

Abstract
Following up the open initiative of anti-malarial drug discovery, a GlaxoSmithKline (GSK) phenotypic screening hit was developed to generate hydroxyethylamine based plasmepsin (Plm) inhibitors exhibiting growth inhibition of the malaria parasite Plasmodium falciparum at nanomolar concentrations. Lead optimization studies were performed with the aim of improving Plm inhibition selectivity versus the related human aspartic protease cathepsin D (Cat D). Optimization studies were performed using Plm IV as a readily accessible model protein, the inhibition of which correlates with anti-malarial activity. Guided by sequence alignment of Plms and Cat D, selectivity-inducing structural motifs were modified in the S3 and S4 sub-pocket occupying substituents of the hydroxyethylamine inhibitors. This resulted in potent anti-malarials with an up to 50-fold Plm IV/Cat D selectivity factor. More detailed investigation of the mechanism of action of the selected compounds revealed that they inhibit maturation of the P. falciparum subtilisin-like protease SUB1, and also inhibit parasite egress from erythrocytes. Our results indicate that the anti-malarial activity of the compounds is linked to inhibition of the SUB1 maturase plasmepsin subtype Plm X.
Keywords: Plasmepsins, Malaria, Plasmodium falciparum, Cathepsin D, Inhibitors, Hydroxyethylamine
Graphical abstract

Highlights
-
•
Peptidomimimetic plasmepsin inhibitors are developed using Plm IV as a model enzyme.
-
•
Up to 50-fold selectivity against Cathepsin D is reached.
-
•
Compounds show growth inhibition of P. falciparum at nanomolar concentrations.
-
•
Inhibition of SUB1 maturation and parasite egress imply (co)inhibition of Plm X.
1. Introduction
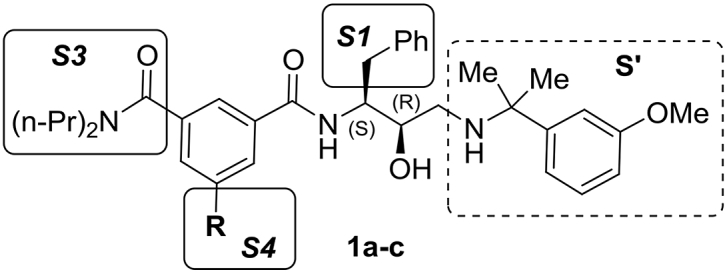
Malaria is a life-threatening disease caused by Plasmodium parasites which are transmitted by mosquitoes [1]. More than half of the earth's population lives in malaria endemic areas, rendering the disease a global health problem. Extensive eradication campaigns have been implemented, leading to considerably reduced malaria morbidity [2]. A key future goal, according to the Global Technical Strategy for Malaria 2016–2030, is a 90% reduction in clinical cases and deaths by 2030 as compared with 2015 [3]. However, these efforts are impeded by widespread resistance of the parasite to all currently used drugs, including artemisinins, the current front line drug class [[4], [5], [6]]. Consequently, new antimalarial drugs with new modes of action are urgently needed. Their development faces notable hurdles, one of which is a low expected profit after market approval. This has prompted several open innovation initiatives by private and academic organizations, including the disclosure of preclinical research data to the scientific community [[7], [8], [9], [10]]. To support one such initiative, GlaxoSmithKline (GSK) recently published the results of a large-scale cell-based (phenotypic) HTS screening campaign that provided a number of starting points for anti-malarial drug discovery [7]. From the pool of parasite growth inhibitory compounds we selected hydroxyethylamine derivative 1a for further development (Table 1) [11]. In our previous studies we showed that compound 1a is an inhibitor of the Plasmodium falciparum aspartic proteases - plasmepsin subtypes Plm I, Plm II and Plm IV with particularly high potency against Plm IV. Structurally simplified potent Plm IV inhibitors 1b,c were developed as compound 1a analogues, retaining high potency in P. falciparum growth assays (see Table 1).
Table 1.
Representative Plm inhibitors 1a-c from previous studies [11]
| Comp. | R | IC50 Plm IV, μΜ |
IC50 Cat D, μΜ |
EC50 Pf growth, μΜ |
|---|---|---|---|---|
| (S,R)-1a | 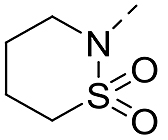 |
0.029 | 0.043 | 0.002 |
| (S,R)-1b | 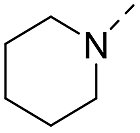 |
0.024 | 0.042 | 0.006 |
| (S,R)-1c | Ph | 0.006 | 0.054 | 0.002 |
It is important to note that despite more than two decades of research on plasmepsin inhibitor discovery, only a few compounds (including inhibitors 1a-c) exhibiting parasite growth inhibition at low nanomolar concentration have been identified [11,12]. This is likely attributable to the fact that most of the efforts so far have been devoted to inhibiting plasmepsin subtypes involved in hemoglobin digestion (Plms I-IV) [[13], [14], [15], [16], [17], [18], [19], [20], [21], [22]]. Gene disruption studies have revealed that none of these hemoglobinase Plms are essential in the parasite asexual blood stage lifecycle, indicating a high degree of redundancy in the hemoglobin catabolic pathway [[23], [24], [25]]. In contrast, the three other plasmepsin subtypes expressed in the P. falciparum blood stages, Plms V [[26], [27], [28]], IX, and X [[29], [30], [31], [32]], all appear to be essential for parasite viability. The hemoglobinase plasmepsins (Plm I, II, IV) share high sequence homology with Plms IX and X, but not Plm V. It might therefore be expected that inhibitors developed to target the hemoglobinase Plms would exhibit activity in cell-based assays only if they additionally target Plms IX and/or Plm X. Recombinant expression of both Plms IX and X has been recently reported [29,30], but this could be achieved only in higher eukaryotic protein expression systems, such as insect or mammalian cells. For our further work to develop the hydroxyethylamine based inhibitors (S,R)-1a-c as anti-malarials we therefore used Plm IV as a readily accessible model plasmepsin, an approach also supported by the previously observed good correlation between inhibition of this enzyme and potency in parasite growth assays in erythrocytes [11,20].
The successful development of protease inhibitors as drugs requires optimization of on-target potency and minimization of undesirable off-target activity, particularly against related host proteases. The human lysosomal aspartic protease cathepsin D (Cat D) plays critical roles in protein catabolism and retinal function [33]. Recent work focused on development of inhibitors of the human aspartic protease β-secretase (BACE1) revealed the importance of ensuring selectivity against Cat D in order to avoid off-target ocular toxicity [34,35]. In view of this, we decided that the next step for optimization of the hydroxyethylamine based Plm inhibitors 1 should aim to improve their selectivity for their malarial target(s) over human Cat D.
2. Results and discussion
2.1. Structural factors determining the selectivity of inhibitor binding to Plms vs Cat D
Our previous SAR investigations revealed that the substituents of the inhibitor (S,R)-1 occupying the prime sub-pockets (part S’, Table 1) are optimal for Plm inhibition [11]. Therefore, we focused our efforts on optimisation of selectivity inducing motifs in the substituents occupying the non-prime sub-pockets (Table 1). To assess the differences in inhibitor recognition between Plms IV, IX, X and Cat D, we generated a structure-based sequence alignment of these proteins and compared their interactions with inhibitor 1b in docking models (Fig. 1). Since Plm IX and X lack experimentally determined structures and in order to avoid possible inaccuracies associated with the use of homology models, the docking studies were performed on the crystal structure of Plm IV (PDB ID 2ANL), which is the closest homologue with an available crystal structure [36]. As can be seen from Fig. 1A, the S3 sub-pocket shows the largest differences in amino acid composition between the Plms and Cat D. Additionally, this revealed that the S3 sub-pocket of Plm IV is wider, more shallow and more hydrophobic than that of Cat D (Fig. 1B). For these reasons, selectivity improvement was first attempted by modifying the N,N-dipropylamide moiety in inhibitors (S,R)-1 which occupies the S3 sub-pocket.
Fig. 1.

(A) Structure-based sequence alignment of the amino acid residues making up the S1′ and S1—S4 pockets of Plm IV, Plm IX, Plm X and Cat D. (B) Surface representation of the S3 and S4 pockets in Plm IV and Cat D docking models with the hydroxyethylamine based inhibitor (S,R)-1b. The figure was prepared in PyMol [37]. Surface oxygen atoms are colored in red, nitrogens in blue, sulfurs in yellow and hydrogens and carbons in grey. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
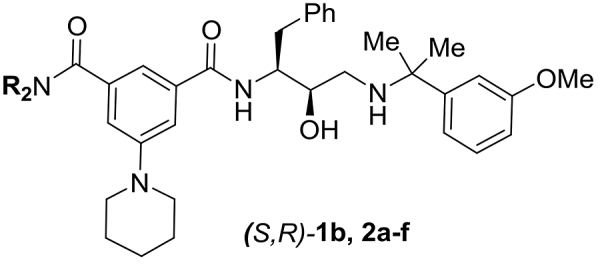
Several N,N-disubstituted amide analogues (S,R)-2a,b,d-f were synthesized (see Section 2.3) bearing hydrophobic groups that would prefer the more hydrophobic S3 sub-pocket of Plm IV. Unexpectedly the N,N-diethyl and N,N-dimethyl substituted analogues (S,R)-2a and (S,R)-2b showed the highest selectivity factor for Plm IV inhibition over Cat D, even though our docking studies indicated that the wider S3 sub-pocket of Plm IV could accommodate bulkier groups. Analogue (S,R)-2c bearing N-hydroxyethyl groups showed 3-fold weaker Plm IV inhibition potency than compound (S,R)-2a. Introduction of larger linear substituents such as N,N-dipropyl (compound (S,R)-1b), N,N-di(methoxyethyl) (compound (S,R)-2d) and N,N-di(3,3,3-trifluoropropyl) (compound (S,R)-2e) resulted in improved Cat D inhibition and correspondingly lower selectivity factors, suggesting that these groups fit well in the deep S3 sub-pocket of Cat D. Analogue (S,R)-2f bearing a N,N-diisobutyl amide showed poor inhibition of both enzymes, indicating that this group is too large to fit into the S3 sub-pocket of both Plm IV and Cat D.
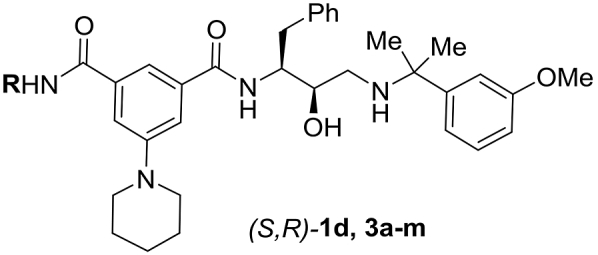
We further explored N-monosubstituted amide analogues (S,R)-3a-m (see Section 2.3 for synthesis). The best compounds in this series showed similar Plm IV inhibition potency compared to the most potent N,N-disubstituted amides (S,R)-2. Gratifyingly, these appeared to be less potent Cat D inhibitors, leading to improved selectivity factors. The removal of one N-substituent was more beneficial for compounds with larger or branched substituents (e.g. (S,R)-2e and (S,R)-2d compared to (S,R)-3d and (S,R)-3f) while for compounds bearing smaller substituents a slight decrease in Plm IV inhibitory activity was observed e.g. (S,R)-1b (Table 2) compared to (S,R)-1d (Table 3) (see Fig. 2 for docking of compounds 2e and 3d into Plm IV and Cat D representing the difference of steric requirements). The docking studies suggested that the introduction of a hydrogen bond donor (inhibitors (S,R)-3c,e,i,j) or hydrogen bond acceptor ((S,R)-3a,l,f) group in the S3 sub-pocket substituents could potentially enable additional electrostatic interactions with electron-rich functional groups in this pocket. Hydrogen bond donor groups could interact with Asn13 and Leu14 backbone carbonyl in Plm IV; and Asp323, Tyr15, Gln14 and Ala13 in CatD, whereas for the hydrogen bond acceptors the most likely interactions are with Asn13 in Plm IV and Gln14 in Cat D (Fig. S1, see supporting information). Although the introduction of hydrogen bond donor or acceptor groups in the S3 sub-pocket occupying substituent reduced Plm IV inhibition activity (up to 2 times if comparing 1b and 3a), it also produced the most selective ligand in this series - (S,R)-3a, as the activity decrease for Cat D is even higher (up to 19 times if comparing (S,R)-1b and (S,R)-3a). The docking studies suggest that the relatively higher drop in activity against Cat D for the compounds bearing the hydrogen bond donor or acceptor groups is due to an unfilled hydrophobic sub-pocket (resulting in an entropic penalty of solvating the non-polar sub-pocket). That is, the position of the amide substituent remains the same in both enzymes due to additional interactions with aforementioned residues (Fig. 3), but by doing so it creates a situation where the Cat D hydrophobic sub-pocket is filled with water resulting in an entropic penalty and reduced Cat D inhibition activity.
Table 2.
SAR of N,N-disubstituted amide analogues (S,R)-1b, 2a-f
| Comp. | R | IC50 Plm IV, μΜ |
IC50 Cat D, μΜ |
Sa |
|---|---|---|---|---|
| (S,R)-1b | n-Pr | 0.024b | 0.042b | 1.8 |
| (S,R)-2a | Et | 0.014 | 0.25 | 17.9 |
| (S,R)-2b | Me | 0.087 | 0.5 | 5.7 |
| (S,R)-2c | HOCH2CH2 | 0.068 | 0.27 | 4.0 |
| (S,R)-2d | MeOCH2CH2 | 0.037 | 0.10 | 2.7 |
| (S,R)-2e | CF3CH2CH2 | 0.21 | 0.12 | 0.57 |
| (S,R)-2f | (CH3)2CHCH2 | 0.5 | 1.3 | 2.6 |
Selectivity factor of Plm IV/Cat D inhibition.
Data from literature [11].
Table 3.
SAR of N-mono-substituted amide analogues (S,R)-1d, 3a-m
| Comp. | R | IC50 Plm IV, μΜ |
IC50 Cat D, μΜ |
Sa |
|---|---|---|---|---|
| (S,R)-1d | n-Pr | 0.038b | 0.11b | 2.9 |
| (S,R)-3a | MeOC(CH3)2CH2 | 0.048 | 2.1 | 43.8 |
| (S,R)-3b | c-PrCH2 | 0.030 | 0.76 | 25.3 |
| (S,R)-3c | HOCH2CH2CH2 | 0.093 | 2.25 | 24.2 |
| (S,R)-3d | CF3CH2CH2 | 0.024 | 0.58 | 24.2 |
| (S,R)-3e | HOC(CH3)2CH2 | 0.10 | 1.66 | 16.6 |
| (S,R)-3f | MeOCH2CH2 | 0.05 | 0.75 | 15.0 |
| (S,R)-3g | Me2NCH2CH2 | 0.36 | 4.8 | 13.3 |
| (S,R)-3h | t-BuCH2 | 0.027 | 0.40 | 14.8 |
| (S,R)-3i | HOCH2C(CH3)2 | 0.12 | 1.46 | 12.2 |
| (S,R)-3j | HOCH2CH2 | 0.21 | 1.42 | 6.8 |
| (S,R)-3k | PhCH2 | 0.038 | 0.22 | 5.8 |
| (S,R)-3l | t-BuOCH2CH2 | 0.031 | 0.15 | 4.8 |
| (S,R)-3m | c-HexCH2 | 0.09 | 0.15 | 1.7 |
Selectivity factor of Plm IV/Cat D inhibition.
Data from literature [11].
Fig. 2.

Docking models of compounds (S,R)-2e and (S,R)-3d in crystal structures of Plm IV and Cat D.
Fig. 3.

Docking models of compound 3a in complex with Plm IV and Cat D.
Despite varying hydrogen bond donor and acceptor groups in the S3 sub-pocket filling substituents, docking studies also suggest that the main factor affecting Plm IV and Cat D inhibition potency is the size and shape of the substituent; branched and long substituents cannot fit into the narrow S3 recess of Cat D whereas the open S3 sub-pocket of Plm IV can accommodate such substituents. The weakest inhibitor of the N-mono-substituted amide analogues was compound (S,R)-3g. This could be explained by the protonated amine group as the amide substituent which, according to the docking studies, tends to form hydrogen bonds and ionic interactions with residues outside of the S3 sub-pocket. Altogether these results indicate that the selectivity against Cat D can be improved by targeting the S3 sub-pocket with mono-substituted amide moieties containing linear or branched hydrophobic groups.
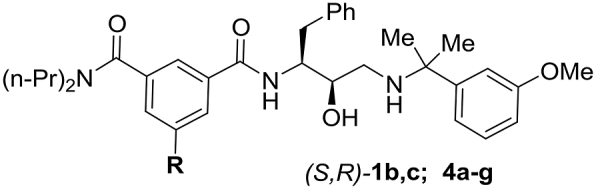
The S4 sub-pocket is another inhibitor binding region which is notably distinct between Plm IV and Cat D (Fig. 1A). Although predominantly hydrophobic in both enzymes, the S4 sub-pocket of Plm IV is flatter and more solvent exposed. Therefore, we investigated SAR for substituents occupying the S4 sub-pocket in the series of compounds (S,R)-4a-g (Table 4, see Section 2.3. for synthesis). Installation of an N,N-dipropylamide group (compound (S,R)-4g) resulted in practically unchanged Plm IV inhibitory activity compared to parent compound 1b, however this was paralleled by a more than 15-fold drop in activity against Cat D resulting in improvement of the selectivity factor. Installation of fluorine or removal of the S4 filling substituent provided compounds 4b,c with slightly decreased Plm IV inhibitory potency, whereas inhibition of Cat D was considerably lower which again improved the selectivity factor. Installation of chlorine in the benzene ring (compound 4d) improved the activity for both Plm IV and Cat D. Cyano, methyl and trifluoromethyl groups in the S4 sub-pocket (compounds 4a,e-f) made less difference for inhibitor binding to Plm IV and Cat D. Collectively, these effects of S4 occupying substituents are difficult to explain from our molecular modelling data, and presumably arise from an interplay of hydrophobic and polar non-covalent interactions.
Table 4.
SAR of phenylgroup substitution in analogues 1b,c, 4a-g
| Comp. | R | IC50 Plm IV, μΜ |
IC50 Cat D, μΜ |
Sa |
|---|---|---|---|---|
| (S,R)-1b | 1-piperidinyl | 0.024b | 0.042b | 1.8 |
| (S,R)-1c | Ph | 0.006b | 0.054b | 9.0 |
| (S,R)-4a | Me | 0.023 | 0.21 | 9.1 |
| (S,R)-4b | H | 0.058 | 1.15 | 19.8 |
| (S,R)-4c | F | 0.050 | 1.0 | 20 |
| (S,R)-4d | Cl | 0.008 | 0.096 | 12.0 |
| (S,R)-4e | CF3 | 0.015 | 0.067 | 4.5 |
| (S,R)-4f | CN | 0.059 | 0.56 | 9.5 |
| (S,R)-4g | (n-Pr)2NC(=O) | 0.018 | 0.7 | 38.9 |
Selectivity factor of Plm IV/Cat D inhibition.
Data from literature [11].
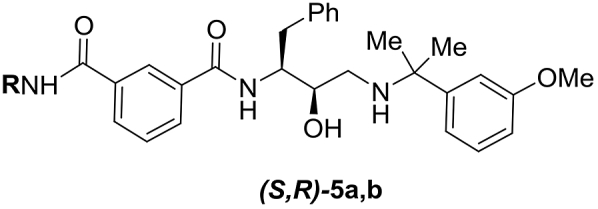
Considering that the lack of an S4 occupying substituent considerably improves selectivity against Cat D, analogues (S,R)-5a,b of the most potent N-mono-substituted amide inhibitors (S,R)-3d,h were prepared and tested (Table 5, see Section 2.3. for synthesis). As expected, a slight drop in Plm IV inhibitory potency was observed; however the modifications appeared to be additive for a considerable improvement in the selectivity factor.
Table 5.
Combining selectivity inducing structural motives in analogues (S,R)-5a,b
| Comp. | R | IC50 Plm IV, μΜ |
IC50 Cat D, μΜ |
Sa |
|---|---|---|---|---|
| (S,R)-5a | t-BuCH2 | 0.076 | 3.8 | 50.0 |
| (S,R)-5b | CF3CH2CH2 | 0.15 | 4.9 | 32.6 |
Selectivity factor of Plm IV/Cat D inhibition.
2.2. Relation of Plm subtype inhibition with P. falciparum growth inhibition
The capacity to inhibit growth in vitro of asexual blood stage P. falciparum was determined for selected compounds (S,R)-2a,3a,b,d,h,4b,c,g,5a,b (Table 6). In all cases, the compounds showed strong growth inhibitory potency with EC50 values in the low nanomolar range. Inhibitory activity against Plm I, Plm II and Plm IV was determined for the same compounds. This revealed no correlation between inhibitory potency in the parasite growth assay and inhibition of Plm I and II enzyme activity. In contrast, there was a much better correlation between parasite growth inhibition potency and Plm IV inhibition potency, although even here there were some notable exceptions; for instance, the most potent growth inhibitory compound, (S,R)-4b, was a 3-fold weaker inhibitor of Plm IV than compound (S,R)-4g, yet showed a 20-fold better inhibition of parasite growth than (S,R)-4g.
Table 6.
Plm I, II, IV inhibition and P. falciparum growth inhibition activity of selected compounds.
| Comp. | IC50 Plm I, μΜ |
IC50 Plm II, μΜ |
IC50 Plm IV, μΜ |
Sa | EC50b Pf Growth, nΜ |
|---|---|---|---|---|---|
| (S,R)-2a | 0.8 | 0.16 | 0.014 | 17.9 | 1.5 |
| (S,R)-3a | 7.4 | 5.4 | 0.048 | 43.8 | 2.0 |
| (S,R)-3b | 1.8 | 0.5 | 0.030 | 25.3 | 1.8 |
| (S,R)-3d | 2.5 | 2.2 | 0.024 | 24.2 | 2.0 |
| (S,R)-3h | 2.0 | 0.85 | 0.027 | 14.8 | 6.0 |
| (S,R)-4b | 3.1 | 1.7 | 0.058 | 19.8 | 0.3 |
| (S,R)-4c | 1.1 | 1.1 | 0.050 | 20 | 1.5 |
| (S,R)-4g | 0.78 | 0.27 | 0.018 | 38.9 | 6.0 |
| (S,R)-5a | 5.6 | 7.1 | 0.076 | 50.0 | 2.0 |
| (S,R)-5b | 10.3 | 10.4 | 0.15 | 32.6 | 6.0 |
Selectivity factor of Plm IV/Cat D inhibition.
The EC50 values for P. falciparum growth were determined using a SYBR Green-based assay with an incubation time of 96 h (2 erythrocytic cycles). Samples were each measured in triplicate, in 2 separate biological assays. Compound TCMDC-13467411,20 was used as a positive control (see Supporting Information).
These results implied that the important parasite target(s) engaged by the growth inhibitory compounds are not the hemoglobinase plasmepsins. Recent reports have shown that inhibitors of the non-hemoglobinase plasmepsins Plm IX and Plm X (which are structurally similar to Plm I, II and IV) can potently block parasite replication [29,30]. A key biological function of Plm X is the proteolytic maturation of SUB1, a parasite subtilisin-like serine protease that plays an essential role in regulating parasite release (egress) from the infected host erythrocyte [38]. SUB1 maturation comprises 2 steps in which the initial ∼82 kDa pre-proenzyme is cleaved to form first a 54 kDa protein (p54) then a 47 kDa terminal product (p47) which accumulates during the latter ∼12 h of intra-erythrocytic parasite development. Whilst the first SUB1 processing step is autocatalytic, the second p54-to-p47 step is believed to be mediated by Plm X [29,30]. We used a Western blot-based assay to examine the effects of selected compounds (S,R)-2a, (S,R)-4b and (S,R)-4c on both SUB1 maturation and parasite egress (Fig. 4).
Fig. 4.

Inhibition of P. falciparum SUB1 maturation and egress by selected compounds indicates that they target Plm X. (A) Synchronous cultures of immature intracellular parasites were treated for ∼8 h with compounds (S,R)-2a or (S,R)-4b,c (10 nM), or vehicle only (DMSO, 1% v/v), or the cGMP-dependent protein kinase inhibitor (4-[7-[(dimethylamino)methyl]-2-(4-fluorphenyl)imidazo[1,2-α]pyridine-3-yl]pyrimidin-2-amine (compound 2; C2, 2 μM) which inhibits egress but not SUB1 maturation. Extracts of the parasites were then analyzed by Western blot, probing with an antibody to SUB1. The positions of migration of the SUB1 p54 and p47 forms (green arrow) are indicated. The schematic below indicates the mode by which Plm X converts SUB1 p54 to the terminal p47 form. (B) Parasites treated for ∼ 24 h as in (A) were transferred to fresh medium containing the various compounds and allowed to undergo egress for 4 h before the culture supernatants were analysed by Western blot, probing with antibodies to the parasite protein SERA5. The positions of migration of the SERA5 precursor, a processing intermediate and the terminal P50 form (green arrow) are indicated. The schematic below indicates the mode by which SUB1 converts the SERA5 precursor to the P50 form. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Egress was quantified by measuring the release of a soluble parasite protein called SERA5 into parasite culture supernatants. SERA5 is also a SUB1 substrate, and is generally released in a P50 form that results from SUB1-mediated cleavage of a larger precursor. Release of correctly processed SERA5 P50 is therefore an indicator of both SUB1 activity and efficiency of egress. As shown in Fig. 4A, treatment of developing intracellular parasites with the three Plm inhibitors (S,R)-2a, (S,R)-4b and (S,R)-4c resulted in a relative enrichment of the p54 form of SUB1, indicating inhibition of conversion of p54 to the terminal p47 form. Analysis of culture supernatants from treated parasites allowed to proceed to egress (Fig. 4B) showed that compounds (S,R)-4b and (S,R)-4c both produced a clear reduction in SERA5 P50 release, whilst all three compounds produced an increase in the release of SERA5 precursor or processing intermediates, indicating defects in egress and SERA5 processing. These results strongly suggest that the mechanism of parasite growth inhibition by compounds (S,R)-4b and (S,R)-4c (as well as possibly (S,R)-2a) involves Plm X inhibition, since the effects on egress were linked to inhibition of SUB1 maturation and its enzymatic activity against an endogenous substrate. Given their structural similarity, it is likely that the other compounds 3a,b,d,h, 4g and 5a,b exerting potency in the parasite growth assay (Table 6) also target Plm X.
2.3. Synthesis of inhibitors (S,R)–2-5
Plm inhibitors (S,R)–2-5 were synthesized from substituted benzoic acids 15, 17 and 19 and either enantiomerically pure amino alcohol (R,S)–11 or the corresponding racemate rac-11 (Scheme 1, Scheme 2, Scheme 3, Scheme 4, Scheme 5). Inhibitors (S,R)–2b-e, (S,R)–3a,c,e-g,i,j,l,m, (S,R)–4c,f,g and (S,R)–5a,b were obtained from enantiomerically pure amino alcohol (R,S)–11, whereas targets 2a,f, 3b,d,h,k and 4a,b,d,e were obtained as mixtures of stereoisomers from racemic alcohol rac-11. Enantiomerically pure inhibitors (S,R)–2a,f, (S,R)–3b,d,h,k and (S,R)–4a,b,d,e were obtained by separation of diastereomers using chromatography on silica gel, followed by separation of enantiomers using the chromatography on a chiral stationary phase.
Scheme 1.

Synthesis of amino alcohol intermediate (R,S)–11. Reagents and conditions: a) Boc2O (1.25 equiv), NEt3 (2 equiv), DCM, rt, 2 h, 87%. b) AD-mix-α (1 equiv), 1:1 (v/v) t-BuOH:water, rt, 20 h c) preparative HPLC on chiral stationary phase (Chiralpak-ID), 25% in two steps. d) Ph3P (1.1 equiv), DEAD (1.1 equiv), CHCl3, 85 °C, 48 h, 64%. e) 2-(3-Methoxyphenyl)propan-2-amine [11] (1.05 equiv), i-PrOH, 70 °C, 40 h, 73%. f) 4 M HCl in dioxane, rt, 6 h, 99%.
Scheme 2.

Synthesis of Plm inhibitors (S,R)–2a-f and (S,R)–3a-m. Reagents and conditions: a) piperidine (1 equiv), Pd(OAc)2 (5 mol%), rac-BINAP (5 mol%), Cs2CO3 (1.5 equiv), toluene, 100 °C, 18 h, 88%. b) General procedure A: aqueous 1 M NaOH (1 equiv), MeOH, rt, 16 h c) General procedure B: R1R2NH (1.2 equiv), HBTU (1 equiv), NEt3 (2 equiv), DMF, rt, 2 h d) General procedure C: aqueous 1 M NaOH (1.5 equiv), MeOH, 50 °C, 18 h e) General procedure D: amino alcohol (R,S)–11 (1.0 equiv), HBTU (1.0 equiv), NEt3 (4 equiv), DMF, rt, 16 h f) amino alcohol rac–11 (1.0 equiv), HBTU (1.0 equiv), NEt3 (4 equiv), DMF, rt, 16 h; then separation of enantiomers by HPLC on chiral stationary phase (Chiralpak-ID).
Scheme 3.

Synthesis of Plm inhibitors (S,R)–4a-f. Reagents and conditions: a) MeB(OH)2 (1.2 equiv), Pd(dppf)Cl2xCH2Cl2 (5 mol%), K3PO4 (3 equiv), toluene, 90 °C, 18 h b) General procedure A: aqueous 1 M NaOH (1 equiv), MeOH, rt, 16 h c) General procedure B: (n-Pr)2NH (1.2 equiv), HBTU (1 equiv), NEt3 (2 equiv), DMF, rt, 2 h d) General procedure C: aqueous 1 M NaOH (1.5 equiv), MeOH, 50 °C, 18 h e) CuCN (2 equiv), NMP, 160 °C, 6 h f) General procedure D: amino alcohol (R,S)–11 (1.0 equiv), HBTU (1.0 equiv), NEt3 (4 equiv), DMF, rt, 16 h g) amino alcohol rac–11 (1.0 equiv), HBTU (1.0 equiv), NEt3 (4 equiv), DMF, rt, 16 h; then separation of enantiomers by HPLC on chiral stationary phase (Chiralpak-ID).
Scheme 4.

Synthesis of Plm inhibitor (S,R)–4g. Reagents and conditions: a) aqueous 1 N NaOH (3 equiv), MeOH, 40 °C, 18 h b) Pd(dppf)Cl2xCH2Cl2 (10 mol%), NEt3 (2.2 equiv), CO (70 psi), MeOH, 100 °C, 18 h c) General procedure B: (n-Pr)2NH (2.2 equiv), HBTU (2 equiv), NEt3 (4 equiv), DMF, rt, 2 h d) General procedure C: aqueous 1 M NaOH (1.5 equiv), MeOH, 50 °C, 18 h e) General procedure D: amino alcohol (R,S)–11 (1.0 equiv), HBTU (1.0 equiv), NEt3 (4 equiv), DMF, rt, 16 h.
Scheme 5.

Synthesis of Plm inhibitors 5a,b. Reagents and conditions: a) General procedure B: (n-Pr)2NH (1.2 equiv), HBTU (1 equiv), NEt3 (2 equiv), DMF, rt, 2 h b) General procedure C: aqueous 1 M NaOH (1.5 equiv), MeOH, 50 °C, 18 h c) General procedure D: amino alcohol (R,S)–11 (1.0 equiv), HBTU (1.0 equiv), NEt3 (4 equiv), DMF, rt, 16 h.
Synthesis of aminoalcohol 11 commenced with N-Boc protection of rac-6 (Scheme 1). Dihydroxylation of the resulting carbamate rac–7 with AD-mix-α afforded diol 8 as a 2:3 mixture of syn:anti diastereomers and a 2:1 mixture of enantiomers. Enantiomerically pure diol (S,S)–8 was obtained by an initial separation of syn/anti diastereomers using chromatography on silica gel, followed by separation of syn–8 enantiomers by chromatography on a chiral stationary phase. The major enantiomer turned out to be the desired diol (S,S)–8 as evidenced by comparison of optical rotation data with that from the literature [39]. Conversion of (S,S)–8 into epoxide (S,S)–9 under Mitsunobu conditions was followed by aminolysis with 2-(3-methoxyphenyl)propan-2-amine [11] to afford N-Boc protected amino alcohol (S,R)–10. Finally, the cleavage of N-Boc protecting group yielded amino alcohol (R,S)–11.
Benzoic acids 15a-s were prepared from dimethyl 5-bromoisophthalate (Scheme 2). Pd-catalyzed amination with piperidine afforded 12a, which was hydrolysed to isophthalic monoester 13a under basic conditions. Subsequent HBTU-mediated condensation with amines afforded amides 14a-s, which were hydrolysed to benzoic acids 15a-s. (see Table 7)
Table 7.
Substitution pattern of compounds in Scheme 2.
| R1 | R2 | No. | No. | No. | Comments |
|---|---|---|---|---|---|
| Et | Et | 14a | 15a | (S,R)-2a | from rac–11 (step f) |
| Me | Me | 14b | 15b | (S,R)-2b | |
| HOCH2CH2 | HOCH2CH2 | 14c | 15c | (S,R)-2c | |
| MeOCH2CH2 | MeOCH2CH2 | 14d | 15d | (S,R)-2d | |
| CF3CH2CH2 | CF3CH2CH2 | 14e | 15e | (S,R)-2e | |
| (CH3)2CHCH2 | (CH3)2CHCH2 | 14f | 15f | (S,R)-2f | from rac–11 (step f) |
| H | MeOC(CH3)2CH2 | 14g | 15g | (S,R)-3a | |
| H | c-PrCH2 | 14h | 15h | (S,R)-3b | from rac–11 (step f) |
| H | HOCH2CH2CH2 | 14i | 15i | (S,R)-3c | |
| H | CF3CH2CH2 | 14j | 15j | (S,R)-3d | from rac–11 (step f) |
| H | HOC(CH3)2CH2 | 14k | 15k | (S,R)-3e | |
| H | MeOCH2CH2 | 14l | 15l | (S,R)-3f | |
| H | Me2NCH2CH2 | 14m | 15m | (S,R)-3g | |
| H | t-BuCH2 | 14n | 15n | (S,R)-3h | from rac–11 (step f) |
| H | HOCH2C(CH3)2 | 14o | 15o | (S,R)-3i | |
| H | HOCH2CH2 | 14p | 15p | (S,R)-3j | |
| H | PhCH2 | 14q | 15q | (S,R)-3k | from rac–11 (step f) |
| H | t-BuOCH2CH2 | 14r | 15r | (S,R)-3l | |
| H | c-HexCH2 | 14s | 15s | (S,R)-3m |
Similar synthetic approach was also used for the preparation of acids 17b-i (Scheme 3). Accordingly, monoesters 13c-f were obtained from commercially available dimethyl isophthates. The synthesis of methyl ester 13b required an initial Pd-catalyzed alkylation of dimethyl 5-bromoisophthalate with methylboronic acid, followed by hydrolysis of one of the two ester moieties. Benzoic acids 13b-f were converted into amides 16b-f and ester moieties were hydrolysed to afford acids 17b-f. Benzoic acid 17h was obtained directly from trifluoromethyl isophthalic acid and n-Pr2NH in the presence of HBTU, whereas the synthesis of 17i was accomplished by Cu(I)-catalyzed substitution of bromide in benzoic acid 17c for cyano group (Scheme 3). (see Table 8)
Table 8.
Substitution pattern of compounds in Scheme 3.
| R | No. | No. | No. | No. | No. | Comments |
|---|---|---|---|---|---|---|
| Me | 12b | 13b | 16b | 17b | (S,R)–4a | from rac–11 (step g) |
| Br | - | 13c | 16c | 17c | – | |
| H | - | 13d | 16d | 17d | (S,R)–4b | from rac–11 (step g) |
| F | - | 13e | 16e | 17e | (S,R)–4c | |
| Cl | - | 13f | 16f | 17f | (S,R)–4d | from rac–11 (step g) |
| CF3 | - | - | - | 17h | (S,R)–4e | from rac–11 (step g) |
| CN | - | - | - | 17i | (S,R)–4f |
Synthesis of benzoic acid 17g (Scheme 4) commenced with hydrolysis of commercially available dimethyl iodo-isophthalate to the corresponding isophthalic acid 18, followed by Pd-catalyzed methoxycarbonylation [40] to afford ester 13g. Subsequent conversion to diamide 16g in the presence of HBTU was followed by ester hydrolysis to form benzoic acid 17g (Scheme 4). The amide bond formation-hydrolysis sequence was also used for the preparation of benzoic acids 19a,b (Scheme 5).
3. Summary
The optimization of hydroxyethylamine based Plm inhibitors was performed with the aim of improving selectivity against the related human aspartic protease Cat D. The studies were performed using Plm IV as a readily accessible model protein, the inhibition of which was previously found to correlate with Plasmodium falciparum growth inhibition. Based on sequence alignment of Plm IV and Cat D, putative selectivity inducing structural motifs were sought in S3 and S4 sub-pocket-occupying substituents of the inhibitors. Installation of an S3 sub-pocket targeting mono-substituted amide moiety in compounds (S,R)-3 containing linear or branched hydrophobic groups resulted in up to 40-fold selectivity (compound (S,R)-3a) against Cat D. Plm IV inhibitors (S,R)-4b,c with no substituents or fluorine targeting the S4 sub-pocket led to 20-fold selectivity against Cat D, though with some loss of Plm IV inhibition potency. Surprisingly, installation of amide as the S4 sub-pocket filling group in compound (S,R)-4g resulted in potent Plm IV inhibition with almost 40-fold selectivity against Cat D. Selectivity-inducing factors in S3 and S4 positions were additive as evidenced by compound (S,R)-5a (50-fold selectivity). Determination of P. falciparum growth inhibition potency for ten of the new Plm inhibitors showed them to display activities in the low nanomolar range. The potent anti-malarial activity did not correlate with the relatively weak inhibition of Plm I and II, whilst in contrast there was a much better correlation with Plm IV inhibition. More detailed investigation of the mechanism of action of the selected compounds showed that they interfered with parasite egress and maturation of the parasite serine protease SUB1, indicating a strong link between anti-malarial activity and inhibition of the non-hemoglobinase plasmepsin and SUB1 maturase Plm X. Future studies should clarify whether cooperative Plm IV and Plm X inhibition or only Plm X inhibition is necessary to achieve optimal anti-malarial activity.
Acknowledgement
This work was supported by the European Regional Development Fund (Agreement No. 1.1.1.1/16/A/290).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2018.11.068.
Contributor Information
Edgars Suna, Email: edgars@osi.lv.
Aigars Jirgensons, Email: aigars@osi.lv.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.WHO . World Health Organ; Geneva: 2016. World Malar. Rep. 2016.http://www.who.int/malaria/publications/world-malaria-report-2016/report/en/ [Google Scholar]
- 2.Wells T.N.C., van Huijsduijnen R.H., Van Voorhis W.C. Malaria medicines: a glass half full? Nat. Rev. Drug Discov. 2015;14(6):424–442. doi: 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- 3.WHO, http://www.Who.Int/Malaria/Publications/Atoz/9789241564991/En/.
- 4.Wells T.N.C., Alonso P.L., Gutteridge W.E. New medicines to improve control and contribute to the eradication of malaria. Nat. Rev. Drug Discov. 2009;8(11):879–891. doi: 10.1038/nrd2972. [DOI] [PubMed] [Google Scholar]
- 5.Hyde J.E. Drug-resistant malaria − an insight. FEBS J. 2007;274(18):4688–4698. doi: 10.1111/j.1742-4658.2007.05999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dondorp A.M., Nosten F., Yi P., Das D., Phyo A.P., Tarning J., Lwin K.M., Ariey F., Hanpithakpong W., Lee S.J. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009;361(5):455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gamo F.-J., Sanz L.M., Vidal J., de Cozar C., Alvarez E., Lavandera J.-L., Vanderwall D.E., Green D.V.S., Kumar V., Hasan S. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465(7296):305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 8.Calderón F., Barros D., Bueno J.M., Coterón J.M., Fernández E., Gamo F.J., Lavandera J.L., León M.L., Macdonald S.J.F., Mallo A. An invitation to open innovation in malaria drug discovery: 47 quality starting points from the TCAMS. ACS Med. Chem. Lett. 2011;2(10):741–746. doi: 10.1021/ml200135p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wells T.N.C., Willis P., Burrows J.N., van Huijsduijnen R.H. Open data in drug discovery and development: lessons from malaria. Nat. Rev. Drug Discov. 2016;15:661. doi: 10.1038/nrd.2016.154. [DOI] [PubMed] [Google Scholar]
- 10.Pieroni M., Azzali E., Basilico N., Parapini S., Zolkiewski M., Beato C., Annunziato G., Bruno A., Vacondio F., Costantino G. Accepting the invitation to open innovation in malaria drug discovery: synthesis, biological evaluation, and investigation on the structure–activity relationships of benzo[b]thiophene-2-carboxamides as antimalarial agents. J. Med. Chem. 2017;60(5):1959–1970. doi: 10.1021/acs.jmedchem.6b01685. [DOI] [PubMed] [Google Scholar]
- 11.Jaudzems K., Tars K., Maurops G., Ivdra N., Otikovs M., Leitans J., Kanepe-Lapsa I., Domraceva I., Mutule I., Trapencieris P. Plasmepsin inhibitory activity and structure-guided optimization of a potent hydroxyethylamine-based antimalarial hit. ACS Med. Chem. Lett. 2014;5(4):373–377. doi: 10.1021/ml4004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciana C.-L., Siegrist R., Aissaoui H., Marx L., Racine S., Meyer S., Binkert C., de Kanter R., Fischli C., Wittlin S. Novel in vivo active anti-malarials based on a hydroxy-ethyl-amine scaffold. Bioorg. Med. Chem. Lett. 2013;23(3):658–662. doi: 10.1016/j.bmcl.2012.11.118. [DOI] [PubMed] [Google Scholar]
- 13.Haque T.S., Skillman A.G., Lee C.E., Habashita H., Gluzman I.Y., Ewing T.J.A., Goldberg D.E., Kuntz I.D., Ellman J.A. Potent, low-molecular-weight non-peptide inhibitors of malarial aspartyl protease plasmepsin II. J. Med. Chem. 1999;42(8):1428–1440. doi: 10.1021/jm980641t. [DOI] [PubMed] [Google Scholar]
- 14.Nöteberg D., Hamelink E., Hultén J., Wahlgren M., Vrang L., Samuelsson B., Hallberg A. Design and synthesis of plasmepsin I and plasmepsin II inhibitors with activity in Plasmodium falciparum-infected cultured human erythrocytes. J. Med. Chem. 2003;46(5):734–746. doi: 10.1021/jm020951i. [DOI] [PubMed] [Google Scholar]
- 15.Ersmark K., Samuelsson B., Hallberg A. Plasmepsins as potential targets for new antimalarial therapy. Med. Res. Rev. 2006;26(5):626–666. doi: 10.1002/med.20082. [DOI] [PubMed] [Google Scholar]
- 16.Ersmark K., Feierberg I., Bjelic S., Hamelink E., Hackett F., Blackman M.J., Hultén J., Samuelsson B., Åqvist J., Hallberg A. Potent inhibitors of the Plasmodium falciparum enzymes plasmepsin I and II devoid of cathepsin D inhibitory activity. J. Med. Chem. 2004;47(1):110–122. doi: 10.1021/jm030933g. [DOI] [PubMed] [Google Scholar]
- 17.Boss C., Corminboeuf O., Grisostomi C., Meyer S., Jones A.F., Prade L., Binkert C., Fischli W., Weller T., Bur D. Achiral, cheap, and potent inhibitors of plasmepsins I, II, and IV. ChemMedChem. 2006;1(12):1341–1345. doi: 10.1002/cmdc.200600223. [DOI] [PubMed] [Google Scholar]
- 18.Meyer M.J., Goldberg D.E. Recent advances in plasmepsin medicinal chemistry and implications for future antimalarial drug discovery efforts - Curr. Top. Med. Chem. 2012;12(5):445–455. doi: 10.2174/156802612799362959. [DOI] [PubMed] [Google Scholar]
- 19.Kumar Singh A., Rajendran V., Singh S., Kumar P., Kumar Y., Singh A., Miller W., Potemkin V., Poonam, Grishina M. Antiplasmodial activity of hydroxyethylamine analogs: synthesis, biological activity and structure activity relationship of plasmepsin inhibitors. Bioorg. Med. Chem. 2018;26(13):3837–3844. doi: 10.1016/j.bmc.2018.06.037. [DOI] [PubMed] [Google Scholar]
- 20.Rasina D., Otikovs M., Leitans J., Recacha R., Borysov O.V., Kanepe-Lapsa I., Domraceva I., Pantelejevs T., Tars K., Blackman M.J. Fragment-based discovery of 2-aminoquinazolin-4(3H)-Ones as novel class nonpeptidomimetic inhibitors of the plasmepsins I, II, and IV. J. Med. Chem. 2016;59(1):374–387. doi: 10.1021/acs.jmedchem.5b01558. [DOI] [PubMed] [Google Scholar]
- 21.Rasina D., Stakanovs G., Borysov O.V., Pantelejevs T., Bobrovs R., Kanepe-Lapsa I., Tars K., Jaudzems K., Jirgensons A. 2-Aminoquinazolin-4(3H)-One based plasmepsin inhibitors with improved hydrophilicity and selectivity. Bioorg. Med. Chem. 2018;26(9):2488–2500. doi: 10.1016/j.bmc.2018.04.012. [DOI] [PubMed] [Google Scholar]
- 22.Kinena L., Leitis G., Kanepe-Lapsa I., Bobrovs R., Jaudzems K., Ozola V., Suna E., Jirgensons A. Azole-based non-peptidomimetic plasmepsin inhibitors. Arch. Pharm. 2018;351(9) doi: 10.1002/ardp.201800151. 1800151. [DOI] [PubMed] [Google Scholar]
- 23.Bonilla J.A., Bonilla T.D., Yowell C.A., Fujioka H., Dame J.B. Critical roles for the digestive vacuole plasmepsins of Plasmodium falciparum in vacuolar function. Mol. Microbiol. 2007;65(1):64–75. doi: 10.1111/j.1365-2958.2007.05768.x. [DOI] [PubMed] [Google Scholar]
- 24.Bonilla J.A., Moura P.A., Bonilla T.D., Yowell C.A., Fidock D.A., Dame J.B. Effects on growth, hemoglobin metabolism and paralogous Gene expression resulting from disruption of genes encoding the digestive vacuole plasmepsins of Plasmodium falciparum. Int. J. Parasitol. 2007;37(3–4):317–327. doi: 10.1016/j.ijpara.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 25.Moura P.A., Dame J.B., Fidock D.A. Role of Plasmodium falciparum digestive vacuole plasmepsins in the specificity and antimalarial mode of action of cysteine and aspartic protease inhibitors. Antimicrob. Agents Chemother. 2009;53(12):4968–4978. doi: 10.1128/AAC.00882-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hodder A.N., Sleebs B.E., Czabotar P.E., Gazdik M., Xu Y., O'Neill M.T., Lopaticki S., Nebl T., Triglia T., Smith B.J. Structural basis for plasmepsin V inhibition that blocks export of malaria proteins to human erythrocytes. Nat. Struct. Mol. Biol. 2015;22(8):590–596. doi: 10.1038/nsmb.3061. [DOI] [PubMed] [Google Scholar]
- 27.Sleebs B.E., Gazdik M., O'Neill M.T., Rajasekaran P., Lopaticki S., Lackovic K., Lowes K., Smith B.J., Cowman A.F., Boddey J.A. Transition state mimetics of the Plasmodium export element are potent inhibitors of plasmepsin V from P. Falciparum and P. Vivax. J. Med. Chem. 2014;57(18):7644–7662. doi: 10.1021/jm500797g. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen W., Hodder A.N., de Lezongard R.B., Czabotar P.E., Jarman K.E., O'Neill M.T., Thompson J.K., Jousset Sabroux H., Cowman A.F., Boddey J.A. Enhanced antimalarial activity of plasmepsin V inhibitors by modification of the P2 position of PEXEL peptidomimetics. Eur. J. Med. Chem. 2018;154:182–198. doi: 10.1016/j.ejmech.2018.05.022. [DOI] [PubMed] [Google Scholar]
- 29.Pino P., Caldelari R., Mukherjee B., Vahokoski J., Klages N., Maco B., Collins C.R., Blackman M.J., Kursula I., Heussler V. A multistage antimalarial targets the plasmepsins IX and X essential for invasion and egress. Science. 2017;358(6362):522. doi: 10.1126/science.aaf8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nasamu A.S., Glushakova S., Russo I., Vaupel B., Oksman A., Kim A.S., Fremont D.H., Tolia N., Beck J.R., Meyers M.J. Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion. Science. 2017;358(6362):518. doi: 10.1126/science.aan1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boddey J.A. Plasmepsins on the antimalarial hit list. Science. 2017;358(6362):445. doi: 10.1126/science.aaq0002. [DOI] [PubMed] [Google Scholar]
- 32.Crunkhorn S. Blocking malaria parasite invasion and egress. Nat. Rev. Drug Discov. 2017;17:17. doi: 10.1038/nrd.2017.253. [DOI] [PubMed] [Google Scholar]
- 33.Steinfeld R., Reinhardt K., Schreiber K., Hillebrand M., Kraetzner R., Brück W., Saftig P., Gärtner J. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am. J. Hum. Genet. 2006;78(6):988–998. doi: 10.1086/504159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zuhl A.M., Nolan C.E., Brodney M.A., Niessen S., Atchison K., Houle C., Karanian D.A., Ambroise C., Brulet J.W., Beck E.M. Chemoproteomic profiling reveals that cathepsin D off-target activity drives ocular toxicity of β-secretase inhibitors. Nat. Commun. 2016;7 doi: 10.1038/ncomms13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghosh A.K., Reddy B.S., Yen Y.-C., Cárdenas E.L., Rao K.V., Downs D., Huang X., Tang J., Mesecar A.D. Design of potent and highly selective inhibitors for human β-secretase 2 (memapsin 1), a target for type 2 diabetes. Chem. Sci. 2016;7(5):3117–3122. doi: 10.1039/c5sc03718b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clemente J.C., Govindasamy L., Madabushi A., Fisher S.Z., Moose R.E., Yowell C.A., Hidaka K., Kimura T., Hayashi Y., Kiso Y., Agbandje-McKenna M., Dame J.B., Dunn B.M., McKenna R. Structure of the aspartic protease plasmepsin 4 from the malarial parasite Plasmodium malariae bound to anallophenylnorstatine-based inhibitor. Acta Crystallogr. 2006;D 62:246–252. doi: 10.1107/S0907444905041260. [DOI] [PubMed] [Google Scholar]
- 37.The PyMOL Molecular Graphics System, Version 1.7.4; Schrödinger, LLC.
- 38.Thomas J.A., Tan M.S.Y., Bisson C., Borg A., Umrekar T.R., Hackett F., Hale V.L., Vizcay-Barrena G., Fleck R.A., Snijders A.P., Saibil H.R., Blackman M.J. A protease cascade regulates release of the human malaria parasite Plasmodium falciparum from host red blood cells. Nat Microbiol. 2018;3(4):447–455. doi: 10.1038/s41564-018-0111-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghosh A.K., Fidanze S. Transition-state mimetics for HIV protease Inhibitors: stereocontrolled synthesis of hydroxyethylene and hydroxyethylamine isosteres by ester-derived titanium enolate syn and anti-aldol reactions. J. Org. Chem. 1998;63(18):6146–6152. doi: 10.1021/jo980159i. [DOI] [PubMed] [Google Scholar]
- 40.Kaganovsky L., Gelman D., Rueck-Braun K. Trans-chelating ligands in palladium-catalyzed carbonylative coupling and methoxycarbonylation of aryl halides. J. Organomet. Chem. 2010;695(2):260–266. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.