

Abstract
Ethylmalonic encephalopathy (EE) is caused by mutations in the ETHE1 gene. ETHE1 is vital for the catabolism of hydrogen sulfide (H2S). Patients with pathogenic mutations in ETHE1 have markedly increased thiosulfate, which is a reliable index of H2S levels. Accumulation of H2S is thought to cause the characteristic metabolic derangement found in EE. Recently introduced treatment strategies in EE, such as combined use of metronidazole (MNZ) and N-acetylcysteine (NAC), are aimed at lowering chronic H2S load. Experience with treatment strategies directed against acute episodes of metabolic decompensation (e.g., hemodialysis) is limited. Here we present an unusually mild, molecularly confirmed, case of EE in a 19-year-old male on chronic treatment with MNZ and NAC. During an acute episode of metabolic decompensation, we employed continuous renal replacement therapy (CRRT) to regain metabolic control. On continuous treatment with NAC and MNZ during the months preceding the acute event, plasma thiosulfate levels ranged from 1.6 to 4 μg/mL (reference range up to 2 μg/mL) and had a mean value of 2.5 μg/mL. During the acute decompensation, thiosulfate levels were 6.7 μg/mL, with hyperlactatemia and perturbed organic acid, acylglycine, and acylcarnitine profiles. CRRT decreased thiosulfate within 24 h to 1.4 μg/mL. Following discontinuation of CRRT, mean thiosulfate levels were 3.2 μg/mL (range, 2.4–3.7 μg/mL) accompanied by clinical improvement with metabolic stabilization of blood gas, acylcarnitine, organic acid, and acylglycine profiles. In conclusion, CRRT may help to regain metabolic control in patients with EE who have an acute metabolic decompensation on chronic treatment with NAC and MNZ.
Electronic supplementary material
The online version of this chapter (10.1007/8904_2018_136) contains supplementary material, which is available to authorized users.
Keywords: Continuous renal replacement therapy, Ethylmalonic encephalopathy, Metronidazole, N-acetylcysteine, Thiosulfate
Introduction
Ethylmalonic encephalopathy (EE; OMIM #602473) is generally considered a rare autosomal recessive neurometabolic disorder of infancy but presents with wide clinical heterogeneity. It is clinically characterized by neurodevelopmental delay and regression, prominent pyramidal and extrapyramidal signs, recurrent petechiae, orthostatic acrocyanosis, and chronic diarrhea (Drousiotou et al. 2011). It was first described by Burlina et al. in 1991, but it was not until 2004 that Tiranti et al. mapped its genomic locus to the ETHE1 gene (OMIM #608451) on chromosome 19q13 (Burlina et al. 1991; Tiranti et al. 2004). The ETHE1 gene encodes a 30-kDa polypeptide (ETHE1), which is a non-heme, iron-dependent, mitochondrial matrix sulfur dioxygenase that is involved in the catabolism of hydrogen sulfide (H2S). It catalyzes the oxidation of glutathione persulfide (GSSH) to give glutathione and persulfite (Kabil and Banerjee 2012; Pettinati et al. 2015).
This reaction is a vital part of the H2S catabolic pathway, as it generates the glutathione (GSH) that is needed for the extraction of sulfur atoms of the intermediately produced thiosulfate (Hildebrandt and Grieshaber 2008; Jackson et al. 2012).
Hydrogen sulfide is a colorless, water-soluble gasotransmitter with physiologic roles in CNS signaling, heart rate regulation, blood pressure regulation, and inflammation (Kimura and Kimura 2004; Nagai et al. 2004; Qingyou et al. 2004; Whiteman et al. 2004; Xu et al. 2008; Bucci et al. 2010; Elsey et al. 2010). In mammals, H2S can be endogenously produced from L-cysteine taken up by diet or synthesized via trans-sulfuration of serine by L-methionine, but most of the H2S load stems from intestinal anaerobes (Viscomi et al. 2010; Di Meo et al. 2015). Under steady-state conditions, tissue concentrations of H2S are generally in the low nanomolar range, but patients harboring biallelic pathogenic mutations in ETHE1 were found to have markedly elevated levels of thiosulfate, which can be used as a stable and readily measurable index of H2S levels (Viscomi et al. 2010). The accumulated H2S inhibits short-chain acyl CoA dehydrogenase (SCAD) and cytochrome c oxidase (COX) activities, causing the characteristic, but far from specific, biochemical changes observed in patients with EE, namely, ethylmalonic acid (EMA) aciduria, sometimes with mild elevations of short-chain acylglycines also detected in the urine organic acid profile (ethylmalonic acid, methylsuccinic acid, isobutyrylglycine, and isovalerylglycine), increased levels of plasma C4- and C5-acylcarnitines, and elevated plasma lactate (Tiranti et al. 2009). The ensuing metabolic derangement is thought to cause damages to the intestinal mucosa and the endothelia and lead to alterations of the blood vessel tone, thereby resulting in the main clinical features of EE (e.g., chronic hemorrhagic and/or mucoid diarrhea, petechial purpura with edematous acrocyanosis, and progressive neurological failure with pyramidal signs).
Treatment
There are several reports in the literature of patients that have been diagnosed with EE through newborn screening, during routine medical referral, or through a known family history, which contributed to our understanding of chronic management of this disease (Grosso et al. 2002; McGowan et al. 2004; Zafeiriou et al. 2007; Mineri et al. 2008; Pigeon et al. 2009; Dionisi-Vici et al. 2016; Tavasoli et al. 2017; Boyer et al. 2018). This has led to the introduction of new treatment strategies, such as the combined use of antibiotics and N-acetylcysteine (NAC), which is aimed at lowering the chronic H2S load (Viscomi et al. 2010). Metronidazole (MNZ) is a commonly used bactericidal nitroimidazole that is broadly active against aerobic and anaerobic bacterial species (Perencevich and Burakoff 2006). N-acetylcysteine, on the other hand, is a cell-permeable precursor of GSH, which is needed as an acceptor of sulfur atoms in H2S catabolism (Atkuri et al. 2007). Notably, a recent report by Boyer et al. suggests that a diet restricted in sulfur-containing amino acids, in addition to medical treatment, results in further improvement in clinical outcomes and biochemical markers in patients with EE identified on newborn screening (Boyer et al. 2018). Despite these advances, our knowledge of chronic management of EE remains scant, due to the rarity of the disease and the fact that many cases of EE described in the literature presented within the first year of life in context of an acute metabolic decompensation with an almost invariably lethal outcome.
Here we describe our experience with the use of continuous renal replacement therapy (CRRT) to lower plasma sulfide levels in an unusually mild clinical course of EE in a 19-year-old male, who nonetheless presented with an acute metabolic decompensation, despite chronic treatment with antibiotics and NAC. To the best of our knowledge, the use of continuous veno-venous hemodialysis was reported only once before in a case of EE to remove EE-associated metabolites during liver transplant surgery (Dionisi-Vici et al. 2016).
Case Report
Our patient is a 19-year-old man with an atypical mild form of EE. He was initially seen at the department of Medical Genetics at 10 years of age because of long-standing spastic paraplegia, dysarthria, and Arnold-Chiari malformation type I of unclear etiology. He was also regularly followed at the department of Pediatric Neurology for treatment of his spastic paraplegia with a baclofen pump. He had no history of diarrhea or acrocyanosis. The clinical features are summarized in Table 1. His parents are a non-consanguineous couple from Serbia, and he has an older healthy brother.
Table 1.
Summary of clinical features
| Clinical features reported in EE | Absent or present (− or +) |
| Failure to thrive | − |
| Retinal lesions with tortuous vessels | n/a |
| Orthostatic acrocyanosis | − |
| Chronic diarrhea | − |
| Petechiae | − |
| Coma/encephalopathy | During metabolic decompensation |
| Developmental regression | − |
| Developmental delay | + |
| Intellectual disability | + |
| Pyramidal symptoms | + |
| Ataxia | + |
| Hypotonia | − |
| Seizures | During metabolic decompensation |
| Hyperintense lesions in the basal ganglia on MRI | + |
| Other clinical features not reported in EE | |
| Trismus | During first hospitalization |
n/a not assessed
First Hospitalization
At 16 years of age, he was admitted to the pediatric ICU for generalized spasticity and trismus in the context of an upper respiratory tract infection. Metabolic work-up revealed elevated C4-carnitine on plasma acylcarnitine profile, although without substantial elevation of C5-carnitine, and moderately elevated EMA on urine organic acid profile on three consecutive measurements (72, 29, and 42 mmol/mol creatinine, reference range <11) along with slightly elevated methylsuccinic acid levels and variable slight elevations of short-chain acylglycines. Plasma lactate was within the normal range on multiple samples. The rest of the metabolic work-up was unremarkable. An MRI of the brain showed bilateral and symmetric increased T2 signaling in the basal ganglia and cerebellum (Fig. 1a, b). Magnetic resonance spectroscopy (MRS) of the basal ganglia demonstrated a corresponding high lactate peak not shown. Sanger sequencing of the ETHE1 gene identified a homozygous missense variant (c.79C>A; p.Gln27Lys) for which both parents were carriers. Interestingly, the same mutation (in heterozygous status with another known pathogenic variant) was described in two sisters with EE, also coming from former Yugoslavia (Pigeon et al. 2009). The acute management during this admission was supportive with IV fluids, dextrose, and carnitine, and, based on his biochemical profile, MNZ and NAC were added for treatment of suspected EE. After discharge and upon molecular confirmation, chronic treatment with MNZ 500 mg tid (1 week on, 1 week off) and NAC at 100 mg/kg/day divided tid was initiated, and plasma thiosulfate levels were measured regularly to assess metabolic control. Over the next 12 months, his plasma thiosulfate levels ranged from 1.6 to 4 μg/mL (reference <2 μg/mL), and he demonstrated significant improvement in mobility and speech.
Fig. 1.
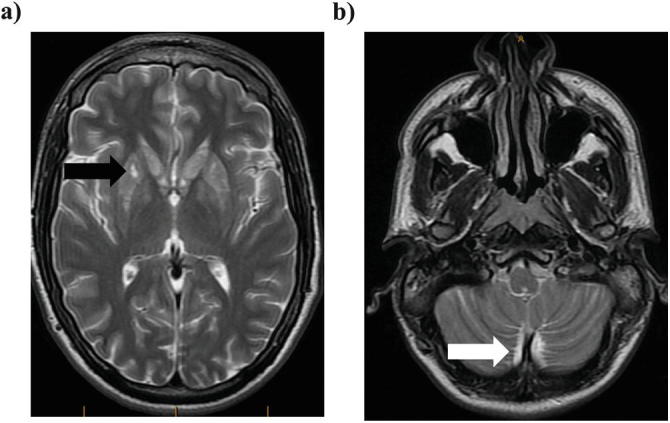
MRI brain images during the first hospitalization. (a) T2-weighted image demonstrating signal hyperintensity at the level of the basal ganglia (black arrow). (b) T2-weighted image demonstrating signal hyperintensity at the level of the cerebellum (white arrow)
Second Hospitalization with Acute Metabolic Decompensation
At 17 years of age, the patient was admitted to the pediatric psychiatric ward for an episode of suicidal ideation. During the admission, he developed a severe metabolic decompensation with encephalopathy, new-onset focal seizures, fever, and high lactate with no identifiable trigger and, while on sufficient caloric intake, required intubation and admission to intensive care. His plasma thiosulfate levels increased from 2.5 to 6.7 μg/mL, and plasma lactate levels were as high as 14.8 mmol/L (reference range 0.6–2.4). Quantitative urine acylglycine profile analysis (Bherer et al. 2015) showed elevations of several C4, C5, and other species, with butyrylglycine (C4) at 16.95 mmol/mol creatinine (reference range, 0.01–0.15 mmol/mol creatinine) being the most markedly elevated during the acute episode. Table 2 summarizes laboratory investigations pre, post, and during the acute events, while Supplementary Table 1 summarizes results for EMA, other organic acids, and acylglycine profile from multiple samples collected during the acute episodes and on other occasions. Plasma acylcarnitine profiling persistently showed C4-carnitine elevation in all samples. Multiple other acylcarnitines were elevated only at the time of this acute episode, although notably C5-carnitine was never substantially elevated (see Table 2). Figure 2 summarizes the MRI images taken during this episode, which demonstrated new evidence of acute and chronic brain injury (Fig. 2a–c). While trying to identify the underlying trigger of this acute episode, it was noted that the subcutaneous baclofen pump was empty, and concerns were raised about the possibility of a baclofen intoxication. Notably, continuous EEG recordings showed moderate to severe disturbance of cerebral activity with patterns suggestive of baclofen toxicity (e.g., triphasic waves, frontal intermittent rhythmic delta activity alternating with faster rhythms in the theta and alpha range) (Sutter et al. 2018). This could not be confirmed by measurement of CSF baclofen levels, which were undetectable. Notably, the CSF sample was drawn only on day 5 of the acute event, 2 days after initiation of CRRT, due to the patient’s clinical instability.
Table 2.
Summary of main biochemical parameters
| Clinical status | Lactate (plasma) (0.6–2.2)a | Thiosulfate (plasma) <2b | Plasma acylcarnitinesc | Urine organic acids d and acylglycines e | ||||
|---|---|---|---|---|---|---|---|---|
| C4 (0.06–0.44) | C5 (0.03–0.24) | EMAf (<11) | Isobutyryl (0.08–1.59) | Butyryl (0.01–0.15) | Isovaleryl (0.14–2.98) | |||
| First hospitalization | 1 | – | 2.4 | 0.47 | 72 | – | – | – |
| First hospitalization | 1.4 | – | 6.71 | 0.92 | – | – | – | – |
| Stable interim period | 2.0 | 1.5 | 1.02 | 0.17 | – | – | – | – |
| DODg – 12 days | 1.7 | 1.6 | – | – | – | – | – | – |
| DOD1g,h | 7.7 | 3.3 | 1.14 | 0.19 | 100 | 7.48 | 16.95 | 8.32 |
| DOD2g,h | 10.6 | 5.4 | – | – | – | – | – | – |
| DOD3g,h | 14.3 | 6.7 | – | – | – | – | – | – |
| DOD4g,h,i | 14.8 | – | – | – | – | – | – | – |
| DOD5g,h,i | 12.9 | 1.4 | – | – | – | – | – | – |
| DOD6g,h,i | 9.7 | 2.7 | 0.71 | 0.19 | 13 | 0.90 | 0.85 | 2.65 |
| DOD7g,h | 5.9 | 3.7 | 0.80 | 0.18 | 18 | – | – | – |
| DOD8g,h | 3.6 | 2.4 | 1.14 | 0.20 | 20 | – | – | – |
| DOD9 | 2.3 | – | – | – | – | – | – | – |
| Stable | <2.2 | 3.6 | – | – | – | – | – | – |
aLactate – highest day level is indicated. Concentrations expressed in mmol/L; reference values shown in parentheses; italic values are above reference range
bThiosulfate, analysis by ion chromatography (NMS labs, USA). Concentrations expressed in mcg/mL; reference values shown; italic values are above reference range
cAcylcarnitines. Concentrations expressed in umol/L; reference values shown in parentheses; italic values are above reference range
dUrine organic acids. Concentrations expressed in mmol/mol creatinine; reference values shown in parentheses; italic values are above reference range
eUrine acylglycines. Concentrations expressed in mmol/mol creatinine; reference values shown in parentheses; italic values are above reference range
fEMA ethylmalonic acid
gDOD day of decompensation
hReceived IV carnitine
iReceived CRRT
Fig. 2.

MRI brain images during the second episode of acute metabolic decompensation. (a) T2-weighted images demonstrated interval atrophy and central necrosis of the known basal ganglia abnormality with a new area of hyperintensity (white arrow) and corresponding diffusion restriction along the posterior putamina. (b) At the level of the cerebellum, there was abnormal symmetric hyperintensity in the dentate nuclei and the inferior cerebellar subcortical white matter (white arrow). (c) Multivoxel MRS demonstrated an inverted lactate peak in the basal ganglia and centrum semiovale at 1.3 ppm. The inverted lactate peak on MRS corresponds to an elevated tissue lactate. At 1.3 ppm, lactate resonates with a characteristic double peak at long echo times but is superimposed on the lipid band. By using an intermediate echo time of 135–145 ms, the lactate peak will be inverted, allowing it to be distinguished
Despite intensive supportive management (IV fluids, dextrose, MNZ, NAC, carnitine, and appropriate caloric intake) over 3 days, there was worsening of the patient’s clinical and metabolic state, reflected by increasing lactate levels. Although his renal function remained intact (serum creatinine 63 mmol/L with good urine output), additional treatment with CRRT was initiated on day 4 and continued for 96 h in the form of continuous venovenous hemodiafiltration (CVVHDF). The CVVHDF prescription included a blood flow of 200 mL/min, dialysate flow of 1.2 L/h, and replacement flow of 1.2 L/h. This rapidly lowered thiosulfate, lactate, and other biochemical parameters to baseline levels and allowed us to regain metabolic control (Fig. 3 and Supplementary Table 1). The improved metabolic state was accompanied by a reversal of the encephalopathy and restoration of consciousness. Over the following weeks, the patient continued his recovery and could regain much of his functionality.
Fig. 3.
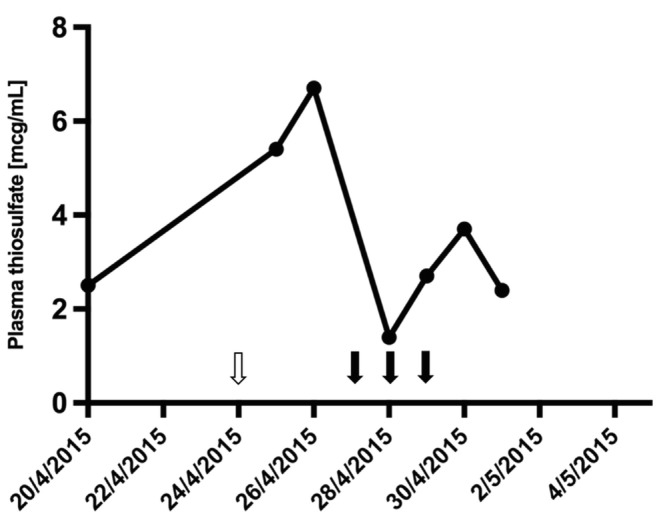
Plasma thiosulfate levels during the second episode of acute metabolic decompensation. On continuous treatment with NAC and MNZ during the months preceding the acute event, plasma thiosulfate levels ranged from 1.6 to 4 μg/mL (reference range up to 2 μg/mL) and had a mean value of 2.5 μg/mL. During the acute decompensation, thiosulfate levels increased to 6.7 μg/mL. After initiation of CRRT over 3 days, plasma thiosulfate levels rapidly returned to near-normal values. The white arrow indicates the onset of the acute event. Black arrows indicate the days when CRRT was given
Discussion
To this day, more than 80 patients have received a molecular diagnosis of EE (Tiranti et al. 2004, 2006; Mineri et al. 2008; Drousiotou et al. 2011; Tiranti and Zeviani 2013). The natural history of the disease was thought to be invariably fatal with onset in the first 2–4 months of life, which is followed by progressive clinical deterioration with psychomotor regression and complete absence of neurological improvement. However, the two sisters (monochorionic twins) reported by Pigeon et al., harboring one of the same mutations as our patient, had a milder disease course suggesting a genotype-phenotype correlation (Pigeon et al. 2009). Like our patient, both sisters lacked the characteristic features of EE, such as petechiae, orthostatic acrocyanosis, or chronic diarrhea. Of note, despite them being monochorionic twins, their clinical courses differed markedly; one had an episode of coma at 3 years of age, had spastic quadriparesis at 10 years of age, and could not speak, while the other sister had pyramidal symptoms mostly limited to the lower extremities and was able to speak two languages. As mentioned by Pigeon et al., this observation highlights the marked clinical heterogeneity displayed in EE. Insight into the biochemical nature of EE has led to the advent of new treatment strategies aimed at lowering the chronic H2S load (e.g., MNZ and NAC). Recently, Dionisi-Vici et al. convincingly reported the successful use of liver transplantation as an effective therapeutic approach in reverting the natural course of an otherwise fatal neurological disease (Dionisi-Vici et al. 2016). However, experience with treatment strategies directed against acute episodes of metabolic decompensation (e.g., hemodialysis) in patients with milder disease forms, who may not qualify for liver transplantation, is limited, except for a recent report of the use of IV NAC in a patient with an acute episode of EE in the context of meningococcemia (Kilic et al. 2017). Here we share our experience with an unusually mild clinical course of EE in a 19-year-old male, who presented with two acute episodes, one being an acute metabolic decompensation. His second episode was characterized by increasingly elevated lactate levels and metabolic deterioration, as reflected by levels of plasma thiosulfate and other biochemical parameters, despite intensive supportive management. To regain control of his metabolic state, we successfully employed CRRT to lower plasma sulfide levels. Given the small size of thiosulfate (112 D), these anions are readily removed by convection from hemofiltration and/or diffusion from dialysis; therefore, our patient received both modalities delivered as CVVHDF. Importantly, CRRT removes NAC but has no appreciable effect on the removal of MNZ (Hernandez et al. 2015; Roberts et al. 2015). It is suggested that the dose of intravenous NAC should be doubled during dialytic therapies. Notably, when we measured baclofen in CSF, levels were undetectable. It is important to point out that the CSF sample could only be obtained on day 5 of the acute episode. Around 70–80% of baclofen is excreted in the urine, and about 15% is metabolized in the liver. The elimination half-life is about 5 h in CSF but has been reported to increase up to 35 h in overdose (Meulendijks et al. 2015). However, given that CRRT is known to improve baclofen clearance, it is conceivable that, after 5 days, most of the baclofen may have been eliminated (Roberts et al. 2015).
In conclusion, CRRT may help to regain metabolic control in patients with milder courses of EE with an acute metabolic decompensation who are on chronic treatment with NAC and MNZ and fail to respond to conventional supportive measures.
Electronic Supplementary Material
Sequential results for urinary ethylmalonic acid, other organic acids, and acylglycines (DOCX 21 kb)
Acknowledgments
We would like to thank the patient and his family for giving us permission to publish this case report. We also thank Patrick Bherer (of the CHUS biochemical genetics laboratory) for analysis of urine acylglycine profiles and Dr. Massimo Zeviani for his advice during the second acute decompensation.
Take-Home Message
By reading this case report on a patient with an unusually mild form of ethylmalonic encephalopathy (EE), readers will learn about the acute and chronic management of this rare condition and about the importance of keeping metabolic causes, such as EE, in mind in patients presenting with a purely neurological phenotype. Finally, our report illustrates the importance of identifying all possible triggers of a metabolic decompensation in patients with inborn errors of metabolism.
Author Contributions
- TMK:
Data analysis and interpretation and planning and drafting most of the article.
- IRG:
Article contribution and revision.
- BO:
Article contribution and revision.
- CP:
Article contribution and revision.
- YT:
Data analysis and interpretation and article contribution and revision.
- PJW:
Data analysis and interpretation and article contribution and revision.
- DCB:
Data analysis and interpretation and planning and drafting of article.
Competing Interests
The authors have no competing interests to declare.
Funding
This work has not received any funding.
Patient Consent
The patient and his parents provided informed consent for publication of this case report.
Ethical Standards
The authors declare that the experiments comply with the current laws of Canada, the country in which they were performed.
References
- Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA. N-Acetylcysteine – a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol. 2007;7:355–359. doi: 10.1016/j.coph.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bherer P, Cyr D, Buhas D, Al-Hertani W, Maranda B, Waters PJ. Acylglycine profiling: a new liquid chromatography-tandem mass spectrometry (LC-MS/MS) method, applied to disorders of organic acid, fatty acid, and ketone metabolism. J Inherit Metab Dis. 2015;38:S35–S378. doi: 10.1007/s10545-015-9877-x. [DOI] [Google Scholar]
- Boyer M, Sowa M, Di Meo I, et al. Response to medical and a novel dietary treatment in newborn screen identified patients with ethylmalonic encephalopathy. Mol Genet Metab. 2018;124:57–63. doi: 10.1016/j.ymgme.2018.02.008. [DOI] [PubMed] [Google Scholar]
- Bucci M, Papapetropoulos A, Vellecco V, et al. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler Thromb Vasc Biol. 2010;30:1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- Burlina A, Zacchello F, Dionisi-Vici C, et al. New clinical phenotype of branched-chain acyl-CoA oxidation defect. Lancet. 1991;338:1522–1523. doi: 10.1016/0140-6736(91)92338-3. [DOI] [PubMed] [Google Scholar]
- Di Meo I, Lamperti C, Tiranti V. Mitochondrial diseases caused by toxic compound accumulation: from etiopathology to therapeutic approaches. EMBO Mol Med. 2015;7:1257–1266. doi: 10.15252/emmm.201505040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionisi-Vici C, Diodato D, Torre G, et al. Liver transplant in ethylmalonic encephalopathy: a new treatment for an otherwise fatal disease. Brain. 2016;139:1045–1051. doi: 10.1093/brain/aww013. [DOI] [PubMed] [Google Scholar]
- Drousiotou A, DiMeo I, Mineri R, Georgiou T, Stylianidou G, Tiranti V. Ethylmalonic encephalopathy: application of improved biochemical and molecular diagnostic approaches. Clin Genet. 2011;79:385–390. doi: 10.1111/j.1399-0004.2010.01457.x. [DOI] [PubMed] [Google Scholar]
- Elsey DJ, Fowkes RC, Baxter GF. Regulation of cardiovascular cell function by hydrogen sulfide (H(2)S) Cell Biochem Funct. 2010;28:95–106. doi: 10.1002/cbf.1618. [DOI] [PubMed] [Google Scholar]
- Grosso S, Mostardini R, Farnetani MA, et al. Ethylmalonic encephalopathy: further clinical and neuroradiological characterization. J Neurol. 2002;249:1446–1450. doi: 10.1007/s00415-002-0880-4. [DOI] [PubMed] [Google Scholar]
- Hernandez SH, Howland M, Schiano TD, Hoffman RS. The pharmacokinetics and extracorporeal removal of N-acetylcysteine during renal replacement therapies. Clin Toxicol (Phila) 2015;53:941–949. doi: 10.3109/15563650.2015.1100305. [DOI] [PubMed] [Google Scholar]
- Hildebrandt TM, Grieshaber MK. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- Jackson MR, Melideo SL, Jorns MS. Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry. 2012;51:6804–6815. doi: 10.1021/bi300778t. [DOI] [PubMed] [Google Scholar]
- Kabil O, Banerjee R. Characterization of patient mutations in human persulfide dioxygenase (ETHE1) involved in H2S catabolism. J Biol Chem. 2012;287:44561–44567. doi: 10.1074/jbc.M112.407411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic M, Dedeoglu O, Gocmen R, Kesici S, Yuksel D. Successful treatment of a patient with ethylmalonic encephalopathy by intravenous N-acetylcysteine. Metab Brain Dis. 2017;32:293–296. doi: 10.1007/s11011-016-9928-5. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18:1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- McGowan KA, Nyhan WL, Barshop BA, et al. The role of methionine in ethylmalonic encephalopathy with petechiae. Arch Neurol. 2004;61:570–574. doi: 10.1001/archneur.61.4.570. [DOI] [PubMed] [Google Scholar]
- Meulendijks D, Khan S, Koks CH, Huitema AD, Schellens JH, Beijnen JH. Baclofen overdose treated with continuous venovenous hemofiltration. Eur J Clin Pharmacol. 2015;71:357–361. doi: 10.1007/s00228-014-1802-y. [DOI] [PubMed] [Google Scholar]
- Mineri R, Rimoldi M, Burlina AB, et al. Identification of new mutations in the ETHE1 gene in a cohort of 14 patients presenting with ethylmalonic encephalopathy. J Med Genet. 2008;45:473–478. doi: 10.1136/jmg.2008.058271. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Tsugane M, Oka J, Kimura H. Hydrogen sulfide induces calcium waves in astrocytes. FASEB J. 2004;18:557–559. doi: 10.1096/fj.03-1052fje. [DOI] [PubMed] [Google Scholar]
- Perencevich M, Burakoff R. Use of antibiotics in the treatment of inflammatory bowel disease. Inflamm Bowel Dis. 2006;12:651–664. doi: 10.1097/01.MIB.0000225330.38119.c7. [DOI] [PubMed] [Google Scholar]
- Pettinati I, Brem J, McDonough MA, Schofield CJ. Crystal structure of human persulfide dioxygenase: structural basis of ethylmalonic encephalopathy. Hum Mol Genet. 2015;24:2458–2469. doi: 10.1093/hmg/ddv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigeon N, Campeau PM, Cyr D, Lemieux B, Clarke JT. Clinical heterogeneity in ethylmalonic encephalopathy. J Child Neurol. 2009;24:991–996. doi: 10.1177/0883073808331359. [DOI] [PubMed] [Google Scholar]
- Qingyou Z, Junbao D, Weijin Z, Hui Y, Chaoshu T, Chunyu Z. Impact of hydrogen sulfide on carbon monoxide/heme oxygenase pathway in the pathogenesis of hypoxic pulmonary hypertension. Biochem Biophys Res Commun. 2004;317:30–37. doi: 10.1016/j.bbrc.2004.02.176. [DOI] [PubMed] [Google Scholar]
- Roberts JK, Westphal S, Sparks MA. Iatrogenic baclofen neurotoxicity in ESRD: recognition and management. Semin Dial. 2015;28:525–529. doi: 10.1111/sdi.12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter R, Pang T, Kaplan PW. Metabolic, toxic, and epileptic encephalopathies. Alphen aan den Rijn: Wolters Kluwer; 2018. [Google Scholar]
- Tavasoli AR, Rostami P, Ashrafi MR, Karimzadeh P. Neurological and vascular manifestations of ethylmalonic encephalopathy. Iran J Child Neurol. 2017;11:57–60. [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, Zeviani M. Altered sulfide (H(2)S) metabolism in ethylmalonic encephalopathy. Cold Spring Harb Perspect Biol. 2013;5:a011437. doi: 10.1101/cshperspect.a011437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, D’Adamo P, Briem E, et al. Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am J Hum Genet. 2004;74:239–252. doi: 10.1086/381653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, Briem E, Lamantea E, et al. ETHE1 mutations are specific to ethylmalonic encephalopathy. J Med Genet. 2006;43:340–346. doi: 10.1136/jmg.2005.036210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, Viscomi C, Hildebrandt T, et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med. 2009;15:200–205. doi: 10.1038/nm.1907. [DOI] [PubMed] [Google Scholar]
- Viscomi C, Burlina AB, Dweikat I, et al. Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat Med. 2010;16:869–871. doi: 10.1038/nm.2188. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Armstrong JS, Chu SH, et al. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J Neurochem. 2004;90:765–768. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- Xu M, Wu YM, Li Q, Wang X, He RR. Electrophysiological effects of hydrogen sulfide on pacemaker cells in sinoatrial nodes of rabbits. Sheng Li Xue Bao. 2008;60:175–180. [PubMed] [Google Scholar]
- Zafeiriou DI, Augoustides-Savvopoulou P, Haas D, et al. Ethylmalonic encephalopathy: clinical and biochemical observations. Neuropediatrics. 2007;38:78–82. doi: 10.1055/s-2007-984447. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequential results for urinary ethylmalonic acid, other organic acids, and acylglycines (DOCX 21 kb)