

SUMMARY
Lyme disease, caused by the spirochete Borrelia burgdorferi, is the most common vector-borne disease in the US and Europe. The spirochetes are transmitted from mammalian and avian reservoir hosts to humans via ticks. Following tick bites, spirochetes colonize the host skin and then disseminate hematogenously to various organs, a process that requires this pathogen to evade host complement, an innate immune defense system. CspZ, a spirochete surface protein facilitates resistance to complement-mediated killing in vitro by binding to the complement regulator, factor H (FH). Low expression levels of CspZ in spirochetes cultivated in vitro or during initiation of infection in vivo has been a major hurdle in delineating the role of this protein in pathogenesis. Here we show that treatment of B. burgdorferi with human blood induces CspZ production and enhances resistance to complement. By contrast, a cspZ-deficient mutant and a strain that expressed a FH-nonbinding CspZ variant were impaired in their ability to cause bacteremia and colonize tissues of mice or quail; virulence of these mutants was however restored in complement C3-deficient mice. These novel findings suggest that FH-binding to CspZ facilitates B. burgdorferi complement evasion in vivo and promotes systemic infection in vertebrate hosts.
Keywords: Lyme disease, CspZ, Borrelia, Factor H, Complement
INTRODUCTION
Lyme disease (LD) is the most common vector-borne illness in North America and Europe, with an estimated 300,000 new human cases occurring annually in the US (Hinckley et al., 2014; Steere et al., 2016). In North America, LD is primarily caused by the spirochete Borrelia burgdorferi sensu stricto (hereafter B. burgdorferi), which is transmitted by Ixodes ticks and maintained in various vertebrate reservoir hosts (mainly mammals and birds) (Brisson, Drecktrah, Eggers, & Samuels, 2012; Eisen & Eisen, 2018). Following tick bites, the spirochetes spread from the skin of inoculation site to multiple tissues and organs via the bloodstream (Steere et al., 2016). In humans, infection with Lyme borreliae can result in long lasting, and debilitating symptoms including arthritis, carditis, and neuroborreliosis (Steere et al., 2016). Thus, dissemination and systemic infection require spirochetes to survive in the bloodstream of their different hosts (Radolf, Caimano, Stevenson, & Hu, 2012).
Complement is one of the key host innate immune defense mechanisms (Meri, 2016; Zipfel & Skerka, 2009). In general, complement can be activated on the surface of invading pathogens by the classical, lectin, and/or alternative pathways. The classical pathway is initiated on pathogens when antibodies bind to target antigens and engage the C1-complex. The lectin pathway is activated when recognition molecules (mannan binding lectin (MBL), collectins or ficolins) bind to select carbohydrates. The alternative pathway is initiated when activated C3b binds to the pathogen surface. Activation of all three pathways leads to the formation of C3 convertases (C4b2a by the classical and lectin pathway, or C3bBb by the alternative pathway), and further activation and deposition of C3b on C3 convertases lead to the formation of C5 convertases. These multi-protein complexes ultimately promote the release of proinflammatory peptides, the deposition of opsonins (C3b and iC3b), and insertion of the pore-forming membrane attack complex (MAC) (Merle, Church, Fremeaux-Bacchi, & Roumenina, 2015; Merle, Noe, Halbwachs-Mecarelli, Fremeaux-Bacchi, & Roumenina, 2015; Zipfel & Skerka, 2009). Complement regulators such as Factor H (FH) and FH-like protein 1 (FHL-1, the alternatively spliced form of FH) that bind to C3b to promote its degradation into iC3b (Sjoberg, Trouw, & Blom, 2009; Zipfel & Skerka, 2009) and prevent excessive complement activation and host cell damage in the absence of pathogens or tissue injury.
Pathogens have developed multiple mechanisms to escape killing by complement (Lambris, Ricklin, & Geisbrecht, 2008). One such mechanism is the production of complement-binding proteins to block the formation of complement complexes (Blom, Hallstrom, & Riesbeck, 2009; Kraiczy, 2016a; A. Marcinkiewicz, Kraiczy, & Lin, 2017; Meri, 2016). Another mechanism is to express complement regulator-binding proteins, which recruit host complement regulators to the cell surface to degrade active complement complexes (Blom et al., 2009; Meri, 2016). For example, Lyme borreliae produce OspC and BBK32, which respectively bind C4b and C1q to inhibit complement (Caine et al., 2017; Garcia, Zhi, Wager, Hook, & Skare, 2016). This pathogen also produces at least five distinct complement regulator-acquiring surface proteins (CRASPs): CspA (CRASP-1), CspZ (CRASP-2), ErpP (CRASP-3), ErpC (CRASP-4), ErpA (CRASP-5) (Kraiczy & Stevenson, 2013). These proteins bind to FH and/or FHL-1 (CspA and CspZ only) to inhibit the alternative pathway (Kraiczy & Stevenson, 2013). One of these FH-binding CRASPs, CspZ, promotes survival of otherwise serum-sensitive, non-pathogenic Lyme borreliae in human serum when overexpressed in these strains (Hartmann et al., 2006; Siegel et al., 2008). This protein is produced during mouse infection but not when the spirochetes reside in unfed or feeding ticks (Bykowski et al., 2007), suggesting a role of CspZ in providing bacteria the ability to survive in the hosts. However, a cspZ-deficient strain is fully infectious via needle infection (Coleman et al., 2008). Additionally, when mice were subcutaneously inoculated with a library of B. burgdorferi mutants carrying transposon insertions, mutants with transposon insertions in cspZ displayed only minor defects in colonization of mice (T. Lin et al., 2012). Interestingly, B. burgdorferi produces extremely low levels of CspZ when cultivated in vitro (Bykowski et al., 2007). This finding raises a possibility that low production of CspZ in in vitro-cultivated wild type B. burgdorferi does not permit elucidation of differences in serum survival or infectivity between the wild type and the cspZ-deficient mutant strain.
Spirochetes produce distinct protein profiles when cultivated in different conditions in vitro or while infecting vertebrate animals (Brooks, Hefty, Jolliff, & Akins, 2003; Hyde, Trzeciakowski, & Skare, 2007; Ojaimi et al., 2003; Revel, Talaat, & Norgard, 2002; Seshu, Boylan, Gherardini, & Skare, 2004; Tokarz, Anderton, Katona, & Benach, 2004). Incubating B. burgdorferi with mammalian blood simulates the conditions in hosts that leads to upregulation of genes that are normally expressed when spirochetes are in vertebrate hosts (Tokarz et al., 2004). In fact, blood treatment of spirochetes has been used to delineate the roles of several spirochete genes in vivo (Caine & Coburn, 2015; Caine et al., 2017; Y. P. Lin et al., 2015; Moriarty et al., 2012; Norman et al., 2008). Therefore, we hypothesized that blood treatment of spirochetes would enhance the production of CspZ thereby permitting examination of this protein in facilitating serum survival and infectivity in diverse hosts. In this study, we tested this hypothesis and elucidated a novel role for CspZ-FH interactions in bacterial pathogenesis in vivo.
RESULTS
Treating B. burgdorferi with human blood enhances CspZ production.
To define CspZ’s role in vitro and in vivo, we obtained wild type (WT) infectious B. burgdorferi B31-A3 and its isogenic cspZ-deficient mutant, B31-A3ΔcspZ (Coleman et al., 2008). This mutant strain carrying shuttle vector pKFSS (B31-A3ΔcspZ-V) or complemented with a plasmid encoding WT cspZ were generated. We also constructed a shuttle vector encoding this gene’s promoter from B. burgdorferi strain B31-A3 to drive cspZ-Y207A/Y211A, which produces the point mutant of CspZ defective in FH-binding activity (Siegel et al., 2008). This plasmid was transformed into B31-A3ΔcspZ as to generate complemented strain as a negative control. Note that CspZ-Y207A/Y211A was chosen to specifically eliminate FH binding; a previous study showed that CspZ-Y207A/Y211A binds neither to human nor mouse FH (A. L. Marcinkiewicz et al., 2018; Siegel et al., 2008). This is because a hydrogen bond between FH and tyrosine-207 of CspZ is abolished in this mutant protein (PDB#6ATG, http://www.rcsb.org/structure/6ATG). We also found that CspZ-Y207A/Y211A does not bind to FH from Coturnix quail, the avian model of LD (Isogai et al., 1994), whereas WT CspZ bound (Fig. S1 bottom and Table 1). Further, recombinant CspZ-Y207A/Y211A protein maintained its secondary structure, similar to the recombinant CspZ, (Fig. S2) and displayed similar levels of fibronectin, laminin, and plasminogen binding as WT CspZ (Hallstrom et al., 2010) (Fig. S3 and Table S1).
Table 1.
CspZ-Y207A/Y211A does not bind to Factor H from human or quail.
| Recombinant CspZ proteins | Factor H source | KD (μM) |
|---|---|---|
| GST-CspZ | Human | 0.29±0.07 |
| Quail | 0.91±0.14 | |
| GST-CspZ-Y207A/Y211A | Human | n.b.a |
| Quail | n.b. | |
| GSTb | Human | n.b. |
| Quail | n.b. |
All values represent the mean ± SEM of three experiments determined by ELISA.
No binding activity was detected.
GST was included as a negative control.
Hypothesizing that CspZ production is upregulated by vertebrate animals’ blood that simulates the host conditions, we first treated WT B. burgdorferi strain B31-A3 with human blood, and cspZ expression was quantitated by RT-PCR (qRT-PCR). Mid-log phase B31-A3 were incubated with that blood for 48 hours as previously described (Tokarz et al., 2004). We found that untreated and blood-treated spirochetes expressed similar levels of recA, a constitutively expressed gene (Fig. 1 A). Untreated B. burgdorferi expressed low but detectable of cspZ, consistent with previous reports (Coleman et al., 2008; Hartmann et al., 2006; Rogers, Abdunnur, McDowell, & Marconi, 2009) (Fig. 1A). Blood-treated B. burgdorferi expressed 4.1-fold greater levels of cspZ compared to untreated spirochetes (Fig. 1 A). We then sought to determine if increased cspZ expression translates to greater amounts of CspZ on the spirochete surface using flow cytometry (Fig. 1B). The periplasmic flagellin protein FlaB and the FH-binding protein CspA were used as controls. As expected, the production of FlaB was detectable in methanol-permeabilized but not in unpermeablized B. burgdorferi cells, consistent with the periplasmic location of this protein (Kumru, Schulze, Slusser, & Zuckert, 2010; Limberger, 2004) (Fig. 1C). The levels of FlaB were indistinguishable between untreated and blood-treated cells (Fig. 1C). The production of CspA was indistinguishable between permeabilized and unpermeabilized cells, in agreement with CspA’s surface localization (Fig. 1C) (Kraiczy et al., 2004). Consistent to previous work (Brooks et al., 2003; Bykowski et al., 2007; Tokarz et al., 2004), CspA levels decreased 3.3-fold in blood-treated compared to untreated cells (Fig. 1C). Finally, similar levels of CspZ was detected on permeabilized and unpermeablized strain B31-A3 (Fig. 1C), in agreement with this protein localized on the surface (Bykowski et al., 2007; Dowdell et al., 2017; Hartmann et al., 2006). Strikingly, the 5.3-fold enhancement of CspZ production in the presence of human blood compared to untreated strain B31-A3 (Fig. 1B and C). Further, we incubated blood with B31-A3ΔcspZ-V or B31-A3ΔcspZ producing WT cspZ or cspZ-Y207A Y211A. Similarly, the strain B31-A3ΔcspZ producing WT cspZ or cspZ-Y207A/Y211A exhibited enhanced CspZ production whereas these strains and the B31-A3ΔcspZ-V displayed decreased CspA production after blood treatment (Fig. 1D to F). Note that all these strains displayed indistinguishable levels of viability and generation time in either untreated or blood treated conditions. These results suggest that spirochete growth is not altered, and there is no difference among strains regarding to viability after blood treatment (Fig. S4 and Table S2 and S3). Taken together, these findings indicate that blood treatment induces CspZ production and down-regulates CspA production in the B. burgdorferi strains that encode the genes to produce these proteins.
Figure 1. The surface production of CspZ was enhanced in human blood-treated B. burgdorferi compared to untreated spirochetes.

Approximately 5×106 cells of the B. burgdorferi strain B31-A3 were cultivated in BSK-II medium with or without (“Untreated”) 5% blood (“Blood”) for 48 hours. (A) RNA from blood-treated or untreated spirochetes was extracted. The expression levels of cspZ, the constitutively expressed genes 16s rRNA, and recA were determined using qRT-PCR. The expression levels of recA (control) and cspZ are presented by normalizing to the expression levels of the gene encoding 16s rRNA. Each bar represents the mean of four independent determinations ± SEM. The asterisk (“*”) indicates significant differences (p < 0.05; Mann-Whitney test) in the normalized expression levels of cspZ in blood-treated spirochetes relative to that of untreated spirochetes. (B) Representative histograms of flow cytometry analysis showing the levels of CspZ surface production to blood-treated or untreated spirochetes. The shaded histograms are derived from untreated or blood-treated spirochetes incubated only with Alexa 647-conjugated goat anti-mouse IgG as control. (C to F) The production of F1aB (negative control), CspA or CspZ on the surface of blood-treated or untreated spirochetes was detected by flow cytometry. The mean fluorescence index (“MFI”) represents the production levels of FlaB, CspA, or CspZ in unpermeablized (filled bars) or methanol-permeabilized (opened bars) (C) B. burgdorferi strain B31-A3, (D) B31-A3ΔcspZ harboring the vector pKFSS-1 (“B31-A3ΔcspZ/Vector”), or (E) this cspZ mutant strain producing CspZ (“B31-A3ΔcspZ/pCspZ”) or (F) CspZ-Y207A/Y211A (“B31-A3ΔcspZ/pCspZ-Y207A/Y211A”). Each bar represents the mean of four independent determinations ± the standard deviation. The asterisk (“*”) indicates significantly different protein production (p < 0.05 by Kruskal-Wallis test with Dunn’s multiple comparison) between blood-treated and untreated indicated B. burgdorferi strains. Note that the production of F1aB, CspA, or CspZ among different spirochete strains when these strains were in blood-treated or untreated conditions is no different (p > 0.05 by Kruskal-Wallis test with Dunn’s multiple comparison).
CspZ requires Tyrosine-207 and −211 to promote blood-treated B. burgdorferi binding to human, mouse, and quail FH.
To define the role of CspZ in binding to human, mouse, and quail FH when expressed on the spirochete surface, FH purified from these hosts were incubated with in vitro cultivated, untreated (no added blood) B31-A3, B31-A3ΔcspZ-V, and bacteria-bound FH was detected by flow cytometry. A high passage and non-infectious B. burgdorferi strain B313 was also included as a negative control because this strain lacks the plasmids encoding CspZ and CspA (Hallstrom et al., 2013). We found that the level of these hosts’ FH bound by B31-A3 and B31-A3ΔcspZ-V were greater than that by B313 but undistinguishable between each other (Fig. S5). These findings indicated that in the absence of blood treatment, FH binding to B31-A3 and B31-A3ΔcspZ-V were similar. To enhance CspZ production in the wild-type isolate and downregulate CspA production in both wild-type and mutant, we treated these strains with human blood and assessed binding of human, mouse, and quail FH to each of these spirochete strains. As expected, B313 bound nearly undetectable levels of these hosts’ FH (Fig. 2) (Hart, Nguyen, et al., 2018; Hartmann et al., 2006). Consistent with previous observations (Hart, Nguyen, et al., 2018; Kenedy, Vuppala, Siegel, Kraiczy, & Akins, 2009; McDowell et al., 2003), B31-A3 bound FH from all three species (Fig. 2). Although B31-A3ΔcspZ-V still retained the ability to bind to these hosts’ FH, possibly due to the production of other FH-binding proteins, this CspZ-deficient strain showed over 4-fold reduced levels of FH binding compared to B31-A3 (Fig. 2). These findings indicate that prior blood treatment of spirochetes is necessary to reveal binding of FH to CspZ. We also treated blood with B31-A3ΔcspZ producing WT CspZ or CspZ-Y207A/Y211A and determine their ability to bind to human, mouse or quail FH. Ectopically produced CspZ but not CspZ-Y207A/Y211A in B31-A3ΔcspZ-V restored binding to these hosts’ FH (Fig. 2). The cspZ-Y207A/Y211A-complemented strain bound to these FH molecules 4-fold less than the cspZ-complemented strain (Fig. 2). These findings indicate that tyrosines at positions 207 and 211 of CspZ are required for maximal binding of human, mouse, and quail FH.
Figure 2. Tyrosine-207 and −211 of CspZ were critical for blood-treated B. burgdorferi to bind to human, mouse, and quail FH.

Human blood-treated B. burgdorferi strain B313, B31-A3, B31-A3ΔcspZ harboring the vector pKFSS-1 (“B31-A3ΔcpsZ/Vector”), or this cspZ mutant strain producing CspZ (“B31-A3ΔcspZ/pCspZ”) or CspZ-Y207A/Y211A (“B31-A3ΔcspZ/pCspZ-Y207A/Y211A”) was incubated with either PBS (negative control, data not shown) or FH from human, mouse, or quail. The bacteria were stained with a sheep anti-FH polyclonal IgG (for the spirochetes incubated with human or mouse FH) or a mouse anti-FH monoclonal antibody VIG8 (for the spirochetes incubated with quail FH) followed by an Alexa 647-conjugated donkey anti-sheep IgG or goat anti-mouse IgG prior to flow cytometry analysis. (Left panel) Representative histograms of flow cytometry analysis showing the levels of FH from (A) human, (B) mouse, or (C) quail binding to the indicated B. burgdorferi strains. (Right panel) The levels of B. burgdorferi binding to FH from (A) human, (B) mouse, or (C) quail were measured by flow cytometry and presented as mean fluorescence index (“MFI”). Each bar represents the mean of three independent determinations ± SEM. Significant differences (p < 0.05 by Kruskal-Wallis test with Dunn’s multiple comparison) in the levels of FH binding relative to the strain B313 (“Φ”), the strain ΔcspZ/Vector (“*”), or between two strains relative to each other (“#”) are indicated.
CspZ-mediated FH-binding contributes to reduced MAC deposition on the surface of blood-treated spirochetes.
Next, we asked if the FH-binding activity of CspZ reduced the deposition of complement activation products on the spirochete surface. We incubated WT strain B31-A3, B31-A3ΔcspZ-V, and B313 with 20% of human or mouse serum and quantified the levels of MAC bound to the spirochete surface using flow cytometry (Fig. 3). Quail serum could not be assessed due to the unavailability of antibodies that recognize avian MAC. The levels of MAC deposited on the surface of WT strain B31-A3 and B31-A3ΔcspZ-V were similar, but lower than that on the strain B313 (Fig. S6A and B). To enhance the production of CspZ, we treated these strains with human blood prior to incubation with serum to measure the levels of surface-associated MAC. As expected, a significant amount of human and mouse MAC could be detected on B313 whereas MAC deposition on the WT strain B31-A3 was undetectable (Fig. 3). Strain B31-A3ΔcspZ-V deposited greater amounts of MAC compared to the WT strain B31-A3 but lower levels compared to strain B313 (Fig. 3B). We also treated B31-A3ΔcspZ complemented with cspZ or cspZ Y207A/Y211A with human blood prior to incubating them with serum. MAC deposition was undetectable on the surface of the strain complemented with the WT cspZ gene as expected (Fig. 3B) while significant amounts of human and mouse MAC were deposited on the strain complemented with the cspZ-Y207A/Y211A (Fig. 3B). These results indicate that blood treatment of spirochetes, which enhances CspZ expression and FH binding, translates to inhibition of human and non-human complement deposition.
Figure 3. CspZ-mediated FH-binding activity decreased MAC deposition on the surface of blood-treated B. burgdorferi.
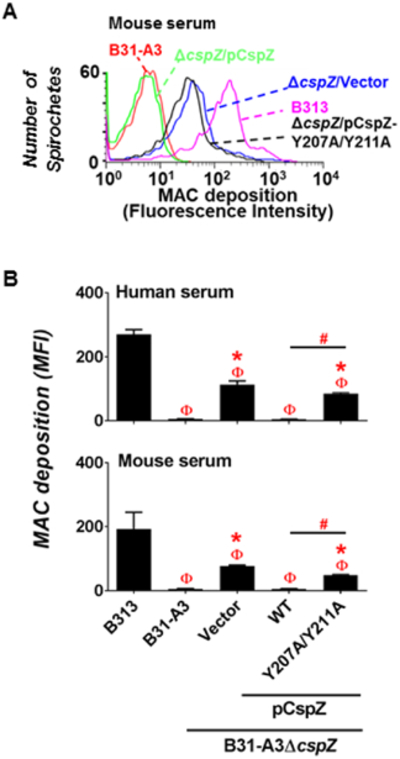
Human blood-treated B. burgdorferi strain B313, B31-A3, B31-A3ΔcspZ harboring the vector pKFSS-1 (“B31-A3ΔcspZ/Vector”), or this cspZ mutant strain producing CspZ (“B31-A3ΔcspZ/pCspZ”) or CspZ-Y207A/Y211A (“B31-A3ΔcspZ/pCspZ-Y207A/Y211A”) was incubated with either PBS (negative control, data not shown) or serum from human or mouse at a final concentration of 20%. The bacteria were stained with a mouse anti-MAC monoclonal antibody aE11 (for spirochetes incubated with human serum), or a rabbit anti-MAC polyclonal IgG (for spirochetes incubated with mouse serum) followed by a goat anti-mouse IgG, or a goat anti-rabbit IgG, prior to flow cytometry analysis. (A) Representative histograms of flow cytometry analysis showing the deposition levels of mouse MAC on the surface of indicated B. burgdorferi strains. (B) The deposition levels of human or mouse MAC on the surface of B. burgdorferi were measured by flow cytometry and presented as mean fluorescence index (“MFI”). Each bar represents the mean of three independent determinations ± SEM. Significant differences (p < 0.05 by Kruskal-Wallis test with Dunn’s multiple comparison) in the deposition levels of MAC relative to the strain B313 (“Φ”), the strain B31-A3ΔcspZ/Vector (“*”), or between two strains relative to each other (“#”) are indicated.
CspZ binding to FH facilitates blood-treated B. burgdorferi to survive in serum.
We aimed to determine CspZ’s role in promoting spirochete survival in serum. The WT strain B31-A3 and B31-A3ΔcspZ-V were incubated with human or quail serum, or negative control sera that were not expected to kill bacteria (C3-depleted human serum or heat inactivated human or quail serum) for four hours. Mouse serum was not tested because mouse complement is highly unstable ex vivo (Caine & Coburn, 2015; Lachmann, 2010; A. Marcinkiewicz et al., 2017; Ristow et al., 2012). Consistent with previous findings (Coleman et al., 2008), we found that nearly 100% of the WT strain B31-A3 and B31-A3ΔcspZ-V survive in human, quail, and negative control sera (Fig. S7). We then treated the WT strain B31-A3 or B31-A3ΔcspZ-V with human blood to enhance expression of cspZ prior to incubation with sera. Over 75% of the WT strain B31-A3 survived in active (Fig. 4 top panel) or C3-depleted human serum (Fig. 4 middle panel). While less than 50% of the strain B31-A3ΔcspZ-V were viable in human serum (Fig. 4 top panel), this strain survived almost 100% in heat-inactivated (Fig. 4 top panel) or C3-depleted human serum (Fig. 4 middle panel). Similarly, approximately 100% of WT strain B31-A3 survived in quail serum (Fig. 4 bottom panel). Although only 36% of B31-A3ΔcspZ-V remained motile in this serum, it showed nearly 100% survival in heat-inactivated quail serum (Fig. 4 bottom panel). These findings indicate that CspZ promotes blood-treated spirochetes to survive in human and quail sera.
Figure 4. CspZ-mediated FH-binding activity contributed to the survival of blood-treated spirochetes in human and quail serum.
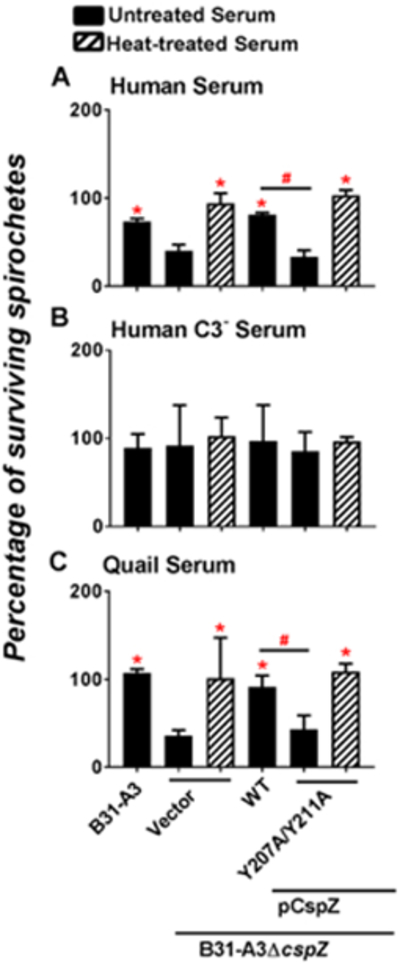
Human blood-treated B. burgdorferi strain B31-A3, B31-A3ΔcspZ harboring the vector pKFSS-1 (“B31-A3ΔcspZ/Vector”), or this cspZ mutant strain producing CspZ (“B31-A3ΔcspZ/pCspZ”) or CspZ-Y207A/Y211A (“B31-A3ΔcspZ/pCspZ-Y207A/Y211A”) was incubated for four hours with untreated (filled bars) or heat-inactivated (“Heat-treated”, hatched bars) serum at a final concentration of 40%. These sera include (A) normal human serum, (B) C3-depleted human serum (“Human C3− serum”), or (C) quail serum. The number of motile spirochetes was assessed microscopically. The percentage of surviving B. burgdorferi was calculated using the number of mobile spirochetes at four hours post incubation normalized to that immediately after incubation with serum. Each bar represents the mean of three independent determinations ± SEM. Significant differences (p < 0.05 by Kruskal-Wallis test with Dunn’s multiple comparison) in the percentage of surviving spirochetes relative to the strain B31-A3ΔcspZ/Vector incubated with untreated serum (“*”) or between two strains relative to each other (“#”) are indicated.
The serum resistance of B31-A3ΔcspZ-V complemented with cspZ or cspZ-Y207A/Y211A was also assessed using identical experimental conditions. Approximately 75% of the cspZ-complemented strain survived in both active or C3-depleted human serum (Fig. 4 top and middle panel). Only 25% of the cspZ-Y207A/Y211A-complemented strain survived in human serum (Fig. 4 top panel) but retained close to 100% viability in heat-inactivated (Fig. 4 top panel) or C3-depleted human serum (Fig. 4 middle panel). Almost 100% of the cspZ-complemented bacteria survived in quail serum while only 36% of the cspZ-Y207A/Y211A-complemented strain was viable (Fig. 4 bottom panel). Moreover, almost 100% of bacteria of this strain remained motile in heat-inactivated quail serum (Fig. 4 bottom panel). These results suggest that CspZ-mediated FH-binding activity provides serum resistance activity for blood-treated B. burgdorferi.
FH binding to CspZ confers bacteremia and tissue colonization of blood-treated B. burgdorferi.
We next asked whether CspZ-mediated FH-binding promotes infectivity in mice. Subcutaneous needle infection of spirochetes with the dose close to ID50 (the dose that infects 50% of animals) often reveals subtle in vivo phenotypes conferred by spirochete genes that are expressed in hosts (Blevins, Hagman, & Norgard, 2008; Hyde et al., 2011; Seshu et al., 2006; Shi, Xu, Seemanaplli, McShan, & Liang, 2008; Weening et al., 2008). We thus introduced 103 cells of WT strain B31-A3 or B31-A3ΔcspZ-V into BALB/c mice (the ID50 of strain B31-A3 is ∼103 cells (Showman, Aranjuez, Adams, & Jewett, 2016; Tilly et al., 2006)). We then used quantitative PCR (qPCR) to quantify spirochete burdens in the bloodstream and tissues. The infection of WT strain B31-A3 or strain B31-A3ΔcspZ-V resulted in indistinguishable levels of bacteremia at 7 days post-infection (dpi) and tissue colonization at both 7 and 14 dpi (Fig. S8A left panel and 8B to E). Note that we were unable to detect spirochetes in the blood at 14 dpi (Fig. S8A right panel), in agreement with kinetics of bacteremia present only at extremely early stages of murine infection (Caine et al., 2017).
In order to delineate the role of CspZ during the course of infection, we treated WT strain B31-A3 and B31-A3ΔcspZ-V with human blood to upregulate cspZ expression. BALB/c mice were then infected with blood-treated B31-A3 or B31-A3ΔcspZ-V. Bacteremia triggered by the former strain was observed at 7 dpi but not at 14 dpi (Fig. 5A). Colonization was detected for WT strain B31-A3 in all tested tissues at 7 and 14 dpi (Fig. 5B to E). In contrast, the strain B31-A3ΔcspZ-V induced six-fold less levels of bacteremia at 7 dpi (p = 0.021, Fig. 5A left panel) and displayed 2600 and 9-fold lower spirochete burden at the inoculation site than the WT strain at 7 and 14 dpi, respectively (p < 0.05, Fig. 5B). The cspZ-deficient strain also exhibited a lower, although not statistically significant, level of colonization of the heart, bladder, and joints compared to the WT strain at 7dpi (Fig. 5C to E left panel). At 14 dpi, the cpsZ-deficient mutant colonized these tissues at 6- to 9-fold reduced levels compared to the WT strain (Fig. 5 C to E right panel). We also infected mice with human blood-treated B31-A3ΔcspZ producing WT CspZ or CspZ-Y207A/Y211A. Ectopically producing WT CspZ but not CspZ-Y207A/Y211A in the strain B31-A3ΔcspZ restores the defect of bloodstream survival at 7 dpi (5-fold greater bacterial burdens than the strain B31-A3ΔcspZ-V, Fig. 5A left panel). At 7 and 14dpi, the cspZ-Y207A/Y211A-complemented strain did not colonize the inoculation site while the cspZ-complemented strain did (13 to 259-fold greater levels than the strain B31-A3ΔcspZ-V) (Fig. 5B). These strains did not display different levels of colonization in heart, bladder, and joints at 7dpi (Fig. 5C to E left panel). However, at 14 dpi, the cspZ-complement strain exhibited 2.4 to 13-fold greater levels of colonization at these tissues compared to B31-A3ΔcspZ-V. Spirochetes producing CspZ-Y207A/Y211A colonized these tissues with a bacterial burden similar to the strain B31-A3ΔcspZ-V (Fig. 5C to E right panel). These results demonstrate that CspZ-mediated FH-binding promotes hematogenous dissemination of blood-treated spirochetes in mice.
Figure 5. CspZ-mediated FH-binding activity promoted bacteremia and tissue colonization of blood-treated B. burgdorferi in mice.

BALB/c mice were subcutaneously infected with 103 cells of human blood-treated B. burgdorferi strain B31-A3, B31-A3ΔcspZ harboring the vector pKFSS-1 (“B31-A3ΔcspZ/Vector”), or this cspZ mutant strain producing CspZ (“B31-A3ΔcspZ/pCspZ”) or CspZ-Y207A/Y211A (“B31-A3ΔcspZ/pCspZ-Y207A/Y211A”). These mice were sacrificed at (left panel) 7 or (right panel) 14 days post-infection (“dpi”). The spirochete burdens in the (A) Blood, (B) inoculation site of skin (“Inoc. Site”), (C) heart, (D) bladder, and (E) tibiotarsus joints were determined by qPCR and normalized to 1μg total DNA. Shown are the geometric mean ± geometric standard deviation of 14 (blood from B31-A3-infected mice at 7dpi), 15 (blood from B31-A3ΔcspZ/Vector- or B31-A3ΔcspZ/pCspZ-Y207A/Y211A-infected mice at 7dpi), 12 (blood from B31-A3ΔcspZ/pCspZ-infected mice at 7dpi), 6 (blood from B31-A3-infected mice at 14 dpi), 8 (tibiotarsus joints from B31-A3-infected mice), 16 (Inoc. Site from B31-A3- or B31-A3ΔcspZ/pCspZ-infected mice at 14dpi), 9 (bladder or heart from B31-A3-infected mice at 14dpi), 7 (blood from B31-A3ΔcspZ/Vector-infected mice or bladder or heart from B31-A3ΔcspZ/pCspZ-infected mice at 14dpi) or 10 (all others) mice per group. Significant differences (p < 0.05 by Kruskal-Wallis test with Dunn’s multiple comparison) in the spirochete burdens relative to the strain B31-A3–cspZ/Vector (“*”) or between two strains relative to each other (“#”) are indicated, (“n.d.”): not determined.
Complement evasion mediated by CspZ-FH interactions facilitates blood-treated spirochete to survive in mouse bloodstream and tissues.
We next sought to investigate whether CspZ-mediated FH-binding activity, by evading the complement, facilitates blood-treated spirochetes to survive in mouse bloodstream and tissues. We first subcutaneously inoculated C3-deficient mice in a BALB/c background (C3−/− mice) with 103 cells of each of the four following spirochete strains after treating these strains with human blood. These strains include WT strain B31-A3, B31-A3ΔcspZ-V, B31-A3ΔcspZ complemented with cspZ, or cspZ Y207A/Y211A mutant. qPCR was used to measure spirochete loads in the bloodstream and tissues. In contrast to the findings in WT mice, we found no differences in bacterial burdens across all four strains in bloodstream at 7 dpi (Fig. 6A) and the inoculation site, heart, bladder, and joints of C3−/− mice at 7 and 14 dpi (Fig. 6B to E). These results indicate that that FH binding to CspZ facilitates blood-treated B. burgdorferi to evade mouse complement and thus renders them pathogenic.
Figure 6. CspZ-mediated FH-binding activity facilitated bacteremia and tissue colonization of blood-treated spirochetes by evading the mouse complement.

BALB/c C3−/− mice were subcutaneously infected with 103 cells of human blood-treated B. burgdorferi strain B31-A3, B31-A3ΔcspZ harboring the vector pKFSS-1 (“B31-A3ΔcspZ/Vector”), or this cspZ mutant strain producing CspZ (“B31-A3ΔcspZ/pCspZ”) or CspZ-Y207A/Y211A (“B31-A3ΔcspZ/pCspZ-Y207A/Y211A”). The mice were sacrificed at (left panel) 7 or (right panel) 14 days post-infection (“dpi”). The spirochete burdens in the (A) blood, (B) inoculation site of skin (“Inoc Site”), (C) heart, (D) bladder, and (E) tibiotarsus joints were determined by qPCR and normalized to 1 μg total DNA. Shown are the geometric mean ± geometric standard deviation of 5 (tibiotarsus joints from B31-A3ΔcspZ/pCspZ-Y207A/Y211A-infected mice at 7dpi) or 6 (all others) mice per group. There were no significant differences (p < 0.05 by Kruskal-Wallis test with Dunn’s multiple comparison) in the spirochete burdens relative to the strain B31-A3ΔcspZ/Vector or between two strains relative to each other.
FH binding activity of CspZ promotes spirochete colonization in quail.
We aimed to test whether CspZ-mediated FH-binding confers infectivity of B. burgdorferi in avian hosts such as the quail. We thus treated WT strain B31-A3 with human blood to enhance the production of CspZ, subcutaneously inoculated 106 cells of B31-A3 cells into quail, and determined bacterial burdens using qPCR in the blood and tissues. We could not detect WT strain B31-A3 in the blood or any tested tissues at 3 dpi (detection limit: one bacterium per one microgram of DNA, Fig. S9). Although we were still unable to detect spirochetes in the blood, live or heart at 7 dpi, spirochetes were detectable at the inoculation site and the brain at this time point (Fig. 7 and S9). The viability of spirochetes in these tissues was verified microscopically after culturing the tissues collected from the quail at 7 dpi: five out of six quail inoculation sites and brain tissues were culture positive. We also subcutaneously infected quail with human blood-treated B31-A3ΔcspZ-V, or this strain producing CspZ or CspZ-Y207A/Y211A. At 7 dpi, the strain B31-A3ΔcspZ-V colonized the inoculation site and brain approximately five to eight-fold less, respectively, compared to WT strain B31-A3 (Fig. 7). Ectopically producing CspZ in B31-A3ΔcspZ restored the colonization defects in these tissues (Fig. 7), indicating CspZ contributes to B. burgdorferi colonization at quail tissues. However, ectopic production of CspZ-Y207A/Y211A did not restore colonization levels in the inoculation site and brain (similar spirochete burdens to mutant B31-A3ΔcspZ-V) (Fig. 7). These results suggest that CspZ binds to FH to facilitate spirochetes to establish infection and promote dissemination in quail.
Figure 7. CspZ-mediated FH-binding activity promoted B. burgdorferi colonization in quail.

Coturnix coturnix quail were subcutaneously infected with 106 cells of human blood-treated B. burgdorferi strain B31-A3, B31-A3ΔcspZ harboring the vector pKFSS-1 (“B31-A3ΔcspZ/Vector”), or this cspZ mutant strain producing CspZ (“B31-A3ΔcspZ/pCspZ”) or CspZ-Y207A/Y211A (“B31-A3ΔcspZ/pCspZ-Y207A/Y211A”). The quail were sacrificed at seven days post-infection (“dpi”). The spirochete burdens in the (A) inoculation site of skin (“Inoc. Site”) and (B) brain were determined by qPCR and normalized to 1 μg total DNA. Shown are the geometric mean ± geometric standard deviation of 8 (B31-A3-infected quail), 14 (Inoc. Site from B31-A3ΔcspZ/pCspZ-infected quail), 9 (brain from B31-A3ΔcspZ/pCspZ-infected quail), 6 (all others) quail per group. Significant differences (p < 0.05 by Kruskal-Wallis test with Dunn’s multiple comparison) in the spirochete burdens relative to the strain B31-A3ΔAcspZ/Vector (“*”) or between two strains relative to each other (“#”) are indicated.
DISCUSSION
B. burgdorferi, the causative agent of LD, produces at least 86 outer surface lipoproteins (Dowdell et al., 2017). Although some of these proteins are produced in abundance, most are expressed at low levels when cultured in vitro and/or during murine infection (Kenedy, Lenhart, & Akins, 2012), which have been hurdles in studying their roles in pathogenesis. Cultivating B. burgdorferi in specific conditions such as blood supplemented growth media to induce the production of proteins of interest has been utilized to investigate the role of such proteins in vitro and in vivo (Kumar et al., 2015; Moriarty et al., 2012; Parveen & Leong, 2000; Zhi et al., 2015). We used human blood to treat spirochetes and observed induction of CspZ (Fig. 1) as this host’s blood has been used as a host-simulated cue to alter the expression of spirochetes genes (Tokarz et al., 2004). However, the varying molecular compositions of the blood of different animal species (Hamdy, 1977; Sojka et al., 2013; Wickramasekara, Bunikis, Wysocki, & Barbour, 2008), raising the possibility that enhanced cspZ expression is host blood-specific, which warrants further investigation. Additionally, human blood treatment of spirochetes followed by murine infection may not reflect to the nature life cycle of Lyme borreliae in mice. This concern could be eased by treating spirochetes with mouse blood prior to infection for future studies. Further, Tokarz et al. did not report an increased cspZ expression in spirochetes treated with human blood using a microarray, which could be due to the stringent criteria used to define the differential gene expression (Tokarz et al., 2004). Treating B. burgdorferi with proteases has been commonly used to determine a particular spirochete protein’s surface localization (El-Hage et al., 2001; Exner, Wu, Blanco, Miller, & Lovett, 2000; Zuckert, Kerentseva, Lawson, & Barbour, 2001). CspZ remains intact after the treatment of proteinase K and trypsin, suggesting this protein’s resistance to digestion by these proteases (Coleman et al., 2008; Dowdell et al., 2017; Hartmann et al., 2006). However, CspZ is eliminated when spirochetes are treated with pronase (Dowdell et al., 2017), a protease isolated from Streptomyces griseus (Hiramatsu & Ouchi, 1963). This finding indicates this protein’s surface localization, consistent with our and previous observations using fluorescence-based methodologies (Bykowski et al., 2007; Hartmann et al., 2006; Siegel et al., 2008)(Fig. 1B, C, E, and F).
We found that blood treatment is essential to demonstrate the CspZ-mediated FH-binding activity to confer spirochete survival in sera (Fig. 4). It is noteworthy that we count immotile cells as the indicator of cell death by dark-field microscopy, a method which has been commonly used to identify killed cells (Alitalo et al., 2005; Alitalo et al., 2001; Brooks et al., 2005; Kraiczy, Hunfeld, Peters, et al., 2000; McDowell et al., 2011; van Dam et al., 1997). Furthermore, the lack of motility is the most apparent sign of complement-mediated bacterial killing, which allows determination of cell viability microscopically without the need for the extended duration of time to cultivate spirochetes. Further, the ability of pathogens to limit complement activation on their surface often correlates with their ability to survive in the bloodstream and cause systemic infection (Lambris et al., 2008; Roantree & Rantz, 1960). For example, the B. burgdorferi outer surface protein BBK32 inhibits activation of the classical pathway and confers resistance to human serum (Garcia et al., 2016). This protein also promotes spirochete survival in the mouse bloodstream and dissemination (Caine & Cobum, 2015; Hyde et al., 2011; Seshu et al., 2006). Similarly, B. burgdorferi produces OspC that inactivates both classical and lectin pathways and contributes to early stages of bacteremia (Caine & Cobum, 2015; Caine et al., 2017). In this study, we treated a cspZ-deficient B. burgdorferi with blood to demonstrate this CspZ’s ability, by evading complement, to promote bacteremia and/or tissue colonization (Fig. 5 and 6). Note that this finding does not imply that CspZ facilitates spirochete survival in fed ticks during transmission as B. burgdorferi does not produce CspZ when it is in either post molting flat nymphs or feeding nymphs (Bykowski et al. Infect Immun. 2007). However, CspZ is produced at low levels in the biting site of skin after transmission (Bykowski et al. Infect Immun. 2007). Therefore, in the case that a cspZ-deficient spirochete displays defect of infectivity in vertebrate hosts during transmission, CspZ’s role should be dependent on the host environment such as skin. Further, B. burgdorferi has been shown to more efficiently spread to distal tissues in mice deficient of some complement proteins (e.g. C3−/− or Clqα−/− mice) Compared to WT mice (Lawrenz et al., 2003; Woodman et al., 2007; Zhi, Xie, & Skare, 2018). Similarly, we observed increased spirochete burdens in the tissues of C3−/− mice (Fig. 6B to E) relative to the WT mice (Fig. 6B to D) at 14dpi, suggesting the need for B. burgdorferi to evade complement in order to disseminate. Further, the initial skin colonization of B. burgdorferi results in an exuberant inflammatory response at inoculation site of dermis (Antonara, Ristow, McCarthy, & Coburn, 2010; Hovius et al., 2009; Xu, Seemanapalli, Reif, Brown, & Liang, 2007). Complement in the vascular compartment or interstitial fluid would accompany the localized influx of cells at the dermal inoculation site (Wilhelm, 1973). This addresses our finding that spirochetes use the FH-binding activity of CspZ to establish infection at inoculation site (Fig. 5B and 6B).
In addition to mammals, birds also maintain Lyme borreliae as a reservoir host in the enzootic cycle (Brisson et al., 2012; Eisen & Eisen, 2018). However, the molecular mechanisms of spirochete colonization and dissemination in this host remain unclear. This could be due to the cumbersome work of maintaining wild-caught birds and/or the inability to persistently infect some avian hosts with Lyme borreliae experimentally (Bishop, Khan, & Nielsen, 1994; Burgess, 1989; Ginsberg et al., 2005; Isogai et al., 1994; Kurtenbach et al., 1998; Kurtenbach, Schafer, et al., 2002; Olsen, Gylfe, & Bergstrom, 1996; Piesman, Dolan, Schriefer, & Burkot, 1996; Richter, Spielman, Komar, & Matuschka, 2000). Among these avian species, Coturnix quail is capable of harboring Lyme borreliae for at least eight weeks after needle infection (Isogai et al., 1994). Similar to previous findings (Isogai et al., 1994), we did not detect spirochetes in the blood during infection (Fig. S9), reiterating the inability of Lyme borreliae to induce high levels of bacteremia in vertebrate animals (Steere et al., 2016). Unlike a previous study detecting spirochetes in the liver and heart of B. garinii-infected quail (Isogai et al., 1994), we did not observe B. burgdorferi colonization at these sites, possibly reflecting strain-specific tissue tropism (Fig. S9)(Craig-Mylius, Lee, Jones, & Glickstein, 2009; Jones et al., 2006; Wang, van Dam, Schwartz, & Dankert, 1999). We found that CspZ-mediated FH-binding activity facilitates spirochete colonization at the quail inoculation site and brain, which reveals the role of a B. burgdorferi protein in promoting avian host competence at the first time (Fig. 7). Note that Not every strain of Lyme borreliae species encodes CspZ (Kingry et al., 2016; Kraiczy, Skerka, Brade, & Zipfel, 2001; Rogers & Marconi, 2007), and allelic variations confer differential human FH/FHL-1-binding activity (Kraiczy et al., 2008; Kraiczy et al., 2001; Rogers et al., 2009). This raises the possibility that CspZ variants may determine host-specificity of FH-binding and determine the animals’ competence to each strain (Bhide et al., 2005; Kraiczy, 2016b; Kurtenbach, De Michelis, et al., 2002). This could be further investigated using the quail and mouse as Lyme infection models established in this study. Such models can also be applied to study the role of additional proteins of B. burgdorferi or other pathogens that are induced during blood treatment in promoting infectivity. Gaining an understanding of the mechanisms of pathogen infectivity can permit further efforts to find treatments or vaccines to improve human health.
EXPERIMENTAL PROCEDURES
Ethics statement.
All mouse experiments were performed in strict accordance with all provisions of the Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the PHS Policy on Humane Care and Use of Laboratory Animals. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of Wadsworth Center, New York State Department of Health (protocol Docket Number 16-451), and University of Massachusetts Medical School (protocol Docket Number 1930). All efforts were made to minimize animal suffering.
Mouse, quail, bacterial strains, and animal sera.
Swiss Webster mice used to generate anti-serum against CspZ and BALB/c mice were purchased from Charles River (Wilmington, MA) and Taconic (Hudson, NY), respectively. C3−/− mice (C57BL/6) purchased from Jackson Laboratory (Bar Harbor, ME) were backcrossed for 11 generations into a BALB/c background. Mice were genotyped for the C3 alleles by PCR analysis of mouse tail DNA (Table S4). Coturnix coturnix quail were purchased from Cavendish Game Bird Farm (Springfield, VT). The Borrelia and Escherichia coli strains used in this study are described in Table 2. All B. burgdorferi strains were grown at 33°C in BSK-II complete medium supplemented with kanamycin (200μg/mL), streptomycin (50μg/mL), or no antibiotics as required. For blood-treatment, spirochetes were incubated with human blood as described (Tokarz et al., 2004). Approximately 5 × 106 cells of mid-log B. burgdorferi were cultivated in BSK-II complete medium with 5% human blood with the buffy coat removed. This concentration of human blood does not reduce the growth and motility of serum-sensitive strains (Breitner-Ruddock, Wurzner, Schulze, & Brade, 1997; Hart, Nguyen, et al., 2018; Kenedy & Akins, 2011; van Dam et al., 1997). The human blood was supplemented with a cocktail of antibiotics (final concentration: 50μg/mL rifampicin, 20μg/mL phosphomycin and 2.5μg/mL amphotericin) to prevent potential bacterial and fungal contamination. Spirochetes were then incubated with human blood at 33°C at 2% CO2 under a microaerophilic condition for 48 hours prior to use. E. coli strains were grown at 37°C in Luria-Bertani (BD Bioscience, Franklin Lakes, NJ) broth or agar, supplemented with kanamycin (50μg/mL), streptomycin (50 μg/mL), ampicillin (100μg/mL), or no antibiotics (Table 2). Human, mouse, and quail sera were obtained from and MB Biomedical, Inc (Santa Ana, CA), Southern Biotech, Inc (Birmingham, AL), and Canola Live Poultry Market (Brooklyn, NY), respectively. Prior to being used, these sera were screened with the C6 Lyme ELISA kit (Diamedix, Miami Lakes, FL) to determine whether the individual from which it was collected had prior exposure to B. burgdorferi by detecting antibodies against the C6 peptide of the B. burgdorferi protein VlsE (Lawrenz et al., 1999).
Table 2.
Strains and plasmids used in this study.
| Strain or plasmid | Genotype or characteristic | Source |
|---|---|---|
| B. burgdorferi | ||
| B313 | High-passage B. burgdorferi B31 missing lp5, lp 17, lp21, lp25, lp28-1, lp28-2, lp28-3, lp28-4, lp36, lp38, lp54, lp56, cp9, cp32-4, cp32-6, cp32-8, cp32-9 | (Hallstrom et al., 2013) |
| B31-A3 | Clone of B. burgdorferi B31 missing cp9 | (Elias et al., 2002) |
| B31-A3ΔcspZ | B31-A3ΔcspZ::KanRa | (Coleman et al., 2008) |
| B31-A3ΔcspZ/pKFSS-1 | B31-A3ΔcspZ:: KanR carrying plasmid pKFSS-1 | This study |
| B31-A3ΔcspZ/pCspZ-WT | B31-A3ΔcspZ::KanR complemented with intact cspZ | This study |
| B31-A3ΔcspZ/pCspZ-Y207A/Y211A | B31-A3ΔcspZ::KanR complemented with intact cspZ-Y207A/Y211A | This study |
| E. coli | ||
| DH5α | F- Φ801acZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17(rk−, mk+) phoA supE44 thi-1 gyrA96 re1A1 λ- | ThermoFisher |
| BL21(DE3) | F–, ompT hsdSB (rB– mB–) gal dcm (DE3) | Novagen |
| BL21 (DE3)/pGEX4T2-CspZ | BL21(DE3) producing GST-tagged residues 19 to 237 of CspZ | This study |
| BL21 (DE3)/pGEX4T2-CspZ-Y207A/Y211A | BL21(DE3) producing GST-tagged residues 19 to 237 of CspZ-Y207A/Y211A | This study |
| Plasmids | ||
| pJET 1.2/Blunt | AmpRb; PCR cloning vector | Therm oFisher |
| pGEX4T2 | AmpR; GST-tagged protein expression vector | Qiagen |
| pGEX4T2-CspZ | AmpR; pGEX4T2 encoding GST fusion protein residue 19 to 237 of CspZ | This study |
| pGEX4T2-CspZ-Y207A/Y211A | AmpR; pGEX4T2 encoding GST fusion protein residue 19 to 237 of CspZ-Y207A/Y211A | This study |
| pKFSS-1 | StrRc; Borrelia shuttle vector | (Frank et al., 2003) |
| pKFSS-1/pCspZ-WT | StrR; pKFSS-1 encoding intact cspZ | This study |
| pKFSS-1/pCspZ-Y207A/Y211A | StrR; pKFSS-1 encoding intact cspZ-Y207A/Y211A | This study |
Kanamycin resistant
Ampicillin resistant
Streptomycin resistant
Generation of recombinant CspZ proteins and antisera.
The open reading frames lacking the putative signal sequences of bbh06 (cspZ) or encoding CspZ-Y207A/Y211A (CspZ with tyrosine-207 and −211 replaced by alanine residues) from B. burgdorferi strain B31-A3 was amplified using listed plasmids and primers (Table 2 and S4)(Siegel et al., 2008). Amplified fragments were engineered to encode a BamHI site at the 5’ end and a stop codon followed by a SalI site at the 3’ end. PCR products were sequentially digested with BamHI and SalI and then inserted into the BamHI and SalI sites of pGEX4T2 (GE Healthcare, Piscataway, NJ). The plasmids were sequenced and then transformed into E. coli strain BL21(DE3) (Wadsworth ATGC facility). The GST-tagged CspZ proteins were produced and purified by GST affinity chromatography according to the manufacturer’s instructions (GE Healthcare, Piscataway, NJ). Antisera against CspZ were generated by immunizing four-week-old Swiss Webster mice with each of the CspZ proteins as described (Benoit, Fischer, Lin, Parveen, & Leong, 2011).
Quantitative RT-PCR and PCR.
For quantitative RT-PCR (qRT-PCR), RNA was extracted from B. burgdorferi strain B31-A3 using Direct-Zol RNA MiniPrep Plus Kit (Zymo Research, Irvine, CA), and contaminating DNA was removed using RQ1 RNase-Free DNase (Promega, Madison, WI). cDNA was synthesized from 1 μg of RNA using qScript cDNA SuperMix (Quanta Bioscience, Beverly, MA). The quantification of 16s rRNA, cspZ, or recA expression from cDNA using listed primers Table S4 (Hodzic et al., 2013)(Bykowski et al., 2007)(Hodzic, Feng, & Barthold, 2013; Morrison, Ma, Weis, & Weis, 1999). For quantitative PCR (qPCR), DNA was extracted using EZ-10 Spin Column Blood DNA Mini-Prep Kit (BioBasic, Inc., Markham, Ontario, Canada). The quantity and quality of DNA for each tissue sample were assessed by measuring the concentration of DNA and the ratio of the UV absorption at 260 to 280 using a Nanodrop 1000 UV/Vis spectrophotometer (ThermoFisher, Waltham, MA). The 280:260 ratio was between 1.75 to 1.85, indicating the lack of contaminating RNA or proteins. qPCR was performed to quantify spirochete loads through amplification of the recA gene as described (Table S4)(Y. P. Lin et al., 2014). Both qRT-PCR and qPCR were performed using an Applied Biosystems 7500 Real-Time PCR system (ThermoFisher) in conjunction with PowerUp SYBR Green Master Mix (ThermoFisher). Cycling parameters were 50°C for 2 minutes, 95°C for 10 minutes, and 45 cycles of 95°C for 15 seconds, and 60°C for 1 minute. Each biological replicate was run in duplicate and checked for intra-run variation. For qPCR, the number of recA copies was calculated by establishing a threshold cycle (Cq) standard curve of a known number of the recA gene extracted from cultivated B. burgdorferi B31-A3. To assure low signals were not due to the presence of PCR inhibitors, five samples from the blood, tibiotarsal joint, and bladder of mice, or the inoculation site and brain of quail were used in qPCR using mouse nidogen primers or quail β-actin (Table S4)(Hart, Yang, Pal, & Lin, 2018; Y. P. Lin et al., 2014). As predicted, we detected 107 copies of the mouse nidogen or quail β-actin gene from 100ng of each DNA sample, ruling out the presence of PCR inhibitors in these samples. For qRT-PCR, the gene expression of cspZ or recA was normalized to that of 16s rRNA using the ΔCq method, where the relative expression of the target (cspZ or recA), normalized to the expression of 16s rRNA, is given by 2−ΔCq, where Cq is the cycle number of the detection threshold (Equation 1).
| (Equation 1) |
Circular dichroism (CD) spectroscopy.
CD analysis was performed on a Jasco 810 spectropolarimeter (Jasco Analytical Instrument, Easton, MD) under nitrogen. CD spectra were measured at room temperature (RT, 25°C) in a 1 mm path length quartz cell. Spectra of 10μM CspZ or CspZ-Y207A/Y211A were recorded in phosphate based saline buffer (PBS) at RT, and three far-UV CD spectra were recorded from 190 to 250nm in 1 nm increments. The background spectrum of PBS without protein was subtracted from the protein spectra. CD spectra were initially analyzed by the software Spectra Manager Program (Jasco). Analysis of spectra to extrapolate secondary structures were performed by Dichroweb (http://www.cryst.bbk.ac.uk/cdweb/html/home) using the K2D and Selcon 3 analysis programs (Y. P. Lin et al., 2009).
ELISA assays.
A ELISA for FH, fibronectin, plasminogen, and laminin binding by CspZ proteins was performed as described (Y. P. Lin et al., 2009). One microgram of BSA (negative control; Sigma-Aldrich, St. Louis, MO), quail FH previously purified from quail serum (Hart et al. PLoS Pathog. 2018), human FH (ComTech, Tyler, Texas), plasma fibronectin, plasma plasminogen, or mouse laminin (Sigma-Aldrich) was coated onto microtiter plate wells. One hundred microliters of increasing concentrations (0.03125, 0.0625, 0.125, 0.25, 0.5, 1, 2μM) of GST (negative control) or GST-tagged CspZ or CspZ-Y207A/Y211A was then added to the wells. Mouse anti-GST tag (ThermoFisher; 1:200x) and HRP-conjugated goat anti-mouse IgG (ThermoFisher; 1:1,000x) were used as primary and secondary antibodies, respectively, to detect the binding of GST-tagged proteins. The plates were washed three times with PBST (0.05% Tween 20 in PBS), and 100μL of tetramethyl benzidine solution (ThermoFisher) was added to each well and incubated for five minutes. The reaction was stopped by adding 100μL of 0.5% hydrosulfuric acid to each well. Plates were read at 405nm using a Tecan Sunrise Microplate reader (Tecan, Morrisville NC). To determine the dissociation constant (KD), the data were fitted with Equation 2 using GraphPad Prism software (GraphPad, La Jolla, CA).
| (Equation 2) |
Shuttle vector construction and plasmid transformation into B. burgdorferi.
cspZ or cspZ-Y207A/Y211A was first PCR amplified with the addition of a SalI site and a BamH1 site at the 5’ and 3’ ends, respectively, using Taq DNA polymerase (Qiagen) and primers listed in Table S4. The unpaired nucleotides at 5’ and 3’ end of the amplified DNA fragments were removed with an exonuclease from CloneJet PCR cloning kit (ThermoFisher) and then inserted into the vector pJET1.2/blunt (ThermoFisher). The plasmids were then digested with SalI and BamH1 to release cspZ and cspZ-Y207A/Y211A, which were then inserted into the SalI and BamH1 sites of pKFSS-1 (Frank, Bundle, Kresge, Eggers, & Samuels, 2003)(Table 2). The promoter region of cspZ from B. burgdorferi B31, 125bp upstream from the start codon of cspZ, was also PCR amplified (Hartmann et al., 2006), added with SphI and SalI sites at the 5’ and 3’ ends, respectively, using primers listed in Table S4. Promoter fragments were then inserted into the SphI and SalI sites of pKFSS-1 to drive the expression of cspZ and cspZ-Y207A/Y211A. Electrocompetent B. burgdorferi B31-A3ΔcspZ prepared as described (Samuels, 1995) was then transformed separately with at least 44 μg of each of the shuttle plasmids (Table 2) and cultured in BSK-II medium at 33°C for 24 hours. Liquid plating transformations were performed in the presence of antibiotic selection (kanamycin and/or streptomycin) as described (Moriarty et al., 2012; Yang, Pal, Alani, Fikrig, & Norgard, 2004). PCR and Sanger sequencing of resulting spirochete strains were performed with primers specific for colE1 and cspZ, respectively, to verify the presence of pKFSS-1 and the insert in the transformants (Table S4). The plasmid profiles of these spirochetes were examined as described (Purser & Norris, 2000) to ascertain identical profiles between these strains and their parental strain B31-A3.
Determination of the viability and generation time of spirochetes.
To determine spirochete viability after blood treatment, B. burgdorferi strains were cultivated in triplicate in untreated or human blood-treated conditions. After resuspending spirochetes in BSK-II medium without rabbit serum, they were stained for 15 minutes with 1x SYBR Green I (ThermoFisher) and 6μM propidium iodide (ThermoFisher) in 0.5% BSA in PBS as described (Feng, Wang, Zhang, Shi, & Zhang, 2014). The live (green) and dead (red) spirochetes were then visualized under overlaid FITC and Texas Red filters using Olympus BX51 fluorescence microscopy (Olympus Corporation, Waltham, MA). An image of four fields of view were taken to determine the proportion of live to dead spirochetes. Additionally, after growing for 48 hours with or without human blood, each of these strains (106 spirochetes/mL) was cultivated BSK II medium without human blood in triplicate to determine the spirochetes’ generation time. The concentration of the spirochetes was calculated microscopically in triplicate every 24 hours. We then calculated the generation time for each strain in exponential phase (G) as previously described (Heroldova, Nemec, & Hubalek, 1998).
Flow cytometry.
CspZ production, FH binding, and MAC deposition on spirochete surface were determined as described (Hart, Nguyen, et al., 2018). To evaluate the surface localization of CspZ, the spirochetes (1 × 108 cells) were washed with HBSC buffer containing glucose and BSA (HBSC-DB, 25mM Hepes acid, 150mM sodium chloride, 1mM MnCl2, 1mM MgCl2, 0.25mM CaCl2, 0.1% glucose, and 0.2% BSA) and then resuspended into the same buffer. To permeabilize spirochetes, they were incubated with 100% methanol for an hour, followed by washed with HBSC-DB. Mouse antiserum raised against CspZ or CspA, or mouse anti-FlaB monoclonal antibody was used as the primary antibody (1:250x). An Alexa 647-conjugated goat anti-mouse IgG (ThermoFisher) (1:250x) was used as the secondary antibody. To determine the ability of these B. burgdorferi strains to bind to FH, spirochetes (1 × 108 cells) were suspended in PBS and then incubated with 1 μg of human, mouse, or quail FH at 25°C for one hour. The spirochetes were then washed with PBS and resuspended in HBSC-DB. A sheep anti-FH polyclonal IgG (ThermoFisher) (1:250x) or a mouse anti-FH monoclonal antibody VIG8 (1:250x) was used as the primary antibody. An Alexa 647-conjugated donkey anti-sheep IgG (1:250x) or goat anti-mouse IgG (ThermoFisher) (1:250x) was used as the secondary antibody. To measure MAC deposition, spirochetes (1 × 108 cells) were washed by PBS, resuspended in the same buffer, and then incubated with human or mouse sera in a final concentration as 20% at 25°C for one hour. Note that more than 80% of B. burgdorferi strains are capable of surviving in this concentration of serum (Breitner-Ruddock et al., 1997; Kraiczy, Hunfeld, Breitner-Ruddock, et al., 2000). The spirochetes were then washed with PBS and resuspended in HBSC-DB. A mouse anti-MAC monoclonal antibody aE11 (1:250x) (ThermoFisher), or a rabbit anti-MAC polyclonal IgG (1:250x) (ThermoFisher) was used as the primary antibody. An Alexa 647-conjugated goat anti-rabbit (ThermoFisher) or a goat anti-mouse IgG (ThermoFisher) (1:250x) was used as the secondary antibody.
After staining, formalin (0.1%) was then added for fixing. The resulting fluorescence intensity of spirochetes was measured by flow cytometry using a FACSCalibur (BD Bioscience). All flow cytometry experiments were performed within two days of preparing B. burgdorferi samples. Spirochetes in the suspension were distinguished by their distinct light scattering properties in the flow cytometer equipped with a 15mW, 488nm air-cooled argon laser, a standard three-color filter arrangement, and CELLQuest™ Software (BD Bioscience). Unstained B. burgdorferi strain B31-A3 was applied to ensure accurate gating, in which the aggregated spirochetes were eliminated as described (Hart, Nguyen, et al., 2018). Additionally, the spirochetes incubated with only the secondary antibody as control experiment to ascertain the specificity of primary antibody to bind to the cognate antigen. The mean fluorescence index (MFI) of each sample was obtained from FlowJo software (Thee-Star Inc., Ashland, OR) representing the surface production of the indicated proteins. Each standard deviation of mean value was no more than 7% of its mean value.
Serum susceptibility assay.
The serum susceptibility of B. burgdorferi was measured as described (Alitalo et al., 2001). Briefly, triplicate samples of each strain were grown to mid-log phase and diluted to a final concentration of 5 × 106 bacteria per milliliter into BSK-II medium without rabbit serum, plus a final concentration of 40% human or quail serum or C3-depleted human serum (ComTech). We also included heat-inactivated serum from these hosts, which was incubated at 55°C for two hours prior to incubation with spirochetes. Immediately after and four hours after incubation, an aliquot was taken from each replicate and counted by a Petroff- Hausser counting chamber (Hausser Scientific, Horsham, PA) using a Nikon Eclipse E600 darkfield microscope (Nikon, Melville, NY). The percentage of survival for B. burgdorferi was calculated using the number of mobile spirochetes at four hours post incubation normalized to that immediately after adding the serum.
Mouse and quail infection experiments.
Four-week-old female BALB/c mice or C3−/− mice in a BALB/c background or Coturnix coturnix quail were subcutaneously infected with 103 (for mouse infection) or 106 (for quail infection) of B. burgdorferi strains as described (Y. P. Lin et al., 2014). The number of quail and mouse used in each experiment is described in respective figure legends. The plasmid profiles and the presence of the shuttle vector of each of these B. burgdorferi strains were verified prior to infection as described (Purser & Norris, 2000). Mice were sacrificed at 7 dpi to collect the blood, and 7, or 14 dpi to collect the inoculation site of skin, tibiotarsal joints, bladder, and heart. Quail were sacrificed at 3 and 7 dpi to collect the blood, inoculation site of skin, liver, heart, and brain. Animal tissues were used to quantitatively evaluate the levels of colonization during infection (see section “Quantitative RT-PCR and PCR”). Note that quail blood and tissues were also incubated in BSK-II complete medium at 33°C for four weeks to cultivate spirochetes microscopically to verify the viability of spirochetes (Liveris et al., 2002).
Statistical analysis.
Significant differences between samples were determined using the Kruskal-Wallis test with Dunn’s multiple comparison, or the Mann-Whitney test. A p-value < 0.05 was considered to be significant.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Roxie Girardin, John Leong, and Xiuli Yang for valuable technical advice, and Thomas Hart for critical reading of the manuscript. We also thank Utpal Pal for providing B. burgdorferi strain B31-A3ΔcspZ, Jorge Benach for providing the α-FlaB CB1 monoclonal antibody, Susan Madison-Antenucci allowing us to use her fluorescence microscope, and Noel Espina for the assistance with that microscope. Additionally, we thank Leslie Eisele and Renjie Song of Wadsworth Biochemistry and Immunology Core for assistance with circular dichroism and flow cytometry, Karen Chave of the Wadsworth Protein Expression Core for purifying FH proteins, and Media & Tissue Culture Core for preparation of Borrelia and E. coli culture medium. This work was supported by NIH R01AI121401 (to P.K. and Y.L.), NSF IOS1755286 (to Y.L., A.L.M., M.Z.C., L.D.K., A.P.D.), DoD TB170111 (to Y.L., M.Z.C., and A.L.M.), NSF DBI1757170 (to M.Z.C.), and New York State Department of Health Wadsworth Center Start-Up Grant (to Y.L., M.Z.C., and A.L.M.). The authors have no conflict of interest to declare.
REFERENCES
- Alitalo A, Meri T, Comstedt P, Jeffery L, Tornberg J, Strandin T, … Meri S (2005). Expression of complement factor H binding immunoevasion proteins in Borrelia garinii isolated from patients with neuroborreliosis. European journal of immunology, 35 (10), 3043–3053. doi: 10.1002/eji.200526354 [DOI] [PubMed] [Google Scholar]
- Alitalo A, Meri T, Ramo L, Jokiranta TS, Heikkila T, Seppala IJ, … Meri S (2001). Complement evasion by Borrelia burgdorferi: serum-resistant strains promote C3b inactivation. Infection and immunity, 69 (6), 3685–3691. doi: 10.1128/IAI.69.6.3685-3691.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonara S, Ristow L, McCarthy J, & Coburn J (2010). Effect of Borrelia burgdorferi OspC at the site of inoculation in mouse skin. Infection and immunity, 78 (11), 4723–4733. doi: 10.1128/IAI.00464-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit VM, Fischer JR, Lin YP, Parveen N, & Leong JM (2011). Allelic variation of the Lyme disease spirochete adhesin DbpA influences spirochetal binding to decorin, dermatan sulfate, and mammalian cells. Infection and immunity, 79 (9), 3501–3509. doi: 10.1128/IAI.00163-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhide MR, Travnicek M, Levkutova M, Curlik J, Revajova V, & Levkut M (2005). Sensitivity of Borrelia genospecies to serum complement from different animals and human: a host-pathogen relationship. FEMS immunology and medical microbiology, 43 (2), 165–172. doi: 10.1016/j.femsim.2004.07.012 [DOI] [PubMed] [Google Scholar]
- Bishop KL, Khan MI, & Nielsen SW (1994). Experimental infection of northern bobwhite quail with Borrelia burgdorferi. Journal of wildlife diseases, 30 (4), 506–513. doi: 10.7589/0090-3558-30.4.506 [DOI] [PubMed] [Google Scholar]
- Blevins JS, Hagman KE, & Norgard MV (2008). Assessment of decorin-binding protein A to the infectivity of Borrelia burgdorferi in the murine models of needle and tick infection. BMC microbiology, 8, 82. doi: 10.1186/1471-2180-8-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom AM, Hallstrom T, & Riesbeck K (2009). Complement evasion strategies of pathogens-acquisition of inhibitors and beyond. Molecular immunology, 46 (14), 2808–2817. doi: 10.1016/j.molimm.2009.04.025 [DOI] [PubMed] [Google Scholar]
- Breitner-Ruddock S, Wurzner R, Schulze J, & Brade V (1997). Heterogeneity in the complement-dependent bacteriolysis within the species of Borrelia burgdorferi. Medical microbiology and immunology, 185 (4), 253–260. [DOI] [PubMed] [Google Scholar]
- Brisson D, Drecktrah D, Eggers CH, & Samuels DS (2012). Genetics of Borrelia burgdorferi. Annual review of genetics, 46, 515–536. doi: 10.1146/annurev-genet-011112-112140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CS, Hefty PS, Jolliff SE, & Akins DR (2003). Global analysis of Borrelia burgdorferi genes regulated by mammalian host-specific signals. Infection and immunity, 71 (6), 3371–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CS, Vuppala SR, Jett AM, Alitalo A, Meri S, & Akins DR (2005). Complement regulator-acquiring surface protein 1 imparts resistance to human serum in Borrelia burgdorferi. Journal of immunology, 175 (5), 3299–3308. [DOI] [PubMed] [Google Scholar]
- Burgess EC (1989). Experimental inoculation of mallard ducks (Anas platyrhynchos platyrhynchos) with Borrelia burgdorferi. Journal of wildlife diseases, 25 (1), 99–102. doi: 10.7589/0090-3558-25.1.99 [DOI] [PubMed] [Google Scholar]
- Bykowski T, Woodman ME, Cooley AE, Brissette CA, Brade V, Wallich R, … Stevenson B (2007). Coordinated expression of Borrelia burgdorferi complement regulator-acquiring surface proteins during the Lyme disease spirochete’s mammal-tick infection cycle. Infection and immunity, 75 (9), 4227–4236. doi: 10.1128/IAI.00604-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caine JA, & Coburn J (2015). A short-term Borrelia burgdorferi infection model identifies tissue tropisms and bloodstream survival conferred by adhesion proteins. Infection and immunity, 83 (8), 3184–3194. doi: 10.1128/IAI.00349-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caine JA, Lin YP, Kessler JR, Sato H, Leong JM, & Coburn J (2017). Borrelia burgdorferi outer surface protein C (OspC) binds complement component C4b and confers bloodstream survival. Cellular microbiology. doi: 10.1111/cmi.12786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman AS, Yang X, Kumar M, Zhang X, Promnares K, Shroder D, … Pal U (2008). Borrelia burgdorferi complement regulator-acquiring surface protein 2 does not contribute to complement resistance or host infectivity. PloS one, 3 (8), 3010e. doi: 10.1371/journal.pone.0003010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig-Mylius KA, Lee M, Jones KL, & Glickstein LJ (2009). Arthritogenicity of Borrelia burgdorferi and Borrelia garinii: comparison of infection in mice. The American journal of tropical medicine and hygiene, 80 (2), 252–258. [PubMed] [Google Scholar]
- Dowdell AS, Murphy MD, Azodi C, Swanson SK, Florens L, Chen S, & Zuckert WR (2017). Comprehensive Spatial Analysis of the Borrelia burgdorferi Lipoproteome Reveals a Compartmentalization Bias toward the Bacterial Surface. Journal of bacteriology, 199 (6). doi: 10.1128/JB.00658-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen RJ, & Eisen L (2018). The Blacklegged Tick, Ixodes scapularis: An Increasing Public Health Concern. Trends in parasitology, 34 (4), 295–309. doi: 10.1016/j.pt.2017.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Babb K, Carroll JA, Lindstrom N, Fischer ER, Miller JC, … Stevenson B (2001). Surface exposure and protease insensitivity of Borrelia burgdorferi Erp (OspEF- related) lipoproteins. Microbiology, 147 (Pt 4), 821–830. doi: 10.1099/00221287-147-4-821 [DOI] [PubMed] [Google Scholar]
- Elias AF, Stewart PE, Grimm D, Caimano MJ, Eggers CH, Tilly K, … Rosa P (2002). Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infection and immunity, 70 (4), 2139–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exner MM, Wu X, Blanco DR, Miller JN, & Lovett MA (2000). Protection elicited by native outer membrane protein Oms66 (p66) against host-adapted Borrelia burgdorferi: conformational nature of bactericidal epitopes. Infection and immunity, 68 (5), 2647–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Wang T, Zhang S, Shi W, & Zhang Y (2014). An optimized SYBR Green I/PI assay for rapid viability assessment and antibiotic susceptibility testing for Borrelia burgdorferi. PloS one, 9 (11), e111809. doi: 10.1371/journal.pone.0111809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KL, Bundle SF, Kresge ME, Eggers CH, & Samuels DS (2003). aadA confers streptomycin resistance in Borrelia burgdorferi. Journal of bacteriology, 185 (22), 6723–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia BL, Zhi H, Wager B, Hook M, & Skare JT (2016). Borrelia burgdorferi BBK32 Inhibits the Classical Pathway by Blocking Activation of the C1 Complement Complex. PLoS pathogens, 12 (1), e1005404. doi: 10.1371/journal.ppat.1005404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg HS, Buckley PA, Balmforth MG, Zhioua E, Mitra S, & Buckley FG (2005). Reservoir competence of native North American birds for the lyme disease spirochete, Borrelia burgdorfieri. Journal of medical entomology, 42 (3), 445–449. doi: 10.1603/0022-2585(2005)042[0445:RCONNA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Hallstrom T, Haupt K, Kraiczy P, Hortschansky P, Wallich R, Skerka C, & Zipfel PF (2010). Complement regulator-acquiring surface protein 1 of Borrelia burgdorferi binds to human bone morphogenic protein 2, several extracellular matrix proteins, and plasminogen. The Journal of infectious diseases, 202 (3), 490–498. doi: 10.1086/653825 [DOI] [PubMed] [Google Scholar]
- Hallstrom T, Siegel C, Morgelin M, Kraiczy P, Skerka C, & Zipfel PF (2013). CspA from Borrelia burgdorferi inhibits the terminal complement pathway. mBio, 4 (4). doi: 10.1128/mBio.00481-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdy BH (1977). Biochemical and physiological studies of certain ticks (Ixodoidea). Excretion during ixodid feeding. Journal of medical entomology, 14 (1), 15–18. [DOI] [PubMed] [Google Scholar]
- Hart T, Nguyen NTT, Nowak NA, Zhang F, Linhardt RJ, Diuk-Wasser M, … Lin YP (2018). Polymorphic factor H-binding activity of CspA protects Lyme borreliae from the host complement in feeding ticks to facilitate tick-to-host transmission. PLoS pathogens, 14 (5), e1007106. doi: 10.1371/journal.ppat.1007106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Yang X, Pal U, & Lin YP (2018). Identification of Lyme borreliae proteins promoting vertebrate host blood-specific spirochete survival in Ixodes scapularis nymphs using artificial feeding chambers. Ticks and tick-borne diseases. doi: 10.1016/j.ttbdis.2018.03.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann K, Corvey C, Skerka C, Kirschfink M, Karas M, Brade V, … Kraiczy P (2006). Functional characterization of BbCRASP-2, a distinct outer membrane protein of Borrelia burgdorferi that binds host complement regulators factor H and FHL-1. Molecular microbiology, 61 (5), 1220–1236. doi: 10.1111/j.1365-2958.2006.05318.x [DOI] [PubMed] [Google Scholar]
- Heroldova M, Nemec M, & Hubalek Z (1998). Growth parameters of Borrelia burgdorferi sensu stricto at various temperatures. Zentralblatt fur Bakteriologie : international journal of medical microbiology, 288 (4), 451–455. [DOI] [PubMed] [Google Scholar]
- Hinckley AF, Connally NP, Meek JI, Johnson BJ, Kemperman MM, Feldman KA, … Mead PS (2014). Lyme disease testing by large commercial laboratories in the United States. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America, 59 (5), 676–681. doi: 10.1093/cid/ciu397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramatsu A, & Ouchi T (1963). On the Proteolytic Enzymes from the Commercial Protease Preparation of Streptomyces Griseus (Pronase P). Journal of biochemistry, 54, 462–464. [DOI] [PubMed] [Google Scholar]
- Hodzic E, Feng S, & Barthold SW (2013). Assessment of transcriptional activity of Borrelia burgdorferi and host cytokine genes during early and late infection in a mouse model. Vector borne and zoonotic diseases, 13 (10), 694–711. doi: 10.1089/vbz.2012.1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovius JW, Bijlsma MF, van der Windt GJ, Wiersinga WJ, Boukens BJ, Coumou J, … van der Poll T (2009). The urokinase receptor (uPAR) facilitates clearance of Borrelia burgdorferi. PLoS pathogens, 5 (5), e1000447. doi: 10.1371/journal.ppat.1000447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde JA, Trzeciakowski JP, & Skare JT (2007). Borrelia burgdorferi alters its gene expression and antigenic profile in response to CO2 levels. Journal of bacteriology, 189 (2), 437–445. doi: 10.1128/JB.01109-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde JA, Weening EH, Chang M, Trzeciakowski JP, Hook M, Cirillo JD, & Skare JT (2011). Bioluminescent imaging of Borrelia burgdorferi in vivo demonstrates that the fibronectin-binding protein BBK32 is required for optimal infectivity. Molecular microbiology, 82 (1), 99–113. doi: 10.1111/j.1365-2958.2011.07801.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai E, Tanaka S, Braga IS 3rd, Itakura C, Isogai H, Kimura K, & Fujii N (1994). Experimental Borrelia garinii infection of Japanese quail. Infection and immunity, 62 (8), 3580–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KL, Glickstein LJ, Damle N, Sikand VK, McHugh G, & Steere AC (2006). Borrelia burgdorferi genetic markers and disseminated disease in patients with early Lyme disease. Journal of clinical microbiology, 44 (12), 4407–4413. doi: 10.1128/JCM.01077-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenedy MR, & Akins DR (2011). The OspE-related proteins inhibit complement deposition and enhance serum resistance of Borrelia burgdorferi, the lyme disease spirochete. Infection and immunity, 79 (4), 1451–1457. doi: 10.1128/IAI.01274-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenedy MR, Lenhart TR, & Akins DR (2012). The role of Borrelia burgdorferi outer surface proteins. FEMS immunology and medical microbiology, 66 (1), 1–19. doi: 10.1111/j.1574-695X.2012.00980.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenedy MR, Vuppala SR, Siegel C, Kraiczy P, & Akins DR (2009). CspA-mediated binding of human factor H inhibits complement deposition and confers serum resistance in Borrelia burgdorferi. Infection and immunity, 77 (7), 2773–2782. doi: 10.1128/IAI.00318-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingry LC, Batra D, Replogle A, Rowe LA, Pritt BS, & Petersen JM (2016). Whole Genome Sequence and Comparative Genomics of the Novel Lyme Borreliosis Causing Pathogen, Borrelia mayonii. PloS one, 11 (12), e0168994. doi: 10.1371/journal.pone.0168994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraiczy P (2016a). Hide and Seek: How Lyme Disease Spirochetes Overcome Complement Attack. Frontiers in immunology, 7, 385. doi: 10.3389/fimmu.2016.00385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraiczy P (2016b). Travelling between Two Worlds: Complement as a Gatekeeper for an Expanded Host Range of Lyme Disease Spirochetes. Vet. Sci, 3 (2), 12–26. doi: doi: 10.3390/vetsci3020012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraiczy P, Hellwage J, Skerka C, Becker H, Kirschfink M, Simon MM, … Wallich R (2004). Complement resistance of Borrelia burgdorferi correlates with the expression of BbCRASP-1, a novel linear plasmid-encoded surface protein that interacts with human factor H and FHL-1 and is unrelated to Erp proteins. The Journal of biological chemistry, 279 (4), 2421–2429. doi: 10.1074/jbc.M308343200 [DOI] [PubMed] [Google Scholar]
- Kraiczy P, Hunfeld KP, Breitner-Ruddock S, Wurzner R, Acker G, & Brade V (2000). Comparison of two laboratory methods for the determination of serum resistance in Borrelia burgdorferi isolates. Immunobiology, 201 (3-4), 406–419. doi: 10.1016/S0171-2985(00)80094-7 [DOI] [PubMed] [Google Scholar]
- Kraiczy P, Hunfeld KP, Peters S, Wurzner R, Ackert G, Wilske B, & Brade V (2000). Borreliacidal activity of early Lyme disease sera against complement-resistant Borrelia afzelii FEM1 wild-type and an OspC-lacking FEM1 variant. Journal of medical microbiology, 49 (10), 917–928. doi : 10.1099/0022-1317-49-10-917 [DOI] [PubMed] [Google Scholar]
- Kraiczy P, Seling A, Brissette CA, Rossmann E, Hunfeld KP, Bykowski T, … Stevenson B (2008). Borrelia burgdorferi complement regulator-acquiring surface protein 2 (CspZ) as a serological marker of human Lyme disease. Clinical and vaccine immunology : CVI, 15 (3), 484–491. doi: 10.1128/CVI.00415-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraiczy P, Skerka C, Brade V, & Zipfel PF (2001). Further characterization of complement regulator-acquiring surface proteins of Borrelia burgdorferi. Infection and immunity, 69 (12), 7800–7809. doi: 10.1128/IAI.69.12.7800-7809.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraiczy P, & Stevenson B (2013). Complement regulator-acquiring surface proteins of Borrelia burgdorferi: Structure, function and regulation of gene expression. Ticks and tick-borne diseases, 4 (1–2), 26–34. doi: 10.1016/j.ttbdis.2012.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Ristow LC, Shi M, Mukherjee P, Caine JA, Lee WY, … Chaconas G (2015). Intravital Imaging of Vascular Transmigration by the Lyme Spirochete: Requirement for the Integrin Binding Residues of the B. burgdorferi P66 Protein. PLoSpathogens, 11 (12), e1005333. doi: 10.1371/journal.ppat.1005333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumru OS, Schulze RJ, Slusser JG, & Zuckert WR (2010). Development and validation of a FACS-based lipoprotein localization screen in the Lyme disease spirochete Borrelia burgdorferi. BMC microbiology, 10, 277. doi: 10.1186/1471-2180-10-277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtenbach K, De Michelis S, Etti S, Schafer SM, Sewell HS, Brade V, & Kraiczy P (2002). Host association of Borrelia burgdorferi sensu lato--the key role of host complement. Trends in microbiology, 10 (2), 74–79. [DOI] [PubMed] [Google Scholar]
- Kurtenbach K, Peacey M, Rijpkema SG, Hoodless AN, Nuttall PA, & Randolph SE (1998). Differential transmission of the genospecies of Borrelia burgdorferi sensu lato by game birds and small rodents in England. Applied and environmental microbiology, 64 (4), 1169–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtenbach K, Schafer SM, Sewell HS, Peacey M, Hoodless A, Nuttall PA, & Randolph SE (2002). Differential survival of Lyme borreliosis spirochetes in ticks that feed on birds. Infection and immunity, 70 (10), 5893–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann PJ (2010). Preparing serum for functional complement assays. Journal of immunological methods, 352 (1–2), 195–197. doi: 10.1016/j.jim.2009.11.003 [DOI] [PubMed] [Google Scholar]
- Lambris JD, Ricklin D, & Geisbrecht BV (2008). Complement evasion by human pathogens. Nature reviews. Microbiology, 6 (2), 132–142. doi: 10.1038/nrmicro1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrenz MB, Hardham JM, Owens RT, Nowakowski J, Steere AC, Wormser GP, & Norris SJ (1999). Human antibody responses to VlsE antigenic variation protein of Borrelia burgdorferi. Journal of clinical microbiology, 37 (12), 3997–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrenz MB, Wooten RM, Zachary JF, Drouin SM, Weis JJ, Wetsel RA, & Norris SJ (2003). Effect of complement component C3 deficiency on experimental Lyme borreliosis in mice. Infection and immunity, 71 (8), 4432–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limberger RJ (2004). The periplasmic flagellum of spirochetes. Journal of molecular microbiology and biotechnology, 7 (1–2), 30–40. doi: 10.1159/000077867 [DOI] [PubMed] [Google Scholar]
- Lin T, Gao L, Zhang C, Odeh E, Jacobs MB, Coutte L, … Norris SJ (2012). Analysis of an ordered, comprehensive STM mutant library in infectious Borrelia burgdorferi: insights into the genes required for mouse infectivity. PloS one, 7 (10), e47532. doi: 10.1371/journal.pone.0047532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YP, Benoit V, Yang X, Martinez-Herranz R, Pal U, & Leong JM (2014). Strain- specific variation of the decorin-binding adhesin DbpA influences the tissue tropism of the lyme disease spirochete. PLoS pathogens, 10 (7), e1004238. doi: 10.1371/journal.ppat.1004238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YP, Chen Q, Ritchie JA, Dufour NP, Fischer JR, Coburn J, & Leong JM (2015). Glycosaminoglycan binding by Borrelia burgdorferi adhesin BBK32 specifically and uniquely promotes joint colonization. Cellular microbiology, 17 (6), 860–875. doi: 10.1m/cmi.12407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YP, Greenwood A, Nicholson LK, Sharma Y, McDonough SP, & Chang YF (2009). Fibronectin binds to and induces conformational change in a disordered region of leptospiral immunoglobulin-like protein B. The Journal of biological chemistry, 284 (35), 23547–23557. doi: 10.1074/jbc.M109.031369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liveris D, Wang G, Girao G, Byrne DW, Nowakowski J, McKenna D, … Schwartz I (2002). Quantitative detection of Borrelia burgdorferi in 2-millimeter skin samples of erythema migrans lesions: correlation of results with clinical and laboratory findings. Journal of clinical microbiology, 40 (4), 1249–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinkiewicz A, Kraiczy P, & Lin Y-P (2017). There is a method to the madness: Strategies to study host complement evasion by Lyme disease and relapsing fever spirochetes. Frontiers in microbiology, 8 (328). doi: 10.3389/fmicb.2017.00328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinkiewicz AL, Lieknina I, Kotelovica S, Yang X, Kraiczy P, Pal U, … Tars K (2018). Eliminating Factor H-Binding Activity of Borrelia burgdorferi CspZ Combined with Virus-Like Particle Conjugation Enhances Its Efficacy as a Lyme Disease Vaccine. Frontiers in immunology, 9, 181. doi: 10.3389/fimmu.2018.00181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell JV, Frederick J, Miller DP, Goetting-Minesky MP, Goodman H, Fenno JC, & Marconi RT (2011). Identification of the primary mechanism of complement evasion by the periodontal pathogen, Treponema denticola. Molecular oral microbiology, 26 (2), 140–149. doi: 10.1111/j.2041-1014.2010.00598.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell JV, Wolfgang J, Tran E, Metts MS, Hamilton D, & Marconi RT (2003). Comprehensive analysis of the factor h binding capabilities of borrelia species associated with lyme disease: delineation of two distinct classes of factor h binding proteins. Infection and immunity, 71 (6), 3597–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meri S (2016). Self-nonself discrimination by the complement system. FEBS letters, 590 (15), 2418–2434. doi: 10.1002/1873-3468.12284 [DOI] [PubMed] [Google Scholar]
- Merle NS, Church SE, Fremeaux-Bacchi V, & Roumenina LT (2015). Complement System Part I - Molecular Mechanisms of Activation and Regulation. Frontiers in immunology, 6, 262. doi: 10.3389/fimmu.2015.00262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, & Roumenina LT (2015). Complement System Part II: Role in Immunity. Frontiers in immunology, 6, 257. doi: 10.3389/fimmu.2015.00257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty TJ, Shi M, Lin YP, Ebady R, Zhou H, Odisho T, … Chaconas G (2012). Vascular binding of a pathogen under shear force through mechanistically distinct sequential interactions with host macromolecules. Molecular microbiology, 86 (5), 1116–1131. doi: 10.1111/mmi.12045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison TB, Ma Y, Weis JH, & Weis JJ (1999). Rapid and sensitive quantification of Borrelia burgdorferi-infected mouse tissues by continuous fluorescent monitoring of PCR. Journal of clinical microbiology, 37 (4), 987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman MU, Moriarty TJ, Dresser AR, Millen B, Kubes P, & Chaconas G (2008). Molecular mechanisms involved in vascular interactions of the Lyme disease pathogen in a living host. PLoS pathogens, 4 (10), e1000169. doi: 10.1371/journal.ppat.1000169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojaimi C, Brooks C, Casjens S, Rosa P, Elias A, Barbour A, … Schwartz I (2003). Profiling of temperature-induced changes in Borrelia burgdorferi gene expression by using whole genome arrays. Infection and immunity, 71 (4), 1689–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen B, Gylfe A, & Bergstrom S (1996). Canary finches (Serinus canaria) as an avian infection model for Lyme borreliosis. Microbial pathogenesis, 20 (6), 319–324. doi: 10.1006/mpat.1996.0030 [DOI] [PubMed] [Google Scholar]
- Parveen N, & Leong JM (2000). Identification of a candidate glycosaminoglycan-binding adhesin of the Lyme disease spirochete Borrelia burgdorferi. Molecular microbiology, 35 (5), 1220–1234. [DOI] [PubMed] [Google Scholar]
- Piesman J, Dolan MC, Schriefer ME, & Burkot TR (1996). Ability of experimentally infected chickens to infect ticks with the Lyme disease spirochete, Borrelia burgdorferi. The American journal of tropical medicine and hygiene, 54 (3), 294–298. [DOI] [PubMed] [Google Scholar]
- Purser JE, & Norris SJ (2000). Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proceedings of the National Academy of Sciences of the United States of America, 97 (25), 13865–13870. doi: 10.1073/pnas.97.25.13865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radolf JD, Caimano MJ, Stevenson B, & Hu LT (2012). Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nature reviews. Microbiology, 10 (2), 87–99. doi: 10.1038/nrmicro2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revel AT, Talaat AM, & Norgard MV (2002). DNA microarray analysis of differential gene expression in Borrelia burgdorferi, the Lyme disease spirochete. Proceedings of the National Academy of Sciences of the United States of America, 99 (3), 1562–1567. doi: 10.1073/pnas.032667699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter D, Spielman A, Komar N, & Matuschka FR (2000). Competence of American robins as reservoir hosts for Lyme disease spirochetes. Emerging infectious diseases, 6 (2), 133–138. doi: 10.3201/eid0602.000205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow LC, Miller HE, Padmore LJ, Chettri R, Salzman N, Caimano MJ, … Coburn J (2012). The beta(3)-integrin ligand of Borrelia burgdorferi is critical for infection of mice but not ticks. Molecular microbiology, 85 (6), 1105–1118. doi: 10.1111/j.1365-2958.2012.08160.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roantree RJ, & Rantz LA (1960). A Study of the Relationship of the Normal Bactericidal Activity of Human Serum to Bacterial Infection. The Journal of clinical investigation, 39 (1), 72–81. doi: 10.1172/JCI104029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers EA, Abdunnur SV, McDowell JV, & Marconi RT (2009). Comparative analysis of the properties and ligand binding characteristics of CspZ, a factor H binding protein, derived from Borrelia burgdorferi isolates of human origin. Infection and immunity, 77 (10), 4396–4405. doi: 10.1128/IAI.00393-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers EA, & Marconi RT (2007). Delineation of species-specific binding properties of the CspZ protein (BBH06) of Lyme disease spirochetes: evidence for new contributions to the pathogenesis of Borrelia spp. Infection and immunity, 75 (11), 5272–5281. doi: 10.1128/IAI.00850-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels DS (1995). Electrotransformation of the spirochete Borrelia burgdorferi. Methods in molecular biology, 47, 253–259. doi: 10.1385/0-89603-310-4:253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshu J, Boylan JA, Gherardini FC, & Skare JT (2004). Dissolved oxygen levels alter gene expression and antigen profiles in Borrelia burgdorferi. Infection and immunity, 72 (3), 1580–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshu J, Esteve-Gassent MD, Labandeira-Rey M, Kim JH, Trzeciakowski JP, Hook M, & Skare JT (2006). Inactivation of the fibronectin-binding adhesin gene bbk32 significantly attenuates the infectivity potential of Borrelia burgdorferi. Molecular microbiology, 59 (5), 1591–1601. doi: 10.1111/j.1365-2958.2005.05042.x [DOI] [PubMed] [Google Scholar]
- Shi Y, Xu Q, Seemanaplli SV, McShan K, & Liang FT (2008). Common and unique contributions of decorin-binding proteins A and B to the overall virulence of Borrelia burgdorferi. PloS one, 3 (10), e3340. doi: 10.1371/journal.pone.0003340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showman AC, Aranjuez G, Adams PP, & Jewett MW (2016). Gene bb0318 Is Critical for the Oxidative Stress Response and Infectivity of Borrelia burgdorferi. Infection and immunity, 84 (11), 3141–3151. doi: 10.1128/IAI.00430-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel C, Schreiber J, Haupt K, Skerka C, Brade V, Simon MM, … Kraiczy P (2008). Deciphering the ligand-binding sites in the Borrelia burgdorferi complement regulator-acquiring surface protein 2 required for interactions with the human immune regulators factor H and factor H-like protein 1. The Journal of biological chemistry, 283 (50), 34855–34863. doi: 10.1074/jbc.M805844200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoberg AP, Trouw LA, & Blom AM (2009). Complement activation and inhibition: a delicate balance. Trends in immunology, 30 (2), 83–90. doi: 10.1016/j.it.2008.11.003 [DOI] [PubMed] [Google Scholar]
- Sojka D, Franta Z, Horn M, Caffrey CR, Mares M, & Kopacek P (2013). New insights into the machinery of blood digestion by ticks. Trends in parasitology, 29 (6), 276–285. doi: 10.1016/j.pt.2013.04.002 [DOI] [PubMed] [Google Scholar]
- Steere AC, Strle F, Wormser GP, Hu LT, Branda JA, Hovius JW, … Mead PS (2016). Lyme borreliosis. Nature reviews. Disease primers, 2, 16090. doi: 10.1038/nrdp.2016.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly K, Krum JG, Bestor A, Jewett MW, Grimm D, Bueschel D, … Rosa P (2006). Borrelia burgdorferi OspC protein required exclusively in a crucial early stage of mammalian infection. Infection and immunity, 74 (6), 3554–3564. doi: 10.1128/IAI.01950-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarz R, Anderton JM, Katona LI, & Benach JL (2004). Combined effects of blood and temperature shift on Borrelia burgdorferi gene expression as determined by whole genome DNA array. Infection and immunity, 72 (9), 5419–5432. doi: 10.1128/IAI.72.9.5419-5432.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam AP, Oei A, Jaspars R, Fijen C, Wilske B, Spanjaard L, & Dankert J (1997). Complement-mediated serum sensitivity among spirochetes that cause Lyme disease. Infection and immunity, 65 (4), 1228–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, van Dam AP, Schwartz I, & Dankert J (1999). Molecular typing of Borrelia burgdorferi sensu lato: taxonomic, epidemiological, and clinical implications. Clinical microbiology reviews, 12 (4), 633–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weening EH, Parveen N, Trzeciakowski JP, Leong JM, Hook M, & Skare JT (2008). Borrelia burgdorferi lacking DbpBA exhibits an early survival defect during experimental infection. Infection and immunity, 76 (12), 5694–5705. doi: 10.1128/IAI.00690-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasekara S, Bunikis J, Wysocki V, & Barbour AG (2008). Identification of residual blood proteins in ticks by mass spectrometry proteomics. Emerging infectious diseases, 14 (8), 1273–1275. doi: 10.3201/eid1408.080227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm DL (1973). Mechanisms responsible for increased vascular permeability in acute inflammation. Agents and actions, 3 (5), 297–306. [DOI] [PubMed] [Google Scholar]
- Woodman ME, Cooley AE, Miller JC, Lazarus JJ, Tucker K, Bykowski T, … Stevenson B (2007). Borrelia burgdorferi binding of host complement regulator factor H is not required for efficient mammalian infection. Infection and immunity, 75 (6), 3131–3139. doi: 10.1128/IAI.01923-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Seemanapalli SV, Reif KE, Brown CR, & Liang FT (2007). Increasing the recruitment of neutrophils to the site of infection dramatically attenuates Borrelia burgdorferi infectivity. Journal of immunology, 178 (8), 5109–5115. [DOI] [PubMed] [Google Scholar]
- Yang XF, Pal U, Alani SM, Fikrig E, & Norgard MV (2004). Essential role for OspA/B in the life cycle of the Lyme disease spirochete. The Journal of experimental medicine, 199 (5), 641–648. doi: 10.1084/jem.20031960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi H, Weening EH, Barbu EM, Hyde JA, Hook M, & Skare JT (2015). The BBA33 lipoprotein binds collagen and impacts Borrelia burgdorferi pathogenesis. Molecular microbiology. doi: 10.1111/mmi.12921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi H, Xie J, & Skare JT (2018). The Classical Complement Pathway Is Required to Control Borrelia burgdorferi Levels During Experimental Infection. Frontiers in immunology, 9, 959. doi: 10.3389/fimmu.2018.00959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel PF, & Skerka C (2009). Complement regulators and inhibitory proteins. Nature reviews. Immunology, 9 (10), 729–740. doi: 10.1038/nri2620 [DOI] [PubMed] [Google Scholar]
- Zuckert WR, Kerentseva TA, Lawson CL, & Barbour AG (2001). Structural conservation of neurotropism-associated VspA within the variable Borrelia Vsp-OspC lipoprotein family. The Journal of biological chemistry, 276 (1), 457–463. doi: 10.1074/jbc.M008449200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.