-κB modulator in response to multiple stimuli.
The stimuli used to test SNW1 as an NF-κB modulator were PAM3CSK4 (catalog number tlrl-pms; InvivoGen), PGN (catalog number tlrl-pgnb3; InvivoGen), P2C (catalog number tlrl-pm2s-1; InvivoGen), FLG (catalog number tlrl-stfla; InvivoGen), IL-1 (catalog number 201-LB-005; R&D Systems).
Co-IP experiments.
For coimmunoprecipitation (co-IP) experiments, antibodies against p-TEFb (catalog number 2316; Cell Signaling), RNA Pol II (catalog number ab5095; Abcam), c-Myc (catalog number 9402; Cell Signaling), IκBα-p (catalog number 2859S; Cell Signaling), total IκBα (catalog number sc-7218; Santa Cruz), beta-actin, and p65 (catalog number sc-8008; Santa Cruz) were used.
ChIP-PCR experiments.
ChIP-PCR experiments were performed using the Active Motif ChIP-It high-sensitivity kit according to the manufacturer’s protocol. The IL-8 and TNF primers used were previously described (28, 36).
Supplementary Material
ACKNOWLEDGMENTS
We thank Tony Hunter for his help with the manuscript and Nina Tonu and Mark Schmitt for their technical and administrative help.
This work was supported by grants from the Leona B. Helmsley Charitable Trust to I.M.V. and from the NIH (R01 AI105184-04) to S.K.C.
We declare that we have no competing financial interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00415-18.
REFERENCES
- 1.Sen R, Baltimore D. 1986. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 46:705–716. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 2.Lawrence T. 2009. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. 1995. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev 9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 4.Liu F, Xia Y, Parker AS, Verma IM. 2012. IKK biology. Immunol Rev 246:239–253. doi: 10.1111/j.1600-065X.2012.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothwarf DM, Zandi E, Natoli G, Karin M. 1998. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 6.Ducut Sigala JL, Bottero V, Young DB, Shevchenko A, Mercurio F, Verma IM. 2004. Activation of transcription factor NF-kappaB requires ELKS, an IkappaB kinase regulatory subunit. Science 304:1963–1967. doi: 10.1126/science.1098387. [DOI] [PubMed] [Google Scholar]
- 7.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. 1997. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 8.Spencer E, Jiang J, Chen ZJ. 1999. Signal-induced ubiquitination of IkappaBalpha by the F-box protein Slimb/beta-TrCP. Genes Dev 13:284–294. doi: 10.1101/gad.13.3.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baeuerle PA, Baltimore D. 1996. NF-kappa B: ten years after. Cell 87:13–20. doi: 10.1016/S0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 10.Caamaño J, Hunter CA. 2002. NF-kappaB family of transcription factors: central regulators of innate and adaptive immune functions. Clin Microbiol Rev 15:414–429. doi: 10.1128/CMR.15.3.414-429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pasparakis M. 2009. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol 9:778–788. doi: 10.1038/nri2655. [DOI] [PubMed] [Google Scholar]
- 12.Kanegae Y, Tavares AT, Izpisúa Belmonte JC, Verma IM. 1998. Role of Rel/NF-kappaB transcription factors during the outgrowth of the vertebrate limb. Nature 392:611–614. doi: 10.1038/33429. [DOI] [PubMed] [Google Scholar]
- 13.Simmonds RE, Foxwell BM. 2008. Signalling, inflammation and arthritis: NF-κB and its relevance to arthritis and inflammation. Rheumatology (Oxford) 47:584–590. doi: 10.1093/rheumatology/kem298. [DOI] [PubMed] [Google Scholar]
- 14.Xia Y, Shen S, Verma IM. 2014. NF-κB, an active player in human cancers. Cancer Immunol Res 2:823–830. doi: 10.1158/2326-6066.CIR-14-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedmann-Morvinski D, Narasimamurthy R, Xia Y, Myskiw C, Soda Y, Verma IM. 2016. Targeting NF-κB in glioblastoma: a therapeutic approach. Sci Adv 2:e1501292. doi: 10.1126/sciadv.1501292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia Y, Yeddula N, Leblanc M, Ke E, Zhang Y, Oldfield E, Shaw RJ, Verma IM. 2012. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat Cell Biol 14:257–265. doi: 10.1038/ncb2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. 1997. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci U S A 94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park EK, Jung HS, Yang HI, Yoo MC, Kim C, Kim KS. 2007. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm Res 56:45–50. doi: 10.1007/s00011-007-6115-5. [DOI] [PubMed] [Google Scholar]
- 19.Chen ZJ. 2005. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol 7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.König R, Chiang CY, Tu BP, Yan SF, DeJesus PD, Romero A, Bergauer T, Orth A, Krueger U, Zhou Y, Chanda SK. 2007. A probability-based approach for the analysis of large-scale RNAi screens. Nat Methods 4:847–849. doi: 10.1038/nmeth1089. [DOI] [PubMed] [Google Scholar]
- 21.Folk P, Půta F, Skruzný M. 2004. Transcriptional coregulator SNW/SKIP: the concealed tie of dissimilar pathways. Cell Mol Life Sci 61:629–640. doi: 10.1007/s00018-003-3215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang C, Baudino TA, Dowd DR, Tokumaru H, Wang W, MacDonald PN. 2001. Ternary complexes and cooperative interplay between NCoA-62/Ski-interacting protein and steroid receptor coactivators in vitamin D receptor-mediated transcription. J Biol Chem 276:40614–40620. doi: 10.1074/jbc.M106263200. [DOI] [PubMed] [Google Scholar]
- 23.Abankwa D, Millard SM, Martel N, Choong CS, Yang M, Butler LM, Buchanan G, Tilley WD, Ueki N, Hayman MJ, Leong GM. 2013. Ski-interacting protein (SKIP) interacts with androgen receptor in the nucleus and modulates androgen-dependent transcription. BMC Biochem 14:10. doi: 10.1186/1471-2091-14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang MR, Lee SW, Um E, Kang HT, Hwang ES, Kim EJ, Um SJ. 2010. Reciprocal roles of SIRT1 and SKIP in the regulation of RAR activity: implication in the retinoic acid induced neuronal differentiation of P19 cells. Nucleic Acids Res 38:822–831. doi: 10.1093/nar/gkp1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brès V, Gomes N, Pickle L, Jones KA. 2005. A human splicing factor, SKIP, associates with P-TEFb and enhances transcription elongation by HIV-1 Tat. Genes Dev 19:1211–1226. doi: 10.1101/gad.1291705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. 1996. Suppression of TNF-alpha-induced apoptosis by NF-κB. Science 274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 27.Brès V, Yoshida T, Pickle L, Jones KA. 2009. SKIP interacts with c-Myc and Menin to promote HIV-1 Tat transactivation. Mol Cell 36:75–87. doi: 10.1016/j.molcel.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM. 2001. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell 8:327–337. doi: 10.1016/S1097-2765(01)00314-8. [DOI] [PubMed] [Google Scholar]
- 29.Sato N, Maeda M, Sugiyama M, Ito S, Hyodo T, Masuda A, Tsunoda N, Kokuryo T, Hamaguchi M, Nagino M, Senga T. 2015. Inhibition of SNW1 association with spliceosomal proteins promotes apoptosis in breast cancer cells. Cancer Med 4:268–277. doi: 10.1002/cam4.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Zhang L, Jones KA. 2011. SKIP counteracts p53-mediated apoptosis via selective regulation of p21 Cip1 mRNA splicing. Genes Dev 25:701–716. doi: 10.1101/gad.2002611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Du F, Wang X. 2008. TNF-α induces two distinct caspase-8 activation pathways. Cell 133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 32.Parnas O, Jovanovic M, Eisenhaure TM, Herbst R, Dixit A, Ye C, Przybylski D, Platt R, Tirosh I, Sanjana N, Shalem O, Satija R, Raychowdhury R, Mertins P, Carr S, Zhang F, Hacohen N, Regev A. 2015. A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 162:675–686. doi: 10.1016/j.cell.2015.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang C, Dowd DR, Staal A, Gu C, Lian JB, Van Wijnen AJ, Stein GS, MacDonald PN. 2003. Nuclear coactivator-62 kDa/Ski-interacting protein is a nuclear matrix-associated coactivator that may couple vitamin D receptor-mediated transcription and RNA splicing. J Biol Chem 278:35325–35336. doi: 10.1074/jbc.M305191200. [DOI] [PubMed] [Google Scholar]
- 34.Hargreaves DC, Horng T, Medzhitov R. 2009. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138:129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuki K, Yamaguchi N, Iwasawa S, Takakura Y, Aoyama K, Yuki R, Nakayama Y, Kuga T, Hashimoto Y, Tomonaga T, Yamaguchi N. 2017. Enhancement of TGF-β-induced Smad3 activity by c-Abl-mediated tyrosine phosphorylation of its coactivator SKI-interacting protein (SKIP). Biochem Biophys Res Commun 490:1045–1051. doi: 10.1016/j.bbrc.2017.06.163. [DOI] [PubMed] [Google Scholar]
- 36.Coles A, Gannon H, Cerny A, Kurt-Jones E, Jones S. 2010. Inhibitor of growth-4 promotes IκB promoter activation to suppress NF-κB signaling and innate immunity. Proc Natl Acad Sci U S A 107:11423–11428. doi: 10.1073/pnas.0912116107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.