

Abstract
Inborn errors of metabolism comprise a wide array of diseases and complications in the pediatric patient. The rarity of these disorders limits the ability to conduct and review robust literature regarding the disease states, mechanisms of dysfunction, treatments, and outcomes. Often, treatment plans will be based on the pathophysiology associated with the disorder and theoretical agents that may be involved in the metabolic process. Medication therapies usually consist of natural or herbal products. Established efficacious pediatric doses for these products are difficult to find in tertiary resources, and adverse effects are routinely limited to single case reports. This review article attempts to summarize some of the more common inborn errors of metabolism in a manner that is applicable to pharmacists who will provide care for these patients.
Keywords: dietary supplements, drug therapy, inborn errors, metabolism, review
Introduction
Inborn errors of metabolism (IEMs) were first described in 1908 and have subsequently grown to more than 1400 different diagnoses described in the literature.1,2 As the means to identify these disorders has improved, the number of patients receiving a diagnosis of IEM has increased. Each individual disease state is rare, but the combined risk for any form of IEM may be as common as 1 in 800 to 2500 patients.3,4 Inborn errors of metabolism can occur in almost any metabolic pathway, including carbohydrate, phospholipid, or amino acid production or metabolism. Other energy production pathways may be affected as well, including the citric acid cycle, the electron transport chain, or the production or use of various coenzymes and vitamins. These errors result in substrate accumulation, metabolite accumulation, enzyme deficiency, energy steal, or molecular accumulation, which in turn results in the symptoms associated with IEM.1,3
Metabolic errors may present with an array of symptoms. Signs and symptoms of IEM are often non-specific and include a sepsis-like presentation, lethargy, vomiting, acidosis, and developmental delay.4 Acute and chronic presentations may differ. Initial acute episodes may be life-threatening if not adequately treated and present with acute neurologic changes, metabolic crisis, and death, whereas chronic symptoms, such as poor growth and development, may be more manageable. The non-specific nature of these sequelae results in the treatment of several diagnoses at the same time. Thus, multiple medications may be started around the same time and the elucidation of which agent resulted in benefit is difficult to determine. This may exacerbate the condition of the patient. For example, the patient who has been unable to receive adequate enteral nutrition may be initiated on parenteral nutrition. Situations in which the metabolic disorder lies within lipid or amino acid metabolism can result in sequelae and contribute to the complications the patient is experiencing. The ability of the pharmacist to curtail the medication cocktail is limited until a diagnosis is established. An understanding of each disease state may help the pharmacist to make appropriate recommendations and minimize polypharmacy once a diagnosis has been established.
Diagnosis may be prolonged because a multitude of laboratory assessments will need to be evaluated. Most institutions are unable to perform the specific laboratory analysis for IEM. This results in a delay in disease identification as samples are sent for testing. Large blood volumes are required to provide a complete definitive diagnosis. The amount of blood necessary will be dependent on the patient's presentation and differential diagnosis. However, in an effort to avoid diagnostic phlebotomy, only a few lab tests may be drawn each day. Depending on patient status, only the most likely tests or those with lower blood volumes may be sent. Some institutions may not have a genetic specialist within the organization. Thus, additional time may be needed to transmit the information to a genetic specialist and receive a response. Genetic variation can lead to a wide range of symptoms within a single IEM, further complicating the ability to accurately diagnose and to promptly treat.
Institutional policies may also result in a delay in therapy. Medication therapy used in IEM often consists of nutritional or herbal supplements or medications that an institution may not routinely carry. Such therapies may be considered non-formulary, requiring further efforts or time to obtain. Institutional policies may label nutritional and herbal supplements as banned substances because of the lack of primary evidence or concern that the US Food and Drug Administration oversight regarding these agents is not as robust as that for prescription medications. Therefore, the likelihood that a pharmacist will assist in caring for these patients is expanding.
Understanding IEM can provide the pharmacist with ample opportunities to impact the care of the patient with a metabolic disorder. The pharmacist can play a critical role in providing care for these patients. This may range from counseling patients and families, providing dosing information to prescribers, assisting the prescriber and family in brand or medication selection, providing inpatient rounding services, and acquiring the needed medications if not available because of formulary restrictions or lack of availability via the wholesaler. The purpose of this review is to provide the pharmacist with an overview of several IEMs, the rationale for using a particular medication, and important pharmacotherapeutic information regarding each agent. This review will not be all-encompassing but should provide the reader with a better understanding of IEM and its medication management. Figures of the metabolic pathways are not provided, but a review of them will greatly help the reader understand the metabolic abnormality, potential symptoms of each disorder, and the rationale for the use of herbal and nutritional supplements.
Errors in Carbohydrate Metabolism
Carbohydrate Metabolism. Carbohydrate metabolism via monosaccharides provides cells with a source of energy as well as a methodology for energy storage. Most organisms use glycolysis in some fashion as a means of energy production.5 The metabolism of glucose, dextrose, fructose, or mannose initially requires phosphorylation via a molecule of adenosine triphosphate (ATP) and a hexokinase.6 Phosphofructokinase-1 then phosphorylates glucose once again in preparation for cleavage by aldolase. Once cleaved, the 2 new molecules of glyceraldehyde-3-phosphate can progress through carbohydrate metabolic processes. The metabolic process results in a net gain of 2 molecules of ATP and 4 molecules of reduced nicotinamide adenine dinucleotide (NADH). This process also generates acetyl–coenzyme A (acetyl-CoA) from pyruvate via pyruvate dehydrogenase, which is a vital source for energy production. The enzymatic reactions involved in phosphorylation are irreversible; however, alternative enzymatic processes can recreate the metabolic precursor and allow for gluconeogenesis.
Galactose metabolism begins with a phosphorylation step as well; however, the process results in the formation of uridine diphosphate glucose. Uridine diphosphate glucose can then be transformed in 2 ways. The first is conversion to glucose-1-phosphate, which will then enter carbohydrate metabolism and follow a pathway similar to that described above. The second is via glycogen synthase, which results in the creation of glycogen. Glycogen provides a means to store glucose for future energy need during periods of fasting. The overall process is regulated via substrates, metabolites, and enzymes, along with hormones such as insulin, to ensure that glycolysis, glycogenesis, and glycogenolysis are being adequately maintained.
Carbohydrate Metabolism Dysfunction and Treatment. Given the sheer number of reactions in the metabolic pathways described above, the potential for error is significant. Several IEMs have been identified in carbohydrate metabolism and may affect the ability to break down monosaccharides, such as fructose and galactose, into pyruvate precursors. For example, a patient with aldose B deficiency or hereditary fructose intolerance accumulates fructose-1-phosphate, resulting in corresponding symptoms.7 The patient may present with vomiting, hypoglycemia, abdominal pain, lethargy, convulsions, jaundice, and hepatomegaly.8 If the disease is not identified in a timely manner, the patient will continue to experience hypoglycemic events and eventually progress to hepatic failure, renal failure, and death. Symptoms and complications of hereditary fructose intolerance can be avoided by restricting dietary intake of fructose.7,9 Galactose-1-phosphate uridyl transferase deficiency, or galactosemia, will cause accumulation of galactose-1-phosphate, which may result in symptoms similar to those of hereditary fructose deficiency. In addition, patients with galactosemia may develop cataracts, intellectual disability, verbal dyspraxia, hypergonadic hypogonadism, increased risk for Escherichia coli sepsis, and coagulopathies.10,11 Avoidance of lactose and galactose is the mainstay of treatment. Rapid identification and dietary restrictions for galactosemia can reverse some of the complications and prevent intellectual disability.
A defect in the pyruvate dehydrogenase complex can result in a wide spectrum of dysfunctions ranging from ataxia to death.12 Neonates may present more severely, with a profound lactic acidosis and eventual progression to encephalopathy and/or death. Those who present within the first few months of life may have psychomotor retardation and significant lactic acidosis. Patients who present at an older age may retain some enzymatic activity and have less severe complications. In rare cases, pyruvate dehydrogenase complex deficiency may be caused by a mutation that alters the affinity for thiamine phyrophosphate, a coenzyme in the metabolic process that may help facilitate normal function and minimize toxic metabolites.13 Because of this, thiamine is routinely supplemented when pyruvate dehydrogenase complex deficiency is part of the differential diagnosis. Thiamine may be initiated at a dose of 5 to 50 mg/kg/day based on response, physician preference, and adverse effects (Table 1).14–16
Table 1.
Medications Used in Select Inborn Errors of Metabolism (IEMs)
| Medication | IEM | Formulations | Initial Dose | Dose Range | FDA Status | Adverse Effects | Additional Pearls |
|---|---|---|---|---|---|---|---|
| Arginine | Urea cycle disorders (not arginase deficiency) | IV Oral powder |
200–600 mg/kg load | 200–600 mg/kg/day infusion17,18 170–700 mg/kg/day |
Drug Supp |
Hyperkalemia, hyperchloremia, extravasation, flushing, hypotension, HA, nausea, vomiting, cerebral edema, metabolic acidosis19,20 | Contraindicated in arginase deficiency;21 not effective when hyperammonia is 2° to organic acidemias |
| Ascorbic acid (vitamin C) | Glutathione synthetase deficiency Hawkinsuria Tyrosinemia type III22 |
Capsules, tablets, powders, oral liquid, IV | 100 mg | 15–25 mg/kg/day (2 g/day max)23 | Drug | HA, insomnia, GI upset, hyperglycemia, nephrolithiasis, iron overload, zinc deficiency, copper deficiency24,25 | Pediatric adverse effects may be limited to patients with renal dysfunction or those receiving very high doses25,26 |
| Betaine | Homocystinuria | Powder for reconstitution | 50 mg/kg/day27 | 50–250 mg/kg/day (20 g/day max)27,28 | Drug | GI upset, nausea, diarrhea, irritability, agitation, depression, anorexia, body odor, cerebral edema29 | Monitor methionine levels to minimize risk of cerebral edema |
| Biotin | Pyruvate or multiple carboxylase deficiency | Tablets, capsules, liquid | 10–20 mg/day | 5–20 mg/day13,30,31 | Supp | GI upset32 | Large doses may interact with thyroid assays33,34 |
| Coenzyme Q10 | Electron transport chain disorders Primary coenzyme Q10 deficiency |
Tablets, capsules, wafers, powder, syrup | 50 mg/kg/day35 | 1.5–50 mg/kg/day (3 g/day max)35,36 | Supp | GI upset, thrombocytopenia, purpura, sinusitis, depression, anxiety37 | Lipophilic, administer with a high-fat meal to facilitate absorption38 |
| Carglumic acid | N-acetyl glutamine synthetase deficiency | Tablet for suspension (dissolve 200-mg tablet in 2.5 mL of water)39 | 100 mg/kg/day40 | 100–300 mg/kg/day18,30,40 | Drug | Bitter taste, hyperhidrosis, diarrhea, vomiting, abdominal pain, decreased appetite, anemia, fever, HA, somnolence, tachycardia40 | |
| Folic acid | Homocystinuria | Tablet, capsule, IV, compounded solution41 | 5 mg | 1–20 mg27,29,42 | Drug | Loss of appetite, nausea, insomnia, confusion, irritability, depression, eczema, thromboemboli | May interact with hepatically metabolized antiepileptics43,44 |
| Hydroxycobalamin | Homocystinuria Disorders of cobalamin metabolism |
IV, IM Tablets |
1 mg | 1–20 mg/day45,46 or 1 mg/wk47 | Drug Supp |
Red discoloration of urine, hypertension, erythema, rash, GI upset, HA, anaphylaxis48 | |
| Levocarnitine | Carnitine deficiency, Maple syrup urine disease | IV Oral solution, tablets |
400 mg/kg/day49 50–100 mg/kg/day |
100–400 mg/kg/day 100–200 mg/kg/day |
Drug | GI upset, muscle weakness, hypertension, hypotension, tachyarrhythmia, anemia, HA50 | Renally eliminated but toxicity may be related to metabolites51 |
| Nitisinone | Tyrosinemia type I | Tablet, capsule, oral suspension | 1 mg/kg/day52 | 0.6–2 mg/kg/day52 | Drug | Eye pain, eye itching, corneal crystals, intellectual decline, developmental delay, seizures53–55 | Metabolized via CYP2C9 and CYP3A4, may interact with other medications56 |
| Phenylbutyrate | Urea cycle disorders | Tablet, powder, oral liquid, compounded suspension57 | 5 g/m2/day58 | 450–600 mg/kg/day (patients <20 kg) 5–13 g/m2/day (patients >20 kg) |
Drug | Respiratory infections, cough, vomiting, diarrhea, gastroenteritis, decreased appetite, pharyngitis59 | GI intolerances may decline over time59 |
| Pyridoxine | Homocystinuria Pyridoxine-dependent epilepsy, ornithine deficiency, primary hyperoxaluria type 122 |
Capsules, tablets, IV, compounded suspension41,60 | 100 mg | 5–30 mg/kg/day(max 1 g/day)42,61 | Drug | Peripheral neuropathy, respiratory depression, HA, somnolence, dermatitis, seizures, decreased folic acid levels30,31 | If pyridoxine is ineffective, trial combined pyridoxineand folic acid therapy27,29,42 |
| Riboflavin (vitamin B2) | Electron transfer flavoprotein defects, Glutaric aciduria, multiple acyl-CoA dehydrogenase deficiency22 | Tablet, capsule, compounded suspension62 | 100 mg/day30 | 3–20 mg/kg/day (max of 150 mg/day)22,30 | Supp | Yellow-orange urine discoloration, urticaria, anaphylaxis63 | |
| Sapropterin | Phenylketonuria | Tablet, powder for oral solution | 20 mg/kg/day (10 mg/kg/day for children younger than 7 yr) | 5–20 mg/kg/day64–66 | Drug | HA, amnesia, dizziness, memory impairment, dysphonia, rhinorrhea, diarrhea, vomiting, bone pain65,66 | Although sapropterinis a synthetic of BH4, therapy is used for all forms of phenylketonuria64,66 |
| Sodium phenylacetate/sodium benzoate | Urea cycle disorders | IV | 250–500 mg/kg during 1–2 hr | 200–500 mg/kg/day as a continuous infusion17,18 | Drug | Non-pleasant odor, hypernatremia, hypokalemia, hypocalcemia, metabolic acidosis, extravasation, hyperglycemia, anemia | Must be infused via acentral line |
| Thiamine (vitamin B1) | Pyruvate dehydrogenase complex deficiency, Maple syrup urine disease | Tablets, IV, compounded suspension67 | 100 mg IV daily | 5–50 mg/kg/day(max 1 g/day)14–16,22 | Drug | Diaphoresis, pruritus, edema, injections site pain (with IV), anaphylaxis (rarely)16 | Contains aluminum (caution in patients with renal dysfunction) |
GI, gastrointestinal; HA, headache; IM, intramuscular; IV, intravenous; Supp, supplement
Table 1.
Medications Used in Select Inborn Errors of Metabolism (IEMs) (cont.)
| Medication | IEM | Formulations | Initial Dose | Dose Range | FDA Status | Adverse Effects | Additional Pearls |
|---|---|---|---|---|---|---|---|
| Arginine | Urea cycle disorders (not arginase deficiency) | IV Oral powder |
200–600 mg/kg load | 200–600 mg/kg/day infusion17,18 170–700 mg/kg/day |
Drug Supp |
Hyperkalemia, hyperchloremia, extravasation, flushing, hypotension, HA, nausea, vomiting, cerebral edema, metabolic acidosis19,20 | Contraindicated in arginase deficiency;21 not effective when hyperammonia is 2° to organic acidemias |
| Ascorbic acid (vitamin C) | Glutathione synthetase deficiency Hawkinsuria Tyrosinemia type III22 |
Capsules, tablets, powders, oral liquid, IV | 100 mg | 15–25 mg/kg/day (2 g/day max)23 | Drug | HA, insomnia, GI upset, hyperglycemia, nephrolithiasis, iron overload, zinc deficiency, copper deficiency24,25 | Pediatric adverse effects may be limited to patients with renal dysfunction or those receiving very high doses25,26 |
| Betaine | Homocystinuria | Powder for reconstitution | 50 mg/kg/day27 | 50–250 mg/kg/day (20 g/day max)27,28 | Drug | GI upset, nausea, diarrhea, irritability, agitation, depression, anorexia, body odor, cerebral edema29 | Monitor methionine levels to minimize risk of cerebral edema |
| Biotin | Pyruvate or multiple carboxylase deficiency | Tablets, capsules, liquid | 10–20 mg/day | 5–20 mg/day13,30,31 | Supp | GI upset32 | Large doses may interact with thyroid assays33,34 |
| Coenzyme Q10 | Electron transport chain disorders Primary coenzyme Q10 deficiency |
Tablets, capsules, wafers, powder, syrup | 50 mg/kg/day35 | 1.5–50 mg/kg/day (3 g/day max)35,36 | Supp | GI upset, thrombocytopenia, purpura, sinusitis, depression, anxiety37 | Lipophilic, administer with a high-fat meal to facilitate absorption38 |
| Carglumic acid | N-acetyl glutamine synthetase deficiency | Tablet for suspension (dissolve 200-mg tablet in 2.5 mL of water)39 | 100 mg/kg/day40 | 100–300 mg/kg/day18,30,40 | Drug | Bitter taste, hyperhidrosis, diarrhea, vomiting, abdominal pain, decreased appetite, anemia, fever, HA, somnolence, tachycardia40 | |
| Folic acid | Homocystinuria | Tablet, capsule, IV, compounded solution41 | 5 mg | 1–20 mg27,29,42 | Drug | Loss of appetite, nausea, insomnia, confusion, irritability, depression, eczema, thromboemboli | May interact with hepatically metabolized antiepileptics43,44 |
| Hydroxycobalamin | Homocystinuria Disorders of cobalamin metabolism |
IV, IM Tablets |
1 mg | 1–20 mg/day45,46 or 1 mg/wk47 | Drug Supp |
Red discoloration of urine, hypertension, erythema, rash, GI upset, HA, anaphylaxis48 | |
| Levocarnitine | Carnitine deficiency, Maple syrup urine disease | IV Oral solution, tablets |
400 mg/kg/day49 50–100 mg/kg/day |
100–400 mg/kg/day 100–200 mg/kg/day |
Drug | GI upset, muscle weakness, hypertension, hypotension, tachyarrhythmia, anemia, HA50 | Renally eliminated but toxicity may be related to metabolites51 |
| Nitisinone | Tyrosinemia type I | Tablet, capsule, oral suspension | 1 mg/kg/day52 | 0.6–2 mg/kg/day52 | Drug | Eye pain, eye itching, corneal crystals, intellectual decline, developmental delay, seizures53–55 | Metabolized via CYP2C9 and CYP3A4, may interact with other medications56 |
| Phenylbutyrate | Urea cycle disorders | Tablet, powder, oral liquid, compounded suspension57 | 5 g/m2/day58 | 450–600 mg/kg/day (patients <20 kg) 5–13 g/m2/day (patients >20 kg) |
Drug | Respiratory infections, cough, vomiting, diarrhea, gastroenteritis, decreased appetite, pharyngitis59 | GI intolerances may decline over time59 |
| Pyridoxine | Homocystinuria Pyridoxine-dependent epilepsy, ornithine deficiency, primary hyperoxaluria type 122 |
Capsules, tablets, IV, compounded suspension41,60 | 100 mg | 5–30 mg/kg/day(max 1 g/day)42,61 | Drug | Peripheral neuropathy, respiratory depression, HA, somnolence, dermatitis, seizures, decreased folic acid levels30,31 | If pyridoxine is ineffective, trial combined pyridoxineand folic acid therapy27,29,42 |
| Riboflavin (vitamin B2) | Electron transfer flavoprotein defects, Glutaric aciduria, multiple acyl-CoA dehydrogenase deficiency22 | Tablet, capsule, compounded suspension62 | 100 mg/day30 | 3–20 mg/kg/day (max of 150 mg/day)22,30 | Supp | Yellow-orange urine discoloration, urticaria, anaphylaxis63 | |
| Sapropterin | Phenylketonuria | Tablet, powder for oral solution | 20 mg/kg/day (10 mg/kg/day for children younger than 7 yr) | 5–20 mg/kg/day64–66 | Drug | HA, amnesia, dizziness, memory impairment, dysphonia, rhinorrhea, diarrhea, vomiting, bone pain65,66 | Although sapropterinis a synthetic of BH4, therapy is used for all forms of phenylketonuria64,66 |
| Sodium phenylacetate/sodium benzoate | Urea cycle disorders | IV | 250–500 mg/kg during 1–2 hr | 200–500 mg/kg/day as a continuous infusion17,18 | Drug | Non-pleasant odor, hypernatremia, hypokalemia, hypocalcemia, metabolic acidosis, extravasation, hyperglycemia, anemia | Must be infused via acentral line |
| Thiamine (vitamin B1) | Pyruvate dehydrogenase complex deficiency, Maple syrup urine disease | Tablets, IV, compounded suspension67 | 100 mg IV daily | 5–50 mg/kg/day(max 1 g/day)14–16,22 | Drug | Diaphoresis, pruritus, edema, injections site pain (with IV), anaphylaxis (rarely)16 | Contains aluminum (caution in patients with renal dysfunction) |
GI, gastrointestinal; HA, headache; IM, intramuscular; IV, intravenous; Supp, supplement
Alternatively, dysfunction in the pyruvate carboxylase enzyme may be due to defects in the metabolic processes that create biotin, a necessary coenzyme for the reaction. A dysfunction in biotin production results in multiple carboxylase deficiency and poor function in all carboxylation reactions. Pyruvate carboxylase deficiency and multiple carboxylase deficiency can range from mild to severe and can include lactic acidosis, ketoacidosis, hyperammonemia, organic acidurias, ataxia, motor deficits, neurologic deficits, and death.68 Biotin stores can be depleted over time by inadequate consumption and absorption. Subsequent supplementation with biotin can be initiated and is generally well tolerated. More information regarding biotin pharmacotherapy can be found in Table 1.
Metabolic errors can impair pathways for energy storage or glucose regeneration. Twelve different glycogen storage diseases have been identified that prevent glycogenolysis and affect glycogen quality, quantity, or both.69 One of these storage diseases, type II glycogen storage disease, or Pompe disease, is caused by a defect in the lysosomal α-1,4-glucosidase, thus preventing the release of glucose-1-phosphate from glycogen macromolecules in the lysosomes. This disease state results in glycogen accumulation in muscle tissues, and subsequently hypotonia similar to that of muscular dystrophies.70 Additional complications include feeding difficulties, hypertrophic cardiomyopathy, hepatomegaly, and cardiorespiratory failure. Interestingly, Pompe disease does not present with hypoglycemia, because cytoplasmic glycogen metabolism remains unaffected.71
Recently, increased research and drug development for novel therapies has resulted in the creation of several recombinant enzymes for patients with an IEM. A recombinant enzyme has been engineered for Pompe disease. Recombinant therapy may help stabilize the deterioration associated with Pompe disease by improving motor function and preventing pulmonary dysfunction.72 Other recombinant enzymes have been developed for Fabry disease, Gaucher disease, and various mucopolysaccharidoses. Most of the recombinant enzymes are derived from a Chinese hamster ovary cell line. Thus, the most common adverse medication events are infusion related, such as tachypnea, rash, flushing, urticaria, agitation, rigors, and tachycardia. Antibody development is quite common, and many patients will develop immunoglobin G to these enzymes. This may result in anaphylaxis or a decreased therapeutic response to these agents.70 Table 2 lists several recombinant enzymes that are used to treat various IEMs, along with each enzyme's indication, dose, and common adverse events. These products are all given via intravenous infusion every 1 to 2 weeks. Given the concern for anaphylactic reactions, the patient must make routine trips to an acute care center or outpatient infusion center to receive their therapy. The method of obtaining the enzymes will depend on the insurer and may require that the product be obtained from the manufacturer. Initiation of enzyme replacement therapy may be delayed while issues around documentation, medication transport, and insurance coverage are addressed.
Table 2.
Select Recombinant Enzymes Available for Inborn Errors of Metabolism
| Medication | Disease | Mechanism of Action | Dose | Adverse drug events |
|---|---|---|---|---|
| Agalsidase beta | Fabry disease | Recombinant α-galactosidase from CHO | 1 mg/kg every 14 days | Infusion-related reactions, nausea, chest pain, muscle pain, antibody development |
| Alglucosidase alfa | GSD type II | Recombinant form of acid-α-glucosidase | 20 mg/kg every 14 days | Infusion-related reactions, anaphylaxis, IgG antibody development, nephrotic syndrome, infections |
| Elosulfase alfa | MPS IVA | Recombinant N-acetylgalactosamine-6-sulfatase from CHO | 2 mg/kg every 7 days | Infusion-related reactions, nausea, vomiting, headache, abdominal pain, antibody development, fatigue |
| Galsulfase | MPS VI | Recombinant N-acetylgalactosamine from CHO | 1 mg/kg every 7 days | Infusion-related reactions, chest pain, muscle pain, abdominal pain, antibody development |
| Idursulfase | MPS II | Recombinant iduronate-2-sulfatase from human cell line | 0.5 mg/kg every 7 days | Infusion-related reactions, fatigue, musculoskeletal pain, antibody development, ear infections |
| Imiglucerase | Gaucher disease | Recombinant β-glucocerebrosidase from CHO | 30–60 units/kg every 2 weeks | Infusion-related reactions, nausea, antibody development, dizziness, headache |
| Laronidase | MPS I | Recombinant analog of α-l-iduronidase from CHO | 100 units/kg every 7 days | Infusion-related reactions, chest pain, facial edema, antibody development, headache, hyperreflexia |
| Taliglucerase alfa | Gaucher disease | Recombinant analog of glucocerebrosidase from carrot cell culture | 60 units/kg every 14 days | Infusion-related reactions, headache, arthralgia, antibody development, dizziness, fatigue |
| Velaglucerase alfa | Gaucher disease | Recombinant form of glucocerbrosidase from human cell line | 60 units/kg every 14 days | Hypersensitivity reaction, headache, dizziness, pyrexia, abdominal pain, back pain, joint pain, asthenia, fatigue |
CHO, Chinese hamster ovary; GSD, glycogen storage disease; IgG, immunoglobulin G; MPS, mucopolysaccharidosis
Errors in Lipid Metabolism
Fatty Acid Metabolism. Fatty acids provide a vital and efficient source of energy for cells. Most fatty acids are obtained through the diet and used for energy or phospholipid production, stored for later use, or converted into bile acids. As with carbohydrate metabolism, hormones and catecholamines play integral roles in determining the ultimate fate of the fatty acid.
A wide variety of fatty acids exist, and fatty acid composition is dependent on carbon chain length, the number of double bonds, and the position of the double bonds in the chain. Despite this, the metabolic processes that fatty acids undergo follow a similar pathway. All fatty acid metabolism begins by activation via acyl CoA synthetase in the outer mitochondrial membrane. The fatty acid molecule binds with CoA to form an acyl CoA molecule. The acyl CoA then combines with carnitine to enter into the inner mitochondrial membrane for metabolism. Carnitine acyltransferase-I facilitates the binding of acyl CoA in the outer mitochondrial membrane, whereas carnitine acyltransferase-II frees the carnitine molecule in the inner mitochondrial membrane for further acyl CoA metabolism. Various subtypes of carnitine acyltransferase exist, and the type used is dependent on the type of the fatty acid.
The acyl CoA molecule is then oxidized to generate acetyl-CoA for energy production. A major enzyme responsible for this reaction is acyl-CoA dehydrogenase. Much like acyltransferase, there are several subtypes, and the structure of the fatty acid determines which enzyme will continue the metabolic process. After this step, further metabolism results in the release of 1 molecule of acetyl-CoA and an acyl CoA shortened by 2 carbon groups. Eventually, only a 4- or 5-carbon chain will remain. An additional cycle will produce either 2 molecules of acetyl-CoA or a molecule of acetyl-CoA and propionyl-CoA, depending on the remaining length of the carbon chain.5
This process results in the creation of an acetyl-CoA which is a 2-carbon chain. This is known as β-oxidation because the last 2 carbon atoms are cleaved from the fatty acid chain. Fatty acid metabolism can be accomplished via several different mechanisms, such as α-, β-, or ω- oxidation. α-Oxidation results in the shortening of the fatty acid chain via 1 carbon group. β-Oxidation as described is the primary mechanism of fatty acid metabolism in humans. ω-Oxidation results in the terminal carbon group's conversion to succinate or adipic acid. Although they are an infrequent method of fatty acid metabolism, ω-oxidation pathways may become more prevalent in patients with disorders in β-oxidation.
Lipid Metabolism Dysfunction and Treatment. As with carbohydrate metabolism, errors in lipid metabolism have been identified in several steps in the process. Common modalities of dysfunction include dysfunction or deficiency of the carnitine transferase and acyl CoA dehydrogenase enzymes. Because enzymatic function is dependent on the fatty acid structure, dysfunction could occur in any of the various carnitine transferase or acyl CoA dehydrogenase enzymes. In these cases, the most common presentation will be life-threatening coma during periods of fasting and hypoglycemia without ketosis, which is also known as hypoketotic hypoglycemia.13,73 Additional clinical manifestations will primarily affect cells in the liver, skeletal, and cardiac muscle, where β-oxidation is greatly used.
Two types of levocarnitine deficiencies exist. Primary carnitine deficiency is due to an inability of acyl CoA to effectively bind to levocarnitine and enter the inner mitochondrial membrane. This disorder results in lower carnitine levels. A secondary carnitine deficiency occurs when an acyl CoA dehydrogenase enzyme is dysfunctional, resulting in levocarnitine rebinding with acyl CoA and lower levels of free levocarnitine for transport of fatty acids into the inner mitochondrial membrane. In either disorder, patients may present with cardiomyopathy, skeletal muscle weakness, lower carnitine levels, renal tubular acidosis, hyperammonia, myoglobinuria, rhabdomyolysis, or hypoketotic hypoglycemia. Without levocarnitine supplementation, this disease can progress to rhabdomyolysis or death.30,74 Levocarnitine supplementation is common in patients with fatty acid metabolism disorders, although use is controversial outside of primary carnitine deficiency.75 Depending on the severity of presentation, therapy may be initiated with intravenous levocarnitine. Once the patient has stabilized therapy can be transitioned to enteral formulations. More information regarding levocarnitine can be found in Table 1.
Acyl CoA enzymes comprise the small-, medium-, long-, and very long–chain acyl CoA dehydrogenases, and the acyl CoA enzyme used is dependent on the size of the acyl CoA molecule. The most common fatty acid β-oxidation deficit is in the medium-chain acyl CoA dehydrogenase enzyme.76 Medium-chain acyl CoA de-hydrogenase (MCAD) facilitates the degradation of fatty acids between 4 and 12 carbon units in length.73 Patients with MCAD deficiency may present with vomiting, hypoketotic hypoglycemia, and lethargy, which can progress to seizures or coma.76 Secondary carnitine deficiency will develop over time as further acyl CoA metabolism is halted. Patients with very long–chain acyl CoA dehydrogenase deficiency, which metabolizes fatty acids with 14 to 20 carbons, will have symptoms similar to those with MCAD deficiency. However, they will have a greater severity of disease and may also have cardiomyopathy, a Reye-like disease, or rhabdomyolysis.77 Short-chain acyl CoA dehydrogenase is often identified in patients with elevated blood concentrations of ethylmalonic acid.78 As with other metabolic disorders, there is a wide range of disease severity, and this may be similar to the other fatty acid oxidation disorders. Patients with any form of acyl CoA dehydrogenase deficiency will most likely be initiated on a 10% dextrose infusion to curtail the acute event. Those with long- or very long–chain acyl CoA dehydrogenase deficiency may eventually be started on medium-chain triglyceride oil. This will provide the patients with calories while avoiding the toxic metabolic pathways.74,77
Finally, defects have been identified in the processes needed to convert propionyl CoA to substrates used in aerobic metabolism. Dysfunction of propionyl CoA carboxylase can be due to deficiency of the enzyme itself or due to multiple carboxylase deficiency, as previously described. This will result in propionic acidemia. Furthermore, methionine, threonine, isoleucine, and valine produce propionyl CoA through the course of their metabolism. Hypotonia, lethargy, dehydration, and metabolic acidosis may be present. Over time this disorder may lead to mental retardation and additional neurologic complications.79–81 Guidelines exist for the treatment of propionic acidemia.80,82 As with other forms of fatty acid dysfunction, initial support consists of hydration, preferably with a dextrose infusion to halt the use of lipids and amino acids for energy production. Special formulas or parental nutrition may be required to limit the amount of valine, isoleucine, threonine, and methionine given during the crisis. Levocarnitine supplementation may be necessary to help normalize β-oxidation and improve acidosis.81 In the setting of multiple carboxylase deficiency, biotin may be supplemented. Additionally, metronidazole may be used for 10 days to eradicate gastrointestinal flora that produce branched chain amino acids and contribute to the propionic acidemia.80
Errors in the Electron Transport Chain
The Citric Acid Cycle and Electron Transport Chain. The citric acid cycle is a vital component to aerobic metabolism and energy production through the electron transport chain. Acetyl-CoA, generated from either carbohydrate, fatty acid, or amino acid metabolism, is essential to this process. The process begins when acetyl-CoA combines with oxaloacetate to form citrate. During the course of the citric acid cycle approximately 1 molecule of ATP, 1 molecule of reduced flavin adenine dinucleotide, and 3 molecules of NADH are generated and the oxaloacetate is regenerated for further use in the cycle. The molecules of NADH and reduced flavin adenine dinucleotide can then be used for additional energy production in the electron transport chain.
The electron transport chain comprises 4 complexes and ATP synthase. These complexes facilitate the transfer of hydrogen atoms out of the mitochondrial matrix for the creation of an electron gradient. This promotes the flow of hydrogen protons through ATP synthase to generate a vast number of ATPs. Complex I is the NADH dehydrogenase complex. This protein facilitates the transfer of hydrogen atoms from NADH to ubiquinone, or coenzyme Q10 (Figure). Ubiquinone acts as a transport molecule, providing a mechanism for the hydrogen ions to pass across the membrane into the intermembrane space. This also restores the oxidized NAD molecules for further use in carbohydrate metabolism and in the citric acid cycle. Complex II, or the succinate dehydrogenase complex, transfers hydrogen atoms from succinate to reduced flavin adenine dinucleotide, and finally to ubiquinone for transport into the intermembrane space. The hydrogen-laden ubiqui-none molecules from complexes I and II then interact with complex III, or the cytochrome bc1 complex. This protein uses the hydrogen molecules to expel hydrogen from the mitochondrial matrix into the intermembrane space and generate an electron gradient. Complex IV, or cytochrome oxidase, then maintains the hydrogen gradient and facilitates the creation of water molecules from O2 and hydrogen. Finally, ATP synthase uses the protons to spin a central shaft in the molecule and generate molecules of ATP.
Figure.
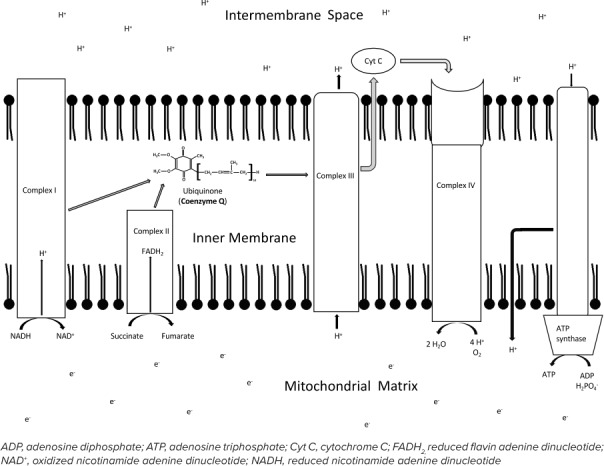
The electron transport chain.
The recycling of hydrogen protons in the process results in the generation of around 2.5 and 1.5 molecules of ATP for each molecule of NADH and reduced flavin adenine dinucleotide, respectively.5 Overall, around 90% of cellular ATP is produced via the electron transport chain and regenerates oxidized NAD without producing lactic acid.83
Electron Transport Dysfunction and Treatment. Defects have been discovered in the electron transport chain and in flavoprotein processes necessary for the transfer of electrons into the mitochondrial matrix. A defect in complexes I through IV or in ATP synthase can result in mitochondrial oxidation dysfunction.84 Because this methodology of energy production is widespread in tissues, the disorder will present with a broad array of complications, including lactic acidosis. One such disorder, Leigh disease, encompasses a variety of metabolic dysfunctions that result in similar characteristics and progressive encephalomyopathies.85,86 Leigh disease is most commonly associated with defects in complex I, complex IV, or in the pyruvate dehydrogenase complex.10 Patients with Leigh disease will present with hypotonia, developmental regression or arrest, demyelination, marked gliosis, and necrotizing encephalomyopathy.83 Other dysfunctions in the electron transport chain include mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes otherwise known as MELAS; myoclonus epilepsy with ragged red fibers, or MERFF; and external ophthalmoplegia, myopathies, retinal degeneration, and heart block or Kearns-Sayre syndrome. Coenzyme Q10 may be started in any of these disorders. Coenzyme Q10 plays an instrumental role in the transfer of hydrogen across the mitochondrial membrane as previously described. When coenzyme Q10 is deficient, protons will remain in the mitochondrial matrix, diminish the electron gradient, and result in an overall decline in ATP production.36 When a defect is suspected in the electron transport chain, coenzyme Q10 will often be administered. Additional information about coenzyme Q10 can be found in Table 1. Levocarnitine supplementation may be initiated as well if there is concern that stores have been depleted.
Complex II contains electron transfer flavoproteins, such as flavin mononucleotide and flavin adenine dinucleotide. Dysfunction in electron transfer flavoprotein dehydrogenase or in the flavoprotein itself can result in inability to transfer hydrogen atoms across the membrane layer. This results in multiple acyl-CoA dehydrogenase deficiency or glutamic aciduria type 2.87,88 Patients with this disorder may present with acidosis, urinary organic acidemia, hypoglycemia, coma, hypotonia, and cardiomyopathy.87 Partial deficiencies may present in a similar manner to MCAD deficiency and subsequent levocarnitine depletion.88,89 Riboflavin, or vitamin B2, is the key component of flavin mononucleotide and flavin adenine dinucleotide.87 Humans are unable to make riboflavin and must rely on dietary intake to maintain levels.87 Supplementation may be used in patients with electron transfer flavoprotein defects or in combination with coenzyme Q10 in Leigh disease.38,88 Providing 100 mg of riboflavin per day in 2 to 3 divided doses to all patients with electron transport flavoprotein defects may limit clinical manifestations of the disease.30 Refer to Table 1 for more information regarding riboflavin.
Errors in Amino Acid Metabolism
Amino acid synthesis and metabolism is an extremely complex topic and much too broad to completely cover in this overview. With 20 different amino acids, the sheer potential for an IEM can be overwhelming. This section will cover 4 of the more common IEMs that occur in amino acid metabolism, such as phenylketonuria, tyrosinemia, homocystinuria, and maple syrup urine disease.
Phenylalanine and Tyrosine. Phenylalanine is an essential amino acid and is a precursor for tyrosine, dopamine, norepinephrine, and epinephrine in the body. Once ingested, phenylalanine is converted to tyrosine by phenylalanine hydroxylase. Tetrahydrobiopterin is a vital cofactor that facilitates the conversion of phenylalanine to tyrosine via phenylalanine hydroxylase. Tyrosine can then progress along 2 different enzymatic pathways. One pathway results in generation of the neurotransmitters L-DOPA, dopamine, norepinephrine, and epinephrine. When there are excessive levels of tyrosine, or the patient is in a catabolic state, tyrosine will undergo enzymatic degradation via a second pathway. This will result in the creation of fumarate and acetoacetate, which can then be used for energy production.
The most common IEM in amino acid metabolism stems from dysfunction of the phenylalanine hydroxylase enzyme or an inadequate supply of tetrahydrobiopterin.90,91 This disorder, known as phenylketonuria, results in excessive amounts of phenylalanine and subsequent shunting of the amino acid to secondary metabolic pathways. This secondary pathway produces phenylpyruvate, a ketone that provides the disease with its name. Phenylpyruvate is then metabolized via unknown enzymes to phenyllactate and phenylacetate, which has a distinctive, musty odor.92,93 The metabolites are excreted in the urine; therefore, patients with an acute bout of phenylketonuria commonly present with malodorous urine. The excessive phenylalanine will negatively impact the ability of other aromatic amino acids to cross the blood-brain barrier, limiting the production of several neurotransmitters and resulting in profound intellectual disability and neurologic abnormalities.90,94 Fortunately, a screening test for phenylketonuria was one of the first neonatal screenings developed for mass testing, and its relatively inexpensive cost has allowed for early identification in several countries.95
Primary treatment for all of these disorders will include dietary restriction of the problematic amino acid and metabolic precursors. By limiting dietary intake of these amino acids and monitoring the amino acid in the blood, disease progression may often be limited or reversed. Warning labels have been added to some food products, such as those with phenylalanine, to alert patients and avoid potential inadvertent ingestion. However, complete avoidance of the problematic amino acid, especially essential amino acids that are not produced within the body, may result in complications as well. Therefore, amino acid level monitoring and maintenance with a goal range is imperative. This goal range for phenylketonuria is between 2 mg/dL and 6 mg/dL. There is a potential for expanding the range when patients enter adolescence; however, dietary restriction should continue because there is concern for deterioration of intelligence and cognitive performance even into adulthood. Supplementation of cofactors in the errant enzyme may also help to provide complete or partial function to the enzyme. Those with a deficiency in tetrahydrobiopterin may not respond to dietary phenylalanine restriction. These patients will benefit from the initiation of sapropterin, a synthetic form of tetrahydrobiopterin. Sapropterin may be initiated at 20 mg/kg/day, although children younger than 7 years may be at a greater risk for hypophenylalaninemia with this dose.64–66 Also, tetrahydrobiopterin is regenerated via dihydrofolate reductase and dihydropteridine reductase. Therefore, patients with defects in tetrahydrobiopterin production should avoid agents such as trimethoprim/sulfamethoxazole, methotrexate, and other antileukemic agents that affect the enzymes that regenerate tetrahydrobiopterin.91 Patients with phenylketonuria not related to a deficit in tetrahydrobiopterin may also benefit from sapropterin supplementation. In these patients, sapropterin may help reduce phenylalanine levels, allowing the patient with phenylketonuria to ingest more phenylalanine while maintaining goal levels.64,66
Tyrosinemia may result from a defect in 1 of 3 different enzymes. Each form of tyrosinemia presents with different manifestations and complications. Tyrosinemia type I is caused by a defect in fumarylacetoacetate hydrolase. This results in degradation to secondary metabolites, including succinylacetone, which is believed to be the primary toxic metabolite.96,97 Patients with tyrosinemia type I, or hepatorenal tyrosinemia, will present with an acute hepatic crisis, fever, irritability, vomiting, hepatomegaly, jaundice, and elevated transaminases. Patients may also experience peripheral neuropathy, renal dysfunction, a Fanconi-like syndrome, and hepatocellular carcinoma.52,96–98 Tyrosinemia type II results from dysfunction at the tyrosine aminotransferase enzyme. This disorder is referred to as oculocutaneous tyrosinemia and produces sequelae such as excessive ocular redness, tearing, and pain; photophobia; herpetiform corneal ulcers; and, eventually, palmar and plantar hyperkeratosis.97,99 Tyrosinemia type III is the least common tyrosinemia and is due to a deficiency in 4-hydroxyphenylpyruvate dioxygenase.47 Patients with tyrosinemia type III may be asymptomatic or present with neurologic manifestations, such as self-destructive behavior, intermittent ataxia, seizures, and developmental delay.47,97 Although 4-hydroxyphenylpyruvate is found in liver and kidney tissue, a deficit in the enzymatic function does not result in injury to these organs.47
Treatment of all three types of tyrosinemia begins with dietary restriction of tyrosine in addition to its precursor, phenylalanine. For type II tyrosinemia, dietary restriction may be the only therapy needed to avoid disease-related complications.99 In other tyrosinemias, additional therapies may be warranted. Nitisinone is the drug of choice for tyrosinemia type I. Nitisinone inhibits 4-hydroxyphenylpyruvate, essentially changing those with type I tyrosinemia to type III. This decreases liver involvement and may negate the need for future liver transplantation, but it does not eliminate the risk for hepatocellular carcinoma.96 Given the conversion from type I to type III, side effects will be similar to the symptoms of type III tyrosinemia. Ascorbic acid supplementation may be attempted for patients with tyrosinemia type III. Ascorbic acid acts as a coenzyme and electron donor in 4-hydroxyphenylpyruvate conversion of 4-hydroxyphenylpyruvate to homogentisate.24 Additional information about these therapies can be found in Table 1.
Methionine. Methionine catabolism begins with the generation of S-adenosylmethionine. S-adenosylmethionine plays an essential role in methylating more than 115 different reactions, including those required for phospholipid, neurotransmitter, and glutathione production.5 Once S-adenosylmethionine has performed its function as a methyl donor, it is converted to homocysteine, where methionine can be regenerated via N5-methyl tetrahydrofolate.29 Alternatively, homocysteine can be degraded in a catabolic state to create the amino acid cysteine. Further metabolism of cysteine results in propionyl-CoA production and eventually molecules that will result in the generation of ATP.
Homocysteine is created in all cells but detoxified in the kidney and liver.29 Dysfunction of homocysteine metabolism results in homocystinuria as the excessive quantities of homocysteine are eliminated in the urine. There are 3 different dysfunctions that can lead to homocystinuria. Deficiency of cystathionine β-synthase is the most common cause. Subluxation of the ocular lens is a common presenting symptom and results in a host of ocular dysfunctions.27 Additional effects include neurodegenerative disorders, seizures, osteoporosis, scoliosis, pectus excavatum, and thromboembolic episodes.27,29 A second form of homocystinuria can occur due to a defect in methylcobalamin formation and subsequent inability to regenerate methionine from homocysteine. This may present with vomiting, poor feeding, lethargy, hypotonia, seizures, poor growth, ocular disorders, and peripheral neuropathy. Patients can develop a pigmentary retinopathy despite appropriate treatment.100 The third deficit occurs due to dysfunction in the N5,10-methylenetetrahydrofolate reductase preventing production of N5-methyltetrahydrofolate and regeneration of methionine from homocysteine. This is the most common metabolic defect of folate metabolism.101 Resultant sequelae include thromboembolic defects, apnea, ataxia, seizure, developmental delay, coma, and death.42,101
Methionine restriction may be warranted in homocystinuria only after supplementation with pyridoxine and folic acid has failed. An initial trial of pyridoxine in this disease will result in dramatic improvement in the responsive patient. An initial dose of 100 mg may be administered regardless of age or size.42,61 Pyridoxine may decrease folic acid levels, and if pyridoxine seems ineffective, concomitant folic acid may be initiated at 5 mg for all patients.27,29,42 Folic acid supplementation is also warranted in homocystinuria secondary to N5,10-methylenetetrahydrofolate reductase deficiency. Both folic acid and pyridoxine are hepatically metabolized to active metabolites. Although the metabolic processes involved in metabolism are unknown, literature suggests that these agents may affect antiepileptic medication concentrations.43,44 Additional monitoring may need to be performed with medications that are significantly cleared by the liver. If both pyridoxine and folic acid are ineffective, dietary restriction may still be avoided if a trial of betaine is successful. Betaine acts as a methyl donor and can subsequently regenerate methionine from homocysteine through an alternative metabolic pathway. Methionine levels should be monitored closely with betaine therapy to avoid the risk for cerebral edema with elevated levels. Hydroxycobalamin may also be used in homocystinuria if there is concern for abnormalities with methylcobalamin formation. See Table 1 for further details regarding dosing, availability, and adverse effects of these agents.
If homocystinuria is suspected, all 4 agents may be initiated at the same time in an effort to resolve the metabolic crisis. An exact knowledge of the metabolic pathways of these medications is unknown, and subsequent information regarding drug interactions is limited. This is not limited to just patients with homocystinuria. Literature regarding many of the herbal and nutritional supplements is sparse. Addition of these agents to a medication regimen may result in drug interactions that go unnoticed until complications arise and warrant further investigation. The pharmacist should be vigilant for drug interactions and monitor therapy appropriately.
Maple Syrup Urine Disease. Branched-chain amino acids comprise valine, isoleucine, and leucine. These amino acids are essential amino acids and play an important role in energy production, non-essential amino acid production, and protein structure.102 The catabolism of these 3 molecules begins very similarly with the creation of a ketone via branched-chain amino acid aminotransferase. The subsequent reaction results in the production of NADH and is enabled by branched-chain α-ketoacid dehydrogenase. Cofactors play an important role in these enzymatic processes; pyridoxine is integral in the first enzymatic step and thiamine in the second. The enzymatic degradation of valine and isoleucine begins to differ from leucine at this point. After several metabolic steps, valine and isoleucine are eventually metabolized to propionyl-CoA. Leucine will eventually be metabolized to acetyl-CoA and acetoacetate.
Maple syrup urine disease is caused by a dysfunction of the branched-chain α-ketoacid dehydrogenase or its coenzyme, thiamine. Five different types of maple syrup urine disease have been discovered, with variations in symptom severity.103 However, all patients present with the disease's namesake, sweet-smelling urine similar to that of maple syrup. Further complications may include poor feeding, irritability, vomiting, hypertonicity, hypotonicity, convulsions, lethargy, mental retardation, coma, and death.79,103 The final metabolic products of valine and isoleucine include propionyl-CoA, which may be of concern in patients with propionic acidemia and has already been described.
Dietary restriction of leucine, valine, and isoleucine is imperative in maple syrup urine disease. In an acute crisis, initial therapy should consist of hydration and cessation of the catabolic state. As with disorders of the fatty acid pathway, this can be facilitated via an infusion of 10% dextrose. Because thiamine is a cofactor in this process, it may be initiated in those with a new diagnosis. Some patients may respond to thiamine supplementation and continue on therapy indefinetly.103 More information regarding thiamine can be found in Table 1.
Errors in the Urea Cycle
The Urea Cycle. Deamination of amino acids results in the production of ammonia which can be extremely toxic to neurologic function. The urea cycle is a 6-step process that converts ammonia molecules to urea for transport in the bloodstream and removal in the kidneys. The first step in the process prepares ammonia for entrance into the urea cycle by combining ammonia and bicarbonate, creating carbamate. Next, carbamoyl phosphate synthetase and ATP convert carbamate to carbamoyl phosphate. This enzymatic reaction is activated by N-acetyl glutamine, which is a metabolite of the amino acid glutamine. The molecule of carbamoyl phosphate will combine with ornithine and enter the urea cycle. After a few intermediary steps, urea and ornithine are produced. Urea is then transported in the bloodstream to the kidneys, where it is removed. Ornithine will combine with another molecule of carbamoyl phosphate and continue the process.
Urea Cycle Disorders and Treatment.Urea cycle disorders can occur in any of the enzymatic processes in the urea cycle except for ornithine α-aminotransferase. Most of these disorders will result in hyperammonemia and subsequent neurologic toxicity, including lethargy, irritability, seizures, and coma.104 Additional generalized sequelae include poor feeding, vomiting, tachypnea, hypothermia, ataxia, intellectual disability, and increased intracranial pressure.21,104,105 Each particular disease state may present with additional manifestations. For example, carbamyl phosphate synthetase and N-acetyl glutamine acid synthetase deficiencies may present with headaches or migraines. Patients with ornithine transcarbamylase deficiency may develop gallstones. A defect in argininosuccinate synthetase or lyase may result in dry, brittle hair in addition to the usual effects of hyperammonemia. These patients are more likely to have developmental delay and intellectual disability.106 Arginase deficiency may present with spastic paraplegia, peripheral neuropathy, choreoathetotic movements, and loss of developmental milestones.17,18,107
Treatment for errors in the urea cycle primarily revolves around stopping the catabolic state and ammonia removal. Frequently, these patients will present with hyperammonemic crisis secondary to an acute process that limited oral intake and resulted in energy production shifting from carbohydrate metabolism to lipid and amino acid metabolism. Therefore, first-line therapy generally consists of an infusion of dextrose.18,21 Once intravenous access has been established and adequate calories provided, attention can shift to the removal of excess ammonia.
Several medications are used to remove excess ammonia. In the acute setting, an intravenous mixture of sodium phenylacetate and sodium benzoate are combined with the 10% dextrose infusion. This provides the patient with the calories to stop the metabolic crisis, along with agents to remove the excess ammonia. Phenylacetate binds to glutamine to create phenylacetylglutamine, which contains 2 moles of nitrogen; benzoate binds with glycine to form hippuric acid, which contains 1 mole of nitrogen.18 The molecular products can then be excreted, eventually resulting in resolution of the hyperammonemia state. When ammonia levels have sufficiently declined, enteral sodium phenylbutyrate, a prodrug for phenylacetate, can be initiated. This may help the patient avoid subsequent acute hyperammonemic crisis. Arginine may also be used to help facilitate ammonia removal and is often combined with sodium phenylacetate and sodium benzoate in the 10% dextrose mixture during the initial crisis. Supplementation with this therapy supplies the urea cycle with ornithine and N-acetyl glutamine. Depending on the metabolic disorder, arginine can facilitate the removal of 1 or 2 moles of ammonia.106 Arginine should not be used in patients with arginase deficiency because the arginine will not be metabolized, resulting in worsening of the metabolic process.21 In patients with N-acetyl glutamine synthetase deficiency, carglumic acid may be supplemented once the acute crisis is controlled. This agent acts as a replacement for N-acetyl glutamine and allows the urea cycle to function normally. Additional agents for treatment or control of hyperammonia include dietary modifications, lactulose, and levocarnitine.108 Levocarnitine can be used in the acute crisis to help mitigate the hyperammonemia and encephalopathy.93 More information regarding the medication therapies used in urea cycle disorders can be found in Table 1.
Overall, the treatment and mitigation of acute hyperammonemic crisis in patients with urea cycle disorders has led to a robust improvement in treatment outcomes. Improvements in screening, standardization of treatment protocols, and other factors have enhanced patient outcomes. Specifically, mortality in the newborn period has declined from 50% before 2002 to 24%.109 Pharmacists can help maximize this benefit by familiarizing themselves with the medications used in an acute crisis, providing children and family members with education regarding their medications, and monitoring for potential adverse effects and drug interactions.
Conclusions
Although a number of dietary supplements and pharmacologic options have been discussed, this has not been all-encompassing. Additional medication therapies, active forms of medications, and metabolic disorders have not been discussed. Hopefully, this document has provided the reader with a baseline level of knowledge necessary to understand the metabolic process associated with IEMs and potential treatment options. Review of the metabolic disorder, normal metabolic processes, and any available literature will be vital to the pharmacist providing care to these patients.
As has been shown, IEMs comprise a vast and complex array of disorders. Literature is often limited because of the rare nature of the diseases. Treatment often comprises a working understanding of the metabolic processes involved and speculation as to what therapies may be beneficial. A pharmacologic cocktail may be initiated to cover several potential metabolic disorders. Because many of these agents are dietary or nutritional supplements, literature regarding efficacy, adverse events, and potential drug interventions is lacking. Therefore, it is necessary that the pharmacist understand the disease process when providing care for patients with IEMs to ensure appropriate medication therapy and minimize the potential risk to patients with these rare disorders.
Acknowledgments
The author would like to thank Kenneth Kurek, PharmD; Amanda Penland, PharmD; and Elizabeth Harthan, PharmD, for their efforts in reviewing this document.
ABBREVIATIONS
- ATP
adenosine triphosphate
- CoA
coenzyme A
- IEM
inborn error of metabolism
- MCAD
medium chain acyl CoA dehydrogenase deficiency
- NADH
reduced nicotinamide adenine dinucleotide
Footnotes
Disclosure The author declares no conflicts or financial interest in any product or service mentioned in the manuscript, including grants, equipment, medications, employment, gifts, and honoraria.
REFERENCES
- 1.Clarke JTR. A Clinical Guide to Inherited Metabolic Diseases. 2nd ed. Cambridge, United Kingdom: Cambridge University Press; 2004. [Google Scholar]
- 2.Cutts C. Pediatric congenital errors of metabolism: the pharmacist's extended role. Am J Health Syst Pharm. 2003;60(2):182–184. doi: 10.1093/ajhp/60.2.182. [DOI] [PubMed] [Google Scholar]
- 3.Rao AN, Cavitha J, Koch M et al. Inborn errors of metabolism: review and data from a tertiary care center. Indian J Clin Biochem. 2009;24(3):215–222. doi: 10.1007/s12291-009-0041-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dietzen DJ, Rinaldo P, Whitley RJ et al. National academy of clinical biochemistry laboratory medicine practice guidelines: follow-up testing for metabolic disease identified by expanded newborn screening using tandem mass spectrometry; executive summary. Clin Chem. 2009;55(9):1615–1626. doi: 10.1373/clinchem.2009.131300. [DOI] [PubMed] [Google Scholar]
- 5.Mckee T, McKee JR. Biochemistry: The Molecular Basis of Life. Oxford, United Kingdom: Oxford University Press; 2014. [Google Scholar]
- 6.Baron DN, McIntyre N. Letter: glucose is dextrose is glucose. Br Med J. 1976;2(6026):41–42. doi: 10.1136/bmj.2.6026.41-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouteldja N, Timson DJ. The biochemical basis of hereditary fructose intolerance. J Inherit Metab Dis. 2010;33(2):105–112. doi: 10.1007/s10545-010-9053-2. [DOI] [PubMed] [Google Scholar]
- 8.Cox TM. Iatrogenic deaths in hereditary fructose intolerance. Arch Dis Child. 1993;69(4):413–415. doi: 10.1136/adc.69.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marcason W. Is medical nutrition therapy (MNT) the same for hereditary vs dietary fructose intolerance. J Am Diet Assoc. 2010;110(7):1128. doi: 10.1016/j.jada.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 10.Kishnani PS, Chen YT. Defects in metabolism of carbohydrates. In: Kliegman RM, Stanton BF, St. Geme JW III, editors. Nelson's Textbook of Pediatrics. 19th ed. Philadelphia, PA: Elsevier; 2011. [Google Scholar]
- 11.Bosch AM. Classical galactosaemia revisited. J Inherit Metab Dis. 2006;29(4):516–525. doi: 10.1007/s10545-006-0382-0. [DOI] [PubMed] [Google Scholar]
- 12.Castiglioni C, Verrigni D, Okuma C et al. Pyruvate dehydrogenase deficiency presenting as isolated paroxysmal exercise induced dystonia successfully reversed with thiamine supplementation: case report and mini-review. Eur J Paediatr Neurol. 2015;19(5):497–503. doi: 10.1016/j.ejpn.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Ogier de Baulny H. Management and emergency treatments of neonates with a suspicion of inborn errors of metabolism. Semin Neonatol. 2002;7(1):17–26. doi: 10.1053/siny.2001.0084. [DOI] [PubMed] [Google Scholar]
- 14.Barnerias C, Saudubray JM, Touati G et al. Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev Med Child Neurol. 2010;52(2):e1–e9. doi: 10.1111/j.1469-8749.2009.03541.x. [DOI] [PubMed] [Google Scholar]
- 15.Sedel F, Challe G, Mayer JM et al. Thiamine responsive pyruvate dehydrogenase deficiency in an adult with peripheral neuropathy and optic neuropathy. J Neurol Neurosurg Psychiatry. 2008;79(7):846–847. doi: 10.1136/jnnp.2007.136630. [DOI] [PubMed] [Google Scholar]
- 16.Osman M, Casey P. Angioneurotic oedema secondary to oral thiamine. BMJ Case Rep. 2013;2013 doi: 10.1136/bcr-2013-200558. pii: bcr2013200558. DOI: 10.1136/bcr-2013-200558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rezvani I, Yudkoff M. Urea cycle and hyperammonemia (arginine, citrulline, ornithine) In: Kliegman RM, Stanton BF, St Geme JW III, editors. Nelson's Textbook of Pediatrics. 19th ed. Philadelphia, PA: Elsevier; 2011. [Google Scholar]
- 18.Häberle J, Boddaert N, Burlina A et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. doi: 10.1186/1750-1172-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abraham MB, van der Westhuyzen J, Khanna V. Arginine extravasation leading to skin necrosis. J Paediatr Child Health. 2012;48(3):E96–E97. doi: 10.1111/j.1440-1754.2011.02074.x. [DOI] [PubMed] [Google Scholar]
- 20.Yeo TW, Lampah DA, Gitawati R et al. Safety profile of L-arginine infusion in moderately severe falciparum malaria. PLoS One. 2008;3(6):e2347. doi: 10.1371/journal.pone.0002347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakamura K, Kido J, Mitsubuchi H et al. Diagnosis and treatment of urea cycle disorder in Japan. Pediatr Int. 2014;56(4):506–509. doi: 10.1111/ped.12439. [DOI] [PubMed] [Google Scholar]
- 22.Alfadhel M, Al-Thihil K, Moubayed H et al. Drug treatment of inborn errors of metabolism. Arch Dis Child. 2013;98(6):454–461. doi: 10.1136/archdischild-2012-303131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The nutrition committee of the Canadian Paediatric Society Vitamin C for prophylaxis of tyrosinemia in the newborn. Can Med Assoc J. 1976;114(5):447. [PMC free article] [PubMed] [Google Scholar]
- 24.Lykkesfeldt J, Michels AJ, Frei B. Vitamin C. Adv Nutr. 2014;5(1):16–18. doi: 10.3945/an.113.005157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X, Shen L, Gu X et al. High-dose supplementation with vitamin C-induced pediatric urolithiasis: the first case report in a child and literature review. Urology. 2014;84(4):922–924. doi: 10.1016/j.urology.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 26.Ferraro PM, Curhan GC, Gambaro G et al. Total, dietary, and supplemental vitamin C intake and risk of incident kidney stones. Am J Kidney Dis. 2016;67(3):400–407. doi: 10.1053/j.ajkd.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sacharow SJ, Picker JD, Levy HL. Homocystinuria caused by cystathionine beta-synthase deficiency. In: Pagon RA, Adam MP, Ardinger HH, editors. GeneReviews. Seattle, WA: University of Washington; 2017. [PubMed] [Google Scholar]
- 28.Ucar SK, Koroglu OA, Berk O et al. Titration of betaine therapy to optimize therapy in an infant with 5,10-methylenetetrahydrofolate reductase deficiency. Eur J Pediatr. 2010;169(2):241–243. doi: 10.1007/s00431-009-0997-x. [DOI] [PubMed] [Google Scholar]
- 29.Kumar T, Sharma GS, Singh LR. Homocystinuria: therapeutic approach. Clin Chim Acta. 2016;458:55–62. doi: 10.1016/j.cca.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Saudubray JM, Sedel F, Walter JH. Clinical approach to treatable inborn metabolic diseases: an introduction. J Inherit Metab Dis. 2006;29(2–3):261–274. doi: 10.1007/s10545-006-0358-0. [DOI] [PubMed] [Google Scholar]
- 31.Rahman S, Footitt EJ, Varadkar S et al. Inborn errors of metabolism causing epilepsy. Dev Med Child Neurol. 2013;55(1):23–36. doi: 10.1111/j.1469-8749.2012.04406.x. [DOI] [PubMed] [Google Scholar]
- 32.Hendriksz CJ, Preece MA, Chakrapani A. Successful pregnancy in a treated patient with biotinidase deficiency. J Inherit Metab Dis. 2005;28(5):791–792. doi: 10.1007/s10545-005-0060-7. [DOI] [PubMed] [Google Scholar]
- 33.Kummer S, Hermsen D, Distelmaier F. Biotin treatment mimicking graves' disease. N Engl J Med. 2016;375(7):704–706. doi: 10.1056/NEJMc1602096. [DOI] [PubMed] [Google Scholar]
- 34.Trambas CM, Sikaris KA, Lu ZX. A caution regarding high-dose biotin therapy: misdiagnosis of hyperthyroidism in euthyroid patients. Med J Aust. 2016;205(4):192. doi: 10.5694/mja16.00544. [DOI] [PubMed] [Google Scholar]
- 35.Baruteau J, Hargreaves I, Krywawych S et al. Successful reversal of propionic acidaemia associated cardiomyopathy: evidence for low myocardial coenzyme Q10 status and secondary mitochondrial dysfunction as an underlying pathophysiological mechanism. Mitochondrion. 2014;17:150–156. doi: 10.1016/j.mito.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 36.Horvath R, Gorman G, Chinnery PF. How can we treat mitochondrial encephalomyopathies?: approaches to therapy. Neurotherapeutics. 2008;5(4):558–568. doi: 10.1016/j.nurt.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hyson HC, Kieburtz K, Shoulson I et al. Safety and tolerability of high-dose coenzyme Q10 in Huntington's disease and healthy subjects. Mov Disord. 2010;25(12):1924–1928. doi: 10.1002/mds.22408. [DOI] [PubMed] [Google Scholar]
- 38.Weis M, Mortensen SA, Rassing MR et al. Bioavailability of four oral coenzyme Q10 formulations in healthy volunteers. Mol Aspects Med. 1994;15(suppl):s273–s280. doi: 10.1016/0098-2997(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 39.Carbaglu (carglumic acid) [package insert] Lebanon, NJ: Recordati Rare Diseases Inc; https://www.carbaglu.net/wp-content/uploads/2016/04/carbaglu-pi.pdf Accessed October 19, 2018. [Google Scholar]
- 40.Haberle J. Role of carglumic acid in the treatment of acute hyperammonemia due to N-acetylglutamate synthase deficiency. Ther Clin Risk Manag. 2011;7:327–332. doi: 10.2147/TCRM.S12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nahata MC, Pai VB, Hipple TF. Pediatric Drug Formulations. 5th ed. Cincinnati, OH: Harvey Whitney Books Co; 2004. [Google Scholar]
- 42.Rezvani I, Rosenblatt DS. Methionine. In: Kliegman RM, Stanton BF, St Geme JW III, editors. Nelson's Textbook of Pediatrics. 19th ed. Philadelphia, PA: Elsevier; 2011. [Google Scholar]
- 43.Hansson O, Sillanpaa M. Pyridoxine and serum concentration of phenytoin and phenobarbitone. Lancet. 1976;1(7953):256. doi: 10.1016/s0140-6736(76)91385-4. [DOI] [PubMed] [Google Scholar]
- 44.Jensen ON, Olesen OV. Subnormal serum folate due to anticonvulsive therapy: a double-blind study of the effect of folic acid treatment in patients with drug-induced subnormal serum folates. Arch Neurol. 1970;22(2):181–182. doi: 10.1001/archneur.1970.00480200087010. [DOI] [PubMed] [Google Scholar]
- 45.Van Hove JL, Van Damme-Lombaerts R, Grünewald S et al. Cobalamin disorder Cbl-C presenting with late-onset thrombotic microangiopathy. Am J Med Genet. 2002;111(2):195–201. doi: 10.1002/ajmg.10499. [DOI] [PubMed] [Google Scholar]
- 46.Carrillo-Carrasco N, Sloan J, Valle D et al. Hydroxocobalamin dose escalation improves metabolic control in cblC. J Inherit Metab Dis. 2009;32(6):728–731. doi: 10.1007/s10545-009-1257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szymanska E, Sredzinska M, Ciara E et al. Tyrosinemia type III in an asymptomatic girl. Mol Genet Metab Rep. 2015;5:48–50. doi: 10.1016/j.ymgmr.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uhl W, Nolting A, Golor G et al. Safety of hydroxocobalamin in healthy volunteers in a randomized, placebo-controlled study. Clin Toxicol (Phila) 2006;44(suppl 1):17–28. doi: 10.1080/15563650600811755. [DOI] [PubMed] [Google Scholar]
- 49.Cano A, Ovaert C, Vianey-Saban C et al. Carnitine membrane transporter deficiency: a rare treatable cause of cardiomyopathy and anemia. Pediatr Cardiol. 2008;29(1):163–165. doi: 10.1007/s00246-007-9051-9. [DOI] [PubMed] [Google Scholar]
- 50.Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7–8):1287–1293. doi: 10.1345/aph.1P135. [DOI] [PubMed] [Google Scholar]
- 51.Eknoyan G, Latos DL, Lindberg J. Practice recommendations for the use of L-carnitine in dialysis-related carnitine disorder: National Kidney Foundation carnitine consensus conference. Am J Kidney Dis. 2003;41(4):868–876. doi: 10.1016/s0272-6386(03)00110-0. [DOI] [PubMed] [Google Scholar]
- 52.Zeybek AC, Kiykim E, Soyucen E et al. Hereditary tyrosinemia type 1 in Turkey: twenty year single-center experience. Pediatr Int. 2015;57(2):281–289. doi: 10.1111/ped.12503. [DOI] [PubMed] [Google Scholar]
- 53.Bendadi F, de Koning TJ, Visser G et al. Impaired cognitive functioning in patients with tyrosinemia type I receiving nitisinone. J Pediatr. 2014;164(2):398–401. doi: 10.1016/j.jpeds.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 54.Mayorandan S, Meyer U, Gokcay G et al. Cross-sectional study of 168 patients with hepatorenal tyrosinaemia and implications for clinical practice. Orphanet J Rare Dis. 2014;9:107. doi: 10.1186/s13023-014-0107-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thimm E, Richter-Werkle R, Kamp G et al. Neurocognitive outcome in patients with hypertyrosinemia type I after long-term treatment with NTBC. J Inherit Metab Dis. 2012;35(2):263–268. doi: 10.1007/s10545-011-9394-5. [DOI] [PubMed] [Google Scholar]
- 56.Orfandin (nitisinone) [package insert] Stockholm, Sweden: Swedish Orphan Biovitrum AB; 2016. https://sobi-northamerica.com/sites/sobi-northamerica.com/files/2018-06/Canada.Orfadin_PM_EN.pdf Accessed October 19, 2018. [Google Scholar]
- 57.Caruthers RL, Johnson CE. Stability of extemporaneously prepared sodium phenylbutyrate oral suspensions. Am J Health Syst Pharm. 2007;64(14):1513–1515. doi: 10.2146/ajhp060450. [DOI] [PubMed] [Google Scholar]
- 58.Ravicti (glycerol phenylbutyrate) [package insert] Lake Forest, IL: Horizon Therapeutics; 2017. https://hznp.azureedge.net/public/ravicti_1-1g-ml_vial_pi_effective_us.PDF Accessed October 19, 2018. [Google Scholar]
- 59.Berry SA, Lichter-Konecki U, Diaz GA et al. Glycerol phenylbutyrate treatment in children with urea cycle disorders: pooled analysis of short and long-term ammonia control and outcomes. Mol Genet Metab. 2014;112(1):17–24. doi: 10.1016/j.ymgme.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ensom MH, Decarie D. Stability of extemporaneously compounded pyridoxine in glass and plastic bottles and plastic syringes. Can J Hosp Pharm. 2014;67(5):394–396. [PMC free article] [PubMed] [Google Scholar]
- 61.Stockler S, Plecko B, Gospe SM, Jr et al. Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment, and follow-up. Mol Genet Metab. 2011;104(1–2):48–60. doi: 10.1016/j.ymgme.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 62.Allen Loyd V., Jr Riboflavin 5 mg/mL oral suspension. Int J Pharm Compd. 2011;15(4):343. [Google Scholar]
- 63.Ou LS, Kuo ML, Huang JL. Anaphylaxis to riboflavin (vitamin B2) Ann Allergy Asthma Immunol. 2001;87(5):430–433. doi: 10.1016/S1081-1206(10)62927-4. [DOI] [PubMed] [Google Scholar]
- 64.Vockley J, Andersson HC, Antshel KM et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med. 2014;16(2):188–200. doi: 10.1038/gim.2013.157. [DOI] [PubMed] [Google Scholar]
- 65.Longo N, Arnold GL, Pridjian G et al. Long-term safety and efficacy of sapropterin: the PKUDOS registry experience. Mol Genet Metab. 2015;114(4):557–563. doi: 10.1016/j.ymgme.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 66.Somaraju UR, Merrin M. Sapropterin dihydrochloride for phenylketonuria. Cochrane Database Syst Rev. 2015;(3) doi: 10.1002/14651858.CD008005.pub4. DC008005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ensom MHH, Decarie D. Stability of thiamine in extemporaneously compounded suspensions. Can J Hosp Pharm. 2005;58:26–30. [Google Scholar]
- 68.Arnold GL, Griebel ML, Porterfield M et al. Pyruvate carboxylase deficiency: report of a case and additional evidence for the “mild” phenotype. Clin Pediatr (Phila) 2001;40(9):519–521. doi: 10.1177/000992280104000909. [DOI] [PubMed] [Google Scholar]
- 69.Mayatepek E, Hoffmann B, Meissner T. Inborn errors of carbohydrate metabolism. Best Pract Res Clin Gastroenterol. 2010;24(5):607–618. doi: 10.1016/j.bpg.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 70.Llerena Junior JC, Nascimento OJ, Oliveira AS et al. Guidelines for the diagnosis, treatment and clinical monitoring of patients with juvenile and adult Pompe disease. Arg Neuropsiquiatr. 2016;74(2):166–176. doi: 10.1590/0004-282X20150194. [DOI] [PubMed] [Google Scholar]
- 71.Burton BK. Inborn errors of metabolism in infancy: a guide to diagnosis. Pediatrics. 1998;102(6):E69. doi: 10.1542/peds.102.6.e69. [DOI] [PubMed] [Google Scholar]
- 72.Schoser B, Stewart A, Kanters S et al. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol. 2017;264(4):621–630. doi: 10.1007/s00415-016-8219-8. [DOI] [PubMed] [Google Scholar]
- 73.Levy PA. Inborn errors of metabolism: part 2: specific disorders. Pediatr Rev. 2009;30(4):e22–e28. doi: 10.1542/pir.30-4-e22. [DOI] [PubMed] [Google Scholar]
- 74.Shima A, Yasuno T, Yamada K et al. First Japanese case of carnitine palmitoyltransferase II deficiency with the homozygous point mutation S113L. Intern Med. 2016;55(18):2659–2661. doi: 10.2169/internalmedicine.55.6288. [DOI] [PubMed] [Google Scholar]
- 75.Potter BK, Little J, Chakraborty P et al. Variability in the clinical management of fatty acid oxidation disorders: results of a survey of Canadian metabolic physicians. J Inherit Metab Dis. 2012;35(1):115–123. doi: 10.1007/s10545-011-9352-2. [DOI] [PubMed] [Google Scholar]
- 76.Couce ML, Sánchez-Pintos P, Diogo L et al. Newborn screening for medium-chain acyl-CoA dehydrogenase deficiency: regional experience and high incidence of carnitine deficiency. Orphanet J Rare Dis. 2013;8:102. doi: 10.1186/1750-1172-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.ØRngreen MC, Nørgaard MG, Sacchetti M et al. Fuel utilization in patients with very long-chain acyl-coa dehydrogenase deficiency. Ann Neurol. 2004;56(2):279–283. doi: 10.1002/ana.20168. [DOI] [PubMed] [Google Scholar]
- 78.Pedersen CB, Kølvraa S, Kølvraa A et al. The ACADS gene variation spectrum in 114 patients with short-chain acyl-CoA dehydrogenase (SCAD) deficiency is dominated by missense variations leading to protein misfolding at the cellular level. Hum Genet. 2008;124(1):43–56. doi: 10.1007/s00439-008-0521-9. [DOI] [PubMed] [Google Scholar]
- 79.Rezvani I, Rosenblatt DS. Valine, leucine, isoleucine, and related organic acidemias. In: Kliegman RM, Stanton BF, St Geme JW III, editors. Nelson's Textbook of Pediatrics. 19th ed. Philadelphia, PA: Elsevier; 2011. [Google Scholar]
- 80.Chapman KA, Gropman A, Macleod E et al. Acute management of propionic academia. Mol Genet Metab. 2012;105(1):16–25. doi: 10.1016/j.ymgme.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ribas GS, Biancini GB, Mescka C et al. Oxidative stress parameters in urine from patients with disorders of propionate metabolism: a beneficial effect of L-carnitine supplementation. Cell Mol Neurobiol. 2012;32(1):77–82. doi: 10.1007/s10571-011-9736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Baumgartner MR, Hörster F, Dionisi-Vici C et al. Proposed guidelines for the diagnosis and management of methyl-malonic and propionic academia. Orphanet J Rare Dis. 2014;9:130. doi: 10.1186/s13023-014-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jain IH, Zazzeron L, Goli R et al. Hypoxia as a therapy for mitochondrial disease. Science. 2016;352(6281):54–61. doi: 10.1126/science.aad9642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Levy RJ, Rios PG, Akman HO et al. Long survival in patients with Leigh syndrome and the m.10191T>C mutation in MT-ND3: a case report and review of the literature. J Child Neurol. 2014;29(10):NP105–NP110. doi: 10.1177/0883073813506783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cakir B, Teksam M, Kosehan D et al. Inborn errors of metabolism presenting in childhood. J Neuroimaging. 2011;21(2):e117–e133. doi: 10.1111/j.1552-6569.2011.00575.x. [DOI] [PubMed] [Google Scholar]
- 86.Baertling F, Rodenburg RJ, Schaper J et al. A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry. 2014;85(3):257–265. doi: 10.1136/jnnp-2012-304426. [DOI] [PubMed] [Google Scholar]
- 87.Izumi R, Suzuki N, Nagata M et al. A case of late onset riboflavin-responsive multiple acyl-coa dehydrogenase deficiency manifesting as recurrent rhabdomyolysis and acute renal failure. Intern Med. 2011;50(21):2663–2668. doi: 10.2169/internalmedicine.50.5172. [DOI] [PubMed] [Google Scholar]
- 88.Olsen RK, Koňaříková E, Giancaspero TA et al. Riboflavin-responsive and –non-responsive mutations in FAD synthase cause multiple acyl-coa dehydrogenase and combined respiratory-chain deficiency. Am J Hum Genet. 2016;98(6):1130–1145. doi: 10.1016/j.ajhg.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stanley CA, Bennett MJ. Defects in metabolism of lipids. In: Kliegman RM, Stanton BF, St Geme JW III, editors. Nelson's Textbook of Pediatrics. 19th ed. Philadelphia, PA: Elsevier; 2011. [Google Scholar]
- 90.Feldmann R, Wolfgart E, Weglage J, Rutsch F. Sapropterin treatment does not enhance the health-related quality of life of patients with phenylketonuria and their parents. Acta Paediatr. 2017;106(6):953–959. doi: 10.1111/apa.13799. [DOI] [PubMed] [Google Scholar]
- 91.Rezvani I, Melvin JJ. Phenylalanine. In: Kliegman RM, Stanton BF, St Geme JW III, editors. Nelson's Textbook of Pediatrics. 19th ed. Philadelphia, PA: Elsevier; 2011. [Google Scholar]
- 92.Kaufman S. A model of human phenylalanine metabolism in normal subjects and in phenylketonuric patients. Proc Natl Acad Sci U S A. 1999;96(6):3160–3164. doi: 10.1073/pnas.96.6.3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zheng Z, Ma C, Gao C et al. Efficient conversion of phenylpyruvic acid to phenyllactic acid by using whole cells of bacillus coagulans SDM. PLoS One. 2011;6(4):e19030. doi: 10.1371/journal.pone.0019030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.ten Hoedt AE, Maurice-Stam H, Boelen CC et al. Parenting a child with phenylketonuria or galactosemia: implications for health-related quality of life. J Inherit Metab Dis. 2011;34(2):391–398. doi: 10.1007/s10545-010-9267-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ozben T. Expanded newborn screening and confirmatory follow-up testing for inborn errors of metabolism detected by tandem mass spectrometry. Clin Chem Lab Med. 2013;51(1):157–176. doi: 10.1515/cclm-2012-0472. [DOI] [PubMed] [Google Scholar]
- 96.McKiernan PJ, Preece MA, Chakrapani A. Outcome of children with hereditary tyrosinaemia following newborn screening. Arch Dis Child. 2015;100(8):738–741. doi: 10.1136/archdischild-2014-306886. [DOI] [PubMed] [Google Scholar]
- 97.Mitchell GA, Rezvani I. Tyrosine. In: Kliegman RM, Stanton BF, St Geme JW III, editors. Nelson's Textbook of Pediatrics. 19th ed. Philadelphia, PA: Elsevier; 2011. [Google Scholar]
- 98.De Lucia S, Pichard S, Ilea A et al. An unfortunate challenge: ketogenic diet for the treatment of lennox-gastaut syndrome in tyrosinemia type 1. Eur J Paediatr Neurol. 2016;20(4):674–677. doi: 10.1016/j.ejpn.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 99.Kocabeyoglu S, Mocan MC, Irkec M. In vivo confocal microscopic features of cornealpseudodendritic lesions in tyrosinemia type II. Cornea. 2014;33(10):1106–1108. doi: 10.1097/ICO.0000000000000226. [DOI] [PubMed] [Google Scholar]
- 100.Carillo-Carrasco N, Venditti CP. Combined methylmalonic acidemia and homocystinuria, cblC type. II: complications, pathophysiology, and outcomes. J Inherit Metab Dis. 2012;35(1):103–114. doi: 10.1007/s10545-011-9365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cappuccio G, Cozzolino C, Frisso G et al. Pearls & oysters: familial epileptic encephalopathy due to methylenetetrahydrofolate reductase deficiency. Neurology. 2014;83(3):e41–e44. doi: 10.1212/WNL.0000000000000591. [DOI] [PubMed] [Google Scholar]
- 102.Vogel KR, Arning E, Wasek BL et al. Brain-blood amino acid correlates following protein restriction in murine maple syrup urine disease. Orphanet J Rare Dis. 2014;9:73. doi: 10.1186/1750-1172-9-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Frazier DM, Allgeier C, Homer C et al. Nutrition management guideline for maple syrup urine disease: an evidence- and consensus-based approach. Mol Genet Metab. 2014;112(3):210–217. doi: 10.1016/j.ymgme.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 104.Yang X, Shi J, Lei H et al. Neonatal-onset carbamoyl phosphate synthetase I deficiency: a case report. Medicine (Baltimore) 2017;96(26):e7365. doi: 10.1097/MD.0000000000007365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fujisawa D, Mitsubuchi H, Matsumoto S et al. Early intervention for late-onset ornithine transcarbamylase deficiency. Pediatr Int. 2015;57(1):e1–e3. doi: 10.1111/ped.12457. [DOI] [PubMed] [Google Scholar]
- 106.Rūegger CM, Lindner M, Ballhausen D et al. Cross-sectional observational study of 208 patients with non-classical urea cycle disorders. J Inherit Metab Dis. 2014;37(1):21–30. doi: 10.1007/s10545-013-9624-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tsang JP, Poon WL, Luk HM et al. Arginase deficiency with new phenotype and a novel mutation: contemporary summary. Pediatr Neurol. 2012;47(4):263–269. doi: 10.1016/j.pediatrneurol.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 108.Jiang Q, Jiang G, Shi KQ et al. Oral acetyl-L-carnitine treatment in hepatic encephalopathy: view of evidence-based medicine. Ann Hepatol. 2013;12(5):803–809. [PubMed] [Google Scholar]
- 109.Batshaw ML, Tuchman M, Summar M et al. A longitudinal study of urea cycle disorders. Mol Genet Metab. 2014;113(1–2):127–130. doi: 10.1016/j.ymgme.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]