

Abstract
Complete mitogenomes from the order Thysanoptera are limited to representatives of the subfamily Thripinae. Therefore, in the present study, we sequenced the mitochondrial genome of Neohydatothrips samayunkur (15,295 bp), a member of subfamily Sericothripinae. The genome possesses the canonical 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), and two ribosomal RNA genes (rRNAs) as well as two putative control regions (CRs). The majority strand was 77.42% A + T content, and 22.58% G + C with weakly positive AT skew (0.04) and negative GC skew (−0.03). The majority of PCGs start with ATN codons as observed in other insect mitochondrial genomes. The GCG codon (Alanine) was not used in N. samayunkur. Most tRNAs have the typical cloverleaf secondary structure, however the DHU stem and loop were absent in trnV and trnS1, while the TΨC loop was absent in trnR and trnT. The two putative control regions (CR1 and CR2) show 99% sequence similarity indicated a possible duplication, and shared 57 bp repeats were identified. N. samayunkur showed extensive gene rearrangements, with 11 PCGs, 22 tRNAs, and two rRNAs translocated when compared to the ancestral insect. The gene trnL2 was separated from the ‘trnL2-cox2’ gene block, which is a conserved, ancestral gene order found in all previously sequenced thrips mitogenomes. Both maximum likelihood (ML) and Bayesian inference (BI) phylogenetic trees resulted in similar topologies. The phylogenetic position of N. samayunkur indicates that subfamily Sericothripinae is sister to subfamily Thripinae. More molecular data from different taxonomic groups is needed to understand thrips phylogeny and evolution.
Introduction
The order Thysanoptera (thrips) includes nine families in two suborders, the Terebrantia and Tubulifera. The family Thripidae is the largest of the Terebrantia and is further subdivided into four subfamilies; Dendrothripinae, Panchaetothripinae, Sericorthripinae, and Thripinae1. The members of Sericothripinae have a worldwide distribution and are usually associated with flowers2,3. This subfamily currently includes 168 species in three genera, Neohydatothrips, Hydatothrips, and Sericothrips. The marigold thrips, Neohydatothrips samayunkur is a pest of marigold (Tagetes spp.) with a worldwide distribution4–6. Recently, N. samayunkur has also been suspected as a vector for tospoviruses7. Integration of molecular data with morphology is required for fast and accurate species identification and to understand phylogenetic relationships2. The mitochondrial genes cox1 and 16S rRNA have been found to be useful in the identification of thrips species and to infer phylogenetic relationships8–10, however, phylogenetic relationships below the family level in thrips are still unclear and require more molecular data2–4.
Insects typically have a single circular mitochondrial genome, 14–19 kb in size, with 37 genes, including 13 protein-coding genes (PCGs), large and small ribosomal RNA genes (rRNAs), 22 transfer RNA genes (tRNAs) and variable number of A + T rich control regions (CRs). The characteristic features of the animal mitochondrial genomes are (i) conserved gene content, (ii) conserved genome size and organization, (iii) lack of extensive recombination, (iv) maternal inheritance, and (v) an accelerated rate of nucleotide substitution11–13. Therefore, this small molecule has been widely used in insect phylogenetic and evolutionary studies14–16.
To date, the highly rearranged mitogenomes of five thrips species (Anaphothrips obscurus, Frankliniella intonsa, Frankliniella occidentalis, Scirtothrips dorsalis and Thrips imaginis) are available17–21. However, the availability of thrips mitogenomes is limited to the subfamily Thripinae. In this study, we sequenced the complete mitochondrial genome of N. samayunkur, a member of subfamily Sericothripinae using next-generation sequencing (NGS) technology and compared it to other thrips mitogenomes, analysing genome organization, codon usage patterns, tRNA secondary structure and strand asymmetry. Phylogenetic relationships were inferred by analysing the 13 PCGs from published thrips mitogenomes using maximum likelihood (ML) and Bayesian inference (BI).
Results and Discussion
Genome structure, organization and composition
The complete sequence of the mitochondrial genome of N. samayunkur (accession number MF991901) is 15,295 base pairs (bp) in length. This is longer than those of A. obscurus, F. intonsa, F. occidentalis, and S. dorsalis South Asia (SA1), but smaller than the genomes of T. imaginis and S. dorsalis East Asia (EA1) (Table S1). As in other insect species, the mitochondrial genome of N. samayunkur included 37 genes: 13 PCGs, large and small rRNAs and 22 tRNAs but two putative CRs (Fig. 1). There are 204 bp intergenic nucleotides in total, across 23 locations, with individual spacer length of 1 to 41 bp. The longest intergenic spacer (41 bp) was located between the trnS2 and cox1 gene, with an extremely high AT content (85.37%). Three pairs of genes overlap with lengths ranging from 1 to 24 bp. Thirty genes are on the majority strand and seven on the minority (Table 1). Nucleotide composition was 77.42% A + T content and 22.58% G + C content (Table 2); similar to other thrips mitogenomes. A + T content was highest at 80.87%, in tRNAs, followed by rRNAs (79.31%), PCGs (77.15%), and CRs (71.12%). The mitogenome showed weakly positive AT (0.04) and negative GC (−0.03) skews (Table 2).
Figure 1.

The mitochondrial genome of the marigold thrips, N. samayunkur. Direction of gene transcription is indicated by arrows in entire complete genome. PCGs are shown as purple arrows, rRNA genes as sea green arrows, tRNA genes as blue arrows and CR regions as red rectangles. The GC content is plotted using a black sliding window, as the deviation from the average GC content of the entire sequence. GC-skew is plotted using a colored sliding window (green and orchid color), as the deviation from the average GC-skew of the entire sequence. The figure was drawn using CGView online server (http://stothard.afns.ualberta.ca/cgview_server/) with default parameters. The species photograph was taken by the second author (KT) using Leica Microscope DM1000 with Leica software application suite (LAS EZ) and edited manually in Adobe Photoshop CS 8.0.
Table 1.
List of annotated mitochondrial genes of Neohydatothrips samayunkur and its characteristic features.
| Gene | Strand | Location | Size (bp) | Anti codon | Start codon | Stop codon | IGN |
|---|---|---|---|---|---|---|---|
| cox1 | + | 1–1536 | 1536 | — | ATA | TAA | 0 |
| nad3 | + | 1537–1890 | 354 | — | ATG | TAA | 25 |
| cox2 | + | 1916–2584 | 669 | — | ATA | TAA | 10 |
| trnD | + | 2595–2661 | 67 | GAC | — | — | 0 |
| trnR | + | 2662–2723 | 62 | CGA | — | — | 3 |
| trnG | + | 2727–2788 | 62 | GGA | — | — | 7 |
| trnK | + | 2796–2858 | 63 | AAA | — | — | 13 |
| cox3 | + | 2872–3702 | 831 | — | ATA | TAA | 14 |
| trnN | + | 3717–3782 | 66 | AAC | — | — | −3 |
| trnE | + | 3780–3842 | 63 | GAA | — | — | 0 |
| trnI | + | 3843–3909 | 67 | ATC | — | — | 4 |
| cytb | + | 3914–5014 | 1101 | — | ATT | TAA | 1 |
| trnY | − | 5016–5079 | 64 | TAC | — | — | 17 |
| nad2 | + | 5097–6087 | 991 | — | ATC | T(AA) | 0 |
| trnW | + | 6088–6152 | 65 | TGA | — | — | 0 |
| nad1 | + | 6153–7089 | 937 | — | ATA | T(AA) | −24 |
| trnM | + | 7066–7126 | 61 | ATG | — | — | 8 |
| trnF | + | 7135–7206 | 72 | TTC | — | — | 6 |
| rrnS | + | 7213–7935 | 723 | — | — | — | 1 |
| atp8 | − | 7937–8152 | 216 | — | ATT | TAA | 13 |
| atp6 | + | 8166–8777 | 612 | — | ATG | TAG | 0 |
| trnL1 | + | 8778–8841 | 64 | CTA | — | — | 5 |
| trnT | + | 8847–8906 | 60 | ACA | — | — | −1 |
| trnQ | + | 8906–8974 | 69 | CAA | — | — | 10 |
| trnP | − | 8985–9050 | 66 | CCA | — | — | 8 |
| trnA | + | 9059–9121 | 63 | GCA | — | — | 10 |
| nad5 | − | 9132–10817 | 1686 | — | ATA | TAA | 0 |
| trnH | − | 10818–10877 | 60 | CAC | — | — | 2 |
| nad4 | − | 10880–12178 | 1299 | — | ATT | TAA | 2 |
| nad4L | − | 12181–12459 | 279 | — | ATG | TAA | 0 |
| CR2 | + | 12460–12759 | 300 | — | — | — | 0 |
| trnV | + | 12760–12819 | 60 | GTA | — | — | 0 |
| CR1 | + | 12820–13447 | 628 | — | — | — | 0 |
| nad6 | + | 13448–13918 | 471 | — | ATT | TAA | 1 |
| rrnL | + | 13920–15004 | 1085 | — | — | — | 1 |
| trnS1 | + | 15006–15069 | 64 | TCA | — | — | 0 |
| trnC | + | 15070–15129 | 60 | TGC | — | — | 0 |
| trnL2 | + | 15130–15194 | 65 | TTA | — | — | 2 |
| trnS2 | + | 15197–15254 | 58 | AGA | — | — | 0 |
The protein coding and ribosomal RNA genes are represented by standard nomenclature, tRNAs are represented as trn followed by the IUPAC-IUB single letter amino acid codes. (+) values in strand represent as heavy (H) and (−) values represent as light (L). IGN represents (+) values as intergenic nucleotides and (−) values as overlapping regions. CR represents the control region.
Table 2.
Composition and skew in different Thysanoptera mitogenomes included for comparative analysis.
| Species | Size(bp) | A% | G% | T% | C% | GC% | AT% | AT skew | GC skew |
|---|---|---|---|---|---|---|---|---|---|
| Whole mtgenome | |||||||||
| N.samayunkur | 15,295 | 40.25 | 10.98 | 37.17 | 11.60 | 22.58 | 77.42 | 0.04 | −0.03 |
| T. imaginis | 15,407 | 43.85 | 10.47 | 32.72 | 12.96 | 23.43 | 76.57 | 0.15 | −0.11 |
| F. intonsa | 15,215 | 41.24 | 11.06 | 34.68 | 13.01 | 24.07 | 75.93 | 0.09 | −0.08 |
| F. occidentalis | 14,889 | 40.98 | 11.35 | 36.62 | 11.06 | 22.41 | 77.59 | 0.06 | 0.01 |
| S. dorsalis EA1 | 15,343 | 39.12 | 11.61 | 36.62 | 12.64 | 24.26 | 75.74 | 0.03 | −0.04 |
| S. dorsalis SA1 | 15,204 | 39.83 | 11.18 | 37.56 | 11.42 | 22.60 | 77.40 | 0.03 | −0.01 |
| A. obscurus | 14,890 | 38.38 | 11.27 | 39.75 | 10.60 | 21.87 | 78.13 | −0.02 | 0.03 |
| PCG | |||||||||
| N. samayunkur | 10,982 | 39.59 | 10.79 | 37.56 | 12.06 | 22.85 | 77.15 | 0.03 | −0.06 |
| T. imaginis | 10,922 | 42.75 | 10.15 | 32.89 | 14.21 | 24.36 | 75.64 | 0.13 | −0.17 |
| F. intonsa | 11,009 | 39.95 | 11.39 | 34.58 | 14.08 | 25.47 | 74.53 | 0.07 | −0.11 |
| F. occidentalis | 10,852 | 39.82 | 11.62 | 36.72 | 11.84 | 23.46 | 76.54 | 0.04 | −0.01 |
| S. dorsalis EA1 | 10,954 | 38.06 | 11.92 | 36.53 | 13.48 | 25.41 | 74.59 | 0.02 | −0.06 |
| S. dorsalis SA1 | 10,973 | 38.94 | 11.36 | 37.67 | 12.03 | 23.38 | 76.62 | 0.02 | −0.03 |
| A. obscurus | 11,167 | 37.36 | 11.46 | 39.93 | 11.25 | 22.71 | 77.29 | −0.03 | 0.01 |
| tRNA | |||||||||
| N. samayunkur | 1,401 | 43.11 | 9.85 | 37.76 | 9.28 | 19.13 | 80.87 | 0.07 | 0.03 |
| T. imaginis | 1,492 | 43.83 | 9.45 | 36.66 | 10.05 | 19.50 | 80.50 | 0.09 | −0.03 |
| F. intonsa | 1,392 | 43.53 | 10.70 | 35.78 | 9.99 | 20.69 | 79.31 | 0.10 | 0.03 |
| F. occidentalis | 1,380 | 42.39 | 10.58 | 37.39 | 9.64 | 20.22 | 79.78 | 0.06 | 0.05 |
| S. dorsalis EA1 | 1,426 | 40.53 | 11.01 | 37.52 | 10.94 | 21.95 | 78.05 | 0.04 | 0.00 |
| S. dorsalis SA1 | 1,429 | 41.36 | 10.43 | 38.21 | 10.01 | 20.43 | 79.57 | 0.04 | 0.02 |
| A. obscurus | 1,430 | 39.79 | 10.63 | 39.86 | 9.72 | 20.35 | 79.65 | 0.00 | 0.04 |
| rRNA | |||||||||
| N. samayunkur | 1,808 | 44.97 | 11.73 | 34.35 | 8.96 | 20.69 | 79.31 | 0.13 | 0.13 |
| T. imaginis | 1,876 | 47.65 | 10.77 | 32.14 | 9.43 | 20.20 | 79.80 | 0.19 | 0.07 |
| F. intonsa | 1,699 | 47.15 | 11.30 | 32.02 | 9.54 | 20.84 | 79.16 | 0.19 | 0.08 |
| F. occidentalis | 1,848 | 45.94 | 12.18 | 33.93 | 7.95 | 20.13 | 79.87 | 0.15 | 0.21 |
| S. dorsalis EA1 | 1,775 | 43.21 | 11.89 | 34.99 | 9.92 | 21.80 | 78.20 | 0.11 | 0.09 |
| S. dorsalis SA1 | 1,777 | 45.36 | 11.65 | 34.44 | 8.55 | 20.20 | 79.80 | 0.14 | 0.15 |
| A. obscurus | 1,812 | 43.16 | 11.70 | 36.59 | 8.55 | 20.25 | 79.75 | 0.08 | 0.16 |
| Control region | |||||||||
| N. samayunkur | 928 | 33.84 | 14.87 | 37.28 | 14.01 | 28.88 | 71.12 | 0.05 | −0.03 |
| T. imaginis | 900 | 47.56 | 16.67 | 25.22 | 10.56 | 27.22 | 72.78 | 0.31 | 0.22 |
| F. intonsa | 942 | 41.72 | 7.86 | 38.22 | 12.21 | 20.06 | 79.94 | 0.04 | −0.22 |
| F. occidentalis | 595 | 40.34 | 7.90 | 43.70 | 8.07 | 15.97 | 84.03 | −0.04 | −0.01 |
| S. dorsalis EA1 | 1,775 | 43.21 | 11.89 | 34.99 | 9.92 | 21.80 | 78.20 | 0.11 | 0.09 |
| S. dorsalis SA1 | 767 | 35.33 | 9.26 | 43.55 | 11.86 | 21.12 | 78.88 | −0.10 | −0.12 |
| A. obscurus | 145 | 25.52 | 8.97 | 62.76 | 2.76 | 11.72 | 88.28 | −0.42 | 0.53 |
Protein-coding genes
All 13 PCGs used ATN start codons (five with ATA, four with ATT, three with ATG and one with ATC) as is observed in most of the insect mitochondrial genomes22,23. The stop codon TAA was used by 10 PCGs, and TAG for atp6, while an incomplete stop codon is present in nad1 and nad2. Comparative analysis of start and stop codons among thrips showed the unique features of N. samayunkur: ATT start codon in cytb and nad6, ATC in nad2, and ATG in atp6. The complete stop codon TAA was used by atp8 in N. samayunkur, while it was terminated by an incomplete stop codon T(AA) in other thrips species (S2 Table). The detection of an incomplete stop codon in atp8 gene may be due to misannotation, as atp8-atp6 is a conserved ancestral gene block with no tRNA between them24.
The entire length of PCGs of N. samayunkur was 10,982 bp. Overall A + T content of 13 PCGs was 77.15% in N. samayunkur, while it ranges from 74.53% to 77.29% across thrips. Codon usage in N. samayunkur shows a significant bias towards A/T rich codons. Relative synonymous codon usage analysis of N. samayunkur revealed that the codon GCG (Alanine) was not present at all. The most frequently utilized amino acids were Lysine (K), Phenylalanine (F), Leucine (L), Isoleucine (I), Tyrosine (Y), and Serine (S) as in other insects (S3 Table).
Ribosomal and transfer RNA genes
N. samayunkur has two rRNAs as in other insects. The large ribosomal gene (16S) was 1085 bp long, and located between nad6 and trnS1; the small (12S) was 723 bp long, located between trnF and atp8 (Table 2). A + T content of two rRNAs was 79.31%, while it ranges from 79.16% (S. dorsalis EA1) to 79.87% (F. occidentalis) observed in other thrips.
N. samayunkur contained a complete set of 22 tRNAs (total length 1,401 bp) individually ranging from 58 to 72 bp in length. Collectively tRNAs have the highest A + T content 80.87% of any gene group (78.05% to 80.87% in thrips) (Table 2). Most tRNAs have the typical cloverleaf secondary structure except trnV, trnS1, trnR, and trnT. The DHU stem and loop were absent in trnV and trnS1 while TΨ C loop absent in trnR and trnT (Fig. S1). The absence of DHU stem and loop in trnV is consistent across all thrips species sequenced to date.
Control regions
Control regions (CRs) in mitogenomes play an important role in transcription and replication25. A CR was found with following conserved elements; a poly T stretch at the 5′ end, a TA(A)n-like stretch, a stem and loop structure, a TATA motif, and a GAT motif26–28. The N. samayunkur mitogenome contains two putative control regions, CR1 (628 bp) and CR2 (300 bp), located between trnV and nad6, and trnV and nad4L respectively. CR2 had 99% sequence similarity with CR1, indicating a possible duplication. Three near tandem repeats (57 bp) were identified in CR1, while one repeat sequence was present in CR2 (Fig. 2). Most thrips species have been documented to have multiple CRs except A. obscurus. Three CRs are present in F. intonsa, F. occidentalis, and S. dorsalis SA1, two in T. imaginis, S. dorsalis EA1, and one in A. obscurus18–21. A location of CR1 upstream of nad5 gene has been suggested to be ancestral condition of thrips17, however, the CR locations in N. samayunkur (subfamily Sericothripinae) differ from those of other thrips.
Figure 2.

Comparison of the nucleotide sequences of two putative control regions of N. samayunkur. Four types of sequences were recognized in the control regions: tandem repeats, Poly T-stretches, A + T-rich sequences, TA(A)n motif, TATA motif, GAT motif and stem and loop. The figure was edited in Adobe Photoshop CS 8.0.
Gene arrangement
The mitogenome gene arrangements have been characterized by following patterns, transpositions, inversions, and inverse transpositions11,29,30. Tandem duplication–random loss (TDRL) is the most widely accepted process to explain transpositions11. The gene arrangement of N. samayunkur was assessed by comparing the common intervals with the ancestral insect gene order as an outgroup17,31. CREx32 analysis identified eight gene rearrangement events in N. samayunkur, including four inversions plus four TDRLs, assignable to two sets of alternative scenarios (Fig. S2). CREx detected inversions of atp8, trnF, trnC and gene block nad1-rrnS in both scenarios. N. samayunkur is a highly rearranged mitogenome with rearrangements of 11 PCGs, 22tRNAs, and two rRNAs as compared with the ancestral insect (Fig. 3). The majority of rearrangements were transpositions, while nine rearrangements (nad1, atp8, trnF, trnL1, trnQ, trnV, trnC, rrnS, and rrnL) were inverse transpositions. Further, when N. samayunkur was compared to other thrips species, the following derived gene blocks: trnG-cox3, trnN-trnE, trnY-nad1, trnF-atp6, and nad5-nad4L were conserved in all thrips species. Within the conserved gene block trnF-atp6, atp8 was subsequently inverted in N. samayunkur. The following tRNAs were inverted in thrips species as compared to the ancestral insect: trnY in S. dorsalis SA1, trnP in both S. dorsalis, trnS1 in T. imaginis, and trnF in all species except S. dorsalis SA1. The gene trnL2 was transposed in N. samayunkur away from the gene block trnL2-cox2, which is conserved in most insects including thrips (Fig. 3). The gene block trnD-cox3 is conserved in five thrips species including N. samayunkur, while trnD and trnR were translocated in T. imaginis and interrupted by the CR2 in S. dorsalis EA1. The gene block nad4L-nad5 is ancestral in insects and conserved in all thrips species. The conserved gene blocks trnY-nad1 and atp6-trnF are separated by trnM and trnA in most of the thrips species except N. samayunkur (trnM alone) and A. obscurus (trnA alone).
Figure 3.

Linearized view of complete mitochondrial genome organization and gene rearrangement, transposition, inversion, and inverse transposition in N. samayunkur compared with the ancestral insect gene order. The green color blocks show the conserve gene blocks. The grey color blocks showed the pseudo genes of T. imaginis. Genes nomenclature: atp6 and atp8; ATP synthase subunits 6 and 8; cytb: cytochrome b; cox1–3: cytochrome c oxidase subunits 1–3; nad1–6 and nad4L: NADH dehydrogenase subunits 1–6 and 4L; rrnS and rrnL: small and large subunit ribosomal RNA (rRNA) genes; Transfer RNA genes are denoted by a one-letter symbol according to the IPUCIUB single-letter amino acid codes. CR indicates the control regions. The figure was edited in Adobe Photoshop CS 8.0.
Strand asymmetry
AT and GC skews on the majority strand are used to measure strand asymmetry33–35. Most insects have positive AT skew (A > T) and negative GC skew (C > G). A reversal of the strand asymmetry (T > A and G > C) has been observed in a few species, and is proposed to be caused by the inversion of the replication origin within the control region26–28,35. N. samayunkur showed weakly positive AT skew (0.04) and negative GC skew (−0.03), similar to most other thrips species (Table 2). AT skew in other thrips species, ranges from −0.02 (A. obscurus) to 0.15 (T. imaginis), while GC skew varies from −0.11 (T. imaginis) to 0.03 (A. obscurus). Insect species with reversal of strand asymmetry have faster rate of gene rearrangements, however, species with faster rate of gene rearrangements do not always show reversal of strand asymmetry35. Two species of thrips (A. obscurus and F. occidentalis) showed weekly positive GC skew value. However, there is no inversion of replication related elements in CR indicating that strand asymmetry is not reversed in thrips.
Phylogenetic analysis
Both Maximum likelihood (ML) and Bayesian Inference (BI) phylogenetic trees resulted in similar topologies (Fig. S3, Fig. 4). BI posterior probabilities (PP) were higher than ML bootstrap support (BS) values. It has been suggested that PP and BS values are not directly comparable and interchangeable, as PP gives higher nodal support than BS36. Species within the same genus, F. intonsa and F. occidentalis were grouped together and closely related to T. imaginis. The two cryptic species of S. dorsalis (EA1 and SA1) also clustered together and were closely related to the Frankliniella + Thrips clade. The four genera in the subfamily Thripinae: Anaphothrips, Frankliniella, Scirtothrips, and Thrips grouped together. N. samayunkur shows a sister relatioship to the Thripinae clade in the present phylogeny. Although, gene order is extensively rearranged among thrips mitogenomes, the branching pattern inferred by MLGO37 is congruent with the PCGs based ML and BI phylogeny (Fig. 5). Previous studies showed a close relationship between T. imaginis with S. dorsalis17, however, our study found that T. imaginis was closer to Frankliniella than to Scirtothrips, congruent with morphological understanding of these taxa38.
Figure 4.
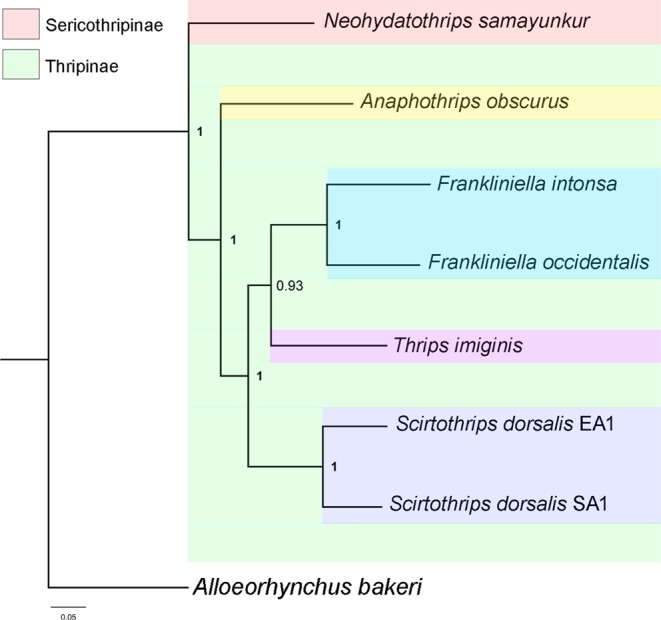
Phylogenetic tree inferred from nucleotide sequences of 13 PCGs using Bayesian Inference method in MrBayes v3.2. The tree is drawn to scale with bayesian posterior probability values indicated along with the branches. The figure was edited in Adobe Photoshop CS 8.0.
Figure 5.

Phylogenetic tree inferred from MLGO web server based on gene arrangements.
Previous studies described a close relationships between Scirtothrips (subfamily Thripinae) and Neohydatothrips (subfamily Sericothripinae) using both morphological6 and molecular data8,9. These two genera share the following morphological characters:6 presence of closely spaced rows of microtrichia on lateral thirds of abdominal tergites; median pair of tergal setae close together; campaniform sensilla absent on tergite IX, tergite X not split longitudinally. The suborder Terebrentia is typically classified into eight families with four subfamilies39; however, an alternative view proposes 28 families based on highly conserved taxonomic characters and elevates the subfamily Sericothripinae to family rank40. According to Bhatti, the proposed family Sericothripidae can be separated from other families of suborder Terebrantia by the presence of sublateral callosities on antecostal line on tergites II to VII and sternites II to VI (II to VIII in male), prominent anteriorly directed sublateral apodeme on each side on female sternites VII, one pair of cervical sclerites, annular rows of microtrichia on femora and tibiae, short and straight hind coxal apodeme, metathoracic furca not elongate and lyre-shaped, metasternellum somewhat to strongly enlarged, forming a transversely striate area on each side of the mid line, mesothorax with sternal coxal process small and inconspicuous; trochantin small and inconspicuous, absence of sclerites anterior to mesoacrotergite, one anteriorly directed apodeme on each side of sternite I. Moreover, previous studies clearly stated that the relationships between Thripinae (1779 species in 234 genera) and Sericothripinae (168 species in 3 genera) were unclear due to the absence of molecular data8,41. The present phylogenetic analysis contradicts a close relationship between Scirtothrips and Neohydatothrips.
The mitogenomes of two cryptic species of S. dorsalis (SA1 and EA1) vary considerably with respect to gene rearrangement and chromosome size20. S. dorsalis was described from “castor and chillies” at Coimbatore, India42. It is a polyphagous pest and a vector of tospoviruses with a global distribution. Earlier studies indicated that this species is a complex, consisting of many morphologically indistinguishable species43,44. Recently, nine cryptic species of S. dorsalis were delimited using multilocus molecular data45, however, these cryptic species has never been morphologically treated to validate and describe these species. Tagging specimens with the correct species name is a major problem as it is difficult to ascertain which of these cryptic species represent the true S. dorsalis.
To date, molecular phylogenetic studies of thrips is in its early stages due to lack of large scale data and taxonomic sampling.The generation of comprehensive molecular data on families/subfamilies is still needed.
Materials and Methods
Sample collection and DNA extraction
Adult specimens of N. samayunkur were collected from Odisha State, India. The studied species are common pests of crops, thus no prior permission was required for collection. Specimens were morphologically identified by the second author (K.T.) with available taxonomic keys2,10, and preserved in absolute ethyl alcohol at −30 °C in Centre for DNA Taxonomy, Molecular Systematics Division, Zoological Survey of India, Kolkata. Genomic DNA was extracted using DNeasy (QIAGEN) following the manufacturer’s standard protocol. Concentration of DNA was determined using a Qubit fluorometer with a dsDNA high-sensitivity kit (Invitrogen), and by agarose gel (0.8%) electrophoresis.
Mitogenome sequencing and assembly
The whole genome library of genomic DNA was sequenced using the Illumina Hiseq2500 (2 × 150 base paired-end reads) (Illumina, USA) platform which yielded ~23 million reads. The paired-end library was constructed according to standard protocols for the TruSeq DNA Library Preparation kit (https://support.illumina.com/downloads/truseq). Raw sequencing reads were trimmed and quality filtered using the NGS-Toolkit46 to removing adapter contamination and low-quality reads (N’s or more than 70% of bases with a quality score < 20). High quality reads were filtered by using the Burrows-Wheeler Alignment (BWA) tool47 and assembled with SPAdes 3.9.048, using default parameters, and the S. dorsalis mitochondrial genome (NC_025241.1) as a reference. Aligned reads were used for de novo mitochondrial genome assembly.
Genome annotation, visualization, and comparative analysis
The assembled mitogenome was annotated using the MITOS web-server (http://mitos.bioinf.uni-leipzig.de/index.py)49. PCGs and rRNAs were confirmed manually by BLASTn, BLASTp and ORF Finder in NCBI50,51 (https://www.ncbi.nlm.nih.gov/orffinder/). Nucleotide sequences from protein coding genes (PCGs) were translated into putative proteins on the basis of the invertebrate mitochondrial genetic code. Initiation and termination codons were identified in ClustalX52 using other thrips reference mitogenome sequences. MEGA653 was used for the alignment of homologous sequences across thrips species. The complete annotated mitogenome was submitted to NCBI GenBank using Sequin tool (http://www.ncbi.nlm.nih.gov/Sequin/). The circular map of the N. samayunkur mitogenome was illustrated by the CGView online server (http://stothard.afns.ualberta.ca/cgview_server/) with default parameters54. MEGA6 was used for estimation of nucleotide composition, codon usages, relative synonymous codon usage (RSCU) and composition of skewness with the following formula: AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C)55. Secondary structures of transfer RNA (tRNA) genes were predicted by MITOS and further confirmed using tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE/)56 and ARWEN 1.257. RNAstructure version 6.0.1 was used to predict possible secondary structure within CRs58. Homology between CR1 and CR2 in N. samayunkur was determined through the ClustalW sequence alignment tool implemented in MEGA6. Gene arrangements pathways in N. samayunkur were evaluated by CREx (Common Interval Rearrangement Explorer)32.
Phylogenetic analysis
Six complete mitogenomes of five thrips species were retrieved from GenBank on 1st November 2017 for phylogenetic inference (S1 Table). The A. bakeri mitogenome was used as an out group31. Each PCG was aligned individually using the MAFFT algorithm in the TranslatorX59 online platform under the L-INS-i strategy based on codon-based multiple alignment. Poorly aligned nucleotides (1652 bp) were removed from the protein alignment using GBlocks (within TranslatorX) with default settings. The resulting alignments were concatenated by using Sequence Matrix1.7.860. Concatenated dataset (9330 bp) was used for Bayesian inference (BI) and maximum likelihood (ML) analysis. PartitionFinder version 2.1.161, with the greedy algorithm was used to find the best substitution models and partition schemes. Partitions were predefined for the codon positions for each PCGs (13 genes X 3 codons = 39 partitions). The BI analysis was performed using Mr. Bayes 3.262 with HKY + I + G, TVM + G, TRN + G, GTR + I + G, HKY + G, GTR + I, TVM + I + G model estimated by PartitionFinder (S4 Table). Two runs each with four chains (three heated and one cold) for 500,000 generations, and trees were sampled every 100 generations. A consensus tree was acquired and visualized after excluding the first 25% trees as burn-in. The ML analysis was performed using the IQ-TREE63 Web Server in W-IQ-TREE64 (http://iqtree.cibiv.univie.ac.at/) with 1,000 replicates of ultrafast likelihood bootstrap65. The phylogenetic tree was visualized and edited using FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/)66. Phylogenetic relationships of studied taxa were also estimated based on gene arrangement patterns in the MLGO web server37 (S5 Table).
Supplementary information
Acknowledgements
The authors are thankful to the Director, Zoological Survey of India, Kolkata, for providing necessary facilities, constant support and encouragement throughout the study. The study is financially supported by Zoological Survey of India, Kolkata, Ministry of Environment Forest and Climate Change under National Faunal Genome Resources (NFGR) Program. This work is a part of the Ph. D thesis of the RC.
Author Contributions
K.T., V.K. and D.S. collected specimens, K.T. and V.K. conceived and designed the experiment, K.T. performed taxonomic identification of the thrips species and captured photographs, V.K. and K.C. contributed chemicals, K.T., S.K., R.C. generated DNA data, V.K., K.T. and S.K., analysed the data, wrote the manuscript text, and prepared the figures, all authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-37889-6.
References
- 1.ThripsWiki. ThripsWiki - providing information on the World’s thrips. Available from: http://thrips.info/wiki/. (Accessed 15 January 2018).
- 2.Wang CL. Hydatothrips and Neohydatothrips (Thysanoptera, Thripidae) of East and South Asia with three new species from Taiwan. Zootaxa. 2007;1575:47–68. [Google Scholar]
- 3.Mound LA, Tree DJ. Identification and host-plant associations of Australian Sericothripinae (Thysanoptera, Thripidae) Zootaxa. 2009;1983:1–22. [Google Scholar]
- 4.Nakahara S. Validation of Neohydatothrips samayunkur (Kudô) (Thysanoptera: Thripidae) for a thrips damaging marigolds (Tagetes spp.) Proc. Entomol. Soc. Wash. 1999;101:458–459. [Google Scholar]
- 5.Abd El-Wahab AS, El-Sheikh MAK, Elnagar S. Marigold thrips Neohydatothrips samayunkur (Kudô), a new thrips species in Egypt associated with the African marigold, Tagetes erecta L. Afr. Entomol. 2015;23:2. [Google Scholar]
- 6.Bhatti, J. S., Varatharajan, R., Vareichung, S. C., Kumar. V. & Tyagi, K. The exotic species Neohydatothrips samayunkur (Kudo 1995) (Terebrantia: Sericothripidae) is now discovered from 6 states in India. In Subrahmanyan, B. & Ramamurthy, V. V. (eds), 666–667, Proc. Natnl. Symp. Frontier Areas Ent. Res. New Delhi, 5–7 (2003).
- 7.Rotenberg D, Jacobson AL, Schneweis DJ, Whitfield AE. Thrips transmission of tospoviruses. Curr Opin Virol. 2015;15:80–89. doi: 10.1016/j.coviro.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Buckman RS, Mound LA, Whiting MF. Phylogeny of thrips (Insecta: Thysanoptera) based on five molecular loci. Syst. Entomol. 2013;38:123–133. doi: 10.1111/j.1365-3113.2012.00650.x. [DOI] [Google Scholar]
- 9.Tyagi K, et al. DNA Barcoding studies on Thrips in India: Cryptic species, Species complexes. Sci. Rep. 2017;7:4898. doi: 10.1038/s41598-017-05112-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lima EFB, Mound LA. Species-richness in Neotropical Sericothripinae (Thysanoptera: Thripidae) Zootaxa. 2016;4162:1–45. doi: 10.11646/zootaxa.4162.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Cameron SL. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 2014;59:95–117. doi: 10.1146/annurev-ento-011613-162007. [DOI] [PubMed] [Google Scholar]
- 12.Curole JP, Kocher TD. Mitogenomics: digging deeper with complete mitochondrial genomes. Trends. Ecol. Evol. 1999;14:394–398. doi: 10.1016/S0169-5347(99)01660-2. [DOI] [PubMed] [Google Scholar]
- 13.Lin CP, Danforth BN. How do insect nuclear and mitochondrial gene substitution patterns differ? Insights from Bayesian analyses of combined datasets. Mol. Phylogen. Evol. 2004;30:686–702. doi: 10.1016/S1055-7903(03)00241-0. [DOI] [PubMed] [Google Scholar]
- 14.Li H, et al. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 2015;5:8527. doi: 10.1038/srep08527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song F, et al. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian Site-Heterogeneous Mixture Models. Genome Biol. Evol. 2016;8:1411–1426. doi: 10.1093/gbe/evw086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh D, et al. The mitochondrial genome of Muga silkworm (Antheraea assamensis) and its comparative analysis with other lepidopteran insects. PLoS ONE. 2017;12:e0188077. doi: 10.1371/journal.pone.0188077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hangrui L, et al. Novel insights into mitochondrial gene rearrangement in thrips (Insecta: Thysanoptera) from the grass thrips, Anaphothrips obscurus. Sci. Rep. 2017;7:4284. doi: 10.1038/s41598-017-04617-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan D, et al. The mitochondrial genome of Frankliniella intonsa: insights into the evolution of mitochondrial genomes at lower taxonomic levels in Thysanoptera. Genomics. 2014;104:306–312. doi: 10.1016/j.ygeno.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Yan D, et al. The complete mitochondrial genome sequence of the western flower thrips Frankliniella occidentalis (Thysanoptera: Thripidae) contains triplicate putative control regions. Gene. 2012;506:117–124. doi: 10.1016/j.gene.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 20.Dickey AM, et al. A novel mitochondrial genome architecture in thrips (Insecta: Thysanoptera): extreme size asymmetry among chromosomes and possible recent control region duplication. BMC Genomics. 2015;16:439. doi: 10.1186/s12864-015-1672-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shao R, Barker SC. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol. Biol. Evol. 2003;20:362–370. doi: 10.1093/molbev/msg045. [DOI] [PubMed] [Google Scholar]
- 22.Crozier RH, Crozier YC. The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics. 1993;133:97–117. doi: 10.1093/genetics/133.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korkmaz EM, Dogan O, Budak M, Basibuyuk HH. Two nearly complete mitogenomes of wheat stem borers, Cephus pygmeus (L.) and Cephus sareptanus Dovnar-Zapolskij (Hymenoptera: Cephidae): An unusual elongation of rrnS gene. Gene. 2015;558:254–264. doi: 10.1016/j.gene.2014.12.069. [DOI] [PubMed] [Google Scholar]
- 24.Cameron SL. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst. Entomol. 2014;39(3):400–411. doi: 10.1111/syen.12071. [DOI] [Google Scholar]
- 25.Zhang DX, Hewitt GM. Insect mitochondrial control region: a review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997;25:99–120. doi: 10.1016/S0305-1978(96)00042-7. [DOI] [Google Scholar]
- 26.Cameron SL, Johnson KP, Whiting MF. The mitochondrial genome of the screamer louse Bothriometopus (Phthiraptera: Ischnocera): effects of extensive gene rearrangements on the evolution of the genome. J. Mol. Evol. 2007;65:589–604. doi: 10.1007/s00239-007-9042-8. [DOI] [PubMed] [Google Scholar]
- 27.Hassanin A, Leger N, Deutsch J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 2005;54:277–298. doi: 10.1080/10635150590947843. [DOI] [PubMed] [Google Scholar]
- 28.Hassanin A. Phylogeny of Arthropoda inferred from mitochondrial sequences: strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol. Phylogenet. Evol. 2006;38:100–116. doi: 10.1016/j.ympev.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 29.Dowton M, Castro LR, Austin AD. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: The examination of genome “morphology”. Invert. Syst. 2002;16:345–356. doi: 10.1071/IS02003. [DOI] [Google Scholar]
- 30.Boore, J. L. The duplication random-loss model for gene rearrangement exemplified by mitochondrial genomes of deuterosome animals. In Comparative Genomics: Empirical and Analytical Approaches to Gene Order Dynamics, Map Alignment and the Evolution of Gene Families, ed. D Sankoff, JH Nadeau, pp. 133–48 Dordrecht, The Neth.: Kluwer Academic Publishers (2000).
- 31.Li H, et al. The complete mitochondrial genome of the damsel bug Alloeorhynchus bakeri (Hemiptera: Nabidae) Int. J. Biol. Sci. 2012;8:93–107. doi: 10.7150/ijbs.8.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernt M, et al. CREx: Inferring Genomic Rearrangements Based on Common Intervals. Bioinformatics. 2007;23:2957–2958. doi: 10.1093/bioinformatics/btm468. [DOI] [PubMed] [Google Scholar]
- 33.Nikolaou C, Almirantis Y. Deviations from Chargaff’s second parity rule in organellar DNA - insights into the evolution of organellar genomes. Gene. 2006;381:34–41. doi: 10.1016/j.gene.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Albrecht-Buehler G. Asymptotically increasing compliance of genomes with Chargaff’s second parity rules through inversions and inverted transpositions. Proc. Natl. Acad. Sci. USA. 2006;103:17828–17833. doi: 10.1073/pnas.0605553103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei SJ, et al. New Views on Strand Asymmetry in Insect Mitochondrial Genomes. PLoS ONE. 2010;5:e12708. doi: 10.1371/journal.pone.0012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki Y, Glazko GV, Nei M. Overcredibility of molecular phylogenies obtained by Bayesian phylogenetics. Proc. Natl. Acad. Sci. USA. 2002;99:16138–16143. doi: 10.1073/pnas.212646199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu F, Lin Y, Tang J. MLGO: phylogeny reconstruction and ancestral inference from gene-order data. BMC Bioinformatics. 2014;15:354. doi: 10.1186/s12859-014-0354-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mound, L. A. The Thrips and Frankliniella genus groups: the phylogenetic significance of ctenidia. 2002; 379–386 in Marullo R & Mound LA [eds] Thrips and Tospoviruses: Proceedings of the 7th International Symposium on Thysanoptera. Australian National Insect Collection, Canberra (2002).
- 39.Mound LA, Heming BS, Palmer JM. Phylogenetic relationships between the families of recent Thysanoptera. Zool. J. Linn. Soc. 1980;69:111–141. doi: 10.1111/j.1096-3642.1980.tb01934.x. [DOI] [Google Scholar]
- 40.Bhatti JS. The classification of Terebrantia (Insecta) into families. Orient. Insects. 2006;40:339–375. doi: 10.1080/00305316.2006.10417487. [DOI] [Google Scholar]
- 41.Mound LA, Morris DC. The insect order Thysanoptera: classification versus systematics. Zootaxa. 2007;1668:395–411. [Google Scholar]
- 42.Hood JD. On some new Thysanoptera from southern India. Insecutor. Inscitiae. menstruus. 1919;7:90–103. [Google Scholar]
- 43.Hoddle MS, Heraty JM, Rugman-Jones PF, Mound LA, Stouthamer R. Relationships among species of Scirtothrips (Thysanoptera: Thripidae, Thripinae) using molecular and morphological data. Ann. Entomol. Soc. Am. 2008;101:491–500. doi: 10.1603/0013-8746(2008)101[491:RASOST]2.0.CO;2. [DOI] [Google Scholar]
- 44.Rugman-Jones PF, Hoddle MS, Mound LA, Stouthamer R. Molecular identification key for pest species of Scirtothrips (Thysanoptera: Thripidae) J. Econ. Entomol. 2006;99:1813–1819. doi: 10.1093/jee/99.5.1813. [DOI] [PubMed] [Google Scholar]
- 45.Dickey AM, et al. The Scirtothrips dorsalis Species Complex: Endemism and Invasion in a Global Pest. PLoS ONE. 2015;10:e0123747. doi: 10.1371/journal.pone.0123747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patel RK, Jain M. NGS QC Toolkit: A Toolkit for Quality Control of Next Generation Sequencing Data. PLos One. 2012;7(2):e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bankevich A, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bernt M, et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013;69:313–319. doi: 10.1016/j.ympev.2012.08.023. [DOI] [PubMed] [Google Scholar]
- 50.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. “Basic local alignment search tool.”. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 51.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2005;33:D34–D38. doi: 10.1093/nar/gki063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson, J. D., Gibson, T. J., Higgins, D. G. Multiple Sequence Alignment Using ClustalW and ClustalX. Curr. Protoc. Bioinformatics. 2.3.1–2.3.22 (2002). [DOI] [PubMed]
- 53.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis Version 6.0. Mol. Biol. Evol. 2013;30:2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grant JR, Stothard P. The CGViewServer: a comparative genomics tool for circular genomes. Nucleic. Acids. Res. 2008;36:W181–W184. doi: 10.1093/nar/gkn179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995;41:353–358. doi: 10.1007/BF01215182. [DOI] [PubMed] [Google Scholar]
- 56.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic. Acids. Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laslett D, Canbäck B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–175. doi: 10.1093/bioinformatics/btm573. [DOI] [PubMed] [Google Scholar]
- 58.Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic. Acids. Res. 2010;38:W7–W13. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vaidya G, Lohman DJ, Meier R. SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics. 2010;27:171–180. doi: 10.1111/j.1096-0031.2010.00329.x. [DOI] [PubMed] [Google Scholar]
- 61.Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017;34:772–773. doi: 10.1093/molbev/msw260. [DOI] [PubMed] [Google Scholar]
- 62.Ronquist F, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012;61:539–42. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016;44:W232–W235. doi: 10.1093/nar/gkw256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Minh BQ, Nguyen MA, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013;30:1188–1195. doi: 10.1093/molbev/mst024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rambaut, A. FigTree. Version 1.4.2, Inst. Evol. Biol., Univ. Edinburgh (2014).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.