

Abstract
Leptin, a hormone primarily produced by adipocytes, contributes to the regulation of bone health by modulating bone density, growth and adiposity. Upon leptin binding, multiple sites of the long form of the leptin receptor (LepRb) are phosphorylated to trigger activation of downstream signaling pathways. To address the role of LepRb signaling pathways in bone health, we compared the effects of three LepRb mutations on bone density, adiposity and growth in male and female mice. The ∆65 mutation, which lacks the known tyrosine phosphorylation sites, caused obesity and the most dramatic bone phenotype marked by excessive bone adiposity, osteoporosis, and decreased growth, consistent with the phenotype of db/db and ob/ob mice that fully lack leptin receptor signaling. Mutation of LepRb Tyr1138, which results in an inability to recruit and phosphorylate STAT3, also caused obesity, but bone loss and adiposity were more dominant in male mice and no growth defect was observed. In contrast, mutation of LepRb Tyr985, which blocks SHP2/SOCS3 recruitment to LepRb and contributes to leptin hypersensitivity, promoted increased femur bone density only in male mice, while marrow adiposity and bone growth were not affected. Additional analyses of vertebral trabecular bone volume indicate that only the Tyr1138 mutant mice exhibit bone loss in vertebrae. Together, our findings suggest that the phosphorylation status of specific sites of the LepRb contribute to the sex and location dependent bone responses to leptin. Unraveling the mechanisms by which leptin responses are sex and location dependent can contribute to the development of uniquely targeted osteoporosis therapies.
Keywords: leptin receptor, bone volume, bone density, marrow adiposity, obesity, male, female, osteoporosis, adipocytes
Leptin is a 16 kD hormone/adipokine produced primarily by adipocytes and is involved in the regulation of body mass and bone density (Hamrick 2004, Reid 2004, Elefteriou, Ahn et al. 2005, Karsenty 2006, Iwaniec, Boghossian et al. 2007, Iwaniec, Boghossian et al. 2011, Motyl and Rosen 2012). Serum leptin levels are positively linked with body fat stores. In the brain, leptin signals through neurons expressing the long form of the leptin receptor (LepRb) to communicate the status of the body’s energy reserves (Villanueva and Myers 2008, Abella, Scotece et al. 2017). When adipose tissue is abundant, increased serum leptin levels lead to increased energy expenditure, decreased food intake and weight loss. Conversely, the absence of leptin causes a reduction in energy expenditure, increased food intake and weight gain. Leptin also directly regulates peripheral cell function by binding to leptin receptors expressed on a variety of cell types including mesenchymal stem cells, which can mature into osteoblasts or adipocytes (Thomas, Gori et al. 1999, Reseland, Syversen et al. 2001, Cornish, Callon et al. 2002, Scheller, Song et al. 2010, Zhou, Yue et al. 2014).
The effects of leptin on bone are complex. Leptin can stimulate or inhibit bone formation depending upon bone location and whether leptin is acting directly via receptors on mesenchymal stem/stromal cells (MSCs) and/or osteoblasts (Reseland, Syversen et al. 2001, Cornish, Callon et al. 2002, Zhou, Yue et al. 2014) or indirectly through LepRb-expressing hypothalamic neurons that can polysynaptically regulate bone (Cornish, Callon et al. 2002, Hamrick 2004, Hamrick, Della Fera et al. 2007). Mice that are completely leptin deficient (ob/ob mice) have age-dependent, site-specific phenotypes including lower femoral bone mineral content (BMC), cortical thickness, bone mineral density (BMD), trabecular bone volume, and decreased bone length (Steppan, Crawford et al. 2000, Hamrick 2004, Iwaniec, Boghossian et al. 2007, Turner, Kalra et al. 2013, Philbrick, Martin et al. 2017). Similarly, leptin receptor deficiency (db/db mice) causes decreased tibial trabecular bone volume, bone length and cortical thickness (Williams, Callon et al. 2011, Turner, Kalra et al. 2013). Absence of leptin or LepRb in mice also increases bone marrow adiposity (Steppan, Crawford et al. 2000, Hamrick 2004, Lindenmaier, Philbrick et al. 2016). The receptor-mediated mechanisms underlying these skeletal and marrow adiposity phenotypes remain unknown.
The leptin receptor (LR) belongs to the class I cytokine receptor superfamily and is encoded by a single gene (Lepr). Several alternatively-spliced LR isoforms exist and can be divided into three classes: secreted, long and short (Gong, Ishida-Takahashi et al. 2007). Leptin initiates its action through binding to the single long form of the leptin receptor (LepRb) which leads to the auto-phosphorylation and activation of the LepRb-associated Jak2 protein. Jak2 in turn phosphorylates three critical LepRb tyrosine residues (Tyr985, Tyr1077, and Tyr1138) located in an intracellular domain that is composed of approximately 300 residues. Each of the phosphorylation sites can recruit specific Src homology 2 (SH2) domain-containing proteins. Specifically, phosphorylation of the first site, Tyr985, recruits Src homology phosphatase-2 (SHP-2) as well as suppressor of cytokine signaling 3 (SOCS3) which leads to an attenuation of LepRb signaling(Gong, Ishida-Takahashi et al. 2007). Phosphorylation of the second site, Tyr1077, recruits the signal transducer, latent transcription factor, and activator of transcription 5 (STAT5)(Bjornholm, Munzberg et al. 2007). Phosphorylation of Tyr1138 results in the recruitment of STAT3 (Banks, Davis et al. 2000, Munzberg, Huo et al. 2003). Thus, each phosphorylation site activates a unique signaling cascade to mediate distinct aspects of leptin action.
Genetically modified mice containing mutations in the LepRb can be used to determine the role of each of the three key phosphorylation sites on mouse physiology. Mice were generated by replacing the LepRb-specific exon 18b of Lepr with a mutant exon 18b. This gene-targeting strategy expresses mutant LepRb molecules from the genomic context of endogenous LepRb so that expression patterns and levels of mutant LepRb mirror those of wild-type LepRb(Bates, Stearns et al. 2003). Mice expressing a mutation in Tyr985 (termed LL mice) have abrogated phosphorylation of the site and blocked SHP2/SOCS3 recruitment. LL mice are lean and display leptin hypersensitivity. Conversely, mice expressing a mutation in Tyr1138 (termed SS mice), are unable to recruit STAT3 to LepRb and display an obese and hyperphagic phenotype marked by decreased energy expenditure with increased growth (Bates, Stearns et al. 2003). Finally, Δ65 animals lack LepRb signaling and are similar to db/db mice (Robertson, Ishida-Takahashi et al. 2010). To understand the role of LepRb and its key phosphorylation sites on bone phenotypes, we carried out an exploratory study examining male and female skeletal parameters (trabecular and cortical; femur and vertebral) in the three LepRb mutant mouse lines and compared our findings to the corresponding WT littermates for each mutant line. Our findings support the complexity of LepRb signaling in the regulation of skeletal health.
Methods:
LepRb Mutant Mouse Models:
This study examined LepRb ∆65, LL and SS mice on the C57/Bl6 background, as previously described (Bates, Stearns et al. 2003, Bjornholm, Munzberg et al. 2007, Robertson, Ishida-Takahashi et al. 2010). Heterozygous mice were intercrossed to generate mice homozygous for each LepRb variant and littermate controls used for subsequent studies, which were identified by genotyping between 2–4 weeks of age. Study mice were group housed until 8–9 weeks of age, then were anesthetized with a lethal dose of pentobarbital and transcardially perfused with 10% neutral buffered formalin. Bones were removed and post-fixed in formalin for 24-hours prior to storage in 70% ethanol. All mice were bred and housed at the University of Michigan and maintained in a 12-hour light/dark cycle with ad libitum access to food and water. All procedures were approved by the University of Michigan University Committee on the Use and Care of Animals in accordance with Association for Assessment and Accreditation of Laboratory Animal Care and National Institutes of Health guidelines.
Bone density measurement:
Femurs and vertebrae (lumbar 3–4) were fixed in 10% formalin and imaged using a GE Explore Locus microcomputed tomography (μCT) system at a voxel of 20 μm obtained from 720 views. The beam angle increment was 0.5 and beam strength was set at 80 peak kV and 450 μA. Each run consisted of control and mutant mouse bones and a calibration phantom to standardize gray scale values and maintain consistency. Bone measurements were blinded. Maximum vertebral height was determined using GE Healthcare Microview software and was consistent with previous reports(Hamrick, Pennington et al. 2004). Maximum femur length was determined as the distance between the most proximal region of the trochanter to the most distal region of the medial condyle. Distal femur trabecular bone analyses were performed in the metaphyseal region defined at 1% of the total length (~ 0.17mm) proximal to the growth plate extending 2 mm toward the diaphysis excluding the outer cortical bone. Trabecular bone volume fraction was computed by GE Healthcare MicroView software application using a threshold of 700. Cortical bone measurements were determined with a 2-mm3 region of interest (ROI) in the mid-diaphysis.
Leptin serum measurements:
Leptin concentration in female mouse serum was determined by ELISA using the Mouse/Rat Leptin Quantikine Kit (R&D Systems).
Adipocyte counts:
Fixed bones were processed on an automated Thermo Electron Excesior tissue processor for dehydration, clearing, and infiltration using a routine overnight processing schedule. Samples were then embedded in Surgipath-embedding paraffin on a Sakura Tissue Tek II-embedding center. Paraffin blocks were sectioned at 5 µm on a Reichert Jung 2030 rotary microtome and were H&E stained. Femur sections were examined by microscopy at 4x optical zoom and digital images obtained. Images were examined blind to the section’s condition. The marrow area starting at 170 µm from the growth plate and extending 2000 µm toward the diaphysis was measured, by outlining the region and quantifying the area using ImagePro software. Adipocytes greater than 30 µm in diameter were counted and expressed relative to the total marrow area. Analyses were done blinded to conditions.
Statistical analyses:
All measurements are presented as the mean ± SE. All groups contained at least 5 mice, except for the ∆65 male mouse group which has 3 mice. Student’s two tail t-tests were used to determine significance by comparing each genotype to its own littermate and sex matched controls. Outliers were identified by ROUT and removed (only 3 identified). Statistical analyses used student t-tests that compared littermate WT to mutant mouse values for each sex. Analyses were performed using GraphPad Prism software version 6 (GraphPad, San Diego, CA, USA). A p-value ≤0.05 was considered significant.
Results
To determine the role of LepRb and its key phosphorylation sites on the male and female mouse skeleton, we examined 8–9-week-old mice that had one of the three LepRb mutations and compared findings to their corresponding sex and age-matched littermates. We first assessed body mass (Figure 1), which is an important parameter that influences bone phenotype. Consistent with previous studies in db/db mice(Villanueva and Myers 2008) which lack LepRb signaling, both male and female ∆65 mice had significantly greater body mass compared to their corresponding littermates, by 49% and 72% respectively. An even greater increase was seen in both the male and female SS mutant mice which displayed significant increases in body mass that were 57% and 90% (respectively) greater than their corresponding littermate counterparts. In contrast, the LL mutant mice weighed less(Bjornholm, Munzberg et al. 2007). While the LL mutant males were on average only 4% less in weight, the female LL mutant mice exhibited a significant 27% decrease in body weight. This is consistent with the Tyr985 mutation promoting leanness specifically in female mice(Bjornholm, Munzberg et al. 2007, Villanueva and Myers 2008). Consistent with changes in adiposity and body mass, serum leptin was significantly elevated in SS (148 +/− 19 ng/ml) and decreased in LL (1.7 +/− 0.45 ng/ml) compared to corresponding control female mice (2.8 +/− 0.76 and 2.76 +/− 0.48).
Figure 1. LepRb mutations ∆65 and SS increase male and female mouse body weight, while the LL mutation causes weight loss in female mice.
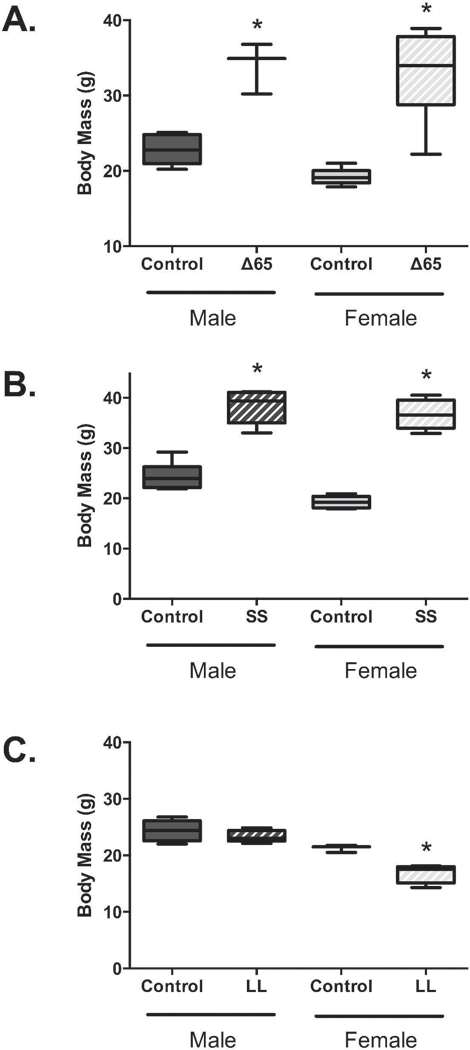
Body weights were obtained from male and female 8–9-week-old mice expressing LepRb mutants: ∆65, SS (Tyr1138) or LL (Tyr985). Mutant mouse data is graphed with data obtained from corresponding littermate control mice. Values represent averages ± SE. n = 3–7 per group. * p < 0.05.
Next, we examined femur bone length to determine if the mutations had any impact on overall bone growth (Figure 2 A–C). Both the male and female ∆65 mice displayed reduced femoral growth compared to littermate controls, by 9% and 12%, respectively. However, no differences were detected for the other genetically modified mice, suggesting that signals beyond Tyr1138 induced Stat3 and Tyr985 recruited SHP2/SOCS3 mediate the control of bone length by leptin. We also examined vertebral bone height (L3–4) but in contrast to another report(Kishida, Hirao et al. 2005) we did not observe differences in vertebral height between any of the conditions (data not shown).
Figure 2. Only LepRb mutation ∆65 effected femur length/growth.
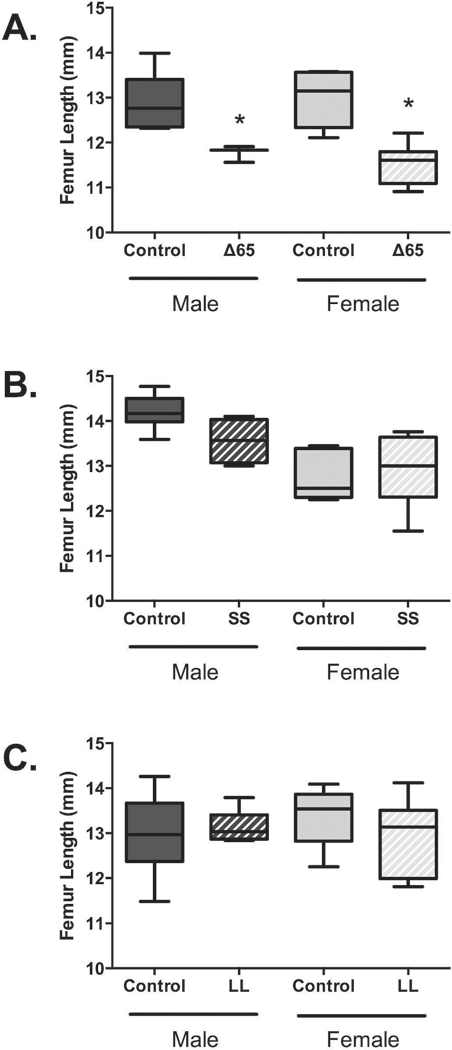
Femur lengths were measured for male and female 8–9-week-old mice expressing LepRb containing mutations ∆65, SS or LL. Mutant mouse data is graphed and analyzed relative to data obtained from corresponding littermate control mice. Values represent averages ± SE, ∆65 mice n ≥3 per group; SS and LL mice n = 6–7 per group. * p < 0.05.
To determine if the LepRb mutations affected bone architecture, distal femur trabecular/cancellous bone was examined by microcomputed tomography. Analyses of bone volume fraction (BV/TV) did not differ between conditions except for male LL mutant mice which displayed higher bone volume and greater trabecular thickness compared to controls (Table 1). Male and female ∆65 mice trended toward a 26% and 50% reduction in trabecular BV/TV. By contrast, SS mice, which were of similar body mass, had a trending, though non-significant 14% decrease and 19% increase in trabecular bone volume fraction. Bone volume was further analyzed relative to body weight since the mice displayed broad differences in body weight (as shown in Figure 1). When corrected for body weight, both male and female ∆65 mice had significantly reduced femur trabecular bone volume when compared to their corresponding control littermates, by 51% and 74% respectively, consistent with previous reports(Hamrick 2004, He, Liu et al. 2004, Ealey, Fonseca et al. 2006, Ramos-Junior, Leite et al. 2016). The SS mutant mice showed a significant 53% decrease for male BV/TV, and a non-significant 43% decrease for females (p = 0.08). In contrast, the LL mutant mice increased bone volume with average significant increase of 52% for males and a non-significant 20% increase for females, relative to littermate controls.
Table 1.
Femoral Trabecular Bone Parameters
| MALE | FEMALE | |||
|---|---|---|---|---|
| Control | Mutant | Control | Mutant | |
| ∆65 | ||||
| BV/TV | 23.9 ± 3.6 | 17.7 ± 3.8 | 19.6 ± 4.1 | 9.8 ± 1.7 |
| Tb. Th. (mm) | 0.032 ± 0.003 | 0.029 ± 0.002 | 0.030 ± 0.002 | 0.026 ± 0.001 |
| Tb. N. (1/mm) | 7.288 ± 0.847 | 5.957 ± 0.964 | 6.150 ± 0.873 | 3.651 ± 0.547 |
| Tb. Sp. (mm) | 0.121 ± 0.029 | 0.164 ± 0.040 | 0.153 ± 0.031 | 0.315 ± 0.085 |
| SS mutant | ||||
| BV/TV | 52.9 ± 5.7 | 45.3 ± 9.5 | 24.3 ± 5.1 | 29.0 ± 1.8 |
| Tb. Th. (mm) | 0.064 ± 0.007 | 0.056 ± 0.011 | 0.029 ± 0.003 | 0.037 ± 0.001 |
| Tb. N. (1/mm) | 8.308 ± 0.342 | 7.973 ± 0.839 | 5.805 ± 0.874 | 7.874 ± 0.199 |
| Tb. Sp. (mm) | 0.058 ± 0.008 | 0.078 ± 0.022 | 0.165 ± 0.040 | 0.091 ± 0.005 |
| LL mutant | ||||
| BV/TV | 33.4 ± 0.7 | 48.2 ± 6.8 * | 32.6 ± 4.7 | 32.7 ± 4.3 |
| Tb. Th. (mm) | 0.038 ± 0.001 | 0.054 ± 0.009 * | 0.039 ± 0.003 | 0.039 ± 0.003 |
| Tb. N. (1/mm) | 8.891 ± 0.509 | 8.862 ± 0.459 | 8.095 ± 0.576 | 8.157 ± 0.687 |
| Tb. Sp. (mm) | 0.076 ± 0.004 | 0.059 ± 0.009 | 0.088 ± 0.0130 | 0.088 ± 0.014 |
Values represent the mean ± standard error (n ≥ 3 per group).
p<0.05
Examination of vertebral BV/TV, without correction to bodyweight, show an increase in BV/TV caused by the ∆65 mutation in male (significant) and female (trend) mice (Table 2). The ∆65 mutation also caused a significant reduction in vertebral trabecular spacing in both males and females. The SS mutation did not cause any notable effects on vertebral trabecular parameters while the LL mutation induced a significant decrease in trabecular BV/TV in females only. Given the significant differences in body weight between the mouse groups, we also analyzed the BV/TV data relative to body weight and found that there was no longer a difference in BV/TV in the ∆65 and LL mutant mice compared to their corresponding controls. However, the SS mutant mice exhibited a 22% decrease in males and a significant 42% decrease in females (Figure 3E), suggesting that the SS mutant could identify a unique signaling pathway that regulates vertebral bone density.
Table 2.
Vetebral Trabecular Bone Parameters
| MALE | FEMALE | |||
|---|---|---|---|---|
| Control | Mutant | Control | Mutant | |
| ∆65 | ||||
| BV/TV | 35.5 ± 4.0 | 57.9 ± 8.7* | 40.5 ± 2.3 | 54.7 ± 5.5 |
| Tb. Th. (mm) | 0.039 ± 0.002 | 0.058 ± 0.016 | 0.044 ± 0.002 | 0.059 ± 0.008 |
| Tb. N. (1/mm) | 8.923 ± 0.360 | 10.145 ± 1.272 | 9.069 ± 0.215 | 9.319 ± 0.312 |
| Tb. Sp. (mm) | 0.073 ± 0.007 | 0.041 ± 0.004* | 0.066 ± 0.004 | 0.049 ± 0.006* |
| SS mutant | ||||
| BV/TV | 47.3 ± 5.7 | 54.5 ± 1.9 | 39.4 ± 3.5 | 43.4 ± 3.5 |
| Tb. Th. (mm) | 0.048 ± 0.004 | 0.055 ± 0.002 | 0.044 ± 0.003 | 0.044 ± 0.003 |
| Tb. N. (1/mm) | 9.570 ± 0.414 | 9.944 ± 0.089 | 8,834 ± 0.290 | 9.706 ± 0.341 |
| Tb. Sp. (mm) | 0.056 ± 0.008 | 0.046 ± 0.002 | 0.070 ± 0.006 | 0.059 ± 0.005 |
| LL mutant | ||||
| BV/TV | 39.6 ± 5.1 | 53.1 ± 4.6 | 44.8 ± 1.6 | 35.2 ± 2.3* |
| Tb. Th. (mm) | 0.040 ± 0.003 | 0.051 ± 0.002* | 0.048 ± 0.001 | 0.036 ± 0.002* |
| Tb. N. (1/mm) | 9.780 ± 0.433 | 10.287 ± 0.522 | 9.190 ± 0.257 | 9.658 ± 0.400 |
| Tb. Sp. (mm) | 0.064 ± 0.007 | 0.048 ± 0.007 | 0.061 ± 0.003 | 0.069 ± 0.005 |
Values represent the mean ± standard error (n ≥ 3 per group).
p<0.05
Figure 3. LepRb mutations have sex and location dependent effects on bone volume fraction.
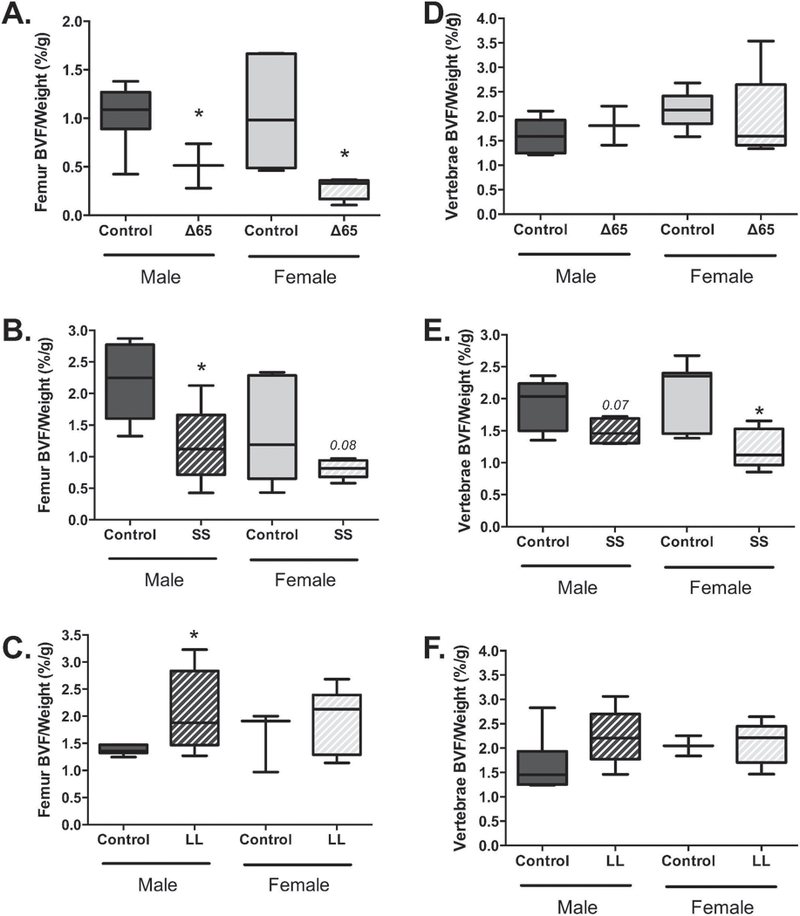
Femur (A–C) and vertebral (D–E) bone volume fraction (BVF) was determined, in 8–9-week-old mice, by microcomputed tomography and expressed relative to mouse body weight. LepRb mutant mouse data (∆65, SS or LL) is grouped with data obtained from corresponding littermate control mice. Values represent averages ± SE, ∆65 mice n ≥3 per group; SS and LL mice n = 6–7 per group. * p < 0.05.
Cortical bone parameters were also examined and revealed differences that were sex and mutation specific (Table 3). Male LL and SS mutant mice did not show significant cortical differences, however the ∆65 mutation displayed less cortical thickness and cortical area compared to WT control mice. Female LepRb mutant mice, on the other hand, displayed several significant differences. Female ∆65 mice had significantly reduced cortical bone density. Female SS mutant mice had greater inner and outer cortical bone perimeter and marrow area as well as lower BMD compared to WT controls. Regarding the LL mice, females had a smaller cortical inner perimeter than controls, but this did not result in greater mean thickness or cortical area (Table 2).
Table 3.
Cortical Bone Parameters
| MALE | FEMALE | |||
|---|---|---|---|---|
| Control | Mutant | Control | Mutant | |
| ∆65 | ||||
| Mean Thickness (mm) | 0.180 ± 0.008 | 0.145 ± 0.003 ** | 0.174 ± 0.004 | 0.169 ± 0.006 |
| Inner (mm) | 3.764 ± 0.047 | 4.071 ± 0.195 | 3.759 ± 0.052 | 3.529 ± 0.181 |
| Outer (mm) | 4.868 ± 0.076 | 4.934 ± 0.230 | 4.774 ± 0.065 | 4.533 ± 0.199 |
| Marrow Area (mm^2) | 0.995 ± 0.022 | 1.034 ± 0.035 | 0.994 ± 0.032 | 0.906 ± 0.056 |
| Cortical Area (mm^2) | 0.687 ± 0.041 | 0.468 ± 0.028 ** | 0.588 ± 0.032 | 0.545 ± 0.042 |
| BMD (mg/cc) | 937 ± 10 | 885 ± 10 | 983 ± 14 | 922 ± 19 ** |
| SS mutant | ||||
| Mean Thickness (mm) | 0.245 ± 0.008 | 0.255 ± 0.010 | 0.200 ± 0.007 | 0.203 ± 0.007 |
| Inner (mm) | 3.720 ± 0.082 | 3.619 ± 0.109 | 3.497 ± 0.056 | 3.775 ± 0.089 * |
| Outer (mm) | 5.246 ± 0.091 | 5.232 ± 0.132 | 4.733 ± 0.055 | 5.007 ± 0.038 * |
| Marrow Area (mm^2) | 0.985 ± 0.043 | 0.942 ± 0.055 | 0.870 ± 0.025 | 0.989 ± 0.041 * |
| Cortical Area (mm^2) | 1.041 ± 0.039 | 1.072 ± 0.060 | 0.752 ± 0.040 | 0.772 ± 0.041 |
| BMD (mg/cc) | 1076 ± 15 | 1081 ± 16 | 1023 ± 12 | 955 ± 18 ** |
| LL mutant | ||||
| Mean Thickness (mm) | 0.211 ± 0.007 | 0.219 ± 0.006 | 0.205 ± 0.007 | 0.201 ± 0.009 |
| Inner (mm) | 3.726 ± 0.042 | 3.780 ± 0.068 | 3.686 ± 0.070 | 3.513 ± 0.039 * |
| Outer (mm) | 5.029 ± 0.030 | 5.126 ± 0.067 | 4.948 ± 0.091 | 4.761 ± 0.072 |
| Marrow Area (mm^2) | 0.983 ± 0.021 | 1.001 ± 0.032 | 0.942 ± 0.026 | 0.895 ± 0.022 |
| Cortical Area (mm^2) | 0.853 ± 0.033 | 0.902 ± 0.038 | 0.780 ± 0.054 | 0.779 ± 0.052 |
| BMD (mg/cc) | 979 ± 28 | 1026 ± 26 | 999 ± 19 | 994 ± 26 |
Values represent the mean ± standard error (n ≥ 3 per group)
p<0.05
p<0.01 compared to corresponding WT control.
Previously, our lab and others (Thomas, Gori et al. 1999, Reseland, Syversen et al. 2001, Cornish, Callon et al. 2002, Motyl and McCabe 2009, Scheller, Song et al. 2010) demonstrated that leptin can influence marrow adiposity. Therefore, we further examined the effect of the leptin receptor mutations on marrow adiposity. Adiposity was markedly increased in the male ∆65 mice by a significant 10.3-fold (to an average of 72 adipocytes/mm2). While adiposity was also significantly increased in the SS male mutants, the increase was modest, 3.5-fold (to an average between 3 and 4 adipocytes per mm2), compared to the effect of ∆65 on marrow fat. The female ∆65 mice exhibited an average increase of 2.7-fold (to 51 adipocytes per mm2) and the SS female mice displayed a 1.5-fold average increase (to approximately 5 adipocytes per mm2) compared to littermate controls, though these results did not reach statistical significance. The LL mutation did not impact marrow adiposity in either the male or female mice.
Discussion:
Past studies demonstrate a role for leptin in the regulation of bone density and adiposity. Using mice expressing different LepRb signaling mutations, we identified distinct roles for leptin receptor signaling pathways in the regulation of bone density, adiposity or growth. By far the strongest phenotype we obtained was from the ∆65 mice, which lack leptin receptor signaling via characterized intracellular tyrosine residues. This mutation made both male and female mice obese while also decreasing bone density and growth and increasing marrow adiposity in mouse femurs. While the male mutant mouse number was underpowered (n=3), our findings are consistent with reports on bone phenotypes caused by either leptin deficiency as seen in ob/ob mice or in db/db mice that lack LepRb (Hamrick 2004, He, Liu et al. 2004, Ealey, Fonseca et al. 2006, Ramos-Junior, Leite et al. 2016). The SS mutation of LepRb Tyr1138, which prevents LepRb-mediated phosphorylation of STAT3, made mice obese comparable to the ∆65 mice, however the bone loss and marrow adiposity was not as great as observed in the ∆65 mice and no changes in growth were observed. In addition, the changes were more evident in the male SS mutant male mice compared to female mice. This suggests that the active signaling occurring in the SS is sufficient for growth and can maintain some normal bone phenotype in males and most of the normal phenotype in females. By contrast, the LL mutant mice did not gain weight and in the case of the female mice we observed reduced body weights. Male LL mutant mice were the only group to show an increase in bone density, which is consistent with leptin hypersensitivity.
Interestingly, when corrected to body weight the ∆65 mutant mice did not experience vertebral bone loss. This is similar to previous reports showing significant bone loss in femur or tibia, with only mild changes in vertebrae, in db/db and ob/ob mice (Ealey, Fonseca et al. 2006, Williams, Callon et al. 2011). This is consistent with leptin having site-specific effects on bone(Hamrick, Pennington et al. 2004). Unexpectedly, we observed a moderate decrease in BVF/body weight in the SS mutant vertebrae. This suggests that a signaling pathway affected by the SS mutation, i.e., STAT3, may contribute to leptin’s location dependent effects in femur but not vertebrae; thus, pathway inhibition allows a response in vertebrae. Future mechanistic studies are needed to better understand the underlying site-specific differences in the regulation of bone.
Analyses of cortical bone indicated that only the ∆65 mutation had an impact on male cortical bone parameters but not density. Whereas, the ∆65 and SS mutation affected female cortical bone density but only the SS mutant had significant negative effects on cortical bone parameters. The LL mutation did not have a major impact on cortical bone in either sex, but females did display a significant decrease in inner cortical perimeter. Thus, the LepRb mutations reveal a sex dependent response. The ∆65 mutation affecting male cortical bone, while the SS (significant) and LL (trend) mutants affect cortical bone only in female mice and in opposite ways. This sex dependent LepRb regulation of cortical parameters needs to be further dissected in a future larger study that identifies the downstream pathways that have opposing influences on cortical bone.
Leptin is a potent regulator of bone marrow adiposity. Increased LepRb signaling can decrease marrow adiposity (Thomas, Gori et al. 1999, Reseland, Syversen et al. 2001, Cornish, Callon et al. 2002, Hamrick, Della-Fera et al. 2005, Scheller, Song et al. 2010), whereas reduced leptin signaling promotes bone marrow adiposity (Steppan, Crawford et al. 2000, Hamrick 2004). The latter is most evident in ob/ob and db/db (Devlin, Cloutier et al. 2010, Ecklund, Vajapeyam et al. 2010). In our study, both the ∆65 mutant and SS mutants had increased femoral bone marrow adiposity. This was more prominent in males than females. In addition, the magnitude of the increase was much greater in ∆65 mutants than in SS, which lack only Tyr1138/STAT3 signaling. The LL mutation had no effect on marrow adiposity. Mechanistically, LepRb signaling can regulate adiposity through its expression and signaling in mesenchymal stromal cells and on 94% of bone marrow derived colonies that mature to make bone, cartilage and adipocytes (Zhou, Yue et al. 2014). In vitro studies demonstrate that leptin promotes bone marrow stromal cell osteogenesis rather than adipogenesis (Thomas, Gori et al. 1999, Reseland, Syversen et al. 2001, Cornish, Callon et al. 2002, Scheller, Song et al. 2010). Leptin treatment has been demonstrated to reduce marrow adiposity and corrects skeletal abnormalities in ob/ob mice, though minimal changes in bone occur in rodents capable of producing leptin (Hamrick, Della-Fera et al. 2005, Iwaniec, Boghossian et al. 2007). Leptin treatment also prevents T1-diabetic induced bone marrow adiposity, but in this model, leptin treatment is unable to prevent T1-diabetic bone loss (Motyl and McCabe 2009).
Leptin has previously been shown to regulate bone growth, in part by its ability to affect chondrocytes (Steppan, Crawford et al. 2000, Kishida, Hirao et al. 2005, Gat-Yablonski and Phillip 2008). Leptin deficient ob/ob mice have fragile growth plates with disturbed columnar structures and increased apoptosis, a phenotype abolished by treatment with leptin (Kishida, Hirao et al. 2005). Interestingly, only the ∆65 mutation, lacking all tyrosine-kinase signaling from LepRb, causes reduced bone length. Neither the LL or SS mutant mice had an observable growth defect in the femur, suggesting that full inhibition of LepRb-mediated tyrosine kinase signaling is required to obtain notable growth stunting. The lack of a growth effect in LL mice is consistent with a previous study that found no difference in snout-anus length in LL mutant versus WT mice (Bjornholm, Munzberg et al. 2007). In our study, we also found no change in vertebral growth as determined by vertebrae height measures. This is consistent with many of the leptin effects being targeted to long-bones rather than axial bones (Hamrick, Pennington et al. 2004).
It should be noted that in contrast to total body LepRb signaling modulation, as in ob/ob, db/db and our studies, the targeted deletion of LepRb in bone marrow mesenchymal stromal cells causes a different phenotype characterized by increased osteogenesis and decreased adipogenesis and increased fracture healing (Scheller, Song et al. 2010, Yue, Zhou et al. 2016). These studies point out the complexity of LepRb signaling in the body which encompasses effects on the brain, immune system, metabolism, and eating behavior (Villanueva and Myers 2008, Turner, Kalra et al. 2013, Lindenmaier, Philbrick et al. 2016, Abella, Scotece et al. 2017, Philbrick, Martin et al. 2017), all of which impact bone. Our study suggests that the modulation of whole body LepRb signaling may outweigh the contribution of mesenchymal stem cell LepRb regulation of bone and thus lead to increased marrow adiposity and decreased bone density. While it is critical to understand the role of LepRb signaling within the bone as well as in other individual tissues such as brain, our studies provide important insight into potential systemic effects of leptin which contribute bone density regulation.
Conclusions
Similar to leptin-receptor deficient db/db mice, truncation of the signaling domain of LepRb in the ∆65 mutant led to increased body mass, decreased femoral trabecular bone volume, bone length and cortical thickness and increased bone marrow adiposity (Table 4). The SS mutant, which lacks Tyr1138/STAT3 signaling, recapitulated the excess body mass phenotype of the db/db and ∆65 mice. However, its impact on bone parameters was reduced. Specifically, femoral length and cortical thickness were normal and marrow adiposity, though marginally increased, was an order of magnitude less than ∆65 animals. This suggests that signals in addition to Tyr1138/STAT3 are necessary to promote maximal bone loss, growth restriction and marrow fat accumulation. In the absence of excess peripheral and bone marrow adiposity, as present in LL mice, bone mass was unchanged (females) or increased (males). These results are relevant to understanding how disruptions in leptin signaling, whether due to monogenetic or diet-induced obesity, may impact bone development and growth. This may be particularly relevant in the context of juvenile obesity, where loss of leptin signaling via LepRb could compromise bone development and growth. Understanding how leptin modifies bone through LepRb will impact the design of strategies to promote bone growth and integrity in such at-risk populations.
Table 4.
Summary of Findings
| Body Weight | Femur BVF/wt | Marrow adiposity | Vertebrae BVF/wt | Cortical parameters | Bone Growth | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LepR mutant: | male | female | male | female | male | female | male | female | male | female | male | female |
| ◧ 65 | ↑ | ↑ | ↓ | ↓ | ↑↑↑ | ▯▯ | - | - | ▯ | ▯ | ↓ | ↓ |
| SS | ↑ | ↑ | ↓ | ▯ | ↑ | ▯ | ▯ | ↓ | - | ▯ | - | - |
| LL | - | ↓ | ↑ | - | - | - | - | - | - | - | - | - |
Changes are releative to littermate sex and age matched controls; ↑, significant increase; ↓ significant decrease; ▯, trend; ▯ = change
Figure 4. LepRb mutations have sex and location dependent effects on bone marrow adipocyte density.
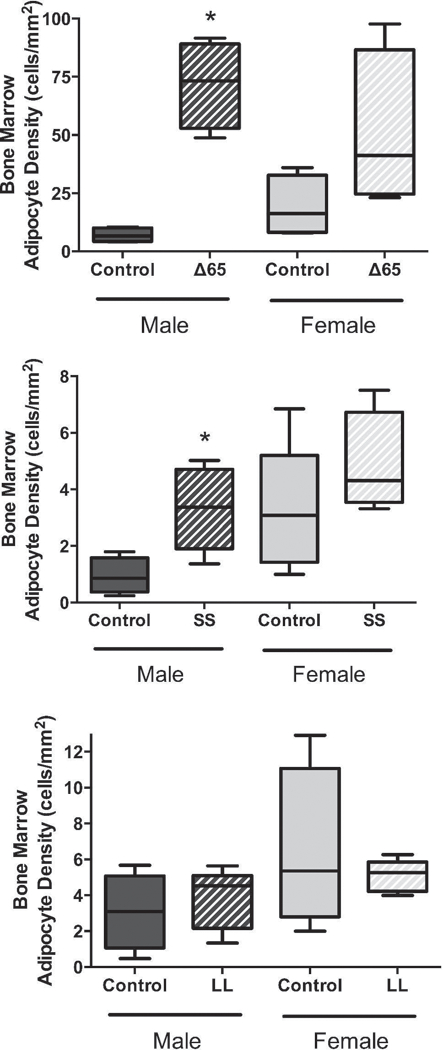
Distal femur bone marrow, proximal to the growth plate, was examined for marrow adipocytes. Adipocytes were counted and expressed relative to the marrow area. LepRb mutant mouse data (∆65, SS or LL) is grouped with data obtained from corresponding littermate control mice. Values represent averages ± SE. n ≥ 4 for all conditions. * p < 0.05.
Acknowledgements
The authors thank the Investigative Histology Laboratory in the Department of Physiology, Division of Human Pathology for their assistance with bone histology as well as the Biomedical Imaging Center for use of the microcomputed tomography imaging. Thank you also to Daniel Barnett for his technical expertise and Drs. Fraser Collins and Natalie Wee for their critical review of the manuscript. These studies were supported by funding from the National Institute of Health, grants RO1 DK101050, RO1 AT007695, R01 DK056731, and R00 DE024178.
Funding: These studies were supported by funding from the National Institute of Health, grants RO1 DK101050 and RO1 AT007695-01 and R00 DE024178.
Footnotes
Disclosure statement: The authors have nothing to disclose and have no conflicts of interest.
References
- Abella V, Scotece M, Conde J, Pino J, Gonzalez-Gay MA, Gomez-Reino JJ, Mera A, Lago F, Gomez R and Gualillo O (2017). “Leptin in the interplay of inflammation, metabolism and immune system disorders.” Nat Rev Rheumatol 13(2): 100–109. [DOI] [PubMed] [Google Scholar]
- Banks AS, Davis SM, Bates SH and Myers MG Jr. (2000). “Activation of downstream signals by the long form of the leptin receptor.” J Biol Chem 275(19): 14563–14572. [DOI] [PubMed] [Google Scholar]
- Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW and Myers MG Jr. (2003). “STAT3 signalling is required for leptin regulation of energy balance but not reproduction.” Nature 421(6925): 856–859. [DOI] [PubMed] [Google Scholar]
- Bjornholm M, Munzberg H, Leshan RL, Villanueva EC, Bates SH, Louis GW, Jones JC, Ishida-Takahashi R, Bjorbaek C and Myers MG Jr. (2007). “Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function.” J Clin Invest 117(5): 1354–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish J, Callon KE, Bava U, Lin C, Naot D, Hill BL, Grey AB, Broom N, Myers DE, Nicholson GC and Reid IR (2002). “Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo.” J Endocrinol 175(2): 405–415. [DOI] [PubMed] [Google Scholar]
- Devlin MJ, Cloutier AM, Thomas NA, Panus DA, Lotinun S, Pinz I, Baron R, Rosen CJ and Bouxsein ML (2010). “Caloric restriction leads to high marrow adiposity and low bone mass in growing mice.” J Bone Miner Res 25(9): 2078–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ealey KN, Fonseca D, Archer MC and Ward WE (2006). “Bone abnormalities in adolescent leptin-deficient mice.” Regul Pept 136(1–3): 9–13. [DOI] [PubMed] [Google Scholar]
- Ecklund K, Vajapeyam S, Feldman HA, Buzney CD, Mulkern RV, Kleinman PK, Rosen CJ and Gordon CM (2010). “Bone marrow changes in adolescent girls with anorexia nervosa.” J Bone Miner Res 25(2): 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elefteriou F, Ahn JD, Takeda S, Starbuck M, Yang X, Liu X, Kondo H, Richards WG, Bannon TW, Noda M, Clement K, Vaisse C and Karsenty G (2005). “Leptin regulation of bone resorption by the sympathetic nervous system and CART.” Nature 434(7032): 514–520. [DOI] [PubMed] [Google Scholar]
- Gat-Yablonski G and Phillip M (2008). “Leptin and regulation of linear growth.” Curr Opin Clin Nutr Metab Care 11(3): 303–308. [DOI] [PubMed] [Google Scholar]
- Gong Y, Ishida-Takahashi R, Villanueva EC, Fingar DC, Munzberg H and Myers MG Jr. (2007). “The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms.” J Biol Chem 282(42): 31019–31027. [DOI] [PubMed] [Google Scholar]
- Hamrick MW (2004). “Leptin, bone mass, and the thrifty phenotype.” J Bone Miner Res 19(10): 1607–1611. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Della Fera MA, Choi YH, Hartzell D, Pennington C and Baile CA (2007). “Injections of leptin into rat ventromedial hypothalamus increase adipocyte apoptosis in peripheral fat and in bone marrow.” Cell Tissue Res 327(1): 133–141. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Della-Fera MA, Choi Y-H, Pennington C, Hartzell D and Baile CA (2005). “Leptin Treatment Induces Loss of Bone Marrow Adipocytes and Increases Bone Formation in Leptin-Deficient ob/ob Mice.” Journal of Bone and Mineral Research 20(6): 994–1001. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Pennington C, Newton D, Xie D and Isales C (2004). “Leptin deficiency produces contrasting phenotypes in bones of the limb and spine.” Bone 34(3): 376–383. [DOI] [PubMed] [Google Scholar]
- He H, Liu R, Desta T, Leone C, Gerstenfeld LC and Graves DT (2004). “Diabetes causes decreased osteoclastogenesis, reduced bone formation, and enhanced apoptosis of osteoblastic cells in bacteria stimulated bone loss.” Endocrinology 145(1): 447–452. [DOI] [PubMed] [Google Scholar]
- Iwaniec UT, Boghossian S, Lapke PD, Turner RT and Kalra SP (2007). “Central leptin gene therapy corrects skeletal abnormalities in leptin-deficient ob/ob mice.” Peptides 28(5): 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaniec UT, Boghossian S, Trevisiol CH, Wronski TJ, Turner RT and Kalra SP (2011). “Hypothalamic leptin gene therapy prevents weight gain without long-term detrimental effects on bone in growing and skeletally mature female rats.” Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 26(7): 1506–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsenty G (2006). “Convergence between bone and energy homeostases: leptin regulation of bone mass.” Cell Metab 4(5): 341–348. [DOI] [PubMed] [Google Scholar]
- Kishida Y, Hirao M, Tamai N, Nampei A, Fujimoto T, Nakase T, Shimizu N, Yoshikawa H and Myoui A (2005). “Leptin regulates chondrocyte differentiation and matrix maturation during endochondral ossification.” Bone 37(5): 607–621. [DOI] [PubMed] [Google Scholar]
- Lindenmaier LB, Philbrick KA, Branscum AJ, Kalra SP, Turner RT and Iwaniec UT (2016). “Hypothalamic Leptin Gene Therapy Reduces Bone Marrow Adiposity in ob/ob Mice Fed Regular and High-Fat Diets.” Front Endocrinol (Lausanne) 7: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyl KJ and McCabe LR (2009). “Leptin treatment prevents type I diabetic marrow adiposity but not bone loss in mice.” Journal of Cellular Physiology 218(2): 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyl KJ and Rosen CJ (2012). “Understanding leptin-dependent regulation of skeletal homeostasis.” Biochimie 94(10): 2089–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzberg H, Huo L, Nillni EA, Hollenberg AN and Bjorbaek C (2003). “Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin.” Endocrinology 144(5): 2121–2131. [DOI] [PubMed] [Google Scholar]
- Philbrick KA, Martin SA, Colagiovanni AR, Branscum AJ, Turner RT and Iwaniec UT (2017). “Effects of hypothalamic leptin gene therapy on osteopetrosis in leptin-deficient mice.” J Endocrinol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Junior ES, Leite GA, Carmo-Silva CC, Taira TM, Neves KB, Colon DF, da Silva LA, Salvador SL, Tostes RC, Cunha FQ and Fukada SY (2016). “Adipokine Chemerin Bridges Metabolic Dyslipidemia and Alveolar Bone Loss in Mice.” J Bone Miner Res [DOI] [PubMed] [Google Scholar]
- Reid IR (2004). “Leptin deficiency—Lessons in regional differences in the regulation of bone mass.” Bone 34(3): 369–371. [DOI] [PubMed] [Google Scholar]
- Reseland JE, Syversen U, Bakke I, Qvigstad G, Eide LG, Hjertner O, Gordeladze JO and Drevon CA (2001). “Leptin is expressed in and secreted from primary cultures of human osteoblasts and promotes bone mineralization.” J Bone Miner Res 16(8): 1426–1433. [DOI] [PubMed] [Google Scholar]
- Robertson S, Ishida-Takahashi R, Tawara I, Hu J, Patterson CM, Jones JC, Kulkarni RN and Myers MG Jr. (2010). “Insufficiency of Janus kinase 2-autonomous leptin receptor signals for most physiologic leptin actions.” Diabetes 59(4): 782–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller EL, Song J, Dishowitz MI, Soki FN, Hankenson KD and Krebsbach PH (2010). “Leptin functions peripherally to regulate differentiation of mesenchymal progenitor cells.” Stem cells 28(6): 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steppan CM, Crawford DT, Chidsey-Frink KL, Ke H and Swick AG (2000). “Leptin is a potent stimulator of bone growth in ob/ob mice.” Regul Pept 92(1–3): 73–78. [DOI] [PubMed] [Google Scholar]
- Thomas T, Gori F, Khosla S, Jensen MD, Burguera B and Riggs BL (1999). “Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes.” Endocrinology 140(4): 1630–1638. [DOI] [PubMed] [Google Scholar]
- Turner RT, Kalra SP, Wong CP, Philbrick KA, Lindenmaier LB, Boghossian S and Iwaniec UT (2013). “Peripheral leptin regulates bone formation.” J Bone Miner Res 28(1): 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva EC and Myers MG Jr. (2008). “Leptin receptor signaling and the regulation of mammalian physiology.” Int J Obes (Lond) 32 Suppl 7: S8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GA, Callon KE, Watson M, Costa JL, Ding Y, Dickinson M, Wang Y, Naot D, Reid IR and Cornish J (2011). “Skeletal phenotype of the leptin receptor-deficient db/db mouse.” J Bone Miner Res 26(8): 1698–1709. [DOI] [PubMed] [Google Scholar]
- Yue R, Zhou BO, Shimada IS, Zhao Z and Morrison SJ (2016). “Leptin Receptor Promotes Adipogenesis and Reduces Osteogenesis by Regulating Mesenchymal Stromal Cells in Adult Bone Marrow.” Cell Stem Cell 18(6): 782–796. [DOI] [PubMed] [Google Scholar]
- Zhou BO, Yue R, Murphy MM, Peyer JG and Morrison SJ (2014). “Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow.” Cell Stem Cell 15(2): 154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]