

Protein synthesis is linked to cell proliferation, and its deregulation contributes to cancer. Eukaryotic translation initiation factor 1A (eIF1A) plays a key role in scanning and AUG selection and differentially affects the translation of distinct mRNAs.
KEYWORDS: 5′ UTR, Rps10, Rps3, translation initiation, eIF1, eIF1A, eIF1AX
ABSTRACT
Protein synthesis is linked to cell proliferation, and its deregulation contributes to cancer. Eukaryotic translation initiation factor 1A (eIF1A) plays a key role in scanning and AUG selection and differentially affects the translation of distinct mRNAs. Its unstructured N-terminal tail (NTT) is frequently mutated in several malignancies. Here we report that eIF1A is essential for cell proliferation and cell cycle progression. Ribosome profiling of eIF1A knockdown cells revealed a substantial enrichment of cell cycle mRNAs among the downregulated genes, which are predominantly characterized by a lengthy 5′ untranslated region (UTR). Conversely, eIF1A depletion caused a broad stimulation of 5′ UTR initiation at a near cognate AUG, unveiling a prominent role of eIF1A in suppressing 5′ UTR translation. In addition, the AUG context-dependent autoregulation of eIF1 was disrupted by eIF1A depletion, suggesting their cooperation in AUG context discrimination and scanning. Importantly, cancer-associated eIF1A NTT mutants augmented the eIF1A positive effect on a long 5′ UTR, while they hardly affected AUG selection. Mechanistically, these mutations diminished the eIF1A interaction with Rps3 and Rps10 implicated in scanning arrest. Our findings suggest that the reduced binding of eIF1A NTT mutants to the ribosome retains its open state and facilitates scanning of long 5′ UTR-containing cell cycle genes.
INTRODUCTION
The process of protein synthesis is one of the most energy-consuming cellular processes and is therefore under tight regulation by nutrient availability and various signal transduction pathways related to stress and cell proliferation (1). Both cell proliferation and mRNA translation are dependent upon the same mitogenic signaling and are therefore strongly linked. Within the eukaryotic translation machinery, the cap-binding eukaryotic translation initiation factor 4E (eIF4E) protein is the major target for these signaling events. The contribution of other initiation factors to the control of cellular growth is largely unknown.
Translation of mRNA requires a complex apparatus consisting of mRNA, ribosomes, tRNAs, and protein factors. mRNA translation is a cyclic process which can be divided into initiation, elongation, termination, and recycling. Within this framework, the initiation stage is considered a major regulatory target. This stage involves different eukaryotic initiation factors (eIFs) and different mRNA features, all of which play important roles in translational control. The predominant form of eukaryotic translation initiation depends on the m7G cap structure, present at the 5′ end of the mRNA. In addition, the translation process involves the 43S preinitiation complex (PIC), consisting of the 40S ribosome and associated factors, attachment of the PIC to the mRNA, ribosomal scanning, start codon selection, and subunit joining (2, 3).
The scanning process, which is critical to translation initiation, is promoted by eIF1 and eIF1A (3, 4). Biochemical, genetic, and structural analyses established that eIF1 and eIF1A bind the 40S subunit near the P and A sites, respectively, and promote an open 40S conformation which is scanning competent (3). The open conformation is supported by the C-terminal tail (CTT) of eIF1A. Base pairing of Met-tRNA interference with an AUG triplet promotes the dissociation of eIF1 from the 40S subunit, rearrangement to a closed conformation, and scanning arrest. The closed arrested conformation of the 40S subunit is facilitated by the N-terminal tail (NTT) of eIF1A. Thus, the two tails of eIF1A play opposing roles in scanning and AUG selection. We have recently shown that the interaction of eIF1A with the 43S subunit is partly mediated by Rps3 and Rps10, which are ribosomal proteins located at the A site and which undergo a conformational change during the transition of the 43S to the 80S subunit (5). How these interactions contribute to the initiation process is presently unknown.
eIF1A is a small 17-kDa initiation factor that is highly conserved among all eukaryotes. Most of our knowledge of eIF1A function and structure was gained from genetic and biochemical studies in Saccharomyces cerevisiae yeast. Interestingly, somatic mutations in eIF1A, especially in the NTT, were recently found to be associated with uveal, thyroid, and ovarian cancers (6–9). A combination of eIF1A and Ras mutations has been reported in different types of epithelial cancer (thyroid carcinoma) and melanomas (7, 8). Presently, very little is known about the biological function of eIF1A in mammalian cells and how perturbations of this function contribute to cancer.
In the present study, we used mammalian cells to investigate the physiological and molecular functions of eIF1A and how eIF1A NTT cancer-associated mutants alter these activities. We report that eIF1A has a critical role in maintaining cell proliferation and cell cycle progression by supporting the G2/M phase of the cell cycle. Using ribosome profiling to explore the translational landscape of cells depleted of the two eIF1A genes, we found 2 major subsets of eIF1A-regulated mRNAs, both of which are related to the function of the eIF1A CTT. The downregulated gene set is enriched with cell cycle genes and predominantly characterized by a lengthy 5′ untranslated region (UTR); thus, it is dependent on the scanning-promoting activity of the eIF1A CTT. The other gene class displays a dramatic stimulation of initiation in 5′ UTRs at near cognate AUG codons, which is unexpectedly linked to translation upregulation, most likely via reinitiation. This observation uncovers those mRNAs that are targeted by eIF1A for suppression of inappropriate initiation. We further demonstrate that the cancer-associated NTT mutants elevate the scanning-promoting activity eIF1A on a long 5′ UTR but have little effect on AUG selection. At the molecular level, these mutations diminish the eIF1A interaction with A-site ribosomal proteins Rps3 and Rps10, which are involved in scanning arrest. Our findings provide a mechanistic basis for the promotion of cell proliferation by eIF1A NTT mutants which involves scanning enhancement of long 5′ UTR-containing cell cycle genes.
RESULTS
eIF1A is essential for cell proliferation and cell cycle progression.
To address the function of eIF1A, we initially attempted to knock out the two paralogs of eIF1A encoded by chromosomes 18 [eIF1A(18)] and X (eIF1AX) that are expressed in mouse embryonic fibroblasts (MEFs) using the CRISPR-Cas9 system. However, we could not obtain stable double knockout of these paralogs. We therefore used small interfering RNA (siRNA) against the two eIF1A-encoding genes, which resulted in the efficient downregulation of eIF1A mRNAs and protein (Fig. 1A). eIF1A knockdown (KD) caused a visible effect on cell number (Fig. 1B), which was validated by live-cell counting (Fig. 1C) and a luminescent cell proliferation assay (Fig. 1D), suggesting that it is required to maintain cell proliferation.
FIG 1.

eIF1A is essential for cell proliferation and cell cycle progression. (A) Mouse embryonic fibroblasts (MEFs) were double transfected with control siRNA (Si-C; nontargeting) or eIF1A(18) siRNA plus eIF1AX siRNA (Si-eIF1A), and at 72 h after the first transfection, RNA and protein lysates were prepared and subjected to real-time PCR and Western blotting for measuring knockdown efficiency. (Top) The levels of eIF1A(18) and eIF1AX mRNAs normalized to the level of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA; (bottom) immunoblot with eIF1A and tubulin antibodies, as indicated. (B) Phase-contrast microscope images showing cells transfected with control siRNA (left) and eIF1A(18) siRNA plus eIF1AX siRNA (right). The pictures were taken at 72 h after transfection. (C and D) The viability of eIF1A KD and control MEFs was analyzed by the trypan blue method (C) and a luminescent cell viability assay (D). (E) Flow cytometric analysis of the cell cycle using propidium iodide (PI) DNA staining dye of control siRNA-transfected MEFs (left) and eIF1A-specific siRNA-transfected MEFs (right). The percentages of cells in the different phases of the cell cycle are indicated. PI-H, propidium iodide emission height.
To investigate whether the reduced cell number seen upon eIF1A depletion is associated with alterations in the cell cycle, we analyzed the partition of cells in G1, S, and G2/M phases of the cell cycle by flow cytometry following DNA staining with propidium iodide. The results revealed that eIF1A KD caused substantial changes in the distribution of cells at the different phases of the cell cycle. Specifically, we observed in eIF1A KD cells the accumulation of cells in the S phase along with a dramatic decrease in cells in G2/M phase of the cell cycle compared with the findings for the control cells (Fig. 1E). In addition, there was an increase in sub-G1 dead cells in the eIF1A KD cells. These results suggest that the growth defect associated with eIF1A depletion can be attributed to a block in S phase or an impaired transition from S to G2/M along with reduced cell survival. Thus, the two eIF1A genes are most likely essential for cell proliferation and survival in MEFs, which can explain the unsuccessful attempts to knock them out.
eIF1A depletion causes extensive changes in translation of cell cycle genes.
To examine the impact of eIF1A on translation, MEFs were transfected with control siRNA or a combination of eIF1AX and eIF1A(18) siRNAs for 72 h (eIF1A KD cells) and then lysed and subjected to sucrose gradient sedimentation. The polysome profile of eIF1A-depleted MEFs was dramatically changed from that of the control cells, as the relative amount of the 80S monoribosome was increased, while the polysomal fractions were substantially decreased (Fig. 2A), indicating the inhibition of translation. To study further the changes in the translation initiation landscape of eIF1A KD cells, we applied the ribosome profiling (RP) approach in the presence of harringtonine (Hrr) and cycloheximide (CHX) as translational inhibitors as described previously (10). We performed deep sequencing of the ribosomal protected RNA fragments along with mRNA sequencing of control and eIF1A-depleted MEFs. Analysis of the length of the ribosome-protected fragments revealed the expected 28- to 31-nucleotide peaks in all samples (Fig. 2B), and comparison of the two biological repeats using a scatter plot indicated their high similarity (Fig. 2D). Inspection of the read coverage of the eIF1AX and eIF1A(18) genes confirmed the efficient knockdown of the two paralogs at the mRNA and translation levels (Fig. 3A). Meta-gene analysis of the distribution of normalized reads in the coding DNA sequence (CDS) of 6,331 genes revealed that the overall coverage of the two eIF1A KD samples was generally lower than that of the control samples (Fig. 2E, left). Considering the expected general role of eIF1A, it was surprising that its depletion resulted in CDS translational downregulation of only 1,014 genes and the upregulation of 439 genes (Fig. 2C), while the vast majority of mRNAs were unaffected. The meta-profiles of the downregulated gene set revealed reduced ribosome occupancy, particularly in the mRNA bodies, in the two KD samples compared to that in their controls (Fig. 2E, right). This finding is consistent with the reduction in the amount of polysomes seen in the polysome profiling following eIF1A KD (Fig. 2A), which together suggest that eIF1A depletion causes a defect in the transition from initiation to elongation in this subset of genes.
FIG 2.

Ribosomal profiling of eIF1A KD cells reveals translation downregulation of cell cycle- and cancer-associated genes. (A) eIF1A depletion dramatically affects global translation. Cell lysates of MEFs treated with control siRNA (black) or eIF1A siRNA (red) were subjected to sucrose gradient sedimentation and fraction collection to obtain polysome profiles. (B) Ribosome-protected fragment length (in nucleotides [nuc]) in control and eIF1A KD samples (n = 2 per sample category). (C) Summary of the number of genes whose ribosomal occupancy was affected (fold change, ≥2 or −2) or unaffected in coding regions (CDS) in response to eIF1A KD. (D) Scatter plot showing the reads per kilobase per million correlations (R2 > 0.9) between the 2 independent replicate experiments with control and eIF1A KD MEFs. (E) Meta-gene analysis of the distribution of normalized reads in the coding region (CDS) of all analyzed genes (n = 6,331) and the downregulated genes (n = 1,014) (down). (F) Control and eIF1A KD cells were analyzed by Western blotting with antibodies against eIF1A, GAPDH, and the indicated eIFs and ribosomal proteins. chrX+18, chromosomes X and 18. (G) Gene enrichment analysis of the biological processes associated with the CDS downregulated genes. The number of genes in each category and the P value are indicated. TGF, tumor growth factor. (H) Gene enrichment analysis of diseases associated with the CDS downregulated gene. The number of genes in each category and the P values are indicated.
FIG 3.

Consequences of eIF1A depletion on several cell cycle-associated mRNAs. (A) Validation of eIF1A knockdown in the ribosome profile. Tracks showing the ribosome profiling reads along eIF1A (18) and eIF1AX mRNAs for replicate samples of each control and eIF1A KD. (B) Tracks showing the ribosome profiling reads along various cell cycle-associated mRNAs for replicate samples of each control and eIF1A KD. Blue bars, the actual translated coding sequence based upon ribosome profiling; red arrows, direction of translation. (C) Western blot analysis of genes from the cell cycle and DNA repair categories that were translationally downregulated upon eIF1A KD. The levels of eIF1A and GAPDH (a control for unaffected genes) are also shown.
The translation of 5′ terminal oligopyrimidine (TOP)-containing mRNAs that included mRNAs for ribosomal proteins and translation initiation factors is known to be effectively downregulated in response to a variety of stresses (11, 12). The RP data revealed that neither ribosomal protein genes nor translation initiation factor genes are among the translationally downregulated genes (Table 1). We confirmed this observation by analyzing the levels of a subset of the associated proteins to which antibodies were available and found them to be largely unchanged (Fig. 2F). Likewise, the translation of several mRNAs known to be upregulated in response to stresses that elevate eIF2α serine 51 phosphorylation, such as ATF4 and CHOP (DDIT3), was unaffected by eIF1A KD (Table 2). These findings rule out the possibility that the translational effect seen upon eIF1A depletion is indirectly related to the translational stress response.
TABLE 1.
Effect of eIF1A knockdown on the mTOR-sensitive TOP mRNAs
| mTOR-affected gene | Effect of eIF1A KD on gene expressiona |
|---|---|
| Rps10 | Up |
| Rps11 | Up |
| Rps12 | Up |
| Rps13 | Up |
| Rps14 | Up |
| Rps15 | Up |
| Rps21 | Up |
| Rps23 | Up |
| Rps24 | Unchanged |
| Rps25 | Up |
| Rps26 | Up |
| Rps27a | Unchanged |
| Rps27l | Unchanged |
| Rps28 | Up |
| Rps29 | Up |
| Rps3 | Unchanged |
| Rps3a1 | Unchanged |
| Rps5 | Up |
| Rps6 | Unchanged |
| Rps6ka1 | Unchanged |
| Rps6ka3 | Down |
| Rps6ka5 | Unchanged |
| Rps6kc1 | Unchanged |
| Rps7 | Up |
| Rps8 | Up |
| Rps9 | Unchanged |
| Rpsa | Up |
| Rpl10 | Up |
| Rpl10a | Up |
| Rpl11 | Unchanged |
| Rpl12 | Unchanged |
| Rpl13 | Up |
| Rpl13a | Unchanged |
| Rpl14 | Up |
| Rpl15 | Up |
| Rpl17 | Unchanged |
| Rpl18 | Up |
| Rpl18a | Up |
| Rpl35a | Up |
| Rpl36 | Up |
| Rpl36a | Unchanged |
| Rpl36al | Up |
| Rpl7l1 | Unchanged |
| Rpl8 | Up |
| Rpl9 | Unchanged |
| Rplp0 | Up |
| Rplp1 | Up |
| Rplp2 | Up |
| Eif1 | Unchanged |
| Eif1ad | Unchanged |
| Eif1b | Unchanged |
| Eif2b1 | Unchanged |
| Eif2b3 | Unchanged |
| Eif2b4 | Unchanged |
| Eif2b5 | Unchanged |
| Eif2d | Unchanged |
| Eif2s2 | Unchanged |
| Eif2s3x | Unchanged |
| Eif3a | Unchanged |
| Eif3b | Unchanged |
| Eif3m | Unchanged |
| Eif4a1 | Unchanged |
| Eif4a3 | Unchanged |
| Eif4b | Unchanged |
| Eif4e | Unchanged |
| Eif4e2 | Unchanged |
| Eif4enif1 | Unchanged |
| Eif4h | Unchanged |
| Eif5 | Unchanged |
| Eif5a | Unchanged |
| Eif6 | Unchanged |
Up, upregulated; Down, downregulated.
TABLE 2.
Effect of eIF1A knockdown on eIF2α serine 51 phosphorylation-regulated mRNAs
| eIF2α-activated gene mRNA | Effect of eIF1A KD on gene expressiona |
|---|---|
| ATF4 | Unchanged |
| ATF5 | Unchanged |
| ATF6 | Unchanged |
| Impact | Unchanged |
| CHOP | Unchanged |
| GCN2 | Down |
| GADD34 | Up |
Up, upregulated; Down, downregulated.
Biological pathway analysis indicated that the most enriched categories associated with the downregulated gene set are related to the cell cycle and include the cell cycle, DNA damage response and repair, mitotic cell division, and G2/M transition of the mitotic cell cycle (Fig. 2G). Translational downregulation of these categories can explain the observed cell proliferation and cell cycle progression defects associated with eIF1A KD (Fig. 1E). Examples of several translationally downregulated cell cycle genes, which included Wee1, Tgfb1, Nedd1, Rad21 Usp8, and Klhl9, can be seen in Fig. 3B. The ribosome occupancy in the CDS of all these representative genes was substantially reduced. Analysis of several proteins from these pathways by Western blotting confirmed that their diminished translation upon eIF1A KD leads to reduced protein levels (Fig. 3C). Analysis of the downregulated genes in a disease database revealed enrichment of genes for various types of cancers more than any other disease (Fig. 2H), highlighting the importance of eIF1A in this pathology.
Translational control by eIF1A is largely determined by the 5′ UTR length.
Considering the role that eIF1A plays in the stringency of start codon selection (13), we compared the context of the start codon of the eIF1A-dependent mRNAs to the AUG flanking sequence of the unaffected mRNAs. As can be seen in Fig. 4A, the logos of the downregulated, upregulated, and unaffected gene sets were almost indistinguishable. Thus, the requirement of eIF1A for the downregulated mRNAs seems to be largely AUG context independent. Next, we compared the 5′ UTR lengths between the different gene sets and found that the downregulated and upregulated mRNAs are significantly longer and shorter, respectively, than those of the unaffected genes (Fig. 4B). To examine whether the 5′ UTR length is relevant to the eIF1A-dependent cell cycle genes, which constitute only 8% of all the downregulated genes, we specifically analyzed the 5′ UTR size and found that they indeed have longer 5′ UTRs than the unaffected or upregulated gene sets (Fig. 4B).
FIG 4.

Translational control by eIF1A is predominantly determined by the 5′ UTR length. (A) Analysis of the nucleotide context, using the MEME program, of the translation start site of the CDS unaffected, downregulated (DOWN), and upregulated (UP) gene sets. (B) Box plot showing the relation between the 5′ UTR length and the translation efficiency of the CDS genes whose expression was unaffected, downregulated, and upregulated as well as the cell cycle gene subset of downregulated genes, as indicated. (C) Schematic diagram of the reporter gene constructs used to examine the role of eIF1A on 5′ UTR length. The Renilla luciferase is preceded by a 5′ UTR with a length of 311 nucleotides (nt), and the 5′ UTR length of firefly luciferase is 111 nucleotides. (D) MEFs were transfected with control or eIF1AX plus eIF1A(18) siRNA, and 48 h later, the cells were cotransfected again with the siRNA pool with the reporter genes described in panel C. Cells were harvested 24 h after the second transfection and analyzed for Renilla and firefly luciferase activities. The graph presents the Renilla and firefly luciferase ratios from 6 independent experiments (average ± standard error). *, statistically significant differences (P < 0.05). (E) Effect of eIF1A KD on the mRNA levels of cotransfected reporter genes described in panel C analyzed by RT-quantitative PCR. RL, Renilla luciferase; FF, firefly luciferase.
To investigate further the association of 5′ UTR length and eIF1A regulation, we used two reporter genes which have an identical strong AUG context but differ in their 5′ UTR lengths, as shown schematically in Fig. 4C. MEFs were transfected with control or eIF1AX plus eIF1A(18) siRNAs and 48 h later were transfected again with these siRNAs together with the two reporters. The results, which are expressed as the ratio between the long (Renilla) and the short (firefly) luciferase activities, revealed the diminished relative activity of the long 5′ UTR in eIF1A KD cells compared to that in control cells (Fig. 4D). The observed effect was not a consequence of the differential expression of the short and long mRNAs in eIF1A KD cells (Fig. 4E). Thus, the ribosomal profiling along with the reporter gene assays suggest that 5′ UTR length is the primary feature involved in the translational control by eIF1A in mammalian cells.
Marked increase in 5′ UTR translation initiation in response to eIF1A KD.
We next analyzed the relative levels of ribosomal occupancies in the 5′ UTR, and we observed that there is an overall increase in 5′ UTR ribosomal footprints upon eIF1A KD. Specifically, 1,907 expressed genes that constituted 28% of all analyzed genes showed upregulation of >2-fold in their 5′ UTR (Fig. 5A). This effect was clearly evident from the meta-gene profile of all genes (Fig. 5B, left) and of this gene set (Fig. 5B, right). As an untranslated open reading frame (uORF) can have an important regulatory role on the translation of the main open reading frame (ORF) (14, 15), we compared the 5′ UTR upregulated gene set to the genes displaying down- and upregulation of translation in their CDS using a Venn diagram (Fig. 5C). Interestingly, there was very little overlap between the genes displaying enhanced 5′ UTR footprints and the CDS downregulated genes, suggesting that elevated 5′ UTR initiation does not significantly contribute to the regulation of these gene sets by eIF1A. On the other hand, a substantial overlap with the CDS upregulated gene set was seen. Interestingly, the 5′ UTR length of this specific overlapping set was significantly shorter than that of the rest of the 5′ UTR upregulated set (Fig. 5D), raising the possibility that increased 5′ UTR translation contributes to enhanced CDS translation, most likely via reinitiation.
FIG 5.

Broad elevation of 5′ UTR initiation upon eIF1A depletion. (A) The number of genes with the indicated alteration in 5′ UTR translation upon eIF1A KD. (B) Meta-gene analysis of the distribution of normalized reads in the 5′ UTR of all analyzed genes (n = 6,331) and upregulated genes (n = 1,907). (C) Venn diagram showing the overlap between CDS upregulated, CDS downregulated, and 5′ UTR upregulated genes. (D) Box plot showing the relation between the 5′ UTR length and the translation efficiency in eIF1A KD versus control samples for 5′ UTR upregulated genes, CDS upregulated genes, and both 5′ UTR and CDS upregulated genes. (E) Comparison of the nucleotide context of the 5′ UTR translation initiation site (TIS) in unaffected and upregulated genes. (F) Pie chart showing the frequency of the start codon in the ribosome footprints in the 5′ UTR in the unaffected and upregulated gene sets. (G) Tracks showing the ribosome profiling reads along various mRNAs with upregulation in ribosome density in the 5′ UTR for replicate samples of each control and eIF1A KD sample. (H) MEFs were transfected with control or eIF1AX plus eIF1A(18) siRNA and 48 h later cells were cotransfected again with the siRNA pool with a firefly luciferase reporter gene driven by AUG, CUG, GUG, and UUG as translation initiation codons, as shown schematically on the top. The Renilla luciferase reporter gene was also cotransfected and served as a normalizing control. The activities of NUG in control and eIF1A KD cells are presented as a percentage of that for AUG for the indicated number of independent experiments (average ± standard error). *, statistically significant differences (P < 0.05).
Translation initiation from the major ORF primarily occurs via AUG, while initiation from the 5′ UTR tends to be more flexible and utilizes near cognate AUGs (16). We examined the sequence context of the translation initiation site (TIS) in the reads of the 5′ UTR in control and eIF1A KD samples and found that the most variable nucleotide is the first nucleotide of the AUG triplet (Fig. 5E). We also determined the percentage of all possible combinations of AUG single substitutions (Fig. 5F). The results confirmed that the first nucleotide is the most diverged, but in the case of eIF1A KD, 13% of the TISs occurred in the ACG triplet, which was very rare in the control sample. Several specific examples that included Jun, Mrps17, Slc35e4, and Atp5g3 can be seen in Fig. 5G.
The features associated with the enhanced 5′ UTR translation suggest that eIF1A depletion enhances the frequency of near cognate AUG utilization in 5′ UTRs. To examine this notion further, we used a series of luciferase reporter genes in which the initiating triplet was either AUG, CUG, GUG, or UUG (Fig. 5H, top). Cells were depleted of eIF1AX plus eIF1A(18) using siRNAs and 48 h later were transfected again with these siRNAs together with the NUG reporters (where N represents any nucleotide). The activities of CUG, GUG, and UUG, which are expressed as a percentage of the activity of AUG, were significantly enhanced by eIF1A KD (Fig. 5H). These findings provide strong support to the idea that eIF1A indeed acts to inhibit 5′ UTR translation at the near cognate AUG in mammalian cells.
eIF1A is an essential component of the eIF1 autoregulatory feedback loop.
eIF1 plays a central role in scanning and AUG selection in an optimal context. eIF1 itself has an evolutionarily conserved poor AUG context that is necessary for an autoregulatory negative feedback loop in which high eIF1 levels inhibit its own translation and prevent its overexpression (17). Considering the overlapping functions of eIF1A CTT and eIF1, we analyzed the effect of eIF1A depletion on eIF1 translation. Remarkably, we found that eIF1 translation is substantially upregulated (Fig. 6A). Consequently, eIF1 protein levels were dramatically elevated in eIF1A KD cells (Fig. 6B), suggesting that the autoregulatory negative feedback loop is severely disrupted. The mRNA levels of eIF1 were also increased (Fig. 6C), presumably due to the effect of translation on mRNA stability. We noticed that the elevation of eIF1 upon eIF1A depletion was associated with the appearance of translation initiation sites in the 5′ UTR at CUG and ACG codons, which were hardly detected in control cells. Thus, the eIF1 ability to restrict its own translation in the 5′ UTR and the main ORF is lost in the absence of eIF1A. Considering the strong scanning-promoting activity of eIF1 (see, for example, references 18 and 19), one would expect the translation of long 5′ UTR mRNAs to be elevated due to this dramatic upregulation upon eIF1A KD. Contrary to this expectation, long 5′ UTR mRNAs were, in fact, downregulated (Fig. 4), suggesting that the scanning-promoting activity of eIF1 requires its cooperation with eIF1A.
FIG 6.
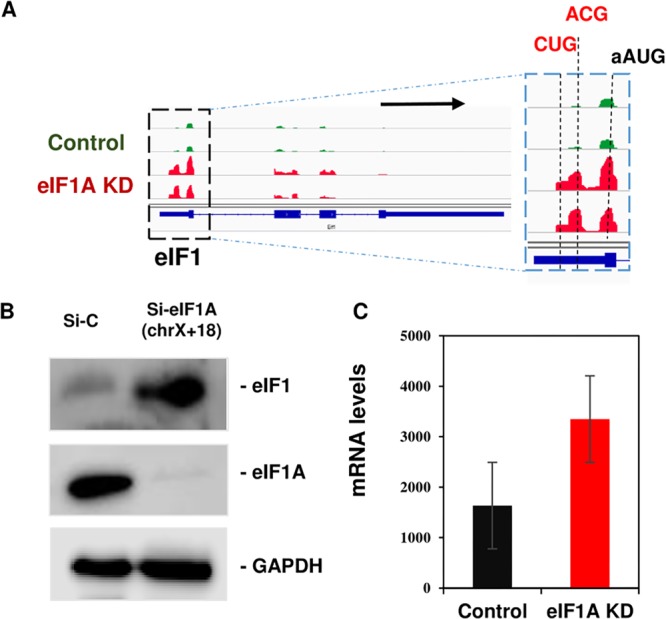
eIF1A is a central player in the eIF1 autoregulatory feedback loop. (A) Tracks showing the ribosome profiling reads along eIF1 mRNA for replicate samples of each control (green) and eIF1A KD (red). (B) eIF1 protein levels in control and eIF1A KD MEFs were analyzed by Western blotting along with those of eIF1A and GAPDH. This image is a representative of images from 3 independent experiments with similar results. (C) eIF1 mRNA levels in control and eIF1A KD MEFs derived from the RNA-seq data.
Cancer-associated eIF1A NTT mutants primarily enhance the translation of long 5′ UTR mRNAs.
Several studies revealed a link between somatic mutations in eIF1A, especially in the NTT, and cancers of the uvea, thyroid, and ovaries (6–9). Given the importance of eIF1A for the translational control of cell cycle genes via their long 5′ UTR and the established role of cell cycle dysregulation in tumorigenesis, we set out to examine the function of cancer-associated NTT mutations. We used studies of gene overexpression, which bear some similarity with cancer cells, in which the eIF1A mutant protein coexists with the wild-type (WT) proteins. MEFs were transfected with the long and short 5′ UTR reporter genes described in Fig. 4C, together with WT and mutant eIF1A plasmids (Fig. 7A). The results revealed that exogenous eIF1A enhanced the activity of the long 5′ UTR reporter compared to that of the short one (Fig. 7B). Interestingly, the effect of all the NTT mutants was significantly greater than that of the WT (Fig. 7B). Analysis of selected mutants in human HEK293T cells revealed similar enhancement of long 5′ UTR translation by NTT mutants and the WT (Fig. 7C), confirming that this effect is not confined to a single cell type. To examine further the effect of the eIF1A NTT mutation on short and long 5′ UTR mRNA, we carried out polysomal profiling following a similar transfection experiment with the eIF1A WT and G8R/G9R mutant together with short and long 5′ UTR reporter genes. Both eIF1A variants caused a modest increase in the heavy polysomal fractions (Fig. 7D). Comparing the effects on short and long 5′ UTR mRNAs revealed a clear differential enhancement of the long 5′ UTR by the G8R/G9R mutant (Fig. 7E and F). We also analyzed the effect of the WT and the NTT mutants on CUG-driven translation, but no significant effect was observed (Fig. 7G). These findings suggest that the effect of these mutations is primarily related to the 5′ UTR length rather than initiation site stringency in mammalian cells.
FIG 7.

eIF1A NTT mutants primarily enhance the translation of long 5′ UTR mRNAs. (A) Schematic view of eIF1A showing the hot spot of NTT mutations reported in various carcinomas. (B) Several of the eIF1A cancer-associated mutations were introduced into an eIF1A expression plasmid. MEFs were transfected with the reporter genes described in Fig. 4C together with control or eIF1A WT and mutant expression plasmids. Cells were harvested 24 h after transfection and analyzed for Renilla and firefly luciferase activities. The graph presents the Renilla (long 5′ UTR) and firefly (short 5′ UTR) luciferase activity ratio of the indicated number of experiments (average ± standard error). *, statistically significant differences (P < 0.05). (C) The same experiment for which the results are presented in panel B was done with the indicated mutants in HEK293T cells. (D) HEK293T cells were transfected with control, WT eIF1A, and eIF1A G8R/G9R mutant plasmids together with the short and long 5′ UTR reporter plasmids that are described in Fig. 4C. At 48 h after transfection, cell lysates were subjected to sucrose gradient sedimentation and fraction collection to obtain the polysome profiles shown. (E) Real-time quantitative PCR analysis of the indicated long (Renilla) and short (firefly) luciferase reporter mRNAs in the free, light, and heavy polysomal fractions of the gradient of control, WT, and mutant eIF1A-expressing cells. (F) The change in the polysomal fraction-to-free fraction ratio of the long and short mRNAs upon eIF1A WT and mutant expression calculated from the data presented in panel E. (G) eIF1A NTT mutants have minor effects on translation through noncognate AUG. The bar graph shows the expression of CUG with respect to that of AUG upon expression of various eIF1A mutants in MEFs, as described in the legend to Fig. 5H. NS, not significant.
NTT mutants are defective in A-site Rps10 and Rps3 binding.
In the PIC, eIF1A is located near A-site ribosomal proteins Rps3 and Rps10 and the mRNA channel (20). Both ribosomal proteins contact AUG downstream nucleotides and are implicated in scanning arrest of short 5′ UTR mRNAs bearing the translation initiator of short 5' UTR (TISU) element (5). In addition, Rps3 and Rps10 were shown to interact with eIF1A in a cell-based split-Renilla assay (5). Considering the potential role of Rps3 and Rps10 in scanning inhibition, we set out to determine the impact of eIF1A NTT mutations on the binding to these ribosomal proteins. We first determined whether these interactions are direct using pulldown assays. His-Rps3, His-Rps10, and eIF1A (tag-less) were each expressed in Escherichia coli. His-Rps3 and His-Rps10 were incubated with nickel-agarose beads in the absence or presence of eIF1A (Fig. 8A and B, lanes 3 and 5, respectively). As a control, eIF1A was incubated with empty nickel beads (Fig. 8A and B, lane 4). The results revealed efficient binding of eIF1A to both ribosomal proteins (Fig. 8A and B, lane 5), confirming their direct specific interaction. Next, a similar analysis was carried out with 3 eIF1A NTT mutants, the G6D mutant, the G9D/R13H double mutant, and the K10E mutant (Fig. 8A, lanes 6 to 18, and Fig. 8B, lanes 6 to 16). Quantifying the relative amount of bound protein revealed a marked reduction in the amount of the bound K10E mutant on Rps3 compared to that for the WT, while the effect of the binding to Rps3 by the G6D and G9D/R13H mutants was not significant (Fig. 8A and C). With Rps10, all mutants, the G6D, G9D/R13H, and K10E mutants, bound less efficiently. Thus, it can be concluded that at least part of the effect of the eIF1A NTT mutations on translation may be attributed to reduced binding to ribosomal proteins.
FIG 8.

NTT mutant binding to Rps10 and Rps3 is defective. (A and B) In vitro binding assays of eIF1A WT and mutants with Rps3 and Rps10. His-tagged Rps3 (A) and His-tagged RPS10 (B) were coupled to Ni-agarose beads and incubated with an untagged eIF1A WT or mutant, as indicated. As a control, eIF1A was incubated with the Ni-agarose beads without Rps3/10. The pulled-down complexes were washed and then run on SDS-PAGE gels, followed by Western blotting with the indicated antibodies. In all the experiments, the input represents 2% of the amount of lysate used for the binding. (C) Quantitation of the bound protein relative to the input. The graph represents the average ± standard error binding level from 3 experiments. *, statistically significant difference (P < 0.05).
DISCUSSION
In this study, we addressed the biological function of eIF1A and the mechanism underlying the translational dysregulation conferred by eIF1A NTT mutations in mammalian cells. By downregulating the two eIF1A paralogs in MEFs, we demonstrated a critical role of this factor in promoting cell proliferation and cell cycle progression, with a particular defect in G2/M phase of the cell cycle. Exploring the translation landscape of cells depleted of both eIF1A paralogs using ribosome profiling, we uncovered 2 major nonoverlapping subsets of eIF1A-regulated mRNAs which are related to known molecular attributes of eIF1A. The first includes 1,014 mRNAs displaying translation downregulation of the major ORF. This set is highly enriched with cell cycle as well as cancer-associated genes, indicating that eIF1A plays a critical role in driving the translation of these mRNAs and highlighting the potential contribution of its deregulation to cancer. Considering the importance of this factor in initiation site selection based on the stringency of the AUG context (2, 4), it was unexpected to observe that the most specific feature characterizing this gene set is a lengthy 5′ UTR rather than differences in the AUG context, a finding that was also validated by the reporter gene assay. This observation raises the possibility that maintaining the stringency of the AUG context in ORFs is fulfilled by other initiation factors with similar activity. Thus, in the downregulated gene set, the most important function conferred by eIF1A is the facilitation of scanning, an activity known to be mediated by the scanning enhancer present in the eIF1A CTT. On the other hand, eIF1A NTT was shown to bear a scanning inhibitor (21). As all the 7 cancer-associated eIF1A NTT mutants analyzed here displayed higher scanning-promoting activity than the WT, it can be concluded that these mutants are defective in scanning inhibition. Thus, it is likely that cancer cells benefit from eIF1A’s ability to enhance scanning to facilitate the translation of cell cycle and tumor promoter genes, such as those identified here.
The second and largest gene collection affected by eIF1A depletion was characterized by elevated translation initiation in the 5′ UTR at a near cognate AUG, suggesting the relaxation of start codon stringency and a higher frequency of scanning arrest at these sites (ribosomal closed conformation). This is clearly exemplified by the dramatic increase in the utilization of the ACG triplet as a start codon in the eIF1A KD samples and the results of the NUG reporter gene assay, which demonstrated heightened near cognate AUG initiation upon eIF1A depletion. Thus, it appears that eIF1A has a major role in preventing initiation from the near cognate AUG in the 5′ UTRs of mammalian genes, to maintain protein homeostasis and avoid energy expenditure. As this inhibitory activity is also attributed to the CTT of eIF1A, it seems that this domain is dominant relative to the NTT in mammalian cells.
Remarkably, 55% of those mRNAs that presented elevated translation in the main ORF overlapped the mRNAs displaying increased 5′ UTR translation (Fig. 5C), suggesting a functional link between these effects. This observation was unexpected, as uORF tend to diminish translation from a downstream main ORF (14, 15). One possibility that can explain this link is the process of translation reinitiation, a mechanism in which a uORF can retain the small ribosomal subunit on the mRNA after termination and initiate translation at a downstream site, as occurs in stress response mRNAs, such as the yeast GCN4 and its mammalian homolog, ATF4 (22–24). Reinitiation efficiency depends on several features and factors, of which a major one is the distance of the uORF from the main ORF. A hint that this mechanism may indeed be involved in the translation upregulation here is the fact that this set is generally characterized by a shorter 5′ UTR length and therefore an increased likelihood for a short distance between the uORF and the main ORF. Thus, inhibition of 5′ UTR initiation by eIF1A serves not only to prevent wasteful uORF synthesis but also to keep the translation of mRNAs with relatively short 5′ UTR under control. Interestingly, in mammalian cells the eIF1A NTT mutants did not show enhanced inhibition of near cognate AUG selection compared to that of WT eIF1A (Fig. 7G), whereas these mutations in yeast eIF1A enhance the stringency of initiation site selection (25). This finding can be explained by a higher degree of redundancy of this activity among mammalian eIFs as well as the fact that both eIF1 and eIF1A are each encoded by at least two genes.
The genes encoding eIF1 in most eukaryotes contain an AUG in a poor context (17, 26), and eIF1 itself inhibits translation from the AUG context that diverges significantly from the Kozak (18, 27). Consequently, it was found that mammalian eIF1 negatively autoregulates its own translation in a manner that is dependent on the poor context of its start codon (17). Thus, eIF1 cannot be overexpressed in cells. Interestingly, our findings revealed that this autoregulatory loop is severely disrupted upon the loss of eIF1A, as eIF1 translation is dramatically elevated and eIF1 protein levels rise substantially. This finding uncovered the important role of eIF1A in limiting eIF1 translation and the cooperation of these factors in discriminating against a poor AUG context. Furthermore, despite the dramatic eIF1 upregulation upon eIF1A KD, the mRNAs that are downregulated tend to have longer 5′ UTR, suggesting that eIF1 cannot compensate for eIF1A loss and, most likely, that their ability to promote scanning is interconnected.
The cryo-electron microscopic structure of the 48S subunit in the presence of eIF1 and eIF1A suggests that eIF1A is located near the A site and the mRNA channel around positions +4 and +5 and is also in proximity to the 18S rRNA, Rps3, and Rps10 (20). Here we show that eIF1A interacts directly with each Rps3 and Rps10 and that both interactions are weakened by several cancer-associated NTT mutants. The effects of these mutations on binding to mRNA and 18S rRNA are yet to be determined. As these mutants also impair the scanning inhibition associated with this domain, we infer that eIF1A-Rps3/10 interactions serve to promote scanning arrest and the closed conformation. These findings nicely fit our recent report of the involvement of Rps3 and Rps10 in the scanning arrest promoted by TISU, a translation regulatory element that directs cap-dependent and scanning-free initiation (18, 19, 28). Both Rps3 and Rps10 were specifically cross-linked to positions +5 and + 6 of TISU in a dynamic manner (5). In this study, TISU was also found to be highly dependent on eIF1A. It was therefore unexpected that TISU genes were not found to be enriched among the eIF1A-affected genes. A possible explanation for this may be related to the dramatic upregulation of eIF1 upon eIF1A KD in MEFs. As TISU activity is highly eIF1 dependent and eIF1 was shown to compensate for the absence of eIF1A in in vitro translation assays (19), it is possible that eIF1 and eIF1A also play a redundant regulatory role in TISU-mediated translation in MEFs. An alternative explanation may relate to the short 5′ UTR length of TISU mRNAs, which may result in a footprint at the initiation site that has a length shorter than 28 nucleotides.
In summary, by combining global translation analysis of cells depleted of the two eIF1A paralogs with complementary studies of the cancer-associated mutants of eIF1A, we uncovered the most important mRNA features underlying the differential regulation of distinct mRNAs by eIF1A in mammalian cells. In particular, our findings point to the essential function of the eIF1A CTT, which has a fundamental and nonredundant role in promoting the scanning of long 5′ UTR mRNAs and inhibiting inappropriate 5′ UTR translation. Thus, eIF1A CTT emerges as a potential target for drugs against cancers with and without eIF1A mutations.
MATERIALS AND METHODS
eIF1A knockdown experiments.
Mouse embryonic fibroblasts were transfected, using the RNAiMAX lipofectamine reagent (catalog no. 13778075), with either 50 nM control siRNA (nontargeting control siRNA d-001810-03-20; Dharmacon, GE Healthcare Life Sciences) or siRNAs targeting chromosome 18 eIF1A (SMARTpool d-0625552-01; Dharmacon, GE Healthcare Life Sciences) and eIF1AX (SMARTpool d-063045-01; Dharmacon, GE Healthcare Life Sciences). The SMARTpool consists of 4 distinct siRNAs for each gene. Twenty-four hours later, the cells were split and after an additional 24 h were transfected again with the siRNAs. At 72 h after the initial transfection, the cells were harvested for the various analyses described in the Results section.
Cell viability experiments.
MEFs in a 6-well plate were transfected with control or eIF1A siRNAs as described above. After 72 h, cells were subjected to cell viability measurement using a CellTiter-Glo luminescent assay (catalog no. G7571; Promega). In addition, eIF1A knockdown and control cells were stained with trypan blue (catalog no. T8154; Sigma), and live cells were counted using a Bio-Rad TC20 cell counter.
Cell cycle analysis.
MEFs were transfected with control or eIF1A siRNA as described above. eIF1A KD and control MEFs were trypsinized and washed twice with ice-cold phosphate-buffered saline (PBS). Subsequently, the cells were fixed overnight in 70% ethanol, washed twice with ice-cold PBS, resuspended in staining buffer (0.1% Triton X-100, 2 mg RNase A, 4% propidium iodide), and incubated at 37°C for 15 min. The cells were monitored by use of a BC LSRII flow cytometer, and the data were analyzed using ModFit LT software.
Ribosome profiling.
Ribosomal footprints in MEFs were generated essentially as described previously (10). Briefly, MEFs in 10-cm plates were transfected with control or eIF1A siRNA as described above. Seventy-two hours later the cells were incubated with 2 µg/ml harringtonin (Hrr; catalog no. H0169; LKT Laboratories) for 2 min, followed by the addition of 100 µg/ml cycloheximide (CHX). Cells were lysed and were used for ribosomal footprinting and library preparation as described previously (10) and for total RNA preparation. The resulting ribosome profiling libraries were sequenced on a HiSeq2500 high-output instrument (Illumina) to yield 60-bp single-end reads. Ribosome sequencing (transcriptome sequencing [RNA-seq]) was performed using a derivation of massively parallel single-cell RNA sequencing (MARS-seq) as described previously (29). The final mRNA libraries were sequenced using a high-throughput 75-bp kit (catalog no. FC404-2005; Illumina) on a NEXTseq 500 instrument to yield 75-bp paired-end reads.
Data analysis of ribosome profile.
(i) Preprocessing and alignment. The number of input RP reads per sample was in the range of 52 million to 62 million single end-of-length 61 bases. The initial analysis steps were similar to those described by Ingolia et al. (10) and consisted of preprocessing of the sequences by removing the first base and adapter and filtering for sequences with lengths of 20 to 50 bases with cutadapt software (http://journal.embnet.org/index.php/embnetjournal/article/view/200/479; parameters, -a CTGTAGGCACCATCAATAGATCGGAAGAGCACACGTCTGAACTCCAGTCAC --discard-untrimmed --times 1 -u 1 -m 20 -M 50), in other words, keeping sequences that had the adapter (and not trimming by the quality of the bases). rRNA was removed by running the bowtie program (30) against a database of rRNA downloaded from http://genome.ucsc.edu/cgi-bin/hgTables and making a Fastq file of non-rRNA sequences (bowtie parameters, -norc –un). The number of reads remaining was in the range 24 million to 30 million. Next, the sequences were aligned to RefSeq mm10 transcripts (downloaded from iGenomes) using bowtie (parameters, --norc -S -l 25 -n 2 -m 100 --best –strata). The proportion plots of the aligned reads to the length of the reads was done using the riboSeqR package (Bioconductor) after converting the sam file to a bam file with SAMtools (31). For gene quantification and RP summit detection, reads with lengths of 28 to 33 were selected using cutadapt (parameters, -m 28 -M 33) and aligned to the mm10 mouse genome using the TopHat2 program (32) (parameters, --no-novel-juncs --library-type fr-firststrand). The bam files were converted to tdf files using the igvtools count, in order to view the files with the Integrative Genomics Viewer (33). The number of reads aligned was in the range 3.5 million to 5 million; out of these, more than half aligned uniquely.
(ii) CDS and 5′ UTR quantification. For quantification of UTR and CDS, gtf files were created from the RefSeq annotation, avoiding regions found in both region types. The HTSeq library was used for the quantification (parameters, -s yes -t exon -m intersection-nonempty). The option intersection-nonempty allows assignment of junction reads to either the CDS or the UTR, depending on where most of the read is placed. The number of reads quantified as 5′ UTR ranged from 107,000 to 135,000, and the number of reads quantified as CDS ranged from 270,000 to 808,000.
(iii) Translation efficiency, 5′ UTR length, and start codon analyses. Counts for 5′ UTR and CDS were unified for each sample, and the quartile distribution was calculated. Since 75% of the counts were below 26, only genes that had at least 26 counts in one of the samples were used in subsequent steps. Variations in sample sizes were normalized using the DEseq2 statistical R language package (34). The translation efficiency for each sample was calculated from the ratio of ribosome profiling counts to the MARS-seq mRNA levels for 5′ UTR and CDS separately. Thereupon, the ratio between samples and controls was calculated on log-transformed data. Genes were categorized into those that were upregulated or those that were downregulated if the sample-to-control ratio of the translation efficiency was over 1 or under −1 for both samples, respectively, or unaffected for all other cases. The mean 5′ UTR length (for all transcripts defined in the iGenomes Illumina mm10 gtf file) per gene distribution was calculated for the aforementioned categories. Differences between gene categories were evaluated using one-way analysis of variance, and the effect size was estimated using Cohen’s D value. Cell cycle genes, which represent a subset of genes that are downregulated in their CDS translation efficiency ratio, were separately compared to upregulated or unaffected genes. Similarly, the mean 5′ UTR length distribution was also performed for categories in a pairwise fashion after mutually excluding genes that belonged to both categories (intersection).
Start codon boundary (±6-bp) sequences were retrieved from a Fasta file containing all mm10 mRNA sequences (downloaded from the NBCI nucleotide page search using “mouse[organism]” and filtered for mRNA and RefSeq using the left panel) using the bedtools getfasta program. Thereupon, logos were created using MEME (-minw 6 -maxw 9 -maxsize 1000000 –rna) (35).
(iv) Finding ribosome profiling summits in the 5′ UTR. Regions enriched with RP within the 5′ UTR were identified using macs (36) (parameters, -g 3e9 --keep-dup all --nomodel --shiftsize = 1) with no model and no filtering of PCR duplicates. Summit peaks were annotated by intersection (IntersectBed [37]) with the RefSeq gtf file, shifted 12 bases, and expanded to 3 bases (awk commands). bed files were derived for peaks appearing on both replicates and are within the 5′ UTR region and can be extended to 9 bases within exon 5′ UTR boundaries (IntersectBed and a Perl script). These peaks bed files were divided according to gene lists (translation efficiency [TE] downregulated UTR, TE upregulated UTR, and TE unaffected) (dedicated Perl script), and their sequences were extracted to Fasta files (bedtools getfasta). A motif enrichment search within 5′ UTR ribosome profiling-extended summit sequences was done with MEME (35) (parameters, -minw 6 -maxw 9 -rna).
(v) 5′ UTR and CDS body coverage plots. The RSeQC (38) geneBody_coverage.py script was used to plot the coverage using the TopHat bam and 5′ UTR and CDS regions bed12 format (long bed) files. A Perl script was used to create bed files for the specific gene lists (which was needed to match the transcript name to the gene and extract the relevant gene bed file).
Plasmids.
To generate eIF1A expression plasmids, the eIF1AX cDNA was isolated by reverse transcription (RT)-PCR from HEK293T cell RNA and then cloned into pCDNA3 via the BamHI and XhoI sites. The seven eIF1A mutants (the G6D, G9D, G9R, G8R/G9R, K10E, R13H, and G15D mutants) were generated by site-directed mutagenesis using a Q5 site-directed mutagenesis kit from New England Biolabs. The NUG firefly luciferase and Renilla luciferase plasmids were kindly provided by Kastura Asano (Kansas State University). The short and the long 5′ UTRs of the firefly and Renilla luciferases were derived from simian virus 40. The primer sequences are shown in Table 3.
TABLE 3.
Primer list
| Primer name | Application | Sequence |
|---|---|---|
| GAPDH Forward | Real-time PCR | AGGTCGGTGTGAACGGATTTG |
| GAPDH Reverse | Real-time PCR | GGGGTCGTTGATGGCAACA |
| eIF1A (Chr18) Forward | Real-time PCR | GTTGCCCTTTCCATCATGCC |
| eIF1A (Chr18) Reverse | Real-time PCR | ACACCGTCAAAGCACATTGC |
| eIF1AX forward | Real-time PCR | GTGACCAGCCTCTCCTGAGC |
| eIF1AX Reverse | Real-time PCR | GGAACTGCGGTGACTCCGA |
| eIF1A G6D forward | Site-directed mutagenesis | CAAGAATAAAGATAAAGGAGGTAAAAAC |
| eIF1A G6D Reverse | Site-directed mutagenesis | GGCATTTAGATGTCATCAATATC |
| eIF1A G9D forward | Site-directed mutagenesis | GGTAAAGGAGATAAAAACAGAC |
| eIF1A G9D Reverse | Site-directed mutagenesis | TTTATTCTTGGGCATTTAG |
| eIF1A G9R forward | Site-directed mutagenesis | AGGTAAAGGACGTAAAAACAGACG |
| eIF1A G9R Reverse | Site-directed mutagenesis | TTATTCTTGGGCATTTAGATG |
| eIF1A G8R_G9R forward | Site-directed mutagenesis | TAAAGGTAAACGACGTAAAAACAGACGCAG |
| eIF1A G8R_G9R Reverse | Site-directed mutagenesis | TTCTTGGGCATTTAGATG |
| eIF1A G15D forward | Site-directed mutagenesis | CAGACGCAGGGATAAGAATGAGA |
| eIF1A G15D Reverse | Site-directed mutagenesis | TTTTTACCTCCTTTACCTTTATTCTTG |
| eIF1A K10E forward | Site-directed mutagenesis | TAAAGGAGGTGAAAACAGACGCAG |
| eIF1A K10E Reverse | Site-directed mutagenesis | CCTTTATTCTTGGGCATTTAG |
| eIF1A R13H forward | Site-directed mutagenesis | TAAAAACAGACACAGGGGTAAGAATG |
| eIF1A R13H Reverse | Site-directed mutagenesis | CCTCCTTTACCTTTATTCTTG |
| eIF1A Forward | PCR cloning | CATCACCATCATCACCACAGCCAGGATCCGATGCCCAAGAATAAAGGTA |
| eIF1A Reverse | PCR cloning | TCGACTTAAGCATTATGCGGCCGCAAGCTTTTAGATGTCATCAATATCTTCATC |
| Rps3 Forward | PCR cloning | ATGGCAGTGCAAATATCCAAG |
| Rps3 Reverse | PCR cloning | TCGACTTAAGCATTATGCGGCCGCAAGCTTTTATGCTGTGGGGACTGGCTGGGGCA |
| Rps10 Forward | PCR cloning | CATCACCATCATCACCACAGCCAGGATCCGATGTTGATGCCTAAGAAGAAC |
| Rps10 Reverse | PCR cloning | TCGACTTAAGCATTATGCGGCCGCAAGCTTTTACTGAGGTGGCTGACCACGTCC |
Binding assay.
The eIF1A WT and mutants (the G6D, G9D/R13H, and K10E mutants), His-tagged Rps3, and His-tagged Rps10 were each cloned into the pRSFDuet-1 vector. The respective protein was expressed using s BL-21 expression system. The binding assay was performed using Ni-nitrilotriacetic acid His-bind resins (Novagen), followed by Western blotting. Western blots were quantified using the Image Studio Lite (v5.2) program.
ACKNOWLEDGMENTS
This work was supported by grants from the Minerva Foundation (number 712278) and the Israel Science Foundation (number 843/17) and an internal grant donated by the Auctoriana Anstalt organization. R.D. is the incumbent of the Ruth and Leonard Simon Chair of Cancer Research.
R.D. and U.S. conceived of and designed the study, analyzed the data, and wrote the paper. U.S. carried out most of the experiments; F.K. and S.A. performed part of the experiments; G.S. and D.L. performed the analysis of the ribosomal profiling data.
We declare that we have no conflict of interest.
REFERENCES
- 1.Chu J, Cargnello M, Topisirovic I, Pelletier J. 2016. Translation initiation factors: reprogramming protein synthesis in cancer. Trends Cell Biol 26:918–933. doi: 10.1016/j.tcb.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Hershey JW, Sonenberg N, Mathews MB. 2012. Principles of translational control: an overview. Cold Spring Harb Perspect Biol 4:a011528. doi: 10.1101/cshperspect.a011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hinnebusch AG. 2014. The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem 83:779–812. doi: 10.1146/annurev-biochem-060713-035802. [DOI] [PubMed] [Google Scholar]
- 4.Hinnebusch AG, Lorsch JR. 2012. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb Perspect Biol 4:a011544. doi: 10.1101/cshperspect.a011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haimov O, Sinvani H, Martin F, Ulitsky I, Emmanuel R, Tamarkin-Ben-Harush A, Vardy A, Dikstein R. 2017. Efficient and accurate translation initiation directed by TISU involves RPS3 and RPS10e binding and differential eukaryotic initiation factor 1A regulation. Mol Cell Biol 37:e00150-17. doi: 10.1128/MCB.00150-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ewens KG, Kanetsky PA, Richards-Yutz J, Purrazzella J, Shields CL, Ganguly T, Ganguly A. 2014. Chromosome 3 status combined with BAP1 and EIF1AX mutation profiles are associated with metastasis in uveal melanoma. Invest Ophthalmol Vis Sci 55:5160–5167. doi: 10.1167/iovs.14-14550. [DOI] [PubMed] [Google Scholar]
- 7.Karunamurthy A, Panebianco F, Hsiao SJ, Vorhauer J, Nikiforova MN, Chiosea S, Nikiforov YE. 2016. Prevalence and phenotypic correlations of EIF1AX mutations in thyroid nodules. Endocr Relat Cancer 23:295–301. doi: 10.1530/ERC-16-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Etemadmoghadam D, Azar WJ, Lei Y, Moujaber T, Garsed DW, Kennedy CJ, Fereday S, Mitchell C, Chiew YE, Hendley J, Sharma R, Harnett PR, Li J, Christie EL, Patch AM, George J, Au-Yeung G, Mir Arnau G, Holloway TP, Semple T, Pearson JV, Waddell N, Grimmond SM, Kobel M, Rizos H, Lomakin IB, Bowtell DDL, deFazio A, Australian Ovarian Cancer Study Group. 2017. EIF1AX and NRAS mutations co-occur and cooperate in low-grade serous ovarian carcinomas. Cancer Res 77:4268–4278. doi: 10.1158/0008-5472.CAN-16-2224. [DOI] [PubMed] [Google Scholar]
- 9.Hunter SM, Anglesio MS, Ryland GL, Sharma R, Chiew YE, Rowley SM, Doyle MA, Li J, Gilks CB, Moss P, Allan PE, Stephens AN, Huntsman DG, deFazio A, Bowtell DD, Australian Ovarian Cancer Study Group, Gorringe KL, Campbell IG. 2015. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget 6:37663–37677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. 2012. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyuhas O. 2000. Synthesis of the translational apparatus is regulated at the translational level. Eur J Biochem 267:6321–6330. doi: 10.1046/j.1432-1327.2000.01719.x. [DOI] [PubMed] [Google Scholar]
- 12.Meyuhas O, Kahan T. 2015. The race to decipher the top secrets of TOP mRNAs. Biochim Biophys Acta 1849:801–811. doi: 10.1016/j.bbagrm.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell SF, Lorsch JR. 2008. Should I stay or should I go? Eukaryotic translation initiation factors 1 and 1A control start codon recognition. J Biol Chem 283:27345–27349. doi: 10.1074/jbc.R800031200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamarkin-Ben-Harush A, Schechtman E, Dikstein R. 2014. Co-occurrence of transcription and translation gene regulatory features underlies coordinated mRNA and protein synthesis. BMC Genomics 15:688. doi: 10.1186/1471-2164-15-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calvo SE, Pagliarini DJ, Mootha VK. 2009. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci U S A 106:7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinnebusch AG, Ivanov IP, Sonenberg N. 2016. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 352:1413–1416. doi: 10.1126/science.aad9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ivanov IP, Loughran G, Sachs MS, Atkins JF. 2010. Initiation context modulates autoregulation of eukaryotic translation initiation factor 1 (eIF1). Proc Natl Acad Sci U S A 107:18056–18060. doi: 10.1073/pnas.1009269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elfakess R, Sinvani H, Haimov O, Svitkin Y, Sonenberg N, Dikstein R. 2011. Unique translation initiation of mRNAs-containing TISU element. Nucleic Acids Res 39:7598–7609. doi: 10.1093/nar/gkr484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinvani H, Haimov O, Svitkin Y, Sonenberg N, Tamarkin-Ben-Harush A, Viollet B, Dikstein R. 2015. Translational tolerance of mitochondrial genes to metabolic energy stress involves TISU and eIF1-eIF4GI cooperation in start codon selection. Cell Metab 21:479–492. doi: 10.1016/j.cmet.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 20.Hussain T, Llacer JL, Fernandez IS, Munoz A, Martin-Marcos P, Savva CG, Lorsch JR, Hinnebusch AG, Ramakrishnan V. 2014. Structural changes enable start codon recognition by the eukaryotic translation initiation complex. Cell 159:597–607. doi: 10.1016/j.cell.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saini AK, Nanda JS, Lorsch JR, Hinnebusch AG. 2010. Regulatory elements in eIF1A control the fidelity of start codon selection by modulating tRNA(i)(Met) binding to the ribosome. Genes Dev 24:97–110. doi: 10.1101/gad.1871910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hinnebusch AG. 2005. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- 23.Kozak M. 2001. Constraints on reinitiation of translation in mammals. Nucleic Acids Res 29:5226–5232. doi: 10.1093/nar/29.24.5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luukkonen BG, Tan W, Schwartz S. 1995. Efficiency of reinitiation of translation on human immunodeficiency virus type 1 mRNAs is determined by the length of the upstream open reading frame and by intercistronic distance. J Virol 69:4086–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin-Marcos P, Zhou F, Karunasiri C, Zhang F, Dong J, Nanda J, Kulkarni SD, Sen ND, Tamame M, Zeschnigk M, Lorsch JR, Hinnebusch AG. 2017. eIF1A residues implicated in cancer stabilize translation preinitiation complexes and favor suboptimal initiation sites in yeast. Elife 6:e31250. doi: 10.7554/eLife.31250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyasaka H, Endo S, Shimizu H. 2010. Eukaryotic translation initiation factor 1 (eIF1), the inspector of good AUG context for translation initiation, has an extremely bad AUG context. J Biosci Bioeng 109:635–637. doi: 10.1016/j.jbiosc.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 27.Pestova TV, Kolupaeva VG. 2002. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev 16:2906–2922. doi: 10.1101/gad.1020902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elfakess R, Dikstein R. 2008. A translation initiation element specific to mRNAs with very short 5′UTR that also regulates transcription. PLoS One 3:e3094. doi: 10.1371/journal.pone.0003094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, Amit I. 2014. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng J, Liu T, Zhang Y. 2011. Using MACS to identify peaks from ChIP-Seq data. Curr Protoc Bioinformatics Chapter 2:Unit 2.14. doi: 10.1002/0471250953.bi0214s34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Wang S, Li W. 2012. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]