

Abstract
Quantitative systems modeling aims to integrate knowledge in different research areas with models describing biological mechanisms and dynamics to gain a better understanding of complex clinical syndromes. Heart failure (HF) is a chronic complex cardiac disease that results from structural or functional disorders impairing the ability of the ventricle to fill with or eject blood. Highly interactive and dynamic changes in mechanical, structural, neurohumoral, metabolic, and electrophysiological properties collectively predispose the failing heart to cardiac arrhythmias, which are responsible for about a half of HF deaths. Multi-scale cardiac modeling and simulation integrates structural and functional data from HF experimental models and patients to improve our mechanistic understanding of this complex arrhythmia syndrome. In particular, it allows investigating how disease-induced remodeling alters the coupling of electrophysiology, Ca2+ and Na+ handling, contraction, and energetics that lead to rhythm derangements. The Ca2+/calmodulin dependent protein kinase II, which expression and activity are enhanced in HF, emerges as a critical hub that modulates the feedbacks between these various subsystems and promotes arrhythmogenesis.
Graphical/Visual Abstract and Caption
Structural and functional data integration via multi-scale quantitative cardiac modeling illuminates arrhythmia mechanisms in heart failure by investigating how disease-induced remodeling alters the coupling of electrophysiology, Ca2+ and Na+ handling, contractile function, and energetics.
Introduction
Complex systems interplay in heart failure
Heart failure (HF) is a rapidly growing health problem in the US. The number of adults diagnosed with HF increased from about 5.7 million (2009–2012) to about 6.5 million (2011–2014) and is projected to rise to more than 8 million people by 2030.6 HF is a pathological state in which the heart is unable to pump blood at a rate commensurate with the requirements of metabolizing tissues, resulting in inadequate perfusion of tissues during exertion, or is able to do so only with an elevated diastolic filling pressure. HF biology involves multiple adaptive and maladaptive pathways that work on multiple biological scales to maintain end-organ perfusion. These include increase in the sympathetic drive, renin-angiotensin-aldosterone system (RAAS), and oxidative stress (Fig. 1, top).
Fig. 1. Systems interplay in HF and multi-scale systems biology approaches.

Top: schematic of systems involved in HF pathophysiology. HF phenotype includes ventricular hypertrophy, ionic and structural remodeling, and neurohormonal dysregulation leading to both impaired contractile function, and increased arrhythmia susceptibility. Bottom: systems biology approaches for data collection, analysis, and functional and structural integration. The data images in the upper panel are reproduced with permission from 12, 135.
While advances in high-throughput experimental approaches have allowed identifying and characterizing many individual actors in this multifaceted disease process, one major challenge is to mine this massive amount of information from high-throughput biological data to generate novel mechanistic insights in HF pathophysiology. Systems biology, enabled by multidisciplinary expertise from different fields, often including mathematics, physics, engineering, and computer science, allowed developing many tools and approaches to (a) integrate data in physiological networks; (b) reveal emergent mechanisms of disease; and (c) facilitate prediction and development of therapeutic intervention. These approaches apply a wide spectrum of mathematical formalisms and integrate multiple data types across interacting biochemical and biophysical functions and multi-scale models that are structurally integrated over physical scales (Fig. 1, bottom).
Alterations in cardiac electrophysiology, contractile function and their neurohormonal regulation, accompanied by ventricular hypertrophy and structural remodeling, all contribute to the HF phenotype (Fig. 1, top). Ca2+/calmodulin-dependent protein kinase (CaMKII) is upregulated and more active in HF,1, 47, 58 and is a key regulator of cellular subsystems contributing to acute mechanical and electrical dysfunction as well as chronic cardiac remodeling in HF. In the context of a complex cardiac disease like HF multiple signaling pathways are likely to be altered, many of which might crosstalk with CaMKII signaling. Among these, enhanced sympathetic activation is one of the most well characterized forms of neural remodeling in HF and has been clearly linked to ventricular arrhythmias in animal models and humans.101 Altered CaMKII signaling has been associated to all major aspects of HF at various spatial and time scales, which makes this kinase an attractive candidate for therapy and the ideal center piece in our systems analysis. CaMKII phosphoregulates many proteins that are important in excitation-contraction (EC) coupling.7, 75 Upregulated expression and activity of CaMKIIδ (the predominant cardiac isoform) is seen in human HF,47 and cardiac-specific overexpression of CaMKIIδC leads to severe HF and arrhythmias in mice,118, 136 whereas mice lacking CaMKIIδ develop ventricular hypertrophy following aortic banding, but do not decompensate into HF.70 However, despite a growing body of work detailing the molecular, cellular and functional aspects of CaMKII-dependent remodeling, key questions remain to be addressed regarding the extent to which CaMKII upregulation itself can explain the individual alterations that contribute to HF, and whether there exist interaction and synergy among the plethora of downstream CaMKII effects and other signaling pathways.
Systems approach to understanding cardiac arrhythmia
Failing hearts are at increased risk of arrhythmias, accounting for half of HF deaths (the other half due to pump failure). While sinus node dysfunction and atrial fibrillation are commonly seen in HF (and associated with increased risk of stroke), ventricular tachyarrhythmia is the most common cause of death in HF patients, and is usually associated to disease-induced ionic and structural remodeling. Derangements in cardiac rhythms emerge from abnormalities in gene or protein expression or function, cell signaling pathways, cell-to-cell coupling, tissue heterogeneity that can promote or exacerbate cardiac disease. There are several challenges in the study of arrhythmia mechanism in HF that still preclude translation of basic research findings to the clinic. Model organisms (large mammals) are expensive, and, while genetically engineered mice are valuable because there are now many mouse models that recapitulate major clinical features of HF with specifically defined molecular triggers, electrophysiologic properties for mouse are quite different than human. Use of human preparations is complicated by procurement issues and confounding clinical variables, including comorbidities and pharmacological therapies. Patient-specific induced pluripotent stem cell-derived cardiomyocytes are a powerful option providing potentially unlimited supplies of human cardiomyocytes, but they have inherent limitations: (a) myocyte immaturity, (b) restricted to known genetic HF causes and (c) lack of adult patient hearts for myocyte and organ-level study. The wealth of multi-scale data on alterations in cell signaling, electrophysiology, Ca2+ handling, myofilament function and tissue structure that can be measured in the adult animal models cannot be obtained in humans, but integrative mathematical models provide a systematic framework to extrapolate findings in the mouse to the clinical setting. Here, we review recent advances in our comprehensive mechanistic understanding of arrhythmia in this complex clinical syndrome through mechanism-based mathematical models.
Arrhythmia mechanisms in HF: abnormalities in impulse formation and conduction
It is generally accepted that lethal ventricular arrhythmias occur when triggering events (e.g., ectopic foci) propagate in tissue as they encounter substrate for sustainability (e.g., reentrant loop). Premature ventricular contractions are prototypical triggers in the myocardium that are thought to emerge from cellular events known as early and delayed afterdepolarizations (EADs and DADs). DADs occur during diastole and arise from a transient inward current Iti carried by the Na+/Ca2+ exchanger (NCX), which is evoked by diastolic increase in intracellular Ca2+ concentration ([Ca2+]i) due to abnormal spontaneous Ca2+ release (SCR) from the sarcoplasmic reticulum (SR) (Fig. 2, bottom left). In contrast, EADs initiate during action potential (AP) repolarization and involve complex dynamical interaction among the currents that are available late in the AP (Fig. 2, middle left). EADs occur predominantly at slow heart rates in the setting of reduced repolarization reserve and prolonged AP duration (APD) due to increased inward Ca2+ and Na+ or decreased outward K+ currents, but can also arise from Ca2+ handling abnormalities that activate depolarizing NCX current (INCX). Regardless the mechanism, in large mammals, the L-type Ca2+ current (ICaL) eventually carries the majority of inward charge during the EAD upstroke – but recovery from inactivation and reactivation of the Na+ current (INa) has been shown to lead to EAD takeoff in specific conditions (and species).20, 84 Ectopic activity could also result from automatic firing, which occurs when an increase in time-dependent depolarizing inward currents carried by Na+ or Ca2+ or a decrease in repolarizing outward K+ currents cause progressive time-dependent cell depolarization. When threshold potential is reached, the cell fires, producing automatic activity (Fig. 2, top left).
Fig. 2. Mechanisms of arrhythmias.

Abnormalities in impulse formation include altered automaticity, early and delayed afterdepolarizations (left). Alterations in impulse conduction include slowing of conduction and increase in spatial APD heterogeneity (right).
Reentry can occur when an electrical impulse is able to re-excite areas that have already recovered, thereby providing a perpetuation of electrical activity. Reentry can be caused by a fixed anatomical obstacle (fibrosis), or be functional reentry, favored by spatially heterogeneous APD and conduction slowing (Fig. 2, right). For reentry to be sustained, all points in the reentrant path need to become excitable before the arrival of the reentrant impulse (termed ‘excitable gap’). When wavelength (i.e., the distance an impulse travels within a single refractory period) decreases due to shortening of the effective refractory period or due to conduction slowing, reentry will be more likely and more reentrant circuits can fit in the same area, making the arrhythmia more stable and less likely to terminate.
HF-induced changes alter both individual myocytes properties (e.g., ion channel dysfunction or expression remodeling) and cell-to-cell coupling, thus affecting impulse formation and propagation and enhancing both arrhythmia triggers and substrate.
Arrhythmia Triggers in HF
Triggered activity is facilitated by HF-induced modifications in ionic currents and intracellular Ca2+ dynamics. Dysregulation of Ca2+ handling is the most established cause of arrhythmias in failing myocytes, and it is due to the progressive change in the expression and function of many proteins involved in Ca2+ cycling. Compared to nonfailing ventricular myocytes, failing cells are characterized by reduced Ca2+ transient (CaT) amplitude and enhanced SR Ca2+ leak, which are not only responsible for hypocontractility and impaired relaxation, but also for increased the propensity for EADs121 and DADs108 (Fig. 3).
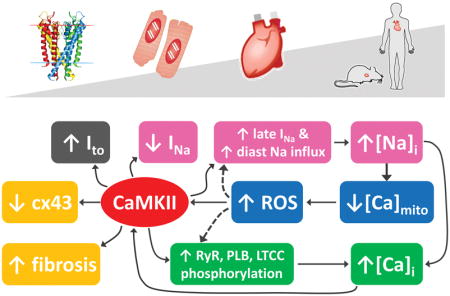
Fig. 3. Arrhythmogenic crosstalk and feedback of Na+-Ca2+-CaMKII-ROS in HF.

Schematic of the main cellular processes linking Na+ and Ca2+ cycling, CaMKII activity and metabolism to increased propensity for arrhythmia in failing myocytes. CaMKII, which expression and activity are enhanced in HF, phosphorylates multiple targets (red symbol “P”), directly influencing membrane electrophysiology and Na+ and Ca2+ signals, thereby facilitating the development of ectopic activity (EADs and DADs). Increased Na+ load enhances [Ca2+]i and reduces [Ca2+]m, leading to further CaMKII activation via both Ca2+/calmodulin- and oxidation-dependent mechanisms. In turn, increased ROS production (oxidative stress) can directly affect membrane electrophysiology, Ca2+ handling, and myofilament function (blue starred symbol). CaMKII is also involved in HF-induced structural remodeling that facilitates the formation of reentrant substrate.
CaMKII has been linked to the triggering of arrhythmias via both EADs and DADs.5 CaMKII activation of ryanodine receptors (RyRs) enhances EC coupling65 and increases SR Ca2+ leak, Ca2+ sparks and waves.42 In HF, where NCX is upregulated and IK1 downregulated, these spontaneous SR Ca2+ release events are more likely to induce DADs.96 CaMKII-dependent phosphorylation of L-type Ca2+ channels (LTCCs) enhances the influx of Ca2+ thereby fueling a positive feedback mechanism that enhances CaMKII activation, and mediates facilitation of ICaL,8, 44 which in turn favors the development of EADs and modulates RyR fluxes.43 All these effects are expected to be more pronounced during sympathetic stimulation, when protein kinase A (PKA)-dependent phosphorylation of LTCC, RyR and phospholamban (PLB) further enhances Ca2+ cycling111 and thereby CaMKII activation. CaMKII-dependent phosphorylation also modulates the voltage-gated Na+ channel (NaV) kinetics similarly to a human mutation (SCN5A 1795InsD) associated with heritable arrhythmias.118 CaMKII-dependent enhancement of late INa could contribute to increased propensity for EADs in HF81 (Fig. 3).
CaMKII activity directly influences Na+ homeostasis, another key regulator of EC coupling that is altered in HF.38 It is well established that intracellular Na+ concentration ([Na+]i) is increased in myocytes from failing hearts compared to nonfailing myocytes by 2–6 mM, but the precise mechanistic and quantitative aspects remain unclear.38 Emerging evidence suggests that perturbed Na+ homeostasis and CaMKII hyperactivity are closely interrelated, and critically involved in electrophysiological and mechanical dysfunction in HF.46, 118, 132 Indeed, it has been suggested that CaMKII and enhanced late INa contribute to [Na+]i elevation in HF, as CaMKIIδC-overexpressing mice with HF exhibit prominent late INa and have increased Na+ content.118 Cellular Na+ loading might cause, via outward shift in INCX, excessive Ca2+ load, which is associated with a number of deleterious cellular consequences, including abnormalities in myocyte shortening, electrical instabilities, and metabolic imbalance.38 It has been proposed that the [Na+]i-induced increase in Ca2+ further activates CaMKII, thus fueling a vicious cycle that favors spontaneous SR Ca2+ release and predisposes to Ca2+-related arrhythmia (Fig. 3). We have confirmed and analyzed quantitatively the extent of this feedback in models of healthy and failing mouse myocytes.82 Similar results were obtained with our model of the human atrial myocyte.91 Nevertheless, further studies are required to assess the details and extent of such feedback in cells characterized by a more positive AP plateau (i.e., human ventricular myocytes). In such condition, increase in late INa would favor AP prolongation (and EAD formation), likely contributing to greater [Na+]i loading.
Altered Na+ homeostasis also impacts mitochondrial function. Increased [Na+]i influences production of reactive oxygen species (ROS) by augmenting mitochondrial Ca2+ efflux via the mitochondrial NCX (mNCX) and thus reducing mitochondrial Ca2+ concentration ([Ca2+]m).60, 73 Notably, higher oxidative stress may further exacerbate [Na+]i loading by enhancing late INa through direct effects on NaV1.5 or secondarily by activating CaMKII21, 119 (Fig. 3). A direct link between oxidative stress and CaMKII-dependent arrhythmias has been suggested by computational studies investigating mechanisms of EAD formation upon H2O2 administration 27, 124, which showed that phosphorylation of NaV1.5 and LTCC (caused by ROS-dependent CaMKII activation) are both required in the generation of these EADs.
Oxidative phosphorylation is the main source of energy for metabolic/contractile works in cardiac myocytes. Mitochondria provide the ATP needed for contractile function and sarcoplasmic and sarcolemmal ion transport, which is responsible for the myocyte electrical activity. Coupling mitochondrial energetics to EC coupling models is therefore needed to investigate the key role of energetics in modulating myocyte mechanical activity and ion concentration gradients, especially during impaired metabolic states in pathological conditions, such as HF. Thus, it is surprising that only a few computational models have been developed to link mitochondrial activity to membrane electrophysiology and cytosolic Ca2+ handling, despite the large number of modeling studies investigating in detail mitochondrial energetics, Ca2+ handling, pH regulation and ROS production,18, 31, 32, 55, 120 and mitochondrial oscillations and waves.89, 130 Cortassa et al. integrated the description of mitochondrial energetics into a model of EC coupling to study the dynamic changes in ADP, NADH, and [Ca2+]m induced by changes in pacing frequency.17 Extensions of this model have been used to investigate the effect of ROS on AP dynamics during ischemia-reperfusion 137 mediated by the sarcolemmal ATP-sensitive K+ (KATP) channels, and the interplay between of local Ca2+ and ROS signaling.45 Recently, Li et al. developed a multi-scale model including a mitochondria-SR microdomain and simulating ROS-dependent modulation of RyR and SR Ca2+-ATPase (SERCA).66 Their simulations showed that elevated ROS production (as in disease) increases cytosolic Ca2+ by stimulating RyRs and inhibiting SERCA. Enhanced Ca2+ signal in turn activates INCX, Ca2+-sensitive nonspecific cationic channels, and Ca2+-induced Ca2+ release, indirectly affecting ICaL and leading to abnormality in AP shape.
Oxidative stress plays an important role in the pathophysiology of cardiac remodeling and HF. ROS production is increased within the mitochondria from failing hearts,60 and directly impairs myocyte function by modifying proteins central to EC coupling.22 Not only does oxidative stress activate CaMKII directly,22 but CaMKII-induced [Na+]i loading can induce an increase in ROS60 (Fig. 3). Studying the deleterious arrhythmogenic consequences of chronic CaMKII activation and oxidative stress is an important next step integrative modeling efforts should tackle.
Arrhythmia Substrates in HF
Development and progression of HF are accompanied by an extensive remodeling of the whole organ, which leads to maladaptive changes in the size of heart chambers and in the mechanical properties of the myocardium. Typical HF features are indeed hypertrophy, increased thickness of the ventricular wall, and abundance of fibrosis. These modifications initially occur as part of a compensatory mechanism in response to pathologic conditions like pressure or volume overload. Number and size of cardiomyocytes increase to accommodate the need of more mechanical work to maintain the cardiac output within the normal range. At the same time, the composition of the connective tissue changes increasing myocardium stiffness. Cardiac hypertrophy is regulated by an intricate web of signaling pathways that influence myocyte growth, including those of CaMKII61 and calcineurin/NFAT.48, 80 Computational models of cardiomyocyte hypertrophic signaling networks have successfully been used to gain a quantitative understanding of the contributions of individual pathways and their interactions, and to predict which signaling nodes are most important for cardiomyocyte hypertrophy,54, 102, 103 and apoptosis.54 Other computational studies have instead investigated the factors responsible for cycling of specific mediators like IP3 and NFAT.15, 16 Along the same lines, computational models have also begun to provide important insights into the development of fibrosis. The Saucerman group has developed a large-scale computational model of the cardiac fibroblast signaling network in order to identify the key drivers of differentiation in myofibroblast and extracellular matrix remodeling.134 Extension of modeling approaches to account for longer time scales is an important step to gain a comprehensive understanding of HF pathophysiology.
Conduction defects
While a decrease in refractoriness seems critical to the development of sustained classical (especially atrial) tachyarrhythmia, reentrant substrate in HF has been rather associated to structural/anatomic abnormalities and conduction delay, whereas refractoriness is actually increased (due to APD prolongation). Thus, structural remodeling including hyperthrophy and enhanced interstitial fibrosis might play a dominant role in sustaining ventricular tachyarrhythmia in HF. However, HF-induced alterations in Na+ and Ca2+ handling (possibly mediated by CaMKII) can functionally affect cell properties and tissue susceptibility to reentry. For example, King et al. recently highlighted novel arrhythmogenic consequences of abnormal Ca2+ handling resulting from catecholaminergic polymorphic ventricular tachycardia (CPVT) mutations, which may play a critical role in arrhythmia maintenance. Specifically, they found a reduced upstroke velocity of monophasic APs, inter-atrial conduction delays and slowed epicardial conduction velocity (CV) in structurally normal RyR2-P2328S hearts, which were more susceptible to atrial arrhythmia triggering than wild type hearts.57 This strongly suggested that Ca2+-dependent alterations in NaVs56 may promote functional reentry also in other disease conditions, such as HF. The increased SR Ca2+ leak in CPVT mice may result in increased CaMKII activity, which is also enhanced during rapid paging13 and can alter cardiac NaV function. Indeed, CaMKII induces loss-of-function effects on peak INa,118 which can explain conduction slowing40 independent of structural and anatomical changes (Fig. 3). Delaying recovery of NaVs from inactivation can increase the slope of the APD restitution curve and hence the likelihood of alternans and reentry,99 and reducing NaV conductance increases the vulnerable window.123 Christensen et al. used a multicellular mathematical model of the cardiac fiber to demonstrate that enhanced CaMKII activity in the infarct border zone, due primarily to increased oxidation, and CaMKII effects on NaVs are associated with reduced CV, prolonged refractoriness, and increased susceptibility to formation of conduction block at the border zone margin, a prerequisite for reentry.14 Heterogeneous Na+ and Ca2+ loading in cardiac tissue can predispose to reentrant arrhythmia, as shown in both ventricular128 and atrial simulations.63 Ca2+-dependent regulation of other ion channels/transporters may also affect CV and APD restitution. For example, Ca2+-dependent inhibition of inward rectifier K+ current (IK1) has been suggested,25 which could indirectly reduce INa availability by depolarizing the resting membrane potential (RMP).
Impaired cell-cell electrical coupling might also affect AP propagation and contribute to the formation of a reentrant substrate. Indeed, connexin43 (cx43), the principal gap-junctional protein in ventricular myocytes, is seen downregulated2, 19 and heterogeneously distributed9 in HF. Dysregulation of cx43 has been associated to reduced electrical coupling among cells, decreased CV, and increased propensity for arrhythmias9, 95, 107 (Fig. 3). Interestingly, a recent study showed that both acute and chronic calmodulin/CaMKII inhibition improved conduction and enhanced localization of cx43 in the intercalated disc.114
Whether they are electrically coupled to myocytes59 or only passive obstacles, myofibroblasts can alter AP propagation in cardiac tissue. The first model coupling a cardiomyocyte to an electrically active fibroblast predicted a depolarizing effect on myocyte MRP and AP shortening.74 Jacquemet and Hernandez first described an opposite effect on APD (and a decrease in CV),50 then determined that the different outcome depends on the fibroblast resting membrane potential, whereby lower resting potentials shorten the AP, while depolarized resting potentials prolong the AP.51 Other computational investigations confirmed that, depending on the context, fibroblasts can differentially affect the electrical activity of myocytes by acting as a current source or sink,76, 86, 104, 105, 126, 127 and increasing/decreasing arrhythmia risk.76, 112 Investigating how different degrees of HF-induced remodeling affect vulnerability to reentry, Gomez et al. showed that, while a moderate amount of fibrosis and cellular uncoupling is sufficient to elicit reentrant activity, very high fibrotic content and/or very low conductivity hinder the development of reentry.35
Spatially heterogeneous APD and Ca2+ signal
Transmural dispersion of repolarization (DOR) and APD gradients are altered in HF.4, 34, 71 Computational studies have shown that electrophysiological and structural HF-induced remodeling (including fibrosis and tissue heterogeneity) increases APD dispersion. For example, while a certain degree of transmural DOR is normal and may be attributed in part to differential expression of Ito (which is larger at the epicardial side), CaMKII-dependent upregulation of Ito shortens APDs in the epicardium but prolongs APDs in the endocardium (where Ito is small) thus amplifying transmural DOR40 (Fig. 3). However, it has been proposed that heterogeneous remodeling modulates repolarization gradients and can even reduce transmural APD dispersion and APD gradients in HF vs. control.36 Notably, HF enhances susceptibility to arrhythmogenic cardiac alternans,62, 122 which amplifies electrical heterogeneities in the heart and has been closely associated with sudden cardiac death.
Subcellular heterogeneity in EC coupling and Ca2+ handling, either anatomical3 or dynamically induced,28, 29, 109, 125 is one very clear contributor to intracellular Ca2+ waves (and triggered activity) and Ca2+-driven AP alternans in ventricular cells. Using a ventricular myocyte model with detailed spatiotemporal Ca2+ cycling, Nivala et al. demonstrated that t-tubular disruption contributes to dyssynchronous Ca2+ release and impaired contraction in HF.90 Comparing results obtained with normal or downregulated SERCA, they also characterized different mechanisms responsible for the development of Ca2+ alternans at different stages of HF-induced remodeling. Studying a mouse model of congestive HF, Louch et al. observed regions of cells characterized by both slow Ca2+ sparks kinetics and slow CaT rise during the AP, suggesting a link between greater variability in spark kinetics and dyssynchronous Ca2+ release in HF.72 A computational analysis showed that alterations in Ca2+ sparks can be explained by rearrangement of RyRs within the Ca2+ release units, and excluded a direct role of t-tubule disruption.72 In a later investigation, the same group showed that synchrony of Ca2+ release in HF is also determined by the interplay between RyR sensitivity and SR Ca2+ loading.92
Fibrosis also affects the formation of alternans. Xie et al. showed that fibroblast-myocyte coupling prolongs the myocyte refractory period, facilitating induction of reentry in cardiac tissue.126 In a more detailed investigation, the same group also showed that such coupling generates a gap junction current that flows into the myocyte and is characterized by two main components: an early transient outward current and a background current present during the repolarizing phase. Depending on the relative prominence of the two, fibroblast-myocyte coupling can either prolong or shorten the AP, promoting or suppressing both voltage-driven and Ca2+-driven alternans.127
It has also been shown that impairment of mitochondrial function adversely affects cardiac Ca2+ cycling leading to proarrhythmic Ca2+ alternans.26 Investigating the mechanisms linking metabolic stress and membrane instabilities, Zhou et al. analyzed how oscillations in mitochondrial inner membrane potential (ΔΨm) affect electrical activity in tissue.138 They showed that ROS-induced regional ΔΨm loss activates KATP channels that cause regional AP shortening, leading to the formation of a metabolic sink. Propensity for reentry and fibrillation resulted increased because of the disparity in refractoriness inside and outside the sink.138
Source-sink
Synchronization mechanisms are required for EADs and DADs to overcome the robust protective effects of electrotonic source-sink mismatch (especially in well-coupled ventricular tissue) and trigger premature ventricular complexes. Structural and electrophysiological remodeling in HF decreases the number of cells displaying EADs or DADs that are required to trigger an arrhythmia in tissue, as elegantly demonstrated in ventricular tissue simulations.129 Notably, whole-heart AP and Ca2+ optical mapping mimicking various HF-related conditions showed results for DADs that were in qualitative agreement with these modeling predictions.85 In failing cardiac tissue, Lang et al. observed a higher than normal (vs. nonfailing cardiac tissue) number of myocytes that did not synchronize with neighboring myocytes, and these asynchronous myocytes also often exhibited independent spontaneous Ca2+ waves. While in normal well-coupled tissue asynchronous Ca2+ waves did not produce detectable depolarizations, poor electrical coupling between failing myocytes enabled local Ca2+-induced inward current of sufficient source strength to overcome a weakened current sink to depolarize the membrane potential in the actual myocyte exhibiting the Ca2+ wave, and even depolarize nearby neighboring myocytes appreciably.64
Influence of mechanical dysfunction on electrophysiology and arrhythmia
Since the myofilaments dynamically and cooperatively buffer large amounts of cytosolic Ca2+,11 developing computational models coupling myofilament contraction to Ca2+ cycling and membrane electrophysiology is critical to study how HF-induced changes in ion channel and Ca2+ dynamically affect contraction, and quantifying how impaired myofilament function in HF feeds back into Ca2+ and AP dynamics.10, 41, 77, 78, 88, 100 We have developed a model that describes electrophysiology, Ca2+ handling, myofilament activation and crossbridge (XB) cycling to understand the effects of β-adrenergic activation on cardiac mechanics in rabbit and mouse heart.87, 113 We showed that distinct changes in myofilament Ca2+ sensitivity and XB cycling influence CaT and contraction responses to β-adrenergic stimulation, but the changes in Ca2+ buffering by the myofilaments have a limited impact on membrane currents and AP morphology in nonfailing ventricular myocytes.87, 113 Zile and Trayanova simulated Ca2+ and mechanical remodeling in a coupled human electromechanical model and showed that HF-induced remodeling of mechanical parameters alters electrical, Ca2+ and mechanical alternans, and myofilament protein dynamics play an important role in EAD formation.139, 140
Myofilament contraction requires ATP, hence it is also important to combine energetics and contraction to understand how the heart adapts the rate of ATP production to energy demand (and HF-associated changes thereof), as for example done in.131 Tewari et al. built a multi-scale model linking myofilament energetics to whole-body cardiovascular function116 and demonstrated that metabolic dysfunction in HF directly impairs myofilament contractile function and, consequently, systolic function.
The expression of ion channels sensitive to mechanical load (in particular to changes in cell strain or volume) creates a feedback mechanism (called mechano-electric feedback) that directly affects the membrane electrical activity. Acting like mechanoreceptors, these channels contribute to the regulation of the cardiac function that has to continuously adapt to the mechanical load. Despite the fact that a large numbers of channels are modulated by mechanical stress,94 modeling studies have primarily focused on stretch-activated channels (SACs), shown to be involved in the generation of arrhythmias induced by mechanical stimulation,30, 67 and in the termination of arrhythmias by precordial thump.68 Recruitment of SACs has also been shown to play a role in the regulation of scroll waves that underlie reentrant arrhythmias,49 though a more recent study showed that both SAC activation and stretch-induced Ca2+ release from myofilaments are associated to triggered propagating contractions, and that both phenomena are required to significantly perturb the AP.117
Using a cell-in-gel system that imposes an afterload during cardiomyocyte contraction, Jian et al. found that nitric oxide synthase (NOS) was involved in transducing mechanical load to potentiate CaTs and Ca2+ sparks.52 Stretch on ventricular cardiomyocytes during diastole also evokes changes in Ca2+ cycling via enhanced X-ROS signaling due to activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2).97, 98 Elevated ROS directly increases RyR open probability and facilitates the formation of Ca2+ sparks and arrhythmogenic waves.97 By modeling compartmentalized ROS signaling and ROS-dependent modulation of RyR gating, Limbu et al. analyzed the role of X-ROS signaling in Ca2+ cycling regulation and arrhythmogenesis.69 They showed that, upon stretch, ROS levels in the dyadic space can be about an order of magnitude higher than the global ROS signal, suggesting the need of a better characterization of ROS signaling compartmentalization. They showed that X-ROS signaling can improve EC coupling in physiological conditions (by enhancing CaT amplitude), but that can also increase the propensity for arrhythmias in response to oxidative stress.
Cardiomyocytes express also mechano-sensitive proteins that are not directly involved in the regulation of membrane potential or Ca2+ handling, but are instead responsible for signaling cascades associated to pathological remodeling and hypertrophy. Tan et al. developed a network model of cardiac mechano-signaling to identify the key sensors involved in the stretch-induced hypertrophic response.115 This large-scale model integrates results from many published studies into one coherent platform that allows the identification of emerging behaviors and crosstalk mechanisms that could not be seen analyzing isolated pathways. The analysis revealed that cooperation between different pathways is necessary to cause growth and remodeling, suggesting potential targets for pharmacological treatment against HF.
Conclusion
Development of computational frameworks for simulation of cardiac electromechanical activity has improved our comprehensive understanding of complex cardiac arrhythmias, such as those occurring in HF patients. By integrating data describing different cell functions (membrane electrophysiology, intracellular signaling, contraction, and metabolism) in different time and spatial scale, these models allow for identification of emergent mechanisms and can potentially facilitate the development of therapeutic strategies. Nevertheless, there exist areas in which our understanding is incomplete and future modeling efforts are needed to advance the field.
We focused our review on CaMKII as a key nodal point in HF pathophysiology. An important extension HF modeling is the inclusion of spatio-temporal characteristics of CaMKII activation in myocytes and of mechanisms of CaMKII activation independent from Ca2+/calmodulin and ROS signaling: O-GlcNAcylation,24 S-nitrosylation,23 and Epac/NOS-dependent pathway.93 This extension will be important to elucidate the potential synergy among the different activation mechanisms, and could be important for the development of CaMKII inhibitors for clinical applications.37
Complex network relationships are emerging through comparative analyses of large-scale genomic, transcriptomic, proteomic, and metabolomic profiles that can be integrated into detailed mechanistic models. Although still rarely done, integrating “omics” approaches with mathematical mechanism-based biology models might be a powerful strategy to both understand the data and improve the predictive ability of the models. While advancements in experimental research will provide modelers with novel data for further improvement of computational frameworks, the parallel systematic use of sensitivity analysis and uncertainty quantification methods will also contribute to enhance the robustness of models and their predictions.53, 79, 83, 106, 110
Finally, an important priority will be to translate the mechanistic insights obtained from the studies to new clinical interventions. The use of clinically-derived patient-specific computer models to optimize catheter ablation strategies133 is a relevant example of the application of systems biology and computational modeling approaches to therapy. The safety screening FDA proposal, Comprehensive in vitro Proarrhythmia Assay (CiPA), also involves the use of computational models for mechanism-based in silico assessment of drug toxicity and overall proarrhythmic risk.33, 39 Indeed, computational models are now poised to deliver breakthroughs at the bedside.
Acknowledgments
Funding. American Heart Association grant 15SDG24910015 (E.G.); National Institutes of Health (NIH): Stimulating Peripheral Activity to Relieve Conditions (SPARC) grant 1OT2OD023848-01 (E.G.), National Heart, Lung, And Blood Institute (NHLBI) grants R01HL131517 (E.G.), R01HL141214 (E.G.), and K99HL138160 award (S.M.); Heart Rhythm Society post-doctoral fellowship 16OA9HRS (S.M.).
Footnotes
COI: none.
Contributor Information
Stefano Morotti, Department of Pharmacology, University of California Davis, Davis, CA, 95616, USA.
Eleonora Grandi, Department of Pharmacology, University of California Davis, Davis, CA, 95616, USA.
References
- 1.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–22. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 2.Ai X, Pogwizd SM. Connexin 43 downregulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A. Circ Res. 2005;96:54–63. doi: 10.1161/01.RES.0000152325.07495.5a. [DOI] [PubMed] [Google Scholar]
- 3.Aistrup GL, Shiferaw Y, Kapur S, Kadish AH, Wasserstrom JA. Mechanisms underlying the formation and dynamics of subcellular calcium alternans in the intact rat heart. Circ Res. 2009;104:639–49. doi: 10.1161/CIRCRESAHA.108.181909. [DOI] [PubMed] [Google Scholar]
- 4.Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003;93:638–45. doi: 10.1161/01.RES.0000092248.59479.AE. [DOI] [PubMed] [Google Scholar]
- 5.Anderson ME. Multiple downstream proarrhythmic targets for calmodulin kinase II: moving beyond an ion channel-centric focus. Cardiovasc Res. 2007;73:657–66. doi: 10.1016/j.cardiores.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 6.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P American Heart Association Statistics C, Stroke Statistics S. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:180–7. doi: 10.1097/FJC.0b013e3181a25078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bers DM, Morotti S. Ca(2+) current facilitation is CaMKII-dependent and has arrhythmogenic consequences. Front Pharmacol. 2014;5:144. doi: 10.3389/fphar.2014.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boulaksil M, Winckels SK, Engelen MA, Stein M, van Veen TA, Jansen JA, Linnenbank AC, Bierhuizen MF, Groenewegen WA, van Oosterhout MF, Kirkels JH, de Jonge N, Varro A, Vos MA, de Bakker JM, van Rijen HV. Heterogeneous Connexin43 distribution in heart failure is associated with dispersed conduction and enhanced susceptibility to ventricular arrhythmias. Eur J Heart Fail. 2010;12:913–21. doi: 10.1093/eurjhf/hfq092. [DOI] [PubMed] [Google Scholar]
- 10.Campbell SG, Flaim SN, Leem CH, McCulloch AD. Mechanisms of transmurally varying myocyte electromechanics in an integrated computational model. Philos Trans A Math Phys Eng Sci. 2008;366:3361–80. doi: 10.1098/rsta.2008.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell SG, Lionetti FV, Campbell KS, McCulloch AD. Coupling of adjacent tropomyosins enhances cross-bridge-mediated cooperative activation in a markov model of the cardiac thin filament. Biophys J. 2010;98:2254–64. doi: 10.1016/j.bpj.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang SN, Chang SH, Yu CC, Wu CK, Lai LP, Chiang FT, Hwang JJ, Lin JL, Tsai CT. Renal Denervation Decreases Susceptibility to Arrhythmogenic Cardiac Alternans and Ventricular Arrhythmia in a Rat Model of Post-Myocardial Infarction Heart Failure. J Am Coll Cardiol Basic Trans Science. 2017;2:9. doi: 10.1016/j.jacbts.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–51. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christensen MD, Dun W, Boyden PA, Anderson ME, Mohler PJ, Hund TJ. Oxidized calmodulin kinase II regulates conduction following myocardial infarction: a computational analysis. PLoS Comput Biol. 2009;5:e1000583. doi: 10.1371/journal.pcbi.1000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooling M, Hunter P, Crampin EJ. Modeling hypertrophic IP3 transients in the cardiac myocyte. Biophys J. 2007;93:3421–33. doi: 10.1529/biophysj.107.110031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooling MT, Hunter P, Crampin EJ. Sensitivity of NFAT cycling to cytosolic calcium concentration: implications for hypertrophic signals in cardiac myocytes. Biophys J. 2009;96:2095–104. doi: 10.1016/j.bpj.2008.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cortassa S, Aon MA, O’Rourke B, Jacques R, Tseng HJ, Marban E, Winslow RL. A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys J. 2006;91:1564–89. doi: 10.1529/biophysj.105.076174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cortassa S, Aon MA, Winslow RL, O’Rourke B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys J. 2004;87:2060–73. doi: 10.1529/biophysj.104.041749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, Kaprielian R, Yacoub MH, Severs NJ. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33:359–71. doi: 10.1006/jmcc.2000.1308. [DOI] [PubMed] [Google Scholar]
- 20.Edwards AG, Grandi E, Hake JE, Patel S, Li P, Miyamoto S, Omens JH, Heller Brown J, Bers DM, McCulloch AD. Nonequilibrium reactivation of Na+ current drives early afterdepolarizations in mouse ventricle. Circ Arrhythm Electrophysiol. 2014;7:1205–13. doi: 10.1161/CIRCEP.113.001666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erickson JR. Mechanisms of CaMKII Activation in the Heart. Front Pharmacol. 2014;5:59. doi: 10.3389/fphar.2014.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–74. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erickson JR, Nichols CB, Uchinoumi H, Stein ML, Bossuyt J, Bers DM. S-Nitrosylation Induces Both Autonomous Activation and Inhibition of Calcium/Calmodulin-dependent Protein Kinase II delta. J Biol Chem. 2015;290:25646–56. doi: 10.1074/jbc.M115.650234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–6. doi: 10.1038/nature12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fauconnier J, Lacampagne A, Rauzier JM, Vassort G, Richard S. Ca2+-dependent reduction of IK1 in rat ventricular cells: a novel paradigm for arrhythmia in heart failure? Cardiovasc Res. 2005;68:204–12. doi: 10.1016/j.cardiores.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 26.Florea SM, Blatter LA. The role of mitochondria for the regulation of cardiac alternans. Front Physiol. 2010;1:141. doi: 10.3389/fphys.2010.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foteinou PT, Greenstein JL, Winslow RL. Mechanistic Investigation of the Arrhythmogenic Role of Oxidized CaMKII in the Heart. Biophys J. 2015;109:838–49. doi: 10.1016/j.bpj.2015.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaeta SA, Bub G, Abbott GW, Christini DJ. Dynamical mechanism for subcellular alternans in cardiac myocytes. Circ Res. 2009;105:335–42. doi: 10.1161/CIRCRESAHA.109.197590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaeta SA, Krogh-Madsen T, Christini DJ. Feedback-control induced pattern formation in cardiac myocytes: a mathematical modeling study. J Theor Biol. 2010;266:408–18. doi: 10.1016/j.jtbi.2010.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garny A, Kohl P. Mechanical induction of arrhythmias during ventricular repolarization: modeling cellular mechanisms and their interaction in two dimensions. Ann N Y Acad Sci. 2004;1015:133–43. doi: 10.1196/annals.1302.011. [DOI] [PubMed] [Google Scholar]
- 31.Gauthier LD, Greenstein JL, Cortassa S, O’Rourke B, Winslow RL. A computational model of reactive oxygen species and redox balance in cardiac mitochondria. Biophys J. 2013;105:1045–56. doi: 10.1016/j.bpj.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gauthier LD, Greenstein JL, O’Rourke B, Winslow RL. An integrated mitochondrial ROS production and scavenging model: implications for heart failure. Biophys J. 2013;105:2832–42. doi: 10.1016/j.bpj.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gintant G, Sager PT, Stockbridge N. Evolution of strategies to improve preclinical cardiac safety testing. Nat Rev Drug Discov. 2016;15:457–71. doi: 10.1038/nrd.2015.34. [DOI] [PubMed] [Google Scholar]
- 34.Glukhov AV, Fedorov VV, Lou Q, Ravikumar VK, Kalish PW, Schuessler RB, Moazami N, Efimov IR. Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Res. 2010;106:981–91. doi: 10.1161/CIRCRESAHA.109.204891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gomez JF, Cardona K, Martinez L, Saiz J, Trenor B. Electrophysiological and structural remodeling in heart failure modulate arrhythmogenesis. 2D simulation study. PLoS One. 2014;9:e103273. doi: 10.1371/journal.pone.0103273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomez JF, Cardona K, Romero L, Ferrero JM, Jr, Trenor B. Electrophysiological and structural remodeling in heart failure modulate arrhythmogenesis. 1D simulation study. PLoS One. 2014;9:e106602. doi: 10.1371/journal.pone.0106602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grandi E, Dobrev D. Non-ion channel therapeutics for heart failure and atrial fibrillation: Are CaMKII inhibitors ready for clinical use? J Mol Cell Cardiol. 2017 doi: 10.1016/j.yjmcc.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grandi E, Herren AW. CaMKII-dependent regulation of cardiac Na(+) homeostasis. Front Pharmacol. 2014;5:41. doi: 10.3389/fphar.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grandi E, Morotti S, Pueyo E, Rodriguez B. Safety Pharmacology - Risk Assessment QT Interval Prolongation and Beyond. Front Physiol. doi: 10.3389/fphys.2018.00678. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grandi E, Puglisi JL, Wagner S, Maier LS, Severi S, Bers DM. Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophys J. 2007;93:3835–47. doi: 10.1529/biophysj.107.114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenstein JL, Hinch R, Winslow RL. Mechanisms of excitation-contraction coupling in an integrative model of the cardiac ventricular myocyte. Biophysical Journal. 2006;90:77–91. doi: 10.1529/biophysj.105.065169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo T, Zhang T, Mestril R, Bers DM. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 43.Hashambhoy YL, Greenstein JL, Winslow RL. Role of CaMKII in RyR leak, EC coupling and action potential duration: a computational model. J Mol Cell Cardiol. 2010;49:617–24. doi: 10.1016/j.yjmcc.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hashambhoy YL, Winslow RL, Greenstein JL. CaMKII-induced shift in modal gating explains L-type Ca(2+) current facilitation: a modeling study. Biophys J. 2009;96:1770–85. doi: 10.1016/j.bpj.2008.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hatano A, Okada J, Washio T, Hisada T, Sugiura S. A three-dimensional simulation model of cardiomyocyte integrating excitation-contraction coupling and metabolism. Biophys J. 2011;101:2601–10. doi: 10.1016/j.bpj.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herren AW, Bers DM, Grandi E. Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am J Physiol Heart Circ Physiol. 2013;305:H431–45. doi: 10.1152/ajpheart.00306.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999;84:713–21. doi: 10.1161/01.res.84.6.713. [DOI] [PubMed] [Google Scholar]
- 48.Hohendanner F, McCulloch AD, Blatter LA, Michailova AP. Calcium and IP3 dynamics in cardiac myocytes: experimental and computational perspectives and approaches. Front Pharmacol. 2014;5:35. doi: 10.3389/fphar.2014.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu Y, Gurev V, Constantino J, Bayer JD, Trayanova NA. Effects of mechano-electric feedback on scroll wave stability in human ventricular fibrillation. PLoS One. 2013;8:e60287. doi: 10.1371/journal.pone.0060287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jacquemet V, Henriquez CS. Modelling cardiac fibroblasts: interactions with myocytes and their impact on impulse propagation. Europace. 2007;9(Suppl 6):vi29–37. doi: 10.1093/europace/eum207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jacquemet V, Henriquez CS. Loading effect of fibroblast-myocyte coupling on resting potential, impulse propagation, and repolarization: insights from a microstructure model. Am J Physiol Heart Circ Physiol. 2008;294:H2040–52. doi: 10.1152/ajpheart.01298.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jian Z, Han H, Zhang T, Puglisi J, Izu LT, Shaw JA, Onofiok E, Erickson JR, Chen YJ, Horvath B, Shimkunas R, Xiao W, Li Y, Pan T, Chan J, Banyasz T, Tardiff JC, Chiamvimonvat N, Bers DM, Lam KS, Chen-Izu Y. Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci Signal. 2014;7:ra27. doi: 10.1126/scisignal.2005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnstone RH, Chang ET, Bardenet R, de Boer TP, Gavaghan DJ, Pathmanathan P, Clayton RH, Mirams GR. Uncertainty and variability in models of the cardiac action potential: Can we build trustworthy models? J Mol Cell Cardiol. 2016;96:49–62. doi: 10.1016/j.yjmcc.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kang JH, Lee HS, Park D, Kang YW, Kim SM, Gong JR, Cho KH. Context-independent essential regulatory interactions for apoptosis and hypertrophy in the cardiac signaling network. Sci Rep. 2017;7:34. doi: 10.1038/s41598-017-00086-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kembro JM, Aon MA, Winslow RL, O’Rourke B, Cortassa S. Integrating mitochondrial energetics, redox and ROS metabolic networks: a two-compartment model. Biophys J. 2013;104:332–43. doi: 10.1016/j.bpj.2012.11.3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.King JH, Wickramarachchi C, Kua K, Du Y, Jeevaratnam K, Matthews HR, Grace AA, Huang CL, Fraser JA. Loss of Nav1.5 expression and function in murine atria containing the RyR2-P2328S gain-of-function mutation. Cardiovasc Res. 2013;99:751–9. doi: 10.1093/cvr/cvt141. [DOI] [PubMed] [Google Scholar]
- 57.King JH, Zhang Y, Lei M, Grace AA, Huang CL, Fraser JA. Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol (Oxf) 2013;207:308–23. doi: 10.1111/apha.12006. [DOI] [PubMed] [Google Scholar]
- 58.Kirchhefer U, Schmitz W, Scholz H, Neumann J. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res. 1999;42:254–61. doi: 10.1016/s0008-6363(98)00296-x. [DOI] [PubMed] [Google Scholar]
- 59.Kohl P, Camelliti P. Cardiac myocyte-nonmyocyte electrotonic coupling: implications for ventricular arrhythmogenesis. Heart Rhythm. 2007;4:233–5. doi: 10.1016/j.hrthm.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 60.Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O’Rourke B, Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–13. doi: 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kreusser MM, Backs J. Integrated mechanisms of CaMKII-dependent ventricular remodeling. Front Pharmacol. 2014;5:36. doi: 10.3389/fphar.2014.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krogh-Madsen T, Christini DJ. Action potential duration dispersion and alternans in simulated heterogeneous cardiac tissue with a structural barrier. Biophys J. 2007;92:1138–49. doi: 10.1529/biophysj.106.090845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krogh-Madsen T, Christini DJ. Slow [Na(+)]i dynamics impacts arrhythmogenesis and spiral wave reentry in cardiac myocyte ionic model. Chaos. 2017;27:093907. doi: 10.1063/1.4999475. [DOI] [PubMed] [Google Scholar]
- 64.Lang D, Sato D, Jiang Y, Ginsburg KS, Ripplinger CM, Bers DM. Calcium-Dependent Arrhythmogenic Foci Created by Weakly Coupled Myocytes in the Failing Heart. Circ Res. 2017 doi: 10.1161/CIRCRESAHA.117.312050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li L, Satoh H, Ginsburg KS, Bers DM. The effect of Ca(2+)-calmodulin-dependent protein kinase II on cardiac excitation-contraction coupling in ferret ventricular myocytes. J Physiol. 1997;501(Pt 1):17–31. doi: 10.1111/j.1469-7793.1997.017bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li Q, Su D, O’Rourke B, Pogwizd SM, Zhou L. Mitochondria-derived ROS bursts disturb Ca(2)(+) cycling and induce abnormal automaticity in guinea pig cardiomyocytes: a theoretical study. Am J Physiol Heart Circ Physiol. 2015;308:H623–36. doi: 10.1152/ajpheart.00493.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li W, Kohl P, Trayanova N. Induction of ventricular arrhythmias following mechanical impact: a simulation study in 3D. J Mol Histol. 2004;35:679–86. doi: 10.1007/s10735-004-2666-8. [DOI] [PubMed] [Google Scholar]
- 68.Li W, Kohl P, Trayanova N. Myocardial ischemia lowers precordial thump efficacy: an inquiry into mechanisms using three-dimensional simulations. Heart Rhythm. 2006;3:179–86. doi: 10.1016/j.hrthm.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 69.Limbu S, Hoang-Trong TM, Prosser BL, Lederer WJ, Jafri MS. Modeling Local X-ROS and Calcium Signaling in the Heart. Biophys J. 2015;109:2037–50. doi: 10.1016/j.bpj.2015.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Brown JH. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–40. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lou Q, Fedorov VV, Glukhov AV, Moazami N, Fast VG, Efimov IR. Transmural heterogeneity and remodeling of ventricular excitation-contraction coupling in human heart failure. Circulation. 2011;123:1881–90. doi: 10.1161/CIRCULATIONAHA.110.989707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Louch WE, Hake J, Mork HK, Hougen K, Skrbic B, Ursu D, Tonnessen T, Sjaastad I, Sejersted OM. Slow Ca(2)(+) sparks de-synchronize Ca(2)(+) release in failing cardiomyocytes: evidence for altered configuration of Ca(2)(+) release units? J Mol Cell Cardiol. 2013;58:41–52. doi: 10.1016/j.yjmcc.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 73.Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–82. doi: 10.1161/01.RES.0000232546.92777.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.MacCannell KA, Bazzazi H, Chilton L, Shibukawa Y, Clark RB, Giles WR. A mathematical model of electrotonic interactions between ventricular myocytes and fibroblasts. Biophys J. 2007;92:4121–32. doi: 10.1529/biophysj.106.101410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maier LS, Bers DM. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc Res. 2007;73:631–40. doi: 10.1016/j.cardiores.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 76.McDowell KS, Arevalo HJ, Maleckar MM, Trayanova NA. Susceptibility to arrhythmia in the infarcted heart depends on myofibroblast density. Biophys J. 2011;101:1307–15. doi: 10.1016/j.bpj.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Michailova A, Spassov V. Computer simulation of excitation-contraction coupling in cardiac muscle. A study of the regulatory role of calcium binding to troponin C. Gen Physiol Biophys. 1997;16:29–38. [PubMed] [Google Scholar]
- 78.Michailova AP, Spassov VZ. Theoretical model and computer simulation of excitation-contraction coupling of mammalian cardiac muscle. J Mol Cell Cardiol. 1992;24:97–104. doi: 10.1016/0022-2828(92)91163-y. [DOI] [PubMed] [Google Scholar]
- 79.Mirams GR, Pathmanathan P, Gray RA, Challenor P, Clayton RH. Uncertainty and variability in computational and mathematical models of cardiac physiology. J Physiol. 2016;594:6833–6847. doi: 10.1113/JP271671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63:467–75. doi: 10.1016/j.cardiores.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 81.Moreno JD, Yang PC, Bankston JR, Grandi E, Bers DM, Kass RS, Clancy CE. Ranolazine for Congenital and Acquired Late INa Linked Arrhythmias: In Silico Pharmacologic Screening. Circ Res. 2013 doi: 10.1161/CIRCRESAHA.113.301971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morotti S, Edwards AG, McCulloch AD, Bers DM, Grandi E. A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J Physiol. 2014;592:1181–97. doi: 10.1113/jphysiol.2013.266676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morotti S, Grandi E. Logistic regression analysis of populations of electrophysiological models to assess proarrythmic risk. MethodsX. 2017;4:25–34. doi: 10.1016/j.mex.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morotti S, McCulloch AD, Bers DM, Edwards AG, Grandi E. Atrial-selective targeting of arrhythmogenic phase-3 early afterdepolarizations in human myocytes. J Mol Cell Cardiol. 2016;96:63–71. doi: 10.1016/j.yjmcc.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local beta-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res. 2012;110:1454–64. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nayak AR, Shajahan TK, Panfilov AV, Pandit R. Spiral-wave dynamics in a mathematical model of human ventricular tissue with myocytes and fibroblasts. PLoS One. 2013;8:e72950. doi: 10.1371/journal.pone.0072950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Negroni JA, Morotti S, Lascano EC, Gomes AV, Grandi E, Puglisi JL, Bers DM. beta-adrenergic effects on cardiac myofilaments and contraction in an integrated rabbit ventricular myocyte model. J Mol Cell Cardiol. 2015;81:162–75. doi: 10.1016/j.yjmcc.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Niederer SA, Smith NP. A mathematical model of the slow force response to stretch in rat ventricular myocytes. Biophys J. 2007;92:4030–44. doi: 10.1529/biophysj.106.095463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nivala M, Korge P, Nivala M, Weiss JN, Qu Z. Linking flickering to waves and whole-cell oscillations in a mitochondrial network model. Biophys J. 2011;101:2102–11. doi: 10.1016/j.bpj.2011.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nivala M, Song Z, Weiss JN, Qu Z. T-tubule disruption promotes calcium alternans in failing ventricular myocytes: mechanistic insights from computational modeling. J Mol Cell Cardiol. 2015;79:32–41. doi: 10.1016/j.yjmcc.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Onal B, Gratz D, Hund TJ. Ca2+/calmodulin kinase II-dependent regulation of atrial myocyte late Na+ current, Ca2+ cycling and excitability: a mathematical modeling study. Am J Physiol Heart Circ Physiol. 2017 doi: 10.1152/ajpheart.00185.2017. ajpheart 00185 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Oyehaug L, Loose KO, Jolle GF, Roe AT, Sjaastad I, Christensen G, Sejersted OM, Louch WE. Synchrony of cardiomyocyte Ca(2+) release is controlled by T-tubule organization, SR Ca(2+) content, and ryanodine receptor Ca(2+) sensitivity. Biophys J. 2013;104:1685–97. doi: 10.1016/j.bpj.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pereira L, Bare DJ, Galice S, Shannon TR, Bers DM. beta-Adrenergic induced SR Ca2+ leak is mediated by an Epac-NOS pathway. J Mol Cell Cardiol. 2017;108:8–16. doi: 10.1016/j.yjmcc.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Peyronnet R, Nerbonne JM, Kohl P. Cardiac Mechano-Gated Ion Channels and Arrhythmias. Circ Res. 2016;118:311–29. doi: 10.1161/CIRCRESAHA.115.305043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Poelzing S, Rosenbaum DS. Altered connexin43 expression produces arrhythmia substrate in heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H1762–70. doi: 10.1152/ajpheart.00346.2004. [DOI] [PubMed] [Google Scholar]
- 96.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–67. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 97.Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–5. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 98.Prosser BL, Ward CW, Lederer WJ. X-ROS signalling is enhanced and graded by cyclic cardiomyocyte stretch. Cardiovasc Res. 2013;98:307–14. doi: 10.1093/cvr/cvt066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Qu Z, Karagueuzian HS, Garfinkel A, Weiss JN. Effects of Na(+) channel and cell coupling abnormalities on vulnerability to reentry: a simulation study. Am J Physiol Heart Circ Physiol. 2004;286:H1310–21. doi: 10.1152/ajpheart.00561.2003. [DOI] [PubMed] [Google Scholar]
- 100.Rice JJ, Wang F, Bers DM, de Tombe PP. Approximate model of cooperative activation and crossbridge cycling in cardiac muscle using ordinary differential equations. Biophys J. 2008;95:2368–90. doi: 10.1529/biophysj.107.119487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ripplinger CM, Noujaim SF, Linz D. The nervous heart. Prog Biophys Mol Biol. 2016;120:199–209. doi: 10.1016/j.pbiomolbio.2015.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ryall KA, Bezzerides VJ, Rosenzweig A, Saucerman JJ. Phenotypic screen quantifying differential regulation of cardiac myocyte hypertrophy identifies CITED4 regulation of myocyte elongation. J Mol Cell Cardiol. 2014;72:74–84. doi: 10.1016/j.yjmcc.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ryall KA, Holland DO, Delaney KA, Kraeutler MJ, Parker AJ, Saucerman JJ. Network reconstruction and systems analysis of cardiac myocyte hypertrophy signaling. J Biol Chem. 2012;287:42259–68. doi: 10.1074/jbc.M112.382937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sachse FB, Moreno AP, Abildskov JA. Electrophysiological modeling of fibroblasts and their interaction with myocytes. Ann Biomed Eng. 2008;36:41–56. doi: 10.1007/s10439-007-9405-8. [DOI] [PubMed] [Google Scholar]
- 105.Sachse FB, Moreno AP, Seemann G, Abildskov JA. A model of electrical conduction in cardiac tissue including fibroblasts. Ann Biomed Eng. 2009;37:874–89. doi: 10.1007/s10439-009-9667-4. [DOI] [PubMed] [Google Scholar]
- 106.Sarkar AX, Christini DJ, Sobie EA. Exploiting mathematical models to illuminate electrophysiological variability between individuals. J Physiol. 2012;590:2555–67. doi: 10.1113/jphysiol.2011.223313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sato T, Ohkusa T, Honjo H, Suzuki S, Yoshida MA, Ishiguro YS, Nakagawa H, Yamazaki M, Yano M, Kodama I, Matsuzaki M. Altered expression of connexin43 contributes to the arrhythmogenic substrate during the development of heart failure in cardiomyopathic hamster. Am J Physiol Heart Circ Physiol. 2008;294:H1164–73. doi: 10.1152/ajpheart.00960.2007. [DOI] [PubMed] [Google Scholar]
- 108.Shannon TR, Wang F, Bers DM. Regulation of cardiac sarcoplasmic reticulum Ca release by luminal [Ca] and altered gating assessed with a mathematical model. Biophys J. 2005;89:4096–110. doi: 10.1529/biophysj.105.068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shiferaw Y, Karma A. Turing instability mediated by voltage and calcium diffusion in paced cardiac cells. Proc Natl Acad Sci U S A. 2006;103:5670–5. doi: 10.1073/pnas.0511061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sobie EA. Parameter sensitivity analysis in electrophysiological models using multivariable regression. Biophys J. 2009;96:1264–74. doi: 10.1016/j.bpj.2008.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Soltis AR, Saucerman JJ. Synergy between CaMKII substrates and beta-adrenergic signaling in regulation of cardiac myocyte Ca(2+) handling. Biophys J. 2010;99:2038–47. doi: 10.1016/j.bpj.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sridhar S, Vandersickel N, Panfilov AV. Effect of myocyte-fibroblast coupling on the onset of pathological dynamics in a model of ventricular tissue. Sci Rep. 2017;7:40985. doi: 10.1038/srep40985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Surdo NC, Berrera M, Koschinski A, Brescia M, Machado MR, Carr C, Wright P, Gorelik J, Morotti S, Grandi E, Bers DM, Pantano S, Zaccolo M. FRET biosensor uncovers cAMP nano-domains at beta-adrenergic targets that dictate precise tuning of cardiac contractility. Nat Commun. 2017;8:15031. doi: 10.1038/ncomms15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Takanari H, Bourgonje VJ, Fontes MS, Raaijmakers AJ, Driessen H, Jansen JA, van der Nagel R, Kok B, van Stuijvenberg L, Boulaksil M, Takemoto Y, Yamazaki M, Tsuji Y, Honjo H, Kamiya K, Kodama I, Anderson ME, van der Heyden MA, van Rijen HV, van Veen TA, Vos MA. Calmodulin/CaMKII inhibition improves intercellular communication and impulse propagation in the heart and is antiarrhythmic under conditions when fibrosis is absent. Cardiovasc Res. 2016;111:410–21. doi: 10.1093/cvr/cvw173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tan PM, Buchholz KS, Omens JH, McCulloch AD, Saucerman JJ. Predictive model identifies key network regulators of cardiomyocyte mechano-signaling. PLoS Comput Biol. 2017;13:e1005854. doi: 10.1371/journal.pcbi.1005854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tewari SG, Bugenhagen SM, Vinnakota KC, Rice JJ, Janssen PML, Beard DA. Influence of metabolic dysfunction on cardiac mechanics in decompensated hypertrophy and heart failure. J Mol Cell Cardiol. 2016;94:162–175. doi: 10.1016/j.yjmcc.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Timmermann V, Dejgaard LA, Haugaa KH, Edwards AG, Sundnes J, McCulloch AD, Wall ST. An integrative appraisal of mechano-electric feedback mechanisms in the heart. Prog Biophys Mol Biol. 2017;130:404–417. doi: 10.1016/j.pbiomolbio.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–38. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555–65. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wei AC, Aon MA, O’Rourke B, Winslow RL, Cortassa S. Mitochondrial energetics, pH regulation, and ion dynamics: a computational-experimental approach. Biophys J. 2011;100:2894–903. doi: 10.1016/j.bpj.2011.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010;7:1891–9. doi: 10.1016/j.hrthm.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wilson LD, Jeyaraj D, Wan X, Hoeker GS, Said TH, Gittinger M, Laurita KR, Rosenbaum DS. Heart failure enhances susceptibility to arrhythmogenic cardiac alternans. Heart Rhythm. 2009;6:251–9. doi: 10.1016/j.hrthm.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xie F, Qu Z, Garfinkel A, Weiss JN. Electrophysiological heterogeneity and stability of reentry in simulated cardiac tissue. Am J Physiol Heart Circ Physiol. 2001;280:H535–45. doi: 10.1152/ajpheart.2001.280.2.H535. [DOI] [PubMed] [Google Scholar]
- 124.Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res. 2009;104:79–86. doi: 10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xie LH, Weiss JN. Arrhythmogenic consequences of intracellular calcium waves. Am J Physiol Heart Circ Physiol. 2009;297:H997–H1002. doi: 10.1152/ajpheart.00390.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Xie Y, Garfinkel A, Camelliti P, Kohl P, Weiss JN, Qu Z. Effects of fibroblast-myocyte coupling on cardiac conduction and vulnerability to reentry: A computational study. Heart Rhythm. 2009;6:1641–9. doi: 10.1016/j.hrthm.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xie Y, Garfinkel A, Weiss JN, Qu Z. Cardiac alternans induced by fibroblast-myocyte coupling: mechanistic insights from computational models. Am J Physiol Heart Circ Physiol. 2009;297:H775–84. doi: 10.1152/ajpheart.00341.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xie Y, Liao Z, Grandi E, Shiferaw Y, Bers DM. Slow [Na]i Changes and Positive Feedback Between Membrane Potential and [Ca]i Underlie Intermittent Early Afterdepolarizations and Arrhythmias. Circ Arrhythm Electrophysiol. 2015;8:1472–80. doi: 10.1161/CIRCEP.115.003085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–15. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang L, Korge P, Weiss JN, Qu Z. Mitochondrial oscillations and waves in cardiac myocytes: insights from computational models. Biophys J. 2010;98:1428–38. doi: 10.1016/j.bpj.2009.12.4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yaniv Y, Stanley WC, Saidel GM, Cabrera ME, Landesberg A. The role of Ca2+ in coupling cardiac metabolism with regulation of contraction: in silico modeling. Ann N Y Acad Sci. 2008;1123:69–78. doi: 10.1196/annals.1420.009. [DOI] [PubMed] [Google Scholar]
- 132.Yao L, Fan P, Jiang Z, Viatchenko-Karpinski S, Wu Y, Kornyeyev D, Hirakawa R, Budas GR, Rajamani S, Shryock JC, Belardinelli L. Nav1.5-dependent persistent Na+ influx activates CaMKII in rat ventricular myocytes and N1325S mice. Am J Physiol Cell Physiol. 2011;301:C577–86. doi: 10.1152/ajpcell.00125.2011. [DOI] [PubMed] [Google Scholar]
- 133.Zahid S, Whyte KN, Schwarz EL, Blake RC, 3rd, Boyle PM, Chrispin J, Prakosa A, Ipek EG, Pashakhanloo F, Halperin HR, Calkins H, Berger RD, Nazarian S, Trayanova NA. Feasibility of using patient-specific models and the “minimum cut” algorithm to predict optimal ablation targets for left atrial flutter. Heart Rhythm. 2016;13:1687–98. doi: 10.1016/j.hrthm.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zeigler AC, Richardson WJ, Holmes JW, Saucerman JJ. A computational model of cardiac fibroblast signaling predicts context-dependent drivers of myofibroblast differentiation. J Mol Cell Cardiol. 2016;94:72–81. doi: 10.1016/j.yjmcc.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena MS, Belke DD, Dillmann WH, Rogers TB, Schulman H, Ross J, Jr, Brown JH. The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem. 2002;277:1261–7. doi: 10.1074/jbc.M108525200. [DOI] [PubMed] [Google Scholar]
- 136.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–9. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 137.Zhou L, Cortassa S, Wei AC, Aon MA, Winslow RL, O’Rourke B. Modeling cardiac action potential shortening driven by oxidative stress-induced mitochondrial oscillations in guinea pig cardiomyocytes. Biophys J. 2009;97:1843–52. doi: 10.1016/j.bpj.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhou L, Solhjoo S, Millare B, Plank G, Abraham MR, Cortassa S, Trayanova N, O’Rourke B. Effects of regional mitochondrial depolarization on electrical propagation: implications for arrhythmogenesis. Circ Arrhythm Electrophysiol. 2014;7:143–51. doi: 10.1161/CIRCEP.113.000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zile MA, Trayanova NA. Rate-dependent force, intracellular calcium, and action potential voltage alternans are modulated by sarcomere length and heart failure induced-remodeling of thin filament regulation in human heart failure: A myocyte modeling study. Prog Biophys Mol Biol. 2016;120:270–80. doi: 10.1016/j.pbiomolbio.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zile MA, Trayanova NA. Myofilament protein dynamics modulate EAD formation in human hypertrophic cardiomyopathy. Prog Biophys Mol Biol. 2017;130:418–428. doi: 10.1016/j.pbiomolbio.2017.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]