

Introduction
McLeod syndrome was originally identified as a hematological variant known as the McLeod hematologic phenotype, which was serologically defined by acanthocytosis and weak antigenicity of Kell antigens associated with the absence of the Kx antigen. McLeod syndrome is now recognized as a rare X‐linked neuroacanthocytosis affecting the peripheral and central nervous systems, red blood cells (RBCs), and internal organs. Loss‐of‐function mutations of the XK gene have been identified as the cause of McLeod hematologic phenotype and syndrome.1 Of 37 causative mutations, most are nonsense mutations, frameshift mutations resulting in a premature stop codon, or splice‐site mutations resulting in mRNA truncation. To date, three missense mutations have been reported for the McLeod hematologic phenotype, but nevertheless, manifestation of McLeod syndrome is observed in only one missense mutation.2, 3, 4 In this report, we describe the clinical features of a patient with McLeod syndrome caused by a novel missense mutation of the XK gene.
Clinical Presentations
A 55‐year‐old male was admitted to our hospital because of long‐lasting undiagnosed distal muscle atrophy/weakness. Neither family history nor medical history was apparent. He developed right arm weakness at age 28, numbness at age 34, and leg weakness at age 40. At age 49, he underwent neurological examination and muscle biopsy at Yokohama Medical Center, but the etiology was not identified. At age 54, he experienced cardiopulmonary arrest due to dilated cardiomyopathy, and a cardioverter defibrillator was implanted. At age 55 on admission, he showed primarily right and distal muscle atrophy/weakness, leg paresthesia, and hyporeflexia. He also exhibited very mild chorea in the tongue (Supporting Information Video 1) and extremities (Supporting Information Video 2). He had neither cognitive impairment nor psychiatric symptoms. Laboratory tests revealed persistent elevation of creatinine kinase (CK; 1042 ± 11.6 IU/l) without hemolytic anemia or abetalipoproteinemia. A peripheral blood smear detected acanthocytes (∼9% in total; Fig. 1A). Brain MRI showed mild atrophy of the bilateral caudate nuclei, primarily on the left side (Fig. 1B). Muscle CT demonstrated low‐density areas, primarily on the right side in the gastrocnemius muscles (Fig. 1C). Electromyography exhibited neurogenic changes in the first dorsal interosseous, biceps, and tibialis anterior muscles. A nerve conduction study showed sensorimotor axonal damage, mainly in the lower extremities. Left sural nerve biopsy revealed moderate demyelination and axonal degeneration (Fig. 1D). Biopsy of his right biceps brachii muscle, performed at age 49, showed mild neurogenic changes with grouped atrophy as well as mild myogenic changes with a few necrotic fibers; no lymphocytic infiltration was observed (Fig. 1E). The Kx antigen was absent on RBCs (Fig. 2A) and Kell blood group antigens were reduced (data not shown), consistent with the essential features of the McLeod hematologic phenotype. Direct DNA sequencing of the XK gene revealed an unreported hemizygous missense mutation in exon 3 (NM_021083: c.665G>C, p.Arg222Pro; Fig. 2B). This Arg222 residue is conserved from zebrafish to human (Fig. 2C), and this novel mutation was predicted to destroy Kx protein using PolyPhen‐2 and MutationTaster2.
Figure 1.
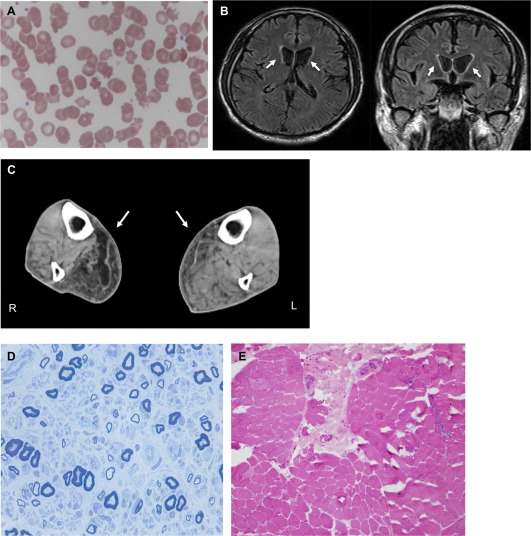
(A) Acanthocytosis in the peripheral blood. A peripheral blood smear shows 9% acanthocytes. (B) Brain MRI images. Axial and coronal sections of fluid attenuated inversion recovery images revealed the bilateral atrophy of caudate nuclei, primarily on the left side (arrows). (C) Muscle CT images. Axial sections of the gastrocnemius muscles demonstrate asymmetrical muscular atrophy with fat replacement (arrows). (D) Toluidine blue staining of a sural nerve section. The section exhibits moderate myelinated fiber loss and axonal degeneration. (E) Hematoxylin and eosin staining of the biceps brachii muscle section. The section shows both mild neurogenic changes with grouped atrophy and mild myogenic changes with a few necrotic fibers.
Figure 2.
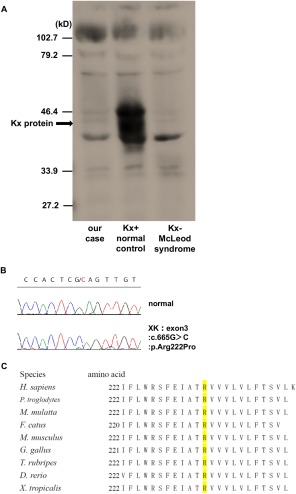
(A) Absence of Kx antigen on RBCs. Western blotting analysis using RBCs reveals an absence of the Kx antigen in our case. (B) Electropherogram from Sanger sequencing of the XK mutation. The c.665G>c (p.Arg222Pro) mutation is identified in our case. (C) Phylogenetic conservation of Arg222 residue in XK.
Discussion
Patients with McLeod syndrome usually develop neurological manifestations, including axonal neuropathy, myopathy with hyperCKemia, cardiomyopathy, Huntington's disease‐like chorea, seizure, behavioral change, and dementia.2, 5 Since the initial symptoms of McLeod syndrome are diverse, early diagnosis is often difficult.6 A previous study reported that 70% of patients developed chorea.2 Because chorea is a key neurological feature of McLeod syndrome, its absence or late onset may lead to a diagnostic delay. This is a rare case with a novel missense mutation of XK in which very mild chorea appeared following featureless distal muscle weakness 27 years after disease onset. The XK gene consists of three exons and encodes XK, a membrane transport protein with 10 transmembrane domains. Impaired XK gene function results in the absence of the Kx antigen on the surface of RBCs. More than 30 XK mutations are currently known to cause McLeod hematologic phenotype and syndrome.1, 2, 3, 4, 7 However, only three missense mutations have been reported: p.Arg222Gly, p.Cys294Arg, and p.Glu327Lys.2, 3, 4 Our patient had the novel missense p.Arg222Pro mutation located in the sixth transmembrane domain. The clinical phenotypes of missense mutations are generally predicted to be milder than those of nonsense or frameshift mutations, and indeed, missense mutations of the genes encoding amino acids Arg222 and Glu327 were reported to cause McLeod hematologic phenotype but not McLeod syndrome, because the p.Arg222Gly and p.Glu327Lys mutations do not impair neuromuscular or cerebral functioning.3, 4, 8 The novel finding in this report is that a missense mutation, p.Arg222Pro, led not only to the McLeod hematologic phenotype, but also to McLeod syndrome. There is only one previous report of a missense mutation, p.Cys294Arg, resulting in McLeod syndrome with severe neurological defects, including muscle weakness, chorea, seizure, cognitive impairment, and involuntary vocalizations, beginning at age 44.9 The clinical features of McLeod syndrome is variable even with the same genotype and within the same family,10 therefore the genotype‐phenotype correlation is not very relevant in this disease. These findings may explain the differential phenotypes resulting from p.Arg222Gly and p.Arg222Pro. Our case might further the understanding of the importance of the Arg222 residue in the function of XK.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
H.K.: 1A, 1C, 3A, 3B
M.T.: 1C
S.H.: 1C
E.U.: 1C
R.F.: 2B
K.T.: 1C
M.T.: 1A, 1C, 3B
H.J.: 1A, 1C
T.T.: 1A, 1C
S.K.: 1A, 1B, 3B
H.D.: 1A, 1B, 3A, 3B
H.T.: 1A, 1B, 3A, 3B
F. T.: 1A, 1B, 3B
Disclosures
Ethical Compliance Statement: This study was performed according to the Helsinki Declaration and approved by the ethical committee of Yokohama City University Hospital. The informed consent was obtained in compliance with the ethical committee of Yokohama City University Hospital. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: No specific funding was received for this work. The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for previous 12 months: The authors declare that there are no additional disclosures to report.
Supporting information
Videos accompanying this article are available in the supporting information here.
Video S1. Chorea in the face and tongue. The patient had mild facial and tongue chorea with motor impersistence.
Video S2. Chorea in the extremities. The patient showed very mild chorea in the extremities.
Acknowledgments
The authors thank the Japan Red Cross Society for evaluating the expression of Kell group antigens on RBCs, and Dr. S. Ikeda, Dr. H. Koike, and Dr. G. Sobue (Department of Neurology, Nagoya University Graduate School of Medicine) for assessing the sural nerve biopsy.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Ho M, Chelly J, Carter N, Danek A, Crocker P, Monaco AP. Isolation of the gene for McLeod syndrome that encodes a novel membrane transport protein. Cell 1994;77(6):869–880. [DOI] [PubMed] [Google Scholar]
- 2. Danek A, Rubio JP, Rampoldi L, et al. McLeod neuroacanthocytosis: Genotype and phenotype. Ann Neurol 2001;50(6):755–764. [DOI] [PubMed] [Google Scholar]
- 3. Jung HH, Hergersberg M, Vogt M, Pahnke J, Treyer V, Röthlisberger B. McLeod phenotype associated with a XK missense mutation without hematologic, neuromuscular, or cerebral involvement. Transfusion 2003;43(July):928–938. [DOI] [PubMed] [Google Scholar]
- 4. Russo DC, Lee S, Reid ME, Redman CM. Point mutations causing the McLeod phenotype. Transfusion 2002;42(3):287–293. [DOI] [PubMed] [Google Scholar]
- 5. Jung HH, Danek A, Walker RH. Neuroacanthocytosis syndromes. Orphanet J Rare Dis 2011;6(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miranda M, Castiglioni C, Frey BM, Hergersberg M, Danek A, Jung HH. Phenotypic variability of a distinct deletion in McLeod syndrome. Mov Disord 2007;22(9):1358–1361. [DOI] [PubMed] [Google Scholar]
- 7. Frey BM, Gassner C, Jung HH. Neurodegeneration in the elderly—when the blood type matters: an overview of the McLeod syndrome with focus on hematological features. Transfus Apher Sci 2015;52(3):277–284. [DOI] [PubMed] [Google Scholar]
- 8. Walker RH, Danek A, Uttner I, Offner R, Reid M, Lee S. McLeod phenotype without the McLeod syndrome. Transfusion 2007;47(2):299–305. [DOI] [PubMed] [Google Scholar]
- 9. Danek A, Tison F, Rubio J, Oechsner M, Kalckreuth W, Monaco AP. The chorea of McLeod syndrome. Mov Disord 2001;16(5):882–889. [DOI] [PubMed] [Google Scholar]
- 10. Walker RH, Jung HH, Tison F, Lee S, Danek A. Phenotypic variation among brothers with the McLeod neuroacanthocytosis syndrome. Mov Disord 2007;22(2):244–248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Videos accompanying this article are available in the supporting information here.
Video S1. Chorea in the face and tongue. The patient had mild facial and tongue chorea with motor impersistence.
Video S2. Chorea in the extremities. The patient showed very mild chorea in the extremities.