

Abstract
LHX3 is an LIM domain transcription factor involved in the early steps of pituitary ontogenesis. We report here functional studies of three allelic variants, including the first heterozygous variant of LHX3 NM_178138.5(LHX3):c.587T>C (p.(Leu196Pro)) that may be responsible for a milder phenotype of hypopituitarism. Our functional studies showed that NM_178138.5(LHX3):c.587T>C (p.(Leu196Pro)) was not able to activate target promoters in vitro, as it did not bind DNA, and likely affected LHX3 function via a mechanism of haplo-insufficiency. Our study demonstrates the possibility that patients with a heterozygous variant of LHX3 may have pituitary deficiencies, with a milder phenotype than patients with homozygous variants. It is thus of vital to propose an optimal follow-up of such patients, who, until now, were considered as not being at risk of presenting pituitary deficiency. The second variant NM_178138.5(LHX3):c.622C>G (p.(Arg208Gly)), present in a homozygous state, displayed decreased transactivating ability without loss of binding capacity in vitro, concordant with in silico analysis; it should thus be considered to affect LHX3 function. In contrast, the NM_178138.5(LHX3):c.929G>C (p.(Arg310Pro)) variant, in a heterozygous state, also predicted as deleterious in silico, proved functionally active in vitro, and should thus still be classified as a variant of unknown significance. Our study emphasizes the need for functional studies due to the limits of software-based predictions of new variants, and the possible association of a pituitary phenotype to heterozygous LHX3 variants.
Subject terms: Development, Genetic predisposition to disease
Introduction
Combined pituitary hormone deficiency (CPHD) is defined as the presence of constitutional deficiency of at least two anterior pituitary hormones, with or without associated syndromic features. Variants of genes involved in late pituitary development (such as PROP1, POU1F1, or TBX19) are associated with endocrine phenotypes without extra-pituitary defects [1], while variants of early-expressed genes, such as LHX3, LHX4, or HESX1, are commonly associated with extra-pituitary malformations including neck rotation abnormalities, hearing loss, or septo-optic dysplasia.
In mice, the normal expression of the LIM (LIN-11, Isl1, and MEC-3)-homedomain transcription factor LHX3 is necessary for pituitary development and function [2–5]. In rodents, LHX3 is necessary for survival, since mice with homozygous inactivation of Lhx3 (Lhx3−/−) die soon after birth; their pituitaries are aplastic and lack the anterior and intermediate lobes [2, 6]. In contrast, Lhx3+/− mice have normal pituitaries and no specific phenotype [7]. The overexpression of two isoforms of LHX3 (LHX3a or LHX3b) leads to a developmental defect of the gonadotroph axis [8]. LHX3a activates the transcription of pituitary genes such as Alpha-Gsu, Prolactin, ß-Tsh, ß-Fsh, and GnRH receptor. LHX3 is also required for the expression of early (HESX1, SF1) and late-acting transcription factors (POU1F1) involved in pituitary development. Finally, LHX3 and POU1F1 interact synergistically to activate the promoters of ß-Tsh and Prolactin [9]. Overall, these data are in favor of a major role of LHX3 during pituitary development. Of note, LHX3 is also involved in the development of motor neurons and the inner ear [10, 11], which explains the complex phenotype of some patients carrying LHX3 variants.
In humans, homozygous variants of LHX3 lead to CPHD. To date, 14 autosomal recessive variants of LHX3 have been reported in patients with CPHD [12–21]. All of these variants were identified in exons, except for one located in an intronic region [18]. Patients bearing LHX3 variants displayed consistent somatotroph, thyrotroph and gonadotroph deficits, and inconsistent corticotroph deficits. On magnetic resonance imaging (MRI), the pituitary was described as hypoplastic or aplastic in 60% of cases, normal in 10%, and hyperplastic in 30% of cases. The pituitary phenotype was frequently associated with a rigid neck (70% of patients), vertebral anomalies and more or less severe hearing loss (50% of cases).
In this study, we report the first heterozygous variant of LHX3 likely responsible for CPHD. We also report the results of functional studies of two newly identified variants of LHX3 presumed to be deleterious in silico, one in a homozygous and the other in a heterozygous state.
Materials and methods
Subjects
GENHYPOPIT is a clinical research network that was launched to investigate the genetic basis of CPHD [22]. In this context, based on previous reports and our own experience, genetic screening of LHX3 variants (RefSeq: NM_178138.5; OMIM 600577) was performed specifically in patients with at least growth hormone (GH), thyrotropin-stimulating hormone (TSH), and luteinizing/follicle-stimulating hormone (LH/FSH) deficiencies. Patients with a known postnatal cause of acquired hypopituitarism were excluded. Hormonal studies and intracranial imaging were performed in all patients at each referring medical center. Malformations were systematically sought on MRI and recorded. Blood samples were collected from patients and, wherever possible, from first-degree relatives. Informed written consent was obtained from the parents, caretakers, or guardians on behalf of minors/children enrolled in the study. The study was approved by the Ethics committee of the University of Aix-Marseille (France).
Screening for LHX3 variants
One hundred and sixty-four patients with features suggesting the presence of LHX3 variants were screened. DNA was extracted from blood lymphocytes. Genomic DNA was PCR-amplified from all index cases using sets of flanking intronic primers for direct sequencing of all coding exons of LHX3 (primer sequences available in Supplemental Data). Amplification was carried out using the Hot Start Taq polymerase kit protocol (Qiagen GmbH, Hilden, Germany). Sequencing was performed with a 3130 XL Genetic analyzer (LifeTechnologies, New York, USA). Sequences were analyzed using Variant Reporter software (LifeTechnologies, New York, USA).
Next-generation sequencing and multiplex ligation-dependent probe amplification (MLPA)
Genomic DNA was extracted from blood lymphocytes (standard EDTA samples) using QiaSymphony DS DNA Midi Kit (Qiagen, France). Exons and 20 bp flanking introns of 16 genes, confirmed as causal in combined pituitary hormones deficits (FGF8, FGFR1, GH1, GLI2, HESX1, IGSF1, LHX3, LHX4, OTX2, PAX6, PROP1, POU1F1, PROKR2, SOX3, SOX2, TBX19, and ARNT2), were amplified using a QIAseq Targeted DNA Custom Panel (Qiagen, France). The preparation of the library was completed according to the manufacturer’s instructions. All sample preparations were then sequenced on MiseqDX (Miseq V2 Reagent Kit 300 cycles) (Illumina®, San Diego, CA, USA). Sequencing data were exported and further analyzed (base calling, read filtering, alignment to the human genome (GRCH37/hg19) and annotated by using Biomedical Genomic Workbench v4.1.1. software (Qiagen, France). Variant calling analysis was checked further on VariantStudio v2.2 (Illumina®, San Diego, CA, USA). Coverage, for a depth exceeding 30×, was 100% in the targeted regions (16 samples in a run). All variants of interest were visually inspected in the Integrative Genomics Viewer (https://www.broadinstitute.org/igv) to evaluate mapping and variant calling quality. The candidate variants were then classified into five main categories using the American College of Medical Genetics and Genomics guidelines—pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign, and benign [23]. Sanger sequencing was performed to validate those variants categorized as pathogenic, likely pathogenic, or VUS.
Multiplex ligation-dependent probe amplification (MLPA) assays were performed, as a partial check of next-generation sequencing (NGS) data to exclude gene copy number alteration (deletion and duplication) in the three patients, by using a commercially available MLPA kit (P216-B1), following the protocol provided by the manufacturer (MRC Holland, Amsterdam, The Netherlands). This MLPA kit includes probes for all coding exons of LHX3 (both transcripts) and of five other genes, included in the NGS panel, and possibly involved in CPHD (POU1F1—except exon 5, PROP1, HESX1, LHX4, and GH1—except exons 1 and 2). MLPA products were run on ABI Prism 3500XL sequencer and analyzed using Coffalyser.Net software (MRC Holland, Amsterdam, The Netherlands).
In silico analysis
Alignment of amino acid sequences was performed using UniProtKB (www.uniprot.org). Polyphen 2 (http://genetics.bwh.harvard.edu/pph2), Predict SNP (http://loschmidt.chemi.muni.cz/predictsnp), and UMD-HTS (www.umd-hts.eu/WHTS9) were used for each allelic variant. Evaluation of variant frequency was performed with GnomAD. ACMG-AMP guidelines for the interpretation of sequence variants were used to determine the functional status of each variant.
Plasmid constructs and mutagenesis
The 550 bp alpha-GSU promoter (pGL3-alphaGSU/Luc) and the 1273 bp beta-TSH promoter (pGL3-betaTSH-1236/Luc) were provided by S. Amselem (Laboratoire de Génétique Médicale, Hôpital Trousseau, Paris, France). The reporter plasmid hPRL-250/Luc, containing 164 bp of the human PRL proximal promoter, was a gift from Dr. J. Martial (Liege University, Belgium). The human LHX3a cDNA, containing an myc tag in its-C-terminal part and inserted into the CMV-driven eukaryotic expression vector pcDNA3.1 (hereafter referred to as pcDNA) (Invitrogen), was provided by S. Rhodes (Indiana University School of Medicine, Indianapolis, USA). Full-length human POU1F1 cDNA was cloned by PCR, using normal pituitary tissues and inserted in the pcDNA expression vector (pcDNA-POU1F1). The NM_178138.5(LHX3):c.587T > C (p.(Leu196Pro)), NM_178138.5(LHX3):c.622C > G (p.(Arg208Gly)), and NM_178138.5(LHX3):c.929G > C (p.(Arg310Pro)) variants in the pcDNA-LHX3a-myc construct, designated LHX3-p.(Leu196Pro), LHX3-p.(Arg208Gly), and LHX3-p.(Arg310Pro), respectively, in the text, were generated, with the QuickChange Mutagenesis Kit (Stratagene, La Jolla, CA, USA) and the following commercially synthesized oligonucleotides (IDT, Leuven, Belgium); variants are indicated in bold and underlined (only the sense strand oligonucleotide is shown):
NM_178138.5(LHX3):c.587T > C (p.(Leu196Pro)): 5′-CTCTCGTCCGAGACGGGCCCGGACATGCGCGTGGTGC-3′;
NM_178138.5(LHX3):c.622C > G (p.(Arg208Gly)): 5′-GCAGGTTTGGTTCCAGAACGGCCGGGCCAAGGAGAAGAGG-3′;
NM_178138.5(LHX3):c.929G > C (p.(Arg310Pro)): 5′-GAGCAGTACCGAGAGCTGCCTCCCGGCAGCCCCTACGG-3′.
The wild-type pcDNA-LHX3a-myc construct is referred to as LHX3-WT.
Cell culture and transfections
HEK293T cells and COS-1 cells were grown in 12-well plates in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Cells were transfected in serum-free medium with Lipofectamine reagent (Invitrogen) according to the manufacturer’s instructions. For the study of the transactivating abilities of LHX3-WT and mutants, the total amount of DNA (600 ng) was kept constant by cotransfection of pcDNA empty vector, and 400 ng promoter (alpha-Gsu, TSHß and prolactin) was transfected for each well. For the study of the dominant-negative effect of LHX3-p.(Leu196Pro), 150 ng of LHX3-WT was transfected with 150 ng of LHX3-p.(Leu196Pro) and compared to 300 ng of LHX3-WT. For the study of synergistic effects of LHX3 and POU1F1 on the prolactin promoter, 300 ng of LHX3-WT or mutant was transfected with 200 ng of POU1F1 and 400 ng of the prolactin promoter. For all experiments, cotransfection with 0.01 ng pCMV-Renilla was performed.
Dual luciferase assay
HEK293T and COS-1 cells were prepared and assayed for luciferase activity 48 h after transfection, using the Dual-luciferase Assay system (Promega). Transfection efficiency was determined using Renilla luciferase luminescence, and luciferase firefly values were normalized to it. Firefly and Renilla luciferase activity was measured 48 h after transfection. All assay points were derived in triplicate. All experiments were performed at least three times. Results are expressed in relative units (fold-increase vs empty vector).
Cell extracts
Forty-eight hours post transfection cells (HEK293T and COS-1) where detached by scraping in ice-cold PB5/5 mM EDTA. Total protein lysates were obtained by RIPA extraction followed by sonication. Nuclear and cytoplasmic proteins were extracted with Thermo Scientific NE-PER Nuclear and Cytoplasmic Extraction Reagents following the manual instructions. For both extractions, Roche complete™ protease inhibitor (1×) and sodium orthovanadate (1 mM) were used to prevent degradation and dephosphorylation. Protein content in the lysates was measured by colorimetric analysis using an improved Bradford assay (Pierce Coomassie Plus Reagent).
Western blot
HEK293T and COS-1 cells transfected with LHX3 expression vectors were analyzed by western blot. Five micrograms of protein extracts were run on a 10% acrylamide gel and transferred to a low fluorescence PVDF membrane (Millipore) using Pierce G2 Fast Blotter and 1-Step transfer buffer. The membrane was then incubated with mouse anti-Myc primary antibody (1/1000, clone 9E10 ThermoFisher Scientific) or anti-LHX3 hybridoma supernatant (clone 67.4E12, 1/200 DSHB, University of Iowa), followed by the secondary antibody (Alexa Fluor 647-coupled donkey anti-mouse monoclonal antibody diluted 1/1000; Invitrogen). ImageJ was used to perform relative quantification by a series of dilutions of LHX3-WT extracts for each membrane.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) experiments were performed using the previously described LUEGO technique (double-labeled Cy5 way) [24] with probes representing the pituitary glycoprotein basal element of the αGSU gene: 5′GGTACTTAGCTAATTAAATGTG3’. Binding was done in {10 mM Tris-HCl, 5 mM DTT, 50 mM NaCl, 7.4 mM MgCl2, 5% saccharose, 500 ng/µl BSA, 0.1% NP40} with 8.3 nM probe and approximately 2 µg protein extract for 10 min at room temperature. The exact quantity of the protein extract was determined to ensure that the same quantity of protein was analyzed for LHX3-WT and all the mutants, as evaluated by quantitation of western blots. Supershift assay was performed with 0.5 μg of mouse anti-6xHis antibody (Thermo Scientific). Depending on the condition, unlabeled wild type (same sequence as labeled probe) or mutated (5′-GGTACTTAGCTAActgacTGTG-3′) probes were used as competitors (50-fold higher concentration). Visualization was performed with a Typhoon FLA 9000 scanner (GE Healthcare).
Statistical analysis
Data points were compared using a one-tailed Student’s t-test for paired samples using XLStat 2013.4.05 (Paris, France). p < 0.05 was considered statistically significant.
Results
Individual data of the patients bearing LHX3 variants
Pedigree A: NM_178138.5(LHX3):c.587T > C (p.(Leu196Pro)) allelic variant (heterozygous) (Chr9(GRCh37):g.139090773A > G), submitted to LOVD (www.lovd.nl/LHX3, individual ID: 00163928) (see Table 1 for phenotypic details).
Table 1.
Phenotypic and biological profiles of the patients bearing the three variants of LHX3
| Variant |
NM_178138.5 c. 587T > C p.(Leu196Pro) |
NM_178138.5 c. 587T > C p.(Leu196Pro) |
NM_178138.5 c. 622C > G p.(Arg208Gly) |
NM_178138.5 c. 929G > C p.(Arg310Pro) |
|---|---|---|---|---|
| Age at evaluation (years) | 2 | 40 | 2 | 3 |
| Status | Propositus A | Mother (Pedigree A) | Propositus B | Propositus C |
| Country | Italy | Italy | Iran | Argentina |
| Medical history of hypopituitarism in family | No | No | No | No |
| Zygosity | Heterozygous | Heterozygous | Homozygous | Heterozygous |
| ACTH deficiency | Yes (neonatal) | No | Yes (neonatal) | Yes (neonatal) |
| TSH deficiency | Yes (neonatal) | No | Yes (neonatal) | Yes (3 years) |
| FSH/LH deficiency | NE | No | NE | NE |
| GH deficiency | Yes (neonatal) | No | Yes (neonatal) | Yes (2 years) |
| Anterior pituitary on MRI | Normal | Normal | Hypoplastic with a cyst | Hypoplastic |
| Posterior pituitary MRI | Normal | Normal | Normal | Not visualized on MRI |
| Pituitary stalk MRI | Normal | Normal | Normal | Thin |
| Associated malformations | No | No | Low-set ears, retinal dystrophy, corpus callosum digenesis | Microcephaly |
For pituitary deficiencies, data between brackets represent age at diagnosis. Complete GH deficiency was defined as GH response after stimulation below 10 mUI/L. Corticotroph deficiency was defined as plasma cortisol value below 500 nmol/L after insulin stimulation test. Gonadotroph axis was investigated only in patients of postpubertal age, i.e., older than 15 years for female and 17 years in male subjects (NE not evaluated). Thyrotroph deficiency was defined as low or normal basal TSH levels associated with low T4 levels. None of the patient was presenting with sensorineural hearing loss, vertebral anomalies, or neck rotation abnormalities
The propositus was an Italian male with neonatal panhypopituitarism and a micropenis. Cerebral MRI appeared normal. The mother of the propositus carried the same variant and had a normal pituitary evaluation up to age 40. The patient also presented with one ARNT2 variant, five GLI2 variants, three IGSF1 variants, two LHX3 variants, one LHX4 variant, one PROKR2 variant, and two PROP1 variants, all considered benign or likely benign, according to the ACMG-AMP guidelines (Supplemental File 1).
Pedigree B: NM_178138.5(LHX3):c.622C > G (p.(Arg208Gly)) allelic variant (homozygous) (Chr9(GRCh37):g.139090651G > C), submitted to LOVD (www.lovd.nl/LHX3, individual ID: 00163929)
The propositus was an Iranian girl presenting with neonatal panhypopituitarism. The phenotype also included retinal dystrophy and low-set ears. Cerebral MRI showed a cystic anterior pituitary and corpus callosum digenesis. Her parents were consanguineous. No genetic analysis was available for any other family member. The patient also presented with five GLI2 variants, four IGSF1 variants, two LHX3 variants, one LHX4 variant, one PAX6 variant, and four PROP1 variants, all considered benign or likely benign, according to the ACMG-AMP guidelines (Supplemental File 1).
Pedigree C: NM_178138.5(LHX3):c.929G > C (p.(Arg310Pro)) allelic variant (heterozygous) (Chr9(GRCh37):g.139089436C > G), submitted to LOVD (www.lovd.nl/LHX3, individual ID: 00163930).
The propositus was a boy from Argentina with neonatal panhypopituitarism associated with microcephaly and a micropenis. He did not show signs of hypoglycemia at birth. Cerebral MRI showed anterior pituitary hypoplasia, a thin pituitary stalk, and a non-visualized posterior pituitary. No genetic analysis was available for any other family member. The patient also presented with five GLI2 variants, one HESX1 variant, five IGSF1, two LHX3 variants, two LHX4 variant, one PROKR2 variant, and three PROP1 variants, all considered benign or likely benign, according to the ACMG-AMP guidelines (Supplemental File 1).
In silico analyses
All of the LHX3 amino acid residues modified by the allelic variations observed in our three index patients are conserved in known Lhx3 sequences in mammals, mice, chicken, and fish (data not shown). Software-based predictions suggested that the three allelic variants would be presumed to alter the function of the protein. In GnomAD, NM_178138.5(LHX3):c.929G > C (p.(Arg310Pro)) variant was reported with a population frequency of 0.064% in a heterozygous state, while the two other variants had never been reported. NM_178138.5(LHX3):c.929G > C (p.(Arg310Pro)) variant was considered as a variant of unknown significance in dbSNP and ClinVar. These three variants were designated VUS, according to the ACMG-AMP guidelines for the interpretation of sequence variants, prior to functional studies.
In vitro analyses
Western blot
We evaluated the expression of WT and mutant LHX3 proteins by western blot using cytoplasmic and nuclear extracts from transfected HEK293T cells. Bands of the expected size for LHX3-WT, LHX3-p.(Leu196Pro), LHX3-p.(Arg208Gly), and LHX3-p.(Arg310Pro) were detected. Of note, a high level of LHX3-p.(Leu196Pro) was found in the cytoplasm of transfected cells (and this was not observed for LHX3-WT and the other mutants) (Fig. 1a). The same result was observed when the western blot was performed with extracts from COS-1 cells (data not shown), and with an anti-LHX3 antibody (data not shown).
Fig. 1.

a Western blot analysis of LHX3 proteins extracted from transfected heterologous HEK293T cells with wild type and mutated pcDNA-LHX3a-myc construct. Immunodetection of LHX3a-myc was performed using an anti-myc mouse monoclonal antibody. Left panel, cytoplasmic extracts; right panel, nuclear extracts. b Western blot analysis to quantify the amount of each LHX3 protein (transfected nuclear extract) for electromobility shift assay experiments (EMSA). c EMSA was performed with a 2xCy5 LUEGO-based probe containing the LHX3-binding site of the alpha-GSU promoter. Supershift (SS) was performed with a monoclonal anti-6xHis antibody. S arrow indicates the position of the LHX3 DNA binding. P, Free probe; pcDNA, pcDNA empty vector; NS, non-specific band; 50-fold higher concentration unlabeled wild-type probe (same sequence as labeled probe) (50x comp WT), mutated probe used as a competitor (50x comp MUT). d EMSA was performed with decreasing (three-fold dilution series) amounts of each mutant LHX3 protein. WT LHX3: proteins from LHX3-WT-transfected HEK293T cells; p.(Leu196Pro): proteins from LHX3-p.(Leu196Pro)-transfected cells; p.(Arg208Gly): proteins from LHX3-p.(Arg208Gly)-transfected cells; p.(Arg310Pro): proteins from LHX3-p.(Arg310Pro)-transfected cells
Electromobility shift assay
To test the binding properties of the mutants, DNA binding was assessed by EMSA analysis using a binding site of the alpha-GSU promoter. The same quantity of each LHX3 protein was used by varying the total amount of transfected nuclear extract (Fig. 1b). We first identified the specific band corresponding to LHX3-WT (Fig. 1c) and confirmed that the binding was specific and sensitive (as shown by competition with the WT and mutant probe) (Fig. 1c). We then compared a three-fold dilution series of each variant vs WT: LHX3-p.(Arg208Gly) and LHX3-p.(Arg310Pro) had the same binding affinity as LHX3-WT. Finally, no binding was detected for LHX3-p.(Leu196Pro) (Fig. 1d). It should be noted that the total amount of protein did not affect the binding properties of LHX3-WT (data not shown) and that the same results were observed when the EMSA was performed with extracts from COS-1 cells (data not shown).
Transfections
The ability of the LHX3 mutants to activate the alpha-GSU promoter was investigated, by cotransfecting HEK293T cells with various amounts of WT and mutant LHX3 expression vectors (Fig. 2a). Both LHX3-WT and LHX3-p.(Arg310Pro) activated the alpha-GSU promoter in a dose-dependent manner, while LHX3-p.(Leu196Pro) and LHX3-p.(Arg208Gly) did not (p < 0.05 for 200 ng, p < 0.005 for 400 ng vs LHX3-WT). Results were comparable when the same expression vectors were tested on the prolactin and beta-TSH promoters (data not shown). As LHX3-p.(Leu196Pro) was deleterious in functional studies, and present in a heterozygous state in our patient, we assessed a possible dominant-negative effect in vitro by cotransfecting LHX3-WT and LHX3-p.(Leu196Pro) along with the three promoters studied. When LHX3-WT was cotransfected with equal amounts of LHX3-p.(Leu196Pro) (150 ng each), a slight (<50%) but significant decrease in the level of activation of each promoter (compared with double the quantity of LHX3-WT, i.e. 300 ng) was observed on the alpha-GSU promoter (p < 0.05, Fig. 2b), as well as on the beta-TSH and prolactin promoters (data not shown), suggesting haplo-insufficiency rather than a dominant-negative effect.
Fig. 2.
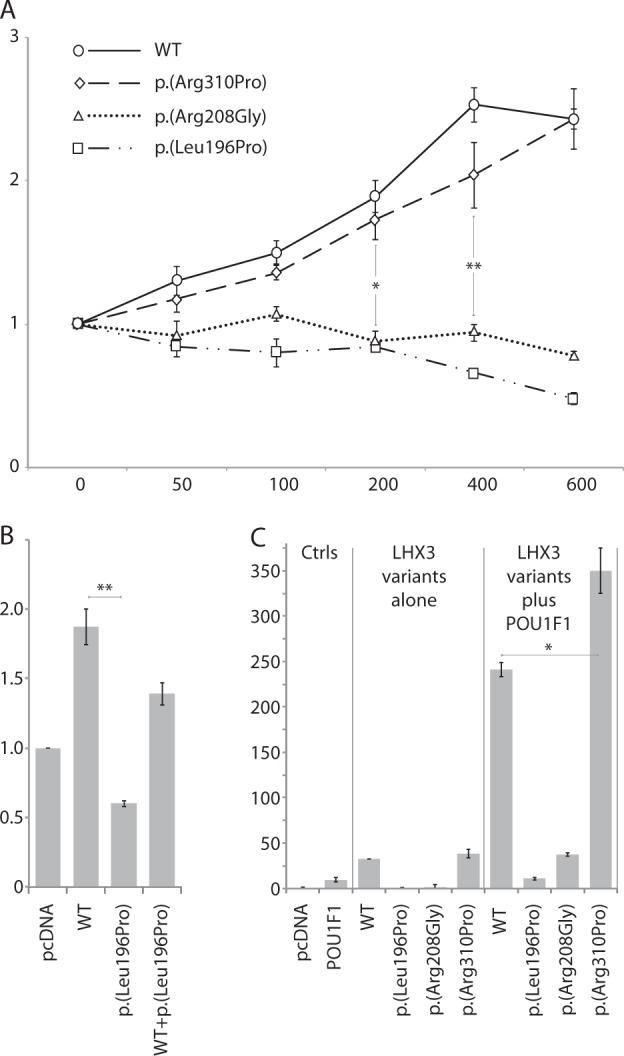
Transfection studies into heterologous HEK293T cells with a luciferase reporter gene under the control of a pituitary promoter. a Dose response Luciferase activity of the alpha-GSU promoter with LHX3-WT and mutants. X-axis, dose of pGL3-alphaGSU/Luc plasmid (in ng). Y-axis, luciferase activity. Empty pcDNA was used as a control. b LHX3-WT (150 ng) and LHX3-p.(Leu196Pro) (150 ng) were cotransfected so that the total amount of DNA was comparable to two-fold LHX3-WT (300 ng). c Synergistic effect of POU1F1 and LHX3-WT on the prolactin promoter (hPRL-250/Luc). Cotransfections were performed with POU1F1 in combination with LHX3-WT or mutant LHX3. *p < 0.05; **p < 0.005. Ctrls: controls; p.(Leu196Pro): LHX3-p.(Leu196Pro)-transfected cells; p.(Arg208Gly): LHX3-p.(Arg208Gly)-transfected cells; p.(Arg310Pro): LHX3-p.(Arg310Pro)-transfected cells; POU1F1:pcDNA-POU1F1, WT: LHX3-WT-transfected HEK293T cells
As LHX3 is known to synergistically interact with POU1F1 to activate the prolactin promoter, we tested whether this synergy was preserved with each LHX3 mutant. Synergy was observed with LHX3-p.(Arg310Pro) (350-fold activation) and was significantly increased compared to the level of activation observed when LHX3-WT and pcDNA-POU1F1 were cotransfected (240-fold activation) (p < 0.05). In contrast, this synergistic activity was lost with LHX3-p.(Leu196Pro) and LHX3-p.(Arg208Gly) (Fig. 2c) (p < 0.05 vs LHX3-WT).
Discussion
We report here the first heterozygous LHX3 variant with likely deleterious consequences in a patient with an isolated neonatal CPHD and lack of an extra-pituitary phenotype. Symptomatic heterozygous LHX3 variants were previously reported in one study, but as compound heterozygous variants in a child presenting with a history of severe respiratory distress, in a context of CPHD and scoliosis [21]. In this previous study, genomic analysis of the patient revealed a NM_178138.5(LHX3):c.252-3C > G p.(?) (Chr9(GRCH37):g.139091729G > C) variant inherited from the father and a NM_178138.5(LHX3):c.353G > A (p.(Cys118Tyr) variant inherited from the mother. Interestingly, the father and the father’s mother, who both had a heterozygous NM_178138.5:c.252-3C > G LHX3 variant, presented with limited neck rotation, a clinical sign that is present in about half of the patients with LHX3 variants, but had normal pituitary evaluation. This suggested that deleterious variants of LHX3, in a heterozygous state, could induce a mild phenotype. Our results support this hypothesis by describing the first case of CPHD due to a heterozygous NM_178138.5(LHX3):c.587T > C (p.(Leu196Pro)) variant. Further supporting this milder phenotype, our patient did not show any extra-pituitary anomalies, such as deafness or pituitary stalk interruption syndrome, characteristics that have been frequently reported in patients with homozygous deleterious variants of LHX3. Such a mild phenotype has not previously been reported in the parents of the propositus carrying other LHX3 variants, and this may be due to a low penetrance of pituitary deficiency in this context, or to a later age at the onset of pituitary deficiency (even though our propositus was diagnosed immediately after birth). Such an incomplete penetrance could also be shown in the mother of our propositus, who had a normal pituitary evaluation by age 40, while she was also carrying the same variant in a heterozygous state. This has already been reported by ourselves and others, for example, in the case of another LIM domain transcription factor, LHX4 [25]. Another possibility could be that this unaffected parent may represent a mosaic. The altered function of LHX3 induced by NM_178138.5(LHX3):c.587T > C (p.(Leu196Pro)), a variant that had not been previously reported, and is in particular absent from the GnomAD database, was elucidated by our functional studies. Amino acid 196 is located in the homeodomain, a functional domain necessary for correct DNA binding: the variant appears to disrupt this domain and strongly diminish binding to DNA, as evidenced by our EMSA analysis. To reproduce the heterozygous state of the mutant in vitro, we performed transient transfections with the same quantities of LHX3-WT and LHX3-p.(Leu196Pro). As compared with double the amount of LHX3-WT, a slight (<50%) but significant decrease of promoter activation was observed in this condition, suggesting the mechanism is haplo-insufficiency rather than a dominant-negative effect. Finally, while murine models of heterozygous inactivation of Lhx3 do not show any pituitary anomaly, it is now well accepted that murine models do not always reproduce the human disease, as has been shown for another LIM domain transcription factor, LHX4, for which heterozygous inactivation in mice does not result in a pituitary phenotype, while heterozygous variants in humans do result in a CPHD phenotype [25]. From a genetic viewpoint, we cannot definitively exclude another etiology for CPHD in this patient. However, the results of NGS confirmed the lack of presumed deleterious variants in 15 other genes known to induce CPHD. As variants in the promoter or the non-coding regions of LHX3 have not previously been described, we sequenced only the exonic and intronic regions immediately flanking LHX3; furthermore, interpretation of variants in distant 5′UTR, 3′UTR, or deep intronic regions is not clear-cut. Finally, while information on copy number variation in the 16 genes, including LHX3, was provided by NGS analysis, excluding whole or multiexon deletion in the analyzed genes, we also performed MLPA for the samples and checked for the absence of any deletion, at least in the exons of genes present in the available MLPA kit dedicated to pituitary deficiencies (GH1, LHX4, POU1F1, PROP1, and LHX3).
The two other variants we report emphasize the difficulties in defining the potential affected function induced by new variants, as has been previously shown in the case of another LIM domain transcription factor, LHX4 [26]. The variant NM_178138.5(LHX3):c.929G > C (p.(Arg310Pro)) was considered to be deleterious in silico and considered as “of unknown significance” in dbSNP. However, in vitro experiments did not highlight any decreased transactivation effect of LHX3-p.(Arg310Pro) compared to LHX3-WT, despite the fact that the amino acid 310 is located in the carboxy-terminal part of the protein, a region containing the major transactivation domain critical for pituitary gene regulation [27, 28]. LHX3-p.(Arg310Pro) was also able to interact synergistically with POU1F1 to activate the PRL promoter, in contrast to LHX3-p.(Leu196Pro). Interestingly, our functional study showed that the synergistic effect observed when LHX3-p.(Arg310Pro) was cotransfected with pcDNA-POU1F1 was superior to the one observed with LHX3-WT. As overexpression of LHX3a can lead to a developmental defect in the pituitary, and persistent expression of PROP1, a late-acting transcription factor, could lead to abnormal pituitary development [29], we cannot rule out the possibility that this increased synergistic activity could be responsible for the phenotype of the patient. We thus still suggest that LHX3-p.(Arg310Pro) should be considered as a VUS.
The third variant, NM_178138.5(LHX3):c.622C > G (p.(Arg208Gly)), has been shown by our functional studies to affect LHX3 function, in agreement with the results of the algorithms of function prediction. The phenotype was novel as it included both retinal dystrophy and corpus callosum dysgenesis. While LHX3 expression has never been demonstrated in mouse midbrain, it has been reported in a subset of bipolar cells in the retina of adult mice [30]. Our western blot data ruled out the possibility that LHX3-p.(Arg208Gly) might lead to defective nuclear localization, despite the fact that the arginine 208 residue is the first amino acid of a RRAK motif, presumed to be a nuclear localization signal [31]. However, complete mutagenesis of the RRAK gene, or a point variant (substitution of alanine 210 by valine), did not modify the localization of LHX3 [19]. As LHX3-p.(Arg208Gly)-binding affinity was similar to LHX3-WT, the lack of target-promoter activation could also be due to at least three alternative mechanisms: a change in the protein conformation as shown for NM_000306.3:c.537C > G (p.(Ser179Arg)) POU1F1 variant [32]; a loss of interaction of LHX3-p.(Arg208Gly) with transcription factors such as ISL1, which are necessary for proper pituitary development [33]; or a loss of interaction of cofactors such as Sp1 or Nuclear Factor 1 which modulate LHX3 expression during development [34]. NM_178138.5(LHX3):c.622C > G (p.(Arg208Gly)) variant should thus be considered to affect the function of LHX3 in the context of pituitary deficiency as we ruled out variants of other genes currently known to be involved in CPHD by NGS. Of note, the atypical extra-pituitary phenotype presented by this patient of Iranian origin might also be due to other genetic etiologies as yet not determined.
In conclusion, this study is important for endocrinologists dealing with CPHD, as it suggests for the first time that a parent carrying a heterozygous variant of LHX3 affecting its function, should be closely evaluated by an endocrinologist as he/she may have pituitary deficiencies that have remained undiagnosed. Such a clinical status, especially when unrecognized, may lead to major comorbidities and could be fatal if left untreated [35]. Another important issue remains the difficulty in determining the functional activity of new variants of genes coding for transcription factors, suggesting that new tools are needed to improve the interpretation of our genetic analyses, in this era of NGS.
Electronic supplementary material
Acknowledgements
We thank all of the clinicians who sent DNA samples of patients with combined pituitary hormone deficiency in the GENHYPOPIT network. Melanie Philippon received funding in the form of a research grant from the French Endocrine Society and Novartis. We thank Ian Darby for editing.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41431-018-0264-6) contains supplementary material, which is available to authorized users.
References
- 1.Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT. Genetic regulation of pituitary gland development in human and mouse. Endocr Rev. 2009;30:790–29. doi: 10.1210/er.2009-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sheng HZ, Zhadanov AB, Mosinger B, Jr., et al. Specification of pituitary cell lineages by the LIM homeobox gene Lhx3. Science. 1996;272:1004–7. doi: 10.1126/science.272.5264.1004. [DOI] [PubMed] [Google Scholar]
- 3.Sloop KW, Parker GE, Hanna KR, Wright HA, Rhodes SJ. LHX3 transcription factor mutations associated with combined pituitary hormone deficiency impair the activation of pituitary target genes. Gene. 2001;265:61–9. doi: 10.1016/S0378-1119(01)00369-9. [DOI] [PubMed] [Google Scholar]
- 4.Yaden BC, Savage JJ, Hunter CS, Rhodes SJ. DNA recognition properties of the LHX3b LIM homeodomain transcription factor. Mol Biol Rep. 2005;32:1–6. doi: 10.1007/s11033-004-4069-z. [DOI] [PubMed] [Google Scholar]
- 5.Tian G, Singh U, Yu Y, et al. Expression and function of the LIM homeobox containing genes Lhx3 and Lhx4 in the mouse placenta. Dev Dyn. 2008;237:1517–25. doi: 10.1002/dvdy.21546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheng HZ, Moriyama K, Yamashita T, et al. Multistep control of pituitary organogenesis. Science. 1997;278:1809–12. doi: 10.1126/science.278.5344.1809. [DOI] [PubMed] [Google Scholar]
- 7.Ellsworth BS, Butts DL, Camper SA. Mechanisms underlying pituitary hypoplasia and failed cell specification in Lhx3-deficient mice. Dev Biol. 2008;313:118–29. doi: 10.1016/j.ydbio.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savage JJ, Mullen RD, Sloop KW, et al. Transgenic mice expressing LHX3 transcription factor isoforms in the pituitary: effects on the gonadotrope axis and sex-specific reproductive disease. J Cell Physiol. 2007;212:105–17. doi: 10.1002/jcp.21010. [DOI] [PubMed] [Google Scholar]
- 9.Girardin SE, Benjannet S, Barale JC, Chretien M, Seidah NG. The LIM homeobox protein mLIM3/Lhx3 induces expression of the prolactin gene by a Pit-1/GHF-1-independent pathway in corticotroph AtT20 cells. FEBS Lett. 1998;431:333–8. doi: 10.1016/S0014-5793(98)00787-X. [DOI] [PubMed] [Google Scholar]
- 10.Hume CR, Bratt DL, Oesterle EC. Expression of LHX3 and SOX2 during mouse inner ear development. Gene Expr Patterns. 2007;7:798–807. doi: 10.1016/j.modgep.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma K, Sheng HZ, Lettieri K, et al. LIM homeodomain factors Lhx3 and Lhx4 assign subtype identities for motor neurons. Cell. 1998;95:817–28. doi: 10.1016/S0092-8674(00)81704-3. [DOI] [PubMed] [Google Scholar]
- 12.Bechtold-Dalla Pozza S, Hiedl S, Roeb J, et al. A recessive mutation resulting in a disabling amino acid substitution (T194R) in the LHX3 homeodomain causes combined pituitary hormone deficiency. Horm Res Paediatr. 2012;77:41–51. doi: 10.1159/000335929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhangoo AP, Hunter CS, Savage JJ, et al. Clinical case seminar: a novel LHX3 mutation presenting as combined pituitary hormonal deficiency. J Clin Endocrinol Metab. 2006;91:747–53. doi: 10.1210/jc.2005-2360. [DOI] [PubMed] [Google Scholar]
- 14.Bonfig W, Krude H, Schmidt H. A novel mutation of LHX3 is associated with combined pituitary hormone deficiency including ACTH deficiency, sensorineural hearing loss, and short neck-a case report and review of the literature. Eur J Pediatr. 2011;170:1017–21. doi: 10.1007/s00431-011-1393-x. [DOI] [PubMed] [Google Scholar]
- 15.Howard PW, Maurer RA. A point mutation in the LIM domain of Lhx3 reduces activation of the glycoprotein hormone alpha-subunit promoter. J Biol Chem. 2001;276:19020–6. doi: 10.1074/jbc.M101782200. [DOI] [PubMed] [Google Scholar]
- 16.Kristrom B, Zdunek AM, Rydh A, Jonsson H, Sehlin P, Escher SA. A novel mutation in the LIM homeobox 3 gene is responsible for combined pituitary hormone deficiency, hearing impairment, and vertebral malformations. J Clin Endocrinol Metab. 2009;94:1154–61. doi: 10.1210/jc.2008-0325. [DOI] [PubMed] [Google Scholar]
- 17.Netchine I, Sobrier ML, Krude H, et al. Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet. 2000;25:182–6. doi: 10.1038/76041. [DOI] [PubMed] [Google Scholar]
- 18.Pfaeffle RW, Savage JJ, Hunter CS, et al. Four novel mutations of the LHX3 gene cause combined pituitary hormone deficiencies with or without limited neck rotation. J Clin Endocrinol Metab. 2007;92:1909–19. doi: 10.1210/jc.2006-2177. [DOI] [PubMed] [Google Scholar]
- 19.Rajab A, Kelberman D, de Castro SC, et al. Novel mutations in LHX3 are associated with hypopituitarism and sensorineural hearing loss. Hum Mol Genet. 2008;17:2150–9. doi: 10.1093/hmg/ddn114. [DOI] [PubMed] [Google Scholar]
- 20.Sobrier ML, Attie-Bitach T, Netchine I, Encha-Razavi F, Vekemans M, Amselem S. Pathophysiology of syndromic combined pituitary hormone deficiency due to a LHX3 defect in light of LHX3 and LHX4 expression during early human development. Gene Expr Patterns. 2004;5:279–84. doi: 10.1016/j.modgep.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 21.Sobrier ML, Brachet C, Vie-Luton MP, et al. Symptomatic heterozygotes and prenatal diagnoses in a nonconsanguineous family with syndromic combined pituitary hormone deficiency resulting from two novel LHX3 mutations. J Clin Endocrinol Metab. 2012;97:E503–9. doi: 10.1210/jc.2011-2095. [DOI] [PubMed] [Google Scholar]
- 22.Reynaud R, Gueydan M, Saveanu A, et al. Genetic screening of combined pituitary hormone deficiency: experience in 195 patients. J Clin Endocrinol Metab. 2006;91:3329–36. doi: 10.1210/jc.2005-2173. [DOI] [PubMed] [Google Scholar]
- 23.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jullien N, Herman JP. LUEGO: a cost and time saving gel shift procedure. Biotechniques. 2011;51:267–9. doi: 10.2144/000113751. [DOI] [PubMed] [Google Scholar]
- 25.Castinetti F, Saveanu A, Reynaud R, et al. A novel dysfunctional LHX4 mutation with high phenotypical variability in patients with hypopituitarism. J Clin Endocrinol Metab. 2008;93:2790–9. doi: 10.1210/jc.2007-2389. [DOI] [PubMed] [Google Scholar]
- 26.Rochette C, Jullien N, Saveanu A, et al. Identifying the deleterious effect of rare LHX4 allelic variants, a challenging issue. PLoS ONE. 2015;10:e0126648. doi: 10.1371/journal.pone.0126648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colvin SC, Malik RE, Showalter AD, Sloop KW, Rhodes SJ. Model of pediatric pituitary hormone deficiency separates the endocrine and neural functions of the LHX3 transcription factor in vivo. Proc Natl Acad Sci USA. 2011;108:173–8. doi: 10.1073/pnas.1009501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sloop KW, Showalter AD, Von Kap-Herr C, Pettenati MJ, Rhodes SJ. Analysis of the human LHX3 neuroendocrine transcription factor gene and mapping to the subtelomeric region of chromosome 9. Gene. 2000;245:237–43. doi: 10.1016/S0378-1119(00)00025-1. [DOI] [PubMed] [Google Scholar]
- 29.Raetzman LT, Ward R, Camper SA. Lhx4 and Prop1 are required for cell survival and expansion of the pituitary primordia. Development. 2002;129:4229–39. doi: 10.1242/dev.129.18.4229. [DOI] [PubMed] [Google Scholar]
- 30.Balasubramanian R, Bui A, Ding Q, Gan L. Expression of LIM-homeodomain transcription factors in the developing and mature mouse retina. Gene Expr Patterns. 2014;14:1–8. doi: 10.1016/j.gep.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parker GE, Sandoval RM, Feister HA, Bidwell JP, Rhodes SJ. The homeodomain coordinates nuclear entry of the Lhx3 neuroendocrine transcription factor and association with the nuclear matrix. J Biol Chem. 2000;275:23891–8. doi: 10.1074/jbc.M000377200. [DOI] [PubMed] [Google Scholar]
- 32.Miyata I, Vallette-Kasic S, Saveanu A, et al. Identification and functional analysis of the novel S179R POU1F1 mutation associated with combined pituitary hormone deficiency. J Clin Endocrinol Metab. 2006;91:4981–7. doi: 10.1210/jc.2005-2289. [DOI] [PubMed] [Google Scholar]
- 33.Granger A, Bleux C, Kottler ML, Rhodes SJ, Counis R, Laverriere JN. The LIM-homeodomain proteins Isl-1 and Lhx3 act with steroidogenic factor 1 to enhance gonadotrope-specific activity of the gonadotropin-releasing hormone receptor gene promoter. Mol Endocrinol. 2006;20:2093–108. doi: 10.1210/me.2005-0184. [DOI] [PubMed] [Google Scholar]
- 34.Yaden BC, Garcia M, 3rd, Smith TP, Rhodes SJ. Two promoters mediate transcription from the human LHX3 gene: involvement of nuclear factor I and specificity protein 1. Endocrinology. 2006;147:324–37. doi: 10.1210/en.2005-0970. [DOI] [PubMed] [Google Scholar]
- 35.Pekic S, Doknic M, Miljic D, et al. Case seminar: a young female with acute hyponatremia and a sellar mass. Endocrine. 2011;40:325–31. doi: 10.1007/s12020-011-9516-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.