

Abstract
Common variable immunodeficiency (CVID) patients have reduced gut microbial diversity compared to healthy controls. The reduced diversity is associated with gut leakage, increased systemic inflammation and ten “key” bacteria that capture the gut dysbiosis (dysbiosis index) in CVID. Rifaximin is a broad-spectrum non-absorbable antibiotic known to reduce gut leakage (lipopolysaccharides, LPS) in liver disease. In this study, we explored as a ‘proof of concept’ that altering gut microbial composition could reduce systemic inflammation, using CVID as a disease model. Forty adult CVID patients were randomized, (1:1) to twice-daily oral rifaximin 550 mg versus no treatment for 2 weeks in an open-label, single-centre study. Primary endpoints were reduction in plasma/serum levels of soluble (s) CD14, sCD25, sCD163, neopterin, CRP, TNF, LPS and selected cytokines measured at 0, 2 and 8 weeks. Secondary endpoint was changes in intra-individual bacterial diversity in stool samples. Rifaximin-use did not significantly change any of the inflammation or gut leakage markers, but decreased gut microbial diversity compared with no treatment (p = 0.002). Importantly, the gut bacteria in the CVID dysbiosis index were not changed by rifaximin. The results suggest that modulating gut microbiota by rifaximin is not the chosen intervention to affect systemic inflammation, at least not in CVID.
Subject terms: Immunological deficiency syndromes, Microbiota
Introduction
Over the last decade, numerous discoveries have highlighted the role of gut microbiota in critical processes to human health including digestion, absorption of nutrients, metabolism, growth, and immune responses1. Compositional and functional changes of the gut microbiome, referred to as dysbiosis, have emerged as an important contributor not only to intestinal diseases, but also systemic metabolic and inflammatory conditions such as obesity2, diabetes3, cardiac disease4, HIV infection5 and brain disorders1. Studies in mice have shown that bacteria and their products (e.g., lipopolysaccharide [LPS]) interact with the immune system both locally in the gut and systemically. The composition of the gut microbiota can also alter the balance between pro- and anti-inflammatory immune mediators and influence important metabolic pathways, sometimes being sufficient to modulate disease development and progression6. However, initial gut microbiota studies in human diseases have been cross-sectional and not able to establish causality, i.e. in principle the dysbiosis could be entirely secondary to disease.
We have recently shown that patients with common variable immunodeficiency (CVID) display a reduced microbial diversity7. CVID is the most common symptomatic primary immunodeficiency among adults, characterized by reduced levels of Immunoglobulin (Ig)G, IgM and/or IgA, and increased susceptibility to respiratory tract infections with capsulated bacteria8. Due to immune dysregulation, 74% of the patients also have one or more inflammatory and/or autoimmune complications, including enteropathy9,10. CVID patients have an underlying B-cell defect, but many patients also display T-cell dysfunction and evidence of systemic immune activation with elevated levels of inflammatory cytokines such as tumor necrosis factor (TNF) and interleukin (IL)-6, and soluble markers of monocyte/macrophage- (e.g., soluble [s]CD14) and T cell activation (i.e., sCD25)11. The reason for this persistent immune activation is not fully elucidated, but could at least partly reflect gut leakage mechanisms with a subsequent stimulation of innate immunity through interaction with Toll like receptors (TLRs) and related molecules12. We have previously reported increased plasma levels of LPS in CVID patients compared to healthy controls, negatively correlated with gut microbial diversity7, suggesting that a less diverse gut microbiota is associated with increased bacterial translocation. We have therefore proposed that chronic inflammation in the gastrointestinal tract can cause a breach of the intestinal barrier with subsequent systemic immune activation, similar to what is observed in HIV infection5.
In humans, interventional studies targeting the gut microbiota could provide evidence of a causal link between the intestine and human disease. In the present study, we used CVID as a disease model to therapeutically manipulate the gut microbiota to test our hypothesis that alteration of the microbiota affects systemic inflammation. The oral non-absorbable antibiotic rifaximin was chosen due to its bactericidal activity against a broad array of enteric pathogens and because it has been shown to reduce plasma levels of LPS in chronic liver disease with reduced risk of developing encephalopathy13. Importantly, there is negligible absorption of rifaximin from the gastrointestinal tract and thereby no direct antibacterial systemic effect which may bias the interpretation of a link between the gut and systemic inflammation14.
Results
All adult CVID patients (n = 124) aged 18–74 years registered at the Section of Clinical Immunology and Infectious Diseases at Oslo University Hospital, Rikshospitalet, Oslo, Norway were pre-screened for inclusion. The Section functions as a national centre for diagnosis and treatment of primary immunodeficiency diseases for adults in Norway, and only a few (<10) patients are followed at other hospitals in Norway. Flowchart for recruitment of patients to the study is shown in Fig. 1. Out of 124 CVID patients, 11 were excluded at pre-screening due to immunosuppressive drugs (n = 3), alcohol abuse (n = 1), type 1 diabetes (n = 2), serious multiple adverse reactions to antibiotics (n = 1), malignancy (n = 1), chronic herpes simplex virus infection (1) and colostomy (n = 2). Of the 113 CVID patients found eligible, 48 volunteered to participate in the study, and of these eight were not included due to recent antibiotic course (n = 6), recent probiotic use (n = 1) and lactation (n = 1). In the end 40 patients, aged 21–69 years (63% women), with CVID were enrolled in this study and randomized to rifaximin 550 mg bd versus no treatment for 14 days.
Figure 1.
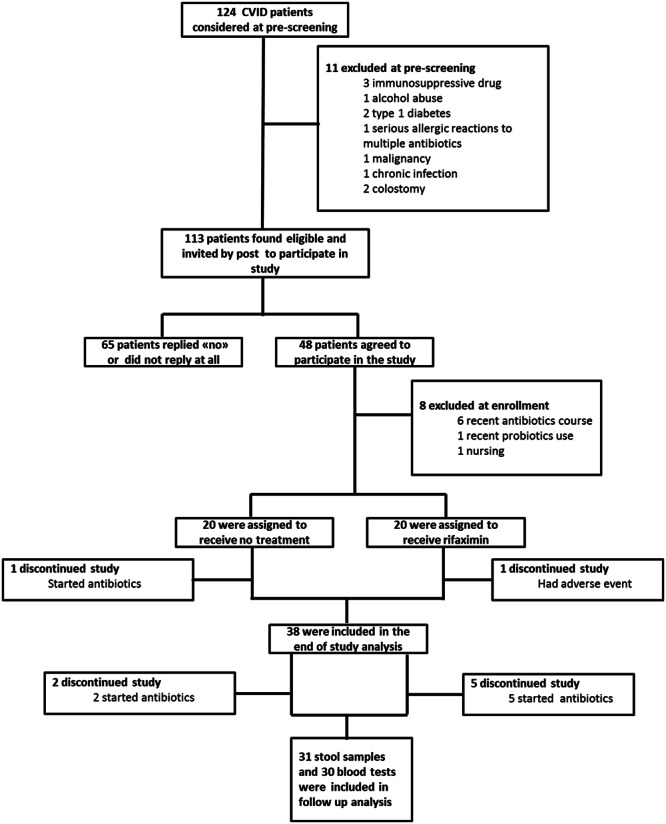
Trial Profile.
Baseline patient characteristics are presented in Table 1. Except for a lower frequency of patients with ‘non-infectious complications’ in the rifaximin group (P = 0.04), there were no significant differences between the rifaximin and the no intervention group at baseline (Table 1).
Table 1.
Baseline characteristics of CVID patients in the Rifaximin study.
| Baseline characteristics | All subjects (n = 40) | Rifaximin arm (n = 20) | No-treatment arm (n = 20) | p-value* |
|---|---|---|---|---|
| Age mean ± SD (range) | 50 ± 12 (21–69) | 47 ± 11 (21–63) | 53 ± 12 (21–69) | 0.07 |
| Male, n (%) | 15 (38) | 8 (40) | 7 (35) | 1.00 |
| Immunoglobulin therapy, n (%): | 0.06 | |||
| s.c.a | 30 (75) | 14 (70) | 16 (80) | |
| i.v.b | 6 (15) | 5 (25) | 1 (5) | |
| s.c + i.v. | 4 (10) | 1 (5) | 3 (15) | |
| Infection only, n (%) | 8 (20) | 7 (35) | 1 (5) | 0.04 |
| Splenomegaly, n (%) | 16 (40) | 7 (35) | 9 (45) | 0.75 |
| Organ-specific autoimmunity | 8 (20) | 5 (25) | 3 (15) | 0.70 |
| Autoimmune cytopenia, n (%) | 8 (20) | 2 (10) | 6 (30) | 0.24 |
| Enteropathy, n (%) | 13 (33) | 8 (40) | 5 (25) | 0.50 |
| Lymphoid hyperplasia, n (%) | 23 (56) | 9 (45) | 14 (70) | 0.20 |
| Granulomas, n (%) | 6 (15) | 3 (15) | 3 (15) | 1.00 |
| Lymphocytic interstitial pneumonitis, n (%) | 1 (2.5) | 0 | 1(95) | 1.00 |
| Nodular regenerative hyperplasia, n (%) | 1 (2.5) | 0 | 1(95) | 1.00 |
| Overall non-infectious complications, n (%) | 32 (80) | 13 (65) | 19 (95) | 0.04 |
as.c., subcutaneously; bi.v., intravenously. *P value for rifaximin arm compared to no-treatment arm
Effect of rifaximin on markers of systemic inflammation and gut leakage
Overall there was no effect of rifaximin use on markers of systemic inflammation or markers of gut leakage (i.e., LPS) (uniANOVA), except for CRP (Table 2). The significant change in CRP was driven by an increase in CRP from baseline to week 8 in the rifaximin group (Wilcoxon, P = 0.015) (Table 2). Furthermore, there was no effect of rifaximin treatment on mRNA levels of selected pro- and anti-inflammatory cytokines and chemokines in PBMC (i.e., IL-6, IL-8/CXCL8, IL-10, IL-12, TNF, TGF-β, MIG/CXCL9, IP-10/CXCL10; Supplementary Table S1). Also, the distribution of B and T cell subpopulations, as measured by 10 readouts, remained unchanged by rifaximin use, except for minor changes in the naive CD4+ T cells reflecting a decrease in the no intervention group (Supplementary Table S2).
Table 2.
Soluble markers of inflammation in plasma in the “rifaximin” (Rif, n = 20) and in the “no intervention” group (No Int, n = 20).
| Marker | Group | Baselinea (n = 40) | 2 weeksa (n = 38) | 8 weeksa (n = 30) | P value interactionb |
|---|---|---|---|---|---|
| CRPmg/L | No Int | 3.5 (2.1–7.1) | 4.7 (1.0–7.9) | 3.2 (2.5–6.9) | 0.029 |
| Rif | 1.9 (0.86–4.80) | 2.20 (1.2–4.8) | 2.8 (1.9–5.4)* | ||
| sCD14ng/mL | No Int | 3949 (3162–4456) | 4032 (3290–4630) | 3826 (2978–4459) | 0.555 |
| Rif | 3378 (3072–4219) | 3639 (3241–4414) | 3552 (2921–4485) | ||
| LPSpg/ml | No Int | 83 (78–91) | 86 (78–92) | 87 (78–94) | 0.955 |
| Rif | 87 (80–96) | 90 (78–98) | 90 (83–102) | ||
| sCD25ng/mL | No Int | 1.44 (0.71–2.10) | 1.54 (0.74–1.89) | 1.33 (0.80–1.86) | 0.636 |
| Rif | 0.65 (0.51–1.43) | 0.63 (0.54–1.18) | 0.99 (0.44–1.55) | ||
| sCD163ng/mL | No Int | 1699 (1350–2001) | 1677 (984–1930) | 1536 (1194–1940) | 0.320 |
| Rif | 971 (647–1353) | 902 (749–1310) | 1151 (793–1549) | ||
| Neopterinnmol/L | No Int | 11.18 (9.40–21.60) | 13.17 (8.32–24.31) | 10.37 (8.11–21.90) | 0.058 |
| Rif | 9.02 (6.71–14.65) | 7.79 (6.37–17.24) | 11.43 (6.66–21.78) |
aData are given in median (25–75 percentile). bThe p value reflects the interaction between time and group from UNIANOVA. *P < 0.05 vs. baseline.
Effect of rifaximin on gut microbiota composition
In contrast to the lack of effect on markers of systemic inflammation, rifaximin treatment was associated with a significant change in microbial alpha diversity (Faith’s PD; UNIANOVA P = 0.0002, Fig. 2). Thus, there was a significant decrease in microbial alpha diversity (Faith’s PD: beta = −0.26, P = 0.002) after the first two weeks of the study, followed by a return to baseline six weeks after rifaximin was terminated (beta: 0.02 linear regression [baseline to week 8] P = 0.82, Fig. 2). Other measures of alpha diversity such as Chao1 (P = 0.03), Shannon index (P = 0.002) and observed OTUs (P = 0.002) were also significantly changed by rifaximin, whereas Simpson index (P = 0.10) was not (linear regression data for baseline to week 2 and baseline to week 8 are given in Supplementary Table S3). Global differences in the gut microbiota measured by beta diversity (i.e., to which degree different taxa are shared between individuals or groups of individuals) are illustrated in Fig. 3 at four time points for the two groups. The beta diversity (as measured by Bray-Curtis distances between paired samples at week 0 and week 2) was significantly higher in the rifaximin group compared to the no intervention group (Mann-Whitney P = 0.014, median 0.34 (IQR 0.32–0.40) and 0.27 (IQR 0.24–0.35), respectively, Fig. 4), but not between week 0 and week 8 (P = 0.46, median 0.32 (IQR 0.28–0.36) and 0.30 (IQR 0.25–0.38), respectively).
Figure 2.
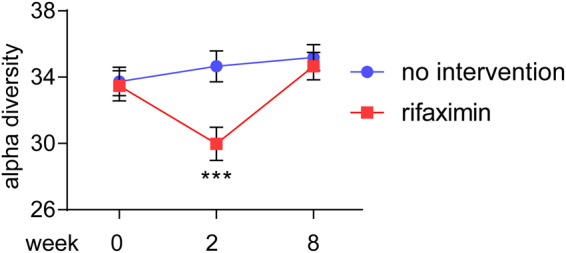
Comparing alpha diversity (phylogenetic diversity [Faith’s PD]) in stool samples before (week 0), after (week 2) and at follow up (week 8) for a 2-week rifaximin course versus no treatment. ***P < 0.001 (UNIANOVA, baseline values at week 0 is corrected for values at week −14 [baseline 2]).
Figure 3.

Beta diversity plot with each patient’s samples at four time points for the rifaximin and no intervention group.
Figure 4.
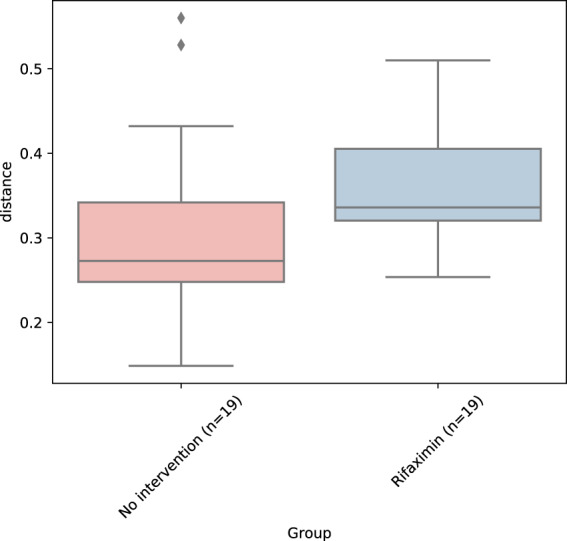
Bray-Curtis distances between (paired) samples at baseline and week 2 in the rifaximin and no intervention group.
Sixteen bacterial taxa, at different taxonomic levels, were significantly changed by rifaximin use (Table 3, Fig. 5). Also, the direction of change (expressed as beta values from the linear regression analysis) for the different taxa from baseline and week 2 and 8 are shown in Table 3. None of the ten main bacteria previously shown to differentiate CVID patients and healthy controls, the CVID specific dysbiosis index7, were significantly changed by rifaximin use (Supplementary Fig. S1), thus the CVID dysbiosis index was unchanged by rifaximin (Supplementary Fig. S2). Consistent with the data from our previous publication7, the dysbiosis index in the present study correlated with the microbial diversity (Faith’s PD) (rho = −0.42, P = 0.007, at week 0 [n = 40]), suggesting that the abundance of these ten bacteria captures the microbial diversity in CVID also in this study.
Figure 5.

On the left are the names of the 16 taxa significantly changed by rifaximin (UNIANOVA) and on the right is the corresponding mean relative abundance of each specific taxa expressed (on a log2 scale) for the no intervention and the rifaximin group at baseline, week 2 and week 8. The colors to the left in the figure illustrate which taxa are in the same taxonomic family.
Table 3.
Taxa that are significantly changed by rifaximin in stool samples from CVID patients.
| Taxa | Group | Baselinea (n = 40) | 2 weeksa (n = 38) | 8 weeksa (n = 31) | P value interactionb | Beta2c (week 2) | Beta2c (week 8) |
|---|---|---|---|---|---|---|---|
| Bacteroidetes | No Int | 0.415 (0.388–0.441) | 0.385 (0.357–0.413) | 0.413 (0.384–0.443) | 0.012 | 0.28* | 0.08 |
| Rif | 0.366 (0.340–0.393) | 0.413 (0.385–0.441) | 0.382 (0.348–0.415) | ||||
| Bacteroidetes.Bacteroidia | No Int | 0.414 (0.388–0.441) | 0.385 (0.357–0.412) | 0.413 (0.383–0.443) | 0.012 | 0.28* | 0.08 |
| Rif | 0.366 (0.340–0.393) | 0.413 (0.385–0.440) | 0.382 (0.348–0.415) | ||||
| Bacteroidetes.Bacteroidia.Bacteroidales | No Int | 0.414 (0.388–0.441) | 0.385 (0.357–0.412) | 0.413 (0.383–0.443) | 0.012 | 0.28* | 0.08 |
| Rif | 0.366 (0.340–0.393) | 0.413 (0.385–0.440) | 0.382 (0.348–0.415) | ||||
| Bacteroidetes.Bacteroidia.Bacteroidales.Bacteroidaceae | No Int | 0.292 (0.267–0.316) | 0.265 (0.239–0.290) | 0.283 (0.255–0.310) | 0.009 | 0.25** | 0.12 |
| Rif | 0.269 (0.245–0.294) | 0.315 (0.289–0.340) | 0.282 (0.251–0.312) | ||||
| Bacteroidetes.Bacteroidia.Bacteroidales.Bacteroidaceae. Bacteroides | No Int | 0.292 (0.267–0.316) | 0.265 (0.239–0.290) | 0.283 (0.255–0.310) | 0.009 | 0.25** | 0.12 |
| Rif | 0.269 (0.245–0.294) | 0.315 (0.289–0.340) | 0.282 (0.251–0.312) | ||||
| Firmicutes.Clostridia.Clostridiales.Family XI.Ezakiella | No Int | −7.8E−20 (−1.3E−5–1.3E−5) | 1.1E−5 (−2.2E−6–2.4E−5) | −4.3E−7 (−1.5E−5–1.4E−5) | 0.029 | −0.09 | 0.43 |
| Rif | 5.3E−6 (−7.5 E−6–1.8E−5) | 5.0E−6 (−8.3E−6–1.8 E−5) | 3.4E−5 (1.8E−5–5.0E−5) | ||||
| Firmicutes.Clostridia.Clostridiales.Family XIII.Family XIII UCG-001 | No Int | 1.1E−4 (−3.9E−6–2.3E−4) | 9.7E−5 (−2.2E−5–2.2E−4) | 1.4E−4 (7.5E−6–2.6E−4) | 0.028 | 0.04 | 0.15 |
| Rif | 2.3E−4 (1.2E−4–3.5E−4) | 2.5E−4 (1.3E−4–3.7E−4) | 0.001 (4.2E−4–0.001) | ||||
| Firmicutes.Clostridia.Clostridiales.Lachnospiraceae. Anaerostipes | No Int | 3.9E−4 (−3.7E−7–0.001) | 0.001 (2.5E−4–0.001) | 0.001 (0.001–0.001) | 0.031 | 0.21 | −0.25 |
| Rif | 4.9E−4 (1.0E−4–0.001) | 0.001 (0.001–0.002) | 4.6E−4 (−3.5E−5–0.001) | ||||
| Firmicutes.Clostridia.Clostridiales.Lachnospiraceae. Lactonifactor | No Int | 2.6E−5 (−9.7E−6–6.2E−5) | 5.9E−5 (2.2E−5–9.6E−5) | 1.2E−5 (−2.9E−5–5.2E−5) | 0.0003 | −0.32* | 0.37* |
| Rif | 1.0E−4 (6.4E−5–1.4E−4) | 4.2E−5 (5.0E−6–8.0E−5) | 1.2 E−4 (7.0E−5–1.6 E−4) | ||||
| Firmicutes.Clostridia.Clostridiales.Peptococcaceae | No Int | 5.3E−5 (−1.6E−5–1.2E−4) | 2.5E−5 (−4.7E−5–9.6E−5) | 3.1E−6 (−7.4E−5–8.0E−5) | 0.023 | −0.15 | 0.08 |
| Rif | 2.6E−4 (1.9 E−4–3.3E−4) | 5.1E−5 (−2–0E−5–1.2 E−4) | 1.8 E−4 (9.6 E−5–2.7 E−4) | ||||
| Firmicutes.Clostridia.Clostridiales.Ruminococcaceae | No Int | 0.003 (0.002–0.005) | 0.005 (0.004–0.007) | 0.003 (0.001–0.005) | 0.012 | −0.19 | −0.01 |
| Rif | 0.007 (0.005–0.009) | 0.003 (0.001–0.005) | 0.006 (0.004–0.008) | ||||
| Firmicutes.Clostridia.Clostridiales.Ruminococcaceae. Anaerotruncus | No Int | 0.003 (0.002–0.005) | 0.005 (0.004–0.007) | 0.003 (0.001–0.005) | 0.013 | −0.28 | 0.12 |
| Rif | 0.007 (0.005–0.009) | 0.003 (0.001–0.005) | 0.006 (0.004–0.008) | ||||
| Firmicutes.Clostridia.Clostridiales.Ruminococcaceae. Oscillibacter | No Int | 1.5E−4 (6.6E−6–2.9E−4) | 4.0E−4 (2.6E−4–0.001) | 1.2E−4 (−3.6E−5–2.8E−4) | 0.028 | −0.22 | 0.16 |
| Rif | 1.3E−4 (−1.4E−5–2.7E−4) | 5.4E−5 (−9.2E−5–2.0E−4) | 1.9E−4 (8.2E−6–3.6E−4) | ||||
| Firmicutes.Clostridia.Clostridiales.Ruminococcaceae. Ruminococcaceae UCG-002 | No Int | 0.011 (0.007–0.015) | 0.017 (0.013–0.021) | 0.015 (0.010–0.019) | 0.014 | −0.28* | −0.22 |
| Rif | 0.025 (0.021–0.029) | 0.017 (0.013–0.021) | 0.019 (0.014–0.025) | ||||
| Firmicutes.Clostridia.Clostridiales.Ruminococcaceae. Ruminococcaceae UCG-014 | No Int | 0.009 (0.005–0.013) | 0.011 (0.006–0.016) | 0.008 (0.003–0.013) | 0.018 | −0.41** | −0.06 |
| Rif | 0.012 (0.008–0.018) | 0.001 (−0.004–0.006) | 0.007 (0.002–0.013) | ||||
| Firmicutes.Clostridia.Clostridiales.Ruminococcaceae. uncultured | No Int | 0.011 (0.009–0.013) | 0.011 (0.009–0.013) | 0.008 (0.005–0.010) | 0.005 | −0.08 | 0.28* |
| Rif | 0.009 (0.007–0.011) | 0.008 (0.001–0.006) | 0.012 (0.010–0.015) |
aData are given in mean (95% confidence interval). bThe p value reflects the interaction between time and group from UNIANOVA. cThe beta coefficient and corresponding asterisk reflects the effect of rifaximin from linear regression with taxa at the indicated time point as dependent and treatment and baseline taxa as independent in a forced model. *P < 0.05, **P < 0.01.
Adverse effects during the study
Out of 20 patients in the rifaximin group, one patient experienced an allergic reaction classified as a moderate adverse event. The patient stopped rifaximin immediately and was treated with betamethasone and the symptoms resolved after treatment. Another eight adverse events [acne (n = 1), nausea (n = 1), dyspepsia (n = 1), bad taste in the mouth (n = 2), abdominal pain (n = 1), loose stools (n = 1) and urticaria (n = 1)] were reported at the end of study visit.
Discussion
In this ‘proof of concept’ study, rifaximin had no significant effect on markers of systemic inflammation or LPS as a marker of gut leakage in CVID. Microbial diversity in the gut and the abundance of multiple bacteria, however, were changed by rifaximin, but returned to baseline after the intervention. In contrast, the previously identified CVID specific dysbiosis index, which has been associated with increased markers of systemic inflammation and gut leakage, was not affected. It could be speculated that this causes the lack of anti-inflammatory effects of rifaximin in these patients.
Multiple studies of rifaximin have been performed for different conditions15,16. However, the majority of these studies have been performed in liver diseases, where rifaximin has been convincingly shown to reduce hepatic encephalopathy17. Studies in cirrhotic patients with minimal encephalopathy, alcoholic cirrhosis and fatty liver disease have all shown reductions in LPS levels after rifaximin treatment13,18,19, also after only 2 weeks of treatment as in the present study20. However, the mechanisms leading to gut leakage in severe liver failure and CVID may differ. In the only previous study in immunodeficient patients (i.e. HIV-infected patients), rifaximin showed very limited effects on neither microbial translocation nor T cell activation compared with placebo in 65 HIV patients, but unfortunately, this study presented no data on gut microbiota composition16.
The lack of effects observed on the primary endpoints cannot be discussed independently from the gut microbiota profile, where antibiotics would be expected to act. In the present study, a transient reduction of alpha diversity was observed together with changes in multiple taxa, including reduction of several Ruminococcaceae and increase of Bacteroides. In contrast, in a study by Ponziani et al. including 20 patients with different gastrointestinal and liver diseases, using 16S rRNA-based gut microbiota profiling, rifaximin increased Lactobacilli and decreased Roseburia, Haemophilus, Veilonella and Streptococcus. Furthermore, in two other studies including patients with liver cirrhosis, rifaximin use reduced Veillonellaceae21, and increased Eubacteriaceae13,21. None of these changes overlap with the results of the current study, but these studies did not report measures of diversity. One important cause of non-overlapping results could be that the effect of rifaximin depends on the initial bacterial composition, which probably differs between CVID and intestinal or hepatic pathologies. Random variation or methodological differences could also in part explain the lack of similarities. In addition to phenotype, comparability between microbiota studies can be influenced by many factors like different baseline lifestyle, diet, geography, host genetics, as well as lab methods including choice of PCR primers and DNA extraction22. Since all samples in the present study were handled exactly the same way and were sequenced in the same MiSeq run, the influence of methodological variation on the results should be minimal.
Several factors could potentially explain the failure of rifaximin to influence the primary end-point. In a previous study from our group, a CVID specific dysbiosis index calculated from key bacteria separated patients from controls7. This CVID specific dysbiosis index has been found to correlate with circulating markers of systemic inflammation and gut leakage, and inversely to alpha diversity. Although rifaximin reduced alpha diversity in the present study, there was no significant impact on the bacteria constituting the CVID specific dysbiosis index, and it could be hypothesized that rifaximin not targeting the putative culprits could explain the lack of effect on systemic inflammation. Of particular interest would be to increase Bifidobacterium, which is reduced in CVID7, and that has been shown to improve gut barrier and reduce systemic LPS and inflammation in mice23. Unfortunately, Bifidobacterium was not increased by rifaximin in our study. Thus, although rifaximin altered alpha diversity, the potential “inflammatory” bacteria were not changed. However, additional studies, including intervention studies, are needed to causally link gut dysbiosis and systemic inflammation in CVID.
The strengths of the present study were a randomized controlled study design, which although open-label had blinded primary end-points. Secondly, we included both data of gut microbiota using metagenomic techniques and systemic inflammation, thereby expanding previous rifaximin studies in humans, irrespective of phenotype. The length of the intervention was an important possible limitation of the present study. Our aim was to administrate a sufficient dose and length of therapy to get the desired effect on the microbiota and at the same time reducing the likelihood of adverse events. Although the intervention was sufficient to induce a significant change in alpha- and beta diversity, and in 16 taxa, we cannot exclude that a longer intervention could result in more subtle changes to the gut microbial composition and levels of systemic inflammation than we were able to detect with a 2-week intervention. This important issue should be investigated in forthcoming studies.
The randomization of CVID patients was not stratified, and by chance, the patients randomized to rifaximin treatment had a smaller proportion of patients with non-infectious complications compared to the no intervention group. This could imply that the rifaximin group was healthier than the no intervention group, which may have influenced the results. In addition, the lack of placebo excluded the use of GI symptom-questionnaire for evaluation of symptomatic relief with rifaximin use.
Even though the alterations in the gut microbiota induced by rifaximin did not change systemic inflammation in CVID, other interventions such as probiotics, prebiotics or fecal transplantation are likely to have different impacts on the gut microbiota and, theoretically, systemic inflammation. There is therefore still an unused potential to manipulate the gut microbiota in CVID preferably targeting the ten bacteria making up the CVID dysbiosis index. The results herein may be of relevance to other diseases with systemic inflammation, and suggest that rifaximin is not the chosen intervention to affect systemic inflammation, although more data is needed to support this finding. In the future, other interventions should be trialled in patients with chronic systemic inflammation, to further explore the ‘proof of concept’ that manipulation of the gut microbiota can influence systemic inflammation also in humans.
Methods
Study design and participants
We did this randomized, open, prospective, single-center, clinical trial at Oslo University Hospital, Rikshospitalet, Oslo, Norway between Oct 8 2013, and Oct 20, 2014. All CVID patients aged 18 to 74 registered at our clinic were initially pre-screened for eligibility. CVID was defined as decreased serum levels of IgG, IgA and/or IgM by at least two standard deviations below the mean for age, and exclusion of other causes of hypogammaglobulinemia24,25. Exclusion criteria were: antibiotics in the last 12 weeks, history of allergic reaction to rifaximin, malignancy, impaired kidney function, pregnancy or lactation, on-going infection, use of probiotics in the last 6 months, immunosuppressive drugs, comorbidity that may influence with the patient’s safety or compromise the study results (e.g., cardiovascular disorders, alcoholism, psychiatric disease, HIV infection), and polypharmacy (patient with an extensive medication list i.e. ten drugs or more). The study was approved by the Regional Committee for Medical and Health Research Ethics of South-Eastern Norway (number 2013/1037) and the Norwegian Medicines Agency (EudraCT number: 2013-000883-27), and conducted according to the Declaration of Helsinki. Written informed consent was obtained from all participants. The trial is registered with clinicaltrials.gov, number NCT01946906 (registration date 20.09.2013).
Randomization and masking
The CVID patients were randomized into two groups (rifaximin and no intervention) using computer-generated randomization (1:1) to receive rifaximin (Norgine, Alanno Scalo, Italy) or no treatment. There was no placebo drug. A statistician, not otherwise involved in the trial, generated the random allocation sequence. A randomization list with a combination of permuted blocks of four (n = 10) and blocks of two (n = 5) was generated. The sequence was concealed by using sealed envelopes containing the group allocation until a decision to enrol a patient was made. Due to the estimated low number of participants in each arm, no stratification between the two treatment arms was performed. The persons conducting the laboratory work related to primary and secondary endpoints were blinded to which samples were receiving the active treatment.
Procedures
The remaining patients after pre-screening were invited to participate in the study (see Results). The trial involved three outpatient visits for all patients. The patients’ eligibility to participate in the study was again evaluated at the first visit based on medical history, physical examination and blood tests (hematology, electrolytes, kidney function tests, liver function tests, human chorion-gonadotropin in fertile women). The patients enrolled in the study gave baseline blood test and fecal samples at visit 1. However, 14 days before this visit a fecal sample was sent by post, leaving two baseline stool assessments (baseline refers to fecal samples taken at visit 1 whereas baseline 2 refers to fecal samples taken 14 days prior to visit 1). Participants allocated to rifaximin were asked to take rifaximin 550 mg every 12 hours for 2 weeks, whereas the participants receiving no intervention were followed in the same way as the group receiving treatment. A written material containing information about potential adverse effects of rifaximin and contact details to the investigators in case of moderate to serious adverse events was handed out to the participants receiving rifaximin at visit 1.
At the second visit, the end of study visit, the participants brought a fecal sample and had blood tests taken. Assessment of compliance was carried out by collecting the compliance diary and counting remaining tablets in the box for the participants in the rifaximin group. Furthermore, the patients were assessed for the development of any new symptoms (blood tests and physical examination) or treatment-related adverse events. The third visit at 8 weeks, follow up visit, included all the activities of the first and second visit except assessment of compliance. The study procedure is outlined in Fig. 6.
Figure 6.

The study procedure.
Primary and secondary outcome
Study endpoints were assessed at 2 weeks (i.e., study end) and at 8 weeks (i.e. follow up). The primary endpoint was time-dependent changes in inflammatory and anti-inflammatory mediators and markers of gut leakage in plasma, serum and leukocytes. This included sCD14, sCD25, sCD163, neopterin, CRP, LPS and selected cytokines. The key secondary end points were changes in gut microbial diversity abundance of specific bacteria as evaluated by 16S rRNA sequencing.
Stool collection and analysis
All stool samples were collected by the participants at home with a standardised collection device26 and transferred to Stool Collection Tubes with Stool DNA Stabilizer (Stratec Biomedical, Birkenfeld, Germany)27. The first sample (2 weeks before visit 1 [baseline 2]) was returned by post together with a questionnaire reporting the sampling time, recent antibiotic and/or probiotic use, and medication. At visit 1 (baseline), 2 and 3, the stool samples (collected at home within the last 24 hours) were immediately stored at −20 °C, according to the manufacturer’s recommendation, until DNA extraction. Bacterial DNA was extracted using the PSP® Spin Stool DNA Plus Kit (Stratec) and subjected to high-throughput sequencing of the 16S ribosomal RNA gene with dual-indexed barcodes according to an established protocol, followed by sequencing on an Illumina MiSeq (Supplementary Methods)28.
Differences in microbial composition between groups were evaluated by measures of alpha diversity (within sample bacterial diversity), beta diversity (between sample bacterial diversity), dysbiosis index and the relative abundance of individual taxa (Supplementary Methods).
CVID dysbiosis index
We have previously published a specific CVID dysbiosis index that consists of the relative abundance of ten bacteria at different taxonomic levels, which captured the change in alpha diversity between CVID and healthy controls7. We, therefore, calculated the dysbiosis index for all samples at all four time points using the formula: Loge [(sum of the relative abundances of bacteria increased in CVID)/(sum of the relative abundances of bacteria reduced in CVID)]. Bacteria which are increased in CVID include Bacilli, Dorea, Roseburia, Gammaproteobacteria and bacteria which are reduced in CVID include Bifidobacterium, Odoribacteracea, Christensenellaceae, Blautia, Sutterella, Desulfovibrionace7.
Blood sampling protocol
Peripheral venous blood was drawn into sterile blood collection tubes without any additives (serum) or with EDTA as anticoagulant (plasma). The tubes were immediately immersed in melting ice, centrifuged within 15 minutes at 2000g for 20 minutes to obtain platelet-poor plasma or were allowed to clot at room temperature before centrifugation at 1000 g for 10 minutes (serum). Plasma and serum were stored at −80 °C in aliquots and thawed only once. Peripheral blood mononuclear cells (PBMC) were isolated from heparinised venous blood isolated by gradient centrifugation using Lymphoprep (Axis Shield, Oslo, Norway) within 1 hour after blood collection and PBMC pellets were immediately stored at −80 °C until mRNA analyses (Supplementary Methods).
Measurements of inflammatory and gut leakage markers
Serum levels of sCD14, neopterin, sCD163 and sCD25 were quantified in duplicate by enzyme immunoassays obtained from R&D Systems (Minneapolis, MN). LPS was analysed by Limulus Amebocyte Lysate chromogenic assay (Lonza, Walkersville, MD) according to the manufacturer’s instructions, with the following modifications: Samples were diluted 5-fold to avoid interference with background colour, and preheated to 67 °C for 12 minutes prior to analysis to dissolve immune complexes. Serum levels of CRP were sampled together with safety blood samples via the routine hospital laboratory. B- and T-cell subpopulations were analysed by flow cytometry (Supplementary Methods).
Subgroup analysis
Clinical subgroups were classified as “Infection only” or “Complications” based on previously defined criteria9 with one modification; CVID enteropathy was defined as persistent diarrhea after exclusion of gastrointestinal infection.
Statistical analysis
The primary objective of the statistical analysis was to compare the effect of rifaximin therapy with no therapy on gut leakage and inflammatory markers. This was an explorative study and we found that strict samples size calculation was inappropriate. However, we estimated an approximate 50% difference in changes in inflammatory markers between the two groups, and with 25 patients in each group we would have 95% power to detect a 50% (0.75) difference with an alpha of 0.05. Since CVID is a rare disease, we were only able to recruit 20 patients in each group. Univariate Repeated measures ANOVA (UNIANOVA) was used to assess the effect of treatment focusing on the interaction between time and treatment group for the different markers, followed by paired samples t-test or Wilcoxon’s Rank-Sum test for paired data if significant. Skewed variables were log transformed prior to regression analysis.
Differences in microbial composition between groups were evaluated by measures of alpha diversity (within individual bacterial richness), dysbiosis index and the abundance of individual taxa. Also, for alpha diversity, dysbiosis index and taxa UNIANOVA was used ‘a priori’. For the significant taxa, linear regression was performed with taxa at 2 or 8 weeks as dependent and baseline taxa and treatment group as independent variables in a forced model. When not specified otherwise, statistical analyses were performed with SPSS (IBM, Armonk, NY, USA). P values were two-sided and considered significant when <0.05.
Electronic supplementary material
Acknowledgements
Liv Wenche Thorbjørnsen, Anne Pharo and other members of Norwegian PSC Research Center as well as Ann Døli at the Research Institute of Internal Medicine are acknowledged for support on sample collection and logistics. We thank the Fougner-Hartmann Familiefond (Denmark) for funding of an Illumina MiSeq sequencer. S.F.J. was funded by a grant from the South-Eastern Norway Regional Health Authority (Project Number 2012063). The project was also supported by grant number 911802 from Western Norway Regional Health Authority. J.R.H. was funded by the Norwegian PSC Research Center and the Norwegian Research Council (Project Number 240787/F20). The funders had no role in any part of the study including study design, data collection, -analysis and -interpretation.
Author Contributions
S.F.J., J.R.H., B.F., P.A. and T.H.K. had the initial idea for the study. S.F.J. developed the protocol and was responsible for the everyday management of the trial. S.F.J., M.E.M., P.A. and B.F. recruited patients. S.F.J., M.E.M., A.R., P.A. and B.F. obtained human samples and clinical data collection. S.F.J., M.E.M., T.B., K.H., M.K., A.E.M., T.L., B.H., M.T., T.E.M., R.K.B., A.Y., J.H.R., B.F. analyzed data and S.F.J., M.E.M., K.H., T.U., M.K., J.R.H. performed statistical analysis. P.A., T.H.K., J.R.H. and B.F. supervised the project. S.F.J., M.E.M., P.A., J.R.H. and B.F. drafted the manuscript. All authors revised the manuscript for critical content and approved the final version.
Data Availability
The datasets generated during and/or analyzed during the current study are not publicly available due to Norwegian legislation about general data protection regulation, but are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1038/s41598-024-54117-6"
J. R. Hov and B. Fevang contributed equally.
Change history
2/14/2024
This article has been retracted. Please see the Retraction Notice for more detail: 10.1038/s41598-024-54117-6
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-35367-7.
References
- 1.Luczynski P, et al. Growing up in a Bubble: Using Germ-Free Animals to Assess the Influence of the Gut Microbiota on Brain and Behavior. Int. J. Neuropsychopharmacol. 2016;19:pyw020. doi: 10.1093/ijnp/pyw020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giongo A, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5:82–91. doi: 10.1038/ismej.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly TN, et al. Gut Microbiome Associates With Lifetime Cardiovascular Disease Risk Profile Among Bogalusa Heart Study Participants. Circ. Res. 2016;119:956–+. doi: 10.1161/Circresaha.116.309219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vazquez-Castellanos JF, et al. Altered metabolism of gut microbiota contributes to chronic immune activation in HIV-infected individuals. Mucosal Immunol. 2015;8:760–772. doi: 10.1038/mi.2014.107. [DOI] [PubMed] [Google Scholar]
- 6.Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA. 2011;108(Suppl 1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jorgensen SF, et al. Altered gut microbiota profile in common variable immunodeficiency associates with levels of lipopolysaccharide and markers of systemic immune activation. Mucosal Immunol. 2016;9:1455–1465. doi: 10.1038/mi.2016.18. [DOI] [PubMed] [Google Scholar]
- 8.Ahn S, Cunningham-Rundles C. Role of B cells in common variable immune deficiency. Expert Rev. Clin. Immunol. 2009;5:557–564. doi: 10.1586/eci.09.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapel H, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–286. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 10.Jorgensen SF, et al. A Cross-Sectional Study of the Prevalence of Gastrointestinal Symptoms and Pathology in Patients With Common Variable Immunodeficiency. Am. J. Gastroenterol. 2016;111:1467–1475. doi: 10.1038/ajg.2016.329. [DOI] [PubMed] [Google Scholar]
- 11.Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is Not CVID common variable immune deficiency (CVID), what do we know in 2011? Adv. Immunol. 2011;111:47–107. doi: 10.1016/B978-0-12-385991-4.00002-7. [DOI] [PubMed] [Google Scholar]
- 12.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 13.Bajaj JS, et al. Modulation of the metabiome by rifaximin in patients with cirrhosis and minimal hepatic encephalopathy. PLoS One. 2013;8:e60042. doi: 10.1371/journal.pone.0060042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calanni F, Renzulli C, Barbanti M, Viscomi GC. Rifaximin: beyond the traditional antibiotic activity. J. Antibiot. (Tokyo) 2014;67:667–670. doi: 10.1038/ja.2014.106. [DOI] [PubMed] [Google Scholar]
- 15.Qayed M, et al. Rifaximin for preventing acute graft-versus-host disease: impact on plasma markers of inflammation and T-cell activation. J. Pediatr. Hematol. Oncol. 2013;35:e149–152. doi: 10.1097/MPH.0b013e31827e56af. [DOI] [PubMed] [Google Scholar]
- 16.Tenorio AR, et al. Rifaximin has a marginal impact on microbial translocation, T-cell activation and inflammation in HIV-positive immune non-responders to antiretroviral therapy - ACTG A5286. J. Infect. Dis. 2015;211:780–790. doi: 10.1093/infdis/jiu515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bass NM, et al. Rifaximin treatment in hepatic encephalopathy. N. Engl. J. Med. 2010;362:1071–1081. doi: 10.1056/NEJMoa0907893. [DOI] [PubMed] [Google Scholar]
- 18.Kalambokis GN, Mouzaki A, Rodi M, Tsianos EV. Rifaximin improves thrombocytopenia in patients with alcoholic cirrhosis in association with reduction of endotoxaemia. Liver Int. 2012;32:467–475. doi: 10.1111/j.1478-3231.2011.02650.x. [DOI] [PubMed] [Google Scholar]
- 19.Gangarapu V, et al. Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2015;27:840–845. doi: 10.1097/meg.0000000000000348. [DOI] [PubMed] [Google Scholar]
- 20.Zeng X, et al. Does low-dose rifaximin ameliorate endotoxemia in patients with liver cirrhosis: a prospective study. J. Dig. Dis. 2015;16:665–674. doi: 10.1111/1751-2980.12294. [DOI] [PubMed] [Google Scholar]
- 21.Kakiyama G, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013;58:949–955. doi: 10.1016/j.jhep.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kevans D, et al. Characterization of Intestinal Microbiota in Ulcerative Colitis Patients with and without Primary Sclerosing Cholangitis. J Crohns Colitis. 2016;10:330–337. doi: 10.1093/ecco-jcc/jjv204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cani PD, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58:1091–1103. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Primary immunodeficiency diseases Report of an IUIS Scientific Committee. International Union of Immunological Societies. Clin. Exp. Immunol. 1999;118(Suppl 1):1–28. doi: 10.1046/j.1365-2249.1999.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Herz W, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front. Immunol. 2014;5:162. doi: 10.3389/fimmu.2014.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahlquist DA, Schwartz S, Isaacson J, Ellefson M. A stool collection device: the first step in occult blood testing. Ann. Intern. Med. 1988;108:609–612. doi: 10.7326/0003-4819-108-4-609. [DOI] [PubMed] [Google Scholar]
- 27.Wu GD, et al. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol. 2010;10:206. doi: 10.1186/1471-2180-10-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013;79:5112–5120. doi: 10.1128/aem.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are not publicly available due to Norwegian legislation about general data protection regulation, but are available from the corresponding author on reasonable request.