

Abstract
Viral infection is a major contributor to the global cancer burden. Recent advances have revealed that seven known oncogenic viruses promote tumorigenesis through shared host cell targets and pathways. A comprehensive understanding of the principles of viral oncogenesis may enable the identification of unknown infectious aetiologies of cancer and the development of therapeutic or preventive strategies for virus-associated cancers. In this Review, we discuss the molecular mechanisms of viral oncogenesis in humans. We highlight recent advances in understanding how viral manipulation of host cellular signalling, DNA damage responses, immunity and microRNA targets promotes the initiation and development of cancer.
When normal cell growth control mechanisms are disrupted, some cells may exhibit uncontrolled proliferation and cease to perform their tissue-specific functions, leading to the development of cancer. Infection by oncogenic viruses is thought to cause ~15–20% of all human cancers1.
The seven known human oncogenic viruses are Epstein–Barr virus (EBV), hepatitis B virus (HBV), human T-lymphotropic virus 1 (HTLV-1), human papillomaviruses (HPVs), hepatitis C virus (HCV), Kaposi sarcoma-associated herpesvirus (KSHV; also known as human herpesvirus 8 (HHV-8)) and Merkel cell polyomavirus (MCPyV) (Table 1 and reviewed in ref.2). EBV and KSHV are large DNA viruses that can cause solid tumours and lymphoid malignancies3,4 (Table 1). HPV and MCPyV (box 1) have smaller DNA genomes than EBV and KSHV. Whereas oncogenic HPVs establish persistent infections in mucosal epithelia5, MCPyV infects and likely persists latently in dermal fibroblasts6. These small DNA oncogenic viruses promote tumorigenesis using relatively few multifunctional oncoproteins7,8. HCV, a positive-sense, single-stranded RNA virus, and HBV, a small DNA virus, both infect hepatocytes and cause chronic liver inflammation, liver cirrhosis and hepatocellular carcinoma (HCC)9,10. Lastly, HTLV-1 is a human oncogenic retrovirus that infects T cells and can cause adult T cell lymphoma11.
Table 1 |.
The global burden of viral cancers at a glance
| Virus | Cancer | Major regions affected | Refs |
|---|---|---|---|
| Epstein–Barr virus | • 40% of Hodgkin lymphoma • >95% of endemic Burkitt lymphoma • 10% gastric carcinoma • Most (type II and III) nasopharyngeal carcinoma • Kaposi sarcoma • Other lymphomas |
• East Asia • East Africa • Regions of the Americas |
140,141 |
| Hepatitis B virus | • 53% of hepatocellular carcinoma | • Asia • Sub-Saharan Africa • Regions of South America |
142 |
| Human T-lymphotropic virus 1 | • >99% of adult T cell leukaemia | • Japan • Australia • Regions of Africa, South America and the Middle East |
143,144 |
| Human papillomavirus | • >95% of cervical carcinoma • 70% of oropharyngeal carcinoma • Other anogenital carcinomas |
• Central America • South America • Sub-Saharan Africa • Regions of Asia |
145,146 |
| Hepatitis C virus | • 25% of hepatocellular carcinoma • Non-Hodgkin B cell lymphomas |
• Regions of Asia, the Americas, North Africa and the Mediterranean | 147,148 |
| Kaposi sarcoma-associated herpesvirus | • >99% of Kaposi sarcoma • >99% of primary effusion lymphoma |
• Regions of Europe and sub-Saharan Africa | 149 |
| Merkel cell polyomavirus | • 80% of Merkel cell carcinoma | • North America • Australia • Europe |
19,150 |
Box 1 |. Merkel cell polyomavirus.
Merkel cell polyomavirus (MCPyv) is the most recently discovered human oncogenic virus and is associated with Merkel cell carcinoma (MCC), an aggressive malignancy of the dermis7. MCPyv belongs to the Polyomaviridae family. it is a small, non-enveloped, double-stranded DNa virus with a genome of ~5,400 base pairs. More than a decade after identifying Kaposi sarcoma-associated herpesvirus (KsHv) as the causative agent of Kaposi sarcoma, Chang and Moore led the next effort to identify an oncogenic virus in humans7. in keeping with the guiding principle of that prior discovery, it was reasoned that because MCC skin cancer disproportionately affects immunosuppressed and elderly individuals, an infectious agent may contribute to its pathogenesis7. in their search, they performed transcriptomic sequencing of human MCC tumours and then compared these sequences with the human genome to subtract background and non-viral sequence reads from the total sequence data. using this approach, they identified an integrated polyomavirus large t antigen transcript with homology to known animal polyomaviruses. they then used 3′ rapid amplification of cDNa ends (raCe) and viral genome walking to retrieve the sequence of this virus — MCPyv. By comparing integrated MCPyv sequence in metastatic tumours between patients, the group also established that MCPyv integrates monoclonally in the host genome before metastasis. this early observation supported the notion that, like other oncogenic viruses, viral integration is a major event in MCC tumorigenesis. since its discovery, MCPyv has been recognized as a ubiquitous virus that asymptomatically infects most individuals during childhood, yet it can be linked to ~80% of MCC cases. MCPyv can productively infect fibroblasts within the dermal layer of human skin6. However, the details of the MCPyv life cycle and the events driving MCC oncogenesis remain unknown.
Human oncogenic viruses have diverse genomes, cellular tropisms, cancer pathologies and disease prevalence (Table 1). However, they share many features that can lead to cancer in humans. They are transmitted between humans and can establish chronic infections that last for years without obvious symptoms. Throughout these prolonged periods, oncogenic viruses co-opt cellular processes for replication and undermine immune recognition. They derail conserved signalling pathways that control cell cycle progression and apoptosis (box 2) to support their propagation. Although tumorigenesis is a unifying pathological feature for oncogenic viruses, it is neither evolutionarily advantageous for the virus nor required for virus propagation. Many of the properties that are shared among the seven oncogenic viruses are also common to other viruses. To identify what makes these seven unique, we must examine the specific mechanisms by which they alter the cellular environment.
Box 2 |. Signalling pathways manipulated by oncogenic viruses.
Pi3K–AKT–mTor signalling pathway
In the phosphatidylinositol 3-kinase–aKt–mechanistic target of rapamycin (Pi3K–aKt–mtOr) pathway, stimulation of a diverse group of growth factor receptors by various stimuli leads to the activation of Pi3K128. activated Pi3K phosphorylates phosphatidylinositol 4,5-bisphosphate to phosphatidylinositol 3,4,5-trisphosphate, which further activates aKt. aKt subsequently triggers the phosphorylation and activation of diverse downstream effectors, including mTOR128. activated mtOr can stimulate the translation of proteins needed for cell cycle progression by inducing the phosphorylation of eukaryotic translation initiation factor 4e-binding protein 1 (4e-BP1)129. By integrating various growth stimuli and acting through multiple cellular effectors, this pathway has an important role in the regulation of cellular growth, proliferation and survival.
MAPK signalling pathway
Upon stimulation by either extracellular signals (for example, growth factors) or stress stimuli (for example, osmotic stress, heat shock, ultraviolet irradiation and oxidative stress), cell surface receptor kinases activate a mitogen-activated protein kinase (MAPK) cascade, ultimately regulating the transcription of diverse genes involved in cell cycle progression, growth, differentiation, programmed cell death and the antiviral immune response34. the three best-characterized subfamilies of MAPKs are the extracellular-signal-regulated kinases (erKs), JUN N-terminal kinases (JNKs) and p38 enzymes34. each of these MAPKs is activated by their cognate kinases, which respond to distinct stimuli34.
Notch signalling pathway
The Notch signalling pathway is present in a variety of cell types. in this pathway, Notch ligand binding promotes proteolysis of the Notch receptor and translocation of the intracellular domain of the receptor to the nucleus, where it activates transcription of downstream genes, including HES1, CCND1, MYC and BCL2 (REF.41). these genes work together to regulate many fundamental cellular processes, including cell fate determination, differentiation, development, cell proliferation, survival, apoptosis, invasion and metastasis41.
WNT/β-catenin signalling pathway
In this pathway, activation of the frizzled family cell surface receptors by WNT ligands prevents the degradation of β-catenin, allowing stabilized β-catenin to engage DNA-bound transcription factors and stimulate the transcription of downstream target genes that control many important biological processes, including cellular proliferation, stem cell renewal, embryonic development and tissue regeneration54. For example, in human skin, WNT ligands released from basal epidermal keratinocytes promote the proliferation of the dermal fibroblasts underneath130. in addition, wNt signalling from epidermal keratinocytes localized in the outer root sheath of hair follicles is essential for stimulating the growth of surrounding dermal fibroblasts to support hair follicle regeneration130.
NF-κB signalling pathway
Nuclear factor-κB (NF-κB), a key family of transcription factors, is normally sequestered in the cytoplasm in an inactive form in complex with members of the inhibitors of NF-κB (iκB) family of proteins61. stimulation of the NF-κB signalling pathway by extracellular signals, including infectious agents, inflammatory cytokines and other pathogenic insults, leads to a cascade of orderly responses that culminate in the activation of the iκB kinase (iKK) complex. activated iKK in turn induces phosphorylation and degradation of iκB. the released NF-κB can translocate into the nucleus and coordinate the expression of a large number of genes involved in inflammation, immunity, cell death and proliferation61.
DNA damage response
The major components in this signalling network are ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) kinases. theATM kinase pathway is primarily activated by double-stranded DNa breaks, whereas the ATR kinase pathway responds mostly to single-stranded breaks70. activated ATM and ATR phosphorylate the downstream kinases checkpoint kinase 2 (CHK2) and CHK1, respectively70. CHK2 and CHK1 phosphorylate downstream effectors, including cellular tumour antigen p53, to activate the checkpoints that stall cell cycle progression while recruiting the necessary proteins to repair DNA damage70. Depending on the severity of the damage, these pathways can also induce senescence or apoptosis70.
Major discoveries in recent years have revealed similar oncogenic mechanisms among these divergent viruses. Advances in omics technologies have resolved a network of genetic and functional changes induced by oncogenic virus infection. In this Review, we discuss recent insights that explain how oncogenic viral factors modulate host cell processes and cellular microenvironments to promote cellular transformation and metastasis. Identifying commonalities among these events may lead to new approaches for preventing and treating cancers caused by viruses.
Targeting tumour suppressor pathways
The activation of tumour suppressor pathways is crucial to defence against cellular transformation that can occur when cells are infected by oncogenic viruses. The resulting cellular responses, including cell cycle arrest, apoptosis and senescence, can inhibit virus replication, repair DNA damage and prevent cancer development12. Cellular tumour antigen p53 and retinoblastoma protein (prb) are at the heart of the two major tumour suppressor pathways, which function to repress tumorigenesis by tightly regulating cell cycle progression, stimulating cellular DNA damage response and inducing apoptosis after irreversible cell damage12. Nearly all the oncogenic viruses encode oncoproteins that dysregulate the p53 and pRB pathways; however, the mechanisms that they employ are distinct2. Viral oncoproteins inhibit the function of p53 and pRB by inducing their degradation, inactivation, repression or dissociation from cognate functional partners (reviewed in refS2,13,14).
Dysregulating the tumour suppressor activities of p53 and pRB can benefit virus propagation. For example, the oncoproteins encoded by small DNA oncogenic viruses (for example, HPV) and large oncogenic herpesviruses (for example, EBV and KSHV) can inactivate the function of pRB and p53 to drive the cell into S phase (that is, the phase of DNA synthesis), granting the virus access to the cellular replication machinery and nucleotides for viral DNA synthesis13. In addition, both HTLV-1 oncoproteins transactivator from X-gene region (Tax) and basic zipper factor (HBZ) can inhibit p53 function through various mechanisms that predispose cells to oncogenesis15. The p53 and pRB pathways are also frequently dysregulated in HBV-associated HCC; the viral HBV-X protein (HBx) forms a complex with p53 and inhibits its DNA binding and transcription factor functions16.
Elimination of virally infected cells through apoptosis represents a principle host defence mechanism against viral infection. Inhibition of apoptotic signalling by oncogenic viruses therefore permits viral replication and propagation before the death of the host cell2. Nearly all oncogenic viruses have evolved complex apoptosis evasion strategies that target the p53 and pRB pathways to evade host responses to infection and to establish a persistent infection16–20. Targeting of cell cycle checkpoints and apoptosis pathways by viruses places host cells at risk of cellular genomic instability and chromosome abnormality2. Compounding genetic mutations that are acquired by cells in this deteriorating environment could ultimately lead to cancer.
Targeting host signalling pathways
Cellular proliferation is regulated by tightly controlled signalling pathways (box 2). Evidence from recent studies has revealed common strategies that are used by oncogenic viruses to subvert these pathways in a manner that promotes viral infection and occasional cellular transformation (box 2; FIG. 1).
Fig. 1 |. Signalling pathways targeted by oncogenic viruses.
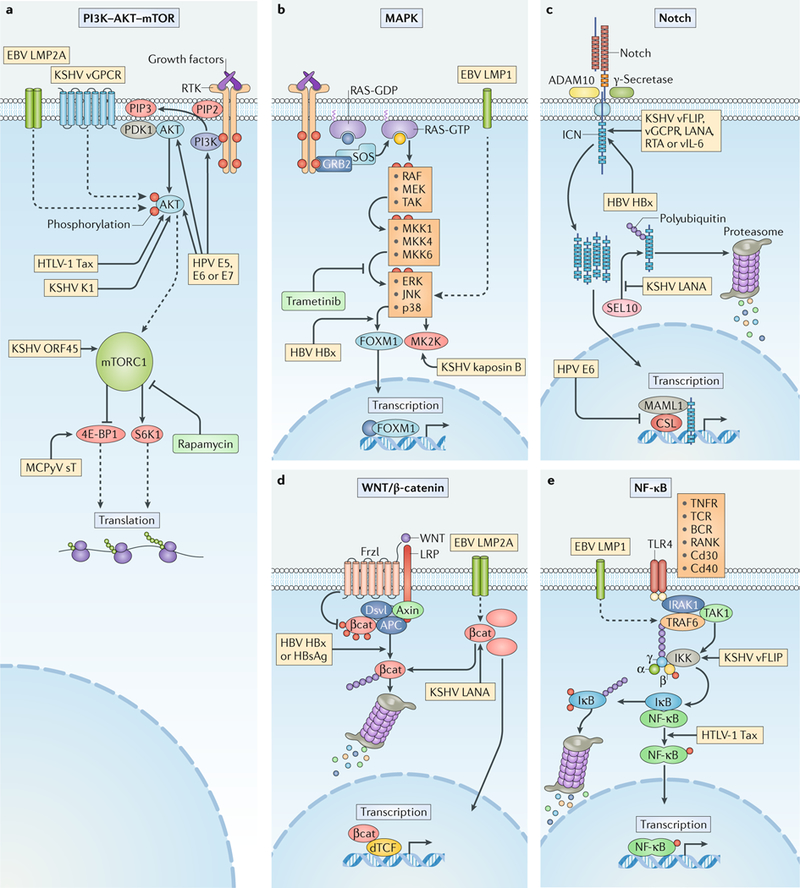
Human oncogenic viruses modulate signal transduction pathways that control cell growth, proliferation and survival to optimize cellular conditions for viral replication, virion assembly and autophagic evasion in the absence of growth or survival signals. Dysregulation of these pathways through mutation or viral factors has been implicated in many cancers. Targeting of critical axes in these pathways by human oncogenic viral factors is indicated by yellow boxes. Arrows represent activation, whereas blocking arrows represent inhibition. Dashed arrows indicate activation or promotion with multiple steps not shown. a | Mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) is a master regulator that coordinates biomolecule availability and stress stimuli to yield tuned responses that promote cell growth and inhibit autophagy. Growth factor binding to receptor tyrosine kinases (RTKs) regulates mTORC1 activity through phosphatidylinositol 3-kinase (PI3K) and the serine/threonine kinase AKT. Ligand-bound RTKs autophosphorylate and recruit PI3K to the plasma membrane, where it converts phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 recruits 3-phosphoinositide-dependent protein kinase 1 (PDK1) and AKT. Multiple viruses modulate the activity of the AKT pathway and downstream components, such as eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1) and ribosomal protein S6 kinase β1 (S6K1). b | The mitogen-activated-protein kinase (MAPK) pathway is also activated by ligand-bound RTKs. Autophosphorylated tyrosine residues bind SH2 domains of growth factor receptor-bound protein 2 (GRB2), which localizes the guanine-exchange factor son-of-sevenless (SOS) to the inner membrane. SOS allows for the exchange of GDP for GTP on RAS. Activated GTP-bound RAS initiates a MAPK cascade, which activates transcription factors such as forkhead box protein M1 (FOXM1) and additional effectors such as MK2 kinase (MK2K). Together, they enhance the expression of pro-survival and pro-inflammatory genes through increased transcription and stabilization of mRNAs, respectively. c | A conformational change in Notch when bound to ligands on neighbouring cells enables sequential cleavages by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and γ-secretase. Cleavage releases intracellular domain of Notch (ICN) into the cytoplasm, where it can translocate to the nucleus and coordinate the transcription of proliferation and differentiation-related genes with DNA-bound CSL protein and the co-activator mastermind-like 1 (MAML1). ICN is downregulated by SEL10 polyubiquitylation-mediated proteasomal degradation. d | β-Catenin (βcat) is inactivated in a complex with adenomatous polyposis coli gene product (APC) and axin, which phosphorylates βcat and targets it for proteasomal degradation. Upon WNT glycolipoprotein binding to extracellular domains of prolow-density lipoprotein receptor related protein 1 (LRP1) and frizzled (Frzl), dishevelled (Dsvl) is recruited to the cytoplasmic domain of Frzl. Subsequent phosphorylation of LRP sequesters axin and prevents degradation of βcat. Accumulating βcat translocates to the nucleus, where it co-activates Drosophila T cell factor (dTCF)-mediated transcription of cell growth genes. e | Several immunity-related cell surface receptors, including Toll-like receptor 4 (TLR4) and tumour necrosis factor receptor (TNFR), activate the canonical nuclear factor-κB (NF-κB) pathway when bound to their respective ligands. TLR4 activation leads to phosphorylation and recruitment of interleukin-1 receptor-associated kinase 1 (IRAK1) to the adaptor protein myeloid differentiation primary response protein MYD88. A complex containing the E3-ubiquitin kinase TNF receptor-associated factor 6 (TRAF6) forms, which generates a scaffold for the polyubiquitin-binding NF-κB essential modulator (NEMO) of inhibitors of NF-κB (IκB) kinase (IKK). Orphan nuclear receptor TAK1 (also known as NR2C2) activates IKK, which then phosphorylates the inhibitory subunit (IκB) and targets it for polyubiquitylation and proteasomal degradation. A conformational change between the NF-κB subunits p50 and p65 allows activating phosphorylation and translocation to the nucleus, where it induces expression of inflammatory and pro-survival genes. BCR, B cell receptor; E5, E6, E7, early proteins 5, 6 and 7; EBV, Epstein–Barr virus; ERK1, extracellular-signal-regulated kinase 1; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; HBx, HBVX protein; HPV, human papilloma virus; HTLV-1, human T-lymphotropic virus 1; JNK, JUN N-terminal kinase; KSHV, Kaposi sarcoma-associated virus; LANA, latency-associated nuclear antigen; LMP, latent membrane protein; MCPyV, Merkel cell polyomavirus; MEK, MAPK/ERK kinase; MKK, mitogen-activated protein kinase kinase; RAF, RAF proto-oncogene serine/threonine-protein kinase; RANK, receptor activator of NF-κB (also known as TNFRSF11A); RTA, replication and transcription activator; sT, small tumour antigen; Tax, transactivator from X-gene region; TCR, T cell receptor; vFLIP, viral FLICE inhibitory protein; vGPCR, viral G protein-coupled receptor.
PI3K–AKT–mTOR signalling.
The phosphatidylinositol 3-kinase–AKT–mechanistic target of rapamycin (PI3K–AKT–mTOR) pathway is a major eukaryotic nutrient-sensing pathway that coordinates macromolecule synthesis and metabolism in response to nutrient abundance (box 2; FIG. 1a). It has an important role in the regulation of cellular growth, cell cycle progression, proliferation, survival, quiescence and longevity by coordinating growth stimuli and regulating downstream effectors, including AKT and mTOR. Dysregulation of the PI3K axis can disrupt normal cellular growth control and result in the survival and proliferation of tumour cells21. Some oncogenic viruses, including HPV, EBV, HTLV-1, KSHV and MCPyV, have evolved mechanisms to engage this pathway in the absence of necessary growth factors and when nutrient levels are low (FIG. 1a).
Activation of PI3K–AKT–mTOR signalling may benefit viral infection by promoting cell proliferation22,23 and inhibiting autophagy, which can impede viral replication24. The most extensively studied case is that of HPV, in which each of the viral oncoproteins E5, E6 and E7 either directly or indirectly target the pathway and promote cell division, predisposing infected cells to tumour initiation and progression25 (FIG. 1a). The EBV latent membrane protein 2A (LMP2A) induces AKT phosphorylation and activates the PI3K–AKT pathway26 (FIG. 1a). This contributes to an anti-apoptotic function that prevents the removal of damaged cells and provides a selective advantage for LMP2A-expressing B cells during the development of EBV-associated malignancies26. LMP2A-mediated activation of the PI3K–AKT pathway also inhibits epithelial cell differentiation in EBV-infected cells, suggesting that the same mechanism contributes to progression of EBV-related carcinomas and lymphomas27. HTLV-1 modulates AKT in CD4+ T cells, promoting a long latent phase28. The HTLV-1 Tax oncoprotein was found to activate the AKT pathway and induce AKT-dependent inactivation of the fork-head box protein O3 (FOXO3), which causes depletion of CD4+ T cells through induction of pro-apoptotic and anti-proliferative target genes28 (FIG. 1a). Inhibition of FOXO3 therefore promotes the survival and proliferation of CD4+ T cells that maintain the capacity to spread infectious HTLV-1 particles28. This Tax protein function enables the long-term maintenance of infected CD4+ T cells during the early phase of HTLV-1 pathogenesis28.
The importance of mTOR signalling in KSHV biology was highlighted by the observation that the mTOR inhibitor rapamycin — but not other immunosuppressants — promotes tumour regression in transplant patients affected by KSHV-induced Kaposi sarcoma29. It was later discovered that expression of KSHV ORF45, a lytic gene expressed in infected lymphatic endothelial cells, selectively upregulates mTOR signalling30 (FIG. 1a). The dependence of KSHV-infected cells on the mTOR signalling pathway for their survival explained their sensitivity to rapamycin-induced apoptosis. Expression of the KSHV G protein-coupled receptor (vGPCR) in a mouse allograft model is sufficient to induce sarcomagenesis through the activation of AKT phosphorylation31. The role of AKT in human Kaposi sarcomagenesis was supported by the observation of robust AKT activation in Kaposi sarcoma biopsy samples taken from individuals with AIDS31. In B cells, the K1 protein of KSHV activates AKT signalling to inhibit apoptosis (FIG. 1a), suggesting that this is a mechanism to protect virus-infected cells from premature cell death during KSHV-induced oncogenesis32. By comparison, the small T oncoprotein of MCPyV targets the PI3K–AKT–mTOR signalling pathway further downstream (FIG. 1a). It promotes the hyper-phosphorylation of eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1), a crucial target of mTOR complex 1 (mTORC1), leading to hyperactivated cap-dependent translation of cellular proteins and cellular transformation33. Infection by each of these evolutionarily distinct viruses leads to a state of anabolism that is caused by targeting mTOR, which ordinarily responds to a network of signals such as amino acid availability and environmental stress.
MAPK signalling.
Mitogen-activated protein kinase (MAPK) pathways regulate the transcription of genes that control cell proliferation and the antiviral immune response34 (box 2). They are involved in the life cycle and propagation of several oncogenic viruses, such as HCV, HPV and MCPyV, by promoting viral assembly, production and release (FIG. 1b). For example, the activity of MAPK-regulated cytosolic phospholipase A2 (PLA2G4A) contributes to the assembly of infectious HCV particles35. Arachidonic acid, the cleavage product of PLA2G4A-catalysed lipolysis, restores the production of infectious HCV particles in the absence of PLA2G4A35. This suggests that PLA2G4A-mediated lipolysis provides a membrane environment for efficient incorporation of core proteins into the lipid envelope of nascent viral particles35. MAPK signalling also enhances non-enveloped virus production, as evidenced by increased HPV virion production upon induction of extracellular-signal-regulated kinase 1 (ERK1) and ERK2 in HPV-infected cells36. In agreement with this finding, inhibition of MAPK/ERK kinase 1 (MeK1) and MEK2 with a cancer drug (trametinib) drastically limits MCPyV infection by blocking MCPyV transcription and/or replication in infected cells, suggesting that activation of the MAPK pathway is needed to support these events in the MCPyV life cycle6. However, whether MAPK pathways also have a role in the development of MCPyV-associated Merkel cell carcinoma (MCC) is unknown.
Oncogenic viruses often manipulate MAPK pathways to promote host cell proliferation, but this process could incidentally give rise to invasive cells that contribute to metastasis. During the switch from the latent to the lytic phase of EBV infection, the p38 MAPK pathway has a crucial role in protecting host cells from apoptosis and in inducing viral reactivation37. The EBV LMP1 that is induced during the latent–lytic transition has been proposed to prevent apoptosis and mediate reactivation, although this hypothesis has not been tested experimentally37. LMP1 expression in epithelial cells activates the ERK–MAPK pathway, promoting cell motility and metastasis38 (FIG. 1b). In this way, LMP1 may contribute to cell invasion in EBV-associated nasopharyngeal carcinoma38.
The molecular mechanism by which KSHV activates MAPK pathways is better understood than in EBV infection39. The KSHV kaposin B protein binds to and activates an effector of the p38 MAPK signalling pathway, MK2 kinase (MK2K), which then stabilizes pro-inflammatory and pro-survival cytokine mRNAs39 (FIG. 1b). The increased cytokine production could promote the growth and survival of tumour cells in KSHV-associated oncogenesis39. As another example, the HBV HBx protein activates the ERK pathway and induces the expression of a master regulator of tumour metastasis, FOXM1 (ref.40) (FIG. 1b). FOXM1 contributes to HBV-induced hepatocarcinogenesis by transactivating the expression of MMP7, RHOC and ROCK1, which promote hepatoma cell invasion and metastasis40. Enhanced invasiveness, dysregulated cell division and elevated cytokine production via hyperactive MAPK signalling may provide the optimal environment for virus propagation, but it also drives cancer pathology and resistance to treatment.
Notch signalling.
Depending on the cellular environment and tissue context, perturbations in the Notch signalling pathway can either promote or suppress tumorigenesis41 (box 2). A role for Notch signalling was found in the development of chronic lymphocytic leukaemia, B cell malignancies and breast cancer42. By contrast, Notch signalling has a tumour suppressor function in skin epithelia and pancreatic cells41. Unlike the pathways explored in the previous sections that are largely upregulated in all cancers, the divergent association of Notch signalling with different cancers is reflected in the variety of approaches through which viruses exploit this pathway (FIG. 1c).
In a systematic analysis of the host interactome and transcriptome networks that are perturbed by oncogenic virus proteins, Notch signalling was identified as a key pathway that is targeted by EBV, HPV and MCPyV oncoproteins, highlighting its importance in viral tumorigenesis43. HPV E6 oncoproteins repress Notch signalling and promote viral persistence in basal epithelial cells. Mastermind-like protein 1 (MAML1) and several other components of the Notch transcription complex are targeted by β-genus HPV E6 proteins to repress Notch transcriptional activation44 (FIG. 1c). E6 proteins of other cutaneous HPVs, such as HPV-8, use a similar strategy to suppress Notch-dependent transcription of the HES1 transcriptional repressor45,46, halting keratinocyte differentiation, a disruption that has been linked to the function of HPV in promoting cell proliferation and oncogenesis45,46. EBV also interferes with Notch signalling to provide a cellular environment for long-term infection47. Epstein–Barr nuclear antigen 2 (EBNA2) and activated Notch both compete for recombining binding protein suppressor of hairless (RBP-Jκ)47, and therefore, activated Notch limits EBNA2-mediated transcription of EBV genes involved in the transformation of infected B cells. Constitutive Notch signalling in the lymphoid microenvironment may lead to EBV latency by downregulating the transcription-promoting function of EBNA247.
HBV and KSHV also activate Notch signalling. The HBV HBx protein stimulates the expression of neurogenic locus Notch homologue protein 1 (NOTCH1), which promotes the proliferation of HCC cells and may thus contribute to the oncogenic mechanism of HBV-associated HCC48 (FIG. 1c). Elevated levels of activated Notch proteins are frequently observed in KSHV-associated Kaposi sarcoma lesions49. KSHV proteins, including viral FLICE inhibitory protein (vFLIP), vGPCR, latency-associated nuclear antigen (LANA), replication and transcription activator (RTA) and viral interleukin-6 (vIL-6), can induce the expression of core Notch receptors and ligands that activate the pathway50,51 (FIG. 1c). Stimulation of Notch signalling by these viral proteins appears to suppress the expression of cell cycle-associated genes in neighbouring uninfected cells, inhibiting their proliferation and potentially providing a growth and survival advantage to infected cells during Kaposi sarcoma pathogenesis50,51. Notch pathway activation induced by vFLIP and vGPCR also results in transcriptional reprogramming of the infected lymphatic endothelial cells to mesenchymal cells through a process called endothelial-to-mesenchymal transition. The growth and migration of infected cells promote viral spread and contribute to Kaposi sarcoma invasiveness52. KSHV LANA competitively inhibits the interaction between the intracellular domain of NOTCH1 (ICN) and an E3 ubiquitin ligase, F-box/WD repeat-containing protein 7 (SEL10; also known as FBXW7), thereby preventing proteasomal degradation of ICN53 (FIG. 1c). Stabilized ICN in turn functions as a proto-oncogene and stimulates the proliferation of KSHV-infected tumour cells, thus promoting virus-mediated transformation53 (FIG. 1c). The observation that positive and negative regulation of Notch signalling can both contribute to viral oncogenesis indicates that transformation depends on the context of the cellular environment and the infected cell type.
WNT/β-catenin signalling.
The WNT/β-catenin signalling pathway regulates diverse physiological processes, such as growth control, stem cell renewal, embryonic development and tissue differentiation54 (box 2). Hyperactivation of the downstream transcription targets of WNT/β-catenin signalling can contribute to many growth-related pathologies, including cancer54. Viral oncoproteins modulate the WNT/β-catenin pathway and contribute to carcinogenesis (FIG. 1d). For example, both KSHV LANA and EBV LMP2A proteins can stabilize β-catenin55–57 (FIG. 1d), which then upregulates downstream genes, such as CCND1 and MYC, to increase cell proliferation and promote tumorigenesis58. HBV encodes multiple proteins, including HBx and hepatitis B surface antigen (HBsAg), that aberrantly activate WNT/β-catenin signalling59 (FIG. 1d). HBx and HBsAg silence antagonists of the pathway or upregulate and stabilize its key components such as β-catenin. Together, these activities stimulate abnormal transcription of target genes that drive cell proliferation, which ultimately contributes to HCC development59. Similarly, continual expression of HTLV-1 HBZ in HTLV-1-induced adult T cell leukaemia cells dysregulates the WNT signalling pathway to promote migration and proliferation60.
The role of WNT/β-catenin in other viral cancers is less clear, though its function in oncogenic virus infection may provide important clues. For example, activation of the pathway stimulates MCPyV infection6. Induction of downstream matrix metalloproteinase (MMP) genes contributes to MCPyV infection by disrupting the extracellular matrix of the host cells6. Skin damage induced by ultraviolet light and ionizing radiation, wounding or ageing processes can lead to the activation of WNT/β-catenin signalling and the expression of MMPs. This suggests that these major risk factors for MCPyV-associated MCC stimulate viral infection and thus promote tumour development through MMP induction6.
NF-κB signalling.
Activation of the nuclear factor-κB (NF-κB) pathway by pathogens and inflammatory cytokines leads to the induction of genes involved in diverse cellular processes, particularly the innate immune and inflammatory responses61 (box 2). Activation of NF-κB and downstream target genes in chronic infection and inflammation also promotes cancer progression by stimulating cell proliferation, inhibiting apoptosis and enhancing invasiveness62. NF-κB activation is part of an appropriate response to acute viral infection, but viruses that establish infections in adaptive immune cells can utilize constitutive NF-κB activation to expand their host environment (FIG. 1e). For instance, the EBV oncoprotein LMP1 drives the development of lymphomas by activating NF-κB downstream target genes63. It does so by mimicking constitutively activated host tumour necrosis factor receptor (TNFR) and engaging interleukin-1 receptor-associated kinase 1 (IRAK1) and TNF receptor-associated factor 6 (TRAF6), the upstream signal transducers of the NF-κB pathway64 (FIG. 1e). This LMP1-induced NF-κB activation promotes the proliferation and survival of infected B cells64,65.
NF-κB is also constitutively activated in the majority of KSHV-induced primary effusion lymphoma (PEL) cells66. In these cells, the KSHV vFLIP protein activates the NF-κB pathway by associating directly with an inhibitor of NF-κB (IκB) kinase (IKK) complex component, inducing a conformational change that renders it constitutively active66,67 (FIG. 1e). In transgenic mice that express KSHV vFLIP, vFLIP-activated NF-κB contributes to enhanced proliferation of lymphocytes and an increased incidence of lymphoma68. Likewise, the HTLV-1 Tax protein is considered the primary factor by which this virus transforms T cells, and part of its function involves activating NF-κB69 (FIG. 1e).
The activation of NF-κB highlights the apparently conflicting roles of inflammation in infection and cancer. NF-κB-mediated inflammation is crucial for proper innate immune responses to acute infection or damaged cells, but also mediates pathology (for example, pain, tissue damage or swelling, and immunosuppression) and cancer progression. The specific situations in which oncogenic viruses evade or induce inflammation can inform our understanding of immunity and disease.
Exploiting the host DNA damage response
The host DNA damage response (DDR) system is a complex network of signalling pathways that collectively monitor and repair DNA damage that results from DNA replication, cellular metabolism and exogenous insults, such as radiation and viral infection70 (box 2). Stimulation of the major components of the DDR signalling network, such as ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related protein (ATR) kinases, can induce a cascade of phosphorylation events that activates downstream effectors (for example, p53) to stall cell cycle progression at checkpoints. Cell cycle check-points allow time to repair damaged DNA or induce senescence or apoptosis70. Cells with disrupted DNA damage recognition and repair systems can accumulate genetic mutations that enhance cell survival and proliferation. Failure to control these populations of cells can ultimately lead to cancer.
Viruses often elicit host DDRs; however, they have evolved mechanisms to undermine these responses and manipulate them to their advantage71,72 (FIG. 2). In the process of engaging the DDR machinery, some viruses optimize the cellular environment for their replication by promoting progression to the S phase and inhibiting apoptosis73–77. In addition, DNA viruses such as HPV and MCPyV activate ATM-related and ATR-related DDR factors and recruit them to viral DNA replication foci, promoting viral DNA replication74–76,78.
Fig. 2 |. Viral oncoproteins and DNA damage responses influence the fate of the host cell.
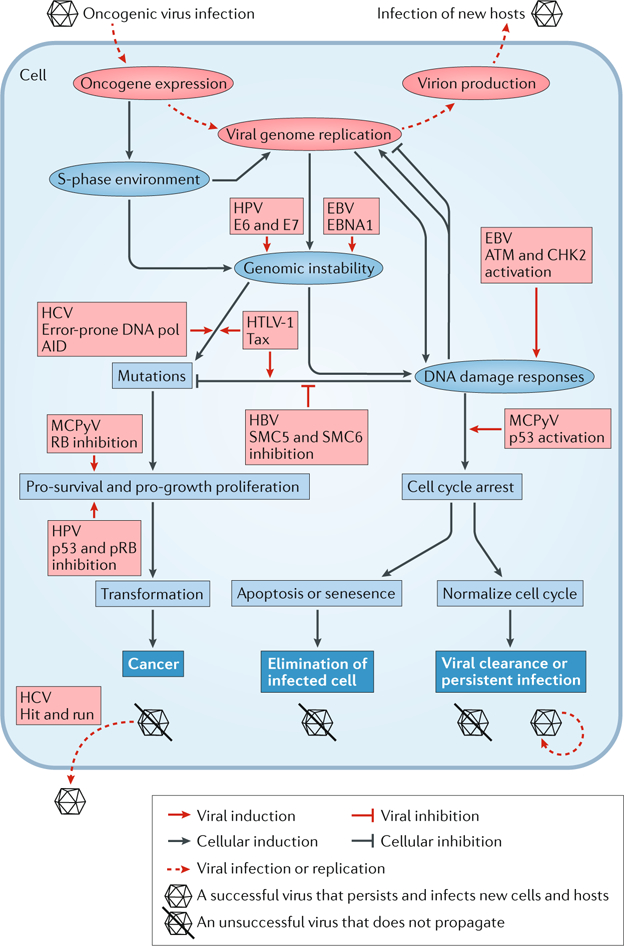
The schematic depicts changes to the cellular environment as a result of oncogenic virus infection. Red ellipses represent stages of the life cycle that are shared by oncogenic viruses; red boxes represent effects caused by the indicated viral effector. Blue ellipses represent the immediate changes to the cellular environment resulting from virus infection; blue boxes represent subsequent effects on the cell; blue boxes with white text are the possible fates of the infected cell. Arrows signify that the factor or status promotes the effect it points to, whereas blocking arrows signify inhibition. For example, genomic instability and viral genome replication can both induce DNA damage responses, which in turn support or hinder viral replication, depending on the viral infection context. Successful viruses avoid abortive fates (virion with a line through it), such as programmed cell death or cancer, to persist and infect new hosts. AID, activation-induced cytidine deaminase; ATM, ataxia telangiectasia mutated; CHK2, checkpoint kinase 2; E6, E7, early proteins 6 and 7; EBNA1, Epstein–Barr virus nuclear antigen 1; EBV, Epstein–Barr virus; HBV, hepatitis B virus; HCV, hepatitis Cvirus; HPV, human papilloma virus; HTLV-1, human T-lymphotropic virus 1; MCPyV, Merkel cell polyomavirus; p53, cellular tumour antigen p53; pol, polymerase; pRB, retinoblastoma protein; SMC5/6, structural maintenance of chromosomes complex 5/6.
The persistent engagement of DDR factors and enforcement of a replicative state by oncogenic viruses results in genomic instability79 (FIG. 2). Generally, oncogenic virus infection increases the rate of DNA breaks while depleting host factors that maintain genome integrity79. Compromised sensing, signalling or repair of damaged DNA may allow cells to acquire mutations that overcome tumour suppressor barriers during oncogenic progression71.
Genomic instability is frequently observed in high-risk HPV-associated cervical neoplasias and is caused by HPV oncoproteins E6 and E7, which induce DNA damage, mitotic defects and centrosome-related mitotic defects80 (FIG. 2). High-risk HPV oncoproteins also hinder DNA repair and destabilize the cellular genome81. By reducing genomic fidelity as cells divide, these viral oncoproteins increase the chances of acquiring additional genetic changes that may contribute to HPV-associated carcinogenesis80,81.
Replication stress, nucleotide deficiency and the production of reactive oxygen species (ROS) during viral infection can also contribute to genomic instability and oncogenesis. For instance, EBNA1 can increase the transcription of NADPH oxidase to induce ROS production, leading to host DNA damage and chromosomal aberrations that contribute to EBV-associated malignancy82 (FIG. 2). During persistent HCV infection, chronically activated inflammatory cells release ROS, which can cause oxidative DNA damage and promote a pro-carcinogenic microenvironment that drives HCC development83.
Although manipulation of the cell cycle and DDR factors can promote a fragile genomic state, appropriate activation of DDRs to viral stressors remains a major barrier for progression to cancer. For example, the metabolic and genotoxic stress that is induced by EBV can trigger cellular senescence84. EBV infection of primary human B cells induces transient hyper-proliferation that activates the ATM–checkpoint kinase 2 (CHK2; also known as CHEK2) DDR pathway, which subsequently suppresses the growth of infected cells. Abrogation of ATM and CHK2 kinase activity, however, results in B cell transformation85.
MCPyV expresses large T antigen carrying carboxy-terminal origin-binding and helicase domains that cause damage to DNA, stimulate host DDRs and activate the p53 pathway to inhibit cellular proliferation86. Unlike MCPyV large T antigen expressed in persistent infection, MCPyV proviruses integrated in malignant MCC cells encode large T antigen truncation mutants that almost invariably delete this DDR-activating domain but retain the aminoterminal pRB-inhibiting motif87. This observation supports the notion that DDRs are an effective barrier to malignant progression, but oncogenic viruses make these defences vulnerable.
As a retrovirus, HTLV-1 undermines genomic integrity as part of its life cycle. HTLV-1 DNA integration into T cell genomes induces a lengthy latency period, in which a polyclonal expansion of the infected cells progresses to an aggressive monoclonal leukaemia in ~5% of infected individuals88. HTLV-1 proviruses preferentially integrate in the vicinity of tumour suppressor genes, which are consequently disrupted by provirus-dependent transcription termination or viral antisense RNA-dependent cis-perturbation88. The same integration pattern was observed in cells at asymptomatic stages as in leukaemia or lymphoma cells, suggesting that provirus-dependent gene perturbations trigger initial polyclonal expansion of the infected clones at non-malignant stages88. Expression of HTLV-1 Tax protein induces further DNA damage and genomic instability by inhibiting DNA repair pathways and causing DNA repair infidelity, allowing the accumulation of somatic mutations in clones that ultimately progress to malignancy89 (FIG. 2).
Whereas the aforementioned viruses promote the accumulation of mutations indirectly, HCV, an oncogenic RNA virus with no apparent oncogenes, directly induces a mutator phenotype90. In B cells, HCV infection induces somatic hypermutations in tumour suppressors and proto-oncogenes, such as p53 and β-catenin. RNAi and antisense targeting experiments revealed that the high mutation frequency in HCV infection is caused by the increased expression of error-prone DNA polymerases and activation-induced cytidine deaminase (AID), which cause the hypermutation of cellular genes90 (FIG. 2). Mutations in the tumour suppressors and proto-oncogenes were amplified and selected for in HCV-associated lymphomas and HCCs but not in similar neoplasias originating from other causes90. Although HCV-related mutations contribute to the development of HCC, HCV RNA is not found in most of the virus-induced HCC cells, suggesting a ‘hit and run’ oncogenic mechanism90 (FIG. 2).
In contrast to the mutator phenotype that is induced by HCV90, HBV engages host DDR pathways in a different manner91,92. The viral protein HBx induces the degradation of the structural maintenance of chromosomes complex 5/6 (SMC5/6), which is a host DNA damage repair regulator that normally binds extrachromosomal HBV genomes to repress viral transcription91,92 (FIG. 2). In doing so, HBx derepresses transcriptional inhibition, allowing productive viral gene expression and replication91,92.
RNA and DNA oncogenic viruses elicit widespread changes to the cellular environment that support the viral infection cycle. This induces both direct and indirect stresses on the integrity of the host genome and the pathways governing cell fate. By repurposing and undermining the mechanisms that protect the host cell from cellular transformation, oncogenic viruses establish a precarious balance between the ideal environment for viral proliferation and termination through cell death or transformation.
Manipulation of host immune responses
Oncogenic viruses interface with host immune systems throughout persistent infections. Epidemiological evidence suggests that their mechanisms to evade detection and elimination are adapted to deal with the constant pressure from the host. Oncogenic viruses maintain persistent infections in immune-competent hosts with few symptoms and are more likely to induce malignancies in immunocompromised individuals18,19,93. Generally, viruses evolve to evade intrinsic restriction, avoid inflammatory responses and prevent targeted killing of their host cells94. Unique immune evasion strategies are used for distinct phases, such as the latent and lytic stages of the viral life cycle. Emerging evidence suggests that viral subversion of immunity potentiates cancer because the same immunomodulatory tactics directed at evading detection or expanding virus number can also prevent adequate surveillance of transformed cells or increase cellular proliferation (FIG. 3).
Fig. 3 |. Modulation of host immune responses by oncogenic viruses.
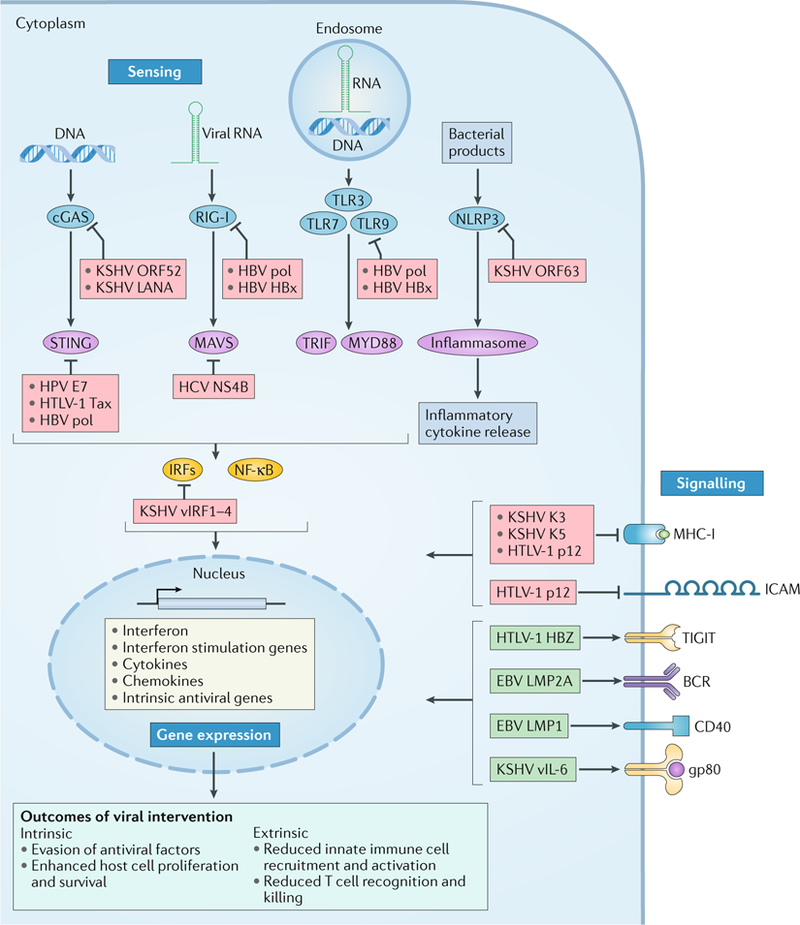
Proteins encoded by oncogenic viruses can target the host immune response (blue boxes with white text), including sensing of pathogen-associated molecular patterns, immune gene expression profiles and intercellular signalling. Arrows indicate activation, whereas blocking arrows indicate inhibition. Viral DNA and RNA structures are detected by pattern recognition receptors (blue ellipses), including cyclic GMP–AMP synthase (cGAS), retinoic acid-inducible gene I (RIG-I) and endosomal Toll-like receptors (TLRs). Activation is transduced through intermediates or adaptors (purple ellipses), such as stimulator of interferon genes protein (STING), mitochondrial antiviral-signalling protein (MAVS), TIR domain-containing adaptor molecule 1 (TRIF; also known as TICAM1) and myeloid differentiation primary response 88 (MYD88). Activated transcription factors, such as interferon regulatory factors (IRFs) and nuclear factor-κB (NF-κB), upregulate expression of immune genes (yellow box). Alternatively, inflammasome activation by NOD-, LRR- and pyrin domains-containing 3 (NLRP3) can mediate proteolytic activation of inflammatory cytokines and inflammatory cell death in response to bacterial effectors or cell damage signals. Oncogenic viruses undermine inflammatory responses at the level of pathogen sensing and signal transduction (red boxes). They also limit recruitment of leukocytes to infected cells by reducing immune modulator intercellular adhesion molecule (ICAM) expression and downregulating the display of viral peptides on major histocompatibility complex I (MHC-I). Oncogenic viruses that infect adaptive immune cells can induce or simulate pro-expansion signals and promote a state that is unresponsive to antigen and endogenous cytokines (green boxes). BCR, B cell receptor; E7, early protein 7; gp80, glycoprotein 80; HBV, hepatitis B virus; HBx, HBV-X protein; HBZ, HTLV-1 basic zipper factor; HCV, hepatitis C virus; HPV, human papilloma virus; HTLV-1, human T-lymphotropic virus 1; KSHV, Kaposi sarcoma-associated virus; LANA, latency-associated nuclear antigen; LMP, latent membrane protein; NS4B, non-structural protein 4B; pol, polymerase; TIGIT, T cell immunoglobulin and ITIM domain; vIL-6, viral interleukin-6; vIRF1–4, viral interferon regulatory factors 1–4.
To initiate a response to infection or to aberrant cells, the host must first sense something atypical to healthy cellular function. Cytosolic DNA represents a danger signal for the cell, whether it originates endogenously or from an invading DNA virus. As DNA is normally compartmentalized within the nucleus and mitochondria, loss of organelle or genomic integrity or the presence of foreign DNA is an ideal signal to trigger an immune response. Cyclic GMP–AMP synthase (cGAS) is a cytosolic DNA sensor that synthesizes a soluble cyclic dinucleotide (cyclic GMP–AMP (cGAMP)) when bound to duplex DNA. cGAMP, in addition to second messengers released by intracellular bacteria, activates endoplasmic reticulum-resident stimulator of interferon genes protein (STING). STING and downstream Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signalling activate interferon-dependent antiviral programmes95 (FIG. 3). Oncogenic DNA viruses antagonize the cGAS–STING pathway to avoid interferon-mediated restriction (FIG. 3). KSHV evolved multiple effectors that inhibit this pathway, including ORF52, LANA and viral interferon regulatory factor 1 (vIRF1)96–98. ORF52 directly binds cGAS and inhibits its enzymatic activity97. LANA, especially its cytoplasmic isoform, also directly associates with cGAS to antagonize the activation of its downstream components98. vIRF1 blocks the interaction between STING and its upstream serine/threonine-protein kinase TBK1, thus preventing STING phosphorylation and activation of downstream signalling96. Inhibition of cGAS–STING by these KSHV oncoproteins contributes not only to the establishment of a latent infection but also to reactivation from latency96–98, which is crucial for both disseminating infectious virus and potentiating tumour growth99.
Blockade of the cGAS–STING axis could be a general feature used by oncogenic viruses to overcome antiviral immune defences (FIG. 3). For instance, HPV E7 binds STING to inhibit downstream signalling and interferon-β (IFNβ) production in tumour cells100. In addition, HTLV-1 oncoprotein Tax suppresses the cGAS–STING pathway to inhibit IRF3 phosphorylation and type I interferon production101. Likewise, HBV polymerase interacts directly with STING to abrogate downstream IRF3 activation102. HCV non-structural protein 4B (NS4B) inhibits this virus-induced interferon signalling pathway by directly interacting with STING to block its interaction with mitochondrial antiviral-signalling protein (MAVS), a member of the retinoic acid-inducible gene-I (RIG-I) viral RNA sensing pathway103. Growing evidence in cancer research suggests that the cGAS–STING pathway is a crucial early detection system for cells that have sustained substantial DNA damage. Cells with unresolved DNA breaks may leak chromosomal DNA into the cytoplasm or exhibit ruptured micronuclei that recruit and activate cGAS104,105. Given the importance of this pathway in defence against cancer, it is possible that inhibition of cGAS–STING compromises an early barrier to viral oncogenesis.
Viral immune evasion extends to other sensory pathways (FIG. 3). HBV polymerase and HBx proteins can abolish interferon production through RIG-I and Toll-like receptor 3 (TLR3), thus blocking IRF3 activation106,107. KSHV also blocks inflammasome activation, which normally facilitates inflammatory cell death programmes and the transition from innate to adaptive response to intracellular pathogens or cell damage. KSHV ORF63 is a viral homologue of human NOD-, LRR- and pyrin domain-containing 1 (NLRP1), a cytosolic sensor that activates the inflammasome in response to infections108. ORF63 binds NLRP1 and inhibits downstream inflammasome-dependent inflammatory cytokine production, contributing to chronic infection108.
Downstream of intracellular threat detection, a compromised cell may activate transcriptional programmes to suppress its growth and survival. Oncogenic viruses express effectors that counteract the anti-proliferative immune response and serve as key drivers of their oncogenic potential. This host–pathogen relationship is typified in KSHV-infected cells. KSHV encodes four homologues of cellular IRFs that mediate broad protection against viral infection and aberrant cellular proliferation109 (FIG. 3). By dimerizing with cellular IRFs and other transcription factors, KSHV vIRFs repress the immune response to infection (by down-regulating interferon signalling) and dysregulate cell growth control (by targeting the NF-κB, MYC and p53 pathways)109. KSHV infection may still induce interferon despite vIRF competitive binding, resulting in p21-mediated cell cycle arrest110. To overcome the growth-limiting effect of interferon, the virus activates an alternative transcriptional programme that allows only vIL-6 expression in response to interferon stimulation110 (FIG. 3). Human IL-6 (hIL-6) normally binds to its receptor membrane glycoprotein 80 (gp80; also known as IL-6R), which forms a functional complex with the transmembrane transducer membrane glycoprotein 130 (gp130; also known as IL6RB) to activate transcription of genes that control cell proliferation111. IFNα was found to specifically downregulate gp80, but this has no effect on gp130 expression110. Unlike hIL-6, vIL-6 can bypass the interferon–gp80 autoregulatory checkpoint by directly binding to and activating gp130, establishing an autocrine feedback circuit to overcome interferon-induced growth inhibition110. KSHV thus provides an example of how oncogenic viruses may subvert innate immunity at the level of transcription for optimal viral propagation.
In addition to cell intrinsic changes, oncogenic viruses also modulate interactions between infected cells and immune cells. Evasion of extrinsic cellular responses can contribute to the pathological expansion of host cells by limiting normal immune clearance (FIG. 3). For instance, many oncogenic viruses have evolved strategies to downregulate major histocompatibility complex class I (MHC-I), which presents peptides derived from intracellular proteins to CD8+ T cells for targeted cell killing112. Viral proteins inhibit MHC-I function by interfering with the synthesis, translocation or assembly of MHC I molecules113. In addition, KSHV K3 and K5 proteins downregulate cell surface MHC-I display by promoting endocytosis and endolysosomal degradation of class I chains.114–116
HTLV-1 manipulates immune cell interactions through a unique set of strategies that have been explored in greater detail (FIG. 3). HTLV-1 p12 down-regulates immune modulator intercellular adhesion molecule 1 (ICAM1), ICAM2 and MHC-I on the cell surface, allowing infected cells to escape killing by natural killer cells and cytotoxic T cells117. HTLV-1 p8 downregulates T cell signalling to induce T cell anergy. At the same time, p8 induces the formation of plasma membrane conduits between infected and uninfected T cells, enabling spread without the virion entering the extracellular space118. HTLV-1 HBZ enhances the immunosuppressive state by upregulating the expression of a T cell co-inhibitory molecule, T cell immunoreceptor with Ig and ITIM domains (TIGIT), in infected CD4+ T cells119. TIGIT activity attenuates T cell responses to another HTLV-1 virus antigen, Tax119. Together, HTLV-1 accessory proteins shape the microenvironment of adaptive and innate immune cell interactions, allowing the virus to escape host immune recognition and achieve efficient propagation.
Similar to HTLV-1 modulation of T cells, EBV exploits the ability of B cells to expand and disseminate continuously in order to propagate and avoid detection120,121 (FIG. 3). LMP1, a key viral protein for EBV-driven human B cell transformation, shares functions with the constitutively active B cell co-stimulatory receptor CD40, and signals through common down-stream pathways, such as JUN N-terminal kinase (JNK), ERK, p38 and NF-κB, to promote B cell survival and proliferation93. EBV LMP2A mimics constitutively activated B cell receptors to stimulate B cell proliferation and associated pathogenesis122,123. By augmenting the natural propensity of B cells to be long-lived, invasive and self-renewing, EBV drives infected populations to a state conducive to malignant lymphoproliferation120,121. The fact that EBV causes solid tumours in addition to lymphomas highlights its capacity to evade detection and promote cellular expansion in different cellular environments.
Cellular immune responses to intracellular pathogens are often similar to responses to nascent transformation, including detection of abnormal molecular signals, cell cycle arrest, cytokine release, inflammation and directed killing of affected cells. Oncogenic viruses employ related strategies to undermine these processes. By enhancing cell survival and proliferation while blocking extrinsic immune destruction, they establish and maintain an optimal environment for viral persistence. In this way, virus immune evasion can contribute to tumorigenesis and associated pathologies. These observations provide support for the anti-antivirus hypothesis, which suggests that, when disabling host antiviral defences, oncogenic viruses incidentally drive infected cells towards cancer2.
Conclusions and outlook
Viruses have evolved an array of tactics to exploit and subvert the host cellular machinery for propagation. In parallel, their hosts evolved mechanisms to maintain the integrity of the cellular environment and perform life-sustaining functions for the organism. As discussed in this Review, the fate of both host and pathogen is decided by the extent to which either one controls growth signalling pathways, genome maintenance machinery and immune surveillance. During persistent and asymptomatic infections of many oncogenic viruses, an equilibrium between these conflicting interests can be achieved. However, cumulative or chance events during infection and outside forces causing immune suppression or DNA damage can disrupt the fragile balance. In these instances, viral strategies that normally support infection instead drive uncontrolled cellular proliferation, accumulation of mutations and evasion of antitumour immunity. Understanding these mechanisms and the contexts in which they promote tumorigenesis is essential to preventing and treating viral cancers.
Oncogenic viruses have been instrumental in divulging key features of normal cellular function and pathology. Recent advances suggest that they remain an effective tool for conducting and guiding basic research. For instance, oncogenic viruses have made it apparent that cellular processes, once thought discrete, are intertwined. It has been proposed that there is overlap between the tumour suppressor and innate immune signalling pathways because both of these pathways can initiate cell cycle arrest and induce host cell death during infection2. It was further suggested that, by targeting key cellular components that are at the interface of these signalling pathways, oncogenic viruses disable both the host antiviral and anticancer mechanisms, priming the infected cells for cancerous transformation. Innate immune responses to intracellular pathogens double as early tumour suppressor measures, supporting the notion that viral oncogenesis is a product of immune evasion mechanisms2. Given that inflammation drives later stages of malignant disease, understanding how and when viral factors engage innate responses may clarify this complicated aspect of cancer. Recent oncogenic virus research has also revealed that double-stranded DNA introduced by viral infection and DNA damage generated during viral proliferation can stimulate innate immune DNA sensing pathways, leading to the production of cytokines that have both antiviral and antitumour function124. It will be particularly exciting to understand how DDRs coordinate with antiviral and antitumour immune signalling pathways throughout oncogenic progression and in the context of viral manipulation.
The seven viruses known to cause cancer in humans employ divergent replication and transmission strategies. Despite their differences, they are all highly adapted to maintain chronic infections in humans. Adaptation to coexist with a single host for prolonged periods requires continuous manipulation of immunity and cell fate decisions. Viruses that cause acute pathology or self-limiting infections, however, do not persist long enough to inflict the changes necessary for metastatic disease.
Although oncogenic viruses have evolved to persist in their host foryears, they are still under selective pressure to propagate to new hosts. The success of this propagation depends on avoiding a terminal fate such as cancer. This helps explain why oncogenic viruses do not cause cancer during most infections and only do so after many years. During years of limited pathology, when the host factors enabling coexistence shift drastically, viral strategies influencing cellular growth and survival can lead to neoplasms. Central to future discussions will be how immune suppression disrupts the interplay between host and pathogen to result in cancer. It may be that inadequate immune surveillance allows unchecked viral replication and expression of viral effectors that dysregulate host cell proliferation. Because immunity to tumours overlaps that of viruses, it may also be that healthy immune systems typically eliminate nascent transformed cells but may fail to do so once compromised.
Each cancer is multifactorial in terms of initiation and progression, making them challenging to treat. Thus, a logical approach to prevent or treat cancers of a viral aetiology is to target the virus. This principle has been given credence by successes in the clinic that have drastically reduced the burden of viral cancers. Innovations in antiviral therapy against the HCV RNA-dependent RNA polymerase have greatly reduced drug toxicity and continue to be effective at clearing HCV infections and preventing HCC125. Vaccinations against HPV and HBV have effectively reduced the incidence of their associated cancers in populations for whom the vaccines are accessible. Beyond preventive measures, reinstating immune activity in ‘cold’ viral tumours (that is, tumours that elicit little to no immune response) has proved to be an effective strategy. A general activator of T cell killing, anti-PD1–PDL1 immune checkpoint blockade in individuals with MCPyV+ MCC improves their survival126,127. Application of this exciting new therapy in MCC and other viral tumours supports the idea that viral factors may dampen immune responses in the tumour microenvironment. If targeted chemotherapies or immunotherapies were developed with specificity to the oncogenic or immune repressive mechanisms induced by viruses, even better clinical outcomes could be expected.
Pursuing novel therapeutics for viral cancers and basic research on virus–host interactions has recently become more practical owing to advances in omics technologies43. For example, deep sequencing and gene expression profiling led to the discovery of MCPyV7 and a better understanding of how the microRNA milieu is affected by oncogenic viruses during oncogenesis (box 3). The combination of high-throughput technologies and big data platforms allows investigators to decipher viral oncogenic mechanisms with the speed and efficiency of omics-level computational biology. These systems-level studies will reveal novel drug targets to advance the development of innovative intervention strategies for viral malignancy and will help resolve the dynamics between host and pathogen during infection and oncogenesis.
Box 3 |. The role of mirNA targeting in viral oncogenesis.
MicrorNas (mirNas) are naturally occurring 20–22 nucleotide single-stranded rNas that can pair to mrNas in higher eukaryotes and repress their translation131. the processing and function of these genetically encoded regulatory molecules are tightly regulated to support timely modulation of gene expression that controls cellular growth, proliferation, development, apoptosis and the stress response132. Dysregulation of miRNA synthesis or processing machinery or the expression of certain individual miRNAs could compromise cellular function and lead to pathological processes, including cancer133. Deep sequencing and gene expression profiling studies have led to the increasing appreciation that interactions between viruses and the miRNA milieu contribute to both viral infection and oncogenesis. the strategies used by oncogenic viruses to contribute to oncogenesis include encoding viral mirNas that target cellular mRNAs and promote a hyperproliferative state; upregulating host miRNAs to stimulate the growth of virus-infected cells; and sequestering host miRNAs with tumour suppressor function.
Herpesviruses, including Kaposi sarcoma-associated herpesvirus (KsHv) and epstein–Barr virus (eBv), establish stable latency programmes in which only a small percentage of their protein-coding OrFs are expressed. the reliance on viral miRNAs rather than viral proteins enables these viruses to escape immune surveillance. viral miRNAs modulate numerous pathways without generating foreign protein antigens that could elicit an immune response. During KsHv and eBv infection, viral miRNAs contribute to the mechanisms by which these viruses affect cell survival or proliferation and ultimately oncogenesis. For example, the seed sequence of KsHv mir-K12–11 miRNA is identical to that of cellular miRNA mir-155, which targets mRNAs involved in the regulation of cell proliferation134. expression of mir-K12–11 leads to downregulation of mir-155 cellular targets and may participate in the induction of B cell transformation134.
A transcriptome-wide analysis of KSHV-induced primary effusion lymphoma (PEL) cells identified hundreds of cellular target mRNAs involved in transcription and cell survival or proliferation as direct targets of KsHv miRNAs135. remarkably, more than half of the host mRNAs identified are also targeted by miRNAs encoded by eBv, which frequently co-infects KSHV-associated PEL cells135. additionally, expression of an extensive array of viral miRNAs in eBv-infected gastric epithelial cells coincides with downregulation of their cellular target genes involved in cellular transformation, suggesting that the eBv-encoded miRNAs function as major contributors to the virus-induced transformation136.
Viruses also dysregulate host miRNAs. For example, human papillomavirus (HPv) e6 or e7 oncogene expression upregulates a cluster of host miRNAs that contribute to the growth of HPv-positive cancer cells through the regulation of cell proliferation, senescence and apoptosis137. The human T-lymphotropic virus 1 (HTLV-1) basic zipper factor (HBZ) protein activates the oncogenic miRNAs mir17 and mir21 to promote host genetic instability and abnormal cell proliferation138. alternatively, hepatitis C virus (HCv) genomic RNA sequesters the liver-specific mirNa mir-122 for viral RNA stabilization and replication, impeding its binding to cellular mRNAs139. as a tumour suppressor function has been observed for mir-122, sequestration of mir-122 by HCv genomic RNA and the resulting derepression of the normal host oncogenic targets of mir-122 may contribute to oncogenesis during chronic HCv infection139.
Oncogenic viruses.
Viruses that cause cancer. Sometimes also called tumour viruses. However, some tumour viruses, such as adenovirus and polyomavirus SV40 promote tumorigenesis in other organisms and infect humans but do not cause human cancers.
Solid tumours.
Masses of transformed and supporting cells that arise in stationary tissues (sarcomas and carcinomas) and not from cells of haematopoietic origin.
Lymphoma.
Tumour arising from a lymphoid cell type that occurs predominantly in the lymphatics, as opposed to leukaemias, in which the cancer cells are found in the blood.
Cellular transformation.
Selective acquisition of cellular traits, such as replicative immortality, increased stemness, growth factor independence, resistance to growth suppressors and alterations to metabolic flux.
Metastasis.
Tumour migration, invasion and colonization of body sites other than the primary site.
p53.
A transcription factor and key tumour suppressor downstream of exogenous signals and DNa damage-sensing pathways that maintains genome integrity and governs cell fate by promoting expression of effectors of DNa repair, cell cycle arrest, senescence and apoptosis.
pRB.
(Retinoblastoma protein). A tumour suppressor that is responsible for a major g1 checkpoint that blocks S-phase entry and cellular growth.
Oncoproteins.
Translated gene products that have the capacity to drive cellular transformation.
Kaposi sarcoma.
A family of endothelial malignancies that are associated with Kaposi sarcoma-associated virus (KSHV) and whose members are classified by the type of immunosuppression that enabled KSHV-mediated oncogenesis.
Carcinomas.
Tumours arising from cells of an epithelial origin, as opposed to sarcomas, which arise from mesenchymal cells.
Rapamycin.
An inhibitor of mechanistic target of rapamycin (mTor)-mediated proliferative function that acts through direct binding of the peptidyl-prolyl cis-trans isomerase fKbP1a–mechanistic target of rapamycin complex and has shown promise as an immunosuppressant and antitumour drug.
Sarcomagenesis.
The seminal event or events leading to cancer progression from mesenchymal-derived cell types.
Cap-dependent translation.
Translation in which initiation is mediated by recognition of the 5′ cap that is specific to eukaryotic mRNAs.
MEK1.
(MAPK/ERK kinase 1 (also known as MaP2K1)). a crucial protein kinase that mediates an intermediate step of the raf–MeK–erK phosphorylation cascade responsible for activating expression of pro-proliferative, survival and differentiation genes in response to external stimuli.
Invasive cells.
Tumour cells with characteristics that enable them to metastasize and invade other tissues.
CHK1.
(Checkpoint kinase 1). A protein kinase essential to normal cell division and development that is activated by ataxia telangiectasia and Rad3-related protein (ATR) in response to single-stranded DNa to facilitate proper DNA replication, cell cycle progression and response to DNA insults.
Endothelial-to-mesenchymal transition.
A process essential to cardiac development and normal angiogenesis by which endothelial cells acquire stem-like, mesenchymal traits including enhanced migration that, in certain cancers, contribute to metastatic ability.
Proto-oncogene.
A type of endogenous gene that, when overexpressed or abnormally activated as a result of mutation, can promote cancer development, at which point it is referred to as an oncogene.
CHK2.
(Checkpoint kinase 2). A tumour suppressor kinase activated by ataxia telangiectasia mutated (ATM) in response to double-stranded breaks in DNA that maintains genomic integrity by mediating cell cycle arrest and DNA repair.
Second messengers.
Soluble small molecules that transduce intracellular signals, which can be secreted by intracellular bacteria to coordinate responses to their environment.
Inflammasome.
A cytoplasmic complex of NoD-, LRR- and pyrin domain-containing proteins (NLRPs), adaptor proteins and caspases that forms in response to cellular damage or bacterial effectors that cause rapid caspase-mediated inflammatory cytokine release and/or a type of lytic cell death called pyroptosis.
T cell anergy.
A process by which CD4+ or CD8+ T cells become tolerant to antigens and functionally inactivated owing to stimulation in the absence of a necessary signal.
Seed sequence.
An eight-nucleotide sequence near the 5′-end of a micro RNA that undergoes Watson–Crick base pairing with a target RNA with high specificity and that is required for efficient targeting.
Acknowledgements
The authors thank E. A. White, C. B. Buck and the members of the You laboratory for valuable comments and suggestions on the manuscript. The authors apologize to all colleagues whose primary research papers could not be cited owing to space constraints. Research on human papilloma virus (HPV) and Merkel cell polyomavirus (MCPyV) in the You laboratory has been supported by National Institutes of Health (NIH) grants (R01CA187718, R01CA148768 and R01CA142723) and the National Cancer Institute (NCI) Cancer Center Support grant (NCI P30 CA016520).
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Microbiology thanks D. Galloway and other anonymous reviewers for their contributions to the peer review of this work.
References
- 1.zur Hausen H & de Villiers EM Cancer “causation” by infections — individual contributions and synergistic networks. Semin. Oncol 41, 860–875 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Moore PS & Chang Y Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 10, 878–889 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Epstein MA, Achong BG & Barr YM Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1, 702–703 (1964). [DOI] [PubMed] [Google Scholar]
- 4.Chang Y et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266, 1865–1869 (1994).In this study, representational difference analysis identifies KSHV as a new human herpesvirus associated with Kaposi sarcoma.
- 5.Doorbar J Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci 110, 525–541 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Liu W et al. Identifying the target cells and mechanisms of Merkel cell polyomavirus infection. Cell Host Microbe 19, 775–787 (2016).This study reveals that human dermal fibroblasts are natural host cells of MCPyV infection in human skin.
- 7.Feng H, Shuda M, Chang Y & Moore PS Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319, 1096–1100 (2008).This study discovers that clonal integration of MCPyV DNA into the genome of Merkel cell carcinoma is a key event for tumour development.
- 8.Durst M, Gissmann L, Ikenberg H & zur Hausen H A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl Acad. Sci. USA 80, 3812–3815 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choo QL et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244, 359–362 (1989). [DOI] [PubMed] [Google Scholar]
- 10.Blumberg BS, Alter HJ & Visnich SA “new” antigen in leukemia sera. JAMA 191, 541–546 (1965). [DOI] [PubMed] [Google Scholar]
- 11.Poiesz BJ et al. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T cell lymphoma. Proc. Natl Acad. Sci. USA 77, 7415–7419 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherr CJ & McCormick F The RB and p53 pathways in cancer. Cancer Cell 2, 103–112 (2002). [DOI] [PubMed] [Google Scholar]
- 13.McLaughlin-Drubin ME & Munger K Viruses associated with human cancer. Biochim. Biophys. Acta 1782, 127–150 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howley PM & Livingston DM Small DNA tumor viruses: large contributors to biomedical sciences. Virology 384, 256–259 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wright DG et al. Human T cell leukemia virus type-1-encoded protein HBZ represses p53 function by inhibiting the acetyltransferase activity of p300/CBP and HBO1. Oncotarget 7, 1687–1706 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ringelhan M, O’Connor T, Protzer U & Heikenwalder M The direct and indirect roles of HBV in liver cancer: prospective markers for HCC screening and potential therapeutic targets. J. Pathol 235, 355–367 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Taylor JM & Nicot C HTLV-1 and apoptosis: role in cellular transformation and recent advances in therapeutic approaches. Apoptosis 13, 733–747 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morales-Sanchez A & Fuentes-Panana EM Human viruses and cancer. Viruses 6, 4047–4079 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu W, MacDonald M & You J Merkel cell polyomavirus infection and Merkel cell carcinoma. Curr. Opin. Virol 20, 20–27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knoll S et al. Dissection of cell context-dependent interactions between HBx and p53 family members in regulation of apoptosis: a role for HBV-induced HCC. Cell Cycle 10, 3554–3565 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Cho DC Targeting the PI3K/Akt/mTOR pathway in malignancy: rationale and clinical outlook. BioDrugs 28, 373–381 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Wong JP & Damania B Modulation of oncogenic signaling networks by Kaposi’s sarcoma-associated herpesvirus. Biol. Chem 398, 911–918 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noch E & Khalili K Oncogenic viruses and tumor glucose metabolism: like kids in a candy store. Mol. Cancer Ther 11, 14–23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Surviladze Z, Sterk RT, DeHaro SA & Ozbun MA Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J. Virol 87, 2508–2517-(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang L, Wu J, Ling MT, Zhao L & Zhao KN The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol. Cancer 14, 87 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukuda M & Longnecker R Latent membrane protein 2A inhibits transforming growth factor-beta 1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J. Virol 78, 1697–1705 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scholle F, Bendt KM & Raab-Traub N Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J. Virol 74, 10681–10689 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olagnier D et al. HTLV-1 Tax-mediated inhibition of FOXO3a activity is critical for the persistence of terminally differentiated CD4+ T cells. PLoS Pathog 10, e1004575 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stallone G et al. Sirolimus for Kaposi’s sarcoma in renal-transplant recipients. N. Engl. J. Med 352, 1317–1323 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Chang HH & Ganem D A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 13, 429–440 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sodhi A et al. Akt plays a central role in sarcomagenesis induced by Kaposi’s sarcoma herpesvirus-encoded G protein-coupled receptor. Proc. Natl Acad. Sci. USA 101, 4821–4826 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomlinson CC & Damania B The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway. J. Virol 78, 1918–1927 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shuda M, Kwun HJ, Feng H, Chang Y & Moore PS Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Invest 121, 3623–3634 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim EK & Choi EJ Compromised MAPK signaling in human diseases: an update. Arch. Toxicol 89, 867–882 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Menzel N et al. MAP-kinase regulated cytosolic phospholipase A2 activity is essential for production of infectious hepatitis C virus particles. PLoS Pathog 8, e1002829 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowser BS, Alam S & Meyers C Treatment of a human papillomavirus type 31b-positive cell line with benzo[a]pyrene increases viral titer through activation of the Erk1/2 signaling pathway. J. Virol 85, 4982–4992 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matusali G, Arena G, De Leo A, Di Renzo L & Mattia E Inhibition of p38 MAP kinase pathway induces apoptosis and prevents Epstein Barr virus reactivation in Raji cells exposed to lytic cycle inducing compounds. Mol. Cancer 8, 18 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dawson CW, Laverick L, Morris MA, Tramoutanis G & Young LS Epstein-Barr virus-encoded LMP1 regulates epithelial cell motility and invasion via the ERK-MAPK pathway. J. Virol 82, 3654–3664 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCormick C & Ganem D The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 307, 739–741 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Xia L et al. Upregulated FoxM1 expression induced by hepatitis B virus X protein promotes tumor metastasis and indicates poor prognosis in hepatitis B virus-related hepatocellular carcinoma. J. Hepatol 57, 600–612 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Ranganathan P, Weaver KL & Capobianco AJ Notch signalling in solid tumours: a little bit of everything but not all the time. Nat. Rev. Cancer 11, 338–351 (2011). [DOI] [PubMed] [Google Scholar]
- 42.South AP, Cho RJ & Aster JC The double-edged sword of Notch signaling in cancer. Semin. Cell Dev. Biol 23, 458–464 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rozenblatt-Rosen O et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487, 491–495 (2012).This study analyses the host interactome and transcriptome networks perturbed by DNA tumour virus proteins, revealing cancer driver genes that contribute to the molecular basis of human cancer.
- 44.Tan MJ et al. Cutaneous β-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc. Natl Acad. Sci. USA 109, E1473–E1480 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brimer N, Lyons C, Wallberg AE & Vande Pol SB Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene 31, 4639–4646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meyers JM, Spangle JM & Munger K The human papillomavirus type 8 E6 protein interferes with NOTCH activation during keratinocyte differentiation. J. Virol 87, 4762–4767 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowe M, Raithatha S & Shannon-Lowe C Counteracting effects of cellular Notch and Epstein-Barr virus EBNA2: implications for stromal effects on virus-host interactions. J. Virol 88, 12065–12076 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao J et al. Hepatitis B virus X protein activates Notch signaling by its effects on Notch1 and Notch4 in human hepatocellular carcinoma. Int. J. Oncol 48, 329–337 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Curry CL et al. Gamma secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi’s sarcoma tumor cells. Oncogene 24, 6333–6344 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Emuss V et al. KSHV manipulates Notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PLoS Pathog 5, e1000616 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu R et al. KSHV-induced notch components render endothelial and mural cell characteristics and cell survival. Blood 115, 887–895 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng F et al. KSHV-initiated notch activation leads to membrane-type-1 matrix metalloproteinase-dependent lymphatic endothelial-to-mesenchymal transition. Cell Host Microbe 10, 577–590 (2011).In this study, KSHV is found to activate the Notch signalling pathway to induce lymphatic endothelial cells to mesenchymal cell transition, which may contribute to Kaposi sarcoma development by promoting the generation of infected cells with invasive mesenchymal cell properties.
- 53.Lan K et al. Kaposi’s sarcoma herpesvirus-encoded latency-associated nuclear antigen stabilizes intracellular activated Notch by targeting the Sel10 protein. Proc. Natl Acad. Sci. USA 104, 16287–16292 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nusse R & Clevers H Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Fujimuro M & Hayward SD The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3β. J. Virol 77, 8019–8030 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morrison JA, Gulley ML, Pathmanathan R & Raab-Traub N Differential signaling pathways are activated in the Epstein-Barr virus-associated malignancies nasopharyngeal carcinoma and Hodgkin lymphoma. Cancer Res 64, 5251–5260 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Morrison JA, Klingelhutz AJ & Raab-Traub N Epstein-Barr virus latent membrane protein 2A activates β-catenin signaling in epithelial cells. J. Virol 77, 12276–12284 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jha HC, Banerjee S & Robertson ES The role of gammaherpesviruses in cancer pathogenesis. Pathogens 5, 42 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Daud M, Rana MA, Husnain T & Ijaz B Modulation of Wnt signaling pathway by hepatitis B virus. Arch. Virol 16, 2937–2947 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Ma G, Yasunaga J, Fan J, Yanagawa S & Matsuoka M HTLV-1 bZIP factor dysregulates the Wnt pathways to support proliferation and migration of adult T cell leukemia cells. Oncogene 32, 4222–4230 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Zhang Q, Lenardo MJ & Baltimore D 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168, 37–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karin M Nuclear factor-kappaB in cancer development and progression. Nature 441, 431–436 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Kulwichit W et al. Expression of the Epstein-Barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proc. Natl Acad. Sci. USA 95, 11963–11968 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luftig M et al. Epstein-Barr virus latent membrane protein 1 activation of NF-κB through IRAK1 and TRAF6. Proc. Natl Acad. Sci. USA 100, 15595–15600 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang LW, Jiang S & Gewurz BE Epstein-Barr virus LMP1-mediated oncogenicity. J. Virol 91, e01718–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gopalakrishnan R, Matta H & Chaudhary PM A purine scaffold HSP90 inhibitor BIIB021 has selective activity against KSHV-associated primary effusion lymphoma and blocks vFLIP K13-induced NF-κB. Clin. Cancer Res 19, 5016–5026 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bagneris C et al. Crystal structure of a vFlip-IKKγ complex: insights into viral activation of the IKK signalosome. Mol. Cell 30, 620–631 (2008). [DOI] [PubMed] [Google Scholar]
- 68.Chugh P et al. Constitutive NF-κB activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice. Proc. Natl Acad. Sci. USA 102, 12885–12890 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lavorgna A & Harhaj EW Regulation of HTLV-1 tax stability, cellular trafficking and NF-κB activation by the ubiquitin-proteasome pathway. Viruses 6, 3925–3943 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ciccia A & Elledge SJ The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lilley CE, Schwartz R. a. & Weitzman MD Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol 15, 119–126 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Weitzman MD, Lilley CE & Chaurushiya MS Genomes in conflict: maintaining genome integrity during virus infection. Annu. Rev. Microbiol 64, 61–81 (2010). [DOI] [PubMed] [Google Scholar]
- 73.Fradet-Turcotte A et al. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol 85, 8996–9012 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gillespie KA, Mehta KP, Laimins LA & Moody CA Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol 86, 9520–9526 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moody CA & Laimins LA Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 5, e1000605 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sakakibara N, Mitra R & McBride AA The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol 85, 8981–8995 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sowd GA, Li NY & Fanning E ATM and ATR activities maintain replication fork integrity during SV40 chromatin replication. PLoS Pathog 9, e1003283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsang SH, Wang X, Li J, Buck CB & You J Host DNA damage response factors localize to Merkel cell polyomavirus DNA replication sites to support efficient viral DNA replication. J. Virol 88, 3285–3297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weitzman MD & Weitzman JB What’s the damage? The impact of pathogens on pathways that maintain host genome integrity. Cell Host Microbe 15, 283–294 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Duensing S & Munger K The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res 62, 7075–7082 (2002). [PubMed] [Google Scholar]
- 81.Wallace NA et al. High-risk alphapapillomavirus oncogenes impair the homologous recombination pathway. J. Virol 91, e01084–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gruhne B et al. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl Acad. Sci. USA 106, 2313–2318 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takeda H, Takai A, Inuzuka T & Marusawa H Genetic basis of hepatitis virus-associated hepatocellular carcinoma: linkage between infection, inflammation, and tumorigenesis. J. Gastroenterol 52, 26–38 (2017). [DOI] [PubMed] [Google Scholar]
- 84.McFadden K et al. Metabolic stress is a barrier to Epstein-Barr virus-mediated B cell immortalization. Proc. Natl Acad. Sci. USA 113, E782–790 (2016).This study discovers that metabolic and genotoxic stress experienced by EBV-infected cells represents a barrier to EBV-mediated transformation.
- 85.Nikitin PA et al. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 8, 510–522 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li J et al. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J. Virol 87, 9173–9188 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shuda M et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl Acad. Sci. USA 105, 16272–16277 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rosewick N et al. Cis-perturbation of cancer drivers by the HTLV-1/BLV proviruses is an early determinant of leukemogenesis. Nat. Commun 8, 15264 (2017).In this study, HTLV-1 and bovine leukaemia virus proviruses are found to integrate preferentially near cancer driver genes and to cause their perturbation, favouring the expansion of the corresponding infected clone, allowing acquisition of additional somatic mutations that lead to the development of leukaemogenesis.
- 89.Nicot C HTLV-I Tax-mediated inactivation of cell cycle checkpoints and DNA repair pathways contribute to cellular transformation: “a random mutagenesis model”. J. Cancer Sci 2, 10.13188/2377-9292.1000009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Machida K et al. Hepatitis C virus induces a mutator phenotype: enhanced mutations of immunoglobulin and protooncogenes. Proc. Natl Acad. Sci. USA 101, 4262–4267 (2004).This study demonstrates that HCV infection induces a mutator phenotype to cause a significant increase in the mutation frequency of several proto-oncogenes, suggesting that HCV transforms cells by a hit-and-run mechanism.
- 91.Decorsiere A et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531, 386–389 (2016). [DOI] [PubMed] [Google Scholar]
- 92.Murphy CM et al. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep 16, 2846–2854 (2016).This study, together with reference 91, identifies the SMC complex SMC5/6 as a host restriction factor for HBV infection.
- 93.Thorley-Lawson DA EBV persistence — introducing the virus. Curr. Top. Microbiol. Immunol 390, 151–209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hopcraft SE & Damania B Tumour viruses and innate immunity. Phil. Trans. R. Soc. B 372, 20160267 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen Q, Sun L & Chen ZJ Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol 17, 1142–1149 (2016). [DOI] [PubMed] [Google Scholar]
- 96.Ma Z et al. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl Acad. Sci. USA 112, E4306–4315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu JJ et al. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18, 333–344 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang G et al. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl Acad. Sci. USA 113, E1034–E1043 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guito J & Lukac DM KSHV reactivation and novel implications of protein isomerization on lytic switch control. Viruses 7, 72–109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lau L, Gray EE, Brunette RL & Stetson DB DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350, 568–571 (2015).This study reveals that the oncogenes of the DNA tumour viruses, including HPV E7 and adenovirus E1A, can specifically inhibit the cGAS–STING pathway.
- 101.Yuen CK et al. Suppression of type i interferon production by human T-cell leukemia virus type 1 oncoprotein Tax through Inhibition of IRF3 phosphorylation. J. Virol 90, 3902–3912 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu Y et al. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J. Virol 89, 2287–2300 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nitta S et al. Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology 57, 46–58 (2013). [DOI] [PubMed] [Google Scholar]
- 104.Mackenzie KJ et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dou Z et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yu S et al. Hepatitis B virus polymerase inhibits RIG-I− and Toll-like receptor 3-mediated β interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKε and DDX3. J. General Virol 91, 2080–2090 (2010). [DOI] [PubMed] [Google Scholar]
- 107.Wang X et al. Hepatitis B virus X protein suppresses virus-triggered IRF3 activation and IFN-β induction by disrupting the VISA-associated complex. Cell. Mol. Immunol 7, 341–348 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gregory SM et al. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331, 330–334 (2011).In this study, KSHV ORF63 is discovered to be a viral homologue that inhibits human NLRP1-mediated innate immune response to promote KSHV reactivation and propagation.
- 109.Jacobs SR & Damania B The viral interferon regulatory factors of KSHV: immunosuppressors or oncogenes? Frontiers Immunol 2, 19 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chatterjee M, Osborne J, Bestetti G, Chang Y & Moore PS Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science 298, 1432–1435 (2002). [DOI] [PubMed] [Google Scholar]
- 111.Taga T & Kishimoto T Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol 15, 797–819 (1997). [DOI] [PubMed] [Google Scholar]
- 112.Hewitt EW The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology 110, 163–169 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Petersen JL, Morris CR & Solheim JC Virus evasion of MHC class I molecule presentation. J. Immunol 171, 4473–4478 (2003). [DOI] [PubMed] [Google Scholar]
- 114.Coscoy L & Ganem D Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl Acad. Sci. USA 97, 8051–8056 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Means RE, Ishido S, Alvarez X & Jung JU Multiple endocytic trafficking pathways of MHC class I molecules induced by a Herpesvirus protein. EMBO J 21, 1638–1649 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ishido S, Wang C, Lee BS, Cohen GB & Jung JU Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol 74, 5300–5309 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Banerjee P, Feuer G & Barker E Human T cell leukemia virus type 1 (HTLV-1) p12I down-modulates ICAM-1 and −2 and reduces adherence of natural killer cells, thereby protecting HTLV-1-infected primary CD4+ T cells from autologous natural killer cell-mediated cytotoxicity despite the reduction of major histocompatibility complex class I molecules on infected cells. J. Virol 81, 9707–9717 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Van Prooyen N et al. Human T cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc. Natl Acad. Sci. USA 107, 20738–20743 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yasuma K et al. HTLV-1 bZIP factor impairs anti-viral immunity by inducing co-inhibitory molecule, T cell immunoglobulin and ITIM domain (TIGIT). PLoS Pathog 12, e1005372 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Price AM & Luftig MA Dynamic Epstein-Barr virus gene expression on the path to B cell transformation. Adv. Virus Res 88, 279–313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hatton OL, Harris-Arnold A, Schaffert S, Krams SM & Martinez OM The interplay between Epstein-Barr virus and B lymphocytes: implications for infection, immunity, and disease. Immunol. Res 58, 268–276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Caldwell RG, Wilson JB, Anderson SJ & Longnecker R Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 9, 405–411 (1998). [DOI] [PubMed] [Google Scholar]
- 123.Minamitani T et al. Evasion of affinity-based selection in germinal centers by Epstein-Barr virus LMP2A. Proc. Natl Acad. Sci. USA 112, 11612–11617 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xia T, Konno H, Ahn J & Barber GN Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep 14, 282–297 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.McQuaid T, Savini C & Seyedkazemi S Sofosbuvir, a significant paradigm change in HCV treatment. J. Clin. Transl Hepatol 3, 27–35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kaufman HL et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet. Oncol 17, 1374–1385 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nghiem PT et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N. Engl. J. Med 374, 2542–2552 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Porta C, Paglino C & Mosca A Targeting PI3K/Akt/mTOR signaling in cancer. Frontiers Oncol 4, 64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dowling RJ et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328, 1172–1176 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chen D, Jarrell A, Guo C, Lang R & Atit R Dermal β-catenin activity in response to epidermal Wnt ligands is required for fibroblast proliferation and hair follicle initiation. Development 139, 1522–1533 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Eulalio A, Huntzinger E & Izaurralde E Getting to the root of miRNA-mediated gene silencing. Cell 132, 9–14 (2008). [DOI] [PubMed] [Google Scholar]
- 132.Yates LA, Norbury CJ & Gilbert RJ The long and short of microRNA. Cell 153, 516–519 (2013). [DOI] [PubMed] [Google Scholar]
- 133.Lujambio A & Lowe SW The microcosmos of cancer. Nature 482, 347–355 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gottwein E et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature 450, 1096–1099 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gottwein E et al. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 10, 515–526 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Marquitz AR, Mathur A, Shair KH & Raab-Traub N Infection of Epstein-Barr virus in a gastric carcinoma cell line induces anchorage independence and global changes in gene expression. Proc. Natl Acad. Sci. USA 109, 9593–9598 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Honegger A et al. Dependence of intracellular and exosomal microRNAs on viral E6/E7 oncogene expression in HPV-positive tumor cells. PLoS Pathog 11, e1004712 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vernin C et al. HTLV-1 bZIP factor HBZ promotes cell proliferation and genetic instability by activating OncomiRs. Cancer Res 74, 6082–6093 (2014). [DOI] [PubMed] [Google Scholar]
- 139.Luna JM et al. Hepatitis C virus RNA functionally sequesters miR-122. Cell 160, 1099–1110 (2015).This study suggests that HCV induced miR-122 sequestration, and subsequent derepression of host miR-122 targets promotes a cellular environment to support HCV-associated oncogenesis.
- 140.Raab-Traub N Novel mechanisms of EBV-induced oncogenesis. Curr. Opin. Virol 2, 453–458 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Young LS & Rickinson AB Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4, 757–768 (2004). [DOI] [PubMed] [Google Scholar]
- 142.Levrero M & Zucman-Rossi J Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol 64, S84–101 (2016). [DOI] [PubMed] [Google Scholar]
- 143.Gessain A & Cassar O Epidemiological aspects and world distribution of HTLV-1 infection. Front. Microbiol 3, 388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Matsuoka M & Jeang KT Human T cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 7, 270–280 (2007). [DOI] [PubMed] [Google Scholar]
- 145.Schiffman M et al. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2, 16086 (2016). [DOI] [PubMed] [Google Scholar]
- 146.Harper DM & DeMars LR HPV vaccines — a review of the first decade. Gynecol. Oncol 146, 196–204 (2017). [DOI] [PubMed] [Google Scholar]
- 147.Mitchell JK, Lemon SM & McGivern DR How do persistent infections with hepatitis C virus cause liver cancer? Curr. Opin. Virol 14, 101–108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Goossens N & Hoshida Y Hepatitis C virus-induced hepatocellular carcinoma. Clin. Mol. Hepatol 21, 105–114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schulz TF & Cesarman E Kaposi Sarcoma-associated Herpesvirus: mechanisms of oncogenesis. Curr. Opin. Virol 14, 116–128 (2015). [DOI] [PubMed] [Google Scholar]
- 150.Wendzicki JA, Moore PS & Chang Y Large T and small T antigens of Merkel cell polyomavirus. Curr. Opin. Virol 11, 38–43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]