

Abstract
Rationale:
Glioblastomas are malignant, infiltrating gliomas classified as grade IV by the World Health Organization. Genetically, most glioblastomas do not exhibit the isocitrate dehydrogenase (IDH) 1/2 gene mutation and rarely harbor the 1p/19q co-deletion. Neuroradiologically, glioblastomas rarely form a cyst with a mural nodule lesion.
Patient concerns:
In this study, a 78-year-old woman, with speech difficulty and forgetfulness, had a cystic tumor with a mural nodule in the right frontoparietal lobe. Therefore, partial tumor resection was performed.
Diagnosis:
Histopathology of the tumor, a glioblastoma, exhibited pseudopapillary features with non-hyalinized capillary cores and rich mini-gemistocytic cells. Genetic analysis of the tumor revealed co-deletion of 1p36/19q13, with wild-type IDH.
Interventions:
The patient underwent a combination of postoperative radiotherapy and temozolomide chemotherapy before leaving the hospital. After discharge, she was treated by 20 courses of temozolomide chemotherapy.
Outcomes:
The patient is free from tumor recurrence 23 months after the operation.
Lessons:
We present a unique case of glioblastoma that exhibited novel neuroradiological, histopathological, and genetic features with a favorable prognosis for the patient. Therefore, a compilation of similar cases with clinicopathological and genetic analyses to characterize this unique glioblastoma is critical. Clinical evidence will help develop effective therapeutic approaches to improve prognosis in patients with glioblastoma.
Keywords: 1p/19q co-deletion, brain tumor, glioblastoma, mural nodule, pseudopapillary structure
1. Introduction
Glioblastomas are malignant, infiltrating gliomas classified as grade IV by the World Health Organization.[1] They are characterized neuroradiologically by an ill-defined, low-density mass with ring enhancement. Neuropathological characteristics include a highly cellular, infiltrating astrocytoma with distinct nuclear anaplasia and high mitotic activity, necrosis, and/or microvascular proliferation. Genetically, most glioblastomas do not exhibit mutations in the isocitrate dehydrogenase (IDH) 1/2 and α-thalassemia/mental retardation syndrome X-linked (ATRX) genes, and are classified as “IDH-wild-type glioblastoma”.[1] Glioblastomas rarely harbor the 1p/19q co-deletion, a characteristic genetic alteration in oligodendroglial neoplasms, such as oligodendroglioma and anaplastic oligodendroglioma.[2] This case report describes a unique case of a cerebral glioblastoma presenting as a neuroradiological cyst with a mural nodule pattern, which was pathologically determined to contain a pseudopapillary structure and harbor the 1p36/19q13 codeletion but not a IDH1/2 mutation.
2. Case presentation
The patient (female; age = 78 years) after experiencing speech difficulty and forgetfulness, for 12 days, visited Nihon University Itabashi Hospital. She was hospitalized after cranial computed tomography (CT) identified a brain tumor. Neurological examination revealed slightly decreased consciousness, myasthenia of the left orbicularis oris muscle, and dysarthria. Brain magnetic resonance imaging (MRI) revealed a mass in the right frontoparietal lobe that was hypo and hyperintense on the T1- and T2-weighted images, respectively. The mass appeared as a heterogeneously enhanced mural nodule with a rim-enhancing cyst (diameter, 45 mm) on the T1-weighted gadolinium-enhanced images (Fig. 1A–C). On day 19 after the onset of symptoms, partial tumor resection was performed, and a combination of postoperative radiotherapy and temozolomide chemotherapy was initiated. Significantly, the patient is alive 23 months after the symptom onset, without tumor recurrence.
Figure 1.
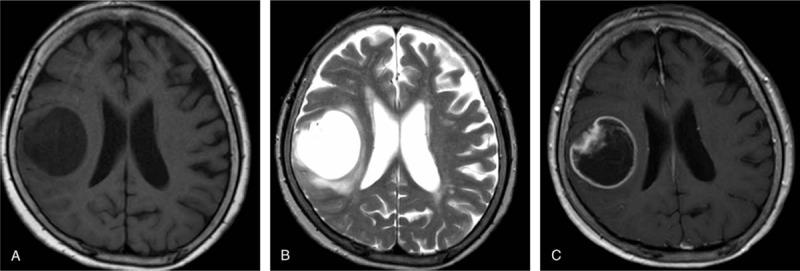
Neuroradiological features. A frontoparietal tumor exhibiting a neuroradiological cyst with mural nodule pattern on magnetic resonance imaging. The mural nodule is hypointense in the T1-weighted (A) and hyperintense in the T2-weighted (B) images. T1-weighted gadolinium-enhanced image reveals the brain tumor as a heterogeneously enhancing mural nodule with a rim-enhancing cyst (C).
Histopathologically, the tumor was a hypercellular neoplasm characterized by the formation of pseudopapillary structures with non-hyalinized capillaries (Fig. 2A and B), and infiltrated the cerebral parenchyma. The tumor comprised medium-sized, naked neoplastic cells that resembled fibrillary astrocytes or mini-gemistocytes. The papillary structures, which were surrounded by naked neoplastic cells, comprised fibrillary and mini-gemistocytic neoplastic cells that tended to proliferate around the non-hyalinized capillaries (Fig. 2C). There was no evidence of oligodendroglia-like cells, ganglionic cells, xanthomatous cells, rosenthal fibers (RFs), and eosinophilic granular bodies (EGBs). Additionally, mitotic figures were frequent, up to 11 per 10 high-power fields. Focal microvascular proliferation was observed in the tumor specimen (Fig. 2D) and lacked necrosis.
Figure 2.
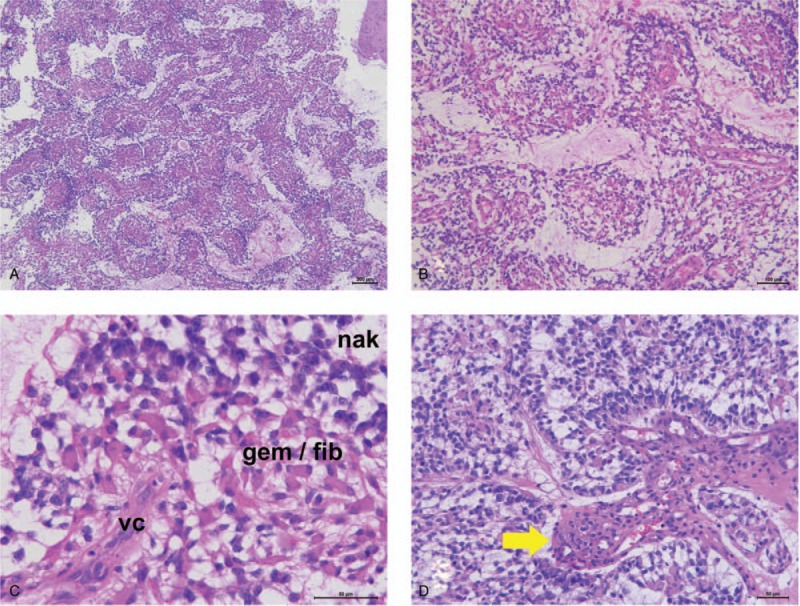
Histopathological features. Histopathologically, the highly cellular tumor exhibits pseudopapillary structures with non-hyalinized central capillaries (A and B; hematoxylin and eosin [H&E], magnification ×40 [A] and ×100 [B]). Non-hyalinized vascular cores (vc) surrounded by both fibrillary (fib) and mini-gemistocytic (gem) neoplastic cells; these pseudopapillary structures are covered by naked neoplastic cells (nkd) (C; H&E, magnification ×400). Focal microvascular proliferation is also observed (D; H&E, magnification ×200).
Immunohistochemically, the neoplastic cells were positive for S-100 protein, glial fibrillary acidic protein (Fig. 3A), and oligodendrocyte transcription factor 2 (Olig2) (Fig. 3B). Neoplastic cells were not positive for the epithelial membrane antigen, D2-40, synaptophysin, neurofilament protein (NFP), neuronal nuclei (NeuN), chromogranin A, and a cluster of differentiation 34 (CD34). However, synaptophysin (Fig. 3C), NeuN (Fig. 3D), and NFP (Fig. 3E) immunoreactivities were observed in both non-neoplastic residual neuronal cells and neuropil. Furthermore, the tumor exhibited negative immunoreactivity to the IDH1-R132H monoclonal antibody (Fig. 3F) and exhibited nuclear expression of the ATRX (Fig. 3G) protein and integrase interactor 1(INI1) protein (Fig. 3H). Additionally, the MIB-1 labeling index was 63.4% (Fig. 3I).
Figure 3.
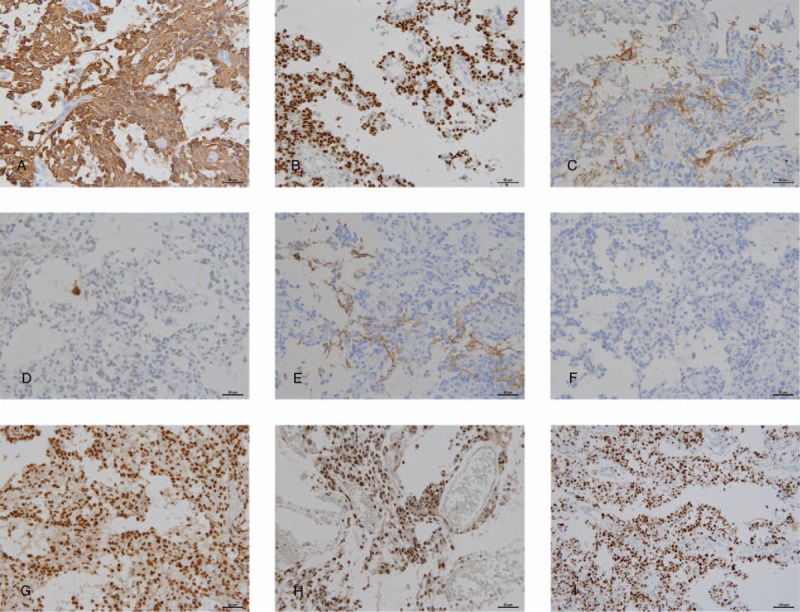
Immunohistochemical features. Immunohistochemically, neoplastic cells express glial fibrillary acidic protein (A; magnification ×200), and oligodendrocyte transcription factor 2 (B; magnification ×200). Tumor stroma reveals immunoreactivity against synaptophysin (C; magnification ×200). A few residual neurons in the tumor show neuronal nuclei (NeuN) immunopositivity (D; ×200), and neurofilament immunoreactivity is focally observed in the tumor stroma (E; magnification ×200). The tumor exhibits no immunoreactivity against mutant isocitrate dehydrogenase (IDH1) R132H (F; magnification ×200) and retains nuclear expression of α-thalassemia/mental retardation syndrome X-linked (G; magnification ×200) and INI-1 (H; magnification ×200). Additionally, MIB-1 labeling index is very high (63.4%) (I; magnification ×100).
Fluorescence in situ hybridization analysis of chromosomes 1p and 19q revealed deletion of 1p36 and 19q13, respectively (Fig. 4A and B). However, tumor mutation analysis revealed no mutations for IDH1, IDH2, BRAF V600E, and SLC44A1-PRKCA fusion.
Figure 4.
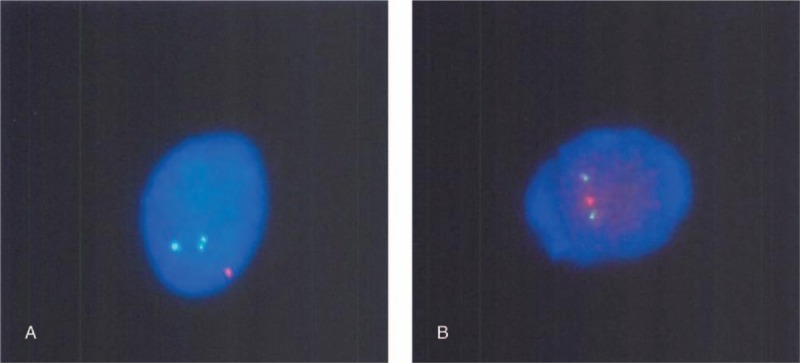
Fluorescence in situ hybridization (FISH) for detecting 1p/19q co-deletion. FISH analysis for chromosomes 1p and 19q reveals co-deletion of 1p36 and 19q13 (A; red dot, 1p36 signal; green dot, 1q25 signal) (B; red dot, 19q13 signal; green dot, 19p13 signal).
Based on the clinicopathological, immunohistochemical, and genetic findings, the neoplasm was diagnosed as an IDH-wild-type, grade IV papillary glioblastoma with 1p36/19q13 co-deletion.
3. Discussion
The brain tumor described in the present report exhibited the following characteristics:
-
1.
frontoparietal cystic tumor with a mural nodule,
-
2.
glioblastoma exhibiting pseudopapillary features with non-hyalinized capillary cores and rich mini-gemistocytic cells,
-
3.
the presence of 1p36/19q13 co-deletion with wild-type IDH.
Brain MRI revealed that the patient had a cystic tumor with a mural nodule. This feature is frequently observed in patients with pleomorphic xanthoastrocytoma (PXA),[3] pilocytic astrocytoma (PA),[4] and papillary glioneuronal tumor (PGNT).[5] Typically, the solid areas in PXA, PA, and PGNT exhibit a heterogeneous enhancement in T1-weighted images with gadolinium enhancement,[3–5] which is consistent with our findings. Interestingly, our patient was an elderly female, whereas the majority of cases of PXA (median age, 17 years [range, 8–65 years]),[3] PA (median age, 17 years [range, 1–74 years]),[6] and PGNT (median age, 25 years [range, 4–75 years])[5] are more frequently reported in younger patients.
PXAs are usually low-grade astrocytomas with pleomorphism, xanthomatous cells, frequent EGBs, and desmoplasia.[3] PAs are characterized by a biphasic pattern comprising piloid astrocytoma cells with RFs and EGBs.[4] Additionally, PGNTs typically consist of neoplastic astrocytic cells forming pseudopapillary structures with hyalinized capillaries and neoplastic neuronal cells in the interpapillary area.[5] Furthermore, mini-gemistocytes are occasionally observed in PGNTs.[5] Anaplastic brain tumors, including anaplastic PXA, anaplastic PA, and anaplastic PGNT, have recently been reported.[4,7,8]
Tumors, including anaplastic PXA, anaplastic PA, and anaplastic PGNT, are considered to be anaplastic when the non-anaplastic components are concurrently observed with the anaplastic features.[3,4,7,8] Interestingly, the patient had an infiltrating, highly cellular, high-grade astrocytoma exhibiting the aforementioned characteristic features, without the non-anaplastic components of PXA,[3] PA,[4] or PGNT.[7] Hence, the histopathology of the current case was different from the anaplastic types of PXA, PA, or PGNT. Our findings are corroborated by several recent cases reporting the histopathological features of spinal glial papillary tumors.[9] However, several immunohistochemical and genetic features in the present case, specifically no epithelial membrane antigen immunoreactivity, and preserved INI-1 nuclear expression are different from those in previous cases.[9]
The BRAF V600E mutation, absent in the present case, is frequently detected in PXA[10] and occasionally in elderly patients with PA.[4,10] Additionally, the SLC44A1-PRKCA fusion gene, encoding a protein kinase C involved in the mitogen-activated protein kinase pathway, is a genetic alteration specifically observed in PGNTs,[11] but was not observed in the present case. Therefore, this case does not belong to any anaplastic types of PXA, PA, or PGNT. Interestingly, although our patient had a 1p36/19q13 co-deletion, considered the genetic characteristic of oligodendroglial neoplasms, particularly classic oligodendroglioma,[2] no oligodendroglial components were observed in the present case. Research suggests that astrocytic neoplasms often, but not always, harbor segmental loss of chromosome 1p or 19q.[2] Therefore, we believe that co-deletion of both 1p36 and 19q13 in our case implies segmental, but not complete, loss as previously reported.[2]
In conclusion, this case report presents an unusual case of primary astrocytic neoplasm exhibiting unique neuroradiological, histopathological, and genetic characteristics. Despite high MIB-1 labeling index, the patient is free from tumor recurrence 23 months after tumor resection. A limitation of this case report is that the detailed neuropathological features of the cyst with mural nodule glioblastoma are unknown. Therefore, it is unclear whether any unique characteristics of this case, including the cystic structure with a mural nodule neuroradiological pattern, pseudopapillary histopathological pattern, and 1p36/19q13 co-deletion, were associated with the prognosis of the patients. Accumulation of similar cases, with clinicopathological and genetic analyses, is warranted to develop therapeutic approaches for this unique glioblastoma subtype and to assess patient prognosis.
Acknowledgments
The authors thank Mrs. Yukari Hirotani at the Division of Oncologic Pathology, Department of Pathology and Microbiology, Nihon University School of Medicine, Tokyo, Japan for their excellent technical assistance.
Author contributions
Conceptualization: Taku Homma.
Data curation: Taku Homma, Yuya Hanashima, Toshiya Maebayashi, and Yoko Nakanishi.
Genetic investigation: Yoko Nakanishi.
Neuroradiological investigation: Toshiya Maebayashi.
Pathological investigation: Taku Homma, Toshiyuki Ishige, and Hiroyuki Hao.
Patient's care including operation and postoperative irradiation therapy: Yuya Hanashima, Toshiya Maebayashi, Takashi Ohta, and Atsuo Yoshino.
Resources: Yuya Hanashima, Takashi Ohta, and Atsuo Yoshino.
Supervision: Atsuo Yoshino and Hiroyuki Hao.
Writing – original draft: Taku Homma.
Writing – review and editing: Taku Homma.
Footnotes
Abbreviations: ATRX gene = α-thalassemia/mental retardation syndrome X-linked genes, CD34 = cluster of differentiation 34, CT = computed tomography, EGBs = eosinophilic granular bodies, IDH 1/2 = isocitrate dehydrogenase 1/2, INI1 = integrase interactor 1, MRI = magnetic resonance imaging, NeuN = neuronal nuclei, NF = neurofilament protein, Olig2 = oligodendrocyte transcription factor 2, PA = pilocytic astrocytoma, PGNT = papillary glioneuronal tumor, PXA = pleomorphic xanthoastrocytoma, RFs = rosenthal fibers.
Informed consent was obtained from the patient described in the report.
The authors have no conflict of interest to declare.
References
- [1].Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016;131:803–20. [DOI] [PubMed] [Google Scholar]
- [2].Barbashina V, Salazar P, Holland EC, et al. Allelic losses at 1p/36 and 19q13 in gliomas: correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene. Clin Cancer Res 2005;11:1119–28. [PubMed] [Google Scholar]
- [3].Crespo-Rodriguez AM, Smimiotopoulos JG, Rushing EJ. MR and CT imaging of 24 pleomorphic xanthoastrocytomas (PXA) and a review of the literature. Neuroradiology 2007;49:307–15. [DOI] [PubMed] [Google Scholar]
- [4].Collins VP, Jones DTW, Giannini C. Pilocytic astrocytoma: pathology, molecular mechanisms, and markers. Acta Neuropathol 2015;129:775–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pimentel J, Barroso C, Miguens J, et al. Papillary glioneuronal tumor. Clin Neuropathol 2009;28:287–94. [PubMed] [Google Scholar]
- [6].Hasselblat M, Riesmeier B, Lechtape B, et al. BRAF-KIAA1549 fusion transcripts are less frequent in pilocytic astrocytomas diagnosed in adults. Neuropathol Appl Neurobiol 2011;37:803–6. [DOI] [PubMed] [Google Scholar]
- [7].Bourekas E, Bell SD, Gandhe AR, et al. Anaplastic papillary glioneuronal tumor with extraneuraal metastases. J Neuropathol Exp Neurol 2014;73:474–6. [DOI] [PubMed] [Google Scholar]
- [8].Schmidt Y, Kleinschmidt-DeMasters BK, Aisner DL, et al. Anaplastic PXA in adults: case series with clinicopathologic and molecular features. J Neurooncol 2013;111:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hasselblatt M, Kurniawan AD, Rozsnoki S, et al. Glial papillary tumor of the spinal cord with MARCB1/INI1-loss and favorable long-term outcome. Neuropathol Appl Neurobiol 2018;44:229–32. [DOI] [PubMed] [Google Scholar]
- [10].Schindler G, Capper D, Meyer J, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma, and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol 2011;121:397–405. [DOI] [PubMed] [Google Scholar]
- [11].Pages M, Lacroix L, Tauziede-Espariat A, et al. Papillary glioneuronal tumors: histological and molecular characteristics and diagnostic value of SLC44A1-PRKCA fusion. Acta Neuropathol Commun 2015;3:85. [DOI] [PMC free article] [PubMed] [Google Scholar]