

Abstract
Multiple acyl-CoA dehydrogenase deficiency (MADD) or glutaric aciduria type II (GAII) is a clinically heterogeneous disorder affecting fatty acid and amino acid metabolism. Presentations range from a severe neonatal form with hypoglycemia, metabolic acidosis, and hepatomegaly with or without congenital anomalies to later-onset lipid storage myopathy. Genetic testing for MADD traditionally comprises analysis of ETFA, ETFB, and ETFDH. Patients may respond to pharmacological doses of riboflavin, particularly those with late-onset MADD due to variants in ETFDH. Increasingly other genes involved in riboflavin transport and flavoprotein biosynthesis are recognized as causing a MADD phenotype. Flavin adenine dinucleotide synthase (FADS) deficiency caused by biallelic variants in FLAD1 has been identified in nine previous cases of MADD. FLAD1 missense mutations have been associated with a riboflavin-responsive phenotype; however the effect of riboflavin with biallelic loss of function FLAD1 mutations required further investigation. Herein we describe a novel, truncating variant in FLAD1 causing MADD in an 8-year-old boy. Fibroblast studies showed a dramatic reduction in FADS protein with corresponding reduction in the FAD synthesis rate and FAD cellular content, beyond that previously documented in FLAD1-related MADD. There was apparent biochemical and clinical response to riboflavin treatment, beyond that previously reported in cases of biallelic loss of function variants in FLAD1. Early riboflavin treatment may have attenuated an otherwise severe phenotype.
Electronic supplementary material
The online version of this chapter (10.1007/8904_2018_139) contains supplementary material, which is available to authorized users.
Keywords: FAD synthase, FLAD1, Multiple acyl-CoA dehydrogenase deficiency (MADD), Myopathy, Riboflavin
Introduction
Multiple acyl-CoA dehydrogenase deficiency (MADD, MIM #231680) or glutaric aciduria type II is an autosomal recessive disorder causing defective electron transport from flavin adenine dinucleotide (FAD)-containing dehydrogenases to coenzyme Q10 in the mitochondrial electron transport chain. The condition is clinically heterogeneous ranging from a severe, neonatal form, presenting with hypoketotic hypoglycemia, metabolic acidosis, cardiomyopathy, and hepatomegaly to a later-onset form characterized by proximal myopathy. The neonatal form is further subdivided based on the presence of congenital anomalies including dysplastic kidneys, cerebral malformations, pulmonary hypoplasia, facial dysmorphism, and abnormal genitalia (Grünert 2014). Episodic vomiting, encephalopathy, liver and renal impairment, rhabdomyolysis, and sudden death may occur at times of catabolic stress (Horvath 2012).
Plasma acylcarnitine profile findings of elevated short-, medium- and long-chain acylcarnitines (C4–C16) may be diagnostic or absent if carnitine depleted (Wen et al. 2015). Urine organic acid analysis may show increased ethylmalonic acid, 2-hydroxyglutarate, glutaric acid, lactate, dicarboxylic acids, and glycine conjugates. Creatine phosphokinase (CPK) and lactate levels are typically raised during metabolic decompensation. Biochemical findings may normalize during periods of stability. Muscle biopsy reveals lipid accumulation in skeletal muscle and reduced activity of mitochondrial respiratory chain complexes I + III and II + III and coenzyme Q10 (Vergani et al. 1999; Olsen et al. 2007; Horvath 2012).
Genetic changes in ETFA (15q23-q25) and ETFB (19q13.3-q13.4) encoding the alpha and beta subunits of electron transport flavoprotein (ETF) and ETFDH (4q32-q35) encoding ETF-ubiquinone oxidoreductase (ETFQO), respectively, were first identified as causing MADD. ETFDH variants are the most common cause of late-onset disease (Grünert 2014). Both ETF and ETFOQ contain FAD prosthetic groups (Henriques et al. 2010). Deficiencies in FAD therefore inhibit activity of ETF and ETFOQ as well as other FAD-dependent enzymes, causing MADD.
Riboflavin (vitamin B2), the precursor for flavin mononucleotide (FMN) and FAD, cannot be synthesized in humans. It is consumed in dietary sources such as meat and dairy, absorbed in the intestine, transported into the blood stream, and taken up by one of three tissue-specific riboflavin transporters thus far identified (RFVT 1, 2, 3), encoded by SLC52A1, SLC52A2, and SLC52A3 (Barile et al. 2016). Inside the cell, riboflavin kinase converts riboflavin into FMN, which is adenylated to FAD, by FAD synthase (FADS), encoded by FLAD1 (Brizio et al. 2006). Activated FADS may chaperone the folding of apoproteins into stable flavoproteins (Giancaspero et al. 2015).
Biochemical features of MADD have been noted in newborns of mothers with riboflavin deficiency and heterozygous mutations in SLC52A1 (Chiong et al. 2007; Ho et al. 2011) and in patients with Brown–Vialetto–Van Laere (BVVL) syndrome, caused by mutations in SLC52A2 or SLC52A3 (Haack et al. 2012). More recently, mutations in the mitochondrial FAD transporter gene (SLC25A32, MIM 610815) (Schiff et al. 2016) and FAD synthase gene (FLAD1, MIM 610595) (Olsen et al. 2016) have been shown to cause MADD. While FLAD1 missense mutations were associated with a riboflavin-responsive phenotype, the effect of riboflavin with biallelic loss of function FLAD1 mutations was less clearly defined. Herein we describe a novel, truncating variant in FLAD1 causing riboflavin-responsive MADD myopathy in an 8-year-old boy.
Case Report
The patient was referred to the metabolic service with a positive newborn screen for medium-chain acyl-CoA dehydrogenase (MCAD) deficiency, due to an elevated C8-acylcarnitine of 0.58 μM (<0.4). He was the first child to consanguineous, first cousins of Palestinian descent. The antenatal history was unremarkable, and he was born by normal vaginal delivery at 40-week gestation, with a birth weight of 3.268 kg. No resuscitation was required, and he fed well 3 h, on a combination of breast milk and formula. Family history was noncontributory and examination normal. The diagnosis of MADD was made when a confirmatory plasma acylcarnitine profile detected elevations in C6–C10 acylcarnitines and urine organic acid analysis detected ethylmalonic acid, 2-hydroxyglutaric acid, glutaric acid, and dicarboxylic aciduria (for biochemical results, see Supplementary Material). Other blood work was normal, including liver enzymes and CPK. Riboflavin (10 mg/kg/day) was initiated at 16 days of age. No pathogenic variants were identified in ETFA, ETFB, or ETFDH (Emory Genetics). An elevated CPK (457) was noted at 3 months of age during his first hospital admission and recurred with subsequent recurrent respiratory infections. Riboflavin was increased to 50 mg/kg/day and carnitine added (100 mg/kg/day). Dietary fat was restricted from 48% of calories from fat at baseline to 32%; however a fat- and protein-restricted diet has never been closely adhered to. Riboflavin was stopped at 29 months of age due to low palatability, while carnitine was continued. A myopathic open mouth appearance was first noted at aged 3 years with nasal speech and open mouth breathing not improved following tonsillectomy and adenoidectomy. On otorhinolaryngology review, aged 6 years, he had hypernasality, weak facial muscles, and a wet cough. The palate did not elevate on phonation, and he displayed significant oral incoordination. Laryngoscopy identified severe velopharyngeal insufficiency, and a wide surgical velopharyngeal flap was recommended.
By 8 years of age, he had an evolving myopathy with fatigue on chewing and worsening exercise intolerance. He was achieving normally in mainstream schooling. Hearing tests performed in the community were normal. He had not attended for ophthalmology review. Annual cardiac evaluation including echocardiogram and electrocardiogram (ECG) were normal. Height was on the 25th centile (122.3 cm) and weight on the 10th (20.5 kg). He had myopathic facies with a tented downturned mouth held in an open position. Dental caries affected the upper dentition, and he had a high-arched palate with no visible cleft. There was no facial asymmetry, and he was able to protrude his tongue in the midline but was unable to raise his eyebrows. Speech was almost unintelligible due to velopharyngeal insufficiency. He had minimal muscle bulk, and tone was generally reduced. Power appeared normal in all four limbs, but reflexes were not obtainable despite reinforcement. Plantars were down-going bilaterally, and there was no clonus. Cardiac, respiratory, and abdominal examinations were all normal.
A microarray identified significant loss of heterozygosity (LOH), consistent with parental consanguinity. The gene for FAD synthase, FLAD1, was noted to be within one such region, and targeted sequencing of FLAD1 identified homozygosity for c.745C > T (p. Arg249*). This nonsense variant results in a premature STOP codon in exon 2 of the molybdopterin-binding (MPTb) domain and is predicted pathogenic (Giancaspero et al. 2015). The variant is seen six times in a heterozygous state but has not previously been reported in the homozygous state (http://gnomad.broadinstitute.org/gene/ENSG00000160688). Functional analyses were performed to better characterize the novel variant. Riboflavin was restarted at 150 mg/day.
Methods
Cell Culturing
Dermal fibroblast cultures from the patient (P) and three healthy, age- and gender-matched controls (C1, C2, C3) (Cambrex #CC-2509, ATCC #CRL-2450, and private donation from Dr. L. Vergani, University of Padua) were incubated at 37°C and humidified atmosphere of 5% (v/v) CO2 in EMEM (Eagle’s minimum essential medium) (Lonza, Basel, Switzerland) supplemented with 2 mmol/L of l-glutamine (Sigma Aldrich, St. Louis, Missouri, USA), 10% fetal calf serum (Sigma Aldrich, St. Louis, Missouri, USA), and 1% penicillin/streptomycin (Sigma Aldrich, St. Louis, Missouri, USA). Following pre-culturing, the cell cultures were transferred to 1 × 75 cm2 culture flask and harvested at sub-confluence by detaching in trypsin-EDTA solution, PBS (pH 7.4), centrifuged for 3 min at 405 × g, and washed twice in PBS. Fibroblast pellets were stored at −80°C after removal of PBS.
Quantification of Cellular Flavin Content and Measurement of FAD Synthesis Rate
Cell pellets were resuspended in 100 μL lysis buffer (50 mmol/L Tris–HCl [pH 7.5], 1% Triton X-100, 5 mmol/L ß-mercaptoethanol [2-ME], 1 mmol/L NaF, 0.1 mmol/L PMSF, and 1 x protease inhibitor cocktail) and passed through a 26G needle. After incubation for 30 min on ice, the cell suspension was centrifuged at 13,000 × g for 10 min at 4°C, and the supernatant was recovered as cell lysate. Protein content was determined by the Bradford protein assay (Bio-Rad, Hercules, California, USA). Riboflavin, FMN, and FAD were measured in neutralized perchloric acid extracts of cell lysates (0.2 mg) by HPLC as previously described (Liuzzi et al. 2012).
The rate of FAD synthesis was measured at 37°C in 600 μL of 50 mM Tris–HCl (pH 7.5) in the presence of 0.2 mg cell lysate, 1 μmol/L FMN, 5 mmol/L ATP, and 5 mmol/L MgCl2. At the appropriate time, 100 μL aliquots were taken, extracted with perchloric acid, and neutralized. Quantitative determination of riboflavin, FMN, and FAD was carried out with a calibration curve made in each experiment with standard solutions diluted in the extraction solution.
Western Blotting
Frozen cell pellets were dissolved in lysis buffer on ice (50 mM Tris–HCl pH 7.8, 5 mM EDTA, 1 mM DTT, 1% Triton X-100, and one protease inhibitor cocktail tablet (cOmplete Mini, Roche, Mannheim, Germany) in 10 mL), followed by 30 s of sonication in ultrasonic bath (Bandelin SONOREX™ SUPER with built-in heating ultrasonic baths) (Sigma Aldrich, St. Louis, Missouri, USA). The lysate was separated into Triton X-100 soluble and insoluble fractions by 15 min of centrifugation at 14,800 × g, 4°C.
20 and 40 μg of the total cell protein extract [determined by the Bradford protein assay (Bio-Rad, Hercules, California, USA)] were analyzed by SDS-PAGE on Criterion™ TGX Stain-free™ Precast Gels (any kD) (Bio-Rad, Hercules, California, USA) in Tris-glycine 0.1% SDS buffer. All Blue Standards (Bio-Rad, Hercules, California, USA) was used as molecular weight (MW) marker. Proteins were blotted onto PVDF membranes [midi format, 0.2 μm (Bio-Rad, Hercules, California, USA)] by semidry electro-blotting [Trans-Blot® Turbo™ Transfer System (Bio-Rad, Hercules, California, USA)] for 30 min. The PVDF membranes were incubated 1 h in 5% nonfat skim milk (VWR, Radnor, Pennsylvania, USA). Transferred proteins were incubated overnight with primary polyclonal rabbit antibodies: (1) anti-VLCAD (very long-chain Acyl-CoA dehydrogenase) antibody (kindly provided by Dr. Arnie Strauss), diluted 1:10,000 (detected at MW 68 kDa); (2) anti-SCAD (short-chain acyl-CoA dehydrogenase) antibody (kindly provided by Dr. Kay Tanaka), diluted 1:15,000 (detected at MW 40 kDa); (3) anti-ETF A and B (electron transfer flavoprotein α and β) antibody (kindly provided by Dr. Kay Tanaka) diluted 1:20,000 (detected at MW 32 and 27 kDa); and (4) anti-FLAD1 antibody (HPA028563) (Sigma Aldrich, St. Louis, Missouri, USA), diluted 1:250 (detected at MW 54 kDa). Polyclonal goat anti-rabbit-HRP antibody (DAKO, Copenhagen, Denmark) at dilution 1:20,000 was used as secondary antibody. ECL plus Western blotting detection system (Amersham Biosciences, Little Chalfont, UK) was used for protein detection, according to manufacturer’s recommendations. Detection was done using the ImageQuant LAS 4000 (GE Healthcare, Little Chalfont, UK). The intensities of bands were quantified using ImageQuant TL (GE Healthcare, Little Chalfont, UK) and normalized to total protein content.
Results
Flavin Content and FAD Synthesis Rate in Fibroblasts
The rate of FAD synthesis in cultured fibroblast from the affected individual was drastically reduced with respect to the control range (0.35 pmol/min mg versus 3.50–4.37 pmol/min mg, student’s t-test p < 0.001) (Fig. 1). FAD synthesis impairment resulted in a significant reduction in FAD content (Table 1) measured in cultured fibroblasts from the affected individual (60.5 pmol/mg, student’s t-test p < 0.05) with respect to that found from controls (ranging from 111.7 to 171.2 pmol/mg). Quite surprisingly, there was also a significant reduction in the cellular content of FMN (5.8 pmol/mg, student’s t-test p < 0.001) and riboflavin (1.0 pmol/mg, student’s t-test p < 0.01) to 44 and to 33%, respectively, that of control values, suggesting a regulatory metabolic response to FADS impairment.
Fig. 1.
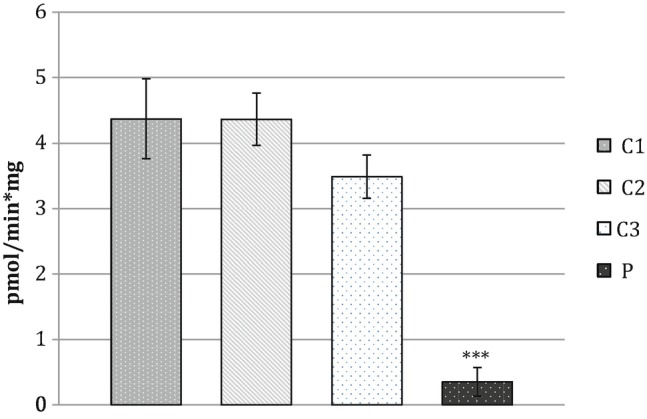
The rate of FAD synthesis was measured in fibroblasts from the patient (P) and from three control individuals (C1, C2, and C3), as described in Sect. 3. Data represent the mean ± SD of two independent cell lysates; student’s t-test: ***p < 0.001
Table 1.
Cellular FAD, FMN, and riboflavin quantification in fibroblast samples
| Cellular flavin content (pmol/mg) | |||
|---|---|---|---|
| FAD | FMN | Rf | |
| C1 | 111.7 ± 2.8 | 13.3 ± 1.4 | 2.6 ± 0.5 |
| C2 | 171.2 ± 18.8 | 12.4 ± 0.3 | 3.1 ± 0.1 |
| C3 | 123.3 ± 15.1 | 14.3 ± 1.3 | 3.5 ± 0.6 |
| P | 60.5 ± 2.6* | 5.8 ± 1.1*** | 1.0 ± 0.4** |
For cellular flavin content, data represent the mean ± SD of two independent cell lysates; student’s t-test: *p<0.05, **p<0.01, and ***p<0.001
FADS and Flavoenzyme Protein Levels in Fibroblasts
FADS and flavoenzyme protein levels in patient and control fibroblasts were determined by Western blot analysis. As expected from the FLAD1 genotype, the MADD patient fibroblasts displayed significantly decreased full-length 50 kDa FADS protein levels compared to control fibroblasts (student’s t-test p < 0.001). However, the 26 kDa FADS band, which was suggested to contain an intact and functional FADS domain (Olsen et al. 2016), seems equally expressed in both patient and control fibroblasts (Fig. 2a, b). Mitochondrial flavoproteins comprising very long-chain acyl-CoA dehydrogenase (VLCAD), short-chain acyl-CoA dehydrogenase (SCAD), and the two ETF subunit proteins showed a tendency to be decreased in the patient as compared to control, although neither of the results was statistically significant (Fig. 3a, b).
Fig. 2.
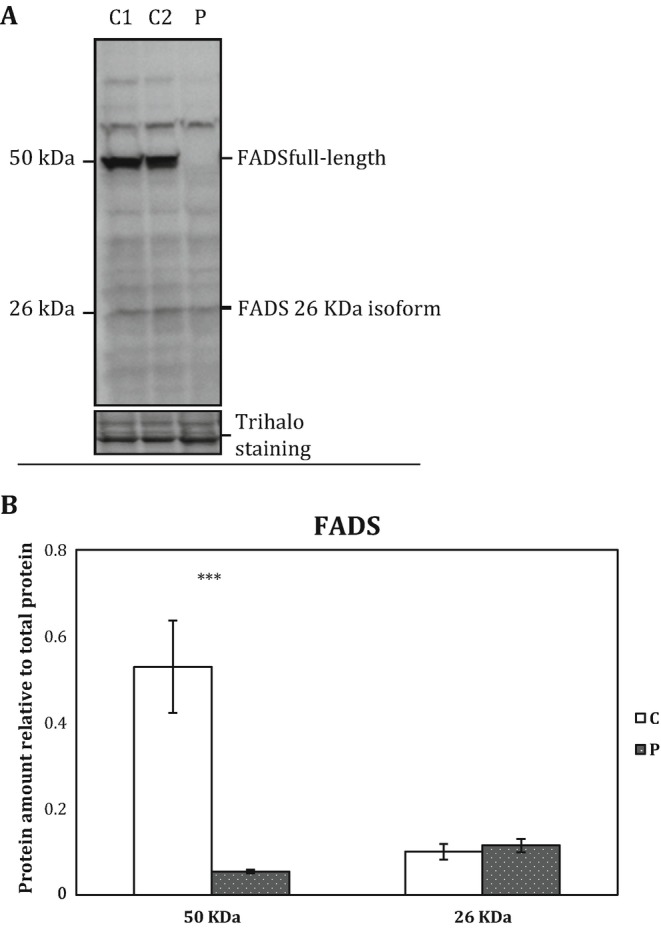
(a) Representative immunoblot analysis of FADS protein. Protein extracts from cultured human dermal fibroblasts were separated by SDS-PAGE and immunoblotted with a polyclonal antibody, raised against the C-terminal part of human FAD synthase protein (FADS). The patient cells were cultured in two separate cultures together with two healthy control individuals (C1 and C2); protein was extracted and 40 μg loaded on Criterion™ TGX Stain-free™ Precast Gels (any kD) (Bio-Rad). The molecular weight of protein bands identified as FADS by mass spectrometry analysis (Olsen et al. 2016) is indicated. (b) FADS protein intensities were quantified relative to total protein content (Trihalo staining). Quantification of patient FADS relative to combined two control individuals as shown. The error bars represent standard error of mean (SEM) of two independent experiments, student’s t-test: ***p < 0.001
Fig. 3.

(a) Representative immunoblot analysis of ACAD proteins. Protein extracts from cultured human dermal fibroblasts were loaded in 20 μg. Samples were separated by SDS-PAGE and immunoblotted with polyclonal antibodies raised against each of the four mitochondrial flavoproteins VLCAD, SCAD, or the two ETF subunit proteins. The patient cells were cultured in two separate cultures together with two healthy control individuals (C1 and C2); protein was extracted and loaded on Criterion™ TGX Stain-free™ Precast Gels (any kD) (Bio-Rad). (b) Protein intensities were quantified relative to total protein content (Trihalo staining). Quantification of patient FADS relative to combined two control individuals as shown. The error bars represent standard error of mean (SEM) of two independent experiments. There was no significant difference in the mitochondrial flavoprotein content between patient and control fibroblasts
Discussion
This is the tenth published case of MADD due to biallelic pathogenic variants in FLAD1. The age of onset in the nine previously reported cases of FLAD1-related MADD ranges from birth to 44 years, however most presented in infancy, with five out of nine dying in the first year of life (Taylor et al. 2014; Olsen et al. 2016). A lipid storage myopathy with respiratory chain deficiency predominates, similar to ETFDH-related riboflavin-responsive forms of MADD. Cardiomyopathy has been reported once, and two patients required pacemaker insertion for tachyarrhythmia/cardiac arrest (Olsen et al. 2016). Neuropathies, hearing or visual impairment as seen in BVVL (Bosch 2011), have not yet been described with FLAD1 variants. Hypotonia, swallowing, and speech difficulties as seen in this case are typical (Olsen et al. 2016).
Two major human FLAD1 transcript products, FADS1 (mitochondrial) and FADS2 (cytosolic), have been characterized in detail (Brizio et al. 2006; Barile et al. 2016); however, only the cytosolic isoform was identified in human fibroblasts (Olsen et al. 2016). FADS contains an N-terminal molybdopterin-binding (MPTb) domain with FAD hydrolase activity and a C-terminal domain, which catalyzes FAD synthesis (FADS domain) (Giancaspero et al. 2015). Patients with biallelic frameshift variants in exon 2, i.e., in the MPTb domain, as found in our case, have had early-onset, severe disease (Olsen et al. 2016).
Consistently, fibroblast studies in our patient revealed a dramatic reduction in FADS protein level with corresponding reduction in both FAD synthesis rate and FAD cellular content. Previous studies measuring residual FADS activity in individuals with FLAD1 variants revealed only a partial defect in FAD synthesis ability of affected fibroblasts, with corresponding cellular FAD, FMN, and riboflavin content within the control range. A low mitochondrial FAD content was observed in one patient (Olsen et al. 2016). While the previously observed residual FAD synthesis activity could be explained by missense mutations in some cases, even previous cases of biallelic frameshift variants in exon 2 in the MPTb domain, predicted to result in complete loss of function, had some expression of FADS. Since the FADS domain can function independently from the MPTb domain to catalyze FAD synthesis (Miccolis et al. 2012), the authors hypothesized that the residual FADS activity could result from the expression of novel FLAD1 isoforms coding for the FADS domain alone (Olsen et al. 2016). It was recently confirmed that one of these truncated isoforms, isoform 6, could produce FAD (Leone et al. 2018). While immunoblotting showed almost no detectable full-length FADS in our case, a 26 kDa protein band, corresponding to a truncated FADS protein, is expressed (Fig. 2). The rate of FAD synthesis in our patient’s fibroblasts, however, was lower than that measured before, only 9% of controls (Fig. 1) as compared to 25% of controls in a previous case with biallelic frameshift FLAD1 variants (Olsen et al. 2016). However, cellular content of FAD was only reduced to 54% that of controls (Table 1). There was a minor, but not significant, decrease in mitochondrial flavoproteins VLCAD, ETF-alpha, and ETF-beta (Fig. 3). It is difficult to relate the significant reduction in FAD synthesis to the milder clinical outcome. FADS activity of 9% that of controls seems to allow cellular FAD supply comparable to heterozygous levels. It is likely that FADS is not the rate-limiting step in this process and that residual synthetic activity, possibly complemented by the 26 kDa truncated FADS protein, is sufficiently active to synthesize half of normal total FAD content. The 26 kDa isoform does not contain a mitochondrial leader peptide; therefore mitochondrial FAD supply in patients with biallelic exon 2 loss of function mutations depends on import by the mitochondrial FAD transporter. This may become limiting during periods of catabolic stress, when the expression of mitochondrial flavoproteins is increased. The clinical course may have been modified by treatment with riboflavin from the neonatal period until 2.5 years of age, and he may have maintained adequate general health and caloric intake, such as to avoid significant catabolic stress.
It should be noted that in our patient’s fibroblasts, there was also an unexpected reduction in the cellular content of FMN and riboflavin rather than the expected increase in substrate levels before the enzymatic block at FADS. A reduction of FAD associated with a reduction of FMN and riboflavin has been also found in the model organism C. elegans with silenced FAD synthase gene (Liuzzi et al. 2012). Feedback control on either riboflavin kinase or SLC52A2/RFVT2 was proposed but has not yet been documented. It is possible that either the riboflavin kinase or the riboflavin transporter step is the rate-limiting step in FAD synthesis overall process, and a secondary deficiency may result from disturbed flavin homeostasis, causing reduced FMN synthesis or riboflavin transport. No reported cases of riboflavin kinase deficiency in humans have yet been described.
Flavoproteins, such as mitochondrial acyl-CoA dehydrogenases, appear to undergo rapid degradation in a state of riboflavin deficiency (Henriques et al. 2010). A trial of high-dose riboflavin supplementation (100–400 mg/day) is recommended in all MADD cases. Riboflavin has low toxicity even at high doses as riboflavin exceeding renal reabsorption is eliminated in the urine (Barile et al. 2016). Side effects are largely limited to non-specific gastrointestinal disturbance. Riboflavin responsiveness has been reported in FLAD1 variants, more often in milder variants affecting a single amino acid in the FADS domain (Auranen et al. 2017). Four out of five patients treated showed a response, including improved cardiac and skeletal muscle function, normalization of urinary organic acids, and reductions in acylcarnitine species (Olsen et al. 2016). Patients with biallelic frameshift variants in exon 2 were more severely affected and less responsive to riboflavin therapy. Based on plasma acylcarnitine profiles and urine organic acid excretion, our patient did appear to partially respond to riboflavin. Off riboflavin his acylcarnitine profile showed more widespread and higher elevations in acylcarnitine species, and urine organic acids showed excretion of ethylmalonic acid (EMA), 2-hydroxyglutaric, glutaric, adipic, and suberic acids. On riboflavin, only urine EMA was detected. Urine EMA on riboflavin was 30% that of urine EMA excretion off riboflavin (see Supplementary Material). Once treatment with 150 mg/day of riboflavin was restarted, his parents reported better muscle endurance and ability to chew food although this has not been quantified objectively.
In summary, this report describes the clinical, biochemical, and molecular findings of an 8-year-old boy with FLAD1-related MADD. Newborn screening was positive for MCAD deficiency, although the pattern was atypical and diagnostic evaluation identified MADD. Disorders of riboflavin metabolism must be considered in the differential diagnosis for MADD to ensure prompt treatment. Although commenced in the neonatal period, riboflavin treatment was not adhered to, and myopathic symptoms became evident by 4 years of age. Recent discovery of homozygous frameshift variants in FLAD1 has been clinically significant as it has provided the necessary motivation for compliance with riboflavin therapy. Fibroblast studies have shown a dramatic reduction in FADS protein with corresponding reduction in FAD synthesis rate and FAD cellular content, beyond that previously documented in FLAD1-related MADD. The presence of biallelic truncating mutations with a significant reduction in cellular flavin content predicts a severe, potentially lethal phenotype. This may have been prevented by early treatment with riboflavin, myopathy developing when riboflavin was ceased, or by relative avoidance of catabolic stress. The residual synthetic activity, possibly complemented by the 26 kDa truncated FADS protein, seems to be sufficient to allow for mitochondrial flavoprotein biogenesis, except at times of catabolic stress. Further studies are planned to clarify the feedback mechanism controlling cellular flavin content.
Electronic Supplementary Material
Biochemical investigations in patient (DOCX 21 kb)
Key Message
Analysis of FLAD1 is recommended in cases of MADD, if no pathological variants are identified in ETFA, ETFB, or ETFDH, and a trial of riboflavin is recommended in all patients with FLAD1 variants.
Author Contributions
Ryder B: primary drafting of the manuscript and clinical care of patient.
Tolomeo M: scientist performing quantification of cellular flavin content and measurement of FAD synthesis rate.
Nochi Z: scientist performing Western blot analysis of FADS and flavoenzymes.
Colella M: senior scientist supervising cell culturing and management.
Barile M: senior scientist supervising quantification of cellular flavin content and measurement of FAD synthesis rate.
Olsen R: senior scientist supervising Western blot analysis of FADS and flavoenzymes.
Inbar-Feigenberg M: primary metabolic physician directing patient care, senior clinician supervising drafting of manuscript.
Corresponding Author
Dr. Bryony Ryder
Conflict of Interest
No conflicts of interest reported.
Acknowledgments and Funding
The study was supported by the Danish Council of Independent Medical Research grant (4004-00548) to Olsen RK.
M. Tolomeo was supported by a postgraduate research fellowship financed by “Fondazione Cassa di Risparmio di Puglia,” Bari, Italy.
The technical assistance in HPLC measurements of Dr. M.L. Defrancesco (University of Bari) and in immunoblotting to Margrethe Kjeldsen is gratefully acknowledged.
Thanks to Stacy Hewson and Michelle Mecija for their clinical care.
Compliance with Ethical Standards
Ethics approval was not required for this paper.
Parental consent was obtained for the publication of the article.
This article does not contain any studies with human or animal subjects performed by the any of the authors.
References
- Auranen M, et al. Patient with multiple acyl-CoA dehydrogenation deficiency disease and FLAD1 mutations benefits from riboflavin therapy. Neuromuscul Disord. 2017;27:581–584. doi: 10.1016/j.nmd.2017.03.003. [DOI] [PubMed] [Google Scholar]
- Barile M, et al. Riboflavin transport and metabolism in humans. J Inherit Metab Dis. 2016;39(4):545–557. doi: 10.1007/s10545-016-9950-0. [DOI] [PubMed] [Google Scholar]
- Bosch AM. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis. 2011;34(1):159–164. doi: 10.1007/s10545-010-9242-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizio C, et al. Over-expression in Escherichia coli and characterization of two recombinant isoforms of human FAD synthetase. Biochem Biophys Res Commun. 2006;344(3):1008–1016. doi: 10.1016/j.bbrc.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Chiong MA, et al. Transient multiple acyl-CoA dehydrogenation deficiency in a newborn female caused by maternal riboflavin deficiency. Mol Genet Metab. 2007;92:109–114. doi: 10.1016/j.ymgme.2007.06.017. [DOI] [PubMed] [Google Scholar]
- Fu HX, et al. Significant clinical heterogeneity with similar ETFDH genotype in three Chinese patients with late-onset multiple acyl-CoA dehydrogenase deficiency. Neurol Sci. 2016;37(7):1099–1105. doi: 10.1007/s10072-016-2549-2. [DOI] [PubMed] [Google Scholar]
- Giancaspero TA, et al. Human FAD synthase is a bi-functional enzyme with a FAD hydrolase activity in the molybdopterin binding domain. Biochem Biophys Res Commun. 2015;465(3):443–449. doi: 10.1016/j.bbrc.2015.08.035. [DOI] [PubMed] [Google Scholar]
- Grünert S. Clinical and genetical heterogeneity of late-onset multiple coenzyme A dehydrogenase deficiency. Orphanet J Rare Dis. 2014;9:117. doi: 10.1186/s13023-014-0117-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, et al. Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J Inherit Metab Dis. 2012;35(6):943–948. doi: 10.1007/s10545-012-9513-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques BJ, Olsen RK, Bross P, Gomes CM. Emerging roles for riboflavin in functional rescue of mitochondrial β-oxidation flavoenzymes. Curr Med Chem. 2010;17(32):3842–3854. doi: 10.2174/092986710793205462. [DOI] [PubMed] [Google Scholar]
- Ho G, et al. Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria Type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum Mutat. 2011;32(1):E1976–E1984. doi: 10.1002/humu.21399. [DOI] [PubMed] [Google Scholar]
- Horvath R. Update on clinical aspects and treatment of selected vitamin responsive disorders II (riboflavin and CoQ10) J Inherit Metab Dis. 2012;35(4):679–687. doi: 10.1007/s10545-011-9434-1. [DOI] [PubMed] [Google Scholar]
- Leone Piero, Galluccio Michele, Barbiroli Alberto, Eberini Ivano, Tolomeo Maria, Vrenna Flavia, Gianazza Elisabetta, Iametti Stefania, Bonomi Francesco, Indiveri Cesare, Barile Maria. Bacterial Production, Characterization and Protein Modeling of a Novel Monofuctional Isoform of FAD Synthase in Humans: An Emergency Protein? Molecules. 2018;23(1):116. doi: 10.3390/molecules23010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liuzzi V, et al. Silencing of FAD synthase gene in Caenorhabditis elegans upsets protein homeostasis and impacts on complex behavioural patterns. Biochim Biophys Acta. 2012;1820:521–531. doi: 10.1016/j.bbagen.2012.01.012. [DOI] [PubMed] [Google Scholar]
- Miccolis Angelica, Galluccio Michele, Giancaspero Teresa, Indiveri Cesare, Barile Maria. Bacterial Over-Expression and Purification of the 3'phosphoadenosine 5'phosphosulfate (PAPS) Reductase Domain of Human FAD Synthase: Functional Characterization and Homology Modeling. International Journal of Molecular Sciences. 2012;13(12):16880–16898. doi: 10.3390/ijms131216880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen R, et al. ETFDH mutations as a major cause of riboflavin responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130(8):2045–2054. doi: 10.1093/brain/awm135. [DOI] [PubMed] [Google Scholar]
- Olsen R, et al. Riboflavin-responsive and -non-responsive mutations in FAD synthase cause multiple Acyl-CoA dehydrogenase and combined respiratory chain deficiency. Am J Hum Genet. 2016;98:1130–1145. doi: 10.1016/j.ajhg.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff M, et al. SLC25A32 mutations and riboflavin-responsive exercise intolerance. N Engl J Med. 2016;374(8):795–797. doi: 10.1056/NEJMc1513610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor R, Pyle A, Griffin H. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA. 2014;312(1):68–77. doi: 10.1001/jama.2014.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani L, et al. Riboflavin therapy. Biochemical heterogeneity in two adult lipid storage myopathies. Brain. 1999;122(Pt 12):2401–2411. doi: 10.1093/brain/122.12.2401. [DOI] [PubMed] [Google Scholar]
- Wen Bing, Li Duoling, Li Wei, Zhao Yuying, Yan Chuanzhu. Multiple acyl-CoA dehydrogenation deficiency as decreased acyl-carnitine profile in serum. Neurological Sciences. 2015;36(6):853–859. doi: 10.1007/s10072-015-2197-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Biochemical investigations in patient (DOCX 21 kb)